

SUMMARY
Most relapsed/refractory large B cell lymphoma (r/rLBCL) patients receiving anti-CD19 chimeric antigen receptor (CAR19) T cells relapse. To characterize determinants of resistance, we profiled over 700 longitudinal specimens from two independent cohorts (n = 65 and n = 73) of r/rLBCL patients treated with axicabtagene ciloleucel. A method for simultaneous profiling of circulating tumor DNA (ctDNA), cell-free CAR19 (cfCAR19) retroviral fragments, and cell-free T cell receptor rearrangements (cfTCR) enabled integration of tumor and both engineered and non-engineered T cell effector-mediated factors for assessing treatment failure and predicting outcomes. Alterations in multiple classes of genes are associated with resistance, including B cell identity (PAX5 and IRF8), immune checkpoints (CD274), and those affecting the microenvironment (TMEM30A). Somatic tumor alterations affect CAR19 therapy at multiple levels, including CAR19 T cell expansion, persistence, and tumor microenvironment. Further, CAR19 T cells play a reciprocal role in shaping tumor genotype and phenotype. We envision these findings will facilitate improved chimeric antigen receptor (CAR) T cells and personalized therapeutic approaches.
In brief
Sworder et al. develop and apply a tool to simultaneously profile tumor and effector-mediated determinants of resistance to anti-CD19 CAR T cells using cell-free DNA. The authors profile two independent cohorts of patients with relapsed/refractory large B cell lymphoma and identify genomic alterations, molecular thresholds, and microenvironmental changes associated with resistance.
Graphical Abstract
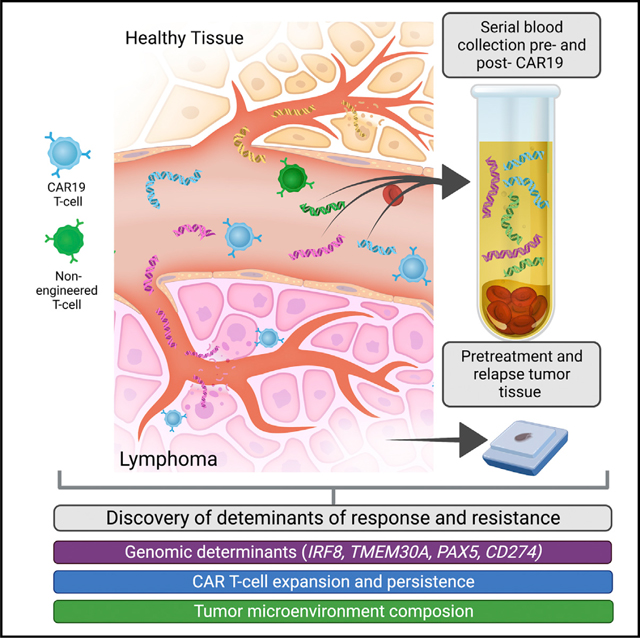
INTRODUCTION
Chimeric antigen receptor (CAR) T cells are a highly potent form of biologically active immunotherapy, with efficacy proved in patients with relapsed/refractory B cell-derived malignancies, including acute lymphoblastic leukemia (ALL), mantle cell lymphoma (MCL), multiple myeloma (MM), and large B cell lymphoma (LBCL).1–4 In LBCL, CAR-modified T cells targeting CD19 on the surface of lymphoma cells (CAR19) have demonstrated significant activity in patients with relapsed/refractory LBCL (r/rLBCL), with the majority achieving a response.1,5,6 Despite these encouraging results, over half of patients receiving CAR T cell therapy will ultimately experience disease progression.7
Available evidence suggests that resistance to CAR T cell therapy is multifactorial and influenced by factors related to both the target and effector cells.8,9 CD19 loss on tumor cells has been observed in both ALL and LBCL at time of relapse after CAR19 therapy.10–13 Hematopoietic lineage switch, induced by CAR19 immune pressure, has been postulated to be a potential mechanism of immune evasion, and tumor genomic factors, such as BCL2 translocations and APOBEC mutational signatures have been associated with treatment failure.14–16 Conversely, higher frequencies of CAR19 cells expressing memory signatures and increased cellular diversity have been associated with superior outcomes, while exhaustion signatures have been associated with inferior survival.17 While these studies elucidate resistance in a subset of patients, resistance mechanisms to CAR19 therapy remain incompletely characterized.
Recently, methods to detect circulating tumor-derived cell-free DNA (ctDNA), from the blood plasma of patients have unlocked significant opportunities to noninvasively study tumor biology during therapy. Targeted sequencing approaches such as cancer personalized profiling by deep sequencing (CAPP-seq) can identify tumor-derived single-nucleotide variants (SNVs), somatic copy-number alterations (SCNAs), small insertions/deletions (indels), and fusions from ctDNA, mitigating the need for tumor tissue for molecular profiling.18–20 We have previously applied this method to patients with LBCL, demonstrating the utility for both non-invasive genotyping and detecting minimal residual disease.21 This tool has led to the identification of robust prognostic factors based on tumor assessments, including the definition of molecular response criteria and the development of dynamic risk assessment tools in lymphoma patients undergoing chemoimmunotherapy.22,23 However, while ctDNA profiling has demonstrated utility for tumor assessments in LBCL, an integrated assessment of tumor cells and non-tumor cells such as native and engineered T cells has not yet been systematically addressed during active cellular therapy.
Here, we extend CAPP-seq through a hybrid-capture strategy to concurrently assess tumor-derived ctDNA as well as CAR19-derived cell-free DNA (cfCAR19) from specific recombinant retroviral sequences in patients undergoing treatment with axicabtagene ciloleucel (axi-cel). This combined strategy allows for the simultaneous characterization of molecular response, identification of genomic alterations associated with treatment failure, and profiling of CAR19 activity, thus facilitating integrative analyses of how these factors work in concert to lead to CAR19 resistance.
RESULTS
Profiling of distinct lymphoid cfDNA compartments during CAR19 therapy
We profiled a total of 719 serial blood and tissue specimens from independent discovery (n = 65) and validation (n = 73) cohorts of subjects receiving axi-cel therapy for r/rLBCL at Stanford University (Tables 1 and S1). Plasma and peripheral blood mononuclear cells (PBMCs) were collected prior to lymphodepletion therapy and before CAR T cell infusion, with serial blood samples longitudinally collected following therapy, as well as at the time of relapse (Figure 1; Table S2). Where available, tumor biopsy specimens were also collected for a subset of patients before therapy and at relapse. When comparing the discovery and validation cohorts, there was no significant difference in the median event-free survival (EFS; 6.18 versus 5.95 months, p = 0.64; Figure S1A), or in the median overall survival (OS; not reached versus 23.2 months, p = 0.55; Figure S1B). Thirty-six patients (55%) in the discovery cohort and 43 patients (58%) in the validation cohort experienced progression following CAR19 therapy, with the remaining patients achieving an ongoing response.
Figure 1. Study overview.

Schematic illustrating the integrative approach to characterize mechanisms of CAR19 resistance undertaken in this study. An r/rLBCL patient undergoing CAR19 therapy is depicted from prior to infusion to relapse. (Above timeline) Plasma samples for simultaneous profiling of ctDNA, cfCAR19, and cfTCR were obtained from pretreatment and multiple post-infusion time points, including relapse, when applicable. Analyses performed at each time point are indicated in colored boxes. Colors reflect the compartment being analyzed: tumor/ctDNA (purple), CAR19/cfCAR19 (blue), immune microenvironment/cfTCR (green). (Below timeline).
To facilitate the concurrent characterization of both tumor B cell responses and anti-tumor T cell dynamics during CAR19 therapy of r/rLBCL, we developed a platform for simultaneous tumor and effector profiling (STEP). Specifically, STEP utilizes non-invasive integrated analysis of cell-free DNA (cfDNA) molecules to profile circulating tumor DNA (ctDNA), CAR19 T cell-derived cfDNA (cfCAR19), and non-engineered T cell receptor cfDNA (cfTCR) molecules (Figure 2A). To identify tumor-derived mutant DNA from malignant lymphoma B cells, we captured 646 kb targeting 187 recurrently mutated protein-coding genes in lymphomas or involved in immunological checkpoints (Table S3). Separately, to profile cfDNA derived from infused CAR19-transduced T cells and from other effector T cells associated with immune reconstitution, we simultaneously applied a 24-kb panel targeting the axi-cel retroviral vector sequence (Figure S2A), as well as the full set of human T cell receptor loci (see STAR Methods).
Figure 2. STEP platform and impact of ctDNA molecular thresholds on outcome.

(A) Illustration summarizing the strategy through which ctDNA, cfCAR19, and cfTCR are simultaneously profiled from a plasma sample using the simultaneous tumor and effector profiling (STEP) platform.
(B) Dynamic changes in ctDNA (top), cfCAR19 (middle), and cfTCR levels (bottom) following CAR19 infusion in patients who progress (red), and those who achieve an ongoing response (blue). Wilcoxon rank-sum test used to compare variables at each noted time point.
(C and D) Kaplan-Meier estimates show EFS for patients stratified by pretreatment (day 0; C) or dynamic (week 4; D) ctDNA levels using optimized molecular thresholds in the discovery cohort. High ctDNA defined as ≥2.5 log10hGE/mL. MMR defined as ≥2.5 log decrease in ctDNA level relative to day 0. hGE, haploid genome equivalent; SABER, sequence affinity capture and analysis by enumeration of cell-free receptors; MMR, major molecular response, GE, genome equivalent. *p < 0.05. See also Figure S1.
Baseline and dynamic ctDNA levels are prognostic for outcome
Using STEP, we quantified ctDNA, cfCAR19, and cfTCR levels prior to CAR19 infusion and at multiple time points following cell therapy in the discovery cohort (Figures 2B and S1C). Higher pre- and post-treatment ctDNA levels have previously been associated with outcomes in patients receiving CAR19 therapy.24 Similarly, we observed that patients ultimately experiencing disease progression had significantly higher pretreatment ctDNA levels (Figure 2B). This adverse prognostic effect of higher ctDNA tumor burden was observed both prior to lymphodepleting chemotherapy (median, 200.1 versus 24.1 haploid genome equivalent [hGE]/mL for progressors versus ongoing responders, p = 0.005) and on the day of CAR19 T cell infusion (median, 540.4 versus 11.8 hGE/mL, p = 0.004). While median ctDNA levels decreased within 1 week after infusion in both progressors and ongoing responders, progressors exhibited higher ctDNA levels throughout the course of therapy. Specifically, higher ctDNA levels were associated with progression 1 week (median. 30.4 versus 0.12 hGE/mL, p = 0.003) and at 4 weeks after CAR19 T cell infusion (median. 7.2 hGE/mL versus not detected, p < 0.001).
Using the discovery cohort, we determined the optimal level of ctDNA to stratify patients for EFS after axi-cel therapy, at both the pretreatment and week +4 time points (Figures S1D and S1E). Interestingly, this exercise resulted in the same pre- and on-treatment ctDNA thresholds previously validated to stratify outcomes in patients with treatment-naive diffuse LBCL (DLBCL) receiving immunochemotherapy.22 On the day of CAR T cell infusion, we observed that high ctDNA levels were strongly predictive of shorter EFS (p < 0.002; Figure 2C). Similarly, at the 4-week landmark, patients that achieved a ctDNA major molecular response (MMR; 2.5log10 ctDNA fold decrease) demonstrated significantly superior outcomes (p < 0.001; Figure 2D).
Having established these molecular thresholds at baseline and early during therapy, we next validated their significance in our independent validation cohort. Importantly, both the pretreatment ctDNA threshold at day 0 (p = 0.003, Figure S1F) and MMR threshold at week 4 (p = 0.028, Figure S1G) stratified EFS.
Profiling of cfCAR19 captures expansion dynamics and bio-distribution
Quantification of cfCAR19 was performed by mapping sequenced reads to an augmented human genome containing the CAR19 retroviral genome construct for axi-cel (STAR Methods; Figure S2A). Although the cell-free concentration per milliliter of blood of cfCAR19 as measured by CAPP-seq was ~1,000 times lower than the cellular PBMC levels as measured by CAR19 fluorescence-activated cell sorting (FACS), these two CAR19 T cell measurements were significantly correlated (Spearman r = 0.69, p < 0.001, Figure 3A). This correlation persisted at early (week 1: Spearman r = 0.60, p < 0.001) and later time points (week 4: r = 0.63, p < 0.001). This correlation was also observed in the validation cohort (Spearman r = 0.67, p < 0.001; Figure S2B), suggesting that CAR19 expansion and functional persistence can be quantified from cfDNA. Interestingly, the levels of cfCAR19 remained similar between patients who had ongoing response or treatment failure, without any significant difference between these two groups (Figure 2B, middle panel, and Figure S2C).
Figure 3. Profiling of engineered and other effector T cells from cfDNA.

(A) Correlation between CAR19 T cell levels as quantified by flow cytometry and STEP (cfCAR19) determined using Spearman’s rank-order method in samples from patients in the discovery cohort. Color depicts outcome and shading depicts time point of blood draw. Dashed line depicts linear regression line.
(B) (Top) Aggregate fragment length profiles of wild-type cfDNA (black), ctDNA (purple), and cfCAR19 (blue) molecules across all evaluable plasma sam-ples.
(Bottom) Enrichment of ctDNA (purple) and cfCAR19 (blue) relative to wild-type cfDNA across the fragment length spectrum.
(C) Illustration demonstrating model in which increased fragmentation of tissue-derived DNA oc-curs in transit to the blood.
(D) Histograms of mean cfCAR19 DNA fragment lengths for samples with low (dark blue) and high (light blue) CAR19 FACS over n = 1,000 random samplings of cfCAR19 DNA fragments.
(E) Cumulative frequency of cfCAR19 fragments shorter than a given length in three temporal bins following CAR19 infusion: week 1, week 4, and > week 4. Bins compared using the Kolmogorov-Smirnov (K-S) test, and p values are indicated in the table.
(F) Correlation between cfTCR levels measured by SABER and cfCAR19 levels 1 week after CAR19 infusion (dark green), and 4 weeks following CAR19 infusion (light green) using samples from patients in the discovery cohort. Correlation coefficients were compared using Fisher’s z-transformation. Prog., progressor; O. Resp., ongoing responder; ND, not detected; FACS, fluorescence-activated cell sorting. *p < 0.05. See also Figure S2.
Consistent with prior studies,20,25 we observed a shorter fragment length profile in mutant ctDNA molecules relative to wild-type cfDNA counterparts (Figure 3B). Retroviral cfCAR19 DNA fragments were also shorter than wild-type non-tumor-derived human cfDNA fragments, with these T cell-derived fragments having a similar size profile to lymphoma-derived mutant ctDNA fragments (Figure 3B). This suggests that a portion of shorter cfCAR19 molecules derive from tumor-infiltrating CAR T cells exposed to tissue nucleases,26 while larger fragments potentially better reflect circulating CAR T cells (Figure 3C). Indeed, the proportion of “long” (>310 bp) cfCAR19 molecules was significantly correlated with circulating CAR19 T cell levels as measured by FACS (Spearman r = 0.36, p < 0.001), while no such correlation was observed between “short” (<150 bp) cfCAR19 molecules (Spearman r = −0.17, p = non-significant; Figure S2D). Moreover, we observed blood samples with high circulating CAR19 T cell levels by FACS (above the median of all post-treatment CARFACS measurements) to have significantly longer cfCAR19 fragments (CAR19 high, mean 209 bp versus CAR 19 low, mean197 bp, p = 0.003; Figure 3D).
This results in a model where shorter cfCAR19 DNA fragments derive from tissue-infiltrating CAR T cells (Figure 3C). Correspondingly, an increased contribution of cfCAR19 from tissue-infiltrating CAR T cells would be expected to left-shift the cfCAR19 fragment length distribution toward smaller fragments. Indeed, we observed a significant shortening of cfCAR19 fragments over time (week 1 versus week 4, p < 0.001; Figure 3E), with cfCAR19 size profiles reaching their nadir and remaining stable after 4 weeks. This likely reflects the early expansion of circulating CAR19 cells after CAR19 infusion, followed by their subsequent infiltration into tumor tissues at later time points.
Non-invasive profiling of T cell expansion and TCR diversification in CAR19 patients
In addition to measuring the CAR T cell transgene, we evaluated T cell receptor (TCR) rearrangements in cfDNA (cfTCR) as a measure of adaptive immune responses during axi-cel therapy. To identify and measure unique cfTCR rearrangements using their distinct complementarity determining regions (CDR3), we developed and applied a method for sequence affinity capture and analysis by enumeration of cell-free receptors (SABER; see STAR Methods).27 cfCAR19 levels were significantly correlated with the number of total rearranged cfTCRs 1 week after CAR19 infusion (r = 0.69, p < 0.001; Figure 3F). While these measurements remained correlated at 4 weeks after infusion, the correlation was weaker (r = 0.33, p = 0.011), suggesting homing of CAR19 T cells to tissue and reconstitution or expansion of other T cells in the periphery (Figure 3F). Similar results were seen in the validation cohort (Figure S2E). Higher levels of cfTCRs at 4 weeks after CAR19 infusion were associated with durable responders compared with patients with disease progression (64.0 versus 31.4, p = 0.004; Figure 2B, bottom panel); however, this result did not replicate in the validation cohort.
Genomic characteristics of relapsed/refractory versus treatment-naive LBCL
In order to compare the genomic characteristics of treatment-naive versus relapsed/refractory LBCL, we profiled tumor and/or pretreatment plasma samples from 115 patients with treatment-naive LBCL (Table S4) and 138 r/rLBCL patients from our discovery and validation cohorts. We identified a similar number of mutations between the treatment-naive and relapsed/refractory cohorts (mean 174 versus 129 mutations per case, p = 0.36; Figure S3A).
We next compared the somatic mutational landscape and subtype distribution of r/r cases prior to CAR19 therapy versus treatment-naive patients, focusing on non-silent mutations in DLBCL cases without a history of antecedent low-grade lymphomas and excluding other LBCL histologies. The mutational profile of DLBCL cases in the relapsed/refractory cohort was largely similar to treatment-naive DLBCL. Nevertheless, we observed more frequent TP53 (p = 0.016), MYC (p = 0.020), and EP300 (p = 0.032) alterations in the relapsed/refractory cohort, and, conversely, fewer non-silent alterations in CD79B (p = 0.040), BCL6 (p = 0.008), and TOX (p = 0.032; Figure S3B). Cell of origin (COO) distributions were also GCB-skewed in the relapsed/refractory cohort (p = 0.03; Figure S3C). Additionally, we observed significant differences in LymphGen subtype distribution (p = 0.02; Figure S3D), with the EZB and A53 subtypes enriched in the relapsed/refractory cohort, and MCD tumors enriched in treatment-naive DLBCL.
Baseline and emergent genomic alterations associated with CAR19 resistance
In addition to quantitating bulk ctDNA levels, STEP provides an opportunity to genotype and assess the biological significance of alterations in specific genes in the context of T cell therapy. To maximize our power for discovery of tumor genotypes associated with outcomes,28 we pooled our discovery and validation cohorts (total n = 138). We then genotyped diverse alterations, including SNVs, indels, fusions, and genome-wide somatic SCNAs using tumor and plasma samples from both pretreatment and relapse time points. Of interest, we did not observe significant differences in outcomes after CAR19 therapy when considering COO or LymphGen DLBCL subtypes (Figures S3E and S3F).
We elucidated both baseline genomic alterations and those that emerged under positive selective evolutionary pressure from CAR19 T cells, and assessed the association of mutations in individual genes with outcomes. We identified mutations in several genes as being significantly associated with inferior EFS (Figures 4A and S4A–S4C). These resistance-associated lesions to CAR19 included mutations in TMEM30A (hazard ratio [HR] = 3.4, 95% confidence interval [CI] 1.5–7.5), IRF8 (HR = 2.9, 95% CI 1.6–5.5), PAX5 (HR = 1.8, 95% CI 1.0–3.0), TP53 (HR = 1.7, 95% CI 1.0–2.7), P2RY8 (HR = 3.3, 95% CI 1.6–7.0), and DTX1 (HR = 1.8, 95% CI 1.1–3.0). When adjusting for multiple hypothesis testing using the Benjamini-Hochberg method, three genes had a q value below 0.1, namely IRF8 (q value = 0.042), P2RY8 (p = 0.049), and TMEM30A (q = 0.057). These genes represent diverse potential mechanisms of resistance, and include genes that are associated with B cell identity (PAX5/IRF8), immune microenvironment modulation (TMEM30A), and adverse treatment outcome in related diseases (TP53).29–33
Figure 4. Genomic determinants of resistance to CAR19 therapy.

(A) (Left) Recurrently mutated genes in patients receiving CAR19 therapy, stratified by ongoing response versus progression among pooled patients from the discovery and validation cohorts. (Right) Effect of mutations in given gene on EFS (HR from proportional hazard model); significant values (p < 0.05) shown in red.
The proportion of emergent mutations is depicted in light red. Genes that remained statistically significant after adjusting for multiple hypothesis testing (Benjamini-Hochberg method, q < 0.1) are annotated. Genes mutated in greater than 5% of patients pre-CAR19 are displayed.
(B) Clonal selection of mutations in specific genes in patients experiencing disease progression shown as a volcano plot. Mutated genes under significant selection are shown on the right in purple; size of dot proportional to number of mutations (also shown in parentheses).
(C and D) Clonal selection of somatic copy-number alterations (SCNAs)—amplifications (C) and deletions (D)—at the level individual genes, in patients experiencing progression shown as a volcano plot. Genes listed in groups that are included in single 500-kb regions. SCNAs under significant selection are shown in purple; size of dot proportional to number of SCNAs (also shown in parentheses). HRSNV, single-nucleotide variant; indel, small insertions/deletions. *p < 0.05.
See also Figure S3.
In order to elucidate genomic alterations under positive selective evolutionary pressure from CAR19 T cells, we assessed for both de novo emergent alterations that first appeared at the time of relapse as well as alterations that were clonally selected for after CAR19 therapy. To assess significance, we developed a statistical framework that considered the clonal hierarchy of mutations for each case over time to quantify therapy-associated changes in relative allele fractions (STAR Methods).
We identified recurrent emergence of mutations in multiple genes accompanying treatment failure, where mutations first appeared in individual patients at disease relapse or arose under evidence of strong selective pressure on clones already present at the pretreatment time point (Figures 4A and 4B). Notably, multiple alterations in CD19 emerged at the time of relapse, which appears to reflect the unique selective pressure placed on the CD19 gene in patients undergoing CD19-directed therapy. Additionally, several emergent mutations in PPM1D, a driver of clonal hematopoiesis of indeterminate potential (CHIP), were noted in relapsing patients.34,35 Further, we observed evidence for positive selection by CAR19 therapy on mutations in TP53, recently implicated in post-CAR19 relapse,33 as well as in PAX5, where mutations were significantly associated with acquired resistance (Figure S4A).
Similarly, we assessed for clonal selection of SCNAs (see STAR Methods), doing so at the level of both genes (Figures 4C and 4D) and cytobands (Figures S4D and S4E). We identified genomic regions housing several key genes relevant to immune evasion and B cell identity to be under selective pressure within these SCNAs. Notably, 9p24.1 amplifications harboring genes encoding immune checkpoint molecules CD274 (PD-L1) and PDCD1LG2 (PD-L2) were under positive selection at relapse. Interestingly genomic regions harboring PAX5 were also among the emergent amplified genomic regions at relapse. While this seems in contrast with our findings regarding the potential role of PAX5 mutations, we believe PAX5 may simply be a “bystander gene” in larger, arm-level, amplifications of chromosome 9p, which also includes CD274. While the identification of the minimally amplified region is desirable, this is difficult to accomplish with the limited allele frequency found in plasma samples. Conversely, we noted that amplifications of the genomic region housing TCF-4 (18q21.2) were selected against at relapse.
Tumor-intrinsic factors influence outcomes and CAR19 interactions via diverse mechanisms
To investigate the mechanisms through which tumor genotypes can influence outcomes, we integrated our genotypic information with cfCAR19 levels assessed over time, and tumor RNA sequencing (RNA-seq) and immune deconvolution from both pre-CAR19 and post-CAR19 samples (Figure 5A). Several emergent mechanisms of immune evasion were evident at the time of relapse in individual patients, including target antigen loss and checkpoint gene copy number alterations. Emergent CD19 mutations were noted in multiple progressing patients (Figure 4B), including a patient that initially achieved undetectable ctDNA levels post-CAR19 but developed an emergent CD19 nonsense mutation at the time of relapse. The allele frequency of the CD19 mutation mirrored the ctDNA molecular relapse, and was associated with re-expansion of CAR19 cells (Figure 5C). When considering patients with available quantification of relapse tumor CD19 protein expression levels, we observed a significant inverse correlation between CD19 H score and cfCAR19 levels (R = −0.66, p = 0.028; Figure 5D). Indeed, CAR19 persistence has previously been associated with CD19 protein loss at relapse in B cell ALL,36 highlighting a potential conserved mechanism of immune of evasion of CAR T cells targeting CD19, wherein persistent CAR19 T cells lead to “immune editing” of the tumor and CD19 loss through either genetic or epigenetic mechanisms.
Figure 5. Tumor-intrinsic factors influence outcomes and CAR19 interactions via diverse mechanisms.

(A) Tumor and CAR19 T cells influence tumor biology, CAR19 expansion and persistence, and the tumor microenvironment via reciprocal interactions. Boxes highlight interactions discussed in this article with references to relevant figures.
(B) Comparison of regulatory T cell levels in pre-CAR19 r/rLBCL tumors (n = 68) as measured using CIBERSORTx in IRF8 altered (light green) or wild-type (dark green) tumors.
(C) Dynamic changes in ctDNA (top) and cfCAR19 (bottom) and an emergent CD19 nonsense mutation in an exemplar patient that relapses following CAR19 therapy. cfCAR19 re-expands along with the re-expansion of ctDNA levels and appearance of CD19 mutation.
(D) Correlation between CD19 membrane expression levels quantified using immunohistochemistry (H score) at relapse (x axis) and relapse cfCAR19 levels (y axis).
(E) Genome-wide heatmap reflecting the copy number profile of three exemplar patients prior to therapy (top), and at the time of relapse (bottom). Each column is representative of a cytoband. Amplifications in cytoband 9p24.1 at the time of relapse in all three patients are highlighted. TME, tumor microenvironment; AF, allele frequency. *p < 0.05.
See also Figure S4.
Amplifications in the genomic region containing the checkpoint gene PD-L1 were under positive clonal selection in patients at the time of relapse (Figures 4C and S4E). This is exemplified in multiple cases with emergence of focal gains in 9p24,1, the cytoband containing PD-L1 (Figure 5E), again reflective of the selective pressure applied by CAR19 T cells and the resultant evolution of the tumor immunophenotype. Here, however, immune escape is mediated not by loss of the target antigen, but by gain of negative regulatory checkpoint signals likely leading to loss of cytotoxic effects of CAR19 T cells.
In order to assess the association between genomic alterations and the composition of the tumor microenvironment, CIBERSORTx was used to deconvolute the intratumoral immune cell composition using bulk RNA-seq data. In particular, we observed that tumors carrying IRF8 mutations—one of the three genes that remained prognostic for outcomes after MHT correction—were enriched for intratumoral regulatory T cells as well as resting memory CD4 T cells (Figures 5B and S5A).
Reciprocal interactions between tumor and CAR T cells influence CAR19 expansion and tumor microenvironment
To explore how the relationship between initial CAR19 expansion and tumor genotype influences outcome, we first integrated cfCAR19 and flow cytometry measurements to calculate a global early CAR19 expansion index (see STAR Methods) for each patient (Figure 6A). The mutational profile was largely similar between patients with high and low expansion; however, TNFRSF14 (p = 0.009), BCL2 (p = 0.02), and BTG (p = 0.05) mutations were more common in patients with low expansion, while IRF4 mutations (p = 0.05) were more common in patients with high expansion (Figures 6B and 6C).
Figure 6. Reciprocal interactions between tumor and CAR T cells influence CAR19 expansion and tumor microenvironment.

(A) Overview of schema used to calculate the early CAR19 expansion index for each patient (see STAR Methods for detail). Patients greater than or equal to the median expansion rank were considered to have a high early CAR19 expansion index, and those below the median were considered to have a low early expansion index. Gray boxes indicate sample time points with no available data.
(B) Enrichment of genomic alterations in individual genes in patients with high and low initial CAR expansion as determined using early CAR19 expansion index,depicted as a volcano plot. Genes significantly associated with expansion are colored blue (enriched in low expansion) or yellow (enriched in high expansion).
(C) Comparison of early CAR19 expansion as quantified using the early CAR19 expansion index in TNFRSF14-mutated (light blue) versus wild-type (dark blue) patients.
(D) Comparison of select immune cell subsets in pre-CAR19 r/rLBCL tumors (n = 68) as inferred using CIBERSORTx in TNFRSF14-mutated (light green) and wild-type (dark green) tumors.
(E) Intratumoral CAR19 T cell levels in post-CAR19 relapse tumors were quantified using CAPP-seq. Gene expression and tumor immune microenvironment composition were compared between tumors with high versus low relapse CAR19 levels.
(F) Correlation between intratumoral CAR19 levels (CAR19 depth/total sequencing depth, x axis) versus matched plasma cfCAR19 levels (CAR19 depth/total sequencing depth, y axis) in six cases with both available matched plasma and tumor samples at the time of relapse.
(G) Comparison of regulatory T cell levels as measured using CIBERSORTx in post-CAR19 relapse tumors (n = 14) with high versus low intratumoral axi-cel levels(stratified to cohort-wise median).
(H–J) Gene set enrichment analysis (GSEA) enrichment plots demonstrating enrichment of (H) inflammatory, (I) interferon gamma, and (J) TGF-β gene sets in post-CAR19 relapsed tumors (n = 14) with high versus low axi-cel content (stratified to cohort-wise median). GSEA*p < 0.05. See also Figures S5 and S6.
To understand how TNFRSF14 mutations may lead to decreased CAR19 expansion, we used CIBERSORTx to deconvolute the tumor immune microenvironment of pre-CAR19 r/rLBCL tumors. Interestingly, we found that TNFRSF14-mutated tumors had increased levels of resting memory CD4 T cells and T follicular helper cells (Figures 6D and S5B), consistent with prior reports that loss of TNFRSF14 can promote lymphomagenesis through facilitation of B cell proliferation, induction of a tumor-supportive microenvironment, and increased recruitment of tumor-supportive T follicular helper cells.37 Additionally, while TNFRSF14 mutations were not associated with outcomes in univariate analyses (Figure 4A), these mutations were associated with inferior outcomes (HR = 2.0, 95% CI 1.5–7.5, p = 0.038) when adjusting for pretreatment ctDNA levels. These data suggest that tumor genotypes can influence CAR19 expansion and outcomes via effects on the tumor microenvironment, although further studies are needed to establish a clear cause-and-effect relationship.
In addition to investigating the relationship between tumor genotypes and CAR19 expansion, we assessed the influence of persistent intratumoral CAR19 cells on tumor biology. We quantified CAR19 levels in tumors at the time of relapse post-CAR19 using CAPP-seq (Figure 6E). Intratumoral CAR19 levels were correlated with matched plasma cfCAR19 levels (r = 0.89, p = 0.033; Figure 6F), indicating not only that cfCAR19 levels are reflective of CAR19 tumor infiltration but that cfCAR19 can potentially be used as a surrogate measurement for tumor CAR19 levels at relapse. Tumors with high CAR19 content at relapse exhibited gene signatures associated with increased inflammation, including inflammatory response (Hallmark, adjusted p = 0.003; Figure 6H) and interferon gamma response (Hallmark, adjusted p = 0.003; Figure 6I) in bulk RNA-seq. Additionally, these tumors demonstrated an enrichment of gene signatures associated with T cell exhaustion (GSE9650 exhausted versus memory CD8 T cell up, adjusted p = 0.002; Figure S6B), which is particularly notable in the context of an ineffective CAR T cell anti-tumor response. Further, transforming growth factor β (TGF- β) signaling (Hallmark, p = 0.003; Figure 6J) and intratumoral regulatory T cells (Tregs; Figures 6G and S6A) were increased in CAR19-high tumors. Overall, these data reflect that tumors with abundant infiltrating CAR T cells demonstrate distinct microenvironmental and inflammatory signatures relative to those with low or absent CAR T cells. This suggests that the mechanisms of resistance to engineered T cell therapy in these two scenarios are likely distinct.
A multivariable model incorporating distinct molecular features predicts outcomes
While we identified several key genomic alterations that influence outcomes after CAR19 therapy, there was no unifying mutational signature in the majority of cases. We thus sought to further quantify the prognostic impact of both tumor-intrinsic and -extrinsic molecular features in order to develop a model predictive of outcomes. We first applied univariate Cox proportional hazards regression models to assess the impact of key features on EFS and OS in the discovery cohort. When considering tumor-intrinsic metrics as continuous variables, pre- and on-treatment ctDNA levels were prognostic for EFS and OS at all time points. Specifically, higher ctDNA levels were associated with inferior EFS outcomes at baseline (EFS, day 0 [HR = 2.2, p = 0.003], at week 1 [HR = 2.0, p = 0.004], and at week 4 [HR = 2.3, p < 0.001]; Figures 7A and S7A). Importantly, higher ctDNA levels also predicted inferior OS outcomes at these same milestones (OS, day 0 [HR = 2.1, p = 0.01], week 1 [HR = 1.7, p = 0.04], week 4 [HR = 2.5, p < 0.001]; Figures 7B and S7B). Among clinical risk factors, high pre-lymphodepletion LDH levels and high international prognostic index (IPI) scores were both associated with inferior outcomes (LDH, EFS HR = 1.3, p = 0.009; OS HR = 1.3, p = 0.02; Figures 7A and 7B; IPI, OS HR = 1.5, p = 0.05; Figure 7B). HRs and p values for additional clinical variables are described in Figures S7A and S7B.
Figure 7. A multivariable model incorporating integrating tumor B cell and effector T cell molecular features predicts outcomes following CAR19 therapy.

(A and B) Results of univariate Cox proportional hazards for EFS (A) and OS (B) and indicated variables in the discovery cohort. Using multivariable stepwise forward selection with Bayesian information criterion (BIC), two variables were selected (week 4 ctDNA and week 1 cfCAR19, emphasized in bold) as the optimal covariates to predict EFS in the discovery cohort. A multivariable Cox model (STEP score) was trained using these variables on EFS in the discovery cohort and applied to the independent validation cohort.
(C and D) Kaplan-Meier estimates show EFS (C) and OS (D) for patients in the validation cohort stratified by high versus low STEP score. EFS, event-free survival; OS, overall survival; ND, not detected; hGE, haploid genome equivalent; Pre-LD, pre-lymphodepletion; IPI, internal prognostic index; TMTV, total metabolic tumor volume.*p < 0.05. See also Figure S7.
We next considered the prognostic significance of immunological features by evaluating the predictive value of CAR19 T cell expansion levels, and higher cfCAR19 levels at week 1 were significantly associated with improved EFS (HR = 0.55, p = 0.011; Figure 7A).
In order to build a multivariable model to predict outcomes, we used the Bayesian information criterion to perform forward stepwise selection on molecular and clinical features, which identified two covariates as best predicting EFS. The selected features combine both tumor B cell and CAR T cell factors (Figure 7A): week 4 ctDNA level (HR = 2.2, p < 0.001) and week 1 cfCAR19 level (HR = 0.6, p = 0.017). They are measured using the STEP analysis platform. A Cox proportional hazards model incorporating these two variables (STEP score) was trained for EFS in the discovery cohort (HR = 2.7, p < 0.001; Figure 7A) and was validated to be prognostic for both EFS and OS in the validation cohort (EFS, HR = 2.7, p < 0.001; OS, HR = 5.4, p < 0.001; Figures 7A–7D). Additionally, the STEP score was able to stratify outcomes in patients with detectable ctDNA at week 4 (Figure S8A), which has been demonstrated to be prognostic in CAR19-treated patients.24 We conclude that distinct tumor-intrinsic and -extrinsic molecular features are complementary for predicting clinical outcomes after CAR19 cell therapy and can be measured noninvasively using cfDNA.
DISCUSSION
Cancers are complex biological ecosystems, requiring integration of multiple factors to resolve biological and clinical heterogeneity38 Molecular, clinical, and genomic features before and during treatment can be integrated to better predict outcomes.23,39 The addition of engineered immunotherapy with CAR19 T cells, a living treatment that must expand and traffic to engage tumor cells, adds further complexity to the tumor-versus-therapy ecosystem.
Features of both tumor and CAR T cells have been demonstrated to effect outcomes after therapy.9,11–13,15,17,40 Additionally, anatomical heterogeneity is known to play a role in resistance to diverse cancer therapies.21,41,42 In this setting, several questions remain unanswered, including identification of the key determinants of treatment resistance, and whether tumor-intrinsic or tumor-extrinsic factors—or both—best explain therapy response. To study the tumor-versus-therapy ecosystem, we developed a strategy to profile both tumor and immunologic compartments noninvasively STEP, capturing tumor, CAR19, and effector T cell DNA from the same plasma or tumor DNA samples using CAPP-seq.
The prognostic role of ctDNA quantitation is well established, where both pretreatment and on-treatment levels are prognostic for outcome in first-line DLBCL and during CAR19 therapy.22,24,43 Consistent with this, we found that both pretreatment and dynamic ctDNA levels are predictive of outcomes following CAR19 therapy, with assessment 4 weeks after treatment best predicting outcomes in multivariate models.
When considering tumor genotypes, we found enrichment in GCB as well as EZB and A53 DLBCL subtypes at the time of CAR19 therapy, while the non-GCB and the MCD subtypes were enriched in untreated patients. These results seemed counter-intuitive given the established poor prognosis of non-GCB and MCD-DLBCL.44 However, this finding is consistent with previous reports of lower rates of the non-GCB subtype in clinical trials evaluating axi-cel,1 potentially suggesting an underlying selection bias where patients with aggressive disease are excluded from clinical studies, which is well established in frontline DLBCL.45,46
We did not observe a difference in outcome when patients receiving CAR19 were stratified according to LymphGen subtype or COO, but we identified specific genotypes associated with adverse outcomes after CAR19 therapy. These included mutations in genes defining B cell identity (IRF8, PAX5), those involved in immunoregulation (CD274), and shaping the immune microenvironment (TMEM30A), and in TP53, which is established to be associated with poor prognosis after CAR19 therapy.33
PAX5 and IRF8 both code for transcription factors that are integral in governing B cell differentiation and surface phenotype.30,31,47 Hemopoietic lineage switch is an established mechanism of resistance to CAR19 therapy in ALL, suggesting a mechanism through which these alterations may lead to resistance in LBCL.14 Additionally, PAX5 is known to directly regulate CD19 gene and protein expression48; however, direct assessment of the effect of PAX5 alterations on CD19 protein expression requires tumor tissue, which was not available in many of our cases. While we observed a tumor microenvironmental signal associated with IRF8 mutations, a definitive explanation of these associations will require future studies of the direct and indirect effects of disruption of PAX5 and IRF8 on lymphoma surface phenotype and T cell function, using validated in vitro and in vivo models for such heterotypic interactions.
Alterations in genes coding for T cell inhibitory molecules, such as CD274, which encodes programmed cell death ligand 1 (PD-L1), were also associated with adverse outcomes after CAR19. We found recurrent amplifications in 9p24.1, which houses CD274, in progressing patients. CD274/PD-L1 amplifications are known to lead to immune evasion in diverse cancers, including B-NHLs, and are targetable with monoclonal antibodies.49–51 Alterations in TMEM30A, which were associated with poor prognosis in our study, have recently been associated with increased tumor associated macrophages (TAMs) in LBCL tumors and increased susceptibility to macrophage checkpoint targeting antibodies.29 Both thus highlight potential avenues through which understanding genomic determinants of resistance can guide future precision medicine trials.
CD19 mutations and decreased CD19 membrane expression emerged at the time of relapse in multiple patients in our cohort, as has been previously demonstrated.10,12,13 However, we demonstrate that somatic mutation-mediated CD19 loss is a relatively uncommon mechanism of resistance in lymphoma, and can accompany CAR19 cell re-expansion. This latter observation confirms that CAR19 cells can persist for prolonged periods and be functional,52 wherein they may select for CD19-altered tumor cell clones through immune editing. Indeed, post CAR19 relapse tumors with high levels of CAR19 cells demonstrated different tumor microenvironment and gene expression profiles than tumors with low CAR19 levels. In particular, tumors with high CAR19 levels demonstrated increased infiltration of regulatory T cells, and it is intriguing to speculate whether the CAR T cells themselves adopt a regulatory T cell phenotype in these cases, thus falling short of an effective anti-tumor response, as has recently been described53,54
Through profiling of emergent resistance mechanisms and the persistence of CAR19 cells at relapse, one can gain clinically relevant information with which to guide subsequent treatment decisions. For example, patients with an emergent CD19 mutation or CD274 amplification, or those with persistent CAR19 cells, would likely not benefit from further T cell-mediated treatments, but would rather need alternative therapies.
Prior CAR T cell tracking methods utilize flow cytometry and qPCR, with a focus on circulating leukocytes.55,56 cfCAR19 levels and flow cytometric quantification of cells were highly correlated, validating quantification using CAR19-specific cfDNA. Further, the cfCAR19 fragment length pattern suggests that focusing on plasma cfDNA may allow for detection of both circulating and tissue-infiltrating CAR19 cells. Our method also allowed assessment of cfCAR19 fragmentation patterns, where we observed a significant shift toward shorter fragments over time, resulting in identical size profiles between CAR19-derived cfDNA and tumor-derived ctDNA. The shift toward shorter fragments over time potentially reflects trafficking of CAR19 cells into tissues as peripheral expansion wanes.
A multivariable model incorporating both tumor-intrinsic and -extrinsic molecular features was validated to be prognostic for outcomes, highlighting the importance of concurrent analysis of both of these compartments when considering CAR19 resistance. Notably, multivariable regression identified cfCAR19 levels and not flow-based CAR19 quantitation, suggesting cfCAR19 as a better analyte for assessment of total body bio-distribution of CAR T cells compared with circulating PBMCs.
These findings suggest several strategies for precision approaches to CAR19 therapy, including (1) decreasing tumor burden using systemic or radiation therapy prior to CAR19, (2) optimizing CAR19 expansion per patient by increasing the dose of infused cells or even the number of infusions, (3) adapting therapy in patients who do not achieve an adequate ctDNA response or ctDNA clearance by 4 weeks after CAR19 infusion, and (4) tailoring next-line therapies based on genomic alterations or CAR19 persistence.
Engineered T cell therapy is rapidly expanding its role in the treatment of r/rLBCL,57,58 as well as in other lymphoma and tumor types.2–4 We show resistance is dynamic and multifactorial, and consideration must be given to both the target tumor cell and effector T cell biology. Future treatment approaches leveraging an integrated approach to understand mechanisms underlying resistance in individual patients should be brought forward in the clinic and may lead to improved outcomes for patients.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ash Alizadeh (arasha@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
DNA and RNA sequencing data from rrLBCL patients have been deposited at dbGaP and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-CD19 | Leica Biosystems | clone BT51E |
| Anti-CD19 specific scFv | Jena et al.59 | clone 136.20.1 |
|
| ||
| Deposited data | ||
|
| ||
| Targeted DNA Sequencing data | dbGaP | Accession Number Pending |
| RNA sequencing data | dbGaP | Accession Number Pending |
| LM22 signature matrix (CIBERSORTx) | Newman et al., 2015 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| xGen Lockdown Probes | IDT | https://www.idtdna.com/ |
| SeqCap EZ Choice Probes | Roche | https://sequencing.roche.com/ |
|
| ||
| Software and algorithms | ||
|
| ||
| CIBERSORTx | Newman et al., 2015 | https://cibersortx.stanford.edu/ |
| FACTERA | Newman et al.18 | https://factera.stanford.edu/ |
| LymphGen Classifier | Wright et al.60 | https://llmpp.nih.gov/lymphgen/index.php |
| METAVOL | Hirata et al.61 | www.metavol.org |
| MATLAB version 2020b | Mathworks | https://www.mathworks.com/products/matlab.html |
| R Statistical Software version 4.0.3 | The R Foundation | https://www.r-project.org |
| survival package | CRAN Repository | https://cran.r-project.org/web/packages/survival/index.html |
| softimpute package | CRAN Repository | https://cran.r-project.org/web/packages/softImpute/index.html |
| maxstat package | CRAN Repository | https://cran.r-project.org/web/packages/maxstat/index.html |
| Fast Gene Set Enrichment Analysis (FGSEA) package | Korotkevich et al.62 | https://github.com/ctlab/fgsea/ |
| afterqc package | Chen et al.63 | https://github.com/OpenGene/AfterQC |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
All samples analyzed in this study were collected with informed consent from participants enrolled on Institutional Review Board-approved protocols complying with ethical regulations at their respective centers: Stanford University, MD Anderson Cancer Center, and Essen University Hospital. A total of 253 patients with a subtype of large B-cell lymphoma (LBCL) were included in this study, including diffuse large B-cell lymphoma (DLBCL), T-cell/histiocytic rich large B-cell lymphoma (THRLBCL), High Grade B-cell Lymphoma (HGBCL) and primary mediastinal B-cell lymphoma (PMBCL). Additionally, patients with an antecedent low-grade lymphoma with histologic transformation were included. Of these patients, 115 patients had newly diagnosed and treatment naïve disease (patient demographics are described in Table S4), while 138 had relapsed/refractory disease after at least 2 prior lines of therapy (patient demographics are described in Table 1). Newly diagnosed patients were enrolled prior to receiving chemoimmunotherapy from within either the phase III multicenter PETAL trial64 (n = 80; , or at MD Anderson (n = 35). Relapsed refractory patients were enrolled as part of two independent cohorts (discovery n = 65, validation = 73). Only pre-treatment patient data and samples were considered for newly diagnosed patients. Relapsed/refractory patients were consented for collection of clinical data as well as blood and lymph node sampling on a clinical outcomes biorepository protocol prior to standard of care axi-cel therapy at the Stanford Cancer Center, CA USA. CAR19 patients were followed longitudinally following therapy and samples were collected prior to, during, and after therapy. An EFS event was defined as disease relapse, death, or unplanned re-treatment of lymphoma. A “progressor” was defined as a subject that experienced an EFS event within 36 months of CAR T-cell infusion. A patient with an ongoing response, or an “ongoing responder” was defined as a subject that did not experience an EFS event at the time of data analysis and had at least 6 months of follow-up after CAR19 infusion. Median follow-up for the r/rLBCL cohorts was calculated based on EFS using the reverse Kaplan-Meier method. The study was approved by the local institutional review board of each institution, and all patients provided written informed consent.
Table 1.
−r/rLBCL patient demographic and clinical characteristics
| Characteristic | CAR19 Discovery cohort, N = 65 | CAR19 Validation cohort, N = 73 |
|---|---|---|
|
| ||
| Outcome | ||
|
| ||
| Ongoing Response | 29 (45%) | 31 (42%) |
| Progression | 36 (55%) | 42 (58%) |
| Follow-up, months; median (95% CI) | 36.9 (30.4–40.8) | 23.2 (17.4 - NA) |
| Age | 58 (49, 68) | 63 (58, 72) |
|
| ||
| Sex | ||
|
| ||
| Female | 22 (34%) | 37 (51%) |
| Male | 43 (66%) | 36 (49%) |
|
| ||
| Histology | ||
|
| ||
| DLBCL | 35 (54%) | 37 (51%) |
| HGBCL | 9 (14%) | 11 (15%) |
| PMBCL | 3 (4.6%) | 3 (4.1%) |
| TFL | 18 (28%) | 21 (29%) |
| THRLBCL | 0 (0%) | 1 (1.4%) |
|
| ||
| Stagea | ||
|
| ||
| 1 | 6 (9.4%) | 15 (21%) |
| 2 | 7 (11%) | 13 (18%) |
| 3 | 6 (9.4%) | 8 (11%) |
| 4 | 45 (70%) | 36 (50%) |
|
| ||
| IPIa | ||
|
| ||
| 0–1 | 18 (28%) | 17 (24%) |
| 2 | 16 (25%) | 24 (33%) |
| 3 | 21 (32%) | 22 (31%) |
| 4–5 | 10 (15%) | 9 (12%) |
|
| ||
| Prior lines of therapy | ||
|
| ||
| 0–1 | 0 (0%) | 0 (0%) |
| 2 | 27 (42%) | 25 (34%) |
| 3 | 18 (28%) | 22 (30%) |
| 4 | 15 (23%) | 18 (25%) |
| 5 or more | 5 (7.7%) | 8 (11%) |
| Prior Auto SCT | 13 (20%) | 19 (26%) |
Median (IQR); n (%).
Stage and IPI reported when available.
DLBCL, diffuse large B cell lymphoma; HGBCL, high-grade B cell lymphoma; TFL, transformed follicular lymphoma; PMBCL, primary mediastinal B cell lymphoma; THRLBCL, T cell/histiocyte-rich large B cell lymphoma.
Relapsed/refractory stage and IPI established prior to CAR19 therapy.
METHOD DETAILS
Sample collection and processing
Peripheral blood samples from Stanford University were collected in K2EDTA or Cell-Free DNA Collection Tubes (Roche, Pleasanton CA, USA) tubes, stored at 4°C and processed within 24hr (K2EDTA) or 7 days (Cell-Free DNA Collection Tubes) by centrifugation at 1,800 g for 10 minutes. Plasma and plasma depleted whole blood were subsequently isolated and cryopreserved at −80°C as previously described.20 When applicable, peripheral blood mononuclear cells (PBMCs) were isolated using SepMate PBMC Isolation Tubes (Stemcell Technologies, Vancouver, BC) according to the manufacturer’s protocol. Peripheral blood samples from the PETAL and MD Anderson cohorts were collected in K2EDTA tubes and processed according to local protocols for the isolation of plasma, which was cryopreserved at −80°C. Cell-free DNA (cfDNA) was extracted from 2–6 mL of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. Cellular DNA from either plasma depleted whole blood, or peripheral blood mononuclear cells, was isolated using the DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer’s protocol. Following isolation, cellular DNA was fragmented to a target size of 170-bp using Covaris S2 sonicator. Tumor derived DNA was isolated from 2–4, 10 micron thick, formalin-fixed, paraffin embedded (FFPE) scrolls of tumor tissue using the RNA Storm/DNA Storm Combination Kit (Cell Data Sciences, Fremont, CA), according to the manufacturer’s protocol. DNA was quantified using the Qubit dsDNA High Sensitivity Kit (Thermo Fisher Scientific, Waltham, MA) and fragment length was assessed using a Fragment Analyzer System (Agilent, Santa Clara, CA).
Library preparation and sequencing
Barcoded DNA sequencing libraries were prepared as described in detail previously,20 followed by hybrid capture. Samples from patients with previously treatment naive LBCL were captured using a custom 608 kb oligonucleotide panel (SeqCap EZ Choice, Roche and IDT, Coralville, IA), designed to target genomic regions known to be recurrently mutated, or of known biologic significance, in B-cell derived neoplasms (Table S3). In addition to 608 kb panel, samples from CAR19 patients were captured with a supplemental custom 62.5 kb oligonucleotide pool (IDT, Coralville, IA) designed to target the axi-cel retroviral vector sequence, T-cell receptor genes, the full sequence of CD19, and genes known to be implicated in immune evasion (Table S3). When comparing treatment naive and relapsed/refractory LBCL, only the shared 608 kb of sequencing space were considered. Hybrid capture was performed as previously described.20 Libraries were sequenced on an Illumina HiSeq4000 with 2 × 150 bp paired-end reads.
Sequencing data analysis and variant calling
FASTQs files were demultiplexed based on 8-bp dual sample barcodes and reads were mapped to a customized version of the human reference genome (hg19) augmented with the retroviral sequence for axi-cel using BWA ALN. Molecules were deduplicated with error-suppression using a custom pipeline as previously described.19,20 Following this, somatic single nucleotide variants (SNV) and insertion-deletion variants (indel) were called using a custom variant algorithm optimized for cfDNA as previously described.19 Somatic variants were identified using paired analysis of pre-treatment plasma or tumor DNA and germline DNA. A minimum allele frequency of 0.5% for plasma samples or 5% for tumor samples was used when calling somatic variants. Missense, frame-shift, nonsense 5’ or 3’UTR, and splice-site variants were considered “non-silent.”
Copy number and structural alterations
Genome-wide somatic copy number alterations (SCNAs) were called using both on-target (~60–80% of reads) and off-target (~20–40%) sequencing reads as previously described.20 Copy number alterations specific to individual genes and at the level of specific cytobands were considered for further analysis. Translocations were identified using The Fusion And Chromosomal Translocation Enumeration and Recovery Algorithm (FACTERA, factera.stanford.edu).18
Cell of origin classification and LymphGen subtype determination
Cell of origin subtypes were inferred from sequencing data as previously described.21,23 LymphGen classification was performed using the LymphGen online tool (https://llmpp.nih.gov/lymphgen/index.php).60
ctDNA quantification and monitoring
ctDNA levels were measured in haploid genome equivalents per milliliter (hGE/mL), calculated as the product of total cell-free DNA concentration and the mean allele-fraction of somatic mutations. ctDNA levels were monitored in individual samples using a previously described Monte-Carlo-based ctDNA detection index.19
Quantification of cfCAR19 levels
120 base-pair hybrid capture oligonucleotides (IDT, Coralville, IA) were designed to tile across the entire 7.022 kb Axi-cel retroviral vector sequence and used in library captures. The region of the vector sequence evaluable for determining axi-cel depth was defined by first considering only the region between the long terminal repeats (LTR), which is the region expected to integrate into host genome. The evaluable region was then refined by comparing the depth in this region between patients known to have received CAR19 therapy and withheld healthy controls, and removing regions with significant depth in controls. Mean depth across this region was then calculated per sample, and axi-cel level was defined as axi-cel genomes per milliliter plasma, which was determined as follows: (axi-cel depth/human depth) * (1 HGE/3e-12 g) * (1e9 ng/1 g) * plasma cfDNA concentration.
Quantification of intratumoral CAR19 T-cell levels
Mean axi-cel depth was determined as described immediately above and normalized to the mean sequencing depth across targeted human regions in the same sample. Normalized axi-cel depth was quantified in all available relapse tumor samples (n = 18), and tumors with normalized CAR19 depth above the median were classified as CAR19 Relapse High, while those below the median were classified as CAR19 Relapse Low. Tumors from 4 patients were excluded from downstream analyses due to intervening therapies.
Cell-free DNA fragment length determination
To determine cfCAR19 DNA fragment length distributions, sequencing reads were aligned to the Axi-cel retroviral reference genome. After excluding non-integrated Axi-cel regions and regions with non-zero depth in healthy controls, reads aligned to remaining evaluable regions with mapping quality >40 were considered for further analysis. Fragment lengths were extracted from field 9 of each BAM file, corresponding to the mapping distance between read 1 and read 2 for each read pair. Fragments with length <50 or >800 were excluded from analysis. ctDNA fragment length distributions in each plasma sample were determined as follows: (A) For each patient, a set of high-confidence somatic mutations was identified by genotyping the pre-treatment plasma sample against a matched normal by CAPP-Seq, as described above. (B) For each plasma timepoint from a given patient, ctDNA fragments were identified by querying the respective BAM file for read pairs in which either mate harbored a mutation identified in (A). (C) After assembling a set of bona fide mutant read pairs, fragment lengths were calculated by the same procedure described above for cfCAR19 DNA. Wild-type cfDNA fragment length distributions were calculated from all remaining human-mapped reads in each plasma sample, after excluding the mutated reads identified in (B). For each cfDNA compartment (wild-type, mutant, and cfCAR19 DNA derived), fragment length distributions were calculated by dividing the number of fragments with a given length by the total number of fragments in that compartment. These distributions were calculated both on a per-sample basis and as an average across all available samples. Distributions were smoothed by using a rolling mean with a sliding window of 11 base pairs. Enrichment of mutant or cfCAR19 DNA fragments over wild-type was calculated by taking the difference between respective fragment length distributions. When correlating cfCAR19 DNA fragment lengths with CARFACS, we controlled for differential abundance of cfCAR19 reads between samples by performing a Monte Carlo simulation, randomly sampling equal numbers of cfCAR19 reads from samples with high or low CAR19 FACS over n = 1,000 iterations.
T-cell receptor profiling from cfDNA
We developed SABER (Sequence Affinity capture & analysis By Enumeration of cell-free Receptors) as a technique for TCR enrichment and analysis of fragmented rearrangements shed in cfDNA.27 TCR enrichment is achieved via Cancer Personalized Profiling by Deep Sequencing (CAPP-Seq) after which raw sequencing data is processed and quantitated into unique clonotypes. Candidate rearrangements within TCR loci are identified by mapping sequencing reads to hg38 and identifying poorly mapped reads. Within this space of potential rearrangements, PCR duplicates are resolved by a strategy that defines the consensus of unique molecular identifiers clustered by Levenshtein distances. For every unique UMI, sequences within a Levenshtein distance threshold of 25 from one another are collapsed into one dominant sequence. These unique cfDNA fragments are probed for CDR3-containing regions by anchoring to IMGT-defined CDR3 start and end motifs. A second filtering stage is employed to remove putative CDR3 reads containing templated DNA by way of remapping to the human genome. Poor quality fragments are also removed at this stage. Fragments where ≥40% of bases have an Illumina quality encoding of 30 or less or ≥30% of bases have a quality encoding of 20 or less are filtered out. This results in a final repertoire of TCR-α, TCR-β, TCR-δ, and TCR-γ clonotypes. SABER leverages information from fragmented TCRs, a critical requirement for cfDNA, to make V gene, CDR3, and J gene assignments.
Establishment of early CAR19 expansion index
To assess the expansion of CAR19 T-cells through the first month of therapy, we developed an “Early CAR19 Expansion Index”, integrating multiple measurements of CAR19 quantity. This allowed us to explore the CAR19 T-cell dynamics for each patient without relying on a single measurement. We integrated both cfCAR19 and flow cytometry assessments over the first four weeks of therapy. The variables incorporated into this index were cfCAR19 from weeks 1 and 4, and CARFACS from weeks 1, 2, 3, and 4. Weeks 2 and 3 cfCAR19 were not included given that only a small proportion of patients had plasma available at these timepoints.
For each variable, patients with available data were ranked based on the specific CAR19 quantification, and ranks were then normalized to a dynamic range of 1 (highest) to 100 (lowest). Patients with missing data for a given variable did not receive a rank for that variable. The average (mean) rank was then calculated for each patient across all variables, and patients were then arranged in descending order based on this cumulative average. A final percent rank was then assigned, which corresponds to the Early CAR19 Expansion Index (Figure 5A).
Total metabolic tumor volume (TMTV) measurement
61 (93.8%) patients in the CAR Discovery Cohort were evaluable for TMTV in having uniform functional imaging by positron emission tomography with 2-deoxy-2-[fluorine-18]fluoro-D-glucose integrated with computed tomography (18F-FDG PET/CT) prior to therapy and available imaging data for analysis. TMTV measurements for patients in the CAR19 Discovery Cohort were performed using METAVOL Software (www.metavol.org).61 TMTV was assessed in pretreatment scans by applying a fixed 41% SUVmax threshold followed by manual segmentation of lesions at or exceeding this threshold.65 We have previously demonstrated that measurements made using this software and criteria are highly correlated [Spearman R = 0.87, p < 0.001].45
CAR T-cell cell flow cytometric analysis (CARFACS)
Immuno-phenotyping flow cytometry was performed in order to quantify chimeric antigen receptor (CAR) positive cells, and to profile T-cell and B-cell lineage-specific surface antigens in patient samples (referred to as CAR-FACS), as previously described.56 CAR positive cells were identified using a monoclonal antibody recognizing the CD19 specific scFv of the CAR construct [clone 136.20.1, DyLight650 Conjugated].59
Immunohistochemistry
Immunohistochemical studies were performed on 4 micron thick formalin-fixed and paraffin-embedded tissue. An automated immunostaining platform (Leica Bond III, Leica Biosystems, Buffalo Grove, IL), and the CD19 monoclonal antibody (BT51E, Leica). Appropriate positive and negative controls were included and evaluated with the specimens tested. The stained slides were reviewed by two pathologists and the percentage of immunoreactive tumor cells (0%–100%) and the average intensity of the staining (0 - negative, 1 - weak staining above background, 2 - moderate staining, 3 - strong staining) was scored. An H-score was calculated by multiplying the percentage of immunoreactive tumor cells by the average stain intensity as previously described.66
Emergent variant analyses
To enable identification of emergent or clonally selected alterations, we assessed cases with paired samples available at both the pretreatment and relapse time-points. In order assess for subclonal selection, variants with an allele frequency of 0.2% or greater were considered in these analyses. To control for differences in disease burden between these samples, we ranked the clonal hierarchy of variants as follows. In a set of n variants , we identified the allele fractions in the pretreatment sample as and the allele fractions in the relapse sample as . Within each sample, we then defined a distribution across all variants representing the selection of each variant by calculating:
for each variant. This value was log-transformed to create an approximately normal distribution. For each case, all SNVs were then assigned a z-score based on this distribution:
This Z-score thus represents the selective pressure placed on each variant in each given case, with variants under positive selection receiving high scores and those under negative selection receiving low scores. We finally assessed the recurrent selective pressure on genes of interest by assessing the set of Z-score for alterations in a gene of interest compared to all other alterations using a Wilcoxon rank-sum test. A similar procedure was performed to assess for clonally selected SCNAs. Here, copy-number alterations were first identified in both the pretreatment and relapse samples, with each alteration assigned a z-score representing the magnitude of the alteration. We then calculated a distribution across all identified variants as:
With a similar distribution for deletions. We then calculated a z-score of these values:
To determine the magnitude of clonal selection for each copy number variant, before performing a Wilcoxon rank-sum test across all variants in a locus of interest as described above.
Determination of optimal ctDNA thresholds – (Pretreatment, week 4)
To determine an optimized threshold to separate patients for event-free survival based on pretreatment and dynamic ctDNA levels, a bootstrap approach was applied as previously described.22 Briefly, data from each timepoint was sampled with replacement 10,000 times and the ctDNA that best separated patients for EFS was determined based on the log-rank test.
RNA-sequencing and deconvolution using CIBERSORTx
RNA was isolated from 2–4 10μM FFPE scrolls using Celldata’s RNAstorm kit and quantified using Nanodrop. Sequencing libraries were prepared from 50ng RNA using the SMARTer Stranded Total RNA-Seq v2 Kit (Takara). Fragmentation steps were omitted for FFPE source RNA as recommended by the vendor. Libraries were evaluated using Qubit and TapeStation, and sequenced on NovaSeq6000 machines targeting ≥35 million paired-end reads per sample. After demultiplexing, reads were quality filtered and trimmed using afterqc (version 0.9.6) using the following flags (-q 30 -u 50 -p 30 -a 3 –trim_front = 9 –trim_tail = 50).63 Transcript abundance was quantified using salmon (version 0.8.2). CIBERSORTx (https://cibersortx.stanford.edu) was used to deconvolute cell types as previously described.67 We used the DESeq2 R-package to normalize transcript abundance and perform differential gene expression. We performed gene set enrichment analyses using the Fast Gene Set Enrichment Analysis (FGSEA) R-package using differentially expressed genes ranked by Wald statistic.62
QUANTIFICATION AND STATISTICAL ANALYSIS
Data imputation and multivariable analysis
To perform multivariable Cox regression analysis in the discovery cohort with a sufficient number of samples (with complete information), we first performed data imputation. We examined several imputation methods and found softImpute (R package ‘softImpute’) as the best performer. We then used softImpute on the log-transformed normalized data matrix (n = 64, p = 21) to create a complete matrix. We then, performed a stepwise forward (covariate) selection with Bayesian Information Criterion (BIC) to determine the optimal covariate set (R package ‘survival’). To do this, we defined the metric for feature set S as where denotes the partial log-likelihood of the Cox model. We then found the covariate set achieving the minimum BIC (Day 0 ctDNA level and Week 4 cfCAR19 level). A Cox proportional hazards model incorporating these two variables was trained on the discovery cohort (STEP Score) and was validated on the validation cohort. Importantly, no data imputation was done in the validation set.
Additional statistical analysis
Statistical analyses were performed using R (v.4.0.3) and MATLAB (R2020b). Analyses for significant differences between two groups were assessed using the Wilcoxon rank-sum test. Survival probabilities were estimated using the Kaplan-Meier method, and survival of two groups was compared using a log-rank test (R package ‘survival’). We considered two survival endpoints in this manuscript: event-free survival (EFS) where an event was defined as disease progression or relapse, unplanned anti-lymphoma treatment or death from any cause, and overall survival (OS) wherein an event was defined as death from any cause. ctDNA detection was determined with our previously described Monte-Carlo-based ctDNA detection index.19 Statistical significance of Pearson or Spearman correlation was determined using t statistics. Regression analysis of covariates was conducted by Cox proportional hazards modeling, with p values assessed using the log-likelihood test (R package ‘survival’). For cox regression analyses continuous variables were used as input and standardized using Fisher’s z-score transformation. Units were defined as follows: ctDNA, log10(hGE/mL), cfCAR19, log10(genomes/mL); CARFACS, log10(CAR19 cells/uL); cfTCR – Total T-cells, log10(T-cell Genomes/mL); cfTCR – Diversity, GINI coefficient; log10 TMTV, log10(cm3); CRP, value/upper limit of normal; Age, log10(age in years); LDH, value/upper limit of normal; Stage, 1–4; IPI, score 1–5. The Kolmogorov–Smirnov test was used to compare cumulative distributions.
Supplementary Material
Highlights.
Tumors and CAR T cell effectors can be simultaneously profiled using cell-free DNA
Alterations in multiple classes of genes influence outcomes after CAR19 therapy
Tumor genotype and phenotype influence CAR19 T cell expansion, and vice versa
A multivariable model incorporating tumor and effector features predicts outcomes
ACKNOWLEDGMENTS
This work was supported by the National Cancer Institute (R01CA233975 and R01CA257655 to A.A.A. and M.D., K08CA241076 to D.M.K., K08CA248968 to M.J.F., 2P01CA049605-29A1 to D.B.M. and C.L.M., and K00CA234954 to N.D.S.), the Leukemia and Lymphoma Society (Fellow Award to B.J.S. and M.H.), the Lymphoma Research Foundation (B.J.S.), Uplifting Athletes (B.J.S.), the Virginia and D.K. Ludwig Fund for Cancer Research (A.A.A. and M.D.), the Bakewell Foundation (A.A.A. and M.D.), the Ellie Guardino Cancer Fellowship (B.J.S.), the Stanford Cancer Institute (B.J.S., D.M.K., and A.A.A.), the Damon Runyon Cancer Research Foundation (PST#09-16 to D.M.K. and DR-CI#71-14 to A.A.A), the American Society of Hematology Scholar Award (A.A.A), V Foundation for Cancer Research Abeloff Scholar Award (A.A.A), Conquer Cancer Foundation of the American Society of Clinical Oncology (D.M.K., B.J.S., and J.S.M.), the Emerson Collective Cancer Research Fund (A.A.A.), the Stinehart/Reed Award (A.A.A.), the CRK Faculty Scholar Fund (M.D.), and the SDW/DT and Shanahan Family Foundations (A.A.A. and D.M.K.). D.B.M. and C.L.M. are members of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program. A.A.A. is a Scholar of The Leukemia & Lymphoma Society.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
B.J.S. reports consultancy for Foresight Diagnostics. D.M.K. reports consultancy for Roche, Adaptive Biotechnologies, and Genentech and equity ownership interest in Foresight Diagnostics S.K.C.A. reports speaker honoraria from Takeda. M.J.F. reports consultancy and research funding from Adaptive Biotechnologies, research funding from Kite/Gilead, stock options from Allogene Therapeutics, and equity in Roche/Genentech. M.S.E. reports consultancy for Foresight Diagnostics. J.H.B. reports research funding from Kite Pharma. S.B. reports employment and stock ownership at Kite-a Gilead company. J.W. has research funding from Kite/Gilead, BMS, Novartis, Genentech/Roche, Morphosys/Incyte, AstraZeneca, and ADC Therapeutics, and consulting funding for Kite/Gilead, BMS, Novartis, Genentech/Roche, Morphosys/Incyte, AstraZeneca, ADC Therapeutics, Merck, MonteRosa, Umoja, and Ikusda. M.S.K. reports research funding from CRISPR Therapeutics and Nutcracker Therapeutics, and advisory committee membership for Myeloid Therapeutics and Daiichi Sankyo. Y.N. reports consulting for Leica Biosystems and Roche, and research funding from Kite Pharma. C.L.M. holds several patents focused on CAR T cells therapies; is a co-founder and holds equity in Lyell Immunopharma, CARGO Therapeutics, and Link Cell Therapies, which are developing CAR-based therapies; and consults for Lyell, CARGO, Link, Apricity, Nektar, Immatics, Mammoth, and Ensoma. R.G.M. is a co-founder of and holds equity in Link Cell Therapies and Syncopation Life Sciences. R.G.M. is a consultant for Lyell Immunopharma, NKarta, Arovella Pharmaceuticals, Innervate Radiopharmaceuticals, GammaDelta Therapeutics, Aptorum Group, Zai Labs, ImmunAI, Gadeta, FATE Therapeutics (DSMB), and Waypoint Bio. M.D. reports research funding from AstraZeneca, Genentech, Varian Medical Systems, and Illumina; ownership interest in CiberMed and Foresight Diagnostics; and consultancy from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Gritstone Oncology, Illumina, Novartis, and Roche. D.B.M. holds a patent with Pharmacyclics supporting ibrutinib for chronic graft-versus-host disease and receives consulting or research fees or serves as an advisor for Pharmacyclics, Kite Pharma, Adaptive Biotechnologies, Novartis, BMS, Janssen Pharmaceuticals, Roche, Genentech, Precision Bioscience, Allogene, Miltenyi Biotec, Fate Therapeutics, 2Seventy, and Adicet. A.A.A. reports consultancy for Celgene, Chugai, Genentech, Gilead, Janssen, Pharmacyclics, and Roche; scientific advisory board membership in the Lymphoma Research Foundation; professional affiliations with the American Society of Hematology, American Society of Clinical Oncology, American Society of Clinical Investigation, and Leukemia & Lymphoma Society; research funding from the National Cancer Institute, National Heart, Lung, and Blood Institute, National Institutes of Health, Celgene, Bristol Myers Squibb, and Pfizer; patent filings, including patent issued, licensed, and with royalties paid from FortySeven, a patent pending and Licensed to Foresight, a patent pending relating to MARIA, a patent issued and licensed to CiberMed, a patent issued, a patent pending to CiberMed, a patent issued to Idio-type Vaccines, and a patent issued, licensed, and with royalties paid From Roche; and equity ownership interests in CiberMed Inc., Foresight Diagnostics, FortySeven Inc., and CARGO Therapeutics. B.J.S., D.M.K., M.S.E., M.D., and A.A.A. also report patent filings related to cancer biomarkers. The remaining authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2022.12.005.
REFERENCES
- 1.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. (2017). Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med 377, 2531–2544. 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. (2018). Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med 378, 439–448. 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, Timmerman JM, Holmes H, Jaglowski S, Flinn IW, et al. (2020). KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med 382, 1331–1342. 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munshi NC, Anderson LD Jr., Shah N, Madduri D, Berdeja J, Lonial S, Raje N, Lin Y, Siegel D, Oriol A, et al. (2021). Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med 384, 705–716. 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 5.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jager U, Jaglowski S, Andreadis C, Westin JR,€ et al. (2019). Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med 380, 45–56. 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 6.Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, Mehta A, Purev E, Maloney DG, Andreadis C, et al. (2020). Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396, 839–852. 10.1016/s0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 7.Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, Lin Y, Braunschweig I, Hill BT, Timmerman JM, et al. (2019). Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 20, 31–42. 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah NN, and Fry TJ (2019). Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol 16, 372–385. 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Locke FL, Rossi JM, Neelapu SS, Jacobson CA, Miklos DB, Ghobadi A, Oluwole OO, Reagan PM, Lekakis LJ, Lin Y, et al. (2020). Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 4, 4898–4911. 10.1182/bloodadvances.2020002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, Levine JE, Qayed M, Grupp SA, Boyer M, et al. (2018). Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med 24, 1504–1506. 10.1038/s41591-018-0146-z. [DOI] [PubMed] [Google Scholar]
- 11.Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH,Frank MJ, Shiraz P, Sahaf B, Craig J, et al. (2021). CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat. Med 27, 1419–1431. 10.1038/s41591-021-01436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plaks V, Rossi JM, Chou J, Wang L, Poddar S, Han G, Wang Z,Kuang SQ, Chu F, Davis RE, et al. (2021). CD19 target evasion as a mechanism of relapse in large B-cell lymphoma treated with axicabtagene ciloleucel. Blood 138, 1081–1085. 10.1182/blood.2021010930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et al. (2015). Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 5, 1282–1295. 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacoby E, Nguyen SM, Fountaine TJ, Welp K, Gryder B, Qin H, Yang Y, Chien CD, Seif AE, Lei H, et al. (2016). CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun 7, 12320. 10.1038/ncomms12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jain MD, Ziccheddu B, Coughlin CA, Faramand R, Griswold AJ, Reid KM, Menges M, Zhang Y, Cen L, Wang X, et al. (2022). Whole-genome sequencing reveals complex genomic features underlying anti-CD19 CAR T-cell treatment failures in lymphoma. Blood 140, 491–503. 10.1182/blood.2021015008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee YG, Guruprasad P, Ghilardi G, Pajarillo R, Sauter CT, Patel R, Ballard HJ, Hong SJ, Chun I, Yang N, et al. (2022). Modulation of BCL-2 in both T cells and tumor cells to enhance chimeric antigen receptor T cell immunotherapy against cancer. Cancer Discov. 12, 2372–2391. 10.1158/2159-8290.CD-21-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, Dai E, Dang M, Jain N, Yang H, et al. (2020). Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med 26, 1878–1887. 10.1038/s41591-020-1061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newman AM, Bratman SV, Stehr H, Lee LJ, Liu CL, Diehn M,and Alizadeh AA (2014). FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics 30, 3390–3393. 10.1093/bioinformatics/btu549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, Stehr H, Liu CL, Bratman SV, Say C, et al. (2016). Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol 34, 547–555. 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chabon JJ, Hamilton EG, Kurtz DM, Esfahani MS, Moding EJ, Stehr H, Schroers-Martin J, Nabet BY, Chen B, Chaudhuri AA, et al. (2020). Integrating genomic features for non-invasive early lung cancer detection. Nature 580, 245–251. 10.1038/s41586-0202140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AFM, Esfahani MS, Lovejoy AF, Chabon JJ, Klass DM, Liu CL, et al. (2016). Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med 8, 364ra155. 10.1126/scitranslmed.aai8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurtz DM, Scherer F, Jin MC, Soo J, Craig AFM, Esfahani MS, Chabon JJ, Stehr H, Liu CL, Tibshirani R, et al. (2018). Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J. Clin. Oncol 36, 2845–2853. 10.1200/JCO.2018.78.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurtz DM, Esfahani MS, Scherer F, Soo J, Jin MC, Liu CL, Newman AM, Dührsen U, Hüttmann A, Casasnovas O, et al. (2019). Dynamic risk profiling using serial tumor biomarkers for personalized outcome prediction. Cell 178, 699–713.e19. 10.1016/j.cell.2019.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frank MJ, Hossain NM, Bukhari A, Dean E, Spiegel JY, Claire GK, Kirsch I, Jacob AP, Mullins CD, Lee LW, et al. (2021). Monitoring of circulating tumor DNA improves early relapse detection after axicabtagene ciloleucel infusion in large B-cell lymphoma: results of a prospective multi-institutional trial. J. Clin. Oncol 39, 3034–3043. 10.1200/jco.21.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang P, Chan CWM, Chan KCA, Cheng SH, Wong J, Wong VWS, Wong GLH, Chan SL, Mok TSK, Chan HLY, et al. (2015). Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 112, E1317–E1325. 10.1073/pnas.1500076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han DSC, Ni M, Chan RWY, Chan VWH, Lui KO, Chiu RWK, and Lo YMD (2020). The biology of cell-free DNA fragmentation and the roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet 106, 202–214. 10.1016/j.ajhg.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shukla ND, Craig AFM, Sworder B, Kurtz DM, Macaulay C, Garofalo A, Frank MJ, Alig S, Duran G, Kim YH, et al. (2020). Profiling T-cell receptor diversity and dynamics during lymphoma immunotherapy using cell-free DNA (cfDNA). Blood 136, 49–50. 10.1182/blood-2020-141655. [DOI] [Google Scholar]
- 28.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ennishi D, Healy S, Bashashati A, Saberi S, Hother C, Mottok A, Chan FC, Chong L, Abraham L, Kridel R, et al. (2020). TMEM30A loss-of-function mutations drive lymphomagenesis and confer therapeutically exploitable vulnerability in B-cell lymphoma. Nat. Med 26, 577–588. 10.1038/s41591-020-0757-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Lee CH, Qi C, Tailor P, Feng J, Abbasi S, Atsumi T, and Morse HC 3rd (2008). IRF8 regulates B-cell lineage specification, commitment, and differentiation. Blood 112, 4028–4038. 10.1182/blood-2008-01-129049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pridans C, Holmes ML, Polli M, Wettenhall JM, Dakic A, Corcoran LM, Smyth GK, and Nutt SL (2008). Identification of Pax5 target genes in early B cell differentiation. J. Immunol 180, 1719–1728. 10.4049/jimmunol.180.3.1719. [DOI] [PubMed] [Google Scholar]
- 32.Klement JD, Paschall AV, Redd PS, Ibrahim ML, Lu C, Yang D, Celis E, Abrams SI, Ozato K, and Liu K (2018). An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Invest 128, 5549–5560. 10.1172/JCI123360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shouval R, Alarcon Tomas A, Fein JA, Flynn JR, Markovits E, Mayer S, Olaide Afuye A, Alperovich A, Anagnostou T, Besser MJ, et al. (2022). Impact of TP53 genomic alterations in large B-cell lymphoma treated with CD19-chimeric antigen receptor T-cell therapy. J. Clin. Oncol 40, 369–381. 10.1200/JCO.21.02143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu JI, Dayaram T, Tovy A, De Braekeleer E, Jeong M, Wang F, Zhang J, Heffernan TP, Gera S, Kovacs JJ, et al. (2018). PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell 23, 700–713.e6. 10.1016/j.stem.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498. 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D,Gooley TA, Cherian S, Chen X, Pender BS, et al. (2019). Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 133, 1652–1663. 10.1182/blood-2018-11-883710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, Jiang M, Mottok A, Denis-Lagache N, Ciriello G, et al. (2016). Loss of the HVEM tumor suppressor in lymphoma and restoration by modified CAR-T cells. Cell 167, 405–418.e13. 10.1016/j.cell.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C, Caldas C, Califano A, Doherty M, Elsner M, et al. (2015). Toward understanding and exploiting tumor heterogeneity. Nat. Med 21, 846–853. 10.1038/nm.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan JCM, White JR, and Diaz LA Jr. (2019). Hey CIRI, what’s my prognosis? Cell 178, 518–520. 10.1016/j.cell.2019.07.005. [DOI] [PubMed] [Google Scholar]
- 40.Singh N, Lee YG, Shestova O, Ravikumar P, Hayer KE, Hong SJ, Lu XM, Pajarillo R, Agarwal S, Kuramitsu S, et al. (2020). Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T-cell dysfunction. Cancer Discov. 10, 552–567. 10.1158/2159-8290.CD-19-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, Kurtz DM, Stehr H, Scherer F, Karlovich CA, et al. (2016). Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun 7, 11815. 10.1038/ncomms11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, et al. (2017). Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med 376, 2109–2121. 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 43.Roschewski M, Dunleavy K, Pittaluga S, Moorhead M, Pepin F, Kong K, Shovlin M, Jaffe ES, Staudt LM, Lai C, et al. (2015). Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: a correlative biomarker study. Lancet Oncol. 16, 541–549. 10.1016/S1470-2045(15)70106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al. (2018). Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. J. Med 378, 1396–1407. 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alig S, Macaulay CW, Kurtz DM, Dührsen U, Hüttmann A, Schmitz C, Jin MC, Sworder BJ, Garofalo A, Shahrokh Esfahani M, et al. (2021). Short diagnosis-to-treatment interval is associated with higher circulating tumor DNA levels in diffuse large B-cell lymphoma. J. Clin. Oncol 39, 2605–2616. 10.1200/JCO.20.02573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maurer MJ, Ghesquières H, Link BK, Jais JP, Habermann TM, Thompson CA, Haioun C, Allmer C, Johnston PB, Delarue R, et al. (2018). Diagnosis-to-Treatment interval is an important clinical factor in newly diagnosed diffuse large B-cell lymphoma and has implication for bias in clinical trials. J. Clin. Oncol 36, 1603–1610. 10.1200/JCO.2017.76.5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nutt SL, Heavey B, Rolink AG, and Busslinger M (1999). Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401, 556–562. 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 48.Mikkola I, Heavey B, Horcher M, and Busslinger M (2002). Reversion of B cell commitment upon loss of Pax5 expression. Science 297, 110–113. 10.1126/science.1067518. [DOI] [PubMed] [Google Scholar]
- 49.Armand P, Rodig S, Melnichenko V, Thieblemont C, Bouabdallah K, Tumyan G, Ö zcan M, Portino S, Fogliatto L, Caballero, et al. (2019). Pembrolizumab in relapsed or refractory primary mediastinal large B-cell lymphoma. J. Clin. Oncol 37, 3291–3299. 10.1200/JCO.19.01389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, Felip E, van den Heuvel MM, Ciuleanu TE, Badin F, et al. (2017). First-line ni-volumab in stage IV or recurrent non-small-cell lung cancer. N. Engl. J. Med 376, 2415–2426. 10.1056/NEJMoa1613493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuruvilla J, Ramchandren R, Santoro A, Paszkiewicz-Kozik E, Gasiorowski R, Johnson NA, Fogliatto LM, Goncalves I, de Oliveira JSR, Buccheri V, et al. (2021). Pembrolizumab versus brentuximab vedotin in relapsed or refractory classical Hodgkin lymphoma (KEYNOTE-204): an interim analysis of a multicentre, randomised, open-label, phase 3 study. Lancet Oncol. 22, 512–524. 10.1016/S1470-2045(21)00005-X. [DOI] [PubMed] [Google Scholar]
- 52.Melenhorst JJ,Chen GM, Wang M,Porter DL,Chen C,Collins MA, Gao P, Bandyopadhyay S, Sun H, Zhao Z, et al. (2022). Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature 602, 503–509. 10.1038/s41586-021-04390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haradhvala NJ, Leick MB, Maurer K, Gohil SH, Larson RC, Yao N, Gallagher KME, Katsis K, Frigault MJ, Southard J, et al. (2022). Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat. Med 28, 1848–1859. 10.1038/s41591-022-01959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Good Z, Spiegel JY, Sahaf B, Malipatlolla MB, Ehlinger ZJ, Kurra S, Desai MH, Reynolds WD, Wong Lin A, Vandris P, et al. (2022). Post-infusion CAR TReg cells identify patients resistant to CD19-CAR therapy. Nat. Med 28, 1860–1871. 10.1038/s41591-022-01960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, et al. (2012). B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimericantigen-receptor-transduced T cells. Blood 119, 2709–2720. 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baird JH, Frank MJ, Craig J, Patel S, Spiegel JY, Sahaf B, Oak JS, Younes SF, Ozawa MG, Yang E, et al. (2021). CD22-directed CAR T-cell therapy induces complete remissions in CD19-directed CAR-refractory large B-cell lymphoma. Blood 137, 2321–2325. 10.1182/blood.2020009432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oluwole OO, Bishop MR, Gisselbrecht C, Gordon LI, Kersten MJ, Maloney DG, Schmitz N, Caballero Barrigon MD, Kuruvilla J, Song KW, et al. (2018). ZUMA-7: a phase 3 randomized trial of axicabtagene ciloleucel (Axi-Cel) versus standard-of-care (SOC) therapy in patients with relapsed/refractory diffuse large B cell lymphoma (R/R DLBCL). J. Clin. Oncol 36, TPS7585. 10.1200/JCO.2018.36.15_suppl.TPS7585. [DOI] [Google Scholar]
- 58.Locke FL, Miklos DB, Jacobson CA, Perales MA, Kersten MJ, Oluwole OO, Ghobadi A, Rapoport AP, McGuirk J, Pagel JM, et al. (2022). Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N. Engl. J. Med 386, 640–654. 10.1056/NEJMoa2116133. [DOI] [PubMed] [Google Scholar]
- 59.Jena B, Maiti S, Huls H, Singh H, Lee DA, Champlin RE, and Cooper LJN (2013). Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PLoS One 8, e57838. 10.1371/journal.pone.0057838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, Wang JQ, Schmitz R, Morin RD, Tang J, et al. (2020). A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell 37, 551–568.e14. 10.1016/j.ccell.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hirata K, Kobayashi K, Wong KP, Manabe O, Surmak A, Tamaki N, and Huang SC (2014). A semi-automated technique determining the liver standardized uptake value reference for tumor delineation in FDG PET-CT. PLoS One 9, e105682. 10.1371/journal.pone.0105682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2021). Fast gene set enrichment analysis. Preprint at bioRxiv. 10.1101/060012. [DOI] [Google Scholar]
- 63.Chen S, Huang T, Zhou Y, Han Y, Xu M, and Gu J (2017). AfterQC: automatic filtering, trimming, error removing and quality control for fastq data. BMC Bioinf. 18, 80. 10.1186/s12859-017-1469-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dührsen U, Müller S, Hertenstein B, Thomssen H, Kotzerke J, Mesters R, Berdel WE, Franzius C, Kroschinsky F, Weckesser M, et al. (2018). Positron emission tomography-guided therapy of aggressive non-hodgkin lymphomas (PETAL): a multicenter, randomized phase III trial. J. Clin. Oncol 36, 2024–2034. 10.1200/jco.2017.76.8093. [DOI] [PubMed] [Google Scholar]
- 65.Meignan M, Sasanelli M, Casasnovas RO, Luminari S, Fioroni F, Coriani C, Masset H, Itti E, Gobbi PG, Merli F, and Versari A (2014). Metabolic tumour volumes measured at staging in lymphoma: methodological evaluation on phantom experiments and patients.Eur. J.Nucl. Med.Mol. Imaging 41, 1113–1122. 10.1007/s00259-014-2705-y. [DOI] [PubMed] [Google Scholar]
- 66.Devereaux KA, Charu V, Zhao S, Charville GW, Bangs CD, van de Rijn M, Cherry AM, and Natkunam Y (2018). Immune checkpoint blockade as a potential therapeutic strategy for undifferentiated malignancies. Hum. Pathol 82, 39–45. 10.1016/j.humpath.2018.06.034. [DOI] [PubMed] [Google Scholar]
- 67.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. (2019). Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol 37, 773–782. 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA and RNA sequencing data from rrLBCL patients have been deposited at dbGaP and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-CD19 | Leica Biosystems | clone BT51E |
| Anti-CD19 specific scFv | Jena et al.59 | clone 136.20.1 |
|
| ||
| Deposited data | ||
|
| ||
| Targeted DNA Sequencing data | dbGaP | Accession Number Pending |
| RNA sequencing data | dbGaP | Accession Number Pending |
| LM22 signature matrix (CIBERSORTx) | Newman et al., 2015 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| xGen Lockdown Probes | IDT | https://www.idtdna.com/ |
| SeqCap EZ Choice Probes | Roche | https://sequencing.roche.com/ |
|
| ||
| Software and algorithms | ||
|
| ||
| CIBERSORTx | Newman et al., 2015 | https://cibersortx.stanford.edu/ |
| FACTERA | Newman et al.18 | https://factera.stanford.edu/ |
| LymphGen Classifier | Wright et al.60 | https://llmpp.nih.gov/lymphgen/index.php |
| METAVOL | Hirata et al.61 | www.metavol.org |
| MATLAB version 2020b | Mathworks | https://www.mathworks.com/products/matlab.html |
| R Statistical Software version 4.0.3 | The R Foundation | https://www.r-project.org |
| survival package | CRAN Repository | https://cran.r-project.org/web/packages/survival/index.html |
| softimpute package | CRAN Repository | https://cran.r-project.org/web/packages/softImpute/index.html |
| maxstat package | CRAN Repository | https://cran.r-project.org/web/packages/maxstat/index.html |
| Fast Gene Set Enrichment Analysis (FGSEA) package | Korotkevich et al.62 | https://github.com/ctlab/fgsea/ |
| afterqc package | Chen et al.63 | https://github.com/OpenGene/AfterQC |