

Abstract
Despite advances in treatment, the prognosis for glioma patients remains poor. Bromodomain-containing protein 4 (BRD4), a member of the bromodomain and extraterminal (BET) protein family, plays an important role in controlling oncogene expression and genome stability. In recent years, numerous BRD4 inhibitors have entered clinical trials and achieved exciting results in tumor treatment. Recent clinical studies have shown that BRD4 expression in glioma is significantly higher than in the adjacent normal brain tissue. BRD4 inhibitors effectively penetrate the blood-brain barrier and target glioma tumor tissues but have little effect on normal brain tissues. Thus, BRD4 is a target for the treatment of glioma. In this study, we discuss the progress in the use of BRD4 inhibitors for glioma treatment, their mechanism of action, and their broad potential clinical application.
Keywords: glioma, bromodomain and extraterminal protein family, BRD4, small molecular inhibitors
Graphical abstract
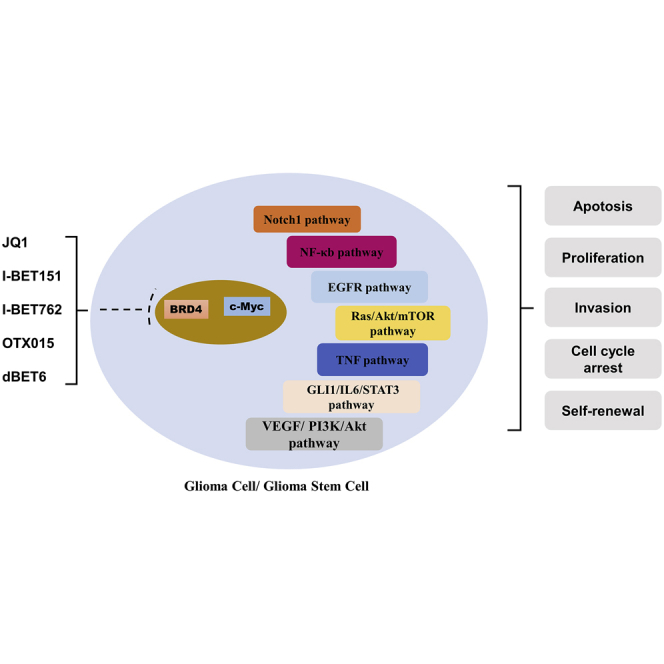
BRD4, a member of BET protein family, has been implicated in cell proliferation, invasion, self-renewal, apoptosis, and cell cycle arrest through multiple pathways. Thus, inhibition of BRD4 seems a prospective therapy. In this review, we discuss the progress of BRD4 inhibitors in glioma treatment and their broad potential clinical application.
Introduction
Gliomas are the most common primary intracranial tumors, accounting for more than 30% of central nervous system neoplasias.1 Gliomas have been traditionally classified by the World Health Organization (WHO) into grades I–IV, which describe them in terms of histopathological lineage as either astrocytoma, oligodendroglioma, or oligoastrocytoma.1 Grades I and II are considered low-grade gliomas, while grades III and IV are considered high-grade gliomas. Most patients have high-grade gliomas, and higher grades generally have a less favorable prognosis.2 The traditional standard management of gliomas involves observation, surgery, chemotherapy, and/or radiotherapy.2
In 2016, a new classification of the WHO recommended molecular diagnosis of the metabolic gene isocitrate dehydrogenase (IDH) mutational status in gliomas. IDH-mutated and IDH wild-type tumors are driven by different oncogenic pathways and respond differently to current therapeutic regimens.1 IDH mutant gliomas are often represented by low-grade cases and have better prognosis. Moreover, IDH mutant tumors are more likely to undergo total resection with low incidence of long-term neurological deficits. Postoperative adjuvant radiotherapy combined with vincristine and/or temozolomide (TMZ)-based chemotherapy is recommended.1 Additionally, the WHO 2016 classification also added a new subtype under grade IV gliomas, that is, H3K27M mutant tumors, which mainly occur in children and younger patients and are characterized by poor prognosis.3
The efficacy of traditional therapeutic approaches is limited, especially for glioblastoma (GBM) cases.4 New treatments have been developed, such as tumor-treating fields (by low-intensity, alternating, electric fields delivered by transducer arrays applied to the scalp for antimitotic therapy),3 immune checkpoint inhibitors (anti-CTLA-4, anti-PD-1, anti-PD-L1),5 angiogenesis inhibitors (bevacizumab),3 IDH inhibitors (enasidenib and ivosidenib),6 oncolytic viruses,7 and tumor vaccines.8
It is unclear whether other genetic changes, apart from IDH mutations, can condition glioma progression; however, epigenetic regulatory mechanisms are gaining a growing interest. Epigenetic modifications are heritable and reversible alterations to DNA, which do not involve changes in the nucleotide sequence. Epigenetics play an important role in maintaining homeostasis in normal cells via changes in CpG island methylation patterns and histone modifications that regulate gene expression. Epigenetic proteins have recently emerged as novel anticancer targets. Among these, the bromodomain and extraterminal (BET) family of proteins has gained increasing attention due to its critical role in recruiting chromatin-regulating enzymes and “reading” chromatin to control gene expression in cancer development.9 In particular, rearrangement or mutations of tumor-associated genes regulated by bromodomain-containing protein 4 (BRD4) have been identified in glioma studies.
This review focuses on the role of BRD4 dysfunction in promoting glioma development. In addition, the application of small-molecule inhibitors targeting specific BET proteins, as a novel therapeutic strategy for glioma patients, is discussed.
BET bromodomain proteins
The BET family consists of four proteins: BRD2, BRD3, BRD4, and BRDT (specifically expressed in testis).9 The BET proteins are characterized by two tandem, conserved, bromodomain motifs (BD1 and BD2), one extraterminal (ET) domain, and a C-terminal motif (CTM).10 The bromodomain structure consists of four reverse parallel α helices and two circular structures, one Za loop and a BC loop, connecting the helices. These structures form a hydrophobic cavity that recognizes acetylated lysine residues. Each BET bromodomain binds to two acetyl-lysine histone markers. In addition, BET proteins interact with the transcription elongation complex and with other transcription factors.11 In summary, BET proteins are key “readers” of epigenetic information in both normal and transformed cells.
BRD4: The most studied BET protein
BRD4 is a double bromodomain (BD1 and BD2)-containing protein. In addition, BRD4 comprises an additional terminal clustering domain (ET), a CTM, and several conserved motifs.12 Multiple studies have indicated a clear role for BRD4 in the regulation of transcription by interaction with acetylated histones (e.g., H3K9ac, H3K14ac, H3K27ac, H4K5ac, H4K8ac, H4K12ac, and H4K16ac)13,14 and non-histonic proteins (e.g., RelA subunit of nuclear factor κB [NF-κB],15,16 TWIST,17 and GATA1).17 Moreover, and possibly most importantly for cancer, BRD4 is involved in the recruitment of the transcriptional machinery to gene targets (such as c-Myc, Tp53, c-Jun) through its interactions with positive transcription elongation factor complex (pTEFb) and RNA polymerase II (RNA Pol II) via its CTM12 (Figure 1). In addition, a non-transcriptional role of BRD4 in regulating DNA replication and repair,18 telomere maintenance,19 and protein kinase activity20 has been reported.
Figure 1.

Interactions with BRD4 domains
BD1 and BD2 bind to acetylated histone and non-histone proteins, an extra-terminal (ET) domain interacts with several chromatin regulators, and a C-terminal motif (CTM) interacts with pTEFb to facilitate transcription factors.
BRD4 is widely expressed in human tissues, cancer cells,13,21 cancer-associated fibroblasts,22 and chronic inflammatory fibroblasts (e.g., liver,23 lung,24 cardiovascular,25 and renal26). In fact, BRD4 is considered the dominant transcriptional regulator within the BET protein family,27 and it plays a significant role in the onset and progression of malignant human diseases, especially by controlling tumor cell proliferation and apoptosis, making it the most studied BET protein in cancer therapy.10,17,28, 29, 30 For example, loss of BRD4, but not of BRD2 or BRD3, causes R-loop-induced DNA damage.31 Clinical studies have validated the efficacy of targeting BRD4, and not BRD2 or BRD3, in glioma,32 breast cancer,33 and prostate cancer34 therapy.
Recent reports have demonstrated that an abnormal expression and/or dysfunction of BRD4 is associated with the development of glioma, lung cancer, melanoma, breast cancer, colorectal cancer, and acute myeloid leukemia. Thus, BRD4 is a potential therapeutic target for many malignant tumors.35
BRD4 inhibitors
BRD4 is not only crucial for the regulation of gene transcription, but it is also a keeper of genome stability. Thus, BET inhibitors have a wide spectrum of prospects for the treatment of cancer.36 Although BRD4 inhibitors have shown exciting antitumor effects, the selectivity of these drugs needs improvement. In fact, since current BRD4 inhibitors all bind to the bromodomain structure (also found in other BET family proteins), none is selective for BRD4 only.
Small-molecule inhibitors of the first generation bind to BET bromodomains as competitors of acetyl-lysine, thus displacing BET protein complexes from chromatin. According to drug selectivity, BRD4 small-molecule inhibitors are subdivided into pan-bromodomain inhibitors (which do not discriminate between the two bromodomains), BD1-selective inhibitors, and BD2-selective inhibitors27 (Figure 2). The pan-bromodomain inhibitors belonging to the triazoloazepine or isoxazole derivative classes are the most studied. JQ1 was the first BRD4 bivalent triazoloazepine inhibitor developed in 2010. JQ1 has strong inhibitory effects on both BD1 and BD2, with a 50% inhibitory concentration (IC50) of 77 nM and a diffusion constant (Kd) of 50 nM.37 Two other well-known BRD4 pan-bromodomain inhibitors are OTX01538 and TEN-010.35 OTX015 has shown efficacy not only in primary hematological and solid cancers but also in refractory or relapsed hematological cancers.39 While the latter inhibitors are triazoloazepines, I-BET151 is the most promising isoxazole-based BRD4 inhibitor (IC50 of 794 nM). I-BET151 showed beneficial effects in the treatment of GBM and leukemia.35,40,41 In addition, CPI-0610, PLX51107, and INCB0543294 belong to isoxazole-based BRD4 inhibitors.35
Figure 2.

Drug selectivity of BRD4 small-molecule inhibitors
As the inhibition of individual BET bromodomains will lead to different transcriptional and functional outcomes, chemists have continued to optimize BRD4 inhibitors with the aim to improve their selectivity. BD1 selective inhibitors, such as GSK778, MS-436, Olinone, and BI-2536, as well as the BD2 selective inhibitors RVX-208, RVX-297, GSK046, and ABBV-744 have been produced.27,42
The second-generation BRD4 inhibitors are mainly synthesized by proteolysis targeting chimera (PROTAC) technology. This approach implicates the use of a molecule that binds both the protein to be degraded and the E3 ubiquitin ligase. Such bridging between the target protein and E3 ligase mediated by PROTAC promotes its ubiquitination and proteasomal degradation.35 For example, BET degraders, called dBETs, bind to both the E3 ubiquitin ligase cereblon (similar to the drug thalidomide) and BET proteins.43 As a second-generation BET degrader, dBET6 is a highly cell-permeable inhibitor of BET bromodomains. Compared to JQ144 and other BET bromodomain inhibitors (BBIs),14 dBET6 is substantially more active.
A variety of BRD4 inhibitors have entered clinical trials. We summarize all clinical trials with BRD4 inhibitors, including one for glioma patients, in Table 1. Actually, the half-life of JQ1 is relatively short, which has hindered its further application in vivo.18 dBet6 is a newly developed drug, which has not been used in clinical trials yet. In addition to these, a series of new drugs, including I-BET762 (GSK525762),45 I-BET151 (GSK2820151),40,41,45 and OTX015/MK-8628,46 have improved risk-benefit profiles in multiple cancers. However, most of the BRD4 inhibitors listed in Table 1 have been tested in GBM cell lines and mouse tumor models. Only one clinical trial was conducted (ClinicalTrials.gov: NCT02296476) by the application of OTX015 in recurrent GBM, after standard front-line therapy failure. Participants received 80, 120, or 160 mg of oral OTX015 administered once daily every day in a 28-day cycle for up to six cycles. Unfortunately, this study was terminated due to lack of clinical efficacy and not due to safety reasons.
Table 1.
Clinical trials of BRD4 inhibitors in oncology
| No. | Name | BRD4 inhibitors/degraders | Binding site | Structure features | Condition | Clinical trials identifier (ClinicalTrials.gov) |
|---|---|---|---|---|---|---|
| 1 | I-BET151 (GSK2820151) | small-molecule inhibitors | BD1 and BD2 | isoxazoles | solid tumors | NCT02630251 (I-BET151 was terminated due to development of GSK525762 with a better understanding of the risk-benefit profile) |
| 2 | I-BET762 (GSK525762) | small-molecule inhibitors | BD1 and BD2 | triazoloazepines | solid tumors and blood tumors | NCT02964507 (active, not recruiting) |
| NCT03266159 (withdrawn before active to fully evaluate the impact of changing practice in the target population) | ||||||
| NCT03150056 (active, not recruiting) | ||||||
| NCT03702036 (no longer available) | ||||||
| NCT03925428 (not yet recruiting) | ||||||
| NCT01943851 (completed) | ||||||
| NCT01587703 (completed) | ||||||
| NCT02706535 (completed) | ||||||
| NCT03702036 (no longer available) | ||||||
| 3 | OTX015/MK-8628 | small-molecule inhibitors | BD1 and BD2 | triazoloazepines | acute leukemia, B cell lymphoma, multiple myeloma, glioblastoma multiforme | NCT01713582 (completed) |
| NCT02259114 (completed) | ||||||
| NCT02296476 (terminated due to lack of clinical activity and not due to safety reasons) | ||||||
| NCT02303782 (withdrawn) | ||||||
| NCT02698176 (terminated due to limited efficacy and not due to safety reasons) | ||||||
| 4 | JQ1 | small-molecule inhibitors | BD1 and BD2 | triazoloazepines | solid tumors and blood tumors | no studies |
| 5 | dBET6 | degraders | BD1 and BD2 | triazoloazepines | solid tumors | no studies |
Clinical trial data are from https://clinicaltrials.gov/.
The side effects and drug resistance profiles of BRD4 inhibitors still deserve attention. In clinical studies, BRD4 inhibitors have shown adverse reactions, including thrombocytopenia, fatigue, headache, and hyperbilirubinemia. Additionally, low expression of BRD4 may be associated with a decreased number of hematopoietic cells, skin hyperplasia with abnormal hair follicles, and intestinal crypt rupture with loss of secretory cells. Moreover, BRD4 inhibitors target BRD2/3 simultaneously, and low expression of BRD2/3 resulting from BRD4 inhibition therapy may relate to neuronal defects and obesity.20 Mechanisms of BRD4 inhibitor resistance were identified in a preclinical model, which may be related to activation of oncogenic pathways, such as WNT/β-catenin/MYC (acute myeloid leukemia [AML]), Hedgehog/GLI2/MYC (pancreatic cancer), mitogen-activated protein kinase (MAPK) (colorectal cancer), or RAS/BCL2 (lymphoma).20
BET proteins in glioma
The dysregulation of BET proteins, in particular BRD4, has been implicated in the development of cancers, including gliomas.36 Numerous BRD4 inhibitors have been studied in recent years, and some are currently in clinical trials. Recent clinical data demonstrated that BRD4 is a valuable target for treating glioma.47 Interestingly, BRD4 inhibitors effectively penetrate the blood-brain barrier (BBB)46,48,49 and target glioma tumor tissues, while having have little effects on normal brain tissue.46
BRD4 expression in glioma
The expression of BRD4 in glioma tissues and cell lines is significantly higher than in normal tissues and cells.36 In 2014, Pastori et al.40 first discovered that BRD4 mRNA is significantly overexpressed in glioma. NanoString technology was used to determine the expression of 40 bromodomain proteins in 27 gliomas compared to 9 control samples isolated from epilepsy patients. BRD4 expression was 1.8-fold higher in glioma samples relative to controls, and the p value was less than 0.05. Wang et al.50 also found that BRD4 expression is increased in glioma compared to adjacent tissues. Analyses of online datasets indicated that BRD4 expression was negatively correlated with the overall survival of glioma patients. Many subsequent studies have explored the role of BRD4 in glioma.50, 51, 52 Genome-wide transcriptional analysis of BRD4-regulated genes and pathways included multiple cellular processes, such as cell cycle and apoptosis in human glioma cells.52
BRD4 inhibitor therapy for glioma
Knockdown of BRD4 by lentivirus or small interfering RNA (siRNA) has been proven to affect the occurrence and development of glioma.50, 51, 52 The use of BRD4 inhibitor monotherapy for glioma shows satisfactory results in both glioma cell lines or glioma stem cells (GSCs) (Table 2).
Table 2.
Summary of the progress of BRD4 inhibitor monotherapy in glioma
| No. | Author | Research objects | Mutation of cell line | Treatment regimen | Mechanisms | Related pathways | Genes/targets | Conclusion |
|---|---|---|---|---|---|---|---|---|
| 1 | Cheng et al.53 | T4105, T4302, and T4597 primary GBM xenograft cell lines | N/A | JQ1 | G1 cell cycle arrest (+), apoptosis (+) | N/A | c-Myc, p21 (CIP1/WAF1), hTERT, Bcl-2, and Bcl-xL | (1) JQ1 induced G1 cell cycle arrest and apoptosis |
| (2) JQ1 treatment resulted in changes in expression of c-Myc, p21 (CIP1/WAF1), hTERT, Bcl-2, and Bcl-xL | ||||||||
| 2 | Liu et al.54 | U87, U87 EGFRvIII, LN229, U373, GBM6 cell lines | EGFRvIII mutations | JQ1 | apoptosis (+) | SOX9 and FOXG1 | EGFRvIII sensitizes GBM cells to JQ1-induced cell death through SOX9 and FOXG1 | |
| 3 | Piunti et al.55 | SF8628, SF7761 and SU-DIPG-IV, pcGBM2, SF9402, and SF9427 primary pediatric human cell lines and mice | H3K27M mutations | JQ1 | proliferation (−) | N/A | c-Myc, CDKN1A (p21), TUBB3 (Tuj1), and MAP2 | (1) JQ1 induces growth arrest through the canonical reduction in c-Myc transcription |
| (2) mice treated with JQ1 for 10 days exhibited significantly reduced tumor size and prolonged animal survival | ||||||||
| 4 | Fahey et al.56 | U87 and U251 GBM cell lines | N/A | JQ1 | proliferation (−), invasion (−) | NF-κB p65/RelA pathway | iNOS, survivin, and Bcl-xL | JQ1 showed better inhibition of iNOS expression, endogenous NO production, and malignant biological behavior of GBM tumor cells than did iNOS inhibitors |
| 5 | Wen et al.51 | CSC2078 and CSC1589 primary murine GSC lines and mice | N/A | JQ1 | proliferation (−), self-renewal (−) | VEGF/PI3K/AKT pathway | MMP, AKT downstream target genes | (1) JQ1 inhibited the proliferation and self-renewal of GSCs |
| (2) JQ1 has notable anti-tumor effects against GBM, which may be mediated via the VEGF/PI3K/AKT signaling pathway | ||||||||
| (3) JQ1 significantly inhibited the growth of GSC tumors in vivo | ||||||||
| 6 | Pastori et al.40 | U87MG, A172, SW1783, and UM20 (from patient) GBM cell lines and mice | N/A | I-BET151 | Cell proliferation (−) | N/A | p21cipl, HEXIM-1 | (1) I-BET151 inhibits the proliferation of GBM cells by reducing cell cycle progression at the G1/S phase |
| G1/S cell cycle arrest (+) | (2) I-BET151 is as potent at inhibiting GBM cell proliferation as TMZ | |||||||
| 7 | Pastori et al.41 | LN18, U87MG, A172, and T98G GBM cell lines and mice | N/A | I-BET151 | proliferation (−) | N/A | HOTAIR, MEG3, NEAT1, DGRR5 | (1) I-BET151 can directly reduce expression levels of lncRNA HOTAIR |
| (2) GBM cells overexpressing HOTAIR could eliminate the anti-proliferative effect of I-BET151 | ||||||||
| 8 | Tao et al.32 | U87, U251 GBM cell line and GSCs (CD133+) and mice | N/A | I-BET-151 | proliferation (−) | Notch1/NICD/Hes1 pathway | Notch1, NICD, Hes1 | (1) I-BET151 is able to reduce proliferation and self-renewal of GSCs |
| self-renewal (−) | (2) BRD4 is mainly located at the promoter region of Notch1 and may be involved in the process of tumor metabolism | |||||||
| (3) I-BET151 eliminated GSC tumorigenicity in the mouse intracranial models | ||||||||
| 9 | Berenguer-Daizé et al.46 | U87MG, T98G, UI18 cell lines and mice | N/A | OTX015 | cell cycle arrest (+) | Ras/Akt/mTOR pathway | c-MYC, CDKN1A, BRD2, BRD3, SESN3, HEXIM-1, HIST2H2BE, HIST1H2BK, MTHFDIL, HIST2H4A, and HIST1H2BJ | (1) OTX015 inhibits the proliferation of GBM cells |
| (2) OTX015 significantly increases survival in GBM mice | ||||||||
| (3) OTX015 can effectively penetrate the blood-brain barrier | ||||||||
| 10 | Xu et al.14 | A172, T98G, U87, U138, U251, and U343 GMB cell lines and mice | N/A | dBET6 | G2/M cell cycle arrest (+) | N/A | CEBPB, RUNX1, FOSL2, and STAT3 | (1) dBET6 interfered with GBM transcriptional regulation, which was controlled by BET and E2F1 |
| self-renewal (−) | (2) dBET6 inhibits GBM cell proliferation better than JQ1, I-BET151, and OTX015 | |||||||
| tumorigenic ability (−) |
NO, nitric oxide; iNOS, inducible NO synthase; GSC, glioma stem cell; DIPG, diffuse intrinsic pontine glioma; N/A, not applicable.
BRD4 inhibitor treatment of glioma cells
Many studies have investigated the effects of JQ1 in glioma. In fact, the therapeutic use of JQ1 for treating glioma was first reported in 2013. Cheng et al.53 discovered the antitumor effects of JQ1 by treating ex vivo cultures derived from xenografts of primary GBM cells. JQ1 induced G1 cell-cycle arrest and apoptosis by affecting the expression of c-Myc, p21 (CIP1/WAF1), hTERT, Bcl-2, and Bcl-xL. JQ1 is effective on glioma cell lines with different mutational status. Liu et al.54 demonstrated that JQ1 suppressed aggressive growth of GBM cells carrying oncogenic epidermal growth factor receptor (EGFR) mutations. Additionally, since the H3K27M histone mutation is commonly found in diffuse intrinsic pontine glioma (DIPG), BRD4 inhibitor is considered a potential therapeutic target for this disease.55 JQ1 induces growth arrest through a typical decrease of c-Myc transcription in H3K27M DIPG cell lines. In vitro, a xenograft mouse model of DIPG treated with JQ1 for 10 days also exhibited significantly reduced tumor size and prolonged animal survival.
Previous studies reported an association of BRD4 with the expression of inducible nitric oxide (NO) synthase (iNOS) in macrophages57 and microglia.58 In GBM, Fahey et al.56 found that JQ1 could greatly improve the clinical outcome of photodynamic therapy (PDT) by inhibiting iNOS expression. In fact, PDT is an effective approach to treat GBM. However, PDT oxidative challenge results in prolonged upregulation of iNOS in GBM U87 cells, and endogenous NO generated by iNOS promotes proliferation, invasion, and resistance to apoptosis in GBM cells. JQ1 can inhibit iNOS expression, endogenous NO production, and malignant biological behavior of GBM cells better than iNOS inhibitors. In mechanistic terms, NF-κB–regulated iNOS expression in GBM cells, and BRD4 can act as a NF-κB co-activator, which explains JQ1-induced enhancement of PDT cytotoxicity. In fact, JQ1 at minimally toxic concentrations significantly suppressed the upregulation of iNOS, survivin (a potent inhibitor of apoptosis), and Bcl-xL (a NF-κB-regulated anti-apoptotic protein) in response to PDT.56
Additional BRD4 inhibitors appear to be useful in the treatment of glioma.
Pastori et al. suggested that inhibition of BET protein is an effective way to reduce GBM cell proliferation. The disruption of BRD4 expression in GBM cells impaired cell cycle progression at the G1/S transition point. Similarly, I-BET151 inhibited glioma cell proliferation in vitro and in vivo. Importantly, I-BET151 is as potent an inhibitor of GBM cell proliferation in vitro as TMZ, which is the main treatment currently administered to GBM patients. Therefore, BRD4 protein inhibitors might be considered as an alternative therapy for TMZ-resistant GBM patients.40 BRD4 can also promote tumorigenesis and development of GBM cells in vitro and in vivo via interfering noncoding RNA. Researchers analyzed the differential expression of long noncoding RNAs (lncRNAs) in GBM using Helicos single-molecule sequencing. A subset of glioma-specific, BET-regulated lncRNAs, including HOX transcript antisense RNA (HOTAIR), was identified. I-BET151 significantly reduced HOTAIR levels and restored the expression of several other lncRNAs downregulated in GBM. In contrast, the overexpression of HOTAIR in GBM cells treated with I-BET151 abrogated the anti-proliferative effects of the BET inhibitor. In addition, chromatin immunoprecipitation analysis demonstrated that BRD4 directly bound to the HOTAIR promoter region, suggesting that BET proteins can directly regulate lncRNA expression.41
In 2016, Berenguer-Daizé et al.46 found that the novel BRD4 inhibitor, OTX015 (MK-8628), showed better anti-proliferative effects than its analog JQ1. Results in three additional glioma cell lines supported the anti-proliferative effects of OTX015. Furthermore, single oral administration of OTX015 significantly increased survival in mice bearing in situ or ectopic U87MG xenografts. Surprisingly, OTX015 effectively penetrated the BBB, and the concentration of OTX015 in glioma tumor tissue was 7- to 15-fold higher than in surrounding normal tissue.
In 2018, Xu et al.14 explored the effects of dBET6, a new generation BRD4 inhibitor that chemically degrades BET proteins. G2/M cell cycle transition, cancer cell self-renewal, and the tumorigenic potential of GCSs were significantly inhibited by dBET6. In addition, dBET6 drastically reduced BET protein genomic occupancy, RNA Pol II activity, and permissive chromatin marks. Mechanistic research showed that dBET6 interfered with glioma transcriptional regulation, which is controlled by BET and E2F1. Moreover, dBET6 inhibits glioma cell proliferation better than traditional BET inhibitors (e.g., JQ1, I-BET151, OTX015) and overcomes both intrinsic and acquired resistance to traditional BBIs. More than 70% of the glioma cell lines showed intrinsic refractoriness to at least one BBI. Moreover, glioma cells can develop adaptive tolerance and cross-resistance to BBIs after prolonged treatment. BBI resistance of glioma cells may relate to 5p15 amplicon or TERT expression being significantly upregulated. Since glioma cells with either intrinsic or acquired BBI resistance still relied on the presence of BET proteins, cell lines with acquired resistance to BBIs may still be sensitive to the activity of the BET protein degraders. Notably, RNA sequencing (RNA-seq) uncovered that dBET6 and JQ1 elicited different transcriptomic responses in glioma cells. dBET6-responsive events in acquired resistant cell lines to BBIs were largely unaffected, indicating that dBET6 can overcome acquired insensitiveness to BBIs by degrading the BET protein in glioma cells.14
BRD4 inhibitor therapy for GSCs
Multiple studies have demonstrate that BRD4 regulates stem cell differentiation. In particular, Wen et al.51 focused on the antitumor effects of JQ1 on GSCs. JQ1 and siRNAs targeting BRD4 (siBRD4) could inhibit GSC proliferation and self-renewal in vitro. In addition, JQ1 significantly inhibited the growth of GSC tumor xenografts in mice. Mechanistic studies suggest that JQ1 inhibited the development of GBM by interfering with the VEGF/phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway. In fact, JQ1 inhibited VEGF receptor-2 phosphorylation in GSCs, thereby reducing PI3K and AKT activity. In addition, JQ1 inhibits the expression of MMP, thereby reducing extracellular matrix degradation and angiogenesis in GBM. Additionally, the treatment with JQ1 induced apoptosis by activating AKT downstream target genes.
A signaling pathway involving BRD4 and Notch1 was also found to reduce GSC proliferation and self-renewal.32 In vitro, I-BET151 reduces the proliferation and self-renewal of GSCs. Mechanistic studies showed that BRD4 is largely associated with the promoter region of Notch1, which may be involved in tumor metabolism. Finally, I-BET151 obstructed the tumorigenic potential of GSCs in intracranial injection mouse models.
In conclusion, BRD4 inhibitors inhibit the development of glioma or GSCs by interfering with cell cycle progression, proliferation, and/or apoptosis in vivo and in vitro. This has been associated with interference with NF-κB, Akt and Notch signaling pathways, transcriptional regulation of oncogenes (such as c-Myc, Bcl-2, and Bcl-xL), lncRNA expression, and inhibition self-renewal of GSCs (Figure 3). Since I-BET151 is as potent at inhibiting GBM cell proliferation as TMZ,40 BRD4 inhibitors might represent an alternative substitute for TMZ in GBM patients.
Figure 3.

Schematic diagram showing the BRD4 inhibitor therapy for GBM
(A–C) The mechanisms of BRD4 inhibitors in the treatment of GBM (A), glioma stem cells (B), and H3K27M mutated glioma cells and EGFRvIII mutated glioma cells (C). HOTAIR, lncRNA HOX transcript antisense RNA; GBM, glioblastoma.
Combination therapy including BRD4 inhibitor for glioma
Cancers are often developing resistance to monotherapies. Notably, recent studies have shown that combinatorial therapeutic regimens including BRD4 inhibitors can show striking effects (Table 3).
Table 3.
Summary of the combination therapy including BRD4 inhibitor for glioma cells
| No. | Author | Research objects | Mutation of cell line | Treatment regimen | Mechanisms | Related pathways | Genes/targets | Conclusions |
|---|---|---|---|---|---|---|---|---|
| 1 | Zhang et al.59 | DIPG cell lines isolated from the dorsal forebrain of mouse embryos and mice | H3K27M mutations | JQ1 + EPZ6438 (EZH2 inhibitor) | proliferation (−) | N/A | p16Ink4a, Igf2bp2, and HOXA10 | (1) in vitro and in vivo, the combination of JQ1 and EPZ6438 exhibited better inhibition of the tumor growth compared to use the inhibitor alone |
| apoptosis (+) | (2) the inhibition was performed by blocking proliferation and promoting cell apoptosis | |||||||
| 2 | Nagaraja et al.60 | SU-DIPG-IV: H3.1-K27M; SU-DIPG-VI/XIII-P, JHH-DIPG1, SF7761: H3.3-K27M patient-derived DIPG cell lines and mice | H3K27M mutations | JQ1 + panobinostat (HDAC inhibitor) | proliferation (−) | N/A | NTRK3, LINGO1, ASCL1, SYT4, SYT17, MYT1, MYRF, and SALL3 | (1) JQ1 together with panobinostat synergistically inhibited cell proliferation and induced apoptosis |
| apoptosis (+) | ||||||||
| 3 | Nagaraja et al.60 | SU-DIPG-IV: H3.1-K27M; SU-DIPG-VI/XIII-P, JHH-DIPG1, SF7761: H3.3-K27M patient-derived DIPG cell lines and mice | H3K27M mutations | JQ1 + THZ1 (CDK7 inhibitor) | proliferation (−) | N/A | ETS1, ELF4, MGA, SOX10, and HES5 | the combination of JQ1 and THZ1 had a synergistic inhibitory effect on cell viability |
| 4 | Wiese et al.61 | VUMC-DIPG-10 DIPG cell line and SF188 pedHGG cell lines | H3K27M mutations | JQ1 + ICG-001 (CREB-binding protein inhibitor) | proliferation (−) | N/A | MYC, MYC-associated factor X, JUND | combined treatment of JQ1 and ICG-001 induced stronger cytotoxic effects than did either drug alone in H3K27M-mutated DIPG cell lines |
| 5 | Meng et al.62 | U87 and U251 GBM cell lines | N/A | JQ1 + panobinostat | proliferation (−) | TNF pathway, PI3K/mTOR pathway, insulin receptor pathway, biosynthesis of antibiotics and FoxO signaling pathway | caspase-3, caspase-7, CCND1, MKI67, TOP2A, FOXO3, p21, and BNIP3 | markedly inhibited cell proliferation and induced apoptosis |
| apoptosis (+) | ||||||||
| 6 | Lam et al.63 | GMB tumor-bearing mice | N/A | JQ1 + TMZ (loaded by Tf-NPs) | DNA damage (+) | N/A | N/A | (1) TMZ plus JQ1 is additive in gliomas |
| apoptosis (+) | (2) treatment of tumor-bearing mice with Tf-NPs loaded with TMZ and the JQ1 leads to increased DNA damage and apoptosis | |||||||
| (3) Tf-NPs loaded with TMZ and JQ1 show a decrease of 1.5- to 2-fold in tumor burden and an increase in survival compared to equivalent free-drug dosing | ||||||||
| 4. Tf-NP therapies protect from systemic drug toxicity | ||||||||
| 7 | Wang et al.64 | CSC2078, CSC1534, and CSC1589 murine GSCs and mice | N/A | JQ1 + RGFP966 (HDAC inhibitor) | proliferation (−) | GLI1/IL-6/STAT3 pathway | c-Myc, cyclin D1, Bcl-2, Bcl-xL, p21, Bim, and Bax | (1) JQ1/RGFP966 combination can suppress GSC growth by blocking the GLI1/IL-6/STAT3 signaling axis in vitro |
| (2) in vivo, the JQ1/RGFP966 combination caused stronger tumor growth suppression than did either drug alone | ||||||||
| 8 | Meng et al.62 | U87 and U251 GBM cell lines | N/A | OTX015 + panobinostat | proliferation (−) | N/A | CCND1, MKI67, TOP2A, FOXO3, p21, and BNIP3 | markedly inhibited cell proliferation and induced apoptosis |
| apoptosis (+) | ||||||||
| 9 | Berenguer-Daizé et al.46 | U87MG, T98G, and UI18 cell lines and mice | N/A | OTX015 + SN38/TMZ/everolimus (cell lines) | G2/M cell cycle arrest (+) | transcriptional regulatory network involving CEBPB, RUNX1, FOSL2, and STAT3 | genes with E2F binding, RNA Pol II, H3K27ac, H3K4me3, H3K27me3, and H3K9me3 | (1) showed synergistic antitumor effect |
| OTX015 + TMZ (mice) | self-renewal (−) | (2) improved survival of mice by suppressing tumorigenic ability over either single agent | ||||||
| tumorigenic ability(−) | ||||||||
| 10 | Ishida et al.65 | NCH644, NCH690, and NCH421K stem-like GBM cells | N/A | OTX015 + imipridones (AKT/ERK inhibitor) | proliferation (−) | mTORC1 pathway | c-Myc, Bcl-2, Bcl-xL, Mcl-1, mTOR, 4EBP1, S6K, S6 | Induced apoptosis in stem-like GBM cells |
| apoptosis (+) | ||||||||
| energy metabolism (−) | ||||||||
| 11 | Zanca et al.45 | U87, mAstr–Ink4a/Arf−/−, U178, and U373 GBM cell lines and mice | EGFRvIII mutations | JQ1/ I-BET-151/ I-BET-762 + EGFR TKIs | proliferation (−) | NF-κB pathway | NF-κB and survivin | IL-6 mediates resistance to EGFR TKIs in GBM and BRD4 inhibitors restores sensitivity of GBM cells to EGFR TKIs |
| 12 | Yao et al.66 | U251 and U87 GBM cell lines and mice | N/A | I-BET151 + TMZ | apoptosis (+) | N/A | PUMA | (1) I-BET151 could augment the effect of TMZ on GBM cells |
| (2) I-BET151 increased the TMZ-induced apoptosis in GBM cells by enhancing the activities of caspase-3 | ||||||||
| I-BET151 increased the amount of reactive oxygen species, superoxide anions with a decrease of activity of SOD, and the anti-oxidative properties of GBM cells; I-BET151 also induced increased PUMA expression |
pedHGG, pediatric high-grade glioma, HDAC: histone deacetylase; Tf-NP, transferrin-functionalized nanoparticle.
Combination therapy including BRD4 inhibitor for glioma cells
JQ1-based combination therapies are the most studied. For glioma cell lines, the association of JQ1 with EPZ6438 (EZH2 inhibitor)59/panobinostat (histone deacetylase inhibitor)60,62/THZ1 (CDK7 inhibitor)60/ICG-001 (CREB-binding protein inhibitor)61 achieved higher inhibition of tumor growth compared to each inhibitor alone. The mechanism may be related to the activation of caspase-3/7 induced by panobinostat plus JQ1.62 Notably, the treatment with ICG-001 or JQ1 alone may lead to the activation of a subset of detrimental DIPG super enhancers (SEs), whereas the combinatorial treatment prevented the unintentional activation of these SEs. Indeed, this approach rescued the effect of single treatments on enhancer-driven oncogenes in H3K27M-mutated DIPG; however, it showed little effect in H3 wild-type DIPG cells.61
Similar to the previous study,62 the combination of OTX015 with panobinostat inhibited proliferation and promoted apoptosis in GBM cell lines as well.
Studies have indicated that BRD4 inhibitors also improve the glioma response to currently used chemotherapy drugs.46,66 For instance, in U87 glioma cells, Berenguer-Daizé et al. showed synergistic antitumor effects of OTX015 in association with SN38 (an active metabolite of irinotecan), with TMZ, or with everolimus. In vivo, OTX015 combined with TMZ improved mouse survival better than either single agent in the treatment of glioma.46 I-BET151 also could augment the effect of TMZ on GBM cells by inducing apoptosis. An increased amount of reactive oxygen species and superoxide anions resulted in caspase-3 activation, possibly associated with decreased superoxide dismutase (SOD) activity and increasing PUMA expression in GBM cells.66 These findings underscore the relevance of approaches tackling GBM by existing chemotherapy drugs in combination with BET inhibitors.
Gene amplification and/or mutation of the EGFR is found in nearly 60% of gliomas. EGFR variant III (EGFRvIII) signaling promotes interleukin (IL)-6 secretion in GBM cells. The crosstalk between the wild-type receptor and its constitutively active mutant form, EGFRvIII, limits sensitivity to EGFR tyrosine kinase inhibitors (TKIs) through an interclonal communication mechanism mediated by IL-6.45 IL-6 activates NF-κB via paracrine and/or autocrine pathways, leading to BRD4-dependent expression of the prosurvival protein survivin (BIRC5) and attenuation of sensitivity to EGFR TKIs. Both in vivo and in vitro experiments confirmed that the treatment with BRD4 inhibitors (JQ1, I-BET-151, and I-BET-762) increases tumor cell sensitivity to EGFR TKIs.45 Thus, BRD4 inhibitors can be combined with EGFR TKIs for the treatment of GBM.
Delivery systems (e.g., nanoparticles) help to improve drug passing across the BBB. Transferrin-functionalized nanoparticles (Tf-NPs) were found capable of crossing the BBB in mice and delivering drugs to GBM tumor tissue. Tf-NPs loaded with TMZ and JQ1 reduced GMB tumor burden and survival rate in mice by 1.5- to 2-fold compared with the same dose of free drugs. Additionally, GMB-mice treated with Tf-NPs loaded with drugs show reduced systemic drug toxicity.63 Thus, nanoscale platforms for delivering combination therapies (e.g., JQ1 or OTX015 + TMZ) have an emerging role in the treatment of gliomas.
Combination therapy including BRD4 inhibitor for GSCs
In recent years, the combination of BRD4 inhibitors and other innovative drugs has been also investigated in GSCs. Wang et al.64 found that a combination of JQ1 and RGFP966 (HDAC3 inhibitor) could suppresses GSC growth by blocking the GLI1/IL-6/STAT3 signaling axis. In fact, HDAC3 inhibition upregulated the acetylation of H3K27, which allowed the recruitment of BRD4 to the GLI1 gene promoter to induce its expression. JQ1, instead, inhibited the transcription of GLI1 gene, thereby blocking the GLI1/IL-6/STAT3 pathway. In vivo, a combination of JQ1 and RGFP966 showed a stronger inhibitory effect on GSC growth than either single drug.
Imipridones are a novel class of AKT/extracellular signal-regulated kinase (ERK) inhibitors that display limited therapeutic efficacy against GBMs. The expression of c-Myc can predict the response to imipramine.65 Since BRD4 is known to regulate the expression of c-Myc to control cancer cell proliferation,67 the effect of a BRD4 inhibitor combined with imipramine in the treatment of GBM was further explored. Ishida et al.65 discovered that the inhibition of c-Myc by OTX015 sensitizes stem-like GBM cells to imipridone-induced apoptosis in vitro and in vivo.
In general, the combination of BRD4 inhibitors can improve the glioma responsiveness not only to current chemotherapy drugs but also to innovative drugs, by impairing cell proliferation and/or promoting apoptosis, through signaling pathways such as STAT3, NF-κB, and mTOR (Figure 4). BRD4 inhibitors showed adverse effects such as thrombocytopenia, fatigue, gastrointestinal symptoms, and hyperbilirubinemia in the clinical trial.39,68 However, there have been few clinical studies using a BRD4 inhibitor for the treatment of glioma or other brain disease; hence, the side effects of BRD4 inhibitors in humans are still largely unknown. Clinical trials with BRD4 inhibitors in combination therapies for glioma are warranted. Moreover, the application of new drug carriers should be assessed in order to help these molecules to pass across the BBB.
Figure 4.

Schematic diagram showing the combination therapy including BRD4 inhibitor for GBM
(A–C) The mechanisms of combination therapy including BRD4 inhibitor in the treatment of GBM (A), glioma stem cells (B), and H3K27M mutated glioma cells and EGFRvIII mutated glioma cells (C). EGFR TKI, epidermal growth factor receptor tyrosine kinase inhibitor; EGFRvIII, epidermal growth factor receptor variant III; TMZ, temozolomide.
Conclusions and future perspectives
Much evidence surveyed in this study shows that BRD4 is a transcriptional and epigenetic regulator that plays a pivotal role in glioma development. BRD4 inhibitors (or the knockdown of BRD4 expression) significantly inhibit the malignant behavior of glioma cells or GSCs, resulting in effective therapeutic targeting of these tumors. Thus, BRD4 inhibitors can significantly delay or even reverse the malignant progression of gliomas. When combined with other innovative drugs, BRD4 inhibitors can induce cell cycle arrest, inhibit oncogenic pathways, activate tumor suppressor genes, and interfere with the transcriptional regulatory activity of BET and E2F1. In particular, the therapy with BRD4 inhibitors, alone or in combination, has an enhanced effect on gliomas bearing H3K27M histone mutations55,59, 60, 61 or the EGFRvIII variant.45,54 Despite the importance of IDH mutations in gliomas, fewer studies have addressed the efficacy of BRD4 inhibitors in IDH-mutated gliomas so far. However, in other models, for example AML or cholangiocarcinoma, IDH mutant cancer cells showed a better response rate to BRD4 inhibitors.69,70
A number of important issues still need to be addressed. First, the molecular mechanisms of action of BRD4 inhibitors need to be further studied. Second, clinical trials applying BRD4 inhibitors in the treatment of glioma need to be carried out to assess their efficacy and side effects in humans. Third, some inhibitors, such as JQ1, can inhibit BRD4, BRD3, and BRD2 at the same time; notably, whether BRD2 or BRD3 plays any role in glioma also needs to be clarified. Finally, the application of delivery systems should be assessed in order to further enhance the transit of BRD4 inhibitors across the BBB. In fact, since BRD4 inhibitors and TMZ achieve similar therapeutic effects on glioma cells in vitro,40 it is exciting to speculate that the treatment with BRD4 inhibitors in association with delivery systems could represent an amenable alternative for the therapy of TMZ-resistant glioma patients.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (nos. 81903088, 81911530169, and 81670180); the Guangdong Basic and Applied Basic Research Foundation (nos. 2020A1515111201 and 2019A1515110495); the University Special Innovative Research Program of Department of Education of Guangdong Province (no. 2020KQNCX073); and by the Medical Research Fund of Guangdong (no. A2020466). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
H. You conceived the hypothesis and revised the manuscript. H. Yang, L. Wei, Y. Xun, and A.P. Yang did the literature search, H. Yang and L.Wei wrote the manuscript. H. Yang and H. You prepared the tables and figures.
Declaration of interests
The authors declare no competing interests.
References
- 1.Miller J.J., Shih H.A., Andronesi O.C., Cahill D.P. Isocitrate dehydrogenase-mutant glioma: Evolving clinical and therapeutic implications. Cancer. 2017;123:4535–4546. doi: 10.1002/cncr.31039. [DOI] [PubMed] [Google Scholar]
- 2.Zang L., Kondengaden S.M., Che F., Wang L., Heng X. Potential epigenetic-based therapeutic targets for glioma. Front. Mol. Neurosci. 2018;11:408. doi: 10.3389/fnmol.2018.00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan A.C., Ashley D.M., López G.Y., Malinzak M., Friedman H.S., Khasraw M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020;70:299–312. doi: 10.3322/caac.21613. [DOI] [PubMed] [Google Scholar]
- 4.Chen R.Q., Liu F., Qiu X.Y., Chen X.Q. The prognostic and therapeutic value of PD-L1 in glioma. Front. Pharmacol. 2019;9:1503. doi: 10.3389/fphar.2018.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan A.C., Heimberger A.B., Khasraw M. Immune checkpoint inhibitors in gliomas. Curr. Oncol. Rep. 2017;19:23. doi: 10.1007/s11912-017-0586-5. [DOI] [PubMed] [Google Scholar]
- 6.Golub D., Iyengar N., Dogra S., Wong T., Bready D., Tang K., Modrek A.S., Placantonakis D.G. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front. Oncol. 2019;9:417. doi: 10.3389/fonc.2019.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foreman P.M., Friedman G.K., Cassady K.A., Markert J.M. Oncolytic virotherapy for the treatment of malignant glioma. Neurotherapeutics. 2017;14:333–344. doi: 10.1007/s13311-017-0516-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srinivasan V.M., Ferguson S.D., Lee S., Weathers S.P., Kerrigan B.C.P., Heimberger A.B. Tumor vaccines for malignant gliomas. Neurotherapeutics. 2017;14:345–357. doi: 10.1007/s13311-017-0522-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White M.E., Fenger J.M., Carson W.E., 3rd Emerging roles of and therapeutic strategies targeting BRD4 in cancer. Cell. Immunol. 2019;337:48–53. doi: 10.1016/j.cellimm.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgado-Pascual J.L., Rayego-Mateos S., Tejedor L., Suarez-Alvarez B., Ruiz-Ortega M. Bromodomain and extraterminal proteins as novel epigenetic targets for renal diseases. Front. Pharmacol. 2019;10:1315. doi: 10.3389/fphar.2019.01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hajmirza A., Emadali A., Gauthier A., Casasnovas O., Gressin R., Callanan M.B. BET family protein BRD4: An emerging actor in NFκB signaling in inflammation and cancer. Biomedicines. 2018;6:E16. doi: 10.3390/biomedicines6010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulikowski E., Rakai B.D., Wong N.C.W. Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med. Res. Rev. 2021;41:223–245. doi: 10.1002/med.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung M., Gelato K.A., Fernández-Montalván A., Siegel S., Haendler B. Targeting BET bromodomains for cancer treatment. Epigenomics. 2015;7:487–501. doi: 10.2217/epi.14.91. [DOI] [PubMed] [Google Scholar]
- 14.Xu L., Chen Y., Mayakonda A., Koh L., Chong Y.K., Buckley D.L., Sandanaraj E., Lim S.W., Lin R.Y., Ke X.Y. Targetable BET proteins- and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma. Proc. Natl. Acad. Sci. USA. 2018;115:E5086–E5095. doi: 10.1073/pnas.1712363115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang B., Yang X.D., Zhou M.M., Ozato K., Chen L.F. Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009;29:1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin Z.Y., Wang T., Su S., Shen L.T., Zhu G.X., Liu Q., Zhang L., Liu K.W., Zhang Y., Zhou Z.H. BRD4 promotes gastric cancer progression and metastasis through acetylation-dependent stabilization of Snail. Cancer Res. 2019;79:4869–4881. doi: 10.1158/0008-5472.CAN-19-0442. [DOI] [PubMed] [Google Scholar]
- 17.Jin X., Yan Y., Wang D., Ding D., Ma T., Ye Z., Jimenez R., Wang L., Wu H., Huang H. DUB3 promotes BET inhibitor resistance and cancer progression by deubiquitinating BRD4. Mol. Cell. 2018;71:592–605.e4. doi: 10.1016/j.molcel.2018.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu T., Lu W., Luo C. A patent review of BRD4 inhibitors (2013–2019) Expert Opin. Ther. Pat. 2020;30:57–81. doi: 10.1080/13543776.2020.1702645. [DOI] [PubMed] [Google Scholar]
- 19.Wang S., Pike A.M., Lee S.S., Strong M.A., Connelly C.J., Greider C.W. BRD4 inhibitors block telomere elongation. Nucleic Acids Res. 2017;45:8403–8410. doi: 10.1093/nar/gkx561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stathis A., Bertoni F. BET proteins as targets for anticancer treatment. Cancer Discov. 2018;8:24–36. doi: 10.1158/2159-8290.CD-17-0605. [DOI] [PubMed] [Google Scholar]
- 21.Gilan O., Rioja I., Knezevic K., Bell M.J., Yeung M.M., Harker N.R., Lam E.Y.N., Chung C.W., Bamborough P., Petretich M. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science. 2020;368:387–394. doi: 10.1126/science.aaz8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noel P., Hussein S., Ng S., Antal C.E., Lin W., Rodela E., Delgado P., Naveed S., Downes M., Lin Y. Triptolide targets super-enhancer networks in pancreatic cancer cells and cancer-associated fibroblasts. Oncogenesis. 2020;9:100. doi: 10.1038/s41389-020-00285-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hassan R., Tammam S.N., Safy S.E., Abdel-Halim M., Asimakopoulou A., Weiskirchen R., Mansour S. Prevention of hepatic stellate cell activation using JQ1- and atorvastatin-loaded chitosan nanoparticles as a promising approach in therapy of liver fibrosis. Eur. J. Pharm. Biopharm. 2019;134:96–106. doi: 10.1016/j.ejpb.2018.11.018. [DOI] [PubMed] [Google Scholar]
- 24.Brasier A.R. Therapeutic targets for inflammation-mediated airway remodeling in chronic lung disease. Expert Rev. Respir. Med. 2018;12:931–939. doi: 10.1080/17476348.2018.1526677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chelladurai P., Boucherat O., Stenmark K., Kracht M., Seeger W., Bauer U.M., Bonnet S., Pullamsetti S.S. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br. J. Pharmacol. 2021;178:54–71. doi: 10.1111/bph.14932. [DOI] [PubMed] [Google Scholar]
- 26.Wang X., Zhou Y., Peng Y., Huang T., Xia F., Yang T., Duan Q., Zhang W. Bromodomain-containing protein 4 contributes to renal fibrosis through the induction of epithelial-mesenchymal transition. Exp. Cell Res. 2019;383:111507. doi: 10.1016/j.yexcr.2019.111507. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y., Vakoc C.R. Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb. Perspect. Med. 2017;7:a026674. doi: 10.1101/cshperspect.a026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z., Wang P., Chen H., Wold E.A., Tian B., Brasier A.R., Zhou J. Drug discovery targeting bromodomain-containing protein 4. J. Med. Chem. 2017;60:4533–4558. doi: 10.1021/acs.jmedchem.6b01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang D., Yu Y., Ma Z. Novel strategies targeting bromodomain-containing protein 4 (BRD4) for cancer drug discovery. Eur. J. Med. Chem. 2020;200:112426. doi: 10.1016/j.ejmech.2020.112426. [DOI] [PubMed] [Google Scholar]
- 30.French C.A. Small-molecule targeting of BET proteins in cancer. Adv. Cancer Res. 2016;131:21–58. doi: 10.1016/bs.acr.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Lam F.C., Kong Y.W., Huang Q., Vu Han T.-L., Maffa A.D., Kasper E.M., Yaffe M.B. BRD4 prevents the accumulation of R-loops and protects against transcription-replication collision events and DNA damage. Nat. Commun. 2020;11:4083. doi: 10.1038/s41467-020-17503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao Z., Li X., Wang H., Chen G., Feng Z., Wu Y., Yin H., Zhao G., Deng Z., Zhao C. BRD4 regulates self-renewal ability and tumorigenicity of glioma-initiating cells by enrichment in the Notch1 promoter region. Clin. Transl. Med. 2020;10:e181. doi: 10.1002/ctm2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrieu G., Tran A.H., Strissel K.J., Denis G.V. BRD4 regulates breast cancer dissemination through Jagged1/Notch1 signaling. Cancer Res. 2016;76:6555–6567. doi: 10.1158/0008-5472.CAN-16-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shafran J.S., Jafari N., Casey A.N., Győrffy B., Denis G.V. BRD4 regulates key transcription factors that drive epithelial-mesenchymal transition in castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2020 doi: 10.1038/s41391-020-0246-y. Published online July 21, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duan Y., Guan Y., Qin W., Zhai X., Yu B., Liu H. Targeting Brd4 for cancer therapy: Inhibitors and degraders. MedChemComm. 2018;9:1779–1802. doi: 10.1039/c8md00198g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donati B., Lorenzini E., Ciarrocchi A. BRD4 and cancer: Going beyond transcriptional regulation. Mol. Cancer. 2018;17:164. doi: 10.1186/s12943-018-0915-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boi M., Gaudio E., Bonetti P., Kwee I., Bernasconi E., Tarantelli C., Rinaldi A., Testoni M., Cascione L., Ponzoni M. The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 2015;21:1628–1638. doi: 10.1158/1078-0432.CCR-14-1561. [DOI] [PubMed] [Google Scholar]
- 39.Amorim S., Stathis A., Gleeson M., Iyengar S., Magarotto V., Leleu X., Morschhauser F., Karlin L., Broussais F., Rezai K. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3:e196–e204. doi: 10.1016/S2352-3026(16)00021-1. [DOI] [PubMed] [Google Scholar]
- 40.Pastori C., Daniel M., Penas C., Volmar C.H., Johnstone A.L., Brothers S.P., Graham R.M., Allen B., Sarkaria J.N., Komotar R.J. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics. 2014;9:611–620. doi: 10.4161/epi.27906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pastori C., Kapranov P., Penas C., Peschansky V., Volmar C.H., Sarkaria J.N., Bregy A., Komotar R., St Laurent G., Ayad N.G., Wahlestedt C. The bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc. Natl. Acad. Sci. USA. 2015;112:8326–8331. doi: 10.1073/pnas.1424220112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh M.B., Sartor G.C. BET bromodomains as novel epigenetic targets for brain health and disease. Neuropharmacology. 2020;181:108306. doi: 10.1016/j.neuropharm.2020.108306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chamberlain P.P., Lopez-Girona A., Miller K., Carmel G., Pagarigan B., Chie-Leon B., Rychak E., Corral L.G., Ren Y.J., Wang M. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 2014;21:803–809. doi: 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- 44.Winter G.E., Mayer A., Buckley D.L., Erb M.A., Roderick J.E., Vittori S., Reyes J.M., di Iulio J., Souza A., Ott C.J. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell. 2017;67:5–18.e19. doi: 10.1016/j.molcel.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zanca C., Villa G.R., Benitez J.A., Thorne A.H., Koga T., D’Antonio M., Ikegami S., Ma J., Boyer A.D., Banisadr A. Glioblastoma cellular cross-talk converges on NF-κB to attenuate EGFR inhibitor sensitivity. Genes Dev. 2017;31:1212–1227. doi: 10.1101/gad.300079.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berenguer-Daizé C., Astorgues-Xerri L., Odore E., Cayol M., Cvitkovic E., Noel K., Bekradda M., MacKenzie S., Rezai K., Lokiec F. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int. J. Cancer. 2016;139:2047–2055. doi: 10.1002/ijc.30256. [DOI] [PubMed] [Google Scholar]
- 47.Deshmukh P., Mathur S., Gangadharan G., Krishnappa G., Dalavaikodihalli Nanjaiah N., Padmanabhan B. Novel pyrano 1,3 oxazine based ligand inhibits the epigenetic reader hBRD2 in glioblastoma. Biochem. J. 2020;477:2263–2279. doi: 10.1042/BCJ20200339. [DOI] [PubMed] [Google Scholar]
- 48.DeMars K.M., Yang C., Candelario-Jalil E. Neuroprotective effects of targeting BET proteins for degradation with dBET1 in aged mice subjected to ischemic stroke. Neurochem. Int. 2019;127:94–102. doi: 10.1016/j.neuint.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 49.Wang Q., Kumar V., Lin F., Sethi B., Coulter D.W., McGuire T.R., Mahato R.I. ApoE mimetic peptide targeted nanoparticles carrying a BRD4 inhibitor for treating medulloblastoma in mice. J. Control. Release. 2020;323:463–474. doi: 10.1016/j.jconrel.2020.04.053. [DOI] [PubMed] [Google Scholar]
- 50.Wang J., Quan Y., Lv J., Gong S., Dong D. BRD4 promotes glioma cell stemness via enhancing miR-142-5p-mediated activation of Wnt/β-catenin signaling. Environ. Toxicol. 2020;35:368–376. doi: 10.1002/tox.22873. [DOI] [PubMed] [Google Scholar]
- 51.Wen N., Guo B., Zheng H., Xu L., Liang H., Wang Q., Wang D., Chen X., Zhang S., Li Y., Zhang L. Bromodomain inhibitor jq1 induces cell cycle arrest and apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling pathway. Int. J. Oncol. 2019;55:879–895. doi: 10.3892/ijo.2019.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Du Z., Song X., Yan F., Wang J., Zhao Y., Liu S. Genome-wide transcriptional analysis of BRD4-regulated genes and pathways in human glioma U251 cells. Int. J. Oncol. 2018;52:1415–1426. doi: 10.3892/ijo.2018.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng Z., Gong Y., Ma Y., Lu K., Lu X., Pierce L.A., Thompson R.C., Muller S., Knapp S., Wang J. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin. Cancer Res. 2013;19:1748–1759. doi: 10.1158/1078-0432.CCR-12-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu F., Hon G.C., Villa G.R., Turner K.M., Ikegami S., Yang H., Ye Z., Li B., Kuan S., Lee A.Y. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol. Cell. 2015;60:307–318. doi: 10.1016/j.molcel.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Piunti A., Hashizume R., Morgan M.A., Bartom E.T., Horbinski C.M., Marshall S.A., Rendleman E.J., Ma Q., Takahashi Y.H., Woodfin A.R. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017;23:493–500. doi: 10.1038/nm.4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fahey J.M., Stancill J.S., Smith B.C., Girotti A.W. Nitric oxide antagonism to glioblastoma photodynamic therapy and mitigation thereof by BET bromodomain inhibitor JQ1. J. Biol. Chem. 2018;293:5345–5359. doi: 10.1074/jbc.RA117.000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu F., Xiong F., He J., Liu K., You Y., Xu Q., Miao J., Du Y., Zhang L., Ren H. Brd4 inhibition ameliorates pyocyanin-mediated macrophage dysfunction via transcriptional repression of reactive oxygen and nitrogen free radical pathways. Cell Death Dis. 2020;11:459. doi: 10.1038/s41419-020-2672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DeMars K.M., Yang C., Castro-Rivera C.I., Candelario-Jalil E. Selective degradation of BET proteins with dBET1, a proteolysis-targeting chimera, potently reduces pro-inflammatory responses in lipopolysaccharide-activated microglia. Biochem. Biophys. Res. Commun. 2018;497:410–415. doi: 10.1016/j.bbrc.2018.02.096. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y., Dong W., Zhu J., Wang L., Wu X., Shan H. Combination of EZH2 inhibitor and BET inhibitor for treatment of diffuse intrinsic pontine glioma. Cell Biosci. 2017;7:56. doi: 10.1186/s13578-017-0184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagaraja S., Vitanza N.A., Woo P.J., Taylor K.R., Liu F., Zhang L., Li M., Meng W., Ponnuswami A., Sun W. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell. 2017;31:635–652.e6. doi: 10.1016/j.ccell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wiese M., Hamdan F.H., Kubiak K., Diederichs C., Gielen G.H., Nussbaumer G., Carcaboso A.M., Hulleman E., Johnsen S.A., Kramm C.M. Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Cell Death Dis. 2020;11:673. doi: 10.1038/s41419-020-02800-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meng W., Wang B., Mao W., Wang J., Zhao Y., Li Q., Zhang C., Tang Y., Ma J. Enhanced efficacy of histone deacetylase inhibitor combined with bromodomain inhibitor in glioblastoma. J. Exp. Clin. Cancer Res. 2018;37:241. doi: 10.1186/s13046-018-0916-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lam F.C., Morton S.W., Wyckoff J., Vu Han T.L., Hwang M.K., Maffa A., Balkanska-Sinclair E., Yaffe M.B., Floyd S.R., Hammond P.T. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat. Commun. 2018;9:1991. doi: 10.1038/s41467-018-04315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Q., Jia S., Wang D., Chen X., Kalvakolanu D.V., Zheng H., Wei X., Wen N., Liang H., Guo B., Zhang L. A combination of BRD4 and HDAC3 inhibitors synergistically suppresses glioma stem cell growth by blocking GLI1/IL6/STAT3 signaling axis. Mol. Cancer Ther. 2020;19:2542–2553. doi: 10.1158/1535-7163.MCT-20-0037. [DOI] [PubMed] [Google Scholar]
- 65.Ishida C.T., Zhang Y., Bianchetti E., Shu C., Nguyen T.T.T., Kleiner G., Sanchez-Quintero M.J., Quinzii C.M., Westhoff M.A., Karpel-Massler G. Metabolic reprogramming by dual AKT/ERK inhibition through imipridones elicits unique vulnerabilities in glioblastoma. Clin. Cancer Res. 2018;24:5392–5406. doi: 10.1158/1078-0432.CCR-18-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yao Z., Yang S., Zhao H., Yang H., Jiang X. BET inhibitor I-BET151 sensitizes GBM cells to temozolomide via PUMA induction. Cancer Gene Ther. 2020;27:226–234. doi: 10.1038/s41417-018-0068-4. [DOI] [PubMed] [Google Scholar]
- 67.Yoo M., Yoo M., Kim J.E., Lee H.K., Lee C.O., Park C.H., Jung K.Y. Synthesis and biological evaluation of indazole-4,7-dione derivatives as novel BRD4 inhibitors. Arch. Pharm. Res. 2018;41:46–56. doi: 10.1007/s12272-017-0978-y. [DOI] [PubMed] [Google Scholar]
- 68.Berthon C., Raffoux E., Thomas X., Vey N., Gomez-Roca C., Yee K., Taussig D.C., Rezai K., Roumier C., Herait P. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016;3:e186–e195. doi: 10.1016/S2352-3026(15)00247-1. [DOI] [PubMed] [Google Scholar]
- 69.Chen C., Liu Y., Lu C., Cross J.R., Morris J.P., 4th, Shroff A.S., Ward P.S., Bradner J.E., Thompson C., Lowe S.W. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. 2013;27:1974–1985. doi: 10.1101/gad.226613.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fujiwara H., Tateishi K., Kato H., Nakatsuka T., Yamamoto K., Tanaka Y., Ijichi H., Takahara N., Mizuno S., Kogure H. Isocitrate dehydrogenase 1 mutation sensitizes intrahepatic cholangiocarcinoma to the BET inhibitor JQ1. Cancer Sci. 2018;109:3602–3610. doi: 10.1111/cas.13784. [DOI] [PMC free article] [PubMed] [Google Scholar]