

Abstract
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a new promising target for prevention and treatment of cardiac hypertrophy and heart failure. There are 3 δ isoforms of CaMKII in the heart and previous studies focused primarily on δB and δC types. Here we report the δA isoform of CaMKII is also critically involved in cardiac hypertrophy. We found that δA was significantly upregulated in pathological cardiac hypertrophy in both neonatal and adult models. Upregulation of δA was accompanied by cell enlargement, sarcomere reorganization and reactivation of various hypertrophic cardiac genes including atrial natriuretic factor (ANF) and β-myocin heavy chain (β-MHC). Studies further indicated the pathological changes were largely blunted by silencing the δA gene. These results provide new evidence for selective interfering cardiac hypertrophy and heart failure when CaMKII is considered as a therapeutic target.
Keywords: Cardiac hypertrophy, Nuclear factor of activated T-cell (NFAT), Histone deacetylase (HDAC), Atrial natriuretic factor (ANF), β-myosin heavy chain (β–MHC)
Introduction
Heart failure remains a leading cause of death and disability in industrialized society and cardiac hypertrophy is a promising target for prevention and treatment of heart failure. A number of signaling pathways have been identified mediating pathological cardiac hypertrophy [1, 2]. Among others, dysregulation of various protein kinases and phosphatases is known to be central to hypertrophic remodeling followed by cell death, heart failure and cardiac arrhythmias [3, 4]. Many prohypertrophic kinases such as Ca2+/calmodulin-dependent kinase II (CaMKII), protein kinase D1 (PKD1), and Ca2+-activated phosphatase calcineurin (CaN), are involved in cardiac hypertrophy by sensing increased intracellular Ca2+. Downstream, CaN dephosphorylate the nuclear factor of activated T-cell (NFAT), followed by its nuclear entry. In parallel, the class II histone deacetylases (HDACs) were phosphorylated by CaMKII and PKD1, and the phosphorylated HDAC4 disinhibits the inert cardiac genome and activates myocyte enhancer factor-2 (MEF2) [1, 2, 4]. Activation of the CaN-NFAT and/or HDAC4-MEF2 signaling pathways initiates hypertrophic genomic programs.
The critical role of CaMKII δ-isoforms in cardiac hypertrophy and failure has been well documented, but the major isoforms extensively investigated are δB and.δC [5–7]. These two isoforms have a broad spectrum of substrates including phospholamban (PLB), ryanodine receptors, HDACs, and the L-type calcium channels [3, 4]. However, CaMKIIδ has an additional isoform termed δA, which is mainly expressed in neonatal cardiac myocytes. Whether δA plays any role in adult cardiac hypertrophy and heart failure remains elusive. A recent study indicated CaMKIIδA is significantly over-expressed along plasma membranes and T-tubules and is required for cardiac hypertrophy in ASF/SF2-deficient animals [8]. These results suggest CaMKIIδA could play an important role in adult cardiac hypertrophy. Here we report CaMKIIδA was significantly upregulated in cardiac hypertrophy in both neonatal and adult models. Upregulation of CaMKIIδA was accompanied by reactivation of fetal cardiac genes such as ANF and β-MHC. These pathological changes were largely blunted by silencing the δA gene and an underlying mechanism indicates selective interference with the HDAC4-MEF2 signaling pathway.
Materials and Methods
Isolation and culture of ventricular myocytes
Sprague-Dawley rats were used throughout experiments and purchased from the Military Academy of the Medical Science Laboratory Animal Center (Beijing, China). The principles governing the care and treatment of animals as described by the American Physiological Society were followed at all times during this study. Isolation of adult rat ventricular myocytes (ARVM) was performed by Langendorff perfusion with a buffer containing low Ca2+, collagenase and protease as described in our previous papers [9, 10]. Isolation and culture of neonatal rat ventricular myocytes (NRVM) were conducted using the overnight trypsin-collagenase digestion method as described [9, 10]. All experiments with NRVMs were performed on 2–4 d cultures when synchronously contracting cells were observed. The purity of the cardiomyocytes was confirmed by anti-α-actin antibody.
Small Interfering RNAs (siRNA)
siRNA was performed using the standard method as described in our recent publication [11]. Two siRNA sequences were designed to target the coding region of CaMKIIδA: 5′-AGCCAACGUGGUAACCAGCTT-3′ and 5′-GCUGGUUACCACGUUGGCUTT-3′. The choice of these sequences was based on high silencing efficacy as verified by reverse transcription-polymerase chain reaction (RT-PCR). A scrambled siRNA 5′-UUCUCCGAACGUGUCACGUTT-3′ was used as negative control (NC). All nucleotides were chemically synthesized and 2′ O-methyl modified by GenePharma Co. (Shanghai, China). Transfection efficiency was estimated using FAM-conjugated NC siRNA and defined as the percentage of FAM-positive cells of the propidium iodine (PI) positive cells. For transient transfection, cells were incubated for 6 h in transfection medium comprising serum-free DMEM, 2 mM glutamine supplemented with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were retained in incubation medium containing serum-free DMEM, 2 mM glutamine, 1x ITS Liquid Media Supplement (Sigma-Aldrich) and penicillin/streptomycin for 36 h. This was followed by treatment with N.S. vehicle or isoproterenol (ISO, 10 μM) for 24 h.
RT-PCR and real-time PCR (qPCR)
Total RNA was isolated from heart tissue or ventricular myocytes using Trizol reagent (Invitrogen). For cDNA synthesis 1.0 μg RNA was used and reactions were carried out using a reverse transcription system (Promega). PCR was performed in a Genemate thermal cycler (Jinge Instr, Hangzhou, China) with the following primer sets for CaMKIIδA: forward, 5′-CGAGAAATTTTTCAGCAGCC-3′; reverse, 5′-ACAGTAGTTTGGGGCTCCAG-3′. 18-S ribosomal RNA (18-S rRNA) was used as an internal control and the primer sets used are: forward, 5′-ACCGCAGCTAGGAATAATGGA-3′; reverse, 5′-GCCTCAGTTCCGAAAACCA-3′. PCR products were subjected to 1.8% agarose gel electrophoresis, stained with ethidium bromide, and visualized with LAS3000.
qPCR was performed using SYBR Green Master Mix (Takara, Japan) in a Bio-Rad IQ5 detection system as described [12]. The primer sets for each gene are as follows: ANF (forward, 5′-GGGGGTAGGATTGACAGGAT-3′; reverse, 5′-CTCCAGGAGGGTATTCACCA-3′); β-MHC (forward, 5′-CCTCGCAATATCAAGGGAAA-3′; reverse, 5′-TACAGGTGCATCA GCTCCAG-3′); MEF2 (forward, 5′-GAGCAGAGCCCCCTGCTGGAGGACA-3′; reverse, 5′-TAGCAGGCCGCTGGGGCAGGCCCGG-3′). 18-S rRNA was used as internal standard. The cycle threshold (CT) values corresponding to the PCR cycle number at which fluorescence emission reaches a threshold above the baseline emission were automatically determined in triplicates and averaged. Abundances of each target gene was normalized to that of 18-S rRNA using the formula of 2−ΔCT, where ΔCT = CT of target genes – CT of 18-S rRNA.
Western blotting
Cells were lysed in RARI buffer (50 mM Tris, pH 7.4, 1.0 mM EDTA, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM sodium orthovanadate, 1 mM sodium pyrophosphate). Cell lysates were resolved in 10% SDS–PAGE and transferred to PVDF membranes (Millipore) as described previously [9]. HDAC4 and HDAC4-p proteins were reacted with polyclonal anti-HDAC4 or anti-HDAC4-p (Santa cruz) followed by incubation with horseradish peroxidase-conjugated secondary antibody (Invitrogen). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was measured as an internal control with appropriate antibody (Santa cruz). Band density was quantified using ImageJ (The US National Institutes of Health, http://rsb.info.nih.gov/ij/) where required.
Indirect Immunofluorescence
Indirect immunofluorescent staining was conducted essentially the same as described previously [9, 11]. Heart cells were grown on laminin-coated glass coverslips and fixed in 4% paraformaldehyde followed by permeabilization with 0.5 % Triton X-100. After blocking in 1 % BSA-containing PBS, cells were incubated with primary antibody and subsequently with secondary antibodies (Invitrogen). Images were collected and analyzed by TCS-SP confocal laser microscopy (Leica, Germany). For surface area determination ImageJ was used and at least 50 individualized cells were analyzed for each experiment.
NFAT–Luciferase Assays
NFAT transcriptional activity was estimated by dual-luciferase reporter assay (Promega, Madison, WI). Neonatal myocytes were co-transfected with NFAT reporter plasmid pGL4.30[luc2P/NFAT-RE/Hygro] and control plasmid pGL7.4 expressing Renilla reniformis luciferase reporter gene (Promega) in the presence or absence of NC siRNA or CaMKIIδA siRNA. Transient transfection was performed with the electroporation nucleofector kit (AMAXA Biosystems). After 36 h incubation in a 12-well plate, cells were treated with N.S. or 10 μM ISO for further 48 h. Cells were then lysed and a luciferase assay was performed with the dual luciferase kit (Promega) according to the manufacturer’s instructions. Luciferase activity was measured using a TR717 microplate luminometer (Applied Biosystems). Data were expressed as fold change (Fold change = average relative light units of induced cells/average relative light units of control cells).
In vivo cardiac hypertrophic model and immunohistochemistry
In vivo cardiac hypertrophy was induced by chronic ISO injection as described [13]. For histology, paraffin-embedded heart tissue sections (4 μm in thickness) were made and stained with standard H&E or immunohistochemistry techniques.
Statistical analysis
Results are expressed as means ± SEM. One-way ANOVA followed by Newman-Keuls test or Student’s t test was performed as implemented in IgorPro (Wavemetrics, Portland, OR) as described [14]. A value of p < 0.05 was accepted as statistically significant.
Results
CaMKIIδA is overexpressed in various cardiac hypertrophy models
Previous studies have shown both δB and δC isoforms of CaMKII are critical to cardiac hypertrophy [5, 15] but the role of the δA isoform in this process is unclear. To investigate function of the latter we first determined how the 3 isoforms were expressed in isolated adult rat myocytes (ARVM) and cultured neonatal myocytes (NRVM) using RT-PCR techniques. As shown in Fig. 1A, neonatal cells expressed all 3 isoforms while adult cells do not have appreciable amounts of CaMKIIδA. Surprisingly, CaMKIIδA was markedly over-expressed in both NRVM and ARVM after incubation with isoproterenol for 24 h, classic in vitro cardiac hypertrophy models (Fig. 1B).
Figure 1. The δA isoform was upregulated in various cardiac hypertrophy models.
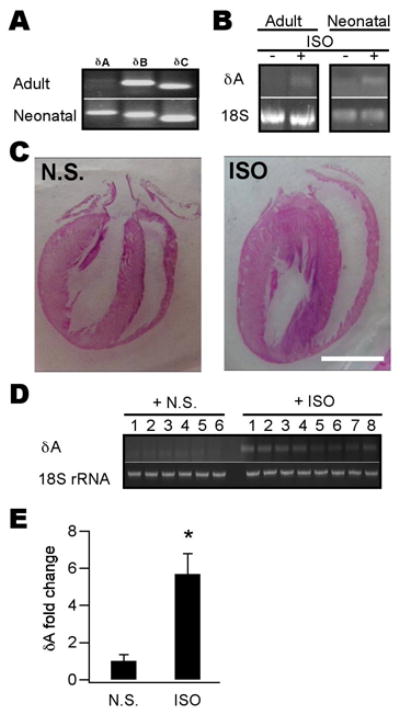
A, mRNAs of 3 CaMKII δ-isoforms were detected using RT-PCR in isolated adult (upper) and cultured neonatal (lower) myocytes. δA was not detectable in adult cells. B, CaMKIIδA mRNA in adult and neonatal myocytes was upregulated by ISO (10 μM) treatment for 24 h. 18-S rRNA was used as an internal control. C, Representative four-chambered heart pictures showing typical cardiac hypertrophy induced by 5-d injection of ISO (30 mg kg−1 d−1, i.p.). The hearts were sectioned and stained by the H&E method. Scale bar = 1 cm. D, CaMKIIδA mRNA (upper) from ISO-injected animal’s heart tissue (n = 8) was upregulated compared to that of NS-injected ones (n = 6). 18-S rRNA was used as a loading control (lower). E, Quantitative analysis of data shown in (D). Band density of CaMKIIδA for each sample was determined using ImageJ and normalized to their respective 18-S rRNA values. (n = 6–8, *p < 0.01, Student’s t test).
We further demonstrated CaMKIIδA was up-regulated in a whole animal hypertrophy model. We induced cardiac hypertrophy by injecting rats with ISO for 5 d and animals were sacrificed on day 7 for examination. Compared to normal saline-injected animals, ISO-treated rats developed typical hypertrophy as evidenced by enlarged heart size (Fig. 1C), over-expression of hypertrophic genes such as ANF and βMHC (see later), and nuclear entry of NFAT (see later). As expected, ISO-treated animal hearts showed significant over-expression of CaMKIIδA (Fig. 1D, E). Thus, CaMKIIδA is upregulated in cardiac hypertrophy of both neonatal and adult models.
Upregulation of CaMKIIδA is accompanied by reactivation of fetal cardiac genes
A salient feature of pathological cardiac hypertrophy is reactivation of fetal cardiac genes such as ANF and βMHC [2, 16]. We investigated the relationship between CaMKIIδA upregulation and fetal cardiac gene reactivation. Using indirect immunofluorescent techniques, we found ISO induced a strong fluorescent signal for ANF along membrane and T-tubule contours in isolated adult myocytes (Fig. 2A). Furthermore, in the ISO-injected animals both ANF and β–MHC mRNAs were increased by ~30-fold and ~25-fold, respectively, in the hypertrophied hearts as detected with qPCR (Fig. 2B). These results demonstrate upregulation of CaMKIIδA in adult cardiac hypertrophy models, is accompanied by reactivation of fetal cardiac genes.
Figure 2. Reactivation of fetal cardiac genes was blunted by CaMKIIδA silencing.

A, induction and immunolocalization of ANF protein in isolated adult myocytes treated by ISO (2 μM) for 48 h. For immunofluorescence cells were first incubated with an anti-ANF antibody followed by reaction with Alexa-488 conjugated second antibody (green). Propidium iodine (PI) was used as counterstain for nuclei (red). Scale bar = 50 μm. B, Upregulation of ANF and β–MHC mRNAs in the hearts from ISO-injected animals. mRNAs were analyzed by qPCR and normalized to 18-S rRNA. The mean normalized value for expression of each gene in NS-injected animals is defined as 1. (n = 3–4, *p < 0.01, Student’s t-test). C, induction and immunolocalization of ANF protein in cultured neonatal myocytes by ISO (10 μM) for 48 h. Note the ANF signal is primarily located in the perinuclear area. Scale bar = 20 μm. D. qPCR assays showing both ANF and βMHC mRNAs were upregulated by ISO (10 μM) application for 48 h and these effects were blunted by δA siRNA. (for both panels n = 3, *p < 0.01 vs NC/NS group, ANOVA).
We further determined whether these upregulated hypertrophic genes were inhibited by silencing CaMKIIδA. Because isolated adult myocytes and whole animals are not feasible for molecular interference, we chose to use neonatal myocytes as an alternative testing model. Depletion of the δA gene was achieved using siRNA. In our system we could achieve 80% transfection efficiency and δA expression was significantly inhibited even after 72 h. Although < 2% cells cultured in regular medium plus NC siRNA for 48 h showed a faint ANF signal as detected with indirect immunofluorescence, nearly 60% cells expressed the protein after incubation with ISO plus NC siRNA for 48 h (Fig. 2C). By contrast, in cells pretreated with siRNA against δA, this effectively prevented the appearance of ANF induced by ISO (Fig. 2C). We further quantified ANF at the mRNA level using qPCR. ISO induced ~6-fold increase in ANF at 48 h and this effect was blunted by pretreatment with siRNA against δA (Fig. 2D). Qualitatively similar results were obtained when βMHC, another well-known hypertrophic marker gene, was compared (Fig. 2D). These results demonstrate reactivation of various fetal cardiac genes requires concomitant CaMKIIδA upregulation.
Silencing CaMKIIδA prevents cell enlargement
In addition to genomic reprogramming, cardiac hypertrophy is accompanied by cell enlargement and sarcomere reorganization [2]. We next determined how the cell morphology is modulated by manipulation of δA in the neonatal hypertrophic model. We employed α-actinin immunofluorescence as a sarcomere marker and cell surface area as a measure of size. Results from these experiments indicated apart from induction of obvious sarcomere reorganization (Fig. 3A), 48-h incubation with ISO increased the cross-sectional areas by ~75% (Fig. 3A and B). This effect was prevented by pre-treatment with siRNA against δA (Fig. 3A and B). Thus, we reveal that silencing CaMKIIδA is sufficient to prevent ISO-induced myocardial hypertrophy.
Figure 3. Morphological alterations were prevented by CaMKIIδA silencing.
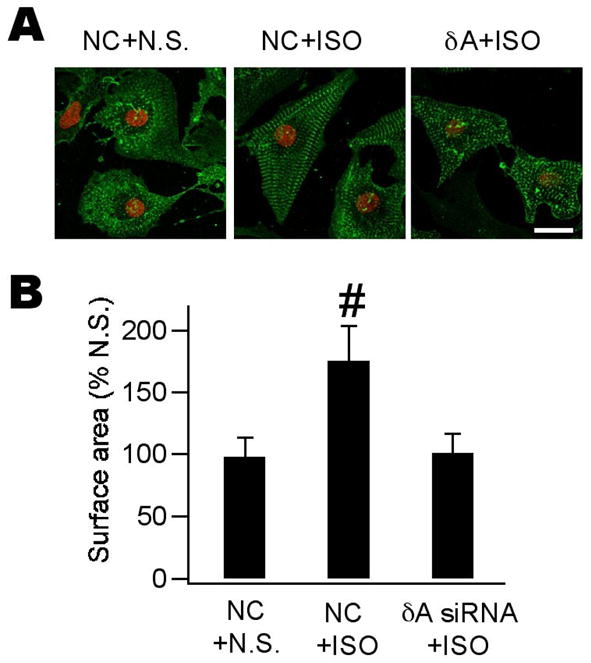
A, confocal images of neonatal myocytes with various treatments as indicated. Cells were treated with N.S. or ISO for 48 h and incubated with anti-α-actinin followed by reaction with Alexa-488 conjugated second antibody (green). Propidium iodine was used as counterstain for nuclei (red). Scale bar = 50 μm. ISO induced cell enlargement accompanied by prominent sarcomere reorganization (middle). These alterations are largely prevented by pretreatment with δA siRNA (right). B, quantitative analysis of data shown in (A). At least 100 cells were quantified for each group and experiments were repeated twice. (n = 3, #p < 0.05, ANOVA).
Silencing CaMKIIδA blunts HDAC4-mediated signaling pathway
Finally we investigated how CaMKIIδA participates in the development of cardiac hypertrophy. Although many hypertrophic stressors exist that induce cardiac hypertrophy, two more general mechanisms seem to operate. The first mechanism involves the transcription factor NFAT [17, 18]. This protein is normally located in the cytoplasm where it is sequestered by the buffering protein 14-3-3. Upon dephosphorylation by CaN, NFAT enters the nucleus and initiates hypertrophic responses by further recruiting other transcription factors such as GATA-4 and Nkx2.5 [19]. In our whole animal hypertrophic model induced by ISO, we found prominent NFAT accumulation in the nucleus (Fig. 4A) as detected using immunohistochemistry methods along with up-regulation of ANF and β-MHC (Fig. 2B) at the mRNA level. GATA-4, a hypertrophic gene closely related to NFAT activation was also significantly upregulated (Fig. 4B). However, when a neonatal hypertrophic model was used to test the effect of δA silencing, we noticed δA silencing exacerbated NFAT transcription activity as estimated using a NFAT-luciferase gene reporter construct (Fig. 4C). Thus, we conclude the pro-hypertrophic effect of CaMKIIδA may be not mainly by interfering with the NFAT-mediated signaling pathway.
Figure 4. CaMKIIdA silencing blunted activation of HDAC4-MEF2 signaling.

A, in situ detection of nuclear NFAT protein (arrows in right panel) using immunohistochemistry in ISO-induced hypertrophy models. In NS-injected animals, no appreciable NFAT signal was revealed (left). Note the larger cell size in right panel. B, GATA-4 mRNA was significantly amplified in heart tissues of ISO-treated animals (n = 6, *p < 0.01, Student’s t test). C, NFAT-mediated transcription activity as reflected by the NFAT-luciferase assay was increased by ISO alone and further enhanced by CaMKIIδA silencing. (n = 3, #p < 0.05 vs N.S.; *p < 0.05 vs ISO alone, ANOVA). These experiments and what follows were performed in neonatal myocytes. D, Western blot showing profound phosphorylation of HDAC4 by 48-h ISO incubation. CaMKIIδA silencing prevented HDAC4 phosphorylation evoked by ISO (left). Plotted on the right is quantitative analysis of data shown in the left panel. Band density was determined and analyzed using ImageJ. (n = 4, *p < 0.01 vs NC/NS group, ANOVA). E, MEF2 mRNA was upregulated by ISO incubation for 48 h and this effect was blunted by CaMKIIδA silencing. (n = 4, *p < 0.01 vs NC/NS group, ANOVA).
We then turned to the second hypertrophic mechanism involving HDAC-MEF2 signaling [20]. HADCs normally remain associated with the cardiac genome, keeping the latter in an inert state. When phosphorylated by HADC kinases such as CaMKII and PKD1, HDACs exit the nucleus and the relatively inactive cardiac genome becomes transcriptionally active. Activation of MEF2 is essential for the HDAC-mediated hypertrophy. As expected, ISO-treated neonatal cells presented a heavily phosphorylated HDAC4 subtype (Fig. 4D) accompanied by significant upregulation of MEF2 mRNA (Fig. 4E). Both pro-hypertrophic effects of ISO were blunted by pre-silencing treatment with δA siRNA (Fig. 4D and E). These results clearly demonstrate silencing CaMKIIδA prevents cardiac hypertrophy by dominant interference of the HDAC4-MEF2 signaling pathway.
Discussion
We have provided clear evidence showing that, in addition to δB and δC, CaMKIIδA is also critically involved in cardiac hypertrophy in both neonatal and adult models. The major immediate target for.δB isoform is HDAC4 [21, 22] while that for δC remains uncertain because the latter has a broad cytoplasmic distribution [15]. Although we showed here that the pro-hypertrophic effect of CaMKIIδA relies on its predominant interference of HDAC-MEF2 signaling, the immediate molecular target does not seem to be HDAC itself because δA is chiefly membrane- and T-tubule-located [8]. Because L-type calcium channels are located in T-tubules and are well-known for their critical involvement in cardiac hypertrophy and heart failure [23, 24] we suspect the channels may be an immediate target for δA. This possibility requires further investigation.
An unexpected finding in this study was that CaMKIIδA negatively regulates the NFAT signaling pathway. A similar finding was also reported with CaMKII in cardiac hypertrophy models although which particular isoform is responsible for such phenomenon remains uncharacterized [25]. The significance of the negative hypertrophic regulation is unknown but may represent a self-protection mechanism against hypertrophy. However, the self-defense mechanism if any is so weak that it could not overcome the pro-hypertrophic effect exerted by the more powerful HDAC4-MEF2 activation. Thus, non-selective inhibition of pan-CaMKIIδ may be detrimental to cardiac hypertrophy and heart failure.
Since both δB and δC isoforms are constitutively expressed in normal adult hearts [5–7], any therapeutic measure targeting these molecules may come up with serious side- effects. In this regard measures directed at the δA isoform apparently have superiority because the latter is only upregulated in diseased hearts. Nanoparticle-mediated siRNA techniques [26], for example, may be implemented as a novel selective therapy toward cardiac hypertrophy and heart failure.
Research Highlights
CaMKIIδA is upregulated in both neonatal and adult cardiac hypertrophy models.
Silencing CaMKIIδA gene blunts cell enlargement and hypertrophic cardiac gene reactivation.
Activation of the HDAC4-MEF2 signaling pathway is inhibited by CaMKIIδA silencing.
Our results provide new evidence for selective interference of cardiac hypertrophy and heart failure.
Acknowledgments
This work was supported in part by grants from Tianjin Science and Technology Support Project (08ZCKFSH04500, X.D.T), NSFC grants (30871011 to X.D.T.; 81072629 to J. L.; 30870988 to X.W.Z.), The Project “973” grant (2010CB945001, X.D.T), MOE Doctorial Training Funds (200800550036 to X.D.T), Tianjin Natural Science Foundation (10JCYBJC14800 to J.L.), and NIH grants (HL55426 and AI058173 to D.L.G.).
The abbreviations used are
- CaMKII
Ca2+/calmodulin-activated kinase II
- HDAC
histone deacetylase
- MEF2
myocyte enhancer factor-2
- CaN
calcineurin
- NFAT
nuclear factor of activated T-cell
- PKD
protein kinase D
- ANF
atrial natriuretic factor
- β•MHC
β-myosin heavy chain
- NRVM
neonatal rat ventricular myocytes
- ARVM
adult rat ventricular myocytes
- siRNA
small RNA interference
- qPCR
quantitative real-time polymerase chain reaction
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- ISO
isoproterenol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sambrano GR, Fraser I, Han H, Ni Y, O’Connell T, Yan Z, Stull JT. Navigating the signalling network in mouse cardiac myocytes. Nature. 2002;420:712–714. doi: 10.1038/nature01306. [DOI] [PubMed] [Google Scholar]
- 2.Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 3.Bers DM. Altered cardiac myocyte Ca2+ regulation in heart failure. Physiology (Bethesda) 2006;21:380–387. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- 4.Anderson ME. CaMKII and a failing strategy for growth in heart. J Clin Invest. 2009;119:1082–1085. doi: 10.1172/JCI39262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bossuyt J, Helmstadter K, Wu X, Clements-Jewery H, Haworth RS, Avkiran M, Martin JL, Pogwizd SM, Bers DM. Ca2+/calmodulin-dependent protein kinase IIδ and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ Res. 2008;102:695–702. doi: 10.1161/CIRCRESAHA.107.169755. [DOI] [PubMed] [Google Scholar]
- 6.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN. The δ isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers DM, Brown JH. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–1240. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, Wang HY, Bermingham JR, Jr, Ye Z, Liu F, Rosenfeld MG, Manley JL, Ross J, Jr, Chen J, Xiao RP, Cheng H, Fu XD. ASF/SF2-regulated CaMKIIδ alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell. 2005;120:59–72. doi: 10.1016/j.cell.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 9.Ma D, Tang XD, Rogers TB, Welling PA. An Andersen-Tawil syndrome mutation in Kir2.1 (V302M) alters the G-loop cytoplasmic K+ conduction pathway. J Biol Chem. 2007;282:5781–5789. doi: 10.1074/jbc.M608776200. [DOI] [PubMed] [Google Scholar]
- 10.Zhou XW, Mudannayake M, Green M, Gigena MS, Wang G, Shen RF, Rogers TB. Proteomic studies of PP2A-B56γ1 phosphatase complexes reveal phosphorylation-regulated partners in cardiac local signaling. J Proteome Res. 2007;6:3433–3442. doi: 10.1021/pr060619l. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, Gill DL. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science. 2010;330:105–109. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ai D, Pang W, Li N, Xu M, Jones PD, Yang J, Zhang Y, Chiamvimonvat N, Shyy JY, Hammock BD, Zhu Y. Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106:564–569. doi: 10.1073/pnas.0811022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heather LC, Catchpole AF, Stuckey DJ, Cole MA, Carr CA, Clarke K. Isoproterenol induces in vivo functional and metabolic abnormalities: similar to those found in the infarcted rat heart. J Physiol Pharmacol. 2009;60:31–39. [PubMed] [Google Scholar]
- 14.Tang XD, Xu R, Reynolds MF, Garcia ML, Heinemann SH, Hoshi T. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature. 2003;425:531–535. doi: 10.1038/nature02003. [DOI] [PubMed] [Google Scholar]
- 15.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, Brown JH. The δC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 16.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63:467–475. doi: 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 18.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 19.Morin S, Charron F, Robitaille L, Nemer M. GATA-dependent recruitment of MEF2 proteins to target promoters. Embo J. 2000;19:2046–2055. doi: 10.1093/emboj/19.9.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson EN, Backs J, McKinsey TA. Control of cardiac hypertrophy and heart failure by histone acetylation/deacetylation. Novartis Found Symp. 2006;274:3–12. discussion 13–19, 152–155, 272–156. [PubMed] [Google Scholar]
- 21.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Little GH, Bai Y, Williams T, Poizat C. Nuclear calcium/calmodulin-dependent protein kinase IIdelta preferentially transmits signals to histone deacetylase 4 in cardiac cells. J Biol Chem. 2007;282:7219–7231. doi: 10.1074/jbc.M604281200. [DOI] [PubMed] [Google Scholar]
- 23.Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005;115:3306–3317. doi: 10.1172/JCI27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koval OM, Guan X, Wu Y, Joiner ML, Gao Z, Chen B, Grumbach IM, Luczak ED, Colbran RJ, Song LS, Hund TJ, Mohler PJ, Anderson ME. CaV1.2 β-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci USA. 2010;107:4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacDonnell SM, Weisser-Thomas J, Kubo H, Hanscome M, Liu Q, Jaleel N, Berretta R, Chen X, Brown JH, Sabri AK, Molkentin JD, Houser SR. CaMKII negatively regulates calcineurin-NFAT signaling in cardiac myocytes. Circ Res. 2009;105:316–325. doi: 10.1161/CIRCRESAHA.109.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leng Q, Woodle MC, Lu PY, Mixson AJ. Advances in Systemic siRNA Delivery. Drugs Future. 2009;34:721. doi: 10.1358/dof.2009.034.09.1413267. [DOI] [PMC free article] [PubMed] [Google Scholar]