

Abstract
Risankizumab is a high‐affinity neutralizing anti‐interleukin (IL)‐23 monoclonal antibody marketed in over 40 countries across the globe to treat several inflammatory diseases, such as plaque psoriasis (PsO), psoriatic arthritis (PsA), and Crohn's disease (CD). This paper reviews the regulatory approval, mechanism of action, pharmacokinetics (PKs)/pharmacodynamics, immunogenicity, and clinical efficacy and safety data for risankizumab, focusing on the three main approved indications. Risankizumab binds to the p19 subunit of IL‐23 and inhibits IL‐23 from interacting with the IL‐23 receptor and subsequent signaling. Biomarker data obtained following treatment with risankizumab in multiple indications provided supportive evidence for downstream blockade of IL‐23 signaling associated with disease pathology. The PKs of risankizumab is linear and time‐independent, consistent with typical IgG1 monoclonal antibodies, across all evaluated indications. Risankizumab exhibited positive exposure‐response relationships for efficacy with no apparent exposure‐dependent worsening in safety. Immunogenicity to risankizumab had no major clinical consequences for either efficacy or safety. Efficacy and safety of risankizumab have been established in PsO, PsA, and CD in the pivotal clinical trials where superior benefit/risk profiles were demonstrated compared to placebo and/or active comparators. Moreover, safety evaluations in open‐label extension studies following long‐term treatment with risankizumab showed stable and favorable safety profiles consistent with shorter‐term studies. These data formed the foundation for risankizumab's marketing approvals to treat multiple inflammatory diseases across the globe.
Clinical and Translational Card for Risankizumab.
Mechanism of action: Binds to the p19 subunit of interleukin‐23 (IL‐23) and inhibits IL‐23 from interacting with the IL‐23 receptor and subsequent signaling.
Indications: Plaque psoriasis (PsO), psoriatic arthritis (PsA), Crohn's disease (CD).
Japan only indications: Generalized pustular psoriasis (GPP) and erythrodermic psoriasis (EP), and palmoplantar pustulosis (PPP).
Dosage and administration: PsO, PsA, GPP, EP, and PPP: 150 mg subcutaneous (s.c.) at weeks 0, 4, and every 12 weeks (q12w) thereafter; CD: 600 mg intravenous (i.v.) at weeks 0, 4, and 8 followed by 360 or 180 mg s.c. q8w starting from week 12 (note: the 180 mg s.c. maintenance dose is only approved in some jurisdictions [e.g., the United States and Puerto Rico]).
Major metabolic pathway: Protein catabolism (not expected to undergo metabolism via hepatic cytochrome P450 [CYP] enzymes or renal elimination).
- Key pharmacokinetic characteristics:
-
○AUC: 466 μg*day/mL during weeks 40‐52 (following dosing regimen in PsO); 2010 μg*day/mL during weeks 8‐12 (following 600 mg i.v. induction regimen in CD) and 985 μg*day/mL during weeks 40‐48 (following 360 mg s.c. maintenance regimen in CD).
-
○Maximum concentration: 12 μg/mL at steady‐state (following dosing regimen in PsO); 156 μg/mL during induction (following 600 mg i.v. induction regimen in CD) and 28.0 μg/mL at steady‐state (following 360 mg s.c. maintenance regimen in CD).
-
○Time to maximum concentration: 3–14 days.
-
○Terminal half‐life: 21‐28 days.
-
○
INTRODUCTION
Interleukin‐23 (IL‐23) plays a critical role in the development and function of T helper 17 cells, which have emerged as an important T‐cell subpopulation involved in the pathogenesis of immune‐mediated disorders. 1 Elevated expression of IL‐23 has been reported in affected skin in plaque psoriasis (PsO), 2 in the synovial tissue of patients with psoriatic arthritis (PsA), 3 and in the gut mucosa in Crohn's disease (CD). 4 There are multiple antibodies marketed that bind IL‐23 with varying affinities and epitopes for treatment of inflammatory diseases, such as PsO, PsA, and inflammatory bowel disease (IBD; i.e., CD and ulcerative colitis [UC]). 5 , 6
Although the exact etiologies of these diseases are unknown, they are all autoimmune‐mediated disorders with periods of waxing and waning disease that leads to progressive damage over time. Additionally, these diseases are hypothesized to be caused by a mix of genetic, environmental, and microbiome factors. Patients with PsO experience marked inflammation and thickening of the epidermis that results in thick, scaly plaques involving the skin. For PsA, patients experience the hallmark features associated with PsO and arthritis, including hyperkeratotic skin lesions, and synovial and entheseal inflammation. CD encompasses a spectrum of clinical and pathological processes manifested by focal, asymmetric, transmural, and occasionally granulomatous inflammation, which can affect any segment of the gastrointestinal tract.
Risankizumab is a high‐affinity neutralizing anti‐IL‐23 antibody that is currently available in multiple presentations globally to treat several inflammatory diseases. 7 In the following sections, the regulatory approval, mechanism of action, pharmacokinetics/pharmacodynamics (PKs/PDs), immunogenicity, and clinical efficacy and safety data for risankizumab focusing on the three main approved indications in adults, PsO, PsA, and CD, will be covered.
DRUG REGULATORY APPROVAL
Risankizumab was approved for adults aged 18 years and above under the tradename SKYRIZI in the United States in April 2019, for the treatment of moderate to severe PsO, in January 2022, for active PsA, and in June 2022, for moderately to severely active CD. To date, risankizumab has been approved in 80 countries for the treatment of PsO, in 70 countries for PsA, and in 44 countries for CD. Moreover, risankizumab has been approved in Japan for the treatment of several PsO‐related indications in adults, including generalized pustular psoriasis (GPP), erythrodermic psoriasis (EP), and palmoplantar pustulosis (PPP). The approved risankizumab dosing regimen for PsO, PsA, GPP, EP, and PPP is 150 mg subcutaneous (s.c.) at weeks 0, 4, and every 12 weeks (q12w) thereafter; and for CD is 600 mg intravenous (i.v.) at weeks 0, 4, and 8 followed by 360 or 180 mg sc q8w starting from week 12 (note: the 180 mg s.c. maintenance dose is only approved in some jurisdictions [e.g., the United States and Puerto Rico]). Finally, risankizumab is currently under review for its marketing application to treat adult patients with moderately to severely active UC (applications submitted to the US Food and Drug Administration and the European Medicines Agency in August 2023).
MECHANISM OF ACTION
Risankizumab is a high‐affinity neutralizing antibody that binds to the p19 subunit of IL‐23 with no binding to IL‐12, a cytokine that shares a p40 subunit with IL‐23. Risankizumab is a human IgG1 kappa antibody containing two mutations in the Fc region (Leu234Ala and Leu235Ala) which reduce Fc gamma receptor interactions.
Via binding to the p19 subunit of IL‐23, risankizumab inhibits IL‐23 from interacting with the IL‐23 receptor and the subsequent signaling that contributes to various inflammatory pathways. A detailed diagram depicting the current understanding of the impact of IL‐23 inhibition, the cell types involved, and the roles of the downstream cytokines are shown in Figure 1, for the three main approved indications.
FIGURE 1.
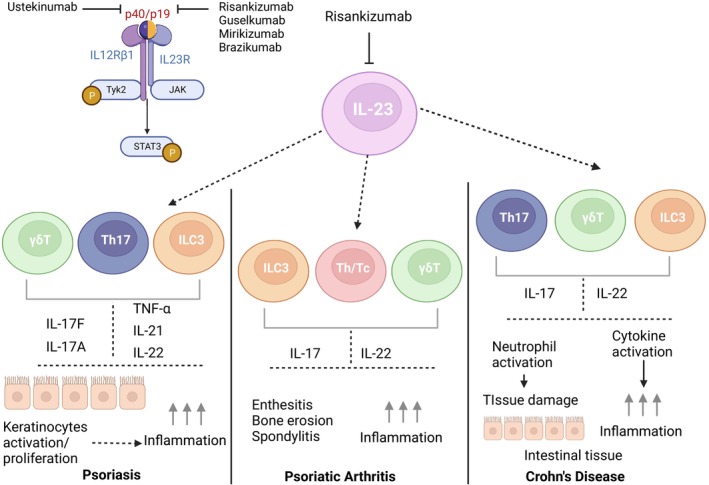
Overview of risankizumab mechanism of action. Created with BioRender.com and adapted from Sanchez et al. 28 In plaque psoriasis, IL‐23 is thought to be required for the maintenance and generation of pathogenic TH17 cells that produce the inflammatory cytokines, IL‐17 and IL‐22, which in turn stimulate keratinocyte responses including proliferation and secretion of psoriasis‐associated mediators. Blocking the activity of IL‐23 with a neutralizing antibody directly reduces the IL‐17–induced inflammation and therefore offers an attractive therapeutic intervention for psoriasis. 29 IL‐23 signaling pathways in the gut involve not only TH17 cells, but also γδTcells 30 and innate lymphoid cells 3 (ILC3s) which are involved in the pathogenesis of IBD. These cells produce IL‐17A, IL‐17F, and IL‐22, which can lead to chronic intestinal inflammation and epithelial damage associated with IBD. 28 Of note, ILC3s also play a role in maintaining gut homeostasis and similarly, IL‐22 is a pleotropic cytokine that can be both inflammatory and protective in the gut depending on the cytokine milieu. IL, interleukin; ILC3, innate lymphoid cells 3; Th, T‐helper cells; Th/Tc, T‐helper and cytotoxic T cells producing IL‐17 or IL‐22.
PHARMACOKINETIC AND PHARMACODYNAMIC CHARACTERISTICS
Pharmacokinetics
The PKs of risankizumab has been well‐characterized following single i.v. or s.c. administration in phase I studies in healthy and PsO participants, 8 and following multiple i.v. or s.c. doses in phase II and phase III studies in participants with PsO, PsA, and CD. 8 , 9 , 10 , 11 Across all of these studies, a dose‐ and dosing frequency‐dependent increase in risankizumab systemic concentrations was observed across the different regimens evaluated with dose levels up to 1800 mg i.v. or 360 mg s.c. Risankizumab exhibited linear and time‐independent PK characteristics consistent with typical IgG1 monoclonal antibodies across all evaluated indications.
Based on population PK (PopPK) analyses of pooled data from phase I–III trials, two‐compartment models with first‐order absorption and elimination best described risankizumab PKs across the indications. Risankizumab systemic clearance, volume of distribution at steady‐state, and terminal phase elimination half‐life (t 1/2) were estimated to be 0.31 L/day, 11.2 L, and 28 days, respectively, for a typical 90 kg participant with PsO. 9 PKs of risankizumab in PsA participants was found to be comparable to PsO. 10 Compared with PsO and PsA, risankizumab appears to be cleared faster from the systemic circulation in participants with CD, with a t 1/2 of ~21 days. 11
Various intrinsic and extrinsic factors were evaluated in the covariate testing of the PopPK analyses for participants with PsO, PsA, and CD. Multiple statistically significant effects were found, but most do not indicate clinically meaningful impact as exposure ratios fell within the range of 0.8–1.25 (Figure 2a,c), except for three factors: bodyweight (for PsO and PsA), high titer antidrug antibodies (ADAs; for PsO), and baseline serum albumin (for CD).
FIGURE 2.
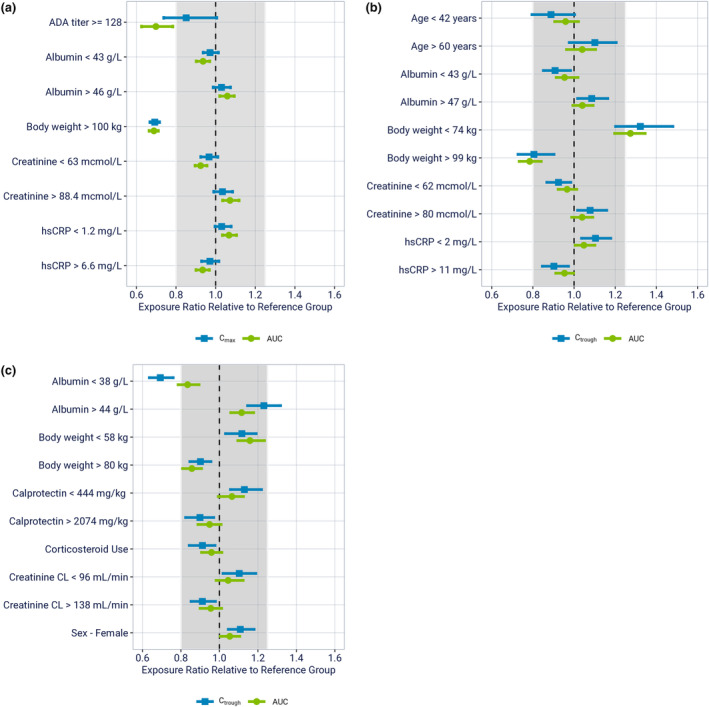
Impact of covariates identified in the population pharmacokinetic analyses on risankizumab exposures. Covariate forest plot per indication (a) psoriasis, 9 (b) psoriatic arthritis, 10 and (c) Crohn's disease. 11 Points represent medians, and error bars represent 95% confidence intervals of the normalized model‐predicted exposure ratios across 200 simulation replicates. The vertical black dashed line shows exposure ratio of 1 relative to the reference group, and the shaded area represents the 0.8–1.25 default equivalence boundaries. ADA, anti‐drug antibody; AUC, area under the concentration–time curve for weeks 40 to 52 (a), weeks 16 to 28 (b) and weeks 0 to 12 (c); C max, maximum concentration between weeks 40 to 52 (a); Creatinine CL: Creatinine clearance; C trough, trough concentration at week 28 (b), week 12 (c); hsCRP, high‐sensitivity C‐reactive protein.
As seen in Figure 2a,b, participants with higher bodyweight (>100 kg in PsO and >99 kg in PsA) were estimated to have moderately lower risankizumab exposures (30% lower in PsO and 20% lower in PsA) compared to those with a typical bodyweight. However, these small differences in exposure were not deemed to be clinically relevant, because bodyweight had no impact on risankizumab efficacy as assessed by the Psoriasis Area and Severity Index (PASI) 90 and Static Physician Global Assessment (sPGA) of clear or almost clear responses (sPGA 0/1) in PsO, 12 and there was lack of exposure–response (ER) relationships for safety and efficacy in PsA over the exposure range associated with the clinical regimen of 150 mg s.c. at week 0, week 4, and q12w thereafter. 10
Besides bodyweight, high ADA titers (≥128) were found to have a significant impact on risankizumab clearance in PsO (Figure 2a) but not in PsA or CD. In the small subset of PsO participants who developed ADA titers greater than or equal to 128 (representing 1.5% of ADA evaluable participants in phase II and phase III studies), risankizumab clearance was estimated to modestly increase by 43%, resulting in ~30% decrease in risankizumab area under the concentration time curve (AUC) on average. 9 Further impact of immunogenicity on efficacy and safety of risankizumab is discussed in the immunogenicity section.
In participants with active CD (but not PsO or PsA), those with low baseline serum albumin (<38 g/L) were predicted to have on average 31% lower week 12 risankizumab trough exposures (C trough) compared with those in the reference range (38–44 g/L; Figure 2c). However, this was not considered clinically relevant because AUC, the main driver for efficacy, was not impacted, and additional ER‐relationship analyses between C trough and week 12 efficacy further confirmed that the difference in C trough caused by low baseline serum albumin is unlikely to be clinically meaningful. 11
Risankizumab is not expected to undergo metabolism via hepatic cytochrome P450 (CYP) enzymes or renal elimination; therefore, no specific studies have been conducted to determine the effect of renal or hepatic impairment on the PKs of risankizumab. Based on PopPK analyses, serum creatinine levels, creatinine clearance, and hepatic function markers (alanine aminotransferase/aspartate aminotransferase/bilirubin) did not have a meaningful impact on risankizumab clearance.
The clearance of risankizumab was not significantly influenced by sex or race/ethnicity (Asian/Japanese or Chinese vs. Whites). Risankizumab exposures in adolescents (16 or 17 year‐old) enrolled in the phase III CD studies were shown to be comparable to those in adults. 11
Risks for drug–drug interactions (DDIs) are considered low for risankizumab, either as victim or perpetrator. PopPK analyses indicated that risankizumab exposure was not impacted in clinical studies by concomitant medications in PsO (metformin, atorvastatin, lisinopril, amlodipine, ibuprofen, acetylsalicylate, and levothyroxine), 9 PsA (methotrexate), 10 or CD (corticosteroids). 11 Given the theoretical risk of a disease‐mediated perpetrator effect of risankizumab on CYP enzymes via downregulation of pro‐inflammatory cytokines, two CYP substrate cocktail drug interaction studies were conducted to evaluate DDI risks of different risankizumab doses in PsO and IBD indications (Study M16‐007, 13 and Study M19‐974/NCT04254783, data to be published). Data from these studies showed no clinically significant changes in exposure of CYP enzyme substrates, including caffeine (CYP1A2), warfarin (CYP2C9), omeprazole (CYP2C19), metoprolol (CYP2D6), or midazolam (CYP3A) were observed when administered concomitantly with risankizumab 150 mg s.c. at weeks 0, 4, 8, and 12 in participants with PsO or risankizumab 1800 mg i.v. at weeks 0, 4, and 8 in participants with CD or UC. 7
Exposure‐response relationships
The ER analyses were conducted in the three main indications to support the final dose recommendations.
For PsO, using combined data from the phase II and III studies, nonlinear regression analyses with maximum effect models demonstrated that the 150 mg s.c. regimen at week 0, week 4, and q12w thereafter achieved the plateau of efficacy for the evaluated end points (including PASI 75, PASI 90, PASI 100, and sPGA 0/1) at week 16 (Figure 3a). Similar results were obtained with the week 52 analyses, and the efficacy plateau was achieved regardless of the bodyweight (e.g., placebo‐adjusted PASI 90 responses at week 16 were 72.1% and 69.2% in patients with bodyweight ≤100 kg and >100 kg, respectively), indicating that the recommended clinical regimen maximized risankizumab efficacy in PsO across the entire expected bodyweight range in this population. 12
FIGURE 3.
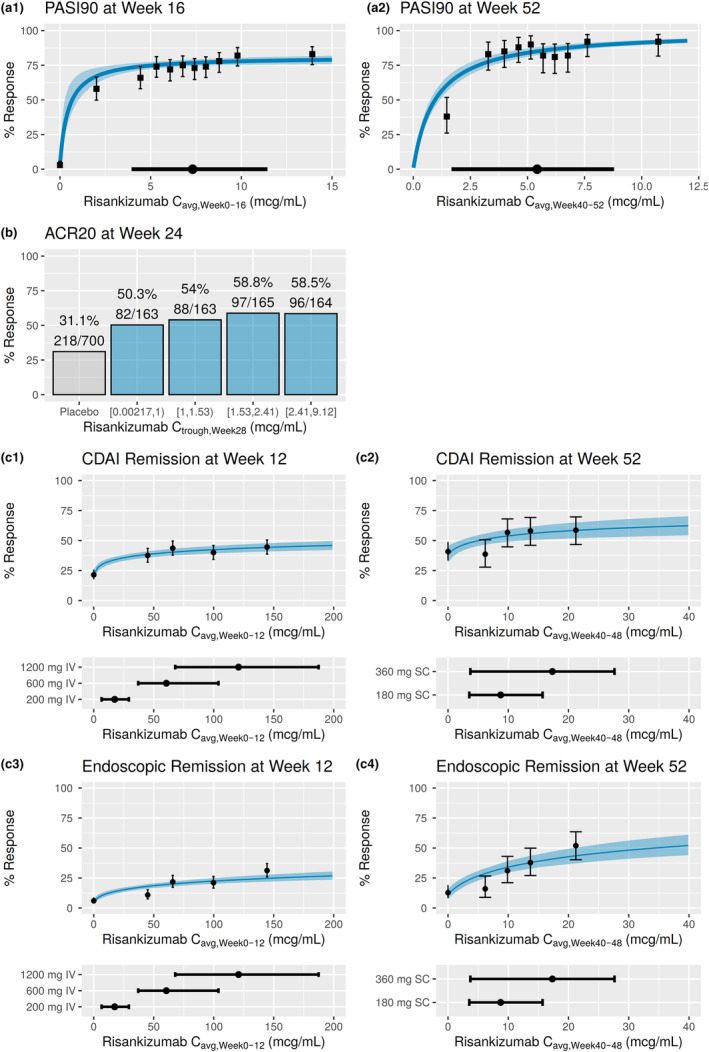
ER analyses for efficacy across indications (a) psoriasis, 12 (b) psoriatic arthritis, 10 (c) Crohn's disease (applying non‐responder imputation). 11 (a1, a2) Observed and model‐estimated exposure–efficacy relationships for PASI and sPGA at weeks 16 and 52 using data from phase II and III trials. Black solid symbols with error bars: observed PASI 90 ([a1] week 16 and [a2] week 52) response and corresponding 95% exact binomial confidence intervals within each C avg decile. Blue solid line and shaded area: model estimated probability of response and 95% confidence interval. Black solid circle and line: median C avg and 5th and 95th percentiles with 150 mg s.c. dose at weeks 0 and 4 and every 12 weeks thereafter, integrated across weeks 0 to 16 (a1) or 40 to 52 (a2). The model estimated probabilities for PASI end points at week 16 are shown at the median value of baseline high‐sensitivity C‐reactive protein for all end points. (b) Exposure‐response relationships for efficacy end point ACR20 at week 24 from phase III studies. Values on the x‐axis represent the range of the observed phase III risankizumab C trough at week 28 for each quartile. Plots show %response and n/N, where n represents number of responders and N represents total number of patients in each exposure‐quartile bin. For ACR analyses at week 24, 52 patients from phase III studies were excluded from these analyses owing to missing C trough values at week 28. Of these 52 patients for ACR, 28 (53.8%) were ACR20. ACR20, at least 20% improvement in American College of Rheumatology response criteria; C trough, concentration at the end of a dosing interval. (c1–c4) ER relationships for efficacy end points CDAI (c1 and c2) and endoscopic remission (c3 and c4), logistic regression models at (c1 and c3) week 12 in the phase II and III induction studies and (c2 and c4) week 52 in the phase III maintenance study. Upper panels represent the model predictions overlaid with the observed response rates (grouped into placebo and exposure quartiles) at (c1 and c2) 12 weeks (left) and (c2 and c4) 52 weeks (right). Description of lines and shaded area in panels (a1) and (a2) applies to panels (c1–c4). ACR, American College of Rheumatology; Cavg, average drug concentration; CDAI, Crohn's disease activity index; C trough, trough plasma concentration; ER, exposure‐response; IV, intravenous; PASI, Psoriasis Area and Severity Index; sPGA, Static Physician Global Assessment.
In participants with PsA, ER quartile analyses on phase III data showed that lower risankizumab exposures had numerically lower response rates compared to the higher exposures for American College of Rheumatology 20%/50%/70% improvement criteria (ACR20/50/70) responses, PASI 100 response, and minimal disease activity response at week 24 (Figure 3b). This suggests that further lowering exposures with doses below 150 mg or a less frequent dosing interval might cause reduced efficacy response rates for these end points. After accounting for treatment effect and study stratification factors, risankizumab systemic exposures showed no statistically significant ER relationships for any end point within the range of exposures evaluated in phase III studies. This indicates that further increasing exposures with doses higher than 150 mg or by more frequent administration than the proposed regimen might not provide additional efficacy. 10
For CD, logistic regression models based on pooled data from phase II and III studies during the 12‐week induction period showed that risankizumab exposures associated with the 600 mg i.v. induction dose at weeks 0, 4, and 8, achieved a near maximal response for all efficacy end points evaluated, with clinically negligible added benefit from the risankizumab 1200 mg i.v. regimen (Figure 3c1, c3). 11 The ER analyses for the maintenance period, including both graphical quartile ER plots and logistic regression models, showed trends of greater response in the higher range of exposures associated with the risankizumab 360 mg s.c. q8w regimen for most of the evaluated efficacy end points, particularly for the more stringent endoscopic end points, such as endoscopic remission (Figure 3c3, c4) and ulcer‐free endoscopy. 11
Analyses of the key safety variables of interest indicated no apparent relationship between risankizumab exposure and any adverse event (AE), serious AE (SAE), infections, or any serious infections through the first 16 weeks or through 52 weeks in participants with PsO 12 ; over the first 24 weeks in the pivotal phase III studies of risankizumab in participants with active PsA 10 ; and over the 12 or 24 weeks of induction, or over 52 weeks of maintenance treatment in participants with CD. 11
Pharmacodynamics
The PD biomarkers were evaluated in participants treated with risankizumab across all three indications, providing supportive evidence for downstream blockade of IL‐23 signaling associated with the pathology of these diseases.
In a phase II study of risankizumab in participants with PsO, reductions in genes associated with the IL‐23/Th17 axis (including IL‐22, IL‐17A, IL21, and DEFB4A) were observed in lesional skin at week 8. 14 Reductions in the protein level of β‐defensin 2 (BD‐2) were also observed in lesional skin after risankizumab treatment. Further, reductions of 80%–90% from baseline in serum BD‐2 levels were observed as early as week 4 and continued through week 12 in patients treated with risankizumab, which correlated with changes in PASI scores and were consistent with changes observed in tissue. 14
Biomarker assessment in participants with active PsA from the phase III KEEPsAKE2 study showed that treatment with risankizumab resulted in significant decreases in the levels of serum IL‐17A, IL‐17F, IL‐6, and BD‐2. 15 In patients with PsA treated with risankizumab, the decrease in IL‐6 correlated with joint based disease improvement (Psoriatic Arthritis Disease Activity Score), whereas the decrease in BD‐2 correlated with PASI improvement.
Moreover, data from the phase II study in CD participants demonstrated that treatment with risankizumab resulted in decreased expression of genes associated with the IL‐23/Th17 axis (including IL‐17 and IL‐22) in gut tissue. 16 Additionally, in the phase II study and phase III induction studies, treatment with risankizumab demonstrated reduction in the levels of serum IL‐22, an IL‐23 pathway engagement and PD biomarker, along with reductions in the inflammatory biomarkers fecal calprotectin (FCP) and serum C‐reactive protein (CRP) at week 12 compared to the baseline of induction. In the phase III maintenance study, treatment with risankizumab maintained inhibition of serum IL‐22, FCP, and CRP levels at week 52 compared to the baseline of induction. Participants in the withdrawal (placebo s.c.) arm, exhibited continued suppression of serum IL‐22 at week 52 compared to the baseline of induction, indicating a robust PD effect of risankizumab i.v. induction on the IL‐23 pathway after 52 weeks of drug withdrawal. 17 , 18 , 19
Immunogenicity
The immunogenicity of risankizumab has been evaluated in numerous phase I–III clinical studies in healthy volunteers as well as in participants with PsO, PsA, or CD. Based on data from the global phase I–III studies in participants with PsO, following 150 mg s.c. at weeks 0 and 4, and q12w thereafter for up to 52 weeks, treatment‐emergent ADA and neutralizing antibodies (NAbs) were detected in 24% (263/1079) and 14% (150/1079) of evaluated participants, respectively. 8 For most participants, antibodies to risankizumab, including NAbs, were of low titer and not associated with changes in risankizumab concentrations, clinical response, or safety. Higher antibody titers in ~1% of participants treated with risankizumab were associated with lower risankizumab concentrations. The median time to appearance of ADA was 16 weeks across all studies. Risankizumab immunogenicity incidences in other indications are generally similar to or lower than those observed in PsO. In PsA participants treated with the risankizumab clinical regimen, by week 28 ~12.1% (79/652) of participants developed treatment‐emergent ADAs but none were classified as neutralizing. 7 In CD phase III studies, by week 64, ~3.4% (2/58) of participants treated with risankizumab at the recommended induction and maintenance dosages developed treatment‐emergent ADAs with none classified as neutralizing. 7
Thorough impact analyses for immunogenicity, including subgroup analyses, PopPK, and ER analyses, generally did not reveal any clinically meaningful impact by ADAs or NAbs on risankizumab exposure, efficacy, or safety. Whereas in PsO and PsA, the incidence of injection site reactions and hypersensitivity reactions was numerically higher in the ADA‐positive groups compared with ADA‐negative groups, these reactions were mild to moderate in severity, and none led to discontinuation of risankizumab. 10 , 12
KEY CLINICAL TRIALS CONDUCTED BY THE SPONSOR
Table 1 provides a summary of key clinical trials conducted by AbbVie for risankizumab across the approved indications and to support future indications of UC and pediatric PsO, CD, and UC, and juvenile PsA.
TABLE 1.
Overview of key clinical studies in patients, all conducted with GCP.
| Protocol/phase/status/NCT | # Participants enrolled (Actual a /planned) | Location | Primary objective | Main findings |
|---|---|---|---|---|
| PsO | ||||
| 20031311.2/phase II/Completed/NCT02054481 | 166/160 | OUS, US | Safety, efficacy, of 3 dose regimens of RZB versus UST in participants with moderate to severe PsO | All RZB doses were safe and well‐tolerated. Pooled analyses for RZB 90 and 180 mg doses were superior to UST for PASI 90 at week 12, with the effect persisting over time. Similar results for PASI 75, PASI 100, and sPGA 0/1. The benefit–risk profile of RZB dose regimens was favorable. |
| M16‐009 (1311.13)/phase II2/Completed/NCT02203851 | 110/100 | OUS, US | OLE of 1311.2 to evaluate long term safety, efficacy of RZB in participants with moderate to severe PsO | RZB was safe and well‐tolerated. Efficacy results were maintained or improved, as demonstrated by PASI and sPGA results at week 48. Overall, the benefit–risk profile for RZB was favorable and consistent with that shown in the lead‐in Study 1311.2. |
| M16‐004 (1311.38) SustaIMM/phase IIb‐III/Completed/NCT03000075 | 171/168 | OUS | Efficacy and safety of 2 dose regimens of RZB compared to PBO in Japanese participants with moderate to severe PsO | RZB was superior to PBO across primary and secondary end points. RZB 75 and 150 mg groups were superior to PBO for PASI 90 at week 16 and a majority of participants who switched from PBO to RZB 150 mg or 75 mg after week 16 achieved PASI 50 and PASI 75 starting at week 22 and maintained achievement through week 52. For most end points, 150 mg showed greater efficacy than 75 mg. RZB was well‐tolerated with no Japanese‐specific safety issues identified. Overall, the benefit–risk profile was favorable. |
| M16‐008 (1311.3) UltIMMa‐1/phase III/Completed/NCT02684370 | 506/500 | OUS, US | Efficacy and safety of RZB versus PBO and UST in participants with moderate to severe PsO | The superior benefits of RZB versus PBO and UST were consistently demonstrated by the primary and secondary end points: The 150 mg s.c. of RZB was superior to PBO and UST for PASI 90 and sPGA 0/1 at week 16 and superior to UST at week 52. RZB was well‐tolerated with comparable safety profile to PBO and UST. Overall, the benefit–risk profile for RZB was favorable. |
| M15‐995 (1311.28) UltIMMa‐2/phase III/Completed/NCT02684357 | 491/500 | OUS, US | Efficacy and safety of RZB versus PBO and UST in participants with moderate to severe PsO | The superior benefits of RZB versus PBO and UST were consistently demonstrated by the primary and secondary end points: The 150 mg s.c. of RZB was superior to PBO and UST for PASI 90 and sPGA 0/1 at week 16 and superior to UST at week 52. RZB was well‐tolerated with comparable safety profile to PBO and UST. Overall, the benefit–risk profile for RZB was favorable. |
| M15‐992 (1311.4) IMMhance/phase III/Completed/NCT02672852 | 507/500 | OUS, US | Efficacy and safety of RZB versus PBO; effect of withdrawal and re‐treatment after week 28 through week 104 in participants with moderate to severe PsO | The superior benefits of RZB versus PBO were consistently demonstrated by the primary and secondary end points: RZB was superior to PBO for PASI 90 and sPGA 0 or 1 at week 16. Continuous RZB treatment was superior to withdrawal of RZB for sPGA 0 or 1 at week 52 with generally similar safety profiles up to week 104. RZB was well‐tolerated with a similar safety profile to PBO. Overall, the benefit–risk profile for RZB was favorable. |
| M15‐994 IMMprint/phase IIIb/Ongoing/NCT04713592 | 173/168 | OUS, US | Efficacy and safety of RZB versus PBO in participants with palmoplantar psoriasis | Ongoing; enrollment complete |
| M16‐010 (1311.30) IMMvent/phase III/Completed/NCT02694523 | 605/600 | OUS, US | Efficacy and safety versus adalimumab; effect of switching from adalimumab to RZB at week 16 in participants with moderate to severe PsO and inadequate response to adalimumab | The superior benefits of RZB versus. adalimumab were consistently demonstrated by the primary and secondary end points: RZB was superior to adalimumab for PASI 90 and sPGA 0 or 1 at week 16. For participants with inadequate response to adalimumab, switching to RZB was superior to continued RZB for PASI 90 at week 44. RZB was well‐tolerated with a similar safety profile to adalimumab. Overall, the benefit–risk profile for RZB was favorable. |
| M16‐178/phase III/Completed/NCT03255382 | 117/110 | OUS | Efficacy and safety of RZB versus oral FUMADERM in participants with moderate to severe PsO who are naïve to and candidates for systemic therapy | The superior benefits of RZB compared with FUMADERM were consistently demonstrated by the primary end points and supported by the secondary end points: RZB was superior to Fumaderm for PASI 90 at week 24. RZB was well‐tolerated. Rates of SAEs and AEs of safety interest reported for RZB were low and were lower than rates in the FUMADERM treatment group. Overall, the benefit–risk profile for RZB was favorable. |
| M15‐997 LIMMitless/phase III/Ongoing/NCT03047395 | 2170/2200 | US, OUS | Long‐term safety, efficacy, tolerability of RZB in participants with moderate to severe PsO |
Ongoing; enrollment complete. Based on interim analyses, long‐term (at least 3 years) RZB was well‐tolerated with high durable efficacy regardless of initial treatment/MOA received in the base study. |
| M16‐176/phase III/Completed/NCT03518047 | 50/50 | OUS | Safety, efficacy, tolerability of RZB versus PBO in participants with moderate to severe chronic PsO in the Russian Federation | The superior benefits of RZB compared with PBO were demonstrated by the primary, secondary, and other end points in participants with moderate to severe chronic PsO. RZB was well‐tolerated with a similar safety profile to that observed with PBO in this study. Overall, the benefit–risk profile for RZB was favorable. |
| M16‐177/phase III/Completed/NCT03219437 | 97b/100 | OUS | Safety and efficacy of RZB versus MTX in participants with moderate to severe PsO in Brazil | RZB demonstrated superiority versus MTX in the co‐primary (PASI 90 and sPGA clear or almost clear at week 28) and first 3 ranked secondary end points. Efficacy was maintained through week 112 for participants who started on RZB, and benefits increased for participants who started on MTX and were switched to RZB at week 28, and this was maintained through week 112. RZB was well‐tolerated with no new safety findings observed. The benefit–risk profile of RZB remains favorable. |
| M16‐766 (IMMerge)/phase III/Completed/NCT03478787 | 327/310 | OUS, US | Efficacy and safety of RZB versus secukinumab for the treatment of adult participants with moderate to severe PsO who are candidates for systemic therapy | RZB was noninferior to secukinumab in proportion of participants achieving PASI 90 at week 16 and superior to secukinumab in proportion of participants achieving PASI 90 at week 52. RZB was well‐tolerated with a similar safety profile to that observed with secukinumab. |
| M15‐999/phase III/Completed/NCT03875482 | 157/150 | US | Efficacy and safety of RZB (new formulation) administered by prefilled syringe versus PBO for the treatment of adult participants with moderate to severe PsO | RZB was superior to PBO for the co‐primary end points (PASI 90 and sPGA at week 16) as well as all ranked secondary end points, with high rates of observer‐measured usability and participant‐reported acceptability of the PFS. The RZB 150 mg/mL formulation was shown to be an effective and well‐tolerated treatment option for participants with moderate to severe PsO. |
| M16‐005/phase III/Completed/NCT03875508 | 108/100 | US | Usability of RZB in an autoinjector, efficacy, safety, and tolerability of new RZB formulation administered by AI for the treatment of adult participants with moderate to severe PsO | The AI usability indicated high rates of success of self‐administration using the AI. Almost all participants had no use related hazards of critical tasks. RZB was well tolerated and efficacious. Overall, self‐administration of RZB 150 mg/mL using the AI showed a favorable benefit–risk profile. |
| M19‐164/aIMM/phase IIIb/Completed/NCT04102007 | 252/250 | US, OUS | Efficacy and safety of switching to RZB in participants with moderate to severe PsO who have had a suboptimal response to secukinumab or ixekizumab | Primary analysis showed clinical benefit following switch to RZB measured by sPGA 0/1 at week 16. >50% participants achieved clear or almost clear skin after switching to RZB at week 16. No new safety risks for RZB were observed by switching without a wash out period from secukinumab or ixekizumab to RZB. The benefit–risk profile of RZB was favorable. |
| M19‐977 OptIMMize1/phase III/Ongoing/NCT04435600 | 107/132 | OUS, US | PK, safety, and efficacy of RZB in pediatric participants 6 to <18 years of age with moderate to severe PsO | Ongoing |
| M19‐973/OptIMMize2/phase III/Ongoing/NCT04862286 | 46/132 | US | Long‐term safety, tolerability, and efficacy of RZB in pediatric participants with moderate to severe chronic PsO who have completed participation in M19‐977 | Ongoing |
| M20‐326/IMMpulse/phase IV/Completed/NCT04908475 | 352/330 | US, OUS | Efficacy and safety of RZB versus apremilast for the treatment of adult participants with moderate PsO who are candidates for systemic therapy | RZB demonstrated superior efficacy compared to apremilast for participants with moderate PsO, including those who did not benefit from prior treatment with apremilast. The safety profile of RZB was similar to prior studies, and no new safety signals were identified. |
| PsA | ||||
| M15‐998/KEEPsAKE2/phase III/Ongoing/NCT03671148 | 443/420 | OUS, US | Efficacy of RZB versus PBO in participants with active PsA including Bio‐IR |
Ongoing; Enrollment complete. Interim lock for the primary efficacy is complete. RZB 150 mg was superior to PBO for ACR20 response at week 24. Efficacy results demonstrated at week 24 were maintained through week 52. RZB was well‐tolerated with no new safety concerns identified. |
| M16‐011/KEEPsAKE1/phase III/Ongoing/NCT03675308 | 964/880 | OUS, US | Efficacy of RZB versus PBO in participants with active PSA with an inadequate response/intolerance to csDMARD therapy |
Ongoing; Enrollment complete. Interim lock for the primary efficacy is complete. RZB 150 mg was superior to PBO for ACR20 response at week 24. These effects were maintained through week 52 among RZB‐to‐RZB participants. RZB was well‐tolerated, and no new safety concerns were identified. The benefit–risk profile of RZB was favorable. |
| CD | ||||
| M20‐259/SEQUENCE/phase III Ongoing/NCT04524611 | 527/508 | OUS, US | Efficacy and safety of RZB versus ustekinumab for the treatment of adult participants with moderate to severe CD who have failed anti‐TNF therapy | Ongoing. Enrollment complete. |
| M15‐993 (1311.6)/phase II/Completed/NCT02513459 | 121/120 | OUS, US | Efficacy of RZB in inducing clinical remission, after 12 weeks of treatment, in participants with moderately to severely active CD | RZB was superior to PBO in achieving improvements in clinical, endoscopic, biomarker and quality of life end points for participants with moderately to severely active CD. RZB was generally safe and well‐tolerated, and no new safety signals were identified. The benefit–risk profile of RZB was favorable. |
| M15‐989 (1311.20)/phase II/Completed/NCT02513459 | 65/60 | OUS, US | OLE study to assess the long‐term safety and efficacy of RZB in participants with moderately to severely active CD | RZB was well‐tolerated, and no safety issues were identified in this trial. Positive trends in rate response for the efficacy end points of clinical remission, clinical response, PRO‐2 remission, PRO‐2 response, CDEIS remission, CDEIS response, mucosal healing, deep remission, IBDQ remission, and IBDQ response were maintained long‐term for the majority of participants by observed case analysis. The benefit–risk profile of RZB was favorable. |
| M15‐991 (1311.7) MOTIVATE/phase III/Completed/NCT03104413 | 618/579 | OUS, US | Efficacy and safety of RZB versus PBO during induction therapy in participants with moderately to severely active CD who have failed a prior biologic | Both dose levels of RZB (600 mg and 1200 mg i.v.) met the co‐primary end points of achieving clinical remission (per CDAI or SF/APS) and endoscopic response at week 12 versus PBO. No new safety risks were identified, and the overall safety profile was consistent with the known safety profile of RZB. The benefit–risk profile of RZB was favorable. |
| M16‐000 (1311.22) FORTIFY/phase III/Ongoing/NCT03105102 | 1336/1250 | OUS, US |
Substudy 1: Efficacy and safety of RZB versus PBO during maintenance therapy versus withdrawing RZB treatment in participants with moderately to severely active CD who responded to i.v. RZB induction treatment. Substudy 2: Exploratory study to evaluate efficacy and safety of 2 different RZB treatment regimens for maintenance therapy. Substudy 3: Long‐term safety, efficacy, and tolerability of RZB. |
Ongoing; Enrollment complete; Substudy 1 met the co‐primary end points of clinical remission (per CDAI or SF/APS) and endoscopic response at week 52 for the RZB 360 mg s.c. and RZB 180 mg s.c. arms for the US protocol and 360 mg s.c. arm for the global protocol compared to the withdrawal (PBO s.c.) arm. RZB was generally well‐tolerated, and no new safety risks were identified. The benefit–risk profile of RZB was favorable. |
| M16‐006 (1311.11) ADVANCE/phase III/Completed/NCT03105128 | 931/940 | OUS, US | Efficacy and safety of RZB versus PBO during induction therapy in participants with moderately to severely active CD | Both dose levels of RZB (600 mg and 1200 mg i.v.) met the co‐primary end points of achieving clinical remission (per CDAI or SF/APS) and endoscopic response at week 12 versus PBO. No new safety risks were identified, and the overall safety profile was consistent with the known safety profile of RZB. The benefit–risk profile of RZB was favorable. |
| UC | ||||
| M16‐067/INSPIRE/phase IIb‐III/Completed/NCT03398135 | 1555/1547 | OUS, US |
Substudy 1 (phase IIb): efficacy, safety, and PK of RZB as induction treatment in participants with moderately to severely active UC and to identify the appropriate induction dose for further evaluation in Substudy 2 Substudy 2 (phase III): efficacy and safety of RZB versus PBO during induction therapy in participants with moderately to severely active UC |
Substudy 1: Primary efficacy results of clinical remission per Adapted Mayo Score at week 12 and exposure‐response analyses supported the selection of RZB 1200 mg i.v. phase III induction dosing regimen to be evaluated in Substudy 2. No new safety risks were identified, and the benefit–risk profile of RZB was favorable. Substudy 2: RZB met the primary end point of clinical remission per Adapted Mayo score at week 12 for RZB 1200 mg i.v. versus PBO. No new safety risks were identified. |
| M16‐066/COMMAND/phase III/Ongoing/NCT03398135 | 1251/942 | OUS, US |
Substudy 1: Efficacy and safety of RZB versus PBO as maintenance therapy in participants with moderately to severely active UC who responded to i.v. RZB induction treatment Substudy 2: Efficacy and safety of 2 different RZB treatment regimens for maintenance therapy Substudy 3: Long‐term safety and efficacy of RZB |
Ongoing; Enrollment complete; Substudy 1 met the primary end point of clinical remission per Adapted Mayo score at week 52 for the RZB 360 mg s.c. and 180 mg s.c. arms versus PBO (withdrawal). No new safety risks were identified, and the benefit–risk profile of RZB was favorable. |
Abbreviations: ACR20, American College of Rheumatology 20% improvement criteria; ADA, anti‐drug antibody; AE, adverse event; AI, autoinjector; APS, abdominal pain score; b.i.d., two times a day; CD, Crohn's disease; CDAI, Crohn's Disease Activity Index; CDEIS, Crohn’s disease endoscopic index of severity; csDMARD, conventional synthetic disease‐modifying antirheumatic drug; DB, double‐blind; EASI, Eczema Area and Severity Index; GCP, Good Clinical Practice; IBDQ, inflammatory bowel disease questionnaire; i.v., intravenous; min, minutes; MOA, mechanism of action; MTX, methotrexate; N/A, not applicable; OBDS, on‐body delivery system; OL, open label; OLE, open‐label extension; OUS, Outside United States; PASI, Psoriasis Area and Severity Index; PBO, placebo; PD, pharmacodynamics; PFS, prefilled syringe; PK, pharmacokinetics; PsA, psoriatic arthritis; PsO, plaque psoriasis; q4w, every 4 weeks; q8w, every 8 weeks; q12w, every 12 weeks; RZB, risankizumab; SAE, serious adverse event; s.c., subcutaneous; SF, stool frequency; SIAQ, Self‐Injection Assessment Questionnaire; sPGA, static Physician Global Assessment; UC, ulcerative colitis; US, United States; UST, ustekinumab; VAS, visual analog scale.
Unless otherwise noted, this number reflects total treated subjects (i.e., including subjects exposed to risankizumab and/or other therapies).
SUMMARY OF CLINICAL EFFICACY AND SAFETY
Efficacy
Efficacy data in the global phase III pivotal studies for treatment of PsO demonstrated that participants treated with risankizumab experienced substantial skin clearance and clinical improvement in the extent and severity of PsO at week 16, after two doses of risankizumab 150 mg administered at weeks 0 and 4. Risankizumab was statistically superior to placebo, ustekinumab, 20 adalimumab, 21 and methotrexate 22 treatment for proportions of participants who achieved PASI 90 and sPGA 0/1. Statistically significant differences in favor of risankizumab were also observed on multiple secondary end points (e.g., PASI 75, PASI 100, sPGA clear, quality of life, and psoriasis symptoms). Efficacy results demonstrated at week 16 were maintained through week 52. Long‐term efficacy has been demonstrated in the 5‐year open‐label extension study, LIMMitless. 23
Efficacy of risankizumab was also demonstrated in PsA in two phase III studies; a statistically significant difference versus placebo was observed in the proportion of participants who achieved at least 20% improvement in ACR20 at week 24. 24 , 25 Efficacy results demonstrated at Week 24 were maintained through Week 52. 24 , 25
Efficacy data in the two global phase III induction studies for treatment of moderately to severely active CD demonstrated that subjects treated with risankizumab achieved statistically significant improvements in the co‐primary end points of clinical remission and endoscopic response for both doses of risankizumab (600 mg or 1200 mg i.v.) versus placebo. 18 In the CD maintenance study (Study M16‐000 Substudy 1), the study met the co‐primary end points of CDAI clinical remission and endoscopic response for both doses of risankizumab (180 mg s.c. or 360 mg s.c.) compared to the withdrawal (placebo s.c.) arm (p values ≤ 0.01). 17
Safety
Across all indications, there were no dose dependent AEs and evaluation of mean changes over time and individual subject changes in laboratory and vital signs data did not reveal any significant safety concerns with risankizumab treatment. In an open‐label extension trial for PsO (LIMMitless), risankizumab's safety profile remained favorable and consistent with profiles in short‐term studies through almost 5 years of continuous therapy. 23 For PsA and CD, open‐label extension trials also showed favorable and consistent profiles with short‐term studies through 76 and 188 weeks of continuous therapy, respectively. 26 , 27
Safety data in subgroups indicated that the safety profile does not differ based on race, gender, age, weight, or disease severity. Across all indications and doses as of March 25, 2023, the most frequently reported AEs (≥5% of participants who received >1 dose of risankizumab) were nasopharyngitis, coronavirus disease 2019 (COVID‐19), upper respiratory tract infection, arthralgia, headache, and hypertension. The rates of AEs, SAEs and AEs of special interest have remained stable with long‐term exposure in PsO, PsA, and CD. Long‐term rates of serious infections, malignancies (malignancies excluding nonmelanoma skin cancer [NMSC] and including NMSC), and adjudicated major adverse cardiovascular events in risankizumab PsO, PsA, and CD clinical trials are consistent with or within the epidemiologic benchmarks for the patients in the respective indications. No safety concerns with regard to opportunistic infections, active tuberculosis, or herpes zoster have been identified. No product complaints that have an impact on patient safety have been identified.
Dose recommendations
The final clinical doses selected for all three indications were based on the totality of data and ER analyses 10 , 11 , 12 (including the phase II and phase III dose ranging data as well as the phase III confirmatory data) to provide the optimal benefit/risk profiles of risankizumab in the target patient populations.
CONCLUSIONS
Based on clinical trial data in healthy participants and those patients with PsO, PsA, or CD, risankizumab has demonstrated linear PKs with slow clearance and long half‐lives, on‐target PD modulation of the IL‐23/Th17 axis, positive ER relationships for efficacy with no apparent exposure‐dependent worsening in safety, an immunogenicity profile with no major clinically meaningful consequences for ADAs or NAbs, and, last but not least, superior benefit/risk profiles compared to placebo and/or active comparators. These data formed the foundation for risankizumab's marketing approvals to treat multiple inflammatory diseases across the globe.
FUNDING INFORMATION
Medical writing support was provided by Mia DeFino, MS, ELS, CMPP, a freelance medical writer under contract with AbbVie. The authors thank Julie Parmentier and Buvana Ravishankar for their critical review of the pharmacodynamics (biomarker) section. AbbVie provided financial support for the studies and participated in the study design, study conduct, and analysis and interpretation of data and the writing, review, and approval of the manuscript.
CONFLICT OF INTEREST STATEMENT
All authors are employees of AbbVie and may hold AbbVie stock.
Pang Y, D’Cunha R, Winzenborg I, Veldman G, Pivorunas V, Wallace K. Risankizumab: Mechanism of action, clinical and translational science. Clin Transl Sci. 2024;17:e13706. doi: 10.1111/cts.13706
DATA AVAILABILITY STATEMENT
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the United States and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/ then select “Home.”
REFERENCES
- 1. Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL‐17A in inflammatory immune responses and host defense against pathogens. Immunol Rev. 2008;226:57‐79. [DOI] [PubMed] [Google Scholar]
- 2. Lee E, Trepicchio WL, Oestreicher JL, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Celis R, Planell N, Fernández‐Sueiro JL, et al. Synovial cytokine expression in psoriatic arthritis and associations with lymphoid neogenesis and clinical features. Arthritis Res Ther. 2012;14:R93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu Z, Yadav PK, Xu X, et al. The increased expression of IL‐23 in inflammatory bowel disease promotes intraepithelial and lamina propria lymphocyte inflammatory responses and cytotoxicity. J Leukoc Biol. 2011;89:597‐606. [DOI] [PubMed] [Google Scholar]
- 5. McDonald BD, Dyer EC, Rubin DT. IL‐23 monoclonal antibodies for IBD: so many, so different? J Crohns Colitis. 2022;16:ii42‐ii53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou L, Wang Y, Wan Q, et al. A non‐clinical comparative study of IL‐23 antibodies in psoriasis. MAbs. 2021;13:1964420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. AbbVie Inc . Skyrizi® (risankizumab‐rzaa). [US Package Insert] 2023.
- 8. Pang Y, Khatri A, Suleiman AA, Othman AA. Clinical pharmacokinetics and pharmacodynamics of Risankizumab in psoriasis patients. Clin Pharmacokinet. 2020;59:311‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suleiman AA, Minocha M, Khatri A, Pang Y, Othman AA. Population pharmacokinetics of Risankizumab in healthy volunteers and subjects with moderate to severe plaque psoriasis: integrated analyses of phase I‐III clinical trials. Clin Pharmacokinet. 2019;58:1309‐1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thakre N, D'Cunha R, Goebel A, Liu W, Pang Y, Suleiman AA. Population pharmacokinetics and exposure‐response analyses for Risankizumab in patients with active psoriatic arthritis. Rheumatol Ther. 2022;9:1587‐1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suleiman AA, Goebel A, Bhatnagar S, D'Cunha R, Liu W, Pang Y. Population pharmacokinetic and exposure‐response analyses for efficacy and safety of Risankizumab in patients with active Crohn's disease. Clin Pharmacol Ther. 2023;113:839‐850. [DOI] [PubMed] [Google Scholar]
- 12. Khatri A, Suleiman AA, Polepally AR, Othman AA. Exposure‐response relationships for efficacy and safety of Risankizumab in phase II and III trials in psoriasis patients. Clin Pharmacol Ther. 2020;107:378‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khatri A, Cheng L, Camez A, Ignatenko S, Pang Y, Othman AA. Lack of effect of 12‐week treatment with Risankizumab on the pharmacokinetics of cytochrome P450 probe substrates in patients with moderate to severe chronic plaque psoriasis. Clin Pharmacokinet. 2019;58:805‐814. [DOI] [PubMed] [Google Scholar]
- 14. Visvanathan S, Baum P, Vinisko R, et al. Psoriatic skin molecular and histopathologic profiles after treatment with risankizumab versus ustekinumab. J Allergy Clin Immunol. 2019;143:2158‐2169. [DOI] [PubMed] [Google Scholar]
- 15. Ravishankar B, Lal P, Sornasse T, et al. POS1545 modulation of serum biomarkers in patients with PSA treated with RISANKIZUMAB in the phase 3 keepsake 2 study. Ann Rheum Dis. 2023;82:1141‐1142. [Google Scholar]
- 16. Visvanathan S, Baum P, Salas A, et al. Selective IL‐23 inhibition by Risankizumab modulates the molecular profile in the colon and Ileum of patients with active Crohn's disease: results from a randomised phase II biopsy sub‐study. J Crohns Colitis. 2018;12:1170‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferrante M, Panaccione R, Baert F, et al. Risankizumab as maintenance therapy for moderately to severely active Crohn's disease: results from the multicentre, randomised, double‐blind, placebo‐controlled, withdrawal phase 3 FORTIFY maintenance trial. Lancet. 2022;399:2031‐2046. [DOI] [PubMed] [Google Scholar]
- 18. D'Haens G, Panaccione R, Baert F, et al. Risankizumab as induction therapy for Crohn's disease: results from the phase 3 ADVANCE and MOTIVATE induction trials. Lancet. 2022;399:2015‐2030. [DOI] [PubMed] [Google Scholar]
- 19. Feagan BG, Sandborn WJ, D'Haens G, et al. Induction therapy with the selective interleukin‐23 inhibitor risankizumab in patients with moderate‐to‐severe Crohn's disease: a randomised, double‐blind, placebo‐controlled phase 2 study. Lancet. 2017;389:1699‐1709. [DOI] [PubMed] [Google Scholar]
- 20. Gordon K, Strober B, Lebwohl M, et al. Efficacy and safety of Risankizumab: results from two double‐blind, randomised, placebo‐ and Ustekinumab‐controlled, phase 3 trials in moderate‐to‐severe plaque psoriasis (UltIMMa‐1 and UltIMMa‐2). Lancet. 2018;392:650‐661. [DOI] [PubMed] [Google Scholar]
- 21. Reich K, Gooderham M, Thaçi D, et al. Risankizumab compared with adalimumab in patients with moderate‐to‐severe plaque psoriasis (IMMvent): a randomised, double‐blind, active‐comparator‐controlled phase 3 trial. Lancet. 2019;394:576‐586. [DOI] [PubMed] [Google Scholar]
- 22. Cestari TF, Souza CD, Azulay‐Abulafia L, et al. 26197 efficacy and safety of risankizumab vs methotrexate in patients with moderate‐to‐severe plaque psoriasis: results from the 28‐week randomized, double‐blind period of an ongoing phase 3 study in Brazil. J Am Acad Dermatol. 2021;85:AB88. [Google Scholar]
- 23. Papp K, Blauvelt A, Puig L, et al. Long‐term safety and efficacy of risankizumab for the treatment of moderate‐to‐severe plaque psoriasis: interim analysis of the limmitless open‐label extension trial up to 5 years of follow‐up. J Am Acad Dermatol. 2023;89(6):1149‐1158. [DOI] [PubMed] [Google Scholar]
- 24. Kristensen LE, Keiserman M, Papp K, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 52‐week results from the KEEPsAKE 1 study. Rheumatology (Oxford). 2023;62:2113‐2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ostor A, Van den Bosch F, Papp K, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 52‐week results from the KEEPsAKE 2 study. Rheumatology (Oxford). 2023;62:2122‐2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mease PJ, Kellner H, Morita A, et al. Long‐term efficacy and safety of Risankizumab in patients with active psoriatic arthritis: results from a 76‐week phase 2 randomized trial. Rheumatol Ther. 2022;9:1361‐1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferrante M, Feagan BG, Panés J, et al. Long‐term safety and efficacy of Risankizumab treatment in patients with Crohn's disease: results from the phase 2 open‐label extension study. J Crohns Colitis. 2021;15:2001‐2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanchez AP, da Costa A, Del Rey C, Silva B, Romiti R. The overview of the immunobiology of Interleukin‐23 associated with immune‐mediated inflammatory disorders: a narrative review. J Drugs Dermatol. 2023;22:375‐385. [DOI] [PubMed] [Google Scholar]
- 29. Chiricozzi A, Saraceno R, Chimenti MS, Guttman‐Yassky E, Krueger JG. Role of IL‐23 in the pathogenesis of psoriasis: a novel potential therapeutic target? Expert Opin Ther Targets. 2014;18:513‐525. [DOI] [PubMed] [Google Scholar]
- 30. Eken A, Singh AK, Oukka M. Interleukin 23 in Crohn's disease. Inflamm Bowel Dis. 2014;20:587‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the United States and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/ then select “Home.”