

Abstract
Objective: Erythropoietin has been shown to have a protective effect in certain models of ischaemia‐reperfusion, and in some cases the protection has been correlated with activation of signalling pathways known to play a role in cell survival and proliferation. We have studied whether erythropoietin would overcome direct toxic effects of hydrogen peroxide (H2O2) treatment to human renal proximal tubular (HK‐2) cells.
Materials and methods: HK‐2 cells were incubated with H2O2 (2 mm) for 2 h with or without erythropoietin at concentrations of 100 and 400 U/ml, and cell viability/proliferation was assessed by chemical reduction of MTT. Changes in phosphorylation state of the kinases Akt, glycogen synthase kinase‐3β (GSK‐3β), mammalian target of rapamycin (mTOR) and extracellular signal‐regulated kinase 1 and 2 (ERK1/ERK2) were also analysed.
Results: Cells incubated with H2O2 alone showed a significant decrease in viability, which did not significantly change by addition of erythropoietin at concentration of 100 U/ml, but was further reduced when concentration of erythropoietin was increased to 400 U/ml. Phosphorylation state of the kinases Akt, GSK‐3β, mTOR and ERK1/ERK2 of H2O2‐treated HK‐2 cells was slightly altered in the presence of erythropoietin at concentration of 100 U/ml, but was significantly less in the presence of erythropoietin at a concentration of 400 U/ml. Phosphorylation of forkhead transcription factor FKHRL1 was diminished in cells incubated with H2O2 and erythropoietin at a concentration of 400 U/ml.
Conclusions: Erythropoietin, at high concentrations, may significantly increase cellular damage in HK‐2 cells subjected to oxidative stress, which may be due in part to decrease in activation of important signalling pathways involved in cell survival and/or cell proliferation.
Introduction
The haematopoietic growth factor erythropoietin promotes proliferation, differentiation and survival of erythroid progenitor cells in the bone marrow resulting in increase in the number of red blood cells. Erythropoietin exerts its actions via erythropoietin receptors to which it binds, resulting in activation of signal transduction pathways. Erythropoiesis has been initially considered to be the only physiological role of erythropoietin, and its use has been particularly useful in management of anaemia in patients with end‐stage renal disease and in patients with cancer and anaemia (1, 2). The discovery of both erythropoietin and erythropoietin receptors in other organs and tissues outside the liver and kidneys (for example, heart, brain and uterus) has led to the conclusion that erythropoietin has other functions in addition to promoting erythropoiesis. Erythropoietin receptors have been found in a wide variety of cell types, including non‐erythroid blood cells (myeloid cells, lymphocytes, megakaryocytes) and non‐haematopoietic cells, such as neurons, astrocytes, vascular smooth muscle cells, cardiac myocytes, mesangial cells, endothelial cells and renal tubular cells (3, 4, 5).
Production and secretion of erythropoietin and expression of erythropoietin receptors seems to be regulated by tissue oxygen supply. Deficiency in tissue oxygen results in production of erythropoietin and expression of erythropoietin receptors in brain, liver and kidneys (6, 7). Such a response to oxygen deficiency suggests that erythropoietin plays a role in preventing ischaemic damage to a number of organs, including kidneys. This has led several research groups to study the effect of erythropoietin in animal models of acute renal injury (8, 9, 10, 11, 12).
In one of these studies, erythropoietin was administered to rats 24 h before bilateral renal artery clamping; at 24 h post‐reperfusion, functional protection was observed, with significant attenuation in rise of serum creatinine induced by ischaemia, which was associated with reduction of proximal tubule cell death (12). In a further study, erythropoietin was given to rats either as pre‐treatment at the time of reperfusion, or post‐reperfusion (9); renal function appeared to be preserved with reduction in number of apoptotic tubule cells; it was evident, however, that administration of erythropoietin after reperfusion resulted in a lesser degree of protection. In another study in Sprague–Dawley rats, erythropoietin appeared effective only when given 30 min before ischaemia, with an increase of tubule cell mitosis (11). In a more recent study, erythropoietin was administered by intraperitoneal injection to Wistar rats, 20 min prior to 45 min of ischaemia. Forty‐eight hours after reperfusion rise of serum creatinine and urea was less in erythropoietin‐treated animals compared to untreated controls; histological tubule damage was similarly reduced by erythropoietin (10).
Up to now few studies have evaluated the direct effect of erythropoietin on human proximal tubule cells subjected to some form of oxidative stress, and found a protective effect of erythropoietin (9, 11). However, Sharples et al. suggest that high doses of erythropoietin (> 100 U/ml) might be ‘associated with failure of cytoprotection in serum starved proximal tubular epithelial cells’ (13).
Given that the mechanisms governing ischaemia‐reperfusion disease are complex, since ischaemia may give rise to reactive oxygen species (ROS) and levels of ROS increase during reperfusion (14), it is possible that erythropoietin counteracts the harmful effects of ROS.
In the present work, we have evaluated whether erythropoietin overcomes the direct toxic effects of hydrogen peroxide (H2O2) in cultures of human renal proximal tubule cells. Since some reports have suggested that the possible mechanism of erythropoietin action relies on activation of serine/threonine kinase Akt (4, 9), we looked at the effects of erythropoietin on the kinases that are known to play a role in cell survival and proliferation.
Materials and methods
Cell culture
HK‐2 cells were obtained from the American Type Culture Collection and grown in 100‐mm culture dishes (Corning, New York, USA) as described by others (15). In brief, they were cultured in Dulbecco's modified Eagle's medium containing Glutamax (Gibco, Invitrogen, Milan, Italy) supplemented with 10% foetal calf serum (FCS), 100 units/ml penicillin and 100 µg/ml streptomycin (Sigma, Milan, Italy) in an atmosphere of 5% CO2 in air at 37 °C, up to approximately 90% confluence (16). All experiments were performed after overnight cell starvation by addition of culture medium not containing FCS.
Cell viability/proliferation
Cell viability and proliferation were measured by ability of viable cells to reduce MTT (3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl‐2H‐tetrazolium bromide) (Sigma) (17). Cells were grown in 6‐well plates. After incubation with H2O2 (2 mm) (Sigma) and after adding erythropoietin (Eprex, Janssen‐Cilag SpA, Milan, Italy) at the same time to the plates, they were washed once with sterile phosphate‐buffered saline (PBS) and incubated with 1 mg/ml MTT (in sterile PBS) for 90 min at 37 °C. Cells were then treated with dimethyl sulphoxide. Measurements of coloured product as a result of chemical reduction of MTT were made at 540 nm using a Beckman DU 800 spectrophotometer (Beckman‐Coulter, Milan, Italy). Each experiment was performed in triplicate.
Western blot analysis
After incubation with (or without) H2O2 (2 mm) and with (or without) erythropoietin (100 or 400 U/ml), HK‐2 cells were washed in cold PBS and then lysed in buffer containing 20 mm HEPES (pH 7.4), 2 mm EGTA, 1 mm DTT, 1 mm NaVO4, 1% (v/v) Triton X‐100, 2 µm leupeptin, 2 µm microcystin, 1.5 µm aprotinin, and 400 µm PMSF. Samples were then centrifuged at 10 000 ×g for 10 min and the supernatant was retained (lysate). A small part of the supernatant was used to determine protein content and the majority was utilized for sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE). Protein concentrations were determined using a modified Bradford protein assay protocol (18) in order to obtain equal loading (approximately 30 µg of protein from each sample were loaded).
Protein extracts were resolved by SDS‐PAGE and transferred to a nitrocellulose membrane (Hybond C®‐Extra, Amersham Biosciences, GE Healthcare, Cologno Monzese, Italy) followed by Western blotting, as previously described (19). Briefly, the membrane was incubated for 1 h at room temperature with 5% (w/v) non‐fat powdered milk in a TBS‐Tween buffer [TBST: 20 mm Tris and 137 mm NaCl, pH 7.6, containing 0.1% (v/v) Tween 20]. Primary antibody, diluted in TBST with 5% (w/v) non‐fat powdered milk, was then added to the membrane and incubated overnight at 4 °C. The membrane was then washed three times, 5 min each, at room temperature with TBST and incubated for 1 h with secondary antibody conjugated with horseradish peroxidase (Dako; Glostrup, Denmark) diluted 1 : 5000 TBST with 1% (w/v) non‐fat powdered milk at room temperature. It was then washed as above (three times). Secondary antibodies, conjugated with horseradish peroxidase, were detected by enhanced chemiluminescence system (Amersham Biosciences, GE Healthcare) according to the manufacturer's instructions. Primary antibodies included the following: anti‐phospho‐ERK1/ERK2 (p44/p42 mitogen‐activated protein kinase, Cell Signaling, Beverly, MA, USA); anti‐phospho‐Akt (Ser 473, Cell Signaling); anti‐phospho‐GSK‐3β (Ser 9, Cell Signaling); anti‐phospho‐FKHRL1 (Thr 32)/anti‐phospho‐FKHR (Thr 24) (Cell Signaling); anti‐phospho‐mTOR (Ser 2448, Cell Signaling); anti‐total‐mTOR (Cell Signaling); and anti‐β‐actin (mouse monoclonal, Sigma).
Statistical analysis
All results were expressed as mean ± standard error. Statistical analysis was performed using analysis of variance for unpaired data (GraphPad 4.0 software). Statistical significance was defined as P < 0.05.
Results
Effect of erythropoietin on cellular damage caused by H2O2
Addition of H2O2 to HK‐2 cells caused a significant decrease in cell viability, as measured by chemical reduction of MTT (Fig. 1a). In the presence of 100 U/ml of alpha‐erythropoietin there was no significant change in cell viability (Fig. 1a). It was therefore decided to increase concentration of alpha‐erythropoietin to 400 U/ml. Surprisingly, at this concentration of erythropoietin, cell viability was further significantly reduced (Fig. 1a). Untreated (no H2O2) cells incubated with erythropoietin alone, at both concentrations of erythropoietin (100 and 400 U/ml), did not exhibit any significant change in cell viability, as shown in Fig. 1(b). This unexpected observation (with erythropoietin at a concentration of 400 U/ml) on viability of H2O2‐treated HK‐2 cells led us to investigate changes in certain kinases that are components of cell signalling pathways, which are believed to play a role in cell survival and proliferation.
Figure 1.

(a) Effect of erythropoietin (EPO) at concentrations of 100 and 400 U/ml on viability of HK‐2 cells treated with hydrogen peroxide (H2O2). Columns represent chemical reduction of MTT by cultured HK‐2 cells either untreated (Control, made equal to 100%) or treated with H2O2 (2 mm) alone (H2O2) or H2O2 (2 mm) and EPO (H2O2 + EPO 100 U) added at the same time to cell cultures, or H2O2 (2 mm) and EPO (H2O2 + EPO 400 U) added also in this case at the same time. H2O2 caused a significant decrease in cell viability. No change was observed when the cells were treated with H2O2 and EPO at concentration of 100 U/ml compared with cells treated with H2O2 alone, indicating no significant effect of EPO (100 U/ml) on cell viability. However, addition of EPO at concentration of 400 U/ml caused further (and significant) decrease in cell viability compared to cells treated with H2O2 alone (as shown by the further and significant decrease in MTT chemical reduction). Data are representative of three separate experiments. H2O2 vs. Control; P < 0.0001 (°). H2O2+ EPO 100 U vs. Control; P < 0.0001 (^). H2O2+ EPO 400 U vs. Control; P < 0.0001 (*). H2O2+ EPO 100 U vs. H2O2; P > 0.05 (+). H2O2+ EPO 400 U vs. H2O2; P < 0.0001 (#). (b) Effect of EPO alone at concentrations of 100 and 400 U/ml on cell viability of H2O2‐untreated HK‐2 cells. Columns represent chemical reduction of MTT by cultured HK‐2 cells either untreated (Control, made equal to 100%) or treated with EPO alone at concentrations of 100 and 400 U/ml (EPO 100 U and EPO 400 U). Addition of EPO alone at concentrations of 100 and 400 U/ml did not cause, in both cases, any significant change in cell viability (P > 0.05). Data are representative of three separate experiments. EPO 100 U vs. Control; P > 0.05 (°). EPO 400 U vs. Control; P > 0.05 (^).
Effect of H2O2 and erythropoietin on phosphorylation of Akt, glycogen synthase kinase‐3β (GSK‐3β), extracellular signal‐regulated kinases (ERK) and mammalian target of rapamycin (mTOR)
Phosphorylation of Akt increased within 10 min of addition of H2O2 and remained so for up to 2 h (Fig. 2). In cells treated with H2O2 in the presence of 100 U /ml erythropoietin, the phosphorylation status of the molecules studied was not so markedly affected as compared with cells treated with H2O2 only, although there were slight decreases in phosphorylation of ERK1/ERK2, GSK‐3β, Akt and FKHRL1.
Figure 2.

Effect of hydrogen peroxide (H2O2) alone and H2O2 + erythropoietin (EPO) at concentrations of 100 and 400 U/ml on phosphorylation of Akt, GSK‐3β, ERKs and mTOR in HK‐2 cells. Samples of lysed HK‐2 cells treated with H2O2 (2 mm) alone and H2O2 (2 mm) and EPO at concentrations of 100 and 400 U/ml added at the same time, were immunoblotted using the appropriate antibody against the phosphorylated forms of the kinases Akt, GSK‐3β, ERK1/ERK2 and mTOR (‘pAkt,’‘pGSK‐3β’, ‘pERK1/2’ and ‘p mTOR’). There was a clear effect of EPO (at a concentration of 400 U/ml) in decreasing phosphorylation levels of all analysed kinases. This same effect of decreasing phosphorylation status was also observed for the transcription factor FKHRL1. Lane for β‐actin also shows that loading was even for all time points. Data are representative of three separate experiments.
When the cells were incubated with H2O2 and erythropoietin at a concentration of 400 U/ml, decrease in phosphorylation of this kinase was observed at all time points studied (Fig. 2). GSK‐3β showed a similar response, but in the case of GSK‐3β, the decrease in phosphorylation level at 10 min was greater (Fig. 2). It is also noteworthy that levels of phospho‐GSK‐3β after 10 min incubation with H2O2 and erythropoietin at 400 U/ml was less than in control untreated cells (‘0’ in Fig. 2), in contrast to phospho‐Akt that showed increase over control untreated cells.
In the case of ERKs, H2O2 induced an increase in both isoforms, which peaked at 30 min and decreased thereafter. However, again in the presence of erythropoietin at a concentration of 400 U/ml, there was a considerable decrease in phosphorylation of ERKs at all time points studied, with maximum level of phosphorylation at 30 min barely more than the control (0) time point (Fig. 2), and with levels of phospho‐ERK1/ERK2 at 10, 60 and 120 min actually lower than in untreated control cells (‘0’ in Fig. 2).
mTOR is a serine/threonine protein kinase that controls cell growth and proliferation in response to nutrient availability and growth factor stimulation (20). mTOR plays a critical role in regulating translation through phosphorylation of inhibitory proteins (i.e. eIF4E‐BP1) that bind the rate‐limiting eukaryotic initiative factor eIF4E. Cells incubated with H2O2 alone showed an increase in levels of phospho‐mTOR over the period studied, whereas levels of phospho‐mTOR were again lower at all corresponding time points when cells were incubated with H2O2 and erythropoietin at 400 U/ml (Fig. 2).
Finally, Forkhead transcription factor FKHRL1 (also known as FOXO3a) was also dephosphorylated at Thr32 when H2O2‐treated cells were incubated with erythropoietin at a concentration of 400 U/ml (Fig. 2). The Thr32 residue is a site that when phosphorylated causes this transcription factor to be rendered ineffective at regulating certain genes involved in cell death. In cells treated with H2O2 alone, there was a gradual increase in its phosphorylation, peaking after 30 min and still more phosphorylated than in control cells, even after 120 min (Fig. 2). This phosphorylation was as expected since it is a substrate for Akt (21).
Effect of erythropoietin alone
A significant increase in ERK1/ERK2 and Akt phosphorylation was observed after 10 min of incubation with erythropoietin alone at both concentrations (100 and 400 U/ml); an increase in GSK‐3β was also observed after 10 min incubation with 400 U/ml erythropoietin (Fig. 3).
Figure 3.
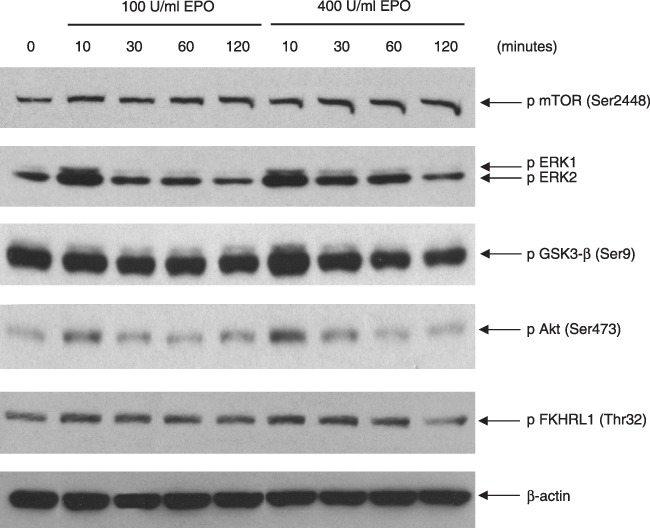
Effect of erythropoietin (EPO) alone at concentrations of 100 and 400 U/ml on phosphorylation of Akt, GSK‐3β, ERK1/ERK2 and mTOR in HK‐2 cells. Samples of lysed HK‐2 cells treated with EPO at concentrations of 100 U/ml and 400 U/ml were immunoblotted using appropriate antibody against phosphorylated forms of the kinases Akt, GSK‐3β, ERK1/ERK2 and mTOR (‘pAkt,’‘pGSK‐3β’, ‘pERK1/2’ and ‘p mTOR’). There was an evident effect of EPO at concentrations of 100 and 400 U/ml on phosphorylation levels of ERKs and Akt, whose phosphorylation increased after 10 min incubation with EPO. Lane for β‐actin shows that loading was even for all time points. Data are representative of three separate experiments.
Discussion
Reactive oxygen species (ROS) have been implicated in pathogenesis of various renal diseases, including ischaemia‐reperfusion and cell death due to toxic acute renal failure (22, 23, 24). ROS include superoxide anion, H2O2, and the hydroxyl radical. Among them, H2O2 is a common mediator and, thus, is widely used to mimic in vitro inflammation and oxidative stress in a large variety of cell lines (25, 26, 27, 28).
We have studied the direct toxic effects of H2O2 in cultures of human renal proximal tubule cells and have evaluated whether erythropoietin overcomes these toxic effects, thereby playing a protective role for human renal proximal tubule cells. Given its properties as a growth factor in promoting proliferation, one would expect that it would help cell proliferation of those cells that have not succumbed to oxidative damage, thereby helping maintain the cell population if not increasing it.
In our in vitro model of oxidative stress, we have observed that erythropoietin, at a concentration of 100 U/ml, did not attenuate cell damage caused by H2O2 on our HK‐2 cells, but at high concentrations (400 U/ml), even exacerbated it. This was surprising since a number of reports have suggested a protective role for erythropoietin in various in vitro and in vivo models of oxidative stress (9, 11). Vesey et al. also observed that concentrations of erythropoietin as high as 400 U/ml were needed to show mitogenic effects (as determined by thymidine incorporation) in human proximal tubule cells subjected to hypoxia followed by reoxygenation; in their model of oxidative stress; however, they were unable to show any effect on lactate dehydrogenase release at any concentration used (up to 400 U/ml) (11).
Some studies have suggested that the protective role of erythropoietin is due to changes in activity of kinases known to be involved in cell survival (4, 9). Indeed, response of a cell to external stress may entail activation of a number of kinases that are components of ‘survival’ pathways that may help the cell resist the particular stress and survive. Thus, we decided to investigate whether there were any changes in such kinases in our experimental model. These kinases include Akt/PKB, mTOR and the ERK subfamily of mitogen‐activated protein kinases; when phosphorylated, these kinases are activated and help the cell to survive and/or proliferate (29, 30, 31). Also, GSK‐3β is inactivated when phosphorylated at Ser9, but when active, it may play a role in cell death and susceptibility to oxidative stress (32, 33). Upon incubation with H2O2, these kinases were phosphorylated as expected since the initial response of cells would be to ‘try’ to upregulate those pathways that would lead to proliferation/survival and decrease in cell death.
The kinetics of ERK phosphorylation upon treatment with H2O2 is transient, peaking at 30 min and then gradually decreasing (Fig. 2); this is in keeping with its role in cell proliferation (31). It has been proposed that ERKs protect mouse proximal tubule cells against H2O2 cytotoxicity (34). Our study shows that in the presence of 400 U/ml erythropoietin, cellular response to H2O2 toxicity in increasing phosphorylation of ERKs was considerably diminished. This observation is more remarkable given that erythropoietin alone (at both concentrations of 100 and 400 U/ml) initially increases levels of phosphorylation of these kinases significantly (Fig. 3).
There are various reports demonstrating activation of Akt in cells upon stimulation with erythropoietin (4, 9, 35). Also in our experimental model, a significant increase in Akt phosphorylation was observed after 10 min incubation with erythropoietin alone at both concentrations of 100 and 400 U/ml; an increase in GSK‐3β was also observed after 10 min incubation with 400 U/ml erythropoietin (Fig. 3). Under the effect of H2O2, phosphorylation of Akt also increased, peaking at 30 min and remaining higher than control levels even after 120 min. Increase in Akt phosphorylation in response to H2O2 has been reported in other cell types (36). However, when H2O2‐treated HK‐2 cells were incubated with erythropoietin at concentration of 400 U/ml, a significant decrease in level of phospho‐Akt was observed, as compared to the effect of H2O2 alone, for all time points of incubation studied (Fig. 2). In fact, for all four kinases, levels of their phosphorylation in cells incubated with H2O2 and 400 U/ml erythropoietin was lower than corresponding time points in cells treated with H2O2 only (Fig. 2).
GSK‐3β is a substrate for Akt (37); thus, it is not surprising that changes in its phosphorylation should follow changes in Akt phosphorylation (Fig. 2). However, decrease in its phosphorylation did not correlate with that of Akt, suggesting phosphorylation by other kinases (37). That phosphorylation of GSK‐3β was less than control untreated cells whereas that of Akt was higher suggests that other kinases, involved in GSK‐3β phosphorylation at Ser9, may also be downregulated under our experimental conditions.
In the case of ERKs and GSK‐3β, levels of phosphorylation upon incubation of HK‐2 cells with H2O2 and erythropoietin at a concentration of 400 U/ml at 10 min was less than that of the control untreated cells (Fig. 2). Given the important role of these kinases in cell survival and proliferation, it is not surprising that a significant decrease in their activity led to decrease in cell viability after 2 h of incubation with H2O2 and erythropoietin at 400 U/ml concentration (Fig. 1a). It has been suggested that there may be cross‐talk between survival pathways that can lead to compensation between them if one or the other is blocked or downregulated (38). For example during reperfusion of rat hearts subjected to a prior period of ischaemia, inhibiting the ERK1/ERK2 pathway at reperfusion resulted in increased activation (phosphorylation) of Akt; conversely, inhibiting the phosphatidyl‐inositol 3‐kinase pathway caused increase in phosphorylation of the ERK1/ERK2 pathway (38). In our experimental model, both pathways were subdued in cells incubated with both H2O2 and erythropoietin at a concentration of 400 U/ml, as compared with cells incubated with H2O2 alone.
The decrease in phosphorylation of these kinases in the presence of H2O2 and high concentration of erythropoietin is difficult to explain. It has been reported that erythropoietin receptors may interact with the common beta receptor (βcR) and it is through this heteromeric complex that erythropoietin exerts its protective effects on the tissues; knockout of the βcR gene abolished this protection (39, 40). The precise interaction between the EpoR, βcR and any other possible component of this complex, however, is not known; thus, it is difficult to predict how such a complex may relay signals under certain conditions.
How important are our observations? Given the frequently observed hyporesponsiveness of patients with anaemia and chronic renal failure to recombinant human erythropoietin treatment due also to inflammation (41), high doses of erythropoietin are sometimes used in uraemic patients. Furthermore, very high doses of erythropoietin are commonly used in patients with neoplastic disease, whose hyporesponsiveness to erythropoietin is almost a rule. Our results suggest that very high doses of erythropoietin may be harmful. Recently, two studies on the use of recombinant human erythropoietin in chronic renal disease have raised the possibility that this treatment is associated with increase in cardiovascular events in those individuals in whom higher haemoglobin concentration is aimed for (42, 43). It has been proposed, however, that it is the dosing regimen itself rather than haemoglobin values that account for the cardiovascular events associated with use of high doses of erythropoietin‐stimulating agents (44). It has also been suggested that action of erythropoietin‐stimulating agents through alternative dose‐dependent pathways may be harmful (45). The results of our study lend support to such suggestions.
In summary, our study demonstrates for the first time that erythropoietin, at high concentrations, can significantly increase cell damage caused by oxidative stress in human renal proximal tubule cells, and that this damage is caused, at least in part, by decrease in activation of important signalling pathways involved in cell survival and proliferation.
Acknowledgements
This research has been supported by the grant ‘Incentivazione alla mobilità di studiosi stranieri e italiani residenti all’estero’ by Ministero dell’Istruzione, dell’Università e della Ricerca (MUIR) (D.M. 26 Gennaio 2001, n. 13), by ‘ERA‐EDTA 2007 Fellowship’ (A. Michael) and partly by the grant of the Italian Ministry of the University and Scientific Research ‘COFIN 2005’ (prot. n. 2005067453_003) to Prof. Michele Andreucci.
References
- 1. Braga M, Gianotti L, Gentilini O, Vignali A, Corizia L, Di Carlo V (1999) Erythropoiesis after therapy with recombinant human erythropoietin: a dose‐response study in anemic cancer surgery patients. Vox Sang. 76, 38–42. [PubMed] [Google Scholar]
- 2. Fisher JW (1998) A quest for erythropoietin over nine decades. Annu. Rev. Pharmacol. Toxicol. 38, 1–20. [DOI] [PubMed] [Google Scholar]
- 3. Anagnostou A, Liu Z, Steiner M, Chin K, Lee ES, Kessimian N, Noguchi CT (1994) Erythropoietin receptor mRNA expression in human endothelial cells. Proc. Natl. Acad. Sci. USA 91, 3974–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chong ZZ, Kang JQ, Maiese K (2002) Angiogenesis and plasticity: role of erythropoietin in vascular systems. J. Hematother. Stem Cell Res. 11, 863–871. [DOI] [PubMed] [Google Scholar]
- 5. Westenfelder C, Biddle DL, Baranowski RL (1999) Human, rat, and mouse kidney cells express functional erythropoietin receptors. Kidney Int. 55, 808–820. [DOI] [PubMed] [Google Scholar]
- 6. Chong ZZ, Kang JQ, Maiese K (2003) Erythropoietin: cytoprotection in vascular and neuronal cells. Curr. Drug Targets Cardiovasc. Haematol. Disord. 3, 141–154. [DOI] [PubMed] [Google Scholar]
- 7. Sasaki R (2003) Pleiotropic functions of erythropoietin. Intern. Med. 42, 142–149. [DOI] [PubMed] [Google Scholar]
- 8. Bagnis C, Beaufils H, Jacquiaud C, Adabra Y, Jouanneau C, Le Nahour G, Jaudon MC, Bourbouze R, Jacobs C, Deray G (2001) Erythropoietin enhances recovery after cisplatin‐induced acute renal failure in the rat. Nephrol. Dial. Transplant. 16, 932–938. [DOI] [PubMed] [Google Scholar]
- 9. Sharples EJ, Patel N, Brown P, Stewart K, Mota‐Philipe H, Sheaff M, Kieswich J, Allen D, Harwood S, Raftery M, Thiemermann C, Yaqoob MM (2004) Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia‐reperfusion. J. Am. Soc. Nephrol. 15, 2115–2124. [DOI] [PubMed] [Google Scholar]
- 10. Spandou E, Tsouchnikas I, Karkavelas G, Dounousi E, Simeonidou C, Guiba‐Tziampiri O, Tsakiris D (2006) Erythropoietin attenuates renal injury in experimental acute renal failure ischaemic/reperfusion model. Nephrol. Dial. Transplant. 21, 330–336. [DOI] [PubMed] [Google Scholar]
- 11. Vesey DA, Cheung C, Pat B, Endre Z, Gobe G, Johnson DW (2004) Erythropoietin protects against ischaemic acute renal injury. Nephrol. Dial. Transplant. 19, 348–355. [DOI] [PubMed] [Google Scholar]
- 12. Yang CW, Li C, Jung JY, Shin SJ, Choi BS, Lim SW, Sun BK, Kim YS, Kim J, Chang YS, Bang BK (2003) Preconditioning with erythropoietin protects against subsequent ischemia‐reperfusion injury in rat kidney. FASEB J. 17, 1754–1755. [DOI] [PubMed] [Google Scholar]
- 13. Sharples EJ, Thiemermann C, Yaqoob MM (2005) Mechanisms of disease: cell death in acute renal failure and emerging evidence for a protective role of erythropoietin. Nat. Clin. Pract. Nephrol. 1, 87–97. [DOI] [PubMed] [Google Scholar]
- 14. Johnson KJ, Weinberg JM (1993) Postischemic renal injury due to oxygen radicals. Curr. Opin. Nephrol. Hypertens. 2, 625–635. [DOI] [PubMed] [Google Scholar]
- 15. Rampino T, Collesi C, Gregorini M, Maggio M, Soccio G, Guallini P, Dal Canton A (2002) Macrophage‐stimulating protein is produced by tubular cells and activates mesangial cells. J. Am. Soc. Nephrol. 13, 2649–2657. [DOI] [PubMed] [Google Scholar]
- 16. Andreucci M, Fuiano G, Presta P, Esposito P, Faga T, Bisesti V, Procino A, Altieri V, Tozzo C, Memoli B, Michael A (2006) Radiocontrast media cause dephosphorylation of Akt and downstream signaling targets in human renal proximal tubular cells. Biochem. Pharmacol. 72, 1334–1342. [DOI] [PubMed] [Google Scholar]
- 17. Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63. [DOI] [PubMed] [Google Scholar]
- 18. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 19. Andreucci M, Michael A, Kramers C, Park KM, Chen A, Matthaeus T, Alessandrini A, Haq S, Force T, Bonventre JV (2003) Renal ischemia/reperfusion and ATP depletion/repletion in LLC‐PK1 cells result in phosphorylation of FKHR and FKHRL1. Kidney Int. 64, 1189–1198. [DOI] [PubMed] [Google Scholar]
- 20. Richter JD, Sonenberg N (2005) Regulation of cap‐dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480. [DOI] [PubMed] [Google Scholar]
- 21. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. [DOI] [PubMed] [Google Scholar]
- 22. Baek SM, Kwon CH, Kim JH, Woo JS, Jung JS, Kim YK (2003) Differential roles of hydrogen peroxide and hydroxyl radical in cisplatin‐induced cell death in renal proximal tubular epithelial cells. J. Lab. Clin. Med. 142, 178–186. [DOI] [PubMed] [Google Scholar]
- 23. Greene EL, Paller MS (1991) Oxygen free radicals in acute renal failure. Miner. Electrolyte Metab. 17, 124–132. [PubMed] [Google Scholar]
- 24. Paller MS (1992) Free radical‐mediated postischemic injury in renal transplantation. Ren. Fail. 14, 257–260. [DOI] [PubMed] [Google Scholar]
- 25. Fatokun AA, Stone TW, Smith RA (2007) Hydrogen peroxide mediates damage by xanthine and xanthine oxidase in cerebellar granule neuronal cultures. Neurosci. Lett. 416, 34–38. [DOI] [PubMed] [Google Scholar]
- 26. Nemoto T, Kawakami S, Yamashita F, Hashida M (2007) Efficient protection by cationized catalase against H2O2 injury in primary cultured alveolar epithelial cells. J. Control. Release 121, 74–80. [DOI] [PubMed] [Google Scholar]
- 27. Sugino N (2007) The role of oxygen radical‐mediated signaling pathways in endometrial function. Placenta 28(Suppl. A), S133–S136. [DOI] [PubMed] [Google Scholar]
- 28. Zhang W, Wang M, Xie HY, Zhou L, Meng XQ, Shi J, Zheng S (2007) Role of reactive oxygen species in mediating hepatic ischemia‐reperfusion injury and its therapeutic applications in liver transplantation. Transplant. Proc. 39, 1332–1337. [DOI] [PubMed] [Google Scholar]
- 29. Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev. 13, 2905–2927. [DOI] [PubMed] [Google Scholar]
- 30. Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945. [DOI] [PubMed] [Google Scholar]
- 31. Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal‐regulated kinase activation. Cell 80, 179–185. [DOI] [PubMed] [Google Scholar]
- 32. Pap M, Cooper GM (1998) Role of glycogen synthase kinase‐3 in the phosphatidylinositol 3‐Kinase/Akt cell survival pathway. J. Biol. Chem. 273, 19929–19932. [DOI] [PubMed] [Google Scholar]
- 33. Salazar M, Rojo AI, Velasco D, De Sagarra RM, Cuadrado A (2006) Glycogen synthase kinase‐3β inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 281, 14841–14851. [DOI] [PubMed] [Google Scholar]
- 34. Arany I, Megyesi JK, Kaneto H, Tanaka S, Safirstein RL (2004) Activation of ERK or inhibition of JNK ameliorates H2O2 cytotoxicity in mouse renal proximal tubule cells. Kidney Int. 65, 1231–1239. [DOI] [PubMed] [Google Scholar]
- 35. Kashii Y, Uchida M, Kirito K, Tanaka M, Nishijima K, Toshima M, Ando T, Koizumi K, Endoh T, Sawada K, Momoi M, Miura Y, Ozawa K, Komatsu N (2000) A member of Forkhead family transcription factor, FKHRL1, is one of the downstream molecules of phosphatidylinositol 3‐kinase‐Akt activation pathway in erythropoietin signal transduction. Blood 96, 941–949. [PubMed] [Google Scholar]
- 36. Radisavljevic ZM, González‐Flecha B (2004) TOR kinase and ran are downstream from PI3K/Akt in H2O2‐induced mitosis. J. Cell Biochem. 91, 1293–1300. [DOI] [PubMed] [Google Scholar]
- 37. Doble BW, Woodgett JR (2003) GSK‐3: tricks of the trade for a multi‐tasking kinase. J. Cell Sci. 116, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hausenloy DJ, Mocanu MM, Yellon DM (2004) Cross‐talk between the survival kinases during early reperfusion: its contribution to ischemic preconditioning. Cardiovasc. Res. 63, 305–312. [DOI] [PubMed] [Google Scholar]
- 39. Brines M, Cerami A (2006) Discovering erythropoietin's extra‐hematopoietic functions: biology and clinical promise. Kidney Int. 70, 246–250. [DOI] [PubMed] [Google Scholar]
- 40. Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, Latini R, Xie QW, Smart J, Su‐Rick CJ, Pobre E, Diaz D, Gomez D, Hand C, Coleman T, Cerami A (2004) Erythropoietin mediates tissue protection through an erythropoietin and common beta‐subunit heteroreceptor. Proc. Natl. Acad. Sci. USA 101, 14907–14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Drüeke T (2001) Hyporesponsiveness to recombinant human erythropoietin. Nephrol. Dial. Transplant. 16(Suppl. 7), 225–228. [DOI] [PubMed] [Google Scholar]
- 42. Drüeke TB, Locatelli F, Clyne N, Eckardt KU, Macdougall IC, Tsakiris D, Burger HU, Scherhag A, CREATE Investigators (2006) Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N. Engl. J. Med. 355, 2071–2084. [DOI] [PubMed] [Google Scholar]
- 43. Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, Reddan D, CHOIR Investigators (2006) Correction of anemia with epoetin alfa in chronic kidney disease. N. Engl. J. Med. 355, 2085–2098. [DOI] [PubMed] [Google Scholar]
- 44. Levin A (2007) Understanding recent haemoglobin trials in CKD: methods and lesson learned from CREATE and CHOIR. Nephrol. Dial. Transplant. 22, 309–312. [DOI] [PubMed] [Google Scholar]
- 45. Strippoli GF, Tognoni G, Navaneethan SD, Nicolucci A, Craig JC (2007) Haemoglobin targets: we were wrong, time to move on. Lancet 369, 346–350. [DOI] [PubMed] [Google Scholar]