
Fig. 1 |. Workflow of STAARpipeline.
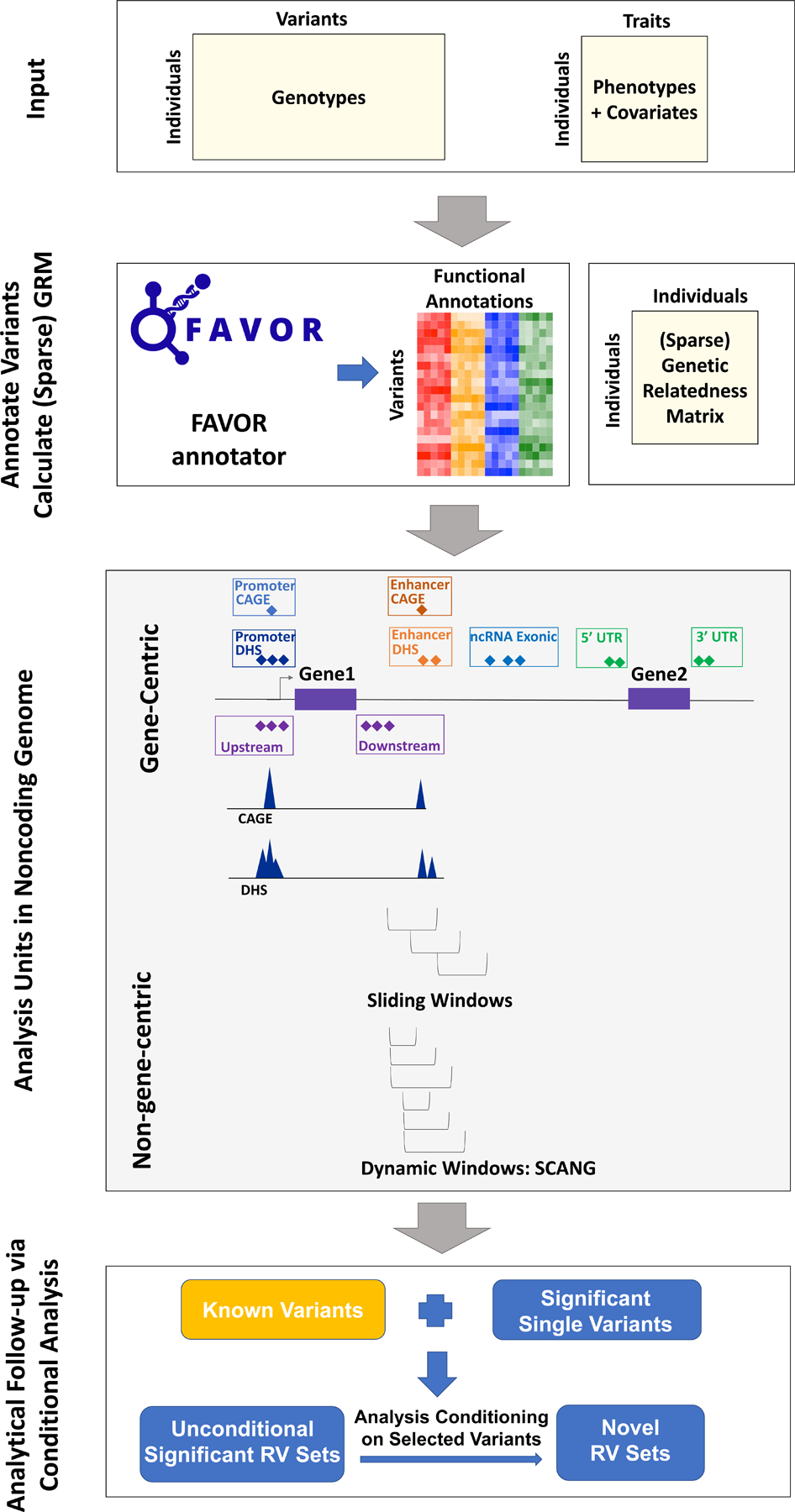
(a) Prepare the input data of STAARpipeline, including genotypes, phenotypes and covariates. (b) Annotate all variants in the genome using FAVORannotator through FAVOR database and calculate the (sparse) genetic relatedness matrix. (c) Define analysis units in the noncoding genome: eight functional categories of regulatory regions, sliding windows and dynamic windows using SCANG. (d) Obtain genome-wide significant associations and perform analytical follow-up via conditional analysis.