

Summary
Obesity is a growing global concern in adults and youth with a parallel rise in associated complications, including cognitive impairment. Obesity induces brain inflammation and activates microglia, which contribute to cognitive impairment by aberrantly phagocytosing synaptic spines. Local and systemic signals, such as inflammatory cytokines and metabolites likely participate in obesity-induced microglial activation. However, the precise mechanisms mediating microglial activation during obesity remain incompletely understood. Herein, we leveraged our mouse model of high-fat diet (HFD)-induced obesity, which mirrors human obesity, and develops hippocampal-dependent cognitive impairment. We assessed hippocampal microglial activation by morphological and single-cell transcriptomic analysis to evaluate this heterogeneous, functionally diverse, and dynamic class of cells over time after 1 and 3 months of HFD. HFD altered cell-to-cell communication, particularly immune modulation and cellular adhesion signaling, and induced a differential gene expression signature of protein processing in the endoplasmic reticulum in a time-dependent manner.
Subject areas: Neuroscience, Physiology, Transcriptomics
Graphical abstract
Highlights
-
•
HFD induces obesity but does not alter microglia morphology by 1 month
-
•
HFD alters microglial endoplasmic reticulum (ER) and ribosome pathways after 1 month
-
•
HFD dysregulates microglia-to-microglia immune signaling, particularly after 3 months
-
•
HFD feeding induces an early ER response followed by a chronic inflammatory response
Neuroscience; Physiology; Transcriptomics
Introduction
The global prevalence of obesity continues to rise. The World Health Organization estimates that global obesity rates tripled from 1975 to 2016, including an increase from 4% to 16% of overweight or obese children and teens.1 This steep rise is concerning as the complications of obesity place an immense strain on patients and healthcare systems. Obesity is associated with cognitive impairment and structural brain changes in adults2,3 and children and adolescents.4,5,6 Midlife obesity also raises the risk of future dementia.7,8 Currently, dementia management is limited to pharmacological and lifestyle interventions, which address only symptoms, not disease pathology. There is a critical need to understand the mechanisms underlying obesity-associated cognitive impairment across the lifespan.
Obesity activates microglia, the resident macrophage-like central nervous system (CNS) immune cells, which constitute a potential target for intervention. Most research has focused on hypothalamic microglial inflammation leading to the loss of regulatory mechanisms controlling satiety.9,10,11,12 However, studies of obese mice demonstrate that microglia are also activated in the hippocampus, a limbic brain structure involved in memory and learning. Murine models of diet-induced obesity exhibit hippocampal inflammation after both acute13,14 and chronic periods of high-fat diet (HFD).15,16,17 Obese mice suffer from hippocampal-dependent cognitive deficits,15,16,18 with hippocampal microglial morphology changes characteristic of pro-inflammatory activation after chronic HFD,15,16 implicating microglia in cognitive impairment secondary to obesity.
In addition to potential morphological changes, obesity increases hippocampal levels of pro-inflammatory cytokines,19e.g. interleukin 1 beta (IL-1β) and tumor necrosis factor alpha (TNFα).20 Inflammatory cytokines may activate microglia,21 with a variety of other local or systemic signals such as dysregulated metabolites,22e.g. saturated fatty acids from HFD. Moreover, obesity increases peripheral immune cell recruitment,23 and impairs blood-brain barrier function, facilitating entry of systemic inflammatory mediators.24
The mechanism of hypothalamic microglial activation and polarization during obesity has been well defined.10,11,25 However, the hypothalamus is in direct contact with the circulation, and thus hypothalamic microglia are more directly exposed to saturated fatty acids. In contrast, hippocampal microglia are more protected from circulation, and therefore, their response to dietary saturated fatty acids and obesity over time may be less pronounced or exhibit a different time response versus hypothalamic microglia. In addition, most studies to date have examined single time points. Thus, there is a need to clarify the evolution of hippocampal microglial activation over time to determine the appropriate temporal window for intervention. There is also a need to address microglial heterogeneity, as a class of functionally diverse cells. Existing studies in the obesity field, such as those examining endoplasmic reticulum (ER) stress,26 are limited by bulk hippocampal tissue analysis, which likely masks microglial-specific findings.
To address microglial heterogeneity, evolution of responses over time, and shortcomings from bulk analysis, we harnessed the power of single-cell RNA sequencing (scRNA-seq). We characterized, for the first time, the hippocampal microglial transcriptomic landscape at fine-grained single-cell resolution in the context of diet-induced obesity after 1 and 3 months (mo) of HFD. scRNA-seq identified obesity-associated dysregulated inflammatory pathways in microglia after 1 month or 3 months of HFD in wild-type C57B/L6J mice as they transitioned from adolescence to adulthood. In addition, we leveraged our single-cell dataset to investigate the dynamic immune cell-to-cell interplay, which is crucial for understanding subtle differences in microglial behavior in obesity. Our characterization of microglial activation states provides the foundation necessary to elucidate the role of microglia in hippocampal pathology in obesity.
Results
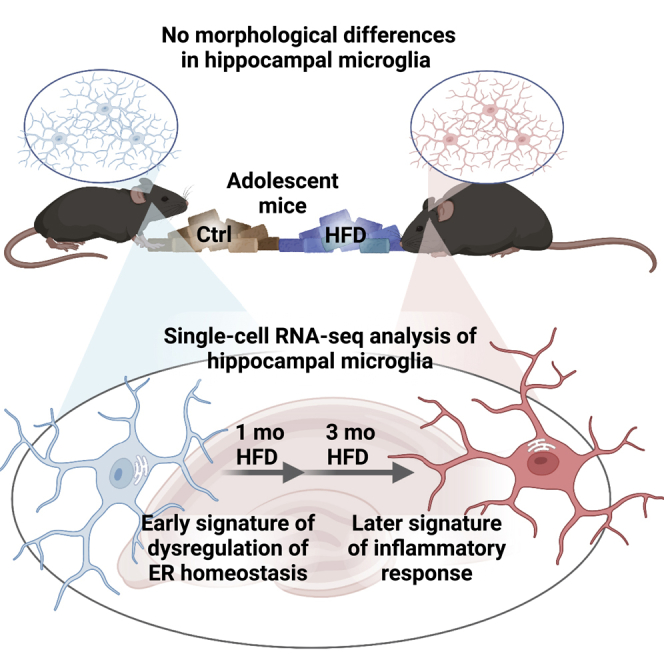
HFD induces obesity but not early hippocampal microglial morphological activation
To determine the effect of obesity on hippocampal microglial activation, we utilized an established and deeply phenotyped mouse model of diet-induced obesity,27,28 which we have previously used in our studies.29 We fed 5-week-old male C57BL/6J mice (n = 10 per group) either a 60% HFD enriched with saturated fatty acids or a control diet containing 10% fat (Figure 1A). To determine time-dependent changes in microglia in response to HFD, mice were divided into two cohorts and fed for 1 month or 3 months. At the study endpoints, we isolated hippocampal microglia and performed scRNA-seq. HFD fed mice were heavier than control fed mice as early as after 2 weeks of diet (Figure 1B). After 1 month and 3 months, HFD mice had deficits in glucose homeostasis versus control mice, characterized by elevated peak blood glucose levels on glucose challenge, alongside a delayed return to baseline (Figure 1C). Baseline, i.e., at 5 weeks of age, GTTs were indistinguishable between HFD and control groups (Figure S1A). Although glucose homeostasis was disrupted after 1 month of HFD, fasting plasma insulin did not increase in HFD mice relative to controls until 3 months (Figure 1D; p = 0.0003), recapitulating hyperinsulinemia in humans with chronic obesity.
Figure 1.

HFD induces obesity and dyslipidemia in mice
(A) Study design. Wild-type C57BL/6J mice aged 5 weeks were randomized to a high-fat diet (HFD) or control standard diet (ctrl). After 1 month (mo) or 3 (mo) of diet, HFD and control mice were sacrificed for microglial isolation and metabolic phenotyping (n = 10 per diet per time point).
(B) Longitudinal body mass; from 0 to 4 weeks (i.e., 1 month time point) n = 20 per diet per time point (HFD, green; ctrl, blue); from 5 weeks onward, n = 10 per diet (HFD, green; ctrl, blue). $p <0.05, $$p <0.01, $$$p <0.001, for HFD versus ctrl 1 month cohort; ∗p <0.05, ∗∗∗p <0.001, ∗∗∗∗p <0.0001 for HFD versus ctrl 3 months cohort; repeated measures two-way ANOVA with Sidak’s multiple comparisons test; data represented as mean ± SD.
(C) Glucose tolerance test (GTT); left panel for HFD (n = 20; light green) versus control (n = 20; light blue) at 1 month (triangles); right panel for HFD (n = 10; dark green) versus control (n = 10; dark blue) at 3 months (circles). Measures above the glucometer’s upper threshold were set to the threshold, 750 mg/dL. Plasma (D) insulin, (E) cholesterol, and (F) phospholipids; top panels for HFD (n = 10; light green) versus control (n = 7; light blue) at 1 month (triangles); bottom panels for HFD (n = 10; dark green) versus control (n = 9; dark blue) at 3 months (circles). C to F, ∗p <0.05, ∗∗p <0.01, ∗∗∗p <0.001, ∗∗∗∗p <0.0001 for HFD versus control, by repeated measures two-way ANOVA and Sidak’s multiple comparisons test for GTTs, by Welch’s t-test for insulin, cholesterol, and phospholipids, and by Mann-Whitney test for insulin 3 months because data were not normally distributed; data represented as mean ± SD.
We also examined basic plasma lipid profiles for cholesterol, triglycerides, phospholipids, and non-esterified fatty acids (NEFAs). Total plasma cholesterol was elevated in HFD mice after 1 month and 3 months diet (Figure 1E; p = 0.0474, p = 0.0021, respectively) and phospholipids after 3 months (Figure 1F; p = 0.0096). Diet did not affect triglyceride (Figure S1B) or NEFA (Figure S1C) levels. Collectively, these metabolic data demonstrate that HFD mice are obese and develop systemic metabolic dysfunction after just 1 month (e.g., glucose intolerance, elevated cholesterol), which becomes more severe after 3 months (i.e., hyperinsulinemia, elevated phospholipids).
We previously reported hippocampal dependent cognitive deficits in short-term memory in a mouse model after 2 weeks, 6 weeks, and 24 weeks of HFD using a novel object recognition task.18 Obesity is associated with activated hippocampal microglia, assessed by morphological activation,15,16 after 3 months of chronic HFD feeding. To address if hippocampal microglial changes occur earlier, like cognitive deficits, we quantified 3-dimensional (3D) microglial morphology after 1 month. Homeostatic microglia display complex branching patterns to surveil their environment, but environmental challenges trigger morphology shifts, characterized by smaller territorial volume and simpler branching patterns.
We performed 3D analyses on tissue sections stained by immunohistochemistry for the microglial marker ionized calcium binding adaptor molecule 1 (Iba1) using a modified 3D-Morph protocol.30 3D z-stack images of hippocampal tissue sections were acquired by confocal microscopy. After pre-processing in Imaris, images were run through 3D-Morph in MATLAB to measure territorial volume, cell volume, and branching parameters based on convex hull analyses and skeletonization30 (representative cells, Figures 2A and 2B). After a 1-month diet, microglia in the CA1 region of the hippocampus from HFD-fed mice were not morphologically distinct from control microglia. Territorial volume (p = 0.16), the 3D space taken up by the cell body and all its branches, cell volume (p = 0.26), defined as the volume of the soma and branches themselves, and ramification index (p = 0.52), defined as the territorial volume divided by the cell volume, did not differ by diet (Figures 2C–2E). There was also no difference in the number of endpoints (p = 0.42) and branchpoints (p = 0.08) (Figures 2F and 2G); however, there was a trend toward fewer branchpoints per cell in HFD microglia.
Figure 2.

Obesity does not alter hippocampal microglial morphology at 1 mo
(A) Representative 3D Morph 30 analysis for a control microglia from the hippocampal CA1 region, confocal microscopy (left), skeletonization (center), and branchpoints (right). Scale bar is 30 μm.
(B) Representative 3D Morph analysis for a HFD microglia from the hippocampal CA1 region. Sections were stained for microglial Iba1 (594 nm, red channel) and with Hoechst nuclear staining (blue channel). Scale bar is 30 μm.
(C–G) Microglial (C) territorial volume, (D) cell volume, (E) ramification index, (F) endpoints per cell, and (G) branchpoints per cell, all at 1 mo; left panels for control (ctrl; n = 3 animals, 3 images/animal; blue) versus HFD (n = 4 animals, 3 images/animal; green), each circle represents an individual cell. There were no significant differences in HFD versus control by linear mixed effects models with random animal-specific intercepts with t-test; data represented as mean ± standard deviation.
Microglial heterogeneity remains constant in HFD and control mice
Next, we investigated the microglial single-cell transcriptome to understand how hippocampal microglia are activated to contribute to obesity associated cognitive impairment. To characterize transcriptomic heterogeneity of the microglial landscape, we performed scRNA-seq on microglia isolated from the hippocampi of HFD and control mice (n = 6 per group) after 1 month and 3 months (Figure 3A). We performed a papain enzymatic digestion followed by serial trituration to prepare a single cell suspension from the hippocampus. We enriched for microglia by applying a 40% Percoll centrifugation to our cells and collecting the cell pellet, and further purified by fluorescence-activated cell sorting (FACS). We sorted CD11b+/CD45low double-positive cells, representing microglia, rather than CD11b+/CD45high cell surface markers, which represent macrophages. Sorted cells were sequenced on the 10X Chromium platform and RNA reads were quality filtered before mapping to the mouse reference genome. We used CellRanger Count to prepare sample files, which were read into Seurat. Cells were excluded from downstream analysis based on criteria outlined in the STAR Methods section. In total, 4,555 HFD and 4,945 control cells were included in the analysis at 1 month, and 1,292 HFD and 1,255 control cells at 3 months. Data were normalized to reduce the effects of differing cell numbers on expression levels in downstream analyses and ratios were reported for changes in cell type rather than changes in absolute number. To determine the success of FACS, we examined expression of the CD11b gene, Itgam, and the CD45 gene, Ptprc, which were expressed by most cells, suggesting a successful cell sort (Figure S2A).
Figure 3.

Isolated cells cluster into eleven distinct populations
(A) Diagram of microglial isolation protocol; cells were isolated from 6 mice per group. Created with BioRender.com.
(B) Uniform Manifold Approximation and Projection (UMAP) shows 11 clusters, which represent homeostatic microglia 1 (HMG1), HMG2, HMG3, macrophages (Mac), uMG (unknown, functionally undescribed), inflammatory microglia (InflamMG), proliferating microglia (ProlifMG), neurons (Neuro), interferon-related microglia (IfnMG), neutrophils (Neu), and monocytes (Mono). UMAP plot represents 4,555 HFD and 4,945 control cells at 1 month, and 1,292 HFD and 1,255 control cells at 3 months.
(C) Dot plot of markers used to assign cluster identity. Dot size represents the percentage of cells from a given cluster expressing the marker, dot color represents average expression relative to all other clusters.
(D) UMAP plots by markers that differentiated various clusters, InflamMG by chemokine Ccl4 (C-C motif chemokine ligand 4), IfnMG by interferon-induced Ifit2 (interferon induced protein with tetratricopeptide repeats 2), ProlifMG by DNA topoisomerase Top2a (DNA topoisomerase II alpha), and macrophages by immunomodulating Mrc1 (mannose receptor C-type 1).
(E) UMAP plots with expression of microglial (Cx3cr1, CX3C chemokine receptor 1; P2ry12, purinergic receptor P2Y12; Tmem119, transmembrane protein 119) versus astrocytic (Gfap, glial fibrillary acidic protein) markers shows a relatively pure microglia isolation. UMAP plot for another microglial marker, Aif1 (allograft inflammatory factor 1), the gene encoding Iba1 protein, is shown in Figure S2.
Next, we performed principal component analysis to reduce data dimensionality and then analyzed and visualized clusters using Uniform Manifold Approximation and Projection (UMAP). We identified eleven cell clusters (Figures 3B andS3 for UMAP plots per mouse), which were characterized by cell-specific markers found using the FindAllMarkers function in Seurat (Figure 3C). We reviewed previously published datasets and leveraged the CellMarker and PanglaoDB databases to assign cell type identities to each cluster. As anticipated, most clusters, seven out of eleven, were microglial subtypes, of which three were homeostatic microglia (HMG), HMG1, 2, 3. The remaining microglial subtypes were inflammatory microglia (InflamMG, expressing C-C motif chemokine ligand 4 [Ccl4] and Ccl3, Figure 3D), interferon-related microglia (IfnMG, expressing interferon induced protein with tetratricopeptide repeats 2 [Ifit2] and Ifit3, Figure 3D), proliferating microglia (ProlifMG, expressing DNA topoisomerase II alpha [Top2a], Figure 3D, and marker of proliferation Ki-67 [Mki67]), and a functionally undescribed subtype labeled “uMG” for ‘undescribed microglia’ (Table S1).
In addition to the microglia subtypes, we also identified macrophages (Mac, expressing mannose receptor C-type 1 [Mrc1], Figure 3D), monocytes (Mono, expressing C-C motif chemokine receptor 2 [Ccr2] and lymphocyte antigen 6 complex, locus C2 [Ly6c2], UMAP in Figure S4, features in Figure 3C), neutrophils (Neu, expressing S100 calcium binding protein A8 [S100a8] and S100a9, UMAP in Figure S4, features in Figure 3C), and neurons (Neuro, expressing calcium/calmodulin dependent protein kinase II alpha [Camk2a] and microtubule-associated protein 2 [Map2], UMAP in Figure S4, features in Figure 3C). Most sequenced cells expressed microglial markers Cx3cr1, P2ry12 (Figures 3E andS5 for Cx3cr1 UMAP plots per mouse), and Aif1 (gene coding for Iba1; Figure S2B) and the microglial-specific gene Tmem119 (Figure 3E), as expected, because we enriched for microglia. Furthermore, the analyzed cells showed almost no expression of the astrocyte specific gene Gfap (Figure 3E) or oligodendrocyte specific gene Olig2 (Figure S2C), indicating the absence of astrocytes and oligodendrocytes among isolated cells, as anticipated following a microglial sort. Cells expressed very low levels of the neuronal specific gene Rbfox3 (Figure S2C), suggestive of a very small neuronal population.
All seven microglial and four additional cell type clusters were present in both HFD and control groups (Figure 4A), and at both 1 month and 3 months. Although all cell clusters were represented in all experimental groups, we assessed the proportion of each cell cluster by diet or age. Cluster proportions were similar between HFD and control groups at 1 month and 3 months (Figure 4B). In all experimental groups, the largest proportion of cells comprised homeostatic microglia, HMG1, 2, and 3 at both 1 month (HFD 77.8% versus control 76.9%) and 3 months (HFD 69.4% versus control 65.3%). Macrophages were the second most abundant cell type after HMGs at 1 month (HFD 7.1% versus control 7.5%) and 3 months (HFD 12.2% versus control 12.4%). Other microglial subtypes (uMG, InflamMG, ProlifMG, and IfnMG) ranged from 0.8% to 8.6% of all cells in HFD and control conditions at 1 month and 3 months.
Figure 4.

Microglial heterogeneity remains constant in HFD versus control mice
(A) UMAP of HFD (5,847 total cells) versus control (ctrl; 6,200 total) at both time points isolated from n = 6 mice per group.
(B) Top panel: Circle chart of all 11 cell types by percentage at 1 month for HFD (left; 4,555 total cells) versus control (right; 4,945 total cells). Bottom panel: Circle chart of all 12 cell types by percentage at 3 months for HFD (left; 1,292 total cells) versus control (right; 1,255 total cells).
Obesity dysregulates microglial inflammatory cell-to-cell signaling
Microglia are immune cells that constantly survey the CNS environment and respond to external signals.31,32,33 Thus, we examined the effect of diet-induced obesity on cell-to-cell communication within our single-cell dataset. To do so, we utilized CellChat,34 a tool that leverages a database of over 2,000 ligand and receptor pairs and scRNA-seq data to infer intercellular communication. We first looked at information flow of intercellular signaling pathways in HFD and control conditions at 1 month and 3 months (Figures 5A and 5D), which is based on a summation of probabilities of pathway communication for all cell type pairs. For each pathway, CellChat also identifies the ligand-receptor gene pairs contributing to pathway signaling, which is visualized in circle plots of cell types that send and receive signals for the given pathway (Figures 5B, 5C, 5E, 5F, andS6 for ligand-receptor expression).
Figure 5.

Cell-to-cell communication analyses reveal HFD specific intercellular communication pathways
(A) Information flow charts at 1 month for HFD (blue) versus control (ctrl; red) generated by CellChat. Teal bars represent information flow in HFD cells (4,555 total cells), red bars represent information flow in control cells (4,945 total cells). Signaling pathways in teal text have significantly higher information flow in HFD cells relative to control, signaling pathways in red text have significantly higher information flow in control cells, and signaling pathways in black text are not significantly different between groups; vertical dashed lines represent information flow equal in both HFD and control.
(B and C) Circle plots of cellular signaling interactions (top) and their top contributing ligand-receptor (L-R) pairs (bottom) for pathway networks involving (B) HSPG for HFD at 1 month and (C) IFN-I for HFD at 1 month. Dots in circle plots represent cell populations with color codes matching UMAP clusters; strokes represent communication between distinct cell populations and loops represent signaling within cell populations. Stroke and loop colors reflect the cluster sending the signal, and thickness reflects strength of the signaling pair.
(D) Information flow chart at 3 months for HFD (blue; 1,292 total cells) versus control (ctrl; red; 1,255 total cells).
(E and F) Circle plots (top) and top contributing L-R pairs (bottom) for pathway networks involving (E) IL6 for HFD at 3 months and (F) PDGF for HFD at 3 months.
Information flow at the 1 month time point was higher in 23 pathways in HFD versus control cells and 12 pathways in control versus HFD cells (Figure 5A). In some instances, signaling pathways were only detected in cells of one diet group, e.g., type-1 interferon (IFN-I) signaling at 1 month in HFD cells. The pathways with microglia-to-microglia signaling that were elevated in HFD at 1 month fell into a few broad categories. ‘CDH’, ‘ICAM1,’ ‘PECAM1’, ‘HSPG, and ‘CD200’ ligands are cell surface glycoproteins with various immune regulatory roles, including T cell co-stimulation,35 cellular adhesion,36 microglial pro-inflammatory activation,37,38,39 as well as inhibition of microglial activation in the case of CD200.40 The ‘IFN-I’ ligand is a cytokine involved in canonical pro-inflammatory signaling. ‘EGF’, ‘GRN’, and ‘TGFb’ ligands are growth factors, and loss of microglial progranulin and granulin signaling has been extensively studied in frontotemporal dementia41 and neurodegeneration.42 Finally, the ‘SEMA6’ ligand is a transmembrane protein with a known role in axon guidance.43
‘HSPG’ and ‘IFN-I’ were the sole pathways driven by microglia-to-microglia signaling with information flow turned on in HFD cells at 1 month (Figures 5B and 5C). For ‘HSPG’ signaling, InflamMG signaled to HMG3, HMG2, and ProlifMG, with autocrine signaling to itself, and IfnMG cells signaled to InflamMG and ProlifMG (Figure 5B; top). Signaling was mediated between the ligand heparan sulfate proteoglycan 2 (Hspg2) and its receptor, dystroglycan 1 (Dag1) (Figure 5B; bottom). The interferon (‘IFN-I’) signaling pathway network in HFD cells at 1 month was more complex and involved connections among multiple cell types, including non-microglia cells. ‘IFN-I’ signaling originated from ProlifMG, Mac, Mono, IfnMG, InflamMG, HMG3, HMG2, and HMG1) (Figure 5C). Because we enriched for microglia using CD11b+/CD45low FACS, the non-microglial immune cells we isolated may comprise a biased sampling of the true populations. Owing to this potential bias and the likelihood that they do not represent the full diversity of monocytes, neutrophils, macrophages, and neurons, it is difficult to draw firm conclusions regarding signaling networks involving these cell types as the senders and/or receivers. However, we can determine that IFN-I signaling involves a dynamic interplay between multiple cell types, highlighting the value of cell-to-cell communication analysis in the context of microglial mediated pathology.
Information flow at the 3 months time point was higher in 27 pathways in HFD versus control cells and 10 pathways in control versus HFD cells (Figure 5D). Again, in some cases, signaling pathways were only detected in cells of one diet group, e.g., ‘IL6’ signaling at 3 months in HFD cells. At 3 months, HFD turned on microglia-to-microglia signaling of ‘IL6’, ‘PDGF’ (Figures 5E and 5F), ‘KIT’, ‘VISTA’ (Figure S7A), ‘HSPG’, and ‘LAMININ’ pathways. The pathways elevated in HFD cells relative to controls containing microglia-to-microglia network signaling included ‘CD86’, ‘ICOS’, ‘CD200’, ‘SEMA6’, ‘PROS’, ‘CD48’, ‘CADM’ (Figures S7B and S7C), ‘TGFb’, and ‘GRN’. Like at 1 month, signaling pathways ligands included growth factors, e.g., ‘PDGF’, ‘KIT’, ‘TGFb’, and ‘GRN’, and glycoproteins with immunomodulatory functions, e.g., ‘HSPG’ and ‘CD200’. ‘IL6’ and ‘KIT’ ligands are cytokines, and other ligands are cell surface immunomodulatory antigens, e.g., ‘CD86’, ‘ICOS’, ‘CD48’, and ‘VISTA’. IL-6, a cytokine with context-dependent pro- or anti-inflammatory effects,44 was expressed by IfnMG and its receptor by HMG2 and HMG3 (Figure 5E). We found HFD turned on PDGF signaling at 3 months (Figure 5F). Several microglia subtypes (InflamMG, HMG1, HMG2, HMG3, ProlifMG, IfnMG, uMG) expressed the platelet-derived growth factor (PDGF) ligand and signaled to other microglia, as well as to macrophages, neutrophils, monocytes, and neurons. PDGF is a mitogen with well-studied roles in development and wound healing, yet has detrimental effects in various disease contexts, such as cancer and atherosclerosis.45,46
Obesity dysregulates microglial ER homeostasis
The results from CellChat yielded insight into the effects of obesity on microglial cell-to-cell communication. However, we were also interested in the dysregulated intracellular processes to identify potential drivers of an activated state. To accomplish this goal, we performed differentially expressed gene (DEG) analysis for all microglia types combined in HFD versus control using DESeq2. There were 89 DEGs after 1 month HFD, and 46 DEGs after 3 months (adjusted P-value <0.05) (Figure 6A, Table S2). To identify the biological significance of these DEGs, we performed Encyclopedia of Genes and Genomes (KEGG) enrichment analysis (Figure 6B). Five pathways were identified at 1 month; ‘ribosome’ and ‘COVID-19’ contained the same genes, which were all ribosomal and included Rpl3, Rplp0, Rpl10, Rps16, Rpl11, among others. ‘Protein processing in the ER contained Hspa8, Dnaja1, Hsp90b1, Pdia6, Calr, Hspa5, and Dnajb1. ‘Mitophagy-animal’ and ‘antigen processing and presentation’ contained only three genes each. Performing KEGG analysis for 3 months was less informative, because of few DEGs, only 46, KEGG enrichment analysis at 3 months yielded ‘lysosome,’ ‘COVID-19,’ ‘pertussis,’ ‘alcoholic liver disease,’ ‘ribosome,’ ‘arachidonic acid metabolism,’ ‘antigen processing and presentation,’‘complement and coagulation cascades,’ and ‘Chagas disease.’ However, there were no more than five genes in each KEGG pathway, including C1qa and C1qb complement genes, which were present in five of the nine pathways.
Figure 6.

HFD dysregulates protein processing in the ER across microglial subtypes
(A) Volcano plots of differentially expressed genes (DEGs) in HFD versus control (ctrl) at 1 month (left) and 3 months (right) for all microglia subtypes combined with top 20 annotated DEGs. Analysis included 4,450 ctrl 1 month, 1,048 ctrl 3 months, 4,121 HFD 1 month, and 1,054 HFD 3 months microglia. Significant DEGs above the horizontal red line based on P-adjusted <0.05, blue dots represent significantly downregulated in HFD, red dots represent significantly upregulated in HFD. FC, fold-change.
(B) KEGG pathway analysis of DEGs in HFD versus control at 1 month (top) and 3 months (bottom). Dot color represents-log10(P-value) from least significant (light pink) to most significant (red); dot size represents gene number, the number of significant DEGs in the KEGG pathway; rich factor represents the fraction of significant DEGs among all genes in the KEGG pathway.
(C) WGCNA modules by color, turquoise, yellow, brown, green, blue, with number of genes assigned to each module (column 1), and corresponding heatmap of median module expression by condition. The gray module comprises genes that did not cluster into a co-expression module. Significant differences in expression of genes in modules between experimental groups by Kruskal-Wallis, ∗p <0.05, ∗∗p <0.01.
(D) Boxplots of yellow and brown module expression. Significant differences by Kruskal-Wallis, ∗p <0.05, ∗∗p <0.01. Boxplot height represents the interquartile range, horizonal line the median, and whiskers the minimum and maximum values of expression.
(E) KEGG pathway analysis of genes in HFD versus control from the yellow (left) and brown (right) modules. Bar color represents-log10(P-value) from least significant (light pink) to most significant (red); bar length represents the number of genes in the KEGG pathway, annotated with a number; rich factor represents the fraction of genes among all genes in the KEGG pathway.
Next, we identified DEGs in HFD versus control for each of the 11 cell types at both 1 month and 3 months. Overall, few DEGs (adjusted P-value <0.05) were identified, with 0 DEGs for most comparisons (Table S3). DEGs for HFD versus control were identified for HMG1 (18 DEGs), HMG2 (8 DEGs), HMG3 (8 DEGs), and Mac (3 DEGs) at 1 month. At 3 months, DEGs were identified for HMG1 (1 DEGs), HMG2 (8 DEGs), and HMG3 (1 DEG). For both the cluster specific and all microglia combined analyses, there were more DEGs at 1 month than 3 months. The cluster specific analysis detected DEGs in HFD versus control only in HMG populations and macrophages. Although there were too few genes to perform KEGG on cluster specific DEGs, we found again, that protein processing in the ER genes were represented. At 1 month, ER related DEGs included Hspa8, Hsp90b1, and Dnaja1 for HMG1, Hspa8 and Calr for HMG2, and Hspa8, Dnaja1, and Pdia6 for HMG3. The DEGs in the macrophage group were H3f3b, Ucp2, and Gm10076. At 3 months, Hsp90ab1 and Hsp90aa1 were DEGs in HMG2.
Owing to the similarity among UMAP clustered HMG subtypes, and because most cluster specific DEGs were identified in HMGs, we next considered the three homeostatic microglia subtypes as a single cluster and performed DESeq2 analysis. We identified 96 DEGs at 1 month and 12 DEGs at 3 months in all three homeostatic microglia subtypes combined (Table S4). KEGG analysis at 1 month in this subset of HMGs again showed enrichment in ‘ribosome,’‘COVID-19,’ and ‘protein processing in the ER’ as well as in ‘prostate cancer’ and ‘salmonella infection.’ Homeostatic microglia make up the largest proportion of all microglia, so it is expected that HMG DEGs reflect the DEGs for the combined microglia analysis.
Because few DEGs were identified between HFD and control groups, we implemented an additional approach to examine differences between groups and infer potential biological significance. We performed weighted gene co-expression network analysis (WGCNA), an unsupervised correlation analysis, to identify co-expressed gene modules across all samples, including genes for all cell types. WGCNA identifies co-expression networks, aka modules, which differ significantly between HFD versus control, rather than individual DEGs by DESeq2 analysis. Analysis of 1 month and 3 months samples yielded five co-expression modules and 1 module of genes (gray module) that were not assigned to a co-expression module (Figure 6C; module dendrogram, Figure S8A). Four modules, turquoise, yellow, brown, and green, differed significantly between HFD and control groups. The turquoise module, containing 867 genes, had reduced expression in HFD at 1 month (p = 0.0087), the yellow module, containing 241 genes, had elevated expression in HFD at 1 month (p = 0.041) and 3 months (p = 0.026), the brown module, containing 331 genes, had elevated expression in HFD after 1 month (p = 0.0022), and the green module, containing 198 genes, had reduced expression in HFD at 3 months (p = 0.041) (Figures 6D and S8B). We next performed KEGG enrichment analysis on each module. The yellow module was enriched for genes in a variety of biological pathways, including ‘Alzheimer’s disease’, containing the genes Psenen, Ndufb5, Atp5o, Gnaq, Ndufs8, Ndufa5, Mapk3, Ndufs4, Atp5g3, Ppp3r1, and Psen1 (Table S5), and ‘B cell receptor signaling pathway’ (Figure 6E). The brown module was enriched for ‘protein processing in the ER’ which contained 12 genes related to the ER stress response, Hspa8, Nfe2l2, Selenos, P4hb, Dnaja1, Ssr1, Edem1, Man1c1, Eif2s1, Ube2j1, Erlec1, and Dnajb1 (Table S5). Some of these genes have been identified as relevant to microglia in obesity by other studies in bulk or functional studies, such as heat shock protein 70 (Hspa8), HSP40 (Dnaja1, Dnajb1),47 and Nrf2 (Nfe2l2).48 The turquoise module was most significantly enriched in ‘ribosome’ and ‘oxidative phosphorylation’ genes, and the green module in ‘protein processing in the ER’ genes (Figures S8C and S8D).
Obesity correlates with distinct transcriptomic signatures in various microglia types
Finally, because our focus was the impact of obesity on microglia transcriptomic signatures, we examined the correlation of obesity, as it progress from 1 month to 3 months, to transcriptomic profiles in distinct microglia clusters. We performed a Spearman correlation analysis of body weights at 1 month and 3 months to transcript levels at 1 month and 3 months in all cell clusters (Figure S9, Table S6). We found several genes whose expression levels correlated positively with body weight in various cell clusters, e.g., C1qc in HMG1 and 2, or negatively, e.g., Hsp90b1 in HMG1, 2, and 3, InflamMG, and Mac. To infer biological meaning, we next performed separate KEGG pathway enrichment analysis of positively (Figure 7A) and negatively (Figure 7B) correlated transcripts (Table S7). The most significant pathways that correlated positively with obesity were ‘ribosome’ and ‘COVID-19’ in HMG1 and 2 and InflamMG. The ‘COVID-19’ pathway contains complement and ribosome genes. Importantly, rich factors were low, so only a few genes in these KEGG pathways were activated with obesity. The most significant pathways that associated negatively with obesity were ‘protein processing in ER’ in HMG3 and InflamMG, ‘spliceosome’ in HMG1, and ‘Fc gamma R−mediated phagocytosis’ in HMG2. Notably, rich factors were low, indicating that only a few genes correlated with body weight of all genes present in the KEGG pathway. The genes (Figure S9) and pathways (Figure 7) that correlated with body weight overlapped with the DEGs and pathways at 1 month and 3 months (Figures 6A and 6B), suggesting that progressive obesity particularly influences these genes.
Figure 7.

Obesity correlates with distinct transcriptomic signatures in various microglia types
KEGG pathway enrichment analysis of transcripts (with Benjamini-Hochberg corrected P-value<0.05) that correlate (A) positively and (B) negatively with body weight in various microglia types [homeostatic microglia 1 (HMG1), HMG2, HMG3, inflammatory microglia (InflamMG), uMG (unknown, functionally undescribed)]. Dot color represents-log10(Padj) from least significant (light pink) to most significant (red); rich factor represents the fraction of significant body weight correlated transcripts among all genes in the KEGG pathway. Padj, adjusted P-value.
Discussion
Rates of obesity are climbing,1 alongside rises in associated complications, such as cognitive impairment.2,8 Thus, there is a need to understand the pathophysiology of cognitive deficits secondary to obesity to intervene. Through their contribution to cognitive impairment in obesity,15 microglia serve as a potential therapeutic target, although the mechanisms of microglial activation in this context remain incompletely understood. In this study, we used scRNA-seq to determine the effect of obesity over time on the hippocampal microglial transcriptome. scRNA-seq allowed us to probe the heterogeneity of microglial populations and identify unique cellular processes and inflammatory pathways that are dysregulated in obesity. We, for the first time in the adolescent and adult C57BL/6J hippocampus, identified seven microglial subtypes, with three other immune cell types and neurons. All cell types were present in HFD and control groups at 1 month and 3 months diet, and diet did not affect cell type proportion. Conversely, cell-to-cell communication analysis revealed a functionally diverse set of signaling pathways, which differed significantly between diet groups. In particular, HFD dysregulated microglial signaling involved in immune modulation. DEG analysis with KEGG pathway enrichment revealed a signature of dysregulated ER genes in HFD microglia, which was independently confirmed by a separate WGNCA analysis.
We studied the effect of obesity on microglial activation in a mouse model of diet-induced obesity,29 which displays hippocampal-dependent cognitive impairment.18 HFD mice were heavier than their control counterparts after only 2 weeks of diet and went on to develop progressive metabolic dysfunction, including hyperinsulinemia and dyslipidemia, as occurs in human obesity. Because we expected that the immune response would evolve over time, we assessed morphological activation early on at 1 month. We found that HFD did not alter microglial morphology in the hippocampal CA1 region at 1 month. This finding is not unexpected at 1 month, because previously published studies showed HFD-induced morphological activation of microglia, quantified in part by fewer primary cell processes, occurs later at 3 months in the hippocampal dentate gyrus and CA1.15,16 Although there was no significant difference in branchpoints per cell between groups, there was a trend toward fewer branchpoints in HFD microglia. Further studies are required to determine whether this trend might represent the start of ramification reduction, which then continues over time.
Although we did not observe hippocampal microglial morphological activation, scRNA-seq enabled us to investigate early transcriptomic changes across diverse microglial subtypes. No studies, to our knowledge, had examined hippocampal microglial heterogeneity in obesity, and no microglial scRNA-seq studies have focused on the healthy hippocampus in the C57BL/6J mouse. Thus, we performed scRNA-seq on microglia isolated from the hippocampi of obese and lean mice after 1 month and 3 months. We sought to determine the effect of obesity on microglial activation and its temporal evolution at the transcriptomic level. In our sequenced CD11b+/CD45low cells, we identified seven microglial subtypes as well as small populations of monocytes, macrophages, neutrophils, and neurons. In addition to three homeostatic populations, other microglia subtypes included inflammatory microglia, interferon-related microglia, proliferative microglia, and a cluster with unknown function, which we termed uMG. As far as we are aware, this uMG cluster has not been previously described. It is characterized by upregulation of mitochondrial genes, and perhaps represents a dying subset, as seen in other scRNA-seq data.49
We found that diet did not affect the cluster proportions. Homeostatic microglia comprised the largest cell population in all experimental groups. In line with our results, microglial scRNA-seq studies have similarly demonstrated that homeostatic microglia are the largest clusters in both healthy and disease contexts in the adolescent and adult brain.50,51,52 The presence of the smaller clusters, InflamMG, IfnMG, ProlifMG, and uMG, is not uniform in the literature, and differs with various factors, including age, brain region, disease state, and study design or perhaps power. We found an InflamMG (Ccl4/3 expressing) population, which has also been identified in small clusters in the healthy cortex at 2 months and 4 months and as a larger microglial percentage in a model of infectious disease.52 This inflammatory population, as well as an interferon-related population like our IfnMG cluster (Ifit3 expressing), are prevalent in the whole brain with aging.50 A proliferative microglia cluster like our ProlifMG (Top2a, Mki67 expressing) are present in the healthy adult brain,53 prevalent in the early developing brain50 and found in the cortex of an HIV infection model.52 The lack of activated morphology at 1 month aligns with our transcriptomic findings; HFD did not alter the proportion of InflamMG and IfnMG and homeostatic microglia dominated all experimental groups. The apparent trending increase in peripheral immune cell types (monocytes, neutrophils) in HFD versus control samples at 3 months could potentially reflect obesity-induced recruitment of immune cells to the brain.23
Our identified populations are unlikely an artifact of processing-associated ex vivo activation, because they have been identified with transcriptional inhibition, which we employed, in the roughly 3-month-old mouse brain.53 Marsh et al. showed that transcriptional inhibition prevented a processing-associated cluster, which has appeared in other microglial scRNA-seq.53 Overall, we showed, for the first time, that hippocampal microglia in the healthy adolescent and adult hippocampus are mostly homeostatic microglia, with small populations of inflammatory, proliferative, and interferon-related microglia. Future work is needed to determine whether young age and/or the chronic, low-grade nature of obesity-induced inflammation may have protected against changes in inflammatory microglia proportions, which are seen in microglial scRNA-seq studies in aging50 and overt transgenic models of disease.51
Immune cells function by sensing and responding to their environment, so cell-to-cell communication is an integral part of microglial function.54 Obesity disrupts the inflammatory milieu in the brain,23,24 so we asked whether obesity alters intercellular microglial signaling. Thus, we next examined whether diet impacted cell-to-cell interactions in the hippocampus by employing CellChat to infer intercellular communication among microglia. Of the signaling that differed by diet, the pathways driving microglia-to-microglia communication serve a diverse set of biological functions, many implicated in immune modulation of microglia or other immune cell types. These pathways contained glycoproteins with known immunomodulatory roles (e.g., ‘HSPG,’‘ICAM1),36,37,38,39 growth factors (e.g., ‘TGFb,’ ‘PDGF’), and immune antigens (e.g., ‘CD86’, ‘ICOS’, ‘CD48’, and ‘VISTA’). Several pathways contained proteins with known pro-inflammatory (e.g., ‘HSPG,’‘IFN-I,’‘IL6’) or anti-inflammatory properties (e.g., ‘VISTA,’‘CD200,’‘IL6’). Heparan sulfate proteoglycans (HSPGs) are immunomodulators39 with pro-inflammatory effects on microglia. HSPGs stimulate tumor necrosis factor alpha cytokine production38 and are involved in the microglial lipopolysaccharide-induced toll-like receptor 4 response.55 V-domain immunoglobulin-containing suppressor of T cell activation (Vista), plays an immune checkpoint role as a negative regulator of T-cells, and is involved in other myeloid cell functions, such as phagocytosis, but its role in microglia is not well characterized.56 Both Vista and cell adhesion molecule 1 (Cadm1), the ligand and receptor for ‘CADM’ signaling, are differentially expressed in Alzheimer’s disease-associated microglia.51,56
Further studies are needed to understand the role of these signaling pathways in modulating microglial behavior in obesity. HFD upregulated more microglial immune and inflammatory signaling at 3 months versus 1 month, which supports the hypothesis that chronic HFD enhances immune responses to obesity. HFD pathways associated with homeostatic and protective microglia (e.g., ‘GRN,’ ‘TGFb,’ ‘CD200’)40,41,57 might reflect a failed attempt to maintain homeostasis under conditions of stress during obesity. Regarding the involvement of signaling pathways related to adhesion molecules, it is possible to speculate these changes may reflect microglial migration as a component of the inflammatory response. In addition to the interactions identified in this study, it is important to consider that microglia in obesity may interact through ligand-receptor pairs that are not in the CellChat database or with other immune cell types. Moreover, our analysis relied on transcriptomic changes in ligand-receptor expression, which may not necessarily reflect protein expression. However, overall, the changes in intercellular microglia-to-microglia signaling revealed by CellChat suggest microglia respond early to an immune challenge, which ramps up over time, leading to a condition of chronic inflammation.
Next, we examined the effect of HFD on intracellular processes in hippocampal microglia by DEG analyses with KEGG enrichment. DEGs between HFD and control microglia at 1 month were enriched in ribosome and protein processing in the ER KEGG pathways. We identified very few cell cluster specific DEGs, but heat shock proteins related to the protein processing in the ER pathway were dysregulated in HFD homeostatic microglia, particularly at 1 month. There were so few DEGs overall at 3 months that we could not infer much biological pathway significance from KEGG enrichment in HFD versus control microglia. However, at 3 months, complement genes (C1qa, C1qb) were upregulated and among the top 20 most significant DEGs in HFD versus control microglia. Many of the DEGs and pathways that were implicated in microglia at 1 month and 3 months overlapped with genes that correlated, either positively or negatively, with progressive body weight gain. These findings encompassed some complement and ribosome genes (positive correlations with body weight), with genes related to protein processing in the ER (negative correlations with body weight). Microglia upregulate C1q in response to insult such as ischemic injury58 or in a model of Alzheimer’s disease.59 The complement cascade’s canonical role is in immune response, but complement is also involved in microglial mediated synaptic pruning in brain development.60 Hippocampal microglia excessively prune synapses in obesity,15 so it is possible that complement proteins contribute to this aberrant pruning. In support of this hypothesis, microglia contribute to aberrant complement mediated synaptic pruning in Alzheimer’s disease.59 HFD at 3 months also upregulated genes such as Trem2, an Alzheimer’s disease associated microglia gene,51,61 as well as genes encoding enzymes for processing pro-inflammatory arachidonic acid derived mediators, prostaglandins (Hpgd, Ptgs1) and leukotrienes (Ltc4s).
Microglia DEG fold-changes were low, but relatively consistent with chronic challenge,62 including chronic HFD.47 Because there were few total DEGs, we also performed WGNCA on all cells, which identified five gene co-expression modules, four of which differed by diet. KEGG analysis of the brown module, elevated at 1 month in HFD versus control, and the green module, reduced at 3 months in HFD, identified genes enriched in protein processing in the ER. The turquoise module, reduced at 1 month in HFD, was enriched in ribosome genes, and the yellow module, elevated at 1 month and 3 months in HFD versus control, identified genes enriched in Alzheimer’s disease and B cell receptor signaling pathways. Overall, DEGs expression in HFD microglia was characterized by an earlier ER response signature at 1 month, followed by a more inflammatory signature at 3 months. These finds align with the correlation analysis of transcript levels with body weight, which was negative for ER response genes, i.e., transcript levels of ER response genes went down with progressive obesity, and positive for complement genes, i.e., transcript levels of complement genes went up with progressive obesity.
Our DEG and WGNCA findings closely align with published bulk RNA-seq data of microglia after 8 weeks of HFD.47 The study found only 77 DEGs that differed in HFD versus control whole brain microglia and an upregulated WGNCA module containing heat shock protein genes, such as Hspa8, Dnaja1, Dnajb1,47 which were identified by our study. We also previously found that chronic HFD after 20 weeks induces hippocampal ER stress, enhancing expression of heat shock protein 5 and a member of the canonical inositol requiring enzyme 1 ER stress response, spliced X-box binding protein 1.63 The link between ER stress and inflammation is a well-studied phenomenon in obesity.64,65 Saturated fatty acids66,67 activate the macrophage ER stress response in obesity, which contributes to a pro-inflammatory phenotype.68 The heat shock proteins upregulated in our study at 1 month, e.g., Hspa8, Dnaja1, and Dnajb1, are protein folding chaperones and co-chaperones, which work to maintain ER homeostasis. Brykczynska et al. proposed that the increase in microglial ER heat shock proteins after 8 weeks HFD might be a protective mechanism.47 By examining DEGs, we identified an inflammatory gene expression signature later at 3 months, which aligns with enhanced immunomodulatory intercellular microglia-to-microglia driven signaling by CellChat. This inflammatory gene expression signature was characterized by HFD upregulated microglial genes, e.g., C1qa, C1qb, Trem2, Hpgd, Ptgs1, and Ltc4s. Possibly, an early microglial ER heat shock protein response combats increased burden which, if misfolded protein aggregates accumulate, may transition to an ER stress response capable of inducing inflammation.
Additional investigations are required to elucidate the mechanistic underpinnings in the evolving microglial response in obesity, which our findings suggest may be characterized by an earlier ER response, followed by a more chronic inflammatory state. Possibly obesity stressors trigger an adaptive ER response in attempt to maintain homeostatic function at 1 month, which evolves to a more activated state by 3 months. Indeed, rats pretreated with a mild ER stress inducer are protected from lipopolysaccharide-induced cognitive impairment and microglial pro-inflammatory activation, suggesting that mild ER stress may mitigate hippocampal sequela of inflammatory challenge.69 Alternatively, obesity may trigger a cascade of processes with increased demand on the ER, followed by an injurious ER stress response, which activates inflammatory pathways, as occurs in macrophages in obesity.68 ER stress has been implicated in microglial mediated inflammation in other disease contexts.70 Further studies that manipulate microglial ER stress are needed to determine whether it mitigates or contributes to microglial pro-inflammatory activation in obesity. Moreover, our findings indicate potential therapeutic targets for microglia-mediated cognitive impairment secondary to obesity. Targeting complement59 and prostaglandin71 in microglia or macrophages slows synapse loss and cognitive decline in aging and Alzheimer’s disease models and may, in addition, constitute promising approaches in obesity-induced cognitive impairment.
In summary, we used morphological and scRNA-seq analyses to determine the effects of obesity on hippocampal microglial activation in mice transitioning from adolescence to adulthood. We found that HFD did not alter hippocampal CA1 microglial morphology after just 1 month. We demonstrated, for the first time, the microglial landscape of the healthy adolescent and adult hippocampus in the C57BL/6J mouse. Although diet did not affect cluster proportions, HFD dysregulated intercellular inflammatory signaling pathways, an effect which was more pronounced at 3 months. Analysis of gene expression revealed a microglial signature of dysregulated ER protein processing and ribosome pathways at 1 month, which transitioned into an inflammatory response at 3 months. Identifying these obesity-associated microglial intercellular and intracellular pathways sets the foundation for further studies to elucidate mechanisms of microglial mediated cognitive deficits in obesity.
Limitations of the study
Although our study had several strengths, such as analyzing two time points using a single-cell method to generate granular transcriptomic information in a deeply phenotyped and well characterized model, we also had several weaknesses. First, significant, DEGs exhibited only small fold-changes and varied by lmicroglia cell subtype, making it difficult to validate these findings by bulk analysis of microglial isolations using qPCR. Second, our study only examined transcriptomic changes, and did not provide information on post-transcriptomic differences related to protein expression. Third, the CellChat analysis was limited to microglia interactions that occur through ligand-receptor pairs present in the database. Lastly, our study only included male mice; however, differences in immune system by sex warrant investigation of hippocampal microglia in female HFD versus control mice.72,73 Future functional studies of an early ER response mediated by hippocampal microglia in HFD mice will be needed to validate these study findings.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TruStain FcX™ (Anti-Mouse CD16/32) | Biolegend | Cat#101320; RRID: AB_1574975 |
| Rat APC anti-mouse CD45 | Biolegend | Cat#103111; RRID: AB_312976 |
| Rat APC-Cy7 anti-mouse/human CD11b | Biolegend | Cat#101226; RRID: AB_830642 |
| TotalSeq™-B0301 Anti-Mouse Hashtag 1 Antibody | BioLegend | Cat#155831; RRID: AB_2814067 |
| TotalSeq™-B0302 Anti-Mouse Hashtag 2 Antibody | BioLegend | Cat#155833; RRID: AB_2814068 |
| TotalSeq™-B0303 Anti-Mouse Hashtag 3 Antibody | BioLegend | Cat#155835; RRID: AB_2814069 |
| Rabbit Anti-IBA1 | Wako | Cat#019-19741; RRID: AB_839504 |
| Goat-Anti Rabbit Alexa-Fluor Plus 594 | Invitrogen, Thermo Fisher Scientific | Cat#A32740; RRID: AB_2762824 |
| Chemicals, peptides, and recombinant proteins | ||
| Glucose | Sigma-Aldrich, Merck | Cat#G8270-1KG |
| Intraperitoneal Pentobarbitol (Fatal-Plus) | Vortech | Cat#NDC0298-9373-68 |
| Hanks Phosphate Buffered Saline (no Calcium, no Magnesium, no Phenol Red) | Gibco, Thermo Fisher Scientific | Cat#14175145 |
| Hanks Phosphate Buffered Saline(10X) (no Calcium, no Magnesium, no Phenol Red) | Gibco, Thermo Fisher Scientific | Cat#14185052 |
| Hanks Buffered Phosphate Saline (Calcium, Magnesium, no Phenol Red) | Gibco, Thermo Fisher Scientific | Cat#14025092 |
| Hoechst 33258 Nuclear Stain | Sigma-Aldrich, Merck | Cat#861405 |
| Triptolide | Sigma-Aldrich, Merck | Cat#T3652 |
| Triptolide | Cayman Chemical | Cat#11973 |
| Anisomycin | Sigma-Aldrich, Merck | Cat#A9789 |
| Hibernate A-Calcium-Magnesium | BrainBits | Cat#HACAMG |
| Glutamax | Gibco, Thermo Fisher Scientific | Cat#35050061 |
| Papain | Worthington | Cat#LS003119 |
| Transcription Inhibitors Actinomycin D | Sigma-Aldrich, Merck | Cat#A1410 |
| Percoll | Cytiva | Cat#17089102 |
| 10XPhosphate-Buffered Saline | Thermo Fisher Scientific | Cat#70011044 |
| 2% Fetal Bovine Serum | Corning | Cat#35-011-CV |
| Ethylenediaminetetraacetic Acid | Sigma-Aldrich, Merck | Cat#E7889 |
| Sucrose | Sigma-Aldrich, Merck | Cat#84097-5KG |
| Paraformaldehyde | Sigma-Aldrich, Merck | Cat#P6148-500G |
| Critical commercial assays | ||
| KAPA Library Quantification Kit | Roche | Cat#07960140001 |
| Deposited data | ||
| scRNA-seq data collected in this study | GEO | NCBI Gene Expression Omnibus, accession ID GSE217464 |
| Data analysis codes for this study | Github | https://github.com/hurlab/HF_iScience |
| PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data | Panglao DB | Franzén et al.74 |
| CellMarker | CellMarker | Zhang et al.75 |
| Kyoto Encyclopedia of Genes and Genomes | Kanehisa Laboratories | https://www.genome.jp/kegg/ |
| Experimental models: Organisms/strains | ||
| C57BL/6J Mice | Jackson Laboratory, Bar Harbor, ME, USA | Cat#000664; RRID:IMSR_JAX:000664 |
| Software and algorithms | ||
| Bcl2fastq2 Conversion Software | Illumina | https://emea.support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html |
| CellRanger Pipeline v4.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Seurat v3 | Stuart et al.76 | https://satijalab.org/seurat/install.html |
| Uniform Manifold Approximation and Projection (UMAP) | Seurat | https://satijalab.org/seurat/reference/runumap |
| DESeq2 version 1.24.0 | Love et al.77 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| richR | Open Source | https://github.com/hurlab/richR |
| CellChat | Jin et al.34 | http://www.cellchat.org/ |
| WGCNA R package | CRAN | https://cran.r-project.org/web/packages/WGCNA/index.html |
| sva R package | Bioconductor | https://bioconductor.org/packages/release/bioc/html/sva.html |
| Imaris Software | Oxford Instruments | https://imaris.oxinst.com/products/imaris-for-cell-biologists?gclid=CjwKCAjwuYWSBhByEiwAKd_n_u39sLI-SVsq6RZTWcYMNDDJFUGacHiJfNo-dOjoTHFHVeNLplaNHBoCIo4QAvD_BwE |
| 3DMorph Script | MATLAB, Elisa M York | https://github.com/ElisaYork/3DMorph |
| Prism 9 | Graph Pad by Dotmatics | https://www.graphpad.com/scientific-software/prism/ |
| ImerTest R package v4.1.1 | CRAN | https://cran.r-project.org/web/packages/lmerTest/lmerTest.pdf |
| SCENIC | Aibar et al.78 | https://scenic.aertslab.org/ |
| GENIE3 | Huynh-Thu et al.79 | https://bioconductor.org/packages/release/bioc/html/GENIE3.html |
| AUCell | Aibar et al.78 | https://github.com/aertslab/AUCell |
| Discriminant Regulon Expression Analysis (DoRothEA) R package | Bioconductor | https://bioconductor.org/packages/release/data/experiment/html/dorothea.html |
| pheatmap R package | CRAN | https://cran.r-project.org/web/packages/pheatmap/index.htm |
| ggplot2 R package | CRAN | https://cran.r-project.org/web/packages/ggplot2/index.html |
| TranscripTools R package | rdrr.io | https://rdrr.io/github/abc-igmm/transcripTools/ |
| Monocle | Trapnell et al.80 | http://cole-trapnell-lab.github.io/monocle-release/ |
| Other | ||
| Control Diet:10% Fat, 70% Carbohydrates, 20% Protein | Research Diets | Cat#D12450J |
| HFD (High-Fat Diet): 60% Fat, 20% Carbohydrates, 20% Protein | Research Diets | Cat#D12492 |
| Glucometers | AlphaTrak, Abbott Laboratories | Cat#ART12303 |
| Blood Glucose Test Stripes | AlphaTrak, Abbott Laboratories | Cat#71681-01 |
| Sterile .5mm Silanized Glass Pasteur Pipette | BrainBits | Cat#NC0319875 |
| 70μm Nylon Cell Strainer | Falcon | Cat#352350 |
| MA900 Multi-Application Cell Sorter | Sony | Ma900 |
| Countess II Automated Cell Counter | Thermo Fisher Scientific | Cat#AMQAX1000 |
| Chromium Next GEM Chip G | 10X Genomics | Cat#2000177 |
| Chromium Controller | 10X Genomics | Cat#PN-120223;120246 |
| Tapestation 4200 | Agilent | Cat#G2991AA |
| NovaSeq 6000 | Illumina | Cat#20013850 |
| Leica Stellaris 8 Falcon Confocal Microscope | Leica | https://www.leica-microsystems.com/ppc/confocal/na/confocal-reimagined/?nlc=20211206-SFDC-013738&utm_source=google&utm_medium=cpc&utm_campaign=Confocal_Stellaris_Generic&utm_content=text_ad&utm_term=confocal%20microscopy&gclid=Cj0KCQjw0PWRBhDKARIsAPKHFGhlshKjfHERcztyilMGquhOJWVh_dLX-vbhDkiVvysxcnuhOYqhugIaAgOPEALw_wcB |
Resource availability
Lead contact
Requests for or questions about resources or reagents can be directed to the lead contact, Eva L. Feldman (efeldman@med.umich.edu).
Materials availability
This study did not generate new materials.
Experimental model and subject details
The HFD model is an established and deeply phenotyped mouse model of diet-induced obesity,27,28 which we have previously used in our studies.29 Male C57BL/6J mice were obtained (Jackson Laboratory, catalog # 000664) at 4 weeks (wk) of age (n=40) and housed in a specific-pathogen-free facility at the Unit for Laboratory Animal Medicine (ULAM) at the University of Michigan. Mice were housed in cages with littermates on paper bedding at 20 ± 2°C and a 12/12-h light/dark cycle and were monitored daily by ULAM staff. Mice were acclimated for 1 wk, and at 5 wk of age were randomized to four groups: Control 1 month (mo) and 3 mo and HFD 1 mo and 3 mo (Figure 1A). Control diet (10% fat, 70% carbohydrates, 20% protein; Research Diets, catalog #D12450J) or HFD (60% fat, 20% carbohydrates, 20% protein; Research Diets, catalog #D12492) were provided ad libitum, and mice also had free access to water. All procedures were performed according to a protocol approved by the University of Michigan’s Institutional Animal Care and Use Committee (IACUC; PRO00008116, approved 2 May 2018).
Method details
Mouse metabolic phenotyping
HFD and control mice were weighed weekly, except at weeks 6 and 9. Baseline glucose tolerance tests (GTTs) were performed for all animals at 5 wk of age. 1 mo control and HFD mice had GTTs at the 1 mo endpoint; 3 mo control and HFD mice had GTTs at both 1 mo and 3 mo endpoints. For GTTs, mice were fasted for 4 hours (h), and blood glucose levels were measured from one drop of tail blood using a glucometer (AlphaTrak, Abbott Laboratories) at baseline and at 15, 30, 60, and 120 minutes (min) after intraperitoneal injection of 1 g/kg body mass glucose in normal saline. Additional metabolic phenotyping included terminal plasma insulin, cholesterol, triglycerides, phospholipids, and non-esterified fatty acids performed by the Mouse Metabolic Phenotyping Center at the University of Cincinnati.
Microglial isolation, sorting, and scRNA-seq
Mice were euthanized using an IACUC approved protocol. Mice were injected with intraperitoneal pentobarbital (Fatal-Plus, Vortech Pharmaceuticals) and perfused with Hanks' balanced salt solution (HBSS; Thermo Fisher, catalog # 14175-145) supplemented with the transcription inhibitors actinomycin D (5 μg/ml; Sigma, catalog # A1410) and triptolide (10 μM; Sigma, catalog #T3652 or Cayman Chemical #11973), an approach described by Marsh et al.53 All subsequent microglial isolation steps were performed on ice or at 4°C when possible and in a laminar flow hood or biological safety cabinet. Transcription inhibitors and ice were used to minimize processing-associated microglial activation to preserve an in vivo transcriptional state. Hippocampi were dissected from HFD and control mice (n=6 per group) and minced on ice. A single cell suspension was prepared by a papain enzymatic digestion at 37°C followed by trituration (1 mg/ml; Worthington catalog # LS003119) in Hibernate A-Calcium-Magnesium (BrainBits, catalog # HACAMG) media with Glutamax (∼0.5 nM; Gibco, catalog # 35050061). Transcription inhibitors actinomycin D, triptolide, and anisomycin (27.1 μg/ml; Sigma, catalog # A9789) were used until the end of the enzymatic digestion step.
Digested tissue was serially triturated at room temperature with a fire-polished pipette twice followed by a smaller diameter salinized ∼0.5 mm fire polished pipette twice (BrainBits, catalog # FPP). The cell suspension was passed through a wet 70 μm strainer on ice and then pelleted. Microglia were enriched by resuspending in 40% Percoll and centrifuging at 500g for 30minat room temperature. The cell pellet was washed in ice-cold HBSS, centrifuged for 10minat 300gat 4°C, and resuspended in ice-cold flow sorting buffer (1X PBS [phosphate buffered saline], 2% fetal bovine serum, 1 mM ethylenediaminetetraacetic acid). Cells were moved to a 96-well plate, blocked with TruStain FcX™ (anti-mouse CD16/32) (Biolegend, catalog # 101320) for 30 min on ice, and then incubated with APC-CD45 (Biolegend, catalog # 103112) and APC-Cy7-CD11b (Biolegend, catalog # 101226) at 1:50 in the presence of 1 μg/100 μl TotalSeq-B anti-mouse Hashtag antibodies (Biolegend, catalog #s 155831, 155833, 155835) for 30 min on ice.
After staining, cells were washed with 200 μl ice-cold flow sorting buffer for 10minat 4°C and resuspended for flow sorting on the Sony MA900 Cell Sorter by the University of Michigan Flow Cytometry Core. The FACS sorting strategy first sorted cells by froward and side scatter, to exclude mostly dead or dying cells along with cellular debris. Gating then selected cells that were CD45low and CD11b+. Tagged control and HFD samples were combined on 10X chips to mitigate batch effects. scRNA-seq was performed by the Advanced Genomics Core at the University of Michigan using the 10X Genomics Chromium system (10X Genomics): An automated counter (Countess II, Thermo Fisher) was used to quantify cells/μl. The single cell suspension was then diluted to a final concentration ranging from 700 to 1000 cells/μl. Using the Chromium Controller, 3’ libraries of single cells were created leveraging 3’ V3.1 chemistry using NextGEM Chip G reagents, according to the manufacturer’s protocol (all from 10X Genomics). The quality of the final library was evaluated by Tapestation 4200 (Agilent) and Kapa qPCR (Roche) was used to quantify libraries. Pooled libraries were sequenced using 150 bp paired-end format (Illumina NovaSeq 6000). De-multiplexed Fastq files were generated (Bcl2fastq2 Conversion Software, Illumina) and reads were aligned and counted (CellRanger Pipeline, 10X Genomics).
scRNA-seq data alignment and sample aggregating
Raw data from 4,644 (HFD 1 mo), 5,024 (control 1 mo), 1,366 (HFD 3 mo), and 1,314 (control 3 mo) cells were obtained from sequencing. Low-quality reads were filtered out (quality less than Q30) and reads were then mapped to the GRCm38 mouse reference genome (CellRanger Pipeline81 version 4.0.0, 10X Genomics). The individual sample output files from CellRanger Count were read into Seurat v3.76 The hashtag oligo (HTO)82 raw counts of each cell were normalized using a centered log ratio transformation across cells. Cells were then demultiplexed by using the HTODemux function in Seurat,76 and droplets with two cells (doublets), more than two cells, or no cell (empty droplet) were subsequently removed. Cells were excluded from downstream analysis based on filtering by the following criteria: unique molecular identifier counts per cell <200, gene count per cell >7500, and the fraction of transcripts mapped to mitochondrial genes >25% (to exclude dead or dying cells). Filtering resulted in 4,555 HFD cells at 1 mo, 4,945 control cells at 1 mo, 1,292 HFD at 3 mo, and 1,255 control cells at 3 mo, which were included in subsequent analyses. The difference in microglia counts at the 1- and 3-mo time points may have arisen to biological differences in the number of brain microglia in mice at 1 versus 3 mo,83,84 or from technical variability, although every effort was made to mitigate contributions from this. To account for differing cell numbers at each time point, data were normalized, and ratios were reported for changes in cell type rather than changes in absolute number. Count data were then normalized using the NormalizeData function in Seurat with the default setting.
Dimension reduction, clustering, and visualization
Principal component analysis (PCA) was performed based on the top 2,000 most variable genes. The optimal principal component (PC) number was selected based on the point where the percent change in variation in consecutive PCs was lower than 0.1%. Then, Uniform Manifold Approximation and Projection (UMAP) was performed on the PCs to visualize cells, and graph-based clustering was performed on the PCA-reduced data.
Celltype annotation and differential expression analysis
To assign a cell type identity to each cluster, the cluster gene markers were identified using the FindAllMarkers function in Seurat. Cell types were assigned based on the cluster gene markers using the CellMarker75 and PanglaoDB74 databases and information available in the relevant literature. DESeq277 was used to identify differentially expressed genes (DEGs) between control and HFD cells for all microglia cell types combined and for each cell type separately. Genes were considered differentially expressed if the adjusted P-value was lower than 0.05. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment was performed using the richR package (https://github.com/hurlab/richR) and an adjusted P-value <0.05 was chosen as the cutoff value to select significant KEGG pathways.
Cell-to-cell communication
CellChat34 was used to examine communication among cells. CellChat uses network analysis and pattern recognition to predict major signaling inputs to cells and signaling outputs from cells. CellChat also predicts how these cells and input and output signals coordinate. First, the software identified the significant ligand-receptor pairs across cell clusters, which were classified into signaling pathways. Next, it predicted incoming signals to specific cell clusters and outgoing signals from specific cell clusters. The global communication pattern was also predicted by pattern recognition approaches. Signaling pathways were then organized by similarity measures and manifold learning from topological perspectives. Finally, CellChat calculated the communication probability of a signaling pathway by summarizing the probabilities of its associated ligand-receptor pairs.
Weighted gene co-expression network analysis
Weighted gene co-expression network analysis (WGCNA)85 was performed to build signed co-expression networks using the WGCNA R package. The co-expression network was built using the top 3,000 most variable genes selected using the “mostVar” function from the transcripTools R package. Batch correction was done using the “ComBat” function from the sva R package. Soft power 6 was chosen by WGCNA’s “pickSoftThreshold” function to calculate the adjacency matrix.
Correlation analysis of body weights to transcript levels
Correlation analysis was performed by Spearman correlation. Body weights at 1 mo and 3 mo were correlated to transcript levels at 1 mo and 3 mo in all cell clusters. Significant correlations were assessed by Spearman’s rank correlation rho. Significant correlated transcripts with Benjamini-Hochberg corrected P-value<0.05 were used as input for KEGG functional enrichment analysis using richR package. The significant pathways with an adjusted P-value<0.05 were selected to generate dot plots.
Immunohistochemistry and microglial morphology analysis
One brain hemisphere from each mouse was dissected at the time of sacrifice and fixed in 4% paraformaldehyde for ∼48 h. Each brain was then placed in 10% sucrose for ∼24 h, followed by 20% sucrose for ∼24 h, and 30% sucrose for a minimum of 24 h. Sections (50 μm) were cut on a cryostat and immunohistochemistry was performed on floating tissue. Sections were stained with rabbit anti-Iba1 (1:1000; Wako, catalog # 019-19741) at 4°C overnight, followed by goat-anti rabbit Alexa fluor Plus 594 secondary antibody (1:2000; Invitrogen, catalog # A32740) for 2hat room temperature followed by a Hoechst nuclear stain for 8 min. Z-stack images were acquired at 40X objective with oil immersion on a Leica Stellaris 8 Falcon Confocal Microscope. Z-stacks (25.5 μm) were pre-processed using Imaris Software (Oxford Instruments). The surface rendering tool was used to identify intact microglial cells, and a mask was created and manually edited to extract the fluorescent signal of full intact cells from the raw data, while eliminating cell branches not associated with cell somas. An open microscopy environment TIF file was then run through an adapted 3DMorph script using MATLAB.30 Objects misidentified as cells and misrepresented cells were manually removed from the final dataset. For each animal (HFD n=4, control n=3), 3 CA1 images were analyzed, and a total of 34 to 46 cells per animal were included in the final analysis.
Quantification and statistical analysis
Statistical analyses of mouse metabolic phenotyping was performed in Prism 9. For HFD versus control comparisons within timepoints, normality was tested using the Shapiro-Wilk test. Welch’s t-test was performed to detect differences between groups for data with normal distributions, and the Mann-Whitney test performed for non-normally distributed data. For repeated measurements (GTTs, body mass) repeated measures 2-way ANOVA with Sidak’s multiple comparisons test was performed. For microglia morphology measures, linear mixed effects models with random animal-specific intercepts were used to detect differences between diet groups. The lmerTest package in R v4.1.1 was used to fit the mixed effects models and model parameter estimates were determined using the maximum likelihood method.86 T-tests calculated using Satterthwaite’s degrees of freedom method were performed to assess differences in morphology measurements between diet groups. Histograms provided visual confirmation of assumptions of normality. The significance cutoff for all comparisons was P<0.05.
Acknowledgments
The authors received funding support from the NIH (U01AG057562, U24DK115255), the Michigan Alzheimer’s Disease Research Center Early Career Investigator Mentorship Program (supported by the NIH/NIA funded by the Michigan Alzheimer’s Disease Research Center (P30AG072931) and the University of Michigan Alzheimer’s Disease Center, Berger Endowment), the NIDDK (T32DK007245), the JDRF (JDRF 5COE-2019-861-S-B), the Edith S. Briskin/SKS Foundation NeuroNetwork Emerging Scholar Fund, the Robert E. Nederlander Sr. Program for Alzheimer's Research, the Andrea and Lawrence A. Wolfe Brain Health Initiative Fund, the A. Alfred Taubman Medical Research Institute, and the NeuroNetwork for Emerging Therapies. The authors thank the Microscopy, Imaging, and Cellular Physiology Core (MICPC) at the University of Michigan and its Imaging Laboratory director, Steve Lentz, PhD, for guidance and support in developing and implementing methods of microglial morphology analysis. They also thank the University of Michigan Flow Cytometry Core for assistance with cell sorting and the Advanced Genomics Core for assistance with 10X Chromium single-cell RNA-sequencing. Finally, they thank the Mouse Metabolic Phenotyping Core (MMPC) at the University of Cincinnati for metabolic phenotyping of plasma.
Author contributions
In author order. Conceptualization, R.E.H., M.G.S., and E.L.F.; methodology R.E.H., K.G., M.G.S., J.H., and E.L.F.; investigation: R.E.H., S.E.E., F.E.M., J.M.H., and D.R.; formal analysis, R.E.H., K.G., and M.H.N.; data curation, K.G.; writing – original draft, R.E.H. and M.G.S.; writing – review and editing, R.E.H., K.G., M.G.S., J.H., and E.L.F.; resources and funding acquisition, E.L.F.; supervision, J.H. and E.L.F.
Declaration of interests
The authors declare no competing interests.
Published: February 8, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106164.
Supplemental information
Document S1. Figures S1–S9
Gene markers used to cluster cells into homeostatic microglia (HMG), HMG1, 2, and 3, inflammatory microglia (InflamMG), interferon-related (IfnMG,), proliferating microglia (ProlifMG), functionally undescribed microglia (uMG), macrophages (Mac), monocytes (Mono), neutrophils (Neu), and neurons (Neuro). Entries arranged by nominal P-value. Adjusted P-value (Padj) based on Bonferroni correction using all features in the dataset. FC, fold-change; pct.1, percentage of cells where the feature is detected in the first group; pct.2, percentage of cells where the feature is detected in the second group.
Differentially expressed genes (DEGs) of all microglia clusters combined from HFD versus control mice at 1 mo (white) and 3 mo (grey) meeting the criteria of nominal P-value>0.05. Entries arranged by nominal P-value. Adjusted P-value based on Bonferroni correction using all features in the dataset. FC, fold-change; pct.1, percentage of cells where the feature is detected in the first group; pct.2, percentage of cells where the feature is detected in the second group.
(A) Summary of the number of significant differentially expressed genes (DEGs) in each cell cluster in HFD versus control mice at 1 mo (white) and 3 mo (grey). (B) DEGs in each cell cluster in HFD versus control mice at 1 mo (white) and 3 mo (grey) meeting the criteria of adjusted P-value (Padj)>0.05 based on Bonferroni correction using all features in the dataset. FC, fold-change; pct.1, percentage of cells where the feature is detected in the first group; pct.2, percentage of cells where the feature is detected in the second group. HMG1, 2, 3, homeostatic microglia 1, 2, 3; InflamMG, inflammatory microglia; IfnMG, interferon-related; ProlifMG, proliferating microglia; uMG, functionally undescribed microglia; Mac, macrophages; Mono, monocytes; Neu, neutrophils; Neuro, neurons.
Differentially expressed genes (DEGs) of all homeostatic microglia (HMG) clusters combined from HFD versus control mice at 1 mo (white) and 3 mo (grey) meeting the criteria of nominal P-value>0.05. Entries arranged by nominal P-value. Adjusted P-value (Padj) based on Bonferroni correction using all features in the dataset. FC, fold-change; pct.1, percentage of cells where the feature is detected in the first group; pct.2, percentage of cells where the feature is detected in the second group.
Unsupervised weighted gene co-expression network analysis (WGCNA) of 1 mo and 3 mo samples identified two significant modules, yellow and brown. KEGG pathway analysis shown in table of yellow (top) and brown (bottom) module. Padj, adjusted P-value. ‘Annot,’ KEGG pathway identification number; ‘Annotated,’ number of genes annotated in KEGG pathway; ‘Significant,’ number of genes in the module that are represented in the KEGG term annotation; ‘Gene,’ gene names for each significant gene.
P-value, significance level of correlation; Padj, Benjamini-Hochberg adjusted significance level of correlation
Sheet 1: KEGG of positively correlated transcripts; sheet 2: KEGG of negatively correlated transcripts. Group, cell cluster; Term, KEGG pathway name; Annotated, number of DEGs within KEGG pathway; Significant, number of significant DEGs within KEGG pathway; P-value, significance level KEGG pathway; Padj, adjusted significance level KEGG pathway; GeneID, gene names for KEGG pathway genes.
Data and code availability
scRNA-seq data generated from this study has been deposited into the NCBI Gene Expression Omnibus with accession ID GSE217464. The data analysis codes can be found at: https://github.com/hurlab/HF_iScience. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Obesity and overweight. https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight.
- 2.Callaghan B.C., Reynolds E.L., Banerjee M., Chant E., Villegas-Umana E., Gardner T.W., Votruba K., Giordani B., Pop-Busui R., Pennathur S., Feldman E.L. The prevalence and determinants of cognitive deficits and traditional diabetic complications in the severely obese. Diabetes Care. 2020;43:683–690. doi: 10.2337/dc19-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dekkers I.A., Jansen P.R., Lamb H.J. Obesity, brain volume, and white matter microstructure at MRI: a cross-sectional UK biobank study. Radiology. 2019;291:763–771. doi: 10.1148/RADIOL.2019181012. [DOI] [PubMed] [Google Scholar]
- 4.Black N., Johnston D.W., Peeters A. Childhood obesity and cognitive achievement. Health Econ. 2015;24:1082–1100. doi: 10.1002/HEC.3211. [DOI] [PubMed] [Google Scholar]
- 5.Alosco M.L., Stanek K.M., Galioto R., Korgaonkar M.S., Grieve S.M., Brickman A.M., Spitznagel M.B., Gunstad J. Body mass index and brain structure in healthy children and adolescents. Int. J. Neurosci. 2013;124:49–55. doi: 10.3109/00207454.2013.817408. [DOI] [PubMed] [Google Scholar]
- 6.Yau P.L., Castro M.G., Tagani A., Tsui W.H., Convit A. Obesity and metabolic syndrome and functional and structural brain impairments in adolescence. Pediatrics. 2012;130:e856–e864. doi: 10.1542/PEDS.2012-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu, W.L., Atti, A.R., Gatz, M., Pedersen, N.L., Johansson, B., and Fratiglioni, L. (2011). Midlife Overweight and Obesity Increase Late-Life Dementia Risk A Population-Based Twin Study. [DOI] [PMC free article] [PubMed]
- 8.Pedditzi E., Peters R., Beckett N. The risk of overweight/obesity in mid-life and late life for the development of dementia: a systematic review and meta-analysis of longitudinal studies. Age Ageing. 2016;45:14–21. doi: 10.1093/AGEING/AFV151. [DOI] [PubMed] [Google Scholar]
- 9.André C., Guzman-Quevedo O., Rey C., Rémus-Borel J., Clark S., Castellanos-Jankiewicz A., Ladeveze E., Leste-Lasserre T., Nadjar A., Abrous D.N., et al. Inhibiting microglia expansion prevents diet-induced hypothalamic and peripheral inflammation. Diabetes. 2017;66:908–919. doi: 10.2337/DB16-0586. [DOI] [PubMed] [Google Scholar]
- 10.Valdearcos M., Myers M.G., Koliwad S.K. Hypothalamic microglia as potential regulators of metabolic physiology. Nat. Metab. 2019;1:314–320. doi: 10.1038/S42255-019-0040-0. [DOI] [PubMed] [Google Scholar]
- 11.Valdearcos M., Douglass J.D., Robblee M.M., Dorfman M.D., Stifler D.R., Bennett M.L., Gerritse I., Fasnacht R., Barres B.A., Thaler J.P., Koliwad S.K. Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metab. 2017;26:185–197.e3. doi: 10.1016/J.CMET.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mendes N.F., Kim Y.B., Velloso L.A., Araújo E.P. Hypothalamic microglial activation in obesity: amini-review. Front. Neurosci. 2018;12:846. doi: 10.3389/FNINS.2018.00846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakandakari S.C.B.R., Muñoz V.R., Kuga G.K., Gaspar R.C., Sant’Ana M.R., Pavan I.C.B., da Silva L.G.S., Morelli A.P., Simabuco F.M., da Silva A.S.R., et al. Short-term high-fat diet modulates several inflammatory, ER stress, and apoptosis markers in the hippocampus of young mice. Brain Behav. Immun. 2019;79:284–293. doi: 10.1016/j.bbi.2019.02.016. [DOI] [PubMed] [Google Scholar]
- 14.Sobesky J.L., D’Angelo H.M., Weber M.D., Anderson N.D., Frank M.G., Watkins L.R., Maier S.F., Barrientos R.M. Glucocorticoids mediate short-term high-fat diet induction of neuroinflammatory priming, the NLRP3 inflammasome, and the danger signal HMGB1. eNeuro. 2016;3 doi: 10.1523/ENEURO.0113-16.2016. ENEURO.0113-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cope E.C., LaMarca E.A., Monari P.K., Olson L.B., Martinez S., Zych A.D., Katchur N.J., Gould E. Microglia play an active role in obesity-associated cognitive decline. J. Neurosci. 2018;38:8889–8904. doi: 10.1523/JNEUROSCI.0789-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao S., Dey A., Yu X., Stranahan A.M. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav. Immun. 2016;51:230–239. doi: 10.1016/j.bbi.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valcarcel-Ares M.N., Tucsek Z., Kiss T., Giles C.B., Tarantini S., Yabluchanskiy A., Balasubramanian P., Gautam T., Galvan V., Ballabh P., et al. Obesity in aging exacerbates neuroinflammation, dysregulating synaptic function-related genes and altering eicosanoid synthesis in the mouse Hippocampus: potential role in impaired synaptic plasticity and cognitive decline. J. Gerontol. A Biol. Sci. Med. Sci. 2019;74:290–298. doi: 10.1093/GERONA/GLY127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sims-Robinson C., Bakeman A., Bruno E., Jackson S., Glasser R., Murphy G.G., Feldman E.L. Dietary reversal ameliorates short- and long-term memory deficits induced by high-fat diet early in life. PLoS One. 2016;11:e0163883. doi: 10.1371/journal.pone.0163883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guillemot-Legris O., Muccioli G.G. Obesity-induced neuroinflammation: beyond the hypothalamus. Trends Neurosci. 2017;40:237–253. doi: 10.1016/J.TINS.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Kang E.B., Koo J.H., Jang Y.C., Yang C.H., Lee Y., Cosio-Lima L.M., Cho J.Y. Neuroprotective effects of endurance exercise against high-fat diet-induced hippocampal neuroinflammation. J. Neuroendocrinol. 2016;28 doi: 10.1111/JNE.12385. [DOI] [PubMed] [Google Scholar]
- 21.Guo D.H., Yamamoto M., Hernandez C.M., Khodadadi H., Baban B., Stranahan A.M. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J. Clin. Invest. 2020;130:1961–1976. doi: 10.1172/JCI126078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melo H.M., Seixas da Silva G.d.S., Sant’Ana M.R., Teixeira C.V.L., Clarke J.R., Miya Coreixas V.S., de Melo B.C., Fortuna J.T.S., Forny-Germano L., Ledo J.H., et al. Palmitate is increased in the cerebrospinal fluid of humans with obesity and induces memory impairment in mice via pro-inflammatory TNF-α. Cell Rep. 2020;30:2180–2194.e8. doi: 10.1016/j.celrep.2020.01.072. [DOI] [PubMed] [Google Scholar]
- 23.Buckman L.B., Hasty A.H., Flaherty D.K., Buckman C.T., Thompson M.M., Matlock B.K., Weller K., Ellacott K.L.J. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav. Immun. 2014;35:33–42. doi: 10.1016/J.BBI.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salas-Venegas V., Flores-Torres R.P., Rodríguez-Cortés Y.M., Rodríguez-Retana D., Ramírez-Carreto R.J., Concepción-Carrillo L.E., Pérez-Flores L.J., Alarcón-Aguilar A., López-Díazguerrero N.E., Gómez-González B., et al. The obese brain: mechanisms of systemic and local inflammation, and interventions to reverse the cognitive deficit. Front. Integr. Neurosci. 2022;16:798995. doi: 10.3389/FNINT.2022.798995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valdearcos M., Robblee M.M., Benjamin D.I., Nomura D.K., Xu A.W., Koliwad S.K. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014;9:2124–2138. doi: 10.1016/j.celrep.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sims-Robinson C., Zhao S., Hur J., Feldman E.L. Central nervous system endoplasmic reticulum stress in a murine model of type 2 diabetes. Diabetologia. 2012;55:2276–2284. doi: 10.1007/s00125-012-2573-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eid S.A., Feldman E.L. Advances in diet-induced rodent models of metabolically acquired peripheral neuropathy. Dis. Model. Mech. 2021;14:dmm049337. doi: 10.1242/DMM.049337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Moura e Dias M., dos Reis S.A., da Conceição L.L., Sediyama C.M.N.d.O., Pereira S.S., de Oliveira L.L., Gouveia Peluzio M.d.C., Martinez J.A., Milagro F.I. Diet-induced obesity in animal models: points to consider and influence on metabolic markers. Diabetol. Metab. Syndr. 2021;13:32. doi: 10.1186/S13098-021-00647-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Brien P.D., Hinder L.M., Rumora A.E., Hayes J.M., Dauch J.R., Backus C., Mendelson F.E., Feldman E.L. Juvenile murine models of prediabetes and type 2 diabetes develop neuropathy. Dis. Model. Mech. 2018;11:dmm037374. doi: 10.1242/dmm.037374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.York E.M., Ledue J.M., Bernier L.P., Macvicar B.A. 3dmorph automatic analysis of microglial morphology in three dimensions from ex vivo and in vivo imaging. eNeuro. 2018;5 doi: 10.1523/ENEURO.0266-18.2018. ENEURO.0266-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colonna M., Butovsky O. Microglia function in the central nervous system during Health and neurodegeneration. Annu. Rev. Immunol. 2017;35:441–468. doi: 10.1146/ANNUREV-IMMUNOL-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.ElAli A., Rivest S. Microglia ontology and signaling. Front. Cell Dev. Biol. 2016;4:72. doi: 10.3389/fcell.2016.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nimmerjahn A., Kirchhoff F., Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/SCIENCE.1110647. [DOI] [PubMed] [Google Scholar]
- 34.Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.-H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wimmer I., Tietz S., Nishihara H., Deutsch U., Sallusto F., Gosselet F., Lyck R., Muller W.A., Lassmann H., Engelhardt B. PECAM-1 stabilizes blood-brain barrier integrity and favors paracellular T-cell diapedesis across the blood-brain barrier during neuroinflammation. Front. Immunol. 2019;10:711. doi: 10.3389/FIMMU.2019.00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van De Stolpe A., Van Der Saag P.T. Intercellular adhesion molecule-1. J. Mol. Med. 1996;74:13–33. doi: 10.1007/BF00202069. [DOI] [PubMed] [Google Scholar]
- 37.Werner A., Kloss C.U., Walter J., Kreutzberg G.W., Raivich G. Intercellular adhesion molecule-1 (ICAM-1) in the mouse facial motor nucleus after axonal injury and during regeneration. J. Neurocytol. 1998;27:219–232. doi: 10.1023/A:1006928830251. [DOI] [PubMed] [Google Scholar]
- 38.Bussini S., Meda L., Scarpini E., Clementi E., Conti G., Tiriticco M., Bresolin N., Baron P. Heparan sulfate proteoglycan induces the production of NO and TNF-α by murine microglia. Immun. Ageing. 2005;2:11. doi: 10.1186/1742-4933-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Callaghan P., Zhang X., Li J.P. Heparan sulfate proteoglycans as relays of neuroinflammation. J. Histochem. Cytochem. 2018;66:305–319. doi: 10.1369/0022155417742147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lyons A., Downer E.J., Crotty S., Nolan Y.M., Mills K.H.G., Lynch M.A. CD200 ligand–receptor interaction modulates microglial activation in vivo and in vitro: arole for IL-4. J. Neurosci. 2007;27:8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Telpoukhovskaia M.A., Liu K., Sayed F.A., Etchegaray J.I., Xie M., Zhan L., Li Y., Zhou Y., Le D., Bahr B.A., et al. Discovery of small molecules that normalize the transcriptome and enhance cysteine cathepsin activity in progranulin-deficient microglia. Sci. Rep. 2020;10:13688. doi: 10.1038/s41598-020-70534-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhinn H., Tatton N., McCaughey S., Kurnellas M., Rosenthal A. Progranulin as a therapeutic target in neurodegenerative diseases. Trends Pharmacol. Sci. 2022;43:641–652. doi: 10.1016/J.TIPS.2021.11.015. [DOI] [PubMed] [Google Scholar]
- 43.Rünker A.E., Little G.E., Suto F., Fujisawa H., Mitchell K.J. Semaphorin-6A controls guidance of corticospinal tract axons at multiple choice points. Neural Dev. 2008;3:1–19. doi: 10.1186/1749-8104-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rothaug M., Becker-Pauly C., Rose-John S. The role of interleukin-6 signaling in nervous tissue. Biochim. Biophys. Acta. 2016;1863:1218–1227. doi: 10.1016/J.BBAMCR.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 45.Andrae J., Gallini R., Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/GAD.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pierce G.F., Mustoe T.A., Altrock B.W., Deuel T.F., Thomason A. Role of platelet-derived growth factor in wound healing. J. Cell. Biochem. 1991;45:319–326. doi: 10.1002/JCB.240450403. [DOI] [PubMed] [Google Scholar]
- 47.Brykczynska U., Geigges M., Wiedemann S.J., Dror E., Böni-Schnetzler M., Hess C., Donath M.Y., Paro R. Distinct transcriptional responses across tissue-resident macrophages to short-term and long-term metabolic challenge. Cell Rep. 2020;30:1627–1643.e7. doi: 10.1016/J.CELREP.2020.01.005. [DOI] [PubMed] [Google Scholar]
- 48.Tarantini S., Valcarcel-Ares M.N., Yabluchanskiy A., Tucsek Z., Hertelendy P., Kiss T., Gautam T., Zhang X.A., Sonntag W.E., de Cabo R., et al. Nrf2 deficiency exacerbates obesity-induced oxidative stress, neurovascular dysfunction, blood-brain barrier disruption, neuroinflammation, amyloidogenic gene expression, and cognitive decline in mice, mimicking the aging phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 2018;73:853–863. doi: 10.1093/GERONA/GLX177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Braeuer R.R., Walker N.M., Misumi K., Mazzoni-Putman S., Aoki Y., Liao R., Vittal R., Kleer G.G., Wheeler D.S., Sexton J.Z., et al. Transcription factor FOXF1 identifies compartmentally distinct mesenchymal cells with a role in lung allograft fibrogenesis. J. Clin. Invest. 2021;131:e147343. doi: 10.1172/JCI147343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hammond T.R., Dufort C., Dissing-Olesen L., Giera S., Young A., Wysoker A., Walker A.J., Gergits F., Segel M., Nemesh J., et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50:253–271.e6. doi: 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T.K., David E., Baruch K., Lara-Astaiso D., Toth B., et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–1290.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 52.Zheng J., Ru W., Adolacion J.R., Spurgat M.S., Liu X., Yuan S., Liang R.X., Dong J., Potter A.S., Potter S.S., et al. Single-cell RNA-seq analysis reveals compartment-specific heterogeneity and plasticity of microglia. iScience. 2021;24:102186. doi: 10.1016/J.ISCI.2021.102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marsh S.E., Walker A.J., Kamath T., Dissing-Olesen L., Hammond T.R., de Soysa T.Y., Young A.M.H., Murphy S., Abdulraouf A., Nadaf N., et al. Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nat. Neurosci. 2022;25:306–316. doi: 10.1038/s41593-022-01022-8. [DOI] [PubMed] [Google Scholar]
- 54.Borst K., Dumas A.A., Prinz M. Microglia: immune and non-immune functions. Immunity. 2021;54:2194–2208. doi: 10.1016/J.IMMUNI.2021.09.014. [DOI] [PubMed] [Google Scholar]
- 55.O’Callaghan P., Li J.P., Lannfelt L., Lindahl U., Zhang X. Microglial heparan sulfate proteoglycans facilitate the cluster-of-differentiation 14 (CD14)/Toll-like receptor 4 (TLR4)-Dependent inflammatory response. J. Biol. Chem. 2015;290:14904–14914. doi: 10.1074/JBC.M114.634337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borggrewe M., Kooistra S.M., Noelle R.J., Eggen B.J.L., Laman J.D. Exploring the VISTA of microglia: immune checkpoints in CNS inflammation. J. Mol. Med. 2020;98:1415–1430. doi: 10.1007/S00109-020-01968-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zöller T., Schneider A., Kleimeyer C., Masuda T., Potru P.S., Pfeifer D., Blank T., Prinz M., Spittau B. Silencing of TGFβ signalling in microglia results in impaired homeostasis. Nat. Commun. 2018;9:4011. doi: 10.1038/s41467-018-06224-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schäfer M.K., Schwaeble W.J., Post C., Salvati P., Calabresi M., Sim R.B., Petry F., Loos M., Weihe E. Complement C1q is dramatically up-regulated in brain microglia in response to transient global cerebral ischemia. J. Immunol. 2000;164:5446–5452. doi: 10.4049/JIMMUNOL.164.10.5446. [DOI] [PubMed] [Google Scholar]
- 59.Hong S., Beja-Glasser V.F., Nfonoyim B.M., Frouin A., Li S., Ramakrishnan S., Merry K.M., Shi Q., Rosenthal A., Barres B.A., et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/SCIENCE.AAD8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schartz N.D., Tenner A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflammation. 2020;17:354. doi: 10.1186/S12974-020-02024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McQuade A., Kang Y.J., Hasselmann J., Jairaman A., Sotelo A., Coburn M., Shabestari S.K., Chadarevian J.P., Fote G., Tu C.H., et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020;11:5370. doi: 10.1038/s41467-020-19227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCarthy G.M., Farris S.P., Blednov Y.A., Harris R.A., Mayfield R.D. Microglial-specific transcriptome changes following chronic alcohol consumption. Neuropharmacology. 2018;128:416–424. doi: 10.1016/J.NEUROPHARM.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sims-Robinson C., Bakeman A., Glasser R., Boggs J., Pacut C., Feldman E.L. The role of endoplasmic reticulum stress in hippocampal insulin resistance. Exp. Neurol. 2016;277:261–267. doi: 10.1016/j.expneurol.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hummasti S., Hotamisligil G.S. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ. Res. 2010;107:579–591. doi: 10.1161/CIRCRESAHA.110.225698. [DOI] [PubMed] [Google Scholar]
- 65.Hotamisligil G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/J.CELL.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korbecki J., Bajdak-Rusinek K. The effect of palmitic acid on inflammatory response in macrophages: an overview of molecular mechanisms. Inflamm. Res. 2019;68:915–932. doi: 10.1007/S00011-019-01273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robblee M.M., Kim C.C., Porter Abate J., Valdearcos M., Sandlund K.L.M., Shenoy M.K., Volmer R., Iwawaki T., Koliwad S.K. Saturated fatty acids engage an IRE1α-dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep. 2016;14:2611–2623. doi: 10.1016/j.celrep.2016.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shan B., Wang X., Wu Y., Xu C., Xia Z., Dai J., Shao M., Zhao F., He S., Yang L., et al. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 2017;18:519–529. doi: 10.1038/ni.3709. [DOI] [PubMed] [Google Scholar]
- 69.Wang Y., Zhou Q., Zhang X., Qian Q., Xu J., Ni P., Qian Y. Mild endoplasmic reticulum stress ameliorates lipopolysaccharide-induced neuroinflammation and cognitive impairment via regulation of microglial polarization. J. Neuroinflammation. 2017;14:233. doi: 10.1186/s12974-017-1002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shi M., Chai Y., Zhang J., Chen X. Endoplasmic reticulum stress-associated neuronal death and innate immune response in neurological diseases. Front. Immunol. 2022;12:5796. doi: 10.3389/FIMMU.2021.794580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Minhas P.S., Latif-Hernandez A., McReynolds M.R., Durairaj A.S., Wang Q., Rubin A., Joshi A.U., He J.Q., Gauba E., Liu L., et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature. 2021;590:122–128. doi: 10.1038/S41586-020-03160-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nelson L.H., Lenz K.M. The immune system as a novel regulator of sex differences in brain and behavioral development. J. Neurosci. Res. 2017;95:447–461. doi: 10.1002/JNR.23821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gubbels Bupp M.R. Sex, the aging immune system, and chronic disease. Cell. Immunol. 2015;294:102–110. doi: 10.1016/J.CELLIMM.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 74.Franzén O., Gan L.M., Björkegren J.L.M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database. 2019;2019:baz046. doi: 10.1093/DATABASE/BAZ046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M., et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47:D721–D728. doi: 10.1093/NAR/GKY900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., R S., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/J.CELL.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/S13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huynh-Thu V.A., Irrthum A., Wehenkel L., Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010;5:e12776. doi: 10.1371/journal.pone.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/NCOMMS14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stoeckius M., Zheng S., Houck-Loomis B., Hao S., Yeung B.Z., Mauck W.M., Smibert P., Satija R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19:224. doi: 10.1186/S13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nikodemova M., Kimyon R.S., De I., Small A.L., Collier L.S., Watters J.J. Microglial numbers attain adult levels after undergoing a rapid decrease in cell number in the third postnatal week. J. Neuroimmunol. 2015;278:280–288. doi: 10.1016/J.JNEUROIM.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tay T.L., Savage J.C., Hui C.W., Bisht K., Tremblay M.È. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J. Physiol. 2017;595:1929–1945. doi: 10.1113/JP272134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang B., Horvath S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 86.Kuznetsova A., Brockhoff P.B., Christensen R.H.B. lmerTest package: tests in linear mixed effects models. J. Stat. Softw. 2017;82:1–26. doi: 10.18637/JSS.V082.I13. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1–S9
Gene markers used to cluster cells into homeostatic microglia (HMG), HMG1, 2, and 3, inflammatory microglia (InflamMG), interferon-related (IfnMG,), proliferating microglia (ProlifMG), functionally undescribed microglia (uMG), macrophages (Mac), monocytes (Mono), neutrophils (Neu), and neurons (Neuro). Entries arranged by nominal P-value. Adjusted P-value (Padj) based on Bonferroni correction using all features in the dataset. FC, fold-change; pct.1, percentage of cells where the feature is detected in the first group; pct.2, percentage of cells where the feature is detected in the second group.
Differentially expressed genes (DEGs) of all microglia clusters combined from HFD versus control mice at 1 mo (white) and 3 mo (grey) meeting the criteria of nominal P-value>0.05. Entries arranged by nominal P-value. Adjusted P-value based on Bonferroni correction using all features in the dataset. FC, fold-change; pct.1, percentage of cells where the feature is detected in the first group; pct.2, percentage of cells where the feature is detected in the second group.
(A) Summary of the number of significant differentially expressed genes (DEGs) in each cell cluster in HFD versus control mice at 1 mo (white) and 3 mo (grey). (B) DEGs in each cell cluster in HFD versus control mice at 1 mo (white) and 3 mo (grey) meeting the criteria of adjusted P-value (Padj)>0.05 based on Bonferroni correction using all features in the dataset. FC, fold-change; pct.1, percentage of cells where the feature is detected in the first group; pct.2, percentage of cells where the feature is detected in the second group. HMG1, 2, 3, homeostatic microglia 1, 2, 3; InflamMG, inflammatory microglia; IfnMG, interferon-related; ProlifMG, proliferating microglia; uMG, functionally undescribed microglia; Mac, macrophages; Mono, monocytes; Neu, neutrophils; Neuro, neurons.
Differentially expressed genes (DEGs) of all homeostatic microglia (HMG) clusters combined from HFD versus control mice at 1 mo (white) and 3 mo (grey) meeting the criteria of nominal P-value>0.05. Entries arranged by nominal P-value. Adjusted P-value (Padj) based on Bonferroni correction using all features in the dataset. FC, fold-change; pct.1, percentage of cells where the feature is detected in the first group; pct.2, percentage of cells where the feature is detected in the second group.
Unsupervised weighted gene co-expression network analysis (WGCNA) of 1 mo and 3 mo samples identified two significant modules, yellow and brown. KEGG pathway analysis shown in table of yellow (top) and brown (bottom) module. Padj, adjusted P-value. ‘Annot,’ KEGG pathway identification number; ‘Annotated,’ number of genes annotated in KEGG pathway; ‘Significant,’ number of genes in the module that are represented in the KEGG term annotation; ‘Gene,’ gene names for each significant gene.
P-value, significance level of correlation; Padj, Benjamini-Hochberg adjusted significance level of correlation
Sheet 1: KEGG of positively correlated transcripts; sheet 2: KEGG of negatively correlated transcripts. Group, cell cluster; Term, KEGG pathway name; Annotated, number of DEGs within KEGG pathway; Significant, number of significant DEGs within KEGG pathway; P-value, significance level KEGG pathway; Padj, adjusted significance level KEGG pathway; GeneID, gene names for KEGG pathway genes.
Data Availability Statement
scRNA-seq data generated from this study has been deposited into the NCBI Gene Expression Omnibus with accession ID GSE217464. The data analysis codes can be found at: https://github.com/hurlab/HF_iScience. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.