

Graphical abstract

Keywords: Systems Bioscience, OMICs, Pathogenicity, Transmission, Adaptation, Data Mining, Big Data, Machine Learning, Association Mining, Host-Pathogen, Interaction, Infectious Disease, Vector-Borne Disease
Abstract
Infectious diseases, including vector-borne diseases transmitted by arthropods, are a leading cause of morbidity and mortality worldwide. In the era of big data, addressing broad-scale, fundamental questions regarding the complex dynamics of these diseases will increasingly require the integration of diverse datasets to produce new biological knowledge. This review provides a current snapshot of the systematic assessment of the relationships between microbial pathogens, arthropod vectors and mammalian hosts using data mining and machine learning. We employ PRISMA to identify 32 key papers relevant to this topic. Our analysis shows an increasing use of data mining and machine learning tasks and techniques, including prediction, classification, clustering, association rules mining, and deep learning, over the last decade. However, it also reveals a number of critical challenges in applying these to the study of vector-host-pathogen interactions at various systems biology levels. Here, relevant studies, current limitations and future directions are discussed. Furthermore, the quality of data in relevant papers was assessed using the FAIR (Findable, Accessible, Interoperable, Reusable) compliance criteria to evaluate and encourage reproducibility and shareability of research outcomes. Although shortcomings in their application remain, data mining and machine learning have significant potential to break new ground in understanding fundamental aspects of vector-host-pathogen relationships and their application in this field should be encouraged. In particular, while predictive modeling, feature engineering and supervised machine learning are already being used in the field, other data mining and machine learning methods such as deep learning and association rules analysis lag behind and should be implemented in combination with established methods to accelerate hypothesis and knowledge generation in the domain.
1. Introduction
1.1. Infectious diseases and vector-borne disease transmission
Infectious diseases have plagued humankind since the dawn of civilization. The control and prevention of such diseases requires a detailed understanding of the intricate interactions between microbial pathogens, their human hosts, and the environments in which they are transmitted. A subset of infectious diseases that are transmitted by arthropods (e.g. ticks, mosquitoes), known as vector-borne diseases, present an added layer of complexity. Vector-borne infections are on the rise globally and include a number of deadly parasitic (e.g. malaria), viral (e.g. Dengue virus), and bacterial (e.g. Lyme disease) diseases with high burdens[1], [2]. The interactions that drive the transmission and progression of these diseases are complicated by the fact that the causative microorganisms not only interphase with human hosts, but also require prolonged contact with an invertebrate vector.
The dynamics of vector-borne diseases are influenced by a number of parameters. Vector competence, which is the innate ability of an arthropod to acquire, maintain, and disseminate a pathogen to a vertebrate host, depends on intrinsic factors such as vector immunity, general feeding behavior, and the microbiome[3]. Meanwhile, intrinsic microbial genetics also factor into the ability of pathogens to adapt to and colonize specific arthropod vectors. On the other hand, vectorial capacity, which is a measure of the efficiency of transmission by a vector in nature, can be impacted by extrinsic and environmental parameters. These include temperature, the availability of pathogen reservoirs and vector habitat, vector lifespan, vector biting rate, and more[4], [5], [6], [7], [8]. Notably, several of the above factors can be manipulated by the microbial pathogens, further confounding the web of interactions[9].
From the human host perspective, the progression of disease is driven not only by intrinsic microbial factors, but also the accompanying immune response. Moreover, there is evidence to suggest that the process of adaptation to particular arthropod vectors can influence microbial pathogenicity in vertebrates. The latter may occur either through microbial genome degradation as a result of the adaptation process and/or as a result of changes in microbial transcriptomes driven by the arthropod host[10], [11], [12].
Although our understanding of the dynamics of infectious diseases, including vector-borne diseases, continues to advance, many unknowns remain. In particular, while much progress has been made towards mechanistic understanding of specific interactions (e.g. the transmission of malaria parasites by Anopheles mosquitoes), fundamental questions with broad, generalizable implications remain difficult to address. For instance, how do pathogens adapt to transmission by specific vectors? Are there molecular signatures to this adaptation? Can we predict the transmissibility and/or pathogenicity of newly identified, potentially vector-borne microbes? Addressing these intriguing questions requires not only large amounts of diverse data from different vector-host-pathogen relationships, but also the ability to integrate this biological information into new knowledge.
Today, there are a growing amount of big data pertaining to vector-borne diseases available in the literature, and structured public repositories for these types of data, such as VectorBase (https://www.vectorbase.org/), are expanding to include more species of interest and data modalities. In addition, data mining and machine learning approaches are already being applied to the study of many infectious diseases in ways that could be adapted to understand vector borne agents. As such, the goal of this review is to explore how data mining and machine learning may be, and in some cases already are, applied to improve understanding of complex vector-host-pathogen relationships.
1.2. Systems driven bioscience and biomedicine investigation
The fast growth of high-throughput technologies at each level of biological organization (i.e. compound, gene, transcript, protein, cell) has provided the bioscience and biomedicine communities with many extensive datasets, leading to several computational challenges, including the systemic integration of different dataset modalities into a reliable and reproducible investigational framework. Systems bioscience and biomedicine are leading a new generation of discovery leveraging the interconnectivity and interdependence of biological processes. Each biological process, such as protein production leading a pathogenic effect, involves additional mechanisms at other systems levels, such as gene expression through Gene Regulatory Networks (GRN), as an example. The integration of datasets from these different biosystems levels poses several data science challenges, including the curse of dimensionality and missing datasets. Leveraging data mining (DM) and machine learning (ML) techniques and methods to tackle these challenges and advance integrative understanding of biological response mechanisms provides exciting new research opportunities. Before discussing the application of DM and ML to vector-host-pathogen relationships, a short overview of DM, knowledge discovery and ML is given for readers with little background in these domains.
1.3. Data mining, machine learning and knowledge discovery
Data mining is a process that applies analysis, management and summarization of data from a large pool of information to obtain insight and discover unknown patterns or relationships in the dataset[13]. It involves six steps according to the Cross-Industry Standard Process for Data Mining (CRISP- DM) of 1996. The CRISP-DM protocol is based on performing procedurals that are universally applicable for data mining methodologies[13]. Knowledge Discovery in Databases (KDD), on the other hand, involves data selection, preprocessing, transformation, mining, interpretation or evaluation, and knowledge discovery[13]. Thus, KDD encompasses data mining as one of the key steps.
Machine Learning can be considered a branch of artificial intelligence that uses a general concept of inference to extract (learn) the solution to a problem from data samples[14]. There are always two phases of learning in machine learning. First is the estimation of the unknown dependencies in a system from a given dataset, and second is the use of estimated dependencies to predict new outputs from the system[14]. Generally, machine learning is divided into two major types, supervised and unsupervised learning.
1.3.1. Supervised learning
In supervised learning, training datasets are labeled, and the machine learns from the labels to assign unknown datasets a label upon encounter. The result is an input dataset mapped to a correct output. Therefore, the term supervised learning refers to supervision by a labeled training dataset to map the input data to a desired output. Supervised learning is further divided into tasks such as regression and classification in which different algorithms, including: Multiple Linear Regression (MLR), Logistic Regression (LR), Support Vector Machine (SVM), Random Forest (RF), Artificial Neural Network (ANN), Decision Tree (DT), and Bayesian Network (BN), are applied to build a model.
1.3.2. Unsupervised learning
In unsupervised learning, there are no labels for the machine to learn from. Hence, it is up to the model to discover patterns in input datasets and group them based on certain rules or associations. Furthermore, unsupervised learning can also be divided into tasks, for example, clustering to which Principal Component Analysis (PCA), Independent Component Analysis (ICA), and k-Means (KM) are applied to make models.
1.3.3. Feature engineering
The success of data science workflows relies on feature engineering. A feature is the basic variable used to capture and represent the knowledge in data for knowledge discovery or machine learning development (e.g. the infection count is 3 on May 2020 in USA, here date and location can be considered as features to analyze and predict the infection count progress/decline). As an additional example, if we assume in a natural setting that the genomic landscape of a given vector or pathogen contains a consistent pattern that can help predict transmission dynamics, a list of variables to measure the genomic landscape should be chosen. Current state of the art tools to measure genomic variables include global gene expression profiling and polymorphism typing. From these two data modalities, we can capture two different feature types: (1) gene expression from vector and pathogen before and after they interact, (2) various genomic markers from the vector and pathogen. In this context, the feature list will include the expressed genes (e.g. 30 k) and the genotypic markers (e.g. 4). The high number of features in this list makes prediction and pattern identification difficult and sometime less meaningful. Furthermore, it stands to reason that all genes from the vector cannot be involved in regulating transmission. In this case, feature engineering will be needed to identify and select/reduce (feature selection, feature reduction) this list of features to the minimal number needed to make relevant predictions. Several approaches have been implemented for diverse data modalities in order to provide data scientist with domain-specific relevant feature identification.
1.4. Strengths and weaknesses of applying data mining and machine learning
DM & ML applications are revolutionizing the field of infectious diseases by contributing to early outbreak detection, surveillance, pathogenicity prediction, diagnostic tools, and more. However, these applications have both strengths and weaknesses. A particular strength is that systems bioscience is producing an abundance of data that machine learning and data mining methods can transform into novel knowledge. However, these omics data come in very heterogenic forms and modalities, creating huge challenges for their use, including the “curse of dimensionality” that pertains to big data. Additionally, problems with missing data, dataset reproducibility, rarity and class imbalance, and big data scalability are among many other challenges. These issues become constraints and cause problems for most machine learning tasks when applied to real-world approaches such as the development of clinical solutions. In turn, these constraints result in most models remaining research tools for non-clinical and academic settings that are useful only in limited ways. Nonetheless, this trend is changing and, in this review, most of the studies discussed addressed important real-world problems.
1.5. Aims of present review
In other bioscience domains, reviews are available about these strengths and challenges of machine learning applications. The two types of machine learning can be used to achieve diverse tasks depending on the discovery goal, application, and the domain of interest. In this review, we will focus on five tasks to tackle several challenges involved in understanding vector-host-pathogen interactions, while assessing the strengths and weakness of machine learning applications in this domain. These tasks include: (1) prediction - to assess the continuous trends or deterministic responses in each relationship; (2) classification – to identify meaningful classes governing the interactions and the responses of each component in the relationship; (3) clustering – to detect functional patterns interesting to the interaction ecosystem; (4) association rules mining / hypothesis generation – to provide formal validation of existing hypotheses, propose new ones, and evaluate their pertinence regarding the vector-host-pathogen relationship; and (5) deep learning – to provide a multi-level organization of different dataset modalities involved in the vector-host-pathogen systemic relationship (Fig. 1).
Fig. 1.

Overview of Systems Bioscience (a) of vector-host-pathogen relationships (b) of Data Mining and Machine Learning processes (c) emphasizing the information flow and interwinding nature of the subject matter in relationship to tools used in the review papers.
2. Methods (PRISMA)
2.1. PRISMA overview
PRISMA workflow was adopted to design our review[15] (Fig. 2). PRISMA is an objective design method for literature reviews in bioscience and biomedicine. PRISMA relies on a rigorous process to improve the relevance of the selected review papers and the reproducibility of the review process. Our design allowed us to query 3 publication databases (PubMed, WoS, Mendeley), to our research questions (Table 1), and yielded 4408 papers related to our query (Table 2). An additional 4 articles were added from other journals and collections not available in the initial databases (e.g. PLOS). After duplicate removal, we obtained 972 unique papers. An independent screening of papers was done by DA, and EG. Following manual screening, we retained 530 qualified papers. For example, a paper that was retrieved because it mentions “clustering”, referring to a group of objects (e.g. population group) and not DM or ML methods was excluded from our list. A deeper assessment followed by group discussion allow us to select 53 papers for further review. During the review process, 32 were found relevant to our problem of interest and were approved by DA, EG, and JP (collection is publicly available at: https://www.ncbi.nlm.nih.gov/sites/myncbi/etienne.gnimpieba z..1/collections/59430212/public/). Although not all of these dealt explicitly with vector-borne pathogens, those that did not had clear implications for the study of vector-borne pathogens.
Fig. 2.
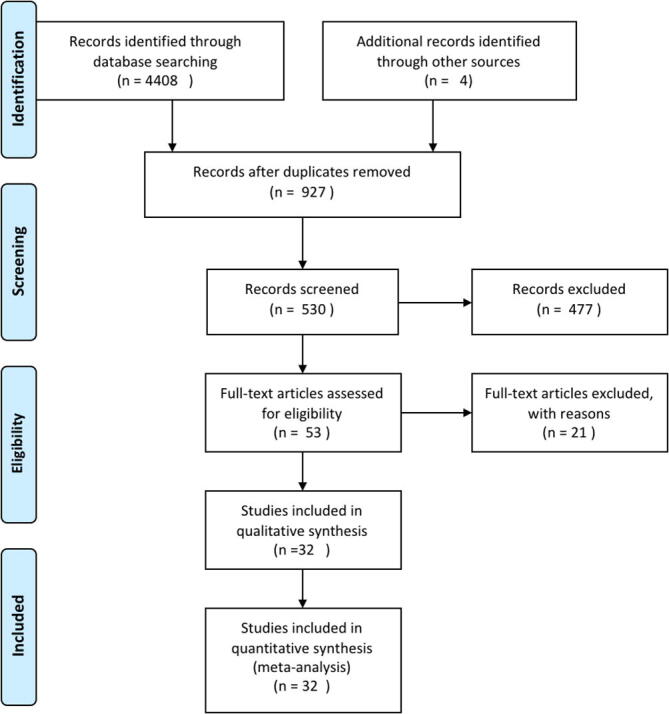
Search workflow (PRISMA) used in article searching, retrieving, processing and inclusion/exclusion decision making.
Table 1.
Research questions mapped to DM and ML tasks. The questions are denoted as Q0 to Q8. Q0 is the main research question involving the overall relationship, Q1 to Q8 are applied to the subtle questions in the relationship with more attention to Q1-Q3 in this review. Tasks are mapped to questions (e.g.Q0-1), which means Vector-Host-Pathogen Interaction (Q0) and use of prediction (1) tasks to answer the question (Q0).
| Query-ID | Research Questions | Prediction (1) | Classification (2) | Clustering (3) | Association Rules (4) | Deep learning (5) |
|---|---|---|---|---|---|---|
| Q0 | To what degree DM & ML have been applied to assess Vector-Host-Pathogen Interactions? | Q0-1 | Q0-2 | Q0-3 | Q0-4 | Q0-5 |
| Q1 | To what degree DM & ML have been applied to assess Pathogen-Vector adaptation? | Q1-1 | Q1-2 | Q1-3 | Q1-4 | Q1-5 |
| Q2 | To what degree DM & ML have been applied to assess Vector-Host transmission? | Q2-1 | Q2-2 | Q2-3 | Q2-4 | Q2-5 |
| Q3 | To what degree DM & ML have been applied to assess Pathogen- Host pathogenicity? | Q3-1 | Q3-2 | Q3-3 | Q3-4 | Q3-5 |
| Q4 | To what degree DM & ML have been applied to assess Pathogen Vector/Host immunity | Q4-1 | Q4-2 | Q4-3 | Q4-4 | Q4-5 |
| Q5 | To what degree DM & ML have been applied to assess Pathogen Vector manipulation of transmission | Q5-1 | Q5-2 | Q5-3 | Q5-4 | Q5-5 |
| Q6 | To what degree DM & ML have been applied to assess Pathogen Vector/arthropod effects on pathogenicity | Q6-1 | Q6-2 | Q6-3 | Q6-4 | Q6-5 |
| Q7 | To what degree DM & ML have been applied to assess Pathogen Vector Reservoir/Host effects | Q7-1 | Q7-2 | Q7-3 | Q7-4 | Q7-5 |
| Q8 | To what degree DM & ML have been applied to assess Pathogen Vector environmental effects | Q8-1 | Q8-2 | Q8-3 | Q8-4 | Q8-5 |
Table 2.
Query formulation and search result count per database. Research questions denoted Q0 to Q8 and tasks denoted 1 to 5 in table 1 were formulated into searchable formatted queries and searched against PubMed, Web of Science, and Mendeley to retrieve the papers of interest. The results are recorded in this table, for example, Q1-3 means question denoted Q1 and task denoted 3 combined. In this example, the search resulted in 70 PubMed, 10 Web of science and 0 Mendeley papers retrieved.
| Query-ID | Query | PubMed | Web of Science | Mendeley (SCOPUS)* |
|---|---|---|---|---|
| Q0 | Pathogen Vector Host AND (Machine Learning OR Data Mining) | 60 | 39 | 18 |
| Q1 | Pathogen Vector Adaptation AND (Machine Learning OR Data Mining) | 12 | 1 | 0 |
| Q2 | Vector Host Transmission AND (Machine Learning OR Data Mining) | 12 | 18 | 9 |
| Q3 | Pathogen Host Pathogenicity AND (Machine Learning OR Data Mining) | 314 | 20 | 11 |
| Q0-1 | Pathogen Vector Host AND Prediction | 251 | 185 | 39 |
| Q0-2 | Pathogen Vector Host interaction AND Classification AND (Learning OR Mining) | 10 | 12 | 2 |
| Q0-3 | Pathogen Vector Host interaction AND Clustering | 269 | 23 | 1 |
| Q0-4 | Pathogen Vector Host interaction AND Association Rule | 3 | 0 | 25 |
| Q0-5 | Pathogen Vector Host interaction AND Deep Learning | 2 | 2 | 16 |
| Q1-1 | Pathogen Vector Adaptation AND Prediction | 128 | 17 | 9 |
| Q1-2 | Pathogen Vector Adaptation AND Classification AND (Learning OR Mining) | 3 | 0 | 0 |
| Q1-3 | Pathogen Vector Adaptation AND Clustering | 70 | 10 | 0 |
| Q1-4 | Pathogen Vector Adaptation AND Association Rule | 0 | 0 | 0 |
| Q1-5 | Pathogen Vector Adaptation AND Deep Learning | 0 | 0 | 0 |
| Q2-1 | Vector Host Transmission AND Prediction | 558 | 240 | 75 |
| Q2-2 | Vector Host Transmission AND Classification AND (Learning OR Mining) | 9 | 4 | 4 |
| Q2-3 | Vector Host Transmission AND Clustering | 212 | 230 | 31 |
| Q2-4 | Vector Host Transmission AND Association rule | 6 | 5 | 3 |
| Q2-5 | Vector Host Transmission AND Deep learning | 0 | 1 | 1 |
| Q3-1 | Pathogen Host Pathogenicity AND Prediction | 219 | 161 | 58 |
| Q3-2 | Pathogen Host Pathogenicity AND Classification AND (Learning OR Mining) | 147 | 4 | 0 |
| Q3-3 | Pathogen Host Pathogenicity AND Clustering | 54 | 550 | 0 |
| Q3-4 | Pathogen Vector Pathogenicity Association Rule | 18 | 0 | 12 |
| Q3-5 | Pathogen Host Pathogenicity AND Deep learning | 11 | 3 | 0 |
| Q4 | Host Pathogen Immunity AND (Machine Learning OR Data Mining) | 112 | 32 | 9 |
| Q5 | Pathogen Vector Manipulation of Transmission AND (Machine Learning OR Data Mining) | 1 | 1 | 0 |
| Q6 | Pathogen Arthropod Pathogenicity AND (Machine Learning OR Data Mining) | 32 | 0 | 0 |
| Q7 | Reservoir Host Adaptation Transmission AND (Machine Learning OR Data Mining) | 1 | 0 | 0 |
| Q8 | Environmental Pathogen Vector Host AND (Machine Learning OR Data Mining) | 7 | 4 | 2 |
| 2521 | 1562 | 325 |
Mendeley queries was augmented with (“”) for source formatting requirements.
2.2. Identification of research questions
In this review, we were inspired by the organization of a similar article on the investigation of air pollution using machine learning and data mining [16]. Here, we undertake a rigorous effort to identify studies that used machine learning (ML) techniques and data mining (DM) methods to assess the complex web of vector-host-pathogen interactions. From the main research question, which we denoted as Q0 – “How to assess vector-host-pathogen relationships using Data Mining and Machine Learning?”, 8 subsequent research questions were formulated to integrate the systemic aspects of the problem (Fig. 1a, b). These research questions were mapped to 5 DM and ML tasks to better understand the systemic depth of ML and DM involvement in this ecosystem during the data mining and knowledge discovery processes (Fig. 1c, Table 1). Focusing on the 4 most pertinent, well-defined questions (Q0, Q1, Q2, Q3), we generated 20 tasks dependent questions, bringing our research question list to a total of 29 questions at 3 systemic levels. These 29 research questions were used to build our queries for database and journal searches. Based on our research questions, we crafted 8 main queries and 20 nested queries to the first 3 questions and machine learning (ML) and data mining (DM) tasks. For example, machine learning (ML) and classification form a query of “vector-host-pathogen interaction AND Classification AND (Learning OR Mining).” A full table of queries is shown below (Table 2).
2.3. Search process design and selection
To perform the actual search process, queries in table 2 were entered into the databases of PubMed, Mendeley (SCOPUS), and Web of Science (Jan/1/ 2020) one at a time to obtain journal articles, conference papers, and other publications relevant to our research questions. (Table 1). With caution not to leave out any important articles, no date or time period constraints were applied. After a successful search, the resulting articles were manually inspected using blinded manual curation to assess their relevance to vector-host-pathogen relationships and use of data mining (DM) and machine learning (ML) techniques. For example, Q0, was manually inspected to exclude articles that did not contain application of machine learning (ML) or data mining (DM) methods on vector-host-pathogen interactions or that indirectly addressed the mentioned subject. Articles or studies that involved simple statistical analyses only were excluded. Also, manual curation was done to identify and merge overlaps resulting from searches of different databases. For instance, if articles were already indexed by PubMed, they were merged with articles from PLOS to consolidate the duplication (Table 2.).
2.4. Data extraction and synthesis
After the raw data (articles) were collected from databases, the following information was extracted from each paper: (a) the study objective summary (b) the findings summary (c) the source and full reference (d) the vector-host-pathogen relationship of interest in the study (e) the machine learning, and data mining methods used to address the study objective, and (f) the data science tasks and systems biology methods leveraged. Furthermore, these annotations were tabulated and used to perform data synthesis to elucidate trends in the research landscape employing machine learning and data mining techniques in vector-host-pathogen relationship studies. The raw datasets from our paper readings were captured using Google Forms and preprocessed and analyzed using Microsoft Excel 2020 (Supplementary file S1: 10.6084/m9.figshare.12053637). The review collection is available at: https://www.ncbi.nlm.nih.gov/sites/myncbi/etienne.gnimpieba z..1/collections/59430212/public/. The raw dataset is freely available in Figshare (10.6084/m9.figshare.12053637) [17].
3. Results
3.1. Summary statistics
PRISMA results overview: The summary statistics provide the numbers of articles obtained from our searches of PubMed, Mendeley and Web of Science databases. The procedures in which an article or publication were excluded or included, and numbers are provided in the PRISMA flow chart in Fig. 2. From the searched databases, the first initial search generated 2521, 325, and 1562 articles from PubMed, Mendeley and Web of Science, respectively (Table 1). In addition to these, 4 articles were suggested and obtained from other sources of publication (home journals) during the review process. After blinded screening and eligibility curation, only 32 articles were included in this review (Fig. 3, Table 3.). As Fig. 3a shows, the distribution of papers increases from past to present (2012 to 2020), illustrating an increasing appreciation of ML and DM in the field of vector-host-pathogen interactions. When considering the subtopics addressed by our research questions posed above, pathogenicity (Q3) leads in the application of machine learning with 27% of papers addressing this issue, followed by transmission (Q2) with 24% and adaptation (Q1) with 17%. The 5 other questions lagged behind. However, all of the 8 questions (Q1 - Q8) were covered to some extent, showing a diverse use of data mining and machine learning in the domain (Fig. 3b).
Fig. 3.

Paper count per year showing (a) a trend increase in MD & ML application in the study of vector-host-pathogen relationships and (b) distribution across research questions and applications of DM & ML.
Table 3.
Overview of key papers involved in the research questions. The 32 papers are listed with their PubMed ID, a short description of the paper objective or goal (s), first authors’ name, ML methods, research questions in Table 1, method accuracy and validations methods, as well as Data Mining key features if available.
| PubMed ID [Ref] | Paper Short Objective | Author | Year | ML Task | ML Method** | Research Problem | SB Levels | Accuracy (%) | Validation Method | DM - Key Features |
|---|---|---|---|---|---|---|---|---|---|---|
| 29,263,245 [56] | Use genomics clustering to identify genomics information transfer during infection | Jani M et al. | 2017 | Clustering, Classification | Q1, Q6 | Epigenomics, Genomics | multiple method comparison | reported genomics island(GI) coordinates and annotation | ||
| 30,744,806 [35] | HRMAn (Host Response to Microbe Analysis), An image analysis program to assess host protein recruitment within general cellular pathogen defense that is based on machine learning algorithms and deep learning. | Fisch D et al | 2019 | Classification, DL | DT, GBT, RF, CNN | Q1, Q3 | Genomics, Transcriptomics, Cellomics (images) | 99.5, 92.1, 69.9 | expert-based cross-validation, Cohen’s kappa values | Nuclei labels, Pathogen labels, Cell labels and vacuoles 1---n |
| 30,579,059 [28] | A potential use of machine learning in prediction of health endpoints in STEC, and risk assessment of microbial infection using whole genome sequencing data | Njage PMK, et al | 2019 | Prediction | RF, SVM-RLk, GBK-LB | Q1, Q2, Q3 | Genomics, Transcriptomics, Proteomics | accuracy of 0.75 (95% CI: 0.60, 0.86), and (Kappa = 0.72). | 10-fold cross validation, bootstrap subsamples | Accessory genes in amino acid sequences |
| 29,448,923 [20] | Distinguishing vector from non-vector to mitigate risk of tick-borne disease transmission | Yang LH, et al | 2018 | Prediction, AR | GBR | Q1, Q2,Q3, Q5, Q6 | Population, Phenomics | 91% accuracy, ID 14 species with 80% probability of causing disease. | 10-fold cross-validation | anatomy, life history metrics, and biomes |
| 30,871,681 [30] | Predict protective antigens or epitopes using data features extracted from protein sequences (Machine Learning: Random Forest, Recursive Feature Elimination (RFE), and minimum redundancy maximum relevance (mRMR)) | Rahman MS, et al | 2019 | Prediction | RF | Q1, Q3, Q4 | Genomics, Transcriptomics | accuracy - sensitivity / specificity values of 78.04%, 78.99% and 77.08% − 10-fold cross-validation testing. In jackknife cross-validation, 80.03%, 80.90% and 79.16% respectively. | accuracy; sensitivity and specificity, 10-fold cross-validation | Relevant features |
| 31,206,514 [43] | Use of ML to identify high risk snail habitats as function of Schistosoma japonium infection control and elimination | Xia C, et al | 2019 | Prediction | RF, CTA, GB | Q1, Q2, Q3, Q7, Q8 | Population, Other SB level | RF Model (AUC = 0.96), ensemble model (AUC = 0.89, sensitivity − 0.79 - specificity = 0.82). | 10 Fold Cross-Validation | climatic, environment and economic factors (very low, low, moderate, high and very high) |
| 29,738,521 [29] | Random forest classifier to identify Salmonella enterica strains associated with extraintestinal disease using measured burden of atypical mutations in protein coding genes across independently evolved lineages. | Wheeler N. E, et al | 2018 | Classification | TD, RF | Q1, Q3, Q4 | Genomics, Transcriptomics | 100% out-of-bag classification accuracy | out-of-bag classification accuracy | atypical mutations in protein coding genes |
| 29,760,095 [22] | Predictive model based on machine learning algorithms to reliably determine malaria infection status in humans based on volatile biomarkers | De Moraes CM, et al | 2018 | Prediction, Classification | RF, RRF, AdaBoost | Q1, Q2, Q3, Q5, Q6 | Proteomics, Metabolomics | 0.95, 80, 92 | 10 Fold cross-validation | 17 (4-hydroxy-4-methylpentan-2-one), multiple compounds (compound 49 , 31, 61, 5, 9, 14, 20, 38) |
| 30,416,498 [34] | Development of an in silico method to predict whether a protein is an effector of type IV secretion system or not based on its sequence information. | Xiong Y, et al | 2018 | Prediction, Classification | NB, KNN, LR, ERT, GBM, XGB, SVM, RF, MC-SGE | Q1, Q3 | Transcriptomics | 73.2, 85.5, 87.9, 89.4, 90.5, 90.1, 90.2, 88.5, metric of F1 | 5-fold cross-validation, independent test for testing the generalization ability | PSSM-composition features |
| 30,682,021 [49] | This study focuses on the best way to use validated effector protein features for effector prediction using three machine learning classifiers, and compares results with those of others to obtain de novo results | Esna Ashari Z, et al | 2019 | Classification, Prediction, Clustering | SVM, E-SVM | Q2, Q3, Q4, Q5 | Transcriptomics, Proteomics | 94.05%, 93.64%, and 92.44%, for Models 1, 2, and 3, respectively. | 10 fold cross-validation | Optimal feature set includes 15 features (i.e, coiled coil domains, hydropath, PSSM composites) |
| 31,146,762 [23] | Enabling rapid assessment of mosquito blood-feeding histories and vectorial capacities using Mid-infrared spectroscopy and supervised machine learning . | Mwanga, E. P., et al | 2019 | Prediction, Classification | KNN, LR, SVM, NB, RF, XGB, MLP | Q1, Q2, Q3, Q4, Q5, Q6 | Proteomics, Fluxomics, Metabolomics, Cellomics, Population, Phenomics | Final model accuracy on hold-out dataset 98.4% | Stratified shuffled split cross-validation | Spectra intensities above 0.11 absorbance units |
| 31,778,355 [50] | The article is a review of recent applications of ML in infection biology, but also discusses the advantages and drawbacks of different techniques. Example Predicting bacterial host attributes by ML using Salmonella enterica serovar Typhimurium genome sequences | Lupolova N, et al | 2019 | Prediction, Clustering | KM, HA, HD-LDA, DL, SVM, RF | Q2, Q3 | Genomics | ~80% accuracy | both cross-validation and leave-one-out | pangenome matrix of predicted proteins |
| 31,835,769 [39] | EpiExploreR provides tools integrating common approaches to analyze spatiotemporal data on animal diseases in Italy, including notified outbreaks, surveillance of vectors, animal movements data and remotely sensed data. EpiExploreR is addressed to scientists and researchers, including public and animal health professionals wishing to test hypotheses and explore data on surveillance activities. | Savini L, et al | 2019 | Clustering, AR | NetA, Clustering | Q1, Q2, Q6, Q7, Q8 | Population | NA | NA | nearly real-time data, including notified outbreaks, surveillance of vectors, animal movements and remotely sensed data; |
| 31,791,409 [45] | The aim of this study was to develop a model based on available observations, climatic and environmental data, and machine learning methods for the prediction of the potential seasonal ranges of Ae. albopictus in China | Zheng, X., et al | 2019 | Prediction | RT | Q2, Q5, Q6, Q8 | Population | accuracy − 98.4% (97.1–99.5%), and AUC − 99.1% (95.6–99.9%) | 10 cross-validation | climatic surface, climatic zone, and regional environmental data |
| 25,521,718 [18] | Understanding and determining host tropism to identify zoonotic influenza virus strains capable of crossing species barrier and infecting humans. | Eng, C. L., et al | 2014 | Prediction, Classification, DL | RF, KNN, NB, SVM, ANN | Q1, Q2, Q3, Q5, Q6, Q7, Q8 | Genomics, Transcriptomics, Proteomics | 99.6, 97.0, 98.3, 97.4, 99.3, Final model(ACC > 96.57; AUC > 0.980; MCC > 0.916) | 10-fold cross-validation | The top 15 features for each protein were selected for inclusion as feature vectors into the dataset for the combined prediction model. |
| 31,881,961 [32] | This study developed a machine learning based classification approach to identify infectious disease associated host genes by integrating sequence and protein interaction network features | Barman, R.K., et al | 2019 | Classification, prediction, DL | DNN, SVM, RF, NB | Q3 | Genomics, Transcriptomics, Proteomics | accuracy of 86.33% sensitivity − 85.61% specificity − 86.57%. DNN classifier accuracy − 83.33% , sensitivity − 83.1% . | 10-fold cross-validation | PAAC_Network properties (10 features) and selected features for PAAC_Network properties (16 features) |
| 31,770,368 [57] | Use of a machine learning framework to determine whether viral virulence can be predicted by ecological traits, including human-to-human transmissibility, transmission routes, tissue tropisms, and host range. | Brierley L, et al | 2019 | Prediction, Classification | DT, RF | Q2, Q3 | Genomics, Transcriptomics | mean accuracy of 89.4% c | cross-validation | taxonomic family, primary tissue tropism, primary transmission route, know vector-borne transmission |
| 29,186,295 [58] | A state-of-the-art T4SE predictor by conducting a comprehensive performance evaluation of different machine learning algorithms along with a detailed analysis of single- and multi-feature selections. | Wang, J., et al | 2019 | Prediction, Classification, Clustering | NB, KNN, LR, RF, SVM, MLP | Q1, Q2, Q3 | Epigenomics, Genomics, Transcriptomics, Proteomics | 5-fold cross-validation | complementary features generally enhance the predictive performance of T4SEs; | |
| 30,385,576 [19] | This study took sequence data from>500 single-stranded RNA viruses and used machine-learning algorithms to extract evolutionary signals imprinted in the virus sequence that offer information about its original hosts and if an arthropod vector, and what type, plays a part in the virus's natural ecology. | Babayan, S. A., et al | 2018 | Prediction, Classification, AR | PN, GLM, GBM | Q1, Q2, Q3, Q5, Q6, Q7 | Genomics, Transcriptomics, Population | 83.5, (bagged accuracy = 97.0%) | (bagged accuracy = 97.0%) | genomic biases can coarsely discriminate viruses, viral codon pair and dinucleotide biases |
| 31,293,540 [33] | The main goal of this study is to predict a set of candidate effectors for the tick-borne pathogen Anaplasma phagocytophilum, the causative agent of anaplasmosis in humans. | Esna Ashari. Z, et al | 2019 | Prediction | SVM-RBF kernel, SVM-L, LR | Q2, Q3 | Genomics, Transcriptomics | Average 94.05, AUC (area under the curve) of 0.98, an average MCC 0.87 | 10-fold cross-validation | |
| 28,051,068 [36] | Development of PaPrBaG: Pathogenicity Prediction for Bacterial Genomes | Deneke. Carlus, et al | 2017 | Prediction, Classification | SVM | Q3 | Genomics, Transcriptomics, Proteomics | 0.88 to 0.93. | 5-fold Cross-validation | trimer, monomer, dimer, tetramer, spaced words, AA Index score, codon frequencies, monopeptides, DNA motifs, amino acid properties and dipeptides |
| 22,285,561 [37] | Development of two new approaches to automatically detect whether the title or abstract of a PubMed publication contains HPI data, and extract the information about organisms and proteins involved in the interaction to build a model that can predict pathogenicity | Thanh Thieu, et al | 2012 | Prediction, Classification | SVM | Q3, Q4 | Genomics, Transcriptomics, Fluxomics | 78 | 10-fold cross-validation | HPI-relevant and HPI-irrelevant |
| 31,288,641 [59] | A Python-based standalone tool, called PyPredT6, was use to predict T6 effector proteins. A total of 873 unique features were extracted from the peptide and nucleotide sequences of the experimentally verified effector proteins. | Sen R, et al | 2019 | Prediction | ANN, SVM, KNN, NB, RF | Q2, Q3 | Genomics, Transcriptomics, Proteomics, Lipidomics | Peptide, nucleotide sequence | ||
| 30,716,030 [51] | Developed a machine learning approach for the prediction of dengue fever severity based solely on human genome data. | Davi C. et al. | 2019 | Prediction, Classification, DL | SVM, ANN | Q3 | Genomics, Phenomics | accuracy>86%, and sensitivity and specificity over 98% and 51%, respectively. | using only genome markers | |
| 29,191,515 [46] | This study present simulated global distribution of Aedes aegypti and Aedes albopictus at a 5 × 5 km spatial resolution with high-dimensional multidisciplinary datasets and machine learning methods | Ding F. et al. | 2018 | Prediction | SVM, GBM, RF | Q2 | Population, Other SB level | RF (AUC) of 0.973 and 0.974, respectively, GBM (AUC of 0.971 and 0.972, respectively) and SVM (AUC of 0.963 and 0.964, respectively) | statistically significant | |
| 31,821,325 [44] | Model tick bite risk using human exposure and tick hazard predictors, represents a step forward in risk modelling by combining a well-known ensemble learning method, Random Forest, with four count data models of the (zero-inflated) Poisson family. | Garcia-Marti I et al. | 2019 | Prediction, Classification | RF, Ensemble | Q2 | Population | stdev = 3.15) | Pearson/Kendall coefficient | Species/organism |
| 29,114,054 [38] | In this study combine techniques in serial block-face scanning-electron microscopy and deep-learning–based image segmentation algorithms to visualize the distribution, abundance, and interactions of Ophiocordyceps unilateralis sensu lato fungus inside the body of its manipulated host. | Fredericksen MA et al. | 2017 | DL | DL | Q1, Q2, Q3 | Cellomics | For simpler stacks, the F1 score is over 96%, and for harder stacks, the F1 score is over 93% (voxel level). | F1-measure | Image features |
| 29,547,915 [60] | Developed the Bastion6: a bioinformatics approach for accurate prediction of type VI secreted effectors | Wang J. et al. | 2018 | Prediction, Classification, Clustering | KM, 2L-SVM-E | Q3 | Genomics, Proteomics | ACC (>0.91), F-value (>0.91), MCC (>0.83) and AUC (>0.96). Independence test ACC (0.943), F-value (0.946), MCC (0.892) and AUC (0.976). | 5-fold cross-validation and independent tests and case studies | integrated into sequence profile, evolutionary information and physicochemical property. |
| 25,113,593 [47] | This study aims to clarify if land use factors other than human settlements, e.g. different types of agricultural land use, water bodies and forest are associated with reported dengue cases from 2008 to 2010 in the state of Selangor, Malaysia. | Cheong. YL, et al | 2014 | Hypothesis Generation/ Association Rules | BRT | Q2 | Population, Other SB level | 81% | cross-validation | land use factors and the reported dengue cases |
| 10.21203/ rs.2.15755/v1 | This study identifies critical climatic risk factors to predict dengue outbreaks with better accuracy | Nejad. FY, et al. | 2019 | Prediction, Association Rules | BN, SVM, NB, DT | Q2 | Population | BN 92.35%, RMSE 0.26 | 10-fold cross-validation | temperature (min, ma, average), minimum humidity and rainfall |
| 10.1109/BigDataCongress.2017.54 [52] | The motivation behind this study is to provide a basic framework for biologists, which is based on big data analytics and deep learning models. | Huaming Chen et al. | 2017 | DL | DL | Q2, Q3 | Proteomics | protein–protein interaction | ||
| 10.1109/ACCESS.2020.2971091 [48] | SMOPredT4SE employed combination features of series correlation pseudo amino acid composition and position-specific scoring matrix to present protein sequences, and employed support vector machines (SVM) to identifying T4SEs | Zihao Yan et al. | 2020 | Prediction Classification | SVM, RF, NB, kNN, Bagging, SGD, LibD3C. | Q2, Q3 | Proteomics | 95.60% | 5-fold cross-validation | composed of 305 T4SEs and 610 non-T4SEs |
Notations: ML-Machine Learning, DM-Data Mining, support vector machines (SVM), and artificial neural networks (ANN), DT:-Decision Tree, RF:-Random Forest, GBR:-Generalized Boosted Regression, NB:-Naïve Bayes, SVM:-Support Vector Machine, KNN:-k-Nearest Neighbors, KM:-k-Means, NetA:-Network Analysis, RT:-Regression Tree, DNN:-Deep Neuron Networks, PN:-Phylogenetic Neighborhood, SVM-RFB-k:-SVM-RBF kernel, ANN:-Artificial Neural Network, DL:-Deep Learning, BRT:-Boosted Regression Tree, BN:-Bayes Network, GB:- Gradient Boosting, GrB:- Generalized Boosted, AdaBoost:-Adaptive Boosting, LR:- Logistic Regression, HD-LDA:- Hierarchical Divisive and Latent Dirichlet Allocation, GBMs:- Gradient Boosting Machines, RBF-t:- RBF tree, GB-t:- gradient boosted tree, SVM-RLK:- support vector machine (radial and linear kernel), CTA:- Classification Tree Analysis, RRF:- Regularized Random Forest, E-SVM:- Ensemble of three SVM, HA:- Hierarchical Agglomerative, C:- Clustering, GLMM:- Generalized Linear Mixed Models, SVM-Lk:- SVM-L kernel, Ens:- Ensemble, 2-L-SVM-E:- two-layer SVM-based ensemble model, CNN:- deep Convolutional Neural Network, ERT:- Extremely Randomized Trees (ERT), DL:- Deep Learning, MLP:-Multilayer Perceptron, XGB:- eXtreme Gradient Boosting, MC-SGE:- Meta-Classifiers (Stacked Generalized Ensemble).
3.2. Current use of data mining and machine learning to understand vector-host-pathogen relationships leveraging systems biology
3.2.1. Q1 – adaptation
Assessing the adaptation of a pathogen to colonize a particular arthropod vector/host can be challenging, due to the complexity of the systems involved and the non-deterministic aspects of many biological processes that control colonization (e.g. environmental factors). Our result show that assessment of pathogen adaptation to certain hosts/vectors using ML and DM techniques have used all 5 machine learning tasks with most interest in prediction-based methods (Fig. 4e). 15 papers that we queried studied adaptation using DM and ML tools. The authors of these used datasets from both the lab and existing databases that involved 9 out of 10 systems biology levels, but not lipidomics datasets. The biological problems tackled in these papers included the prediction of host tropism in order to identify zoonotic influenza virus strains capable of crossing species barriers and infecting humans[18]. Similarly, Babayan et al. sought to predict vectors and reservoirs for RNA viruses based on evolutionary signatures in their genomes[19]. Furthermore, Yang et al. proposed to distinguish vector from non-vector tick species based on a series of traits to determine risk of transmission of infections to humans using a generalized boosted regression model [20]. The model used phenomics and population datasets to reach an accuracy of ~ 91%. Despite the sample size of 14 species, 10-fold cross- validation showed the model has good stability (Table 3).
Fig. 4.

Data analysis results overview (n = 32). Fig. 4 was created with word-cloud by using (a) papers key words, (b) words contained in study objectives (c) scope in systems bioscience covered by the paper (d) and sources of the data in a study, such as databases, lab experiments, or simulations. The visibility of a word among words in their panel emphasize their appearance in a papers' keys words, objective, or scope, and highlight a review papers' focus.
3.2.2. Q2 – transmission
Out of 32 papers, 22 attempted to leverage DM and ML to assess factors that influence the transmission of pathogens to new hosts (Fig. 3b, Table 3). Analysis of these paper shows a diverse use of ML tasks including prediction, classification, clustering, and DL methods (Fig. 4e), by covering 9 systems biology levels out of 10 including: proteomics, fluxomics, metabolomics, cellomics, population, phenomics, transcriptomics, genomics, epigenomics[21] (Table 3). Leveraging datasets from lab experiments and the literature allowed investigators to build ML models with an accuracy range of 79%–100%. Using RF, RRF, and Adaptive Boosting, De Moraes et al. proposed a predictive model to determine the malaria infection status of human patients based on volatile biomarkers. The model used proteomics and metabolomics dataset to reach an accuracy level of 80–95% with a 10-fold cross-validation [22]. In addition, KNN, LR, and SVM were used to assess mosquito blood-feeding histories from multi-OMICs datasets with over 98% accuracy [23]. This knowledge could in turn be useful for identifying anthropophilic species with high potential to transmit pathogens. Supplemented with automated surveillance to detect and classify known vector species based on morphological features[24], [25], [26], [27], such models could facilitate the prediction of risk of new human infections with vector-borne diseases in particular areas.
3.2.3. Q3 – pathogenicity
The use of ML and DM to assess the how the pathogen affects the host toward the development of disease is one of the most popular applications identified in our review (Fig. 4e, Table 3). With 25 papers involved in this question, the community has been able to leverage all systems biology levels to provide a number of data mining processes and machine learning models to predict the pathogenicity of certain microbes and identify meaningful patterns involved in this biological process. Besides association rules mining tasks, all ML tasks have been used, with strong focus placed on prediction and classification tasks. For example, current work allows problems such as prediction of health endpoints in STEC, and risk assessment of microbial pathogenicity using whole genome sequencing data[28], [29], [30]. These modeling approaches are able to reach an accuracy level of 60–95% and similar approaches have been used to predict the diagnosis and clinical prognosis of Dengue in human patients[31] Barman, R.K., et al. also proposed a classification approach to identify infectious disease associated host genes by integrating sequence and protein interaction network features [32]. More directly related to vector-borne diseases, Esna Ashari et al. used DM and ML to identify Type 4 Secretion System effectors that could be involved in the pathogenicity of the tick-borne bacterium Anaplasma phagocytophilum[33], as has been done for non-vector-borne pathogens[34], [35], [36].
3.2.4. Q4 – immunity
The investigation of the level and trend of the host response is critically important to anticipating and preventing infection progress. Our research shows that DM and ML have been used to integrate large scale datasets to provide supervised prediction and pattern identification tools with an accuracy of 78–95% (Table 3). The use of memory-based predictors such as BN (Bayesian Network) and Multilayer Perceptron (MLP) emphasize the complexity of the mechanisms involved in host responses to infection[22], [30], [32], [35], [37], [38].
3.2.5. Q5-Q6 – vector manipulation of transmission and arthropod effects on pathogenicity
Understanding how the change in vector affects transmission efficiency or pathogenicity following infection of a vertebrate host are intriguing questions that were less investigated in the papers we reviewed (8% papers reviewed). The broader usage of ML tasks (prediction, classification, clustering, AR, DL) may critically improve the understanding of these processes (Fig. 1b, Fig. 3b, Fig. 4c). Brierley et al. provide a compelling example by using the random forest approach to predict the virulence of human RNA viruses based on a number of ecological factors, including the host range and transmission route[19]. If applied to agents such as vector-borne bacteria, such approaches may be helpful in addressing important knowledge gaps, such as virulence factor-independent differences in the pathogenicity of Rickettsia and Borrelia species[10].
3.2.6. Q7 – reservoir host effects
Savini et al. proposed to assess reservoir dynamics in the vector-host-pathogen relationship using network analysis clustering, RF, KNN, SVM and ANN, as did Eng et al. and Babayan et al. which are discussed above[18], [19], [39]. Savini et al. developed the EpiExploreR web application by integrating various spatiotemporal data on animal diseases in Italy, including notified outbreaks, surveillance of vectors, animal movements data and remotely sensed data. EpiExploreR is aimed at public health scientists and researchers and facilitates the exploration of complex data and the generation of new hypotheses relevant to a natural setting.
3.2.7. Q8 – environmental effects
The effects of the environment on the various components of host-vector-pathogen relationships are extremely complex and difficult to fully address within a short review. Thus, we focused on the implications of the environment on Q1, Q2, and Q3. We identified several studies using DM and ML in combination with animal movement data, climate data[40], [41], [42], and remotely sensed data to examine the distribution of vectors and vector-borne diseases. Methods used in these contexts included Random Forest, Classification Tree Analysis, Generalized Boosted, k-nearest neighbor (kNN), Naïve Bayes, support vector machines (SVM), and artificial neural networks (ANN). The model accuracy range was 82–97%. The problems addressed included: the identification of high risk snail habitats as a function of Schistosoma japonicum infection[43], modelling of tick bite risk based on ecological factors[44], predicting the global distribution of Aedes mosquitoes and the effects of seasonal changes on their range[45], [46] and the prediction of Dengue virus outbreak risk based on climate[47], [48].
3.3. Knowledge discovery in assessment of vector-host-pathogen relationships: from data to knowledge
From the data science perspective, the state of the art in the field should cover the entire DM and ML process (Fig. 1c). No paper identified in our review was able to cover all step in DM process, but we identified at least one paper using each step in the process (Prediction[28], classification[29], clustering[39], association rules[47], deep learning[38]) (Table 3, Fig. 5, Supplementary File S1). This validates the feasibility of adopting the process to tackle important and challenging biological questions pertaining to vector-host-pathogen relationships. During our investigation, we allowed the community to also assess the quality of the state of available datasets. To achieve this goal, we adopted the FAIR (Findable Accessible Interoperable Reusable) principle, a quality assessment roadmap developed and currently used to improve bioscience dataset shareability and reproducibility. Fig. 1c shows the distribution of the ML tasks adopted to address our research questions (Q1-Q8). Looking at the dataset specifications and key features (Fig. 5), we can observe that each ML task complementarily allows the community to extract highly valuable knowledge from specific datasets in a very restrictive context. This observation raises hope that more ML methods will be adopted in the future, but also reveals many challenges from data quality to good usage of each ML technique.
Fig. 5.

Snapshot of data science perspective on host-pathogen interaction analysis – from raw data to Knowledge Discovery in Databases (KDD), the Data Mining (DM) & Machine Learning (ML) process: (a) data source, (b) dataset annotation, (c) dataset quality using FAIR principle and (d) ML method used.
3.3.1. Quality of data
The increasing need for quality data is driving the data science community toward formal approaches to assess data quality. The complexity of this task in bioscience is significant, however, several initiatives have been successfully tested, including the FAIR principle. Using that principle, we assessed the compliance (or FAIRness) of currently used datasets. The results show a very low adoption of rigorous data good practices (Fig. 5c). The FAIR principle at its core has 4 indicators:(F)-Findable dataset: this criterion checks if the dataset can be located with a persistent identifier. Out of 32 papers, 9 studies provided a URL to access their dataset. These URLs included recognized persistent dataset repositories such as Figshare or GitHub. The remaining studies fell short in allowing their datasets to be found. (A)-Accessible dataset: this criterion measures if the dataset is accessible, even when it not findable (e.g. the identifier is outdated). This criterion usually consists of providing a protocol with an alternative option to allow accessibility to datasets. From our review, only 10 papers provided accessibility protocols in their studies. (I)-Interoperable dataset: this criterion checks if a dataset is annotated enough to be used by others. For example, in our research questions, we used multiple systems biology datasets. A dataset from proteomics measurements should be annotated to emphasize the meaning of the protein values. This allow a genomics expert to use that dataset by integrating gene expression dataset values accordingly for machine learning prediction. Failing to understand that relationship will mislead the feature engineering process, compromising the dataset quality/integrity and therefore, the resulting predictive model. On this criterion, only 9 studies provided interoperable justification. (R) – Reusable dataset: this criterion measures the ability of the community to adopt the dataset and reuse it to achieve similar goals. This is measured by author adherence to community standards for collection, processing and publishing their dataset. There is no doubt that all these published studies followed rigorous scientific protocols, but many papers fell short on reporting the data life cycle to ensure reproducibility.
3.3.2. Prediction decision making
Prediction task adoption in this review ranged from the use of simple linear regression modeling to multiple layer deep neural networks. Prediction is used in all 8 biological research problems more than any other ML task (Fig. 4e). This sometimes led to the misuse of the term. For example, few methods show good assessment of data quality and the assessment of selected features to avoid issues such as overfitting. Boosting approaches are widely used to compensate for poor dataset quality (e.g. small size, Fig. 5d).
3.3.3. Classification and clustering
Classification and clustering ML tasks allow the community to identify meaningful patterns relevant to biological problems. This task was widely used in papers in our review across biological systems levels (e.g. genomics, transcriptomics). Even though adoption of these tasks for addressing Q5-Q8 is still poor, the lack of data availability seems to play a key role in this shortcoming (Fig. 4e). However, the two methods could be effectively extended to a clinical diagnostic problem as applied in[31], where a c4.5 decision tree was used to distinguish dengue from non-dengue febrile illness.
3.3.4. Association rules mining
Association rules mining is one of the fastest growing ML tasks in bioscience and biomedicine. It allows scientists to infer new hypotheses from data and provide rational recommendation for follow-up studies. In our review, very few papers adopted this task (Fig. 4e). Savini et al. show how the use of this method can help the community to integrate multiple systems biology levels to provide a spatiotemporal assessment of a given disease[39], [41].
4. Discussion
4.1. Challenges and limitations
4.1.1. The curse of dimensionality with big data
During the review process, we identified some important challenges to the community. Among these challenges and limitations are the problems of data heterogeneity. Data pertaining to vector-host-pathogen relationships come in many forms and shapes. For example, these could be omics data [28], [34], environmental or clinical samples, or laboratory samples [23]. While some of the surveyed articles address this issue, a number of the articles did not detail the processes they applied to solve said challenge. Only in some articles was ML applied to solve the high dimensional problem of genomics data [28]. However, a lack of better solution to this problem could render models not generalizable or applicable for broadly understanding processes such as pathogenicity, transmission, and adaptation using different data modalities. Furthermore, real-world data must go through preprocessing in data mining and machine learning to normalize the data and remove unwanted or misleading information. The approach may be challenging for domain practitioners as difficulties in implementing one challenging algorithm necessary for their work may force them to use easy to implement, but less appropriate algorithms.
4.1.2. Missing data
Another challenge or limitation is that of missing data. In general, real-word data will always have some missing data points. While building a predicting model, such as one involving a vector-host-pathogen relationship, valuable but very informative data might be missing and difficult to represent. However, the aspiration of model building is to represent all aspects involved in the dataset in addressing the problem. Dealing with these missing data proved difficult and lacking in most of the articles we reviewed. Only in one of the articles [29] did the authors discuss missing data. In this study, the authors marked them as “NA” and imputed. Though imputation is one way to address missing data, this could potentially cause a problem in lower quality omics datasets.
4.1.3. Dataset reproducibility
Data reproducibility is the gold standard of the data mining and machine learning domain. Thus, these considerations should not be taken lightly when conducting a study. From our review, we observed that most studies could be reproducible, with only a few exceptions. Understandably, the topics we queried are highly complicated. Still, for the sake of model usefulness, the data used to make the model should be made available and easily accessible to these who want to reproduce it. Arguably, the datasets used in the papers we reviewed met the standard, but availability was not addressed adequately. Also, the availability of the codes used in the analysis is critically important in that, without it, reproducibility would be near impossible.
4.1.4. Rarity and class imbalance
A further challenge in data mining and machine learning is that of imbalanced data or class. A real-world dataset is not always balanced. This is especially true when it comes to a domain such as vector-host-pathogen interactions, where data collection is multi-faceted and multidisciplinary data modalities are common (e.g. environmental data in map format, OMICs data in sequences format, etc.). Thought this issue was better addressed compared to the missing data issues [32], [34], [49], it was still inadequately addressed by some of the studies identified in our review. In machine learning, imbalanced data input can hamper model performance and contribute to inaccuracy. This is further complicated by the fact that machine learning inputs are always features vectors. Representing systems level data like omics data in features vector forms is important to generate model input, however, it could also introduce class imbalance issues. For example, [34] encoded a protein sequence into informative features vector to use in a model. They used Position-Specific Scoring Matrix (PSSM) to transform their sequence datasets to features vectors. However, being mindful of how they might have introduced imbalance into their datasets, the authors chose not to measure their model performance in Receiver Operating Characteristic curve (ROC) or Matthews Correlation Coefficient (MCC). Instead, they used the metric of F1, because in this case ROC or MCC would perform optimistically in an imbalance dataset, causing model overfitting. This was one way to handle the imbalance effect issues, and failure to resolve that problem could have resulted in misleading model accuracy.
4.1.5. Systems biology and big data scalability
While machine learning and data mining use Systems Biology (SB) generated data to solve important problems at hand, SB big data scalability becomes a limitation at the same time. In this review, most of the models present were limited by SB big data lack of scalability. For example, the model in [23]and others we reviewed were not scalable. In addition, lack of scalability causes most studies to choose a small subset of data from one location or study to focus on as they cannot integrate and scale big data from geospatial, omics, or other forms of big data. This constraint makes most of the models lack generalization and instead built for one particular context or problem. For example, to study the interaction between a pathogen with ~3,000 genes and a human (with ~30,000 genes) at the genomic level, feature engineering is needed to select the most relevant subset of genes for the problem of interest (e.g. 100 genes involved in pathogenicity to host X). Once a predictive model is built from that context, the model must be flexible enough for adoption under different conditions, such as when the host gene list changes. From the systems biology perspective, it is difficult to impossible to scale big data usage without changing the required platforms or increasing the capacity of the available ones. For that reason, only a small subset of big data can be used to address a specific problem at hand, leaving out some datasets, which in turn means leaving out potentially valuable information or insight.
4.2. Future directions
4.2.1. Knowledge discovery
Data mining, knowledge discovery and machine learning are presently revolutionizing every field of biology. Machine learning applications in the medical and public health industry are increasing daily and could become dominant tools in disease prediction and surveillance [23]. In systems biology, machine learning is used in macromolecule structure prediction, gene networks reconstruction, tumor classification, and virtual drug discovery [50]. In bacterial genomics, machine learning has been used in antibiotic resistance prediction, pathogenicity prediction, and the evaluation of host adaptation and zoonotic potential [50]. Potentially, such applications could minimize labor-intensive wet lab assays such as ELISA or PCR. For example, [23] used mid-infrared (MIR) spectroscopy coupled with supervised machine learning to accurately identify blood meals in the guts of mosquitos. This was done to diagnose the propensity of different female mosquitoes to take meals on humans and not other vertebrate hosts. A similar application could be extended to other vectors, such as ticks, to elucidate transmission potential.
4.2.2. Leveraging innovations in DM and ML
4.2.2.1. Deep learning
While application of deep learning increased in other fields, i.e., image classification, it seems to be less applied towards understanding vector-host-pathogen relationships. Among the 32 articles we reviewed, 6 articles used deep learning[32], [35], [38], [50], [51], [52] (Fig. 4e, Table 3). However, the potential of deep learning applications is substantial and should be explored in this context. The ability of deep learning to work well with different types of data modalities could be of value in pathogenicity prediction, as an example. In [35], image-based machine learning was used to define host-pathogen relationships by recognizing, classifying and quantifying host cellular defenses, pathogen killing, and replication with great accuracy This study represents an effective example of the use of artificial intelligence in combination with different data types to assess vector-host-pathogen relationships. Further, several aspects of deep learning were used to identify and classify arthropod vector species such as triatomine bugs and mosquitoes from morphological data[24], [25], [26], [27].
4.2.2.2. Model selection
In data mining and machine learning, selection and evaluation of models are valuable processes in building a useful predictor or classifier. Though these processes are not reported in-depth, they are reasonably important for choosing a proper algorithm that could adequately address the problems of interest. Most of the articles in this review applied the model selection and evaluation process to help them come up with the right machine learning methods for building their models. This shows the importance of these processes in this domain and their use should be encouraged in future applications of machine learning towards predicting vector-host-pathogen relationships.
4.2.2.3. Cross-validation
We observed different types of cross-validation methods in this review. Out of 32 articles we processed, 11 used 10-fold cross-validations, 5 used 5-cross-validation, and 1 article used both 10-fold cross-validation and one-hold-out, while 3 articles did not provide clear information on their validation methods. Because of variability and heterogeneity in the data generated by omics technologies, it is important to select a cross-validation method that takes into consideration data modality and processing and validates applicability. Therefore, in the future, cross-validation selection should be a priority when building vector-host-pathogen interaction models.
4.2.2.4. Association mining
From this review, we observed infrequent use of association mining. However, in a field such as pathogenicity or disease prediction, application of association mining could be very productive. This is because vector-host pathogen relationships are nested interactions in which one aspect effects the other. Thus, involving association mining to produce testable hypotheses needs to be applied in future studies in this domain. For example,[53] built a testable model of future Dengue incidence in the Phillipines.
4.2.2.5. Class imbalance
Though a problem as discussed in the above sections, class imbalance can influence model performance in one direction or another. A positive imbalance will affect a model lightly. However, data are naturally skewed negatively, and this affects specificity gradually [32] which in turn negatively affects model accuracy. In papers identified in this review, several articles addressed model performance on imbalance datasets well, pointing to this as a future direction in the field.
4.2.2.6. Feature engineering
The success of machine learning models relies heavily on feature engineering. In the assessment of vector-host-pathogen relationships, data modality is variable. Hence, engineering of informative features from a variety of data is an important task. Use of unrelated features hurts the accuracy of most classifiers, whereas too many features are computationally time consuming. Striking a balance by feature engineering could save a model time and resources, especially in addressing vector-host-pathogen relationships with high dimensionality data. Therefore, the application of feature engineering in this domain is as important as building a model itself.
4.2.2.7. Explanatory vs predictive ML modeling
The goals of explanatory and predictive ML modelling are different. Explanatory modelling looks for statistically significant relationships, whereas predictive modelling looks for associations that could be valuable in predicting future outcomes. The papers analyzed in this review could be categorized into both explanatory and predictive ML models. Many of them used a combination of algorithms to explore the input for a final model either together or individually selected based on performance. For that reason, both tasks are valuable and applicable in many ways in this field.
4.2.2.8. Crowd sourcing
Finally, the use of crowd sourcing in the vector-host-pathogen domain could be advantageous. Crowd sourcing can fill in the gaps of missing data. It can also enable sharing of methods for big data analysis and model validation and facilitate collaboration on problems and tasks to reduce time and resources constraints. From a biomedicine point of view, it is a valuable tool of great importance and its application here is warranted. Further discussion of the in-depth uses of crowd sourcing is provided by [54], [55].
5. Conclusions
In this review, we explored the concepts of data mining and machine learning as applied towards understanding vector-host-pathogen relationships such as adaptation, transmission, and pathogenicity. From the articles we reviewed, 25 (63.6%) studies involved predictive models using supervised machine learning, while 14 (9.1%, 4.6%) used unsupervised methods and deep learning. In the retrieved articles, prediction and classification were among the most dominant machine learning tasks, which were used to classify and predict relevant features that dictate interaction outcomes (e.g. pathogenicity, adaptation or transmission). Furthermore, the utility of heterogeneous data together with different methods to feature engineer or select proved valuable in many of the reviewed studies. While data mining and machine learning are being increasingly applied in many life science domains (gene networks reconstruction, tumor classification, virtual drug discovery [50], and bacterial genomics), as shown in this review, they have not yet taken roots in the field of vector-borne diseases. In particular, association rules and deep learning lagged behind the other methods of DM & ML, such as classification, and prediction of pathogen vector-host relationships. A future increase in deep learning applications in the field could be valuable, especially when combined with other approaches such as feature engineering, cross-validation, model selection, and supplemented with crow-sourcing. Also, the application of association rules would increase hypothesis generation in the field and reduce the time and resources spent in doing do. In return, this will contribute to more data generated from the field, which also could increase DM & ML use in the domain. Though the application of such methods to specific problems in vector-borne diseases is still in its infancy and faces many expected issues, these approaches have great potential and should be encouraged to bring new perspectives to old problems as large, diverse systems biology datasets become available in the field.
6. Availability of data and materials
The datasets generated or analyzed during our survey are available from the corresponding author upon reasonable request.
Funding
This work was partially funded by the National Science Foundation/Experimental Program to Stimulate Competitive Research (EPSCoR) Grant OIA-1849206 awarded to Gilbert Ustad, and the Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health P20GM103443 (B.E. Goodman).
CRediT authorship contribution statement
Diing D.M. Agany: Conceptualization, Data curation, Writing - original draft. Jose E. Pietri: Conceptualization, Writing - review & editing. Etienne Z. Gnimpieba: Conceptualization, Methodology, Software, Visualization.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.06.031.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Bueno-Marí R., Jiménez-Peydró R. Global change and human vulnerability to vector-borne diseases. Front Physiol. 2013;4:158. doi: 10.3389/fphys.2013.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization A global brief on vector-borne diseases. World Heal Organ. 2014;9 [Google Scholar]
- 3.King J.G. Developmental and comparative perspectives on mosquito immunity. Dev Comp Immunol. 2020;103 doi: 10.1016/j.dci.2019.103458. [DOI] [PubMed] [Google Scholar]
- 4.LaDeau S.L., Allan B.F., Leisnham P.T., Levy M.Z. The ecological foundations of transmission potential and vector-borne disease in urban landscapes. Funct Ecol. 2015;29:889–901. doi: 10.1111/1365-2435.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magori K., Drake J.M. The population dynamics of vector-borne diseases. Nat Educ Knowl. 2013;4(4):14. [Google Scholar]
- 6.Eder M., Cortes F., Teixeira de Siqueira Filha N., Araújo de França G.V., Degroote S., Braga C., Ridde V., Turchi Martelli C.M. Scoping review on vector-borne diseases in urban areas: transmission dynamics, vectorial capacity and co-infection. Infect Dis Poverty. 2018;7(1) doi: 10.1186/s40249-018-0475-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Müller R, Reuss F, Kendrovski V, Montag D. Vector-Borne Diseases. In: Marselle MR, Stadler J, Korn H, Irvine KN, Bonn A, editors. Biodivers. Heal. Face Clim. Chang., Cham: Springer International Publishing; 2019, p. 67–90. doi:10.1007/978-3-030-02318-8_4.
- 8.Kramer L.D., Ciota A.T. Dissecting vectorial capacity for mosquito-borne viruses. Curr Opin Virol. 2015;15:112–118. doi: 10.1016/j.coviro.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murdock C.C., Luckhart S., Cator L.J. Immunity, host physiology, and behaviour in infected vectors. Curr Opin Insect Sci. 2017;20:28–33. doi: 10.1016/j.cois.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lescot M., Audic S., Robert C., Nguyen T.T., Blanc G., Cutler S.J. The genome of Borrelia recurrentis, the agent of deadly louse-borne relapsing fever, is a degraded subset of tick-borne Borrelia duttonii. PLoS Genet. 2008;4 doi: 10.1371/journal.pgen.1000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verhoeve VI, Jirakanwisal K, Utsuki T, Macaluso KR. Differential Rickettsial Transcription in Bloodfeeding and Non-Bloodfeeding Arthropod Hosts. PLoS One n.d.;11:e0163769. doi:10.1371/journal.pone.0163769 [DOI] [PMC free article] [PubMed]
- 12.Abromaitis S., Nelson C.S., Previte D., Yoon K.S., Clark J.M., DeRisi J.L. Bartonella quintana deploys host and vector temperature-specific transcriptomes. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0058773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Worachartcheewan A., Schaduangrat N., Prachayasittikul V., Nantasenamat C. Data mining for the identification of metabolic syndrome status. EXCLI J. 2018;17:72–88. doi: 10.17179/excli2017-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kourou K., Exarchos T.P., Exarchos K.P., Karamouzis M.V., Fotiadis D.I. Machine learning applications in cancer prognosis and prediction. Comput Struct Biotechnol J. 2015;13:8–17. doi: 10.1016/j.csbj.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moher D., Liberati A., Tetzlaff J., Altman D.G. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6 doi: 10.1371/journal.pmed.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellinger C., Mohomed Jabbar M.S., Zaïane O., Osornio-Vargas A. A systematic review of data mining and machine learning for air pollution epidemiology. BMC Public Health. 2017;17 doi: 10.1186/s12889-017-4914-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diing Agany, Jose Pietri, Gnimpieba ZE. Vector-pathogen-Host Machine Learning and Data Mining Review Data. Figshare, Dataset 2020. doi:10.6084/m9.figshare.12053637.v1. [DOI] [PMC free article] [PubMed]
- 18.Eng C.L., Tong J.C., Tan T.W. Predicting host tropism of influenza A virus proteins using random forest. BMC Med Genomics. 2014;7. doi: 10.1186/1755-8794-7-S3-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babayan S.A., Orton R.J., Streicker D.G. Predicting reservoir hosts and arthropod vectors from evolutionary signatures in RNA virus genomes. Science (80-) 2018 doi: 10.1126/science.aap9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang L.H., Han B.A. Data-driven predictions and novel hypotheses about zoonotic tick vectors from the genus Ixodes. BMC Ecol. 2018 doi: 10.1186/s12898-018-0163-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller J.A., Ding S.-L., Sunkin S.M., Smith K.A., Ng L., Szafer A. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Moraes C.M., Wanjiku C., Stanczyk N.M., Pulido H., Sims J.W., Betz H.S. Volatile biomarkers of symptomatic and asymptomatic malaria infection in humans. Proc Natl Acad Sci U S A. 2018 doi: 10.1073/pnas.1801512115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mwanga E.P., Mapua S.A., Siria D.J., Ngowo H.S., Nangacha F., Mgando J. Using mid-infrared spectroscopy and supervised machine-learning to identify vertebrate blood meals in the malaria vector. Anopheles arabiensis. Malar J. 2019 doi: 10.1186/s12936-019-2822-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khalighifar A., Komp E., Ramsey J.M., Gurgel-Gonçalves R., Peterson A.T. Deep learning algorithms improve automated identification of Chagas disease vectors. J Med Entomol. 2019;56:1404–1410. doi: 10.1093/jme/tjz065. [DOI] [PubMed] [Google Scholar]
- 25.Motta D., Santos A.Á.B., Winkler I., Machado B.A.S., Pereira D.A.D.I., Cavalcanti A.M. Application of convolutional neural networks for classification of adult mosquitoes in the field. PLoS ONE. 2019;14 doi: 10.1371/journal.pone.0210829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park J., Kim D.I., Choi B., Kang W., Kwon H.W. Classification and morphological analysis of vector mosquitoes using deep convolutional neural networks. Sci Rep. 2020;10:1012. doi: 10.1038/s41598-020-57875-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorenz C., Ferraudo A.S., Suesdek L. Artificial Neural Network applied as a methodology of mosquito species identification. Acta Trop. 2015;152:165–169. doi: 10.1016/j.actatropica.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 28.Njage P.M.K., Leekitcharoenphon P., Hald T. Improving hazard characterization in microbial risk assessment using next generation sequencing data and machine learning: predicting clinical outcomes in shigatoxigenic Escherichia coli. Int J Food Microbiol. 2019 doi: 10.1016/j.ijfoodmicro.2018.11.016. [DOI] [PubMed] [Google Scholar]
- 29.Wheeler N.E., Gardner P.P., Barquist L. Machine learning identifies signatures of host adaptation in the bacterial pathogen Salmonella enterica. PLoS Genet. 2018;14 doi: 10.1371/journal.pgen.1007333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rahman M.S., Rahman M.K., Saha S., Kaykobad M., Rahman M.S. Antigenic: an improved prediction model of protective antigens. Artif Intell Med. 2019 doi: 10.1016/j.artmed.2018.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Tanner L., Schreiber M., Low J.G.H., Ong A., Tolfvenstam T., Lai Y.L. Decision tree algorithms predict the diagnosis and outcome of dengue fever in the early phase of illness. PLoS Negl Trop Dis. 2008;2 doi: 10.1371/journal.pntd.0000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barman RK, Mukhopadhyay A, Maulik U, Das S, R.K. B, A. M, et al. Identification of infectious disease-associated host genes using machine learning techniques. BMC Bioinformatics 2019;20. doi:10.1186/s12859-019-3317-0. [DOI] [PMC free article] [PubMed]
- 33.Esna Ashari Z., Brayton K.A., Broschat S.L., Ashari Z.E., Brayton K.A., Broschat S.L. Prediction of T4SS effector proteins for anaplasma phagocytophilum using OPT4e. A new software tool. Front Microbiol. 2019;10:1391. doi: 10.3389/fmicb.2019.01391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiong Y., Wang Q., Yang J., Zhu X., Wei D.Q. PredT4SE-stack: Prediction of bacterial type IV secreted effectors from protein sequences using a stacked ensemble method. Front Microbiol. 2018;9 doi: 10.3389/fmicb.2018.02571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fisch D., Yakimovich A., Clough B., Wright J., Bunyan M., Howell M. Defining host–pathogen interactions employing an artificial intelligence workflow. Elife. 2019 doi: 10.7554/eLife.40560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deneke C., Rentzsch R., Renard B.Y. PaPrBaG: a machine learning approach for the detection of novel pathogens from NGS data. Sci Rep. 2017;7 doi: 10.1038/srep39194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thieu T., Joshi S., Warren S., Korkin D. Literature mining of host-pathogen interactions: comparing feature-based supervised learning and language-based approaches. Bioinformatics. 2012;28:867–875. doi: 10.1093/bioinformatics/bts042. [DOI] [PubMed] [Google Scholar]
- 38.Fredericksen M.A., Zhang Y., Hazen M.L., Loreto R.G., Mangold C.A., Chen D.Z. Three-dimensional visualization and a deep-learning model reveal complex fungal parasite networks in behaviorally manipulated ants. Proc Natl Acad Sci U S A. 2017;114:12590–12595. doi: 10.1073/pnas.1711673114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Savini L, Candeloro L, Perticara S, Conte A. EpiExploreR: A Shiny Web Application for the Analysis of Animal Disease Data. Microorganisms 2019;7. doi:10.3390/microorganisms7120680. [DOI] [PMC free article] [PubMed]
- 40.Carvajal T.M., Viacrusis K.M., Hernandez L.F.T., Ho H.T., Amalin D.M., Watanabe K. Machine learning methods reveal the temporal pattern of dengue incidence using meteorological factors in metropolitan Manila, Philippines. BMC Infect Dis. 2018;18:183. doi: 10.1186/s12879-018-3066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flamand C., Fabregue M., Bringay S., Ardillon V., Quenel P., Desenclos J.-C. Mining local climate data to assess spatiotemporal dengue fever epidemic patterns in French Guiana. J Am Med Informatics Assoc. 2014;21:e232–e240. doi: 10.1136/amiajnl-2013-002348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cianci D., Hartemink N., Ibáñez-Justicia A. Modelling the potential spatial distribution of mosquito species using three different techniques. Int J Health Geogr. 2015;14:10. doi: 10.1186/s12942-015-0001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xia C., Hu Y., Ward M.P., Lynn H., Li S.S., Zhang J. Identification of high-risk habitats of oncomelania hupensis, the intermediate host of schistosoma japonium in the poyang lake region, China: A spatial and ecological analysis. PLoS Negl Trop Dis. 2019;13. doi: 10.1371/journal.pntd.0007386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia-Marti I., Zurita-Milla R., Swart A. Modelling tick bite risk by combining random forests and count data regression models. PLoS ONE. 2019;14 doi: 10.1371/journal.pone.0216511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng X., Zhong D., He Y., Zhou G. Seasonality modeling of the distribution of Aedes albopictus in China based on climatic and environmental suitability. Infect Dis Poverty. 2019 doi: 10.1186/s40249-019-0612-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding F., Fu J., Jiang D., Hao M., Lin G. Mapping the spatial distribution of Aedes aegypti and Aedes albopictus. Acta Trop. 2018;178:155–162. doi: 10.1016/j.actatropica.2017.11.020. [DOI] [PubMed] [Google Scholar]
- 47.Cheong Y.L., Leitão P.J., Lakes T. Assessment of land use factors associated with dengue cases in Malaysia using boosted regression trees. Spat Spatiotemporal Epidemiol. 2014 doi: 10.1016/j.sste.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 48.Yan Z., Chen D., Teng Z., Wang D., Li Y. SMOPredT4SE: an effective prediction of bacterial Type IV secreted effectors using SVM training with SMO. IEEE Access. 2020;8:25570–25578. doi: 10.1109/ACCESS.2020.2971091. [DOI] [Google Scholar]
- 49.Ashari Z.E., Brayton K.A., Broschat S.L., Esna Ashari Z., Brayton K.A., Broschat S.L. Using an optimal set of features with a machine learning-based approach to predict effector proteins for Legionella pneumophila. PLoS ONE. 2019;14 doi: 10.1371/journal.pone.0202312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lupolova N., Lycett S.J., Gally D.L. A guide to machine learning for bacterial host attribution using genome sequence data. Microb. Genomics. 2019;5 doi: 10.1099/mgen.0.000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davi C, Pastor A, Oliveira T, Neto FB de L, Braga-Neto U, Bigham AW, et al. Severe Dengue Prognosis Using Human Genome Data and Machine Learning. IEEE Trans Biomed Eng 2019;66:2861–8. doi:10.1109/TBME.2019.2897285. [DOI] [PubMed]
- 52.Chen H, Shen J, Wang L, Song J. Leveraging Stacked Denoising Autoencoder in Prediction of Pathogen-Host Protein-Protein Interactions. 2017 IEEE Int. Congr. Big Data (BigData Congr., IEEE; 2017, p. 368–75. doi:10.1109/BigDataCongress.2017.54.
- 53.Buczak A.L., Baugher B., Babin S.M., Ramac-Thomas L.C., Guven E., Elbert Y. Prediction of high incidence of dengue in the Philippines. PLoS Negl Trop Dis. 2014;8 doi: 10.1371/journal.pntd.0002771. [DOI] [Google Scholar]
- 54.Saez-Rodriguez J., Costello J.C., Friend S.H., Kellen M.R., Mangravite L., Meyer P. Crowdsourcing biomedical research: leveraging communities as innovation engines. Nat Rev Genet. 2016;17:470–486. doi: 10.1038/nrg.2016.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Costello J.C., Heiser L.M., Georgii E., Gönen M., Menden M.P., Wang N.J. A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol. 2014;32:1202–1212. doi: 10.1038/nbt.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jani M., Sengupta S., Hu K., Azad R.K. Deciphering pathogenicity and antibiotic resistance islands in methicillin-resistant Staphylococcus aureus genomes. Open Biol. 2017;7 doi: 10.1098/rsob.170094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brierley L., Pedersen A.B., Woolhouse M.E.J. Tissue tropism and transmission ecology predict virulence of human RNA viruses. PLoS Biol. 2019 doi: 10.1371/journal.pbio.3000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J., Yang B., An Y., Marquez-Lago T., Leier A., Wilksch J. Systematic analysis and prediction of type IV secreted effector proteins by machine learning approaches. Brief Bioinform. 2019;20:931–951. doi: 10.1093/bib/bbx164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sen R., Nayak L., De R.K. PyPredT6: A python-based prediction tool for identification of Type VI effector proteins. J Bioinform Comput Biol. 2019;17:1950019. doi: 10.1142/S0219720019500197. [DOI] [PubMed] [Google Scholar]
- 60.Wang J, Yang B, Leier A, Marquez-Lago TT, Hayashida M, Rocker A, et al. Bastion6: a bioinformatics approach for accurate prediction of type VI secreted effectors. Bioinformatics 2018;34:2546–55. doi:10.1093/bioinformatics/bty155 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated or analyzed during our survey are available from the corresponding author upon reasonable request.