

ABSTRACT
Large-molecule antibody biologics have revolutionized medicine owing to their superior target specificity, pharmacokinetic and pharmacodynamic properties, safety and toxicity profiles, and amenability to versatile engineering. In this review, we focus on preclinical antibody developability, including its definition, scope, and key activities from hit to lead optimization and selection. This includes generation, computational and in silico approaches, molecular engineering, production, analytical and biophysical characterization, stability and forced degradation studies, and process and formulation assessments. More recently, it is apparent these activities not only affect lead selection and manufacturability, but ultimately correlate with clinical progression and success. Emerging developability workflows and strategies are explored as part of a blueprint for developability success that includes an overview of the four major molecular properties that affect all developability outcomes: 1) conformational, 2) chemical, 3) colloidal, and 4) other interactions. We also examine risk assessment and mitigation strategies that increase the likelihood of success for moving the right candidate into the clinic.
KEYWORDS: Antibody, biologics, colloidal, conformational, developability, formulation, manufacturability, specificity, stability
Introduction
The discovery and development of biologicals, especially antibodies and engineered antibody modalities, has expanded in recent decades to the point where more than 100 antibody biologics are marketed and approximately 1000 investigational molecules are currently in the clinic globally. 1, 2 Therapeutic indications successfully targeted by protein and antibody therapeutics span immuno-oncology, cardiovascular, infectious and auto-immune diseases, as well as neurodegenerative indications.3 Moreover, antibodies are amenable to engineering to enhance versatility and produce an extraordinarily diverse range of characteristics, including multi-specific targeting,4 enhanced or decreased effector function,5,6 and increased half-life.7,8 Monoclonal antibodies can be designed to increase target binding and to enhance developability characteristics via selection technologies, screening and engineering.9,10 Successful developability is critical to lead optimization and selection and to ensure robust processing,11 stability,12–14 delivery,15 administration,16 pharmacokinetics (PK)/bioavailability,17 safety,18 and it is associated with clinical success rates.19
Following antibody generation and hit screening, key preclinical developability activities encompass molecular engineering, computational and in silico evaluations, production, analytical and biophysical characterization, stability and forced degradation studies, and process and formulation assessments. We also discuss the appropriate analytical methods and when to use them. Common issues and risks encountered during developability assessments, such as aggregation, self-interaction, hydrophobicity, deamidation and oxidation, are explored. Several non-developability activities are critical to large-molecule discovery and development, such as the development of functional bioassays, biomarker development, PK/pharmacodynamic studies, and in vivo efficacy that may affect lead selection. However, these activities are not the focus of this review. Here, we define developability attributes as anything related to or impacted by the inherent properties of a molecule, distinguished from effects resulting from target biology and mechanism of action, which affect lead selection, development, and even safety and efficacy. The molecular properties that impact all developability attributes and outcomes are herein organized into four major categories: 1) conformational, 2) chemical, 3) colloidal, and 4) other interactions. These molecular categories or properties are defined along with relevant developability attributes, the analytical methods used to interrogate them, and examples of key criteria. It is more recently apparent that these attributes not only affect Chemistry, Manufacturing, and Controls (CMC) attributes and outcomes,20 but also post-administration phenomena.19 We also examine developability risk assessment and mitigation strategies, which are unique to each molecule and are influenced by the nature and number of liabilities and potential mitigations. Overall, this review focuses on large-molecule or protein developability, applied particularly to liquid formulated monoclonal antibodies (mAbs) and engineered versions thereof, the prevailing large molecule of choice for therapeutic indications. Although other modalities combining small molecules, such as antibody-drug conjugates (ADCs) and/or protein-oligonucleotide conjugates, potentially offer additional therapeutic benefits as well as developability and development challenges, they are outside the scope of this review.
Antibody generation
Hit to lead
Preclinical developability of therapeutic antibodies includes initial screening of generated hits using binding and/or activity assessments.20 These molecules are then further screened after transient eukaryotic expression and purification to further evaluate relevant physicochemical characteristics.21 A flowchart of key preclinical activities relating to molecular developability, including hit or antibody generation, is shown in Figure 1. While antibody generation itself is typically not considered a core developability activity, it certainly can affect the resulting range of molecular properties and therefore developability outcomes.12,22 When performing developability assessments, it is important to consider the method of generation used, whether it is mouse immunization (followed by hybridoma screening), phage or yeast display technologies, or synthetic libraries. Different selection technologies may yield different outcomes in terms of antibody properties. For instance, phage display generation can increase the likelihood for developability issues such as reduced solubility,23 increased hydrophobicity,24,25 and increased non-specificity leading to poor half-life.26 These outcomes may result from multiple rounds of phage selection, which are done to yield high affinity molecules but may also build up excessive charge or hydrophobicity in the complementarity-determining regions (CDRs), leading to worse developability outcomes.26 Further, nonspecific binding is not as significant a selection pressure in phage display as it may be in animal immunization approaches, where in the latter, generated antibodies are deselected for autoimmunity.26 To compensate for this lack of natural selection, hydrophobic CDRs may be manually deselected in the phage display screening process27 or additional selection measures can be taken to screen out unfolded or unstable variable domains.28
Figure 1.
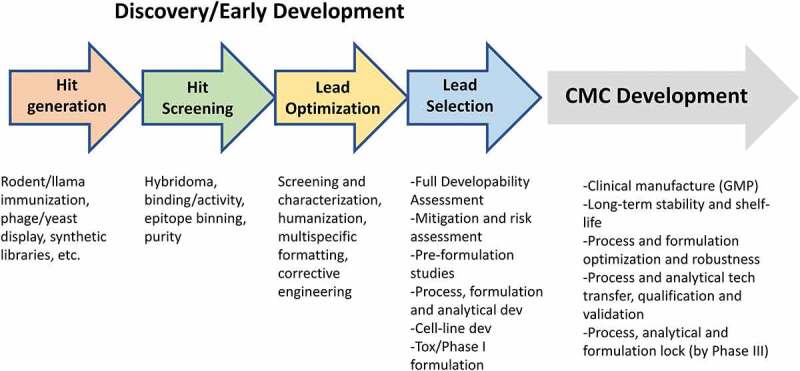
Diagram highlighting various discovery and early development stages for large-molecule therapeutic antibody developability. Later-stage CMC development key deliverables are also listed.
Likewise, yeast display affinity maturation typically increases hydrophobicity, aggregation rates, and self-interaction propensity,19 as binding is iteratively maximized like in phage display. However, a major advantage of using yeast display is the presence of the eukaryotic protein production machinery required to maintain post-translational modifications.29 Further, strategies exist to select for increased thermal stability of yeast display variants.30 To increase candidate diversity, multiple hit generation campaigns may be undertaken, as one approach may generate high redundancy in terms of binding, sequence, and developability profiles.27,31 Ideally, antibodies with a diverse range of binding affinities and antigen epitopes should be generated to maximize the likelihood that a differentiating antibody with desirable efficacy, safety, and mechanism of action will be obtained. Further, from such a diverse pool of antibodies, those with favorable developability profiles can be selected and/or further optimized. Species cross-reactivity of antibody candidates is also an important consideration, and separate generation campaigns may be needed to produce species-specific surrogates for animal studies, especially for rodents.32
Antibodies generated by rodent immunization, typically mouse or rat, necessitates up front engineering and humanization, which will greatly expand the initial number of candidates to screen and characterize. In general, humanization of antibodies presents challenges, such as additional and time-consuming in vitro screening of variants and increased immunogenicity risks.33 Humanization against consensus human germlines in parallel to correcting primary liabilities in silico is recommended to avoid severe liabilities later that will require correction and therefore delay selection efforts.34 Primary liabilities are considered potentially severe chemical or physicochemical liabilities. Primary liability factors include the location, in particular whether the liability resides in the variable framework or more concerningly in the variable CDR regions, and potential severity, where process and formulation measures are unlikely to acceptably mitigate the risk, degradation will be significant post-administration, and/or there are significant effects on activity and/or function. Problematic primary sequence motifs, specifically those containing free cysteines, consensus asparagine glycosylation sites (Asn or N followed by XS/T) other than conserved N297 glycosylation, and NG deamidation motifs, should be initially deselected or corrected in silico.12 This is especially true if the location of the potential modification is in the variable CDR region and is likely to affect target binding or activity if reactive.35 Although evaluating both humanization variants and chemical liability correction variants further expands initial screening efforts, more developable candidates with decreased immunogenicity and fewer liabilities may be later revealed. The number of early candidates to evaluate is further increased if multispecific formatting is required, where usually the monospecific, bivalent antibodies are screened first, followed by rounds of multispecific formatting and customization.36 The desired or available format, e.g., antigen binding fragment (Fab), single-chain variable fragment (scFv), or variable heavy-chain antibody fragment (VHH) derived from llama immunization, is also an important consideration. A plethora of other multispecific formats also exist.36 Such a large toolbox of antibody modalities, in addition to how they can be asymmetrically arranged into multispecifics, can indeed create large numbers of constructs to evaluate. In practice, the developability screening funnel can expand and contract from hit to lead optimization and selection, as shown in Figure 2. To reduce the overall number of antibody variants to screen, immunization against a transgenic, humanized animal, such as Trianni mice, can generate fully human antibodies and bypass the need for in vitro humanization.37 However, a sizable investment in this generation technology is required and fully human mAbs may be generated by other technologies, such as phage display and single human B cell cloning.38 Various isotypes, usually IgG1 or IgG4, or modulated effector function variants thereof, can also be evaluated depending on the desired effector function and mechanism of action.39
Figure 2.
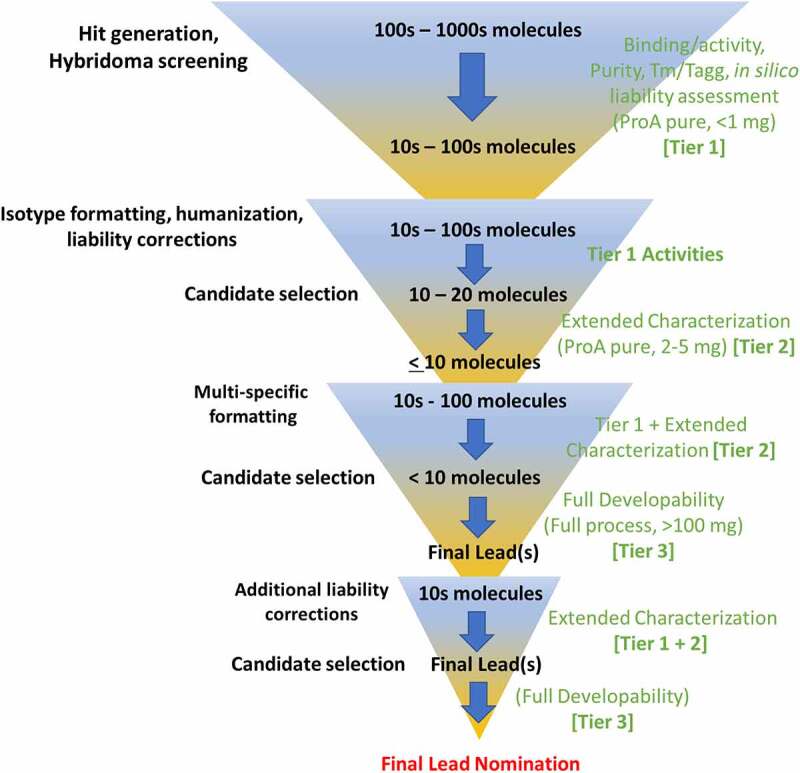
Diagram depicting a hypothetical scenario of various discovery stages of antibody discovery and developability activities. Ultimately, potentially hundreds or thousands of candidates funnel down to relatively few select, favorable candidates. However, along the way, the selection funnel can expand and contract as additional variants, constructs, liability corrections or multispecific scaffolds are created from previously selected candidates in an iterative manner. On the left (black font) are developability activities such as screening, engineering, and selection, which may increase the number of candidates from a preferred subset like in the case of creating humanization or correction variants. On the right (green font) are the developability analytics and assessments employed, which are divided into Tiers. In this example, initial hits are screened to yield a more favorable subset based on limited analytics that include high-throughput binding/activity, purity, Tm/Tagg, and in silico assessments (Tier 1 assays). To funnel to fewer candidates, extended characterization assays should be employed that include cIEF, HIC, DSC and self-interaction (by kDiff and/or AC-SINS depending on throughput needs and material availability), a short, single timepoint, accelerated stability hold to evaluate integrity, particle analysis if available, and non-specificity assays (Tier 2 assays). Subsequently, a full developability assessment is ultimately employed that includes forced degradation, a full purification process, and formulation stability assessments (Tier 3). It should be noted that when variants are created from previous ones (such as humanization, formatting, or correction), or even new lots are tested, the previous-stage analytics, or Tiers, should be repeated so that a complete characterization set (Tiers 1–3) is obtained for any potential lead. For each stage, protein material amounts are proposed, but can vary depending on sample preparation and analytical sample needs.
Lead optimization and selection
Candidate screening
Regardless of the number of generation methods and leads, molecules first and foremost need to be acceptably expressed and have acceptable purity characteristics following affinity capture. A basic workflow to screen molecules typically begins with hybridoma expression followed by Protein A (ProA) capture, where binding characteristics are primarily evaluated along with limited purity and analytical testing on less than one milligram (mg) of material.40 More viable candidates, perhaps on the scale of 10s to 100s, can then be moved forward into transient Chinese hamster ovary (CHO) expression to produce mg quantities followed by ProA capture and screening by high throughput “Tier 1” assays (Figure 2). These include size exclusion high or ultra-performance liquid chromatography (SE-HPLC/UPLC), capillary sodium dodecyl sulfate electrophoresis (cSDS), binding/activity by surface plasmon resonance (SPR), and temperature-dependent melting and aggregation (Tm/Tagg) assessments. While expression and purification can typically be optimized later in CMC development, issues in initial purity and heterogeneity of transiently expressed and purified material can signal an unstable or poorly folded molecule.41,42 Initial purity and heterogeneity assessments on transient leads are vital to identify potential issues, such as aggregation and clipping propensity that occur during expression and/or affinity purification, early in the discovery process. SE-HPLC or UPLC is recommended to initially evaluate aggregation, which is a CMC release and stability quality attribute and can result in immunogenicity or decreased efficacy after administration.43 Online static (multi-angle) light scattering coupled to analytical SEC (or SEC-MALS) can further characterize or confirm the nature of protein aggregate.44 While this is done on unstressed molecules, they may be prone to aggregation in the culture supernatant prior to affinity capture or during the low pH ProA elution step.45 Antibody main-chain clipping can be directly assessed by denaturing reduced-cSDS and domain dissociation directly by non-reduced cSDS, which are CMC critical purity and quality attributes.46 Clipping can be acid or base catalyzed or enzymatic in nature,47 while antibody domain dissociation typically arises from unformed or broken interdomain disulfides.48 Additionally, the presence of clipped products can lead to aggregation and particle formation downstream, which not only affect drug product attributes, but potentially safety and efficacy.49,50 Other common expression impurities elucidated by cSDS are heavy-chain dimers formed during expression or from degradative loss of light chain (LC).51,52 cSDS can also elucidate and quantify homodimers and mismatched impurities in the case of multispecific expression.53 To further refine candidates, it is recommended to also screen temperature-dependent unfolding (Tm), which is often accompanied by complementary temperature-dependent aggregation (Tagg).54 While aggregation propensity at lower process and storage relevant temperatures may or may not be reflected by higher-temperature, kinetics-driven unfolding, there may be a correlation and there are proposed guidelines in selecting a candidate.55 This is because aggregation in solution is a complex phenomenon that is not solely predicted by kinetic Tm measurements.34,56 In vitro binding by SPR or cell-based assays should be conducted on these initially expressed leads or on a preferred subset to verify binding or activity properties, and it should be assumed that various impurity attributes, such as aggregation and clipping, can affect binding or activity.12 Once desirable candidates are selected, their identity should be confirmed by intact mass spectrometry (MS) prior to launching a cascade of engineering and formatting, downstream expression, purification, extended characterization, and formulation and stability studies.
Additional or extended characterization
Prior to selecting final candidates, additional characterization is warranted to maximize the probability of selecting good candidates with desirable quality and developability profiles. It is also critical to factor in non-developability work, such as in vitro functional assays and/or animal model studies, throughout the process. It is important to balance functional properties with developability attributes because changing one property, such as affinity, may affect another, such as stability, and vice versa.57 Commonly, various lead molecules and their variants may be similar in terms of target binding affinity and epitopes, as well as their functional assay outputs. This is particularly true for humanization variants with similar sequences, and multispecific constructs, where similar binding arms are shared, but constructs may vary in linker length, orientation, or format (such as Fab versus scFv). In these cases, additional developability characterization offers an excellent opportunity to further differentiate these molecules based on their physicochemical properties. This may be done on a few to dozens of leads, depending on the generation campaign and whether humanization, liability correction and/or multi-specific formatting is required. A comprehensive summary of the extended characterization developability assays, the attributes they measure, and their justification is provided in Table 1. Further, these assays are also depicted in a hypothetical developability workflow diagram shown in Figure 2 as “Tier 2” assays, which include temperature-dependent unfolding (Tm), preferably by calorimetry, isoelectric point (pI) and charge heterogeneity, hydrophobicity, self-interaction, sub-visible particle evaluation, and polyspecificity or nonspecific interactions. A short stability hold to evaluate integrity is also included as a Tier 2 activity.
Table 1.
Core developability analytical methods, the attributes they describe, and their justification. Many assays describe basic molecular properties, but also are drug product CQAs.
| Method | Attributes | Justification |
|---|---|---|
| SE-HPLC/UPLC | Purity, Aggregate (HMW), fragmentation or dissociation (LMW) | DP release and stability CQA (purity), immunogenicity, may lead to particle formation |
| cSDS (NR- and Red-) | Purity, fragmentation, or dissociation (LMW) | DP release and stability CQA (purity), clipped species may lead to aggregates/particles |
| cIEF | pI, charge heterogeneity | DP release and stability CQA, platform process and formulation fit (pI), PK/bioavailability impact |
| Calorimetry (DSC), Fluorescence (intrinsic or extrinsic) coupled to light scattering (DLS or SLS) | Tm/Tagg transition and onsets (with temperature gradient) | Determine if thermally stable, degree of aggregation upon unfolding, may correlate to aggregation rates on stability or in-process |
| HIC | hydrophobicity, heterogeneity | May correlate to aggregation, nonspecific binding, and/or self-interaction |
| DLS and SLS | kDiff (mL/g), Rh (nm), B22 | Degree of self-interaction. Usually correlates to viscosity at higher concentrations |
| Viscosity | Dynamic viscosity (cP) | Impact to process, injectability, liquid formulation behavior |
| Particles | Particle size, count | DP release and stability CQA, immunogenicity |
| Intact Mass Spectrometry | Identity, impurity detection, heterogeneity | Identity, impurity detection, heterogeneity |
| Peptide Mass Spectrometry | Mapping and quantification of chemical modifications | DP release and stability CQAs, may impact activity and function |
| Glycan analysis | Glycosylation characterization (Fc core) | May impact PK, activity and function |
| Binding/Activity (SPR or cell-based) | Binding, Activity | DP release and stability CQA, activity and potency |
| PSR, BvP, CIC binding | Polyspecificity and nonspecific binding, Cross-interaction metrics | Indicates stickiness, may impact bioavailability and PK |
Temperature-dependent unfolding
To further evaluate conformational stability, differential scanning calorimetry (DSC) may be used as the gold standard to accurately measure melting temperature (Tm) onset and transitions.58 A suitably high Tm may indicate good or acceptable conformational stability at lower temperatures.59 Preferably, the Tm onset of an antibody therapeutic is >50°C, well above the standard accelerated storage temperature of 40°C, as well as physiological temperatures.34 Therefore, the molecule of interest can be expected to be conformationally intact in physiological conditions and subjected to accelerated stress without the confounding effects of excessive temperature-dependent unfolding. As aforementioned, Tm may also correlate to aggregation and even particulate formation propensity in process conditions, on formulated stability, or when subjected to various forced degradation conditions.34 However, this is not necessarily the case, and therefore other properties should be also weighed alongside Tm when attempting to differentiate candidates.40
Isoelectric point
Characterization of the charge heterogeneity, typically by capillary isoelectric focusing (cIEF), is critical to verify the pI is acceptable and will fit platform purification and formulation efforts. Antibodies commonly have basic pIs, which lead to favorable colloidal stability in acidic formulations due to inherent charge repulsions between molecules.60 However, an antibody with an excessively low pI may be difficult to purify from similarly charged host-cell proteins (HCPs),61 as well as formulate at a low, representative formulation pH that balances conformational, colloidal, and chemical stability,21 which typically is approximately pH 6 for commercial mAbs.62,63 Furthermore, differences in charge heterogeneity have been associated with differences in neonatal receptor (FcRn)-dependent PK half-life and efficacy.64 Excessively high pIs have been associated with poor non-FcRn-dependent PK and bioavailability outcomes,65 and further reducing the pI through selective engineering of the variable framework or CDR regions improved half-life in certain cases.17,65,66 Similarly, excessive variable domain charge has correlated to fast clearance in mice, cynomolgus monkeys, and humans.67,68 Since high pI antibodies in formulation typically exhibit excellent colloidal properties while also presenting a PK risk via charge-mediated nonspecific binding and clearance, colloidal properties versus tissue distribution and PK risk should be balanced when nominating a lead candidate, and low pIs that can challenge purification and formulation activities should be avoided. Therefore, a pI in the range of 7–9 is appropriate for antibody-based therapeutics.69
Hydrophobicity and self-interaction
Hydrophobic interaction chromatography (HIC) is a common approach to evaluate the hydrophobicity and heterogeneity of a lead molecule. Highly hydrophobic molecules may have an increased propensity to aggregate,70 nonspecifically interact with other surfaces or molecules,71 or self-interact.72 In particular, hydrophobic patches in the variable domain CDRs of an antibody can manifest in negative developability outcomes. As part of additional characterization, the self-interaction of a molecule, or colloidal stability, should be assessed. Self-interaction can lead to high viscosities, particularly for bivalent antibodies, that impair process steps such as pumping, filling, and filtering as well as injectability, particularly for subcutaneous injectables.73 Typical approaches to evaluate self-interaction at small or medium scale (1–2 mg) involve the use of dynamic light scattering (DLS) to measure hydrodynamic radius (Rh) and the diffusion interaction parameter (kDiff).74 Like DLS, static light scattering (SLS) approaches may be used to obtain the second virial coefficient (B22), another indicator of self-interaction propensity.75 The measurement of zeta potential, or total surface charge, also is an option to characterize potential self-interaction.40 At very small microgram (µg) scales, high-throughput self-interaction assessments have been successful using affinity capture self-interaction nanoparticle spectroscopy (AC-SINS), an antibody-conjugated gold nanoparticle-based assay that reveals self-interaction from UV absorption shifts.76,77 Other nanoparticle-based assays have also been reported to predict viscosity.78 With larger material availability, (dynamic) viscosity can be directly assessed at higher protein concentrations (100 mg/ml or higher) to determine if self-interaction is significant.73
Integrity and targeted forced degradation
How the molecule will maintain integrity during process and storage can be assessed during early screening, which typically involve small-scale efforts with limited amounts of material, by conducting a short, accelerated stability hold at low concentration (1–2 mg/ml), such as holding the sample at 40–50°C for one week in a representative formulation.51 Analytical SEC and cSDS should be used to evaluate basic integrity characteristics such as aggregation and clipping. Further, if in silico predictions show primary chemical liabilities that may be potentially hot, they can be evaluated in a targeted fashion by forced degradation prior to a formal stability and forced degradation assessment. For example, if primary sequence evaluation of the CDRs revealed a N-deamidation motif, such as NS, the molecule could be subjected to high pH stress prior to a more comprehensive assessment that would require more time, material, and a separate scale-up expression. Various other chemical liabilities can also be informed by an in silico sequence or homology model evaluation, and many are listed in Table 2.
Table 2.
Common developability liability risks, conditions for their evaluation and likely occurrence, and mitigation strategies.
| Type | Common sequence motifs/characteristics | Forced degradation/conditions | Common occurrence | Routes of mitigation |
|---|---|---|---|---|
| Deamidation (Asn) | NG, NN, NS, NT | Neutral/high pH, phosphate buffer | Cell culture conditions, physiological pH/temp, higher pH formulations | Corrective engineering, partial mitigation with low formulation pH |
| Isomerization (Asp)79 | DG, DS | Low pH | Low pH conditions, formulations | Adjust formulation pH, corrective engineering |
| Glycation (non-enzymatic)80 | K | Reducing sugars at higher temperatures (glucose, fructose) | Cell culture conditions, physiological pH/temp | Corrective engineering |
| Trp/Met oxidation | M, W | Light, chemical oxidizers (H2O2, tBHP, AAPH) | Light, metals, polysorbate degradation, storage, thermal stress | Corrective engineering, formulate with antioxidants, amber vials, eliminate stainless steel in process |
| N-glycosylation | NXS/T motif, non-consensus | None | Cell culture conditions | Corrective engineering, purification removal |
| Self-interaction | Structural, charge/hydrophobic patches | Concentration dependent | Higher concentrations | Viscosity modifiers, optimize pH |
| Aggregation | Structural, aggregation-prone motifs | Thermal, storage, low/high pH | Cell Culture, process (low pH or pH adjustment), storage, administration | Optimize pH and formulation, stabilizers, removal by purification |
| Clipping | Asp-Pro motif, non-consensus, enzymatic | Low/high pH, thermal | Cell Culture, process (low pH), HCP contamination, storage | Corrective engineering, removal by purification, removal of proteolytic HCPs, formulation pH |
| low/high pI | Excess Glu/Asp or Lys/Arg | N/A | Inherent molecular property (outside 7–9 range) | Corrective pI variants, isotype switching |
Particles
Visible and sub-visible (protein) particle analyses are also critical endeavors in CMC development, and, like aggregation, is a biologics critical quality attribute (CQA) and may lead to unwanted immunogenicity.18,81 Therefore, evaluation of particle formation prior to lead selection on unstressed and stressed samples is valuable. Typical approaches to characterize and quantify sub-visible protein particulates use light obscuration, flow imaging and more recently membrane-based technologies.82,83 However, particle formation is highly process and formulation dependent, and so its evaluation prior to defined, scaled-up downstream process, fill-finish, and formulation activities has limitations.84 Specifically, shear, pumping, mixing, and shaking stresses encountered during manufacture are difficult to mimic prior to downstream process development. Furthermore, the addition of polysorbate (PS)-20 or PS-80 is mainly assessed during early CMC activities and is used to prevent aggregation and particle formation incurred from process- and shipping-related mechanical and agitation stresses.85 In essence, particle analysis may be useful in differentiating lead candidates prior to lead selection and CMC entry, but not in assessing absolute particle counts when final process and formulation variables are unclear. If particle analysis is not obtainable, a simple absorbance-based turbidity assay can serve as a less sensitive surrogate to monitor particle formation.86 Furthermore, earlier evaluation of soluble aggregation rates on thermally stressed samples by SE-UPLC or unfolding propensity by DSC in various formulations may correlate to particle formation propensity for certain therapeutic antibodies.34
Polyspecificity or non-specificity
Lastly, evaluation of stickiness or the propensity of a molecule to interact with process components, formulation excipients, or other biomolecules in vivo, which may affect PK and bioavailability, is of high value. How a molecule may nonspecifically bind to other molecules can be evaluated by polyspecificity reagent (PSR) and baculovirus particle (BvP) assays and cross-interaction chromatography (CIC).19 Notably, favorable PSR and BvP properties on clinical-stage antibodies have correlated with clinical stage progression.19 Along these lines, it is also appropriate to perform a short-term integrity or biomatrix ex vivo stability study on the lead molecule in human serum prior to toxicology and Phase 1 studies.87
Additional characterization
Additional characterization of unstressed leads may also be warranted and of value, such as measuring concentratability (to 150 mg/ml and above), viscosity in various formulation pHs and concentrations, polyethylene glycol (PEG) solubility in various formulation pH conditions, and DLS interaction parameters in various formulation conditions.34,88 This is especially critical if the clinical or commercial target product profile (TPP) warrants a high concentration formulation to support high dosing or subcutaneous injection by pre-filled syringe,89 since these additional activities aim to characterize highly concentration-dependent antibody or protein behavior. Additionally, free thiol content can be measured by reverse phase chromatography, fluorescent-based quantification or MS, particularly for certain multispecifics such as Duobodies, to determine if disulfides are properly formed during assembly or to investigate stability issues that may arise from broken disulfides.90,91
Final lead selection and CMC entry
To confirm a final lead candidate for investigational new drug (IND)-enabling studies, the molecule should be amenable to platform purification and formulation approaches and have acceptable stability and forced degradation attributes. This is highlighted in Figure 2 as “Tier 3” activities. Proper assessment and mitigation of significant developability issues and liabilities helps to ensure control of the process, product quality, clinical supply, and lot comparability throughout years of clinical development. TPP needs and requirements should also be considered to accommodate special circumstances, such as high concentration formulations or alternative drug product presentations.
Purification process
Lead molecules should express acceptably well and ideally be amenable to a platform-based purification strategy.92 While a final lead may be selected based on transiently expressed material, material from stable cell-lines may express at higher purity and yield and have more representative core glycosylation profiles.93 A final lead should have acceptable yield and purity following all platform purification steps, such as ProA affinity, low pH inactivation, anion-exchange (AEX) flow through, and cation-exchange (CEX) bind and elute processes.94 Mixed-modal ion exchange resins are also often used during polishing. If issues arise requiring process improvements such as low yield and/or purity, this can affect lead selection or necessitate reengineering of the molecule. If process mitigation strategies are pursued, ideally the mitigation is straightforward and amenable to manufacture. An example would be the simple addition of a stabilizer to a process step, such as pH adjustment, to prevent aggregation or particle formation.95
Stability and forced degradation
A short-term accelerated stability study, usually performed at 40°C up to one month at higher protein concentrations in solution, and a forced degradation assessment should be performed to determine if a molecule has acceptable developability and CMC risk.96 The (accelerated) stability assessment is performed to determine if the molecule has major integrity or heterogeneity issues, to assess protein concentration-dependent behavior, especially aggregation, to forecast long-term stability in a liquid formulation, and to determine if significant formulation optimization or mitigation will be needed.48 Because degradation mechanisms may follow Arrhenius kinetics, this accelerated study may also forecast long-term normal storage stability and shelf-life.97 However, temperature-dependent phenomena, such as aggregation, may be complex and not follow Arrhenius kinetics,98 yet accelerating this process at higher temperatures is necessary to save time in the discovery stage. Ideally, a platform formulation is assessed on stability that accommodates toxicology study and Phase 1 dose requirements, can be easily translated downstream and uses common “generally regarded as safe” (GRAS) excipients such as histidine and sucrose.63 Sucrose or a similar sugar should be used to further assist in stabilization, particularly for cryoprotection and to achieve isotonicity.99 Another advantage of using sucrose is its amenability to lyophilization if developability efforts fail to adequately stabilize a molecule in the liquid state.100 The analytical methods used to assess the integrity and heterogeneity of these stressed samples should be representative of standard CMC release and stability assays, such as SE-HPLC, cSDS, and cIEF.101 Moreover, peptide mapping MS and binding assessments should be conducted to determine if site-specific chemical modifications occur, where they occur, and their potential effect on binding and activity.101 Therefore, prior to a more thorough formulation assessment that includes optimization of pH and excipients in a representative container closure, usually being glass vials or pre-filled syringe (in the case of direct subcutaneous injection), there is a good understanding of potential issues and how process and formulation efforts can mitigate them. For example, at higher antibody concentrations self-association can result in high viscosity, leading to poor process, stability and/or injectability outcomes.73 If self-interaction issues exist, they may be mitigated through formulation efforts by lowering the pH,102 or using viscosity modifiers such as arginine,103 NaCl,104 glycine,63 and more recently, caffeine.105
Common forced degradation conditions used to evaluate chemical stability are low and high pH, physiological pH, and chemical and/or light stress.106 Evaluating a few formulation conditions prior to lead selection allows the developability scientist to better understand formulation and pH dependent behavior, which can also be leveraged from forced degradation conditions if the various pHs are evaluated at an accelerated, fixed temperature. For example, low pH, representative formulation pH, physiological pH, and high pH can all be evaluated at 40°C for one week. Therefore, if a chemical liability, such as clipping, deamidation or isomerization, is observed, its pH-dependent behavior can be understood from the study results, further suggesting how the liability could affect process, formulation efforts, and post-administration behavior. In addition, most chemical liabilities are pseudo-first order with respect to pH or reactant concentration, and therefore their propensities can be evaluated at lower protein concentrations.107,108 Thus, these forced degradation assessments reveal clues about how to mitigate an issue through formulation pH or whether engineering and/or alternative molecule selection is required. An example of this approach is the evaluation of deamidation mechanisms and rates both experimentally and computationally versus formulation pH.109
Corrective engineering
Although not ideal, a full developability assessment on a final lead, when extensive discovery time and resources have already been invested, may reveal corrective engineering is necessary if significant liabilities such as excessively high or low pI, aggregation, deamidation or self-interaction present concerning risk. In determining the severity of the chemical or physical liability, one must consider its nature, location, and rate of degradation. Further, its dependence on pH, temperature, and antibody concentration are important, and its effect on target binding and other attributes such as immunogenicity or poor PK are critical.38 While the nature and number of liabilities factor heavily into the overall developability risk, the number and nature of potential routes of mitigation are also important. One common example is aggregation, which may be corrected through engineering, but is often improved by purification removal, modulating the formulation pH, or using stabilizers such as arginine or sucrose.110 A summary of major liabilities, how they commonly occur and potential mitigation strategies are summarized in Table 2.
One common liability that may require molecular engineering is Asn deamidation since it is typically pseudo-first order base-catalyzed and is accelerated at higher pH, including physiological pH.108,111 Common hot spot motifs are elucidated by primary N, N + 1 sequence signatures such as NG, NS, NT, and NN.111 The presence of these primary sequence motifs enables the possibility of deamidation, and the potential for deamidation is further enhanced by location in surface-exposed regions, such as CDRs, and main-chain flexibility and conformation.112 Because deamidation is accelerated at higher pH and higher temperatures, it is normally not a storage issue in low pH, liquid formulations at lower temperatures.12,79 However, if deamidation occurs, it is likely unavoidable in physiological cell culture conditions and post-administration at physiological pH and temperatures, where it will persist for weeks at high temperature.107 Furthermore, deamidation is quite prevalent in proteins and is one of the more robust chemical degradation mechanisms.79,112 Therefore, hot spot deamidation sites should be corrected if present in variable regions, such as the heavy chain (HC)-CDR3, which in turn can affect heterogeneity, activity and function. There are instances where therapeutic molecules have been opted for correction when rates of 5–10% deamidation per week at physiological pH and temperature were observed.34 Lower levels can be monitored moving into development, but likely can proceed without correction since overall levels are less likely to reach a significant percentage. If deamidation affects binding or activity, a key critical attribute, the need for correction increases as this also increases the criticality. The Asn deamidation site can also be mutated to Asp, one of the deamidation products, and evaluated for target binding to determine its effect.113 Moreover, one can compare mAb variable site deamidation rates determined by peptide MS to conserved Fc deamidation sites (i.e., NGQPENNYK) to determine the accuracy and relative severity of the deamidation hot spots.114
Like deamidation, another common liability is methionine/tryptophan (Met/Trp) oxidation, which can be efficiently assessed by forced oxidation studies.115 Light oxidation can oxidize both sensitive Met and Trp residues, whereas commonly used chemical oxidants tert-butyl hydrogen peroxide and azobis (2-amidinopropane) dihydrochloride tend to be specific to Met and Trp residues, respectively.106,116 If oxidation is shown to be quite extensive, corrective engineering may be attempted, but usually exposed CDR Trp residues are more difficult to correct and maintain bioactivity.117 Unlike deamidation, oxidation risk can be mitigated more easily via formulation efforts using antioxidants such as ethylenediaminetetraacetic acid, which chelate metal oxidizing agents leeched from process or drug container components, and methionine as a competing peroxide scavenger.118 Additionally, amber or UV-protecting glass vials can be used to further protect the liquid drug product from light oxidation.119 While Met/Trp oxidation is a CQA that warrants proper mitigation and correction attempts should be made as necessary, there are successful commercial products on the market with reactive CDR Met oxidation without antioxidants present in the approved formulation.120
Early CMC development
Formulation development
After a nominated lead is well understood in terms of molecular properties and potentially significant liabilities, a more complete formulation development assessment should be performed. This is typically at the discovery and early CMC interface prior to IND application submission and manufacture and fill-finish efforts. Formulation development activities include optimizing formulation pH, polysorbate/surfactant type and concentration, and evaluating different stresses such as agitation and freeze/thaw in a relevant product presentation.107 In addition, previous developability work, particularly stability and forced degradation assessments, can be confirmed. When considering formulation optimization approaches, previous forced degradation assessments that elucidate pH-dependent behavior can also help determine if changing the formulation to reduce one liability may potentiate an additional unwanted liability, such as aggregation or clipping. For example, lowering the formulation pH to increase colloidal stability and reduce self-association may also promote aggregation, clipping, or Asp (D) isomerization.121 Similarly, raising the pH may promote additional liabilities such as N-deamidation. In Figure 3, this concept is diagrammed for various potential liabilities versus pH. Important antibody properties such as conformational and colloidal stability, along with liabilities such as aggregation, clipping, and deamidation are shown as increasing propensity or risk versus solution pH. However, it should be noted that these phenomena are still antibody-dependent, where certain liabilities may not be significant or exist at all, particularly in the variable domains. Therefore, a final formulation should aim to balance stabilization of the various molecular properties and liabilities for the molecule in question.
Figure 3.
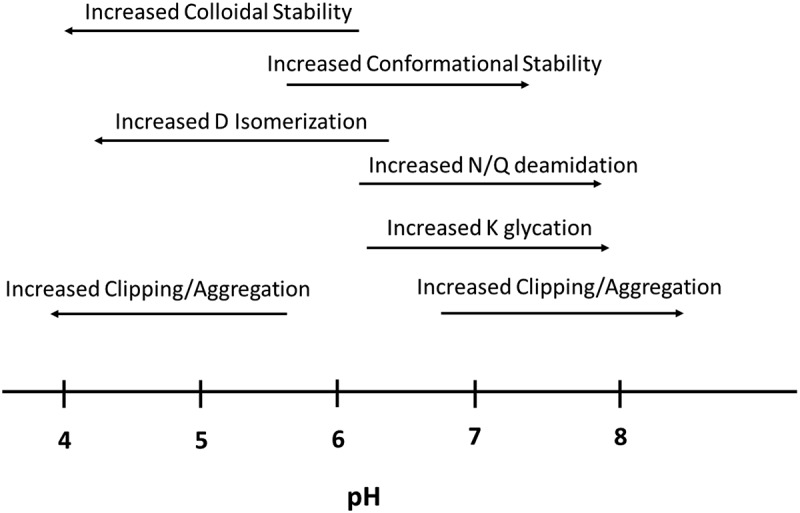
Diagram depicting potential liabilities and their propensity to occur versus solution pH. Arrows depict the direction of increasing propensity for each attribute.
If developability risks are too great or potentially unmanageable even in an optimized liquid formulation, lyophilization can be used to stabilize the molecule in the drug product form,122 but additional processing and steps during administration/reconstitution are then required.123 If long-term storage stability is a concern, a frozen liquid presentation in vials can be considered in early development, pending additional formulation improvement or optimization by Phase 3 process and formulation lock.124 However, additional freeze-thaw cycles can result in unwanted complications such as molecular denaturation and container closure integrity issues.99
Overall, a refrigerated liquid formulation is the most straightforward and versatile presentation in clinical development. A number of factors support this: 1) only one presentation requires evaluation, 2) an additional lyophilization or freezing process is not required, 3) reconstitution or thawing during administration is not required, and 4) a liquid form is easily amenable to vial and pre-filled syringe container closures. However, the therapeutic antibody may be susceptible to in-process, in-storage, and in vivo degradation in the liquid state. Therefore, prior to lead selection, it is paramount to understand the physicochemical behavior of the therapeutic antibody in solution and under different conditions that mimic process, formulation, and physiological-like conditions.
Analytical development
Analytical method development and the qualification of CMC release and stability assays to evaluate CQAs is another key area of deliverables after a final lead is selected. Often, analytical methods used in Discovery can be leveraged into early CMC evaluation and qualification since they are typically platform driven.125 Examples include SE-HPLC and cSDS, where some modifications or optimization may be needed, but common separation columns, mobile phases, sample prep procedures, and capillaries can be leveraged. Later in CMC development, more is understood in terms of impurity identification and method validation.126 However, as a lead is selected prior to CMC Development, assumptions are made regarding how analytical methods can detect and quantify impurities and/or heterogeneity. Fortunately, the analytical methods used to evaluate CQAs can sufficiently detect and quantify aggregation, clipping, pI and charge heterogeneity across various therapeutic antibodies, often without extensive optimization or customization.125 Furthermore, a thorough Discovery-stage developability assessment should evaluate other molecular properties such as Tm, hydrophobicity, and self-interaction, so that a therapeutic’s molecular properties are well understood and therefore downstream development risks are more clear.
Process development
Another key deliverable for large-molecule biologics is stable cell line development (CLD). This can follow final lead selection from thoroughly characterized and transiently expressed candidates, where stable CLD aims to optimize titer expression while maintaining desirable purity attributes and not introducing new liabilities. In doing so, stable cell lines are developed and selected single clones, typically derived from CHO-K1 or CHO-S, are optimized for cell growth and viability to result in antibody product of high quality and titer.127 CLD activities can begin earlier on multiple lead candidates at risk to shorten timelines during the final stage of lead selection, particularly up to the stage of cell-line generation of stable pools, and advances in specific gene transposase targeting may further accelerate stable clone selection.128 Later in the CLD stable clone selection process, efforts should localize on a final lead candidate. This approach of evaluating multiple lead candidates for stable CLD production has particular advantages for multispecific antibodies, where subunit stoichiometry and correct assembly can be optimized beyond transient expression efforts.128 While transient expression of multispecific variants followed by purity characterization may select for variants with improved assembly and reduced formation of common impurities such as homodimer or half-molecule formation, stable CLD may improve upon this or be entirely relied upon to assemble the desired product at reasonable titers and purity to facilitate downstream processing efforts. For instance, Wang and coworkers demonstrated improved bispecific expression with reduced homodimer impurities through stable cell-line generation and selection of optimal clones.129 Such high-quality expression reduces the burden and risk to downstream purification and fill/finish activities to produce a product of acceptable yield and purity. Moreover, core Fc N-glycosylation heterogeneity is typically defined by cell-line expression parameters and conditions.130 Stable CLD can confirm if this is controlled and of desirable heterogeneity, but also determine if molecular variants affect this heterogeneity if CLD is applied to them prior to final lead selection, especially for Fc-engineered variants.131
While lead candidates surveyed for basic molecular properties usually only need affinity purification, final leads that are evaluated by definitive storage stability and forced degradation studies should be purified by a complete purification process analogous to manufacture.132 As aforementioned, this would include ProA affinity, low pH hold, AEX flow through and CEX bind and elute processes.94 These purification steps should purify the molecule of interest to high purity and acceptable heterogeneity. The low pH hold step, typically performed at pH 3.5–4.0, primarily inactivates endogenous and adventitious viruses, but also may cause antibody clipping or aggregation.47,133 Alternatively, viral inactivation can be performed using detergents if the molecule is unstable at low pH.134 The AEX flowthrough step primarily removes negatively charged molecules such as HCPs, DNA, and RNA that basic purity characterization assays (such as SE-UPLC and cSDS) are not sensitive to.135 The CEX step aims to fully polish the molecule of interest and further remove aggregates, misassembled or fragmented molecules, or undesirable heterogeneity including excess glycosylation or charge heterogeneity.135 The selection of CEX resin for purification of a particular antibody is usually empirical, but a default or platform approach is advisable until further optimization is needed.136 To formulate drug substance, ultrafiltration/diafiltration (UF/DF) is a key activity, where the molecule of interest is buffer-exchanged and concentrated to a desired formulation, usually prior to polysorbate addition.137 During developability and early development efforts, product quality attributes focus on bulk purity characteristics defined by assays such as SE-HPLC/UPLC, cSDS, cIEF, and particle analyses, particularly when deciding on a lead molecule or formulation. However, to supply material for animal studies, good laboratory practice toxicology studies and subsequent clinical studies, which require drug produced under good manufacturing practice conditions, other in-process and release analyses to evaluate potential contaminants are critical, such as CHO DNA levels, bioburden and endotoxin.138
The four developability properties
The four major properties that affect all developability outcomes are conformational, colloidal, chemical, and other interactions.12,20,22,139 Many attributes, such as aggregation, are clearly understood to be primarily driven by unfolding and are therefore categorized in the “conformational” category. Other developability outcomes are not as obvious or trivial to categorize. Other issues, such as problematic mechanical issues in manufacture or excipient quality issues, can affect development outcomes, but are herein not considered developability issues per se if they are independent of the therapeutic molecule. Problematic interactions between the therapeutic molecule and excipients or drug product containers are molecular developability issues, as they relate to the inherent properties of a molecule. For example, a molecule may have process failures during filtering or UF/DF operations due to high viscosity caused by self-association. In Table 3, we have summarized many common developability and/or CQAs that affect lead selection and CMC development, as well as potentially safety and efficacy, and categorized each attribute into one or more of the four major categories. Examples of key criteria to ensure successful developability outcomes are also included. It should be noted that while each category can be independently described, they are not wholly independent, as one property may affect another. For instance, chemical deamidation and/or oxidation may arise in conformational instability or aggregation, and vice versa.140
Table 3.
The four major developability properties of large-molecule antibody therapeutics tabulated along with the attributes they impact and examples of key criteria to be considered.
| Property | Relevant Attributes | Examples of Key Developability Criteria |
|---|---|---|
| Conformational and physical stability (Unfolding/refolding) | Initial aggregation (during expression), Aggregation propensity, particle formation (insoluble aggregates), Tm/Tm onset/Tagg | Low initial (or release) aggregation and good accelerated storage stability, Tm onset>50C, low particle levels34 |
| Chemical characteristics (liabilities, properties) | All chemical liabilities (N-deamidation, D-isomerization, M/W-oxidation, N-glycosylation, etc.), clipping or fragmentation, pI, hydrophobicity, surface characteristics, etc. | Desire low or acceptable initial levels, acceptable increases on forced degradation/accelerated stability, behavior at physiological pH/Temp |
| Colloidal stability (Self-association) | Viscosity and self-interaction parameters (kDiff, DLS, AC-SINS, second virial, zeta potential), processability (filtration, pumping), injectability, solubility | Ideally 5–10 cp at 100 mg/ml, no more than 15cP for final target formulation; kDiff (DLS) −15 mL/g or higher is preferred)34 |
| Other interactions | Process, formulation, and container closure interactions; immunogenicity, polyspecificity, biomatrix stability, chaperone assembly and ER secretion (expression), incomplete leader cleavage | No significant expression or process-related impurities, well behaved drug product, acceptable hydrophobicity, favorable polyspecificity, PK, bioavailability |
Conformational stability
Conformational stability attributes can influence aggregation rates, temperature-dependent unfolding by DSC (or Tm), chemical denaturation propensity, and insoluble particle formation.141 CMC stability and release analytical assays such as SEC and sub-visible particle analyses clearly evaluate soluble and insoluble aggregation, respectively, which are mainly driven by protein unfolding, although they can also be influenced by other factors such as native surface electrostatics and hydrophobicity.142–144 Aggregation usually results from a mixture of covalent and non-covalent associations between partially unfolded molecules and is pH, temperature, and concentration dependent.145 If problematic, the nature of aggregation can be further explored by circular dichroism or Fourier transform infrared spectroscopy to evaluate changes in secondary and tertiary structure upon denaturation or unfolding.146 Further, reversibility of unfolding can be analyzed by DSC to gauge how reversible aggregation may be in formulation.147 Conformational stability can also affect many other outcomes aside from aggregation and temperature-dependent unfolding. For example, poor expression or purification yield could be due to incomplete folding upon expression or unfolding in various pH or ionic strength environments encountered during purification.42,133,148,149 The presence of highly conformational states can lead to increased heterogeneity determined by various methods such as HIC.150 Soluble aggregates and insoluble particles formed by partially unfolded antibodies present not only CQA issues, but also immunogenicity risks.151 Post-administration, a poorly folded or conformationally diverse molecule can lead to increased immunogenicity and/or poor PK/bioavailability, as a combinatorial array of surfaces displayed in the unfolded/aggregated state can trigger immune responses or simply be cleared quickly.141
Chemical stability/characteristics
Chemical stability is affected by post-translational modifications (PTMs). Asn deamidation and Met/Trp oxidation are two of the most common PTM that affect quality and functional attributes, as discussed above. Typically, location, solvent accessibility and local main chain conformation and/or flexibility govern inherent molecular reactivity.152 These modifications and others are assessed site-specifically by MS, but other methods such as reverse phase (RP)-HPLC or HIC may be useful to monitor abnormal changes in oxidation and likewise, cIEF to monitor deamidation.106,153 Many other modifications exist, such as pyro-glutamate formation, non-enzymatic lysine glycation, Asp isomerization, main-chain clipping or fragmentation, and disulfide reduction and scrambling.106 Other modifications, such as Asn glycosylation, are enzymatic and driven both by expression conditions and the inherent molecular sequence and structural properties.154
CQAs include many chemical modifications, notably Asn deamidation, Met/Trp oxidation, and Asp isomerization, that are essential to monitor and control for successful CMC development because they also can affect activity, safety and efficacy.155 In one recent study,156 Asp isomerization of a mAb was correlated with increased immunogenicity. Further, antibody Met oxidation has been shown to negatively impact half-life in vivo by affecting binding to FcRn receptors.157 Therapeutic antibody N-deamidation in the CDR region has been shown to affect target binding orders of magnitude113 and conserved Fc deamidation (at N325) has been shown to affect antibody-dependent cell-mediated cytotoxicity by reducing FcγRIIIa binding.3 This category also includes attributes defined by surface characteristics, such as pI and hydrophobicity, although both properties can lead to other undesirable outcomes, notably aggregation, self-interaction, or nonspecific interactions.22,34,139
Colloidal stability
Colloidal stability refers to a molecule’s ability to non-covalently self-associate and form “colloids”, or larger complexes, that affect bulk solution rheology and viscosity.158 Here, we distinguish self-interaction from aggregation, as the latter is composed of a complex network of (partially or fully) unfolded molecules.43 Like self-interaction, aggregation can be reversible, but it is predominantly irreversible, particularly in higher order aggregates.50 Therefore, aggregates are long-lived and can be evaluated by chromatographic techniques such as size-exclusion chromatography, analytical ultracentrifugation or even field-flow fractionation.159,160
In the case of self-interaction or self-association, folded, native molecules bind reversibly in a highly transient and concentration-dependent fashion that cannot be directly examined by chromatographic techniques using analytical sample amounts at dilute concentrations.161 Particularly in bivalent antibodies, self-association can lead to large oligomeric complexes at higher formulation concentrations.72,74,162 Because bivalency enhances antibody oligomerization that leads to higher viscosity, it should be noted that self-interaction risks can change when formatting to multispecific antibodies, where Fab valency is likely to change.117 Moreover, antibody self-interaction leading to decreased stability and solubility has been reported,163 and even phase separation and gelation164,165 can occur as a result. In one report, removal of a hydrophobic surface spanning both the HC-CDR1 and HC-CDR2 corrected self-interaction in vitro and improved serum half-life and nonspecific tissue binding, implying that a high propensity to self-interact may also correlate with nonspecific interactions with other molecules via the same interface.166 Since self-interaction is typically mediated by CDR residues in at least one of the interacting molecules, which also mediate target binding, self-interaction may be difficult to correct via engineering or mutagenesis while maintaining desirable activity.117 Additionally, it may be difficult to predict structurally how two molecules will self-interact or self-bind. Recently, via both crystallographic and modeling efforts, antibodies have been shown to self-interact in a symmetrical “face-to-face” fashion mediated by the CDR apparatus.117,167 Other reports have demonstrated Fab-Fc self-interactions, which can be mitigated by IgG1/IgG4 isotype switching.168 Notably, formulation excipients and modulation of formulation pH have been shown to reduce viscosity by reducing protein-protein interactions in several instances.74,169 Heavy-chain dimer formation, which is driven by the unique self-interaction of the heavy chains during expression, has also been recently reported as a developability issue.51
Other interactions
Other interactions include those involving the therapeutic molecule that can affect the process, drug product or in vivo behavior. Examples include interactions between the therapeutic molecule and process components, formulation excipients, the drug product container, and interactions with expression HCPs and biomolecules in vivo. 170 The therapeutic molecule of interest may unfavorably interact with or adsorb to process components such as process pumps71,171 and filters,172 or help promote leaching of metals such as iron, chromium, copper, and nickel from stainless steel, which may lead to product oxidation and instability.173 Other common interactions may be with analytical column resins, such as size-exclusion HPLC, which may complicate direct analysis of CQAs.174 The therapeutic molecule may also interact unfavorably with formulation excipients, most notably polysorbates, which can lead to instability, such as the formation of aggregates or oxidative degradation.175 Interestingly, enzymatic HCPs, presumably surviving purification removal through interactions with the therapeutic antibody, have been reported to degrade polysorbate, which in turn led to antibody stability and particle issues.176 If not removed during purification, enzymatic or proteolytic HCPs also can directly degrade the antibody of interest, in addition to presenting a general immunogenicity risk.177 Other contamination risks such as lipopolysaccharide endotoxins may survive removal through interactions with the antibody of interest.178 Antibody interactions with the drug product container, specifically with vial rubber stoppers or syringe stopper silicone coatings, can lead to particle formation, often in combination with foreign particles or silicone oil droplets, although formulation is a major driver for these phenomena.179,180 Antibody interactions or adsorption can occur during product administration with various ancillaries such as IV bags or pumping devices.181
Following administration, the target molecule may interact with various biomolecules specifically or nonspecifically, potentially leading to poor PK, bioavailability and efficacy.26,139 One example includes removing charge “patchiness” on an IgG1 and IgG4 antibody without affecting pI, resulting in improved half-life and distribution in mice, which correlated to reduced nonspecific binding in vitro (to CHO cells and heparin).182 Other examples exist where highly specific interactions with other biomolecules led to negative clinical outcomes, such as the strong binding interaction between an anti-programmed cell death protein 1 (PD-1 camrelizumab with vascular endothelial growth factor receptor 2 (VEGFR2) and two other molecules, leading to potent VEGFR2 agonism and hemangioma.26 It should be noted that other anti-PD-1 therapies, such as nivolumab and pembrolizumab, do not result in VEGFR2 agonism, and therefore this side effect was unique to the nonspecific (agonistic) effects of camrelizumab.26
While many of these unwanted interactions are empirically determined and nearly impossible to completely forecast in the discovery stage, various analytical methods have been recently developed that may help predict their likelihood of occurrence. The baculovirus particle assay (BvP) assesses nonspecific binding to a pool of various proteins by ELISA or flow cytometry and has been correlated to clearance rates in humans.183 The PSR assay is a similar approach using CHO membrane extracts to evaluate nonspecific binding to a molecule of interest and has been correlated to other assays used to predict non-specificity.184 Heparin chromatography has been used to correlate in vitro retention times to antibody clearance rates in mice and physiological models.185,186 In a study by Jain and coworkers,19 developability flags associated with these predictive non-specificity assays decrease for therapeutic antibodies further along in development. The correlation to advanced clinical stages indicates these non-specificity assays may predict in vivo interactions that affect clinical endpoints such as safety and efficacy. This appears to introduce an emerging trend to evaluate complex nonspecific interactions at the pre-IND stage to increase the probability of success in the clinic. Developability has long been known to be especially useful to predict CMC development outcomes and manufacturing success. However, since no literature references cite CMC failures as the direct reason for clinical failures, it was generally thought that clinical outcomes could not be predicted by developability assessments. However, the incorporation of non-specificity into developability assessments and their recent correlation to clinical success suggests otherwise. In hindsight, the correlation of improved polyspecificity as a developability attribute with clinical advancement implies that clinical failures may have previously arisen from substandard developability attributes.
Other more commonly used characterization tools, such as HIC, can assess hydrophobicity, which is commonly associated with nonspecific binding and overall “stickiness”.19 Other methods reported to assess potential nonspecific interactions are CIC and standup monolayer adsorption chromatography.187,188 If assays recently shown to be predictive of nonspecific interactions have not been implemented into a developability workflow, standard process and analytical approaches may offer clues. For example, the therapeutic molecule may bind nonspecifically to purification and analytical resins or possess high surface hydrophobicity revealed computationally or experimentally by HIC.34 This information can then be considered when deciding a lead.
Advancement of in silico computational tools
Predictive computation
In recent years, the use of in silico tools is increasingly more common to predict antibody developability and quality attributes.189 Often, these are based on sequences and/or structure-derived parameters to screen and/or funnel initial hits or candidates into more promising leads. Correlations to developability attributes such as aggregation,190–192 viscosity,65 chemical instability,193 and PK clearance194 have been made using antibody homology models and the assessment of calculated surface properties. In essence, these approaches can be used to computationally funnel down hundreds of candidates to relatively smaller subsets prior to expression, purification and characterization. To computationally predict concentration-dependent viscosity, net variable domain or total antibody charge along with surface hydrophobicity indices are commonly evaluated.195 Interestingly, Tomar and coworkers predicted total antibody viscosity curves for 16 mAbs from hydrophobic and electrostatic parameters obtained from full-length homology models.196 They found the greatest statistical significance with respect to total variable heavy (VH) and variable light (VL) charge, hydrophobic surface area, zeta potential and surprisingly, aggregation propensity by WALTZ. In a study of 60 IgG1 mAbs, molecular dynamics simulations of antibody homology models correlated viscosity with hydrophobicity, net charge, and charge distribution.197 Further, fast in vivo clearance was associated with highly charged and/or hydrophobic CDRs. In this same study, molecular dynamic simulation outputs such as solvent accessibility of potential reactive residues also correlated to experimental tryptophan oxidation and aspartic acid isomerization rates. Notably, a study of over 100 clinical stage antibody therapeutics demonstrated that average HC-CDR3 lengths were lower compared to thousands of preclinical antibody sequences evaluated, suggesting that shorter lengths are selected moving from preclinical stages to the clinic because they lead to improved developability outcomes.198 In the same study, similar findings were reported for hydrophobic patchiness metrics with respect to CDR regions. Many web-based tools using structure-based calculations of surface behavior to predict protein stability or developability profiles are available, such as Therapeutic Antibody Profiler (TAP),198 SAP (Spatial Aggregation Propensity) for aggregation,70,191 Abpred (The Antibody Prediction Database) for hydrophobicity,199 and SAbPred (The Structural Antibody Prediction Database), which includes tools for modeling, annotation, humanization and developability prediction.200 To date, there are a plethora of examples where in silico models have successfully predicted experimental outcomes for aggregation, viscosity, chemical stability, and in vivo behavior of large sets of antibodies.
Machine learning and artificial intelligence
Expanding upon this is the use of machine learning (ML) and artificial intelligence (AI) to predict developability properties, such as viscosity and aggregation propensity.201,202 Sequences, structures and molecular dynamics have been used to train machine learning algorithms, which then attempt to predict molecular properties.203 Recent examples include the use of ML to predict affinity and specificity,204 deamidation,205 methionine oxidation,206 hydrophobicity,19 and viscosity.207 Recently, AI has been explored to reveal optimal antibody and protein formulations by predicting protein-solvent interactions.208 In terms of protein structure prediction, AlphaFold and AlphaFold2 have been recently launched commercially to predict protein structures and even their complexes.209,210 ABlooper (Antibody “Looper”) was developed as a deep learning tool to readily predict CDR structures from antibody sequences.211 While their use may not be widespread in the pharmaceutical industry, the emergence of AI/ML may become routine as part of initial in silico efforts to screen and assess molecular properties and interactions prior to any experimental efforts.
Conclusions
Developability of antibody and protein therapeutics is a critical activity to drive and advance the optimization and selection of lead candidates into IND enabling and clinical studies. Proper and thorough developability studies not only examine potential CQAs that will represent the fitness of the process and drug product, but also elucidate the molecular properties that may affect post-administration safety and efficacy and ultimately clinical success. Recent advances in in silico computational approaches and the evaluation of non-specificity have also helped to streamline and enable more informative developability assessments. We propose these four major properties of large-molecule antibody and protein therapeutics as those that affect all developability outcomes: 1) conformational, 2) colloidal, 3) chemical, and 4) other interactions. A complete examination of these properties and their risks during discovery-stage developability and early development activities offers the best likelihood that robust and quality lead candidates are promoted and potential CMC and clinical development risks are manageable.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Abbreviations
ABlooper – Antibody Looper; Abpred – The Antibody Prediction Database; AC-SINS – Affinity Capture Self-Interaction Nanoparticle Spectroscopy; ADC – Antibody-Drug Conjugate; AEX – Anion Exchange Chromatography; AI – Artificial Intelligence; B22 – Second Virial Coefficient; BvP – Baculovirus Particle; CDR – Complementarity-Determining Region; CEX – Cation Exchange Chromatography; CHO – Chinese Hamster Ovary; CIC – Cross-Interaction Chromatography; CLD – Cell-Line Development; CMC – Chemistry, Manufacturing and Controls; CQA – Critical Quality Attribute; cSDS – Capillary Sodium Dodecyl Sulfate Electrophoresis; DLS – Dynamic Light Scattering; DSC – Differential Scanning Calorimetry; Fab – Fragment of Antigen Binding; Fc – Fragment Crystallizable; FcγRIIIa – Fcγ Receptor IIIa; FcRn – Fragment Crystallizable Neonatal Receptor; GRAS – Generally Regarded As Safe; HC – Heavy Chain; HCP – Host Cell Protein; HIC – Hydrophobic interaction chromatography; HPLC – High Performance Liquid Chromatography; IgG – Immunoglobulin G; IND – Investigational New Drug; kDiff – Diffusion Interaction Parameter; LC – Light Chain; mAbs – Monoclonal Antibodies; MS – Mass Spectrometry; PD1 - Programmed cell death protein 1; PEG – Polyethylene Glycol; pI – Isoelectric point; PK – Pharmacokinetics; PK/PD – Pharmacokinetic/Pharmacodynamic; ProA – Protein A; PS-20/80 – Polysorbate-20/80; PSR – Polyspecificity Reagent; PTMs – post-translational modifications; Rh – Hydrodynamic Radius; RP – Reverse Phase; SAP – Spatial Aggregation Propensity; ScFv - (Single-Chain Variable Fragment; SEC – Size Exclusion Chromatography; SEC-MALS – Size Exclusion Chromatography with Multi-angle Light Scattering; SPR – Surface Plasmon Resonance; TAP – Therapeutic Antibody Profiler; Tm/Tagg – Temperature of melting and aggregation; TPP – Target Product Profile; UF/DF – Ultrafiltration/Diafiltration; VEGFR2 - Vascular endothelial growth factor receptor 2; VHH - Variable Heavy-chain Antibody Fragment
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Kaplon H, Chenoweth A, Crescioli S, Reichert JM.. Antibodies to watch in 2022. MAbs. 2022;14:2014296. doi: 10.1080/19420862.2021.2014296. PMID: 35030985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullard A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov. 2021;20:491–20. doi: 10.1038/d41573-021-00079-7. PMID: 33953368. [DOI] [PubMed] [Google Scholar]
- 3.Lu X, Machiesky LA, De Mel N, Du Q, Xu W, Washabaugh M, Jiang XR, Wang J. Characterization of IgG1 Fc deamidation at asparagine 325 and its impact on antibody-dependent cell-mediated cytotoxicity and FcgammaRIIIa binding. Sci Rep. 2020;10:383. doi: 10.1038/s41598-019-57184-2. PMID: 31941950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiu ML, Goulet DR, Teplyakov A, Gilliland GL. Antibody structure and function: the basis for engineering therapeutics. Antibodies (Basel). 2019;8 PMID: 31816964. doi: 10.3390/antib8040055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9:63–73. doi: 10.1007/s13238-017-0473-8. PMID: 28986820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang XR, Song A, Bergelson S, Arroll T, Parekh B, May K, Chung S, Strouse R, Mire-Sluis A, Schenerman M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. 2011;10:101–11. doi: 10.1038/nrd3365. PMID: 21283105. [DOI] [PubMed] [Google Scholar]
- 7.Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-Life. Front Immunol. 2019;10:1296. doi: 10.3389/fimmu.2019.01296. PMID: 31231397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fonseca MHG, Furtado GP, Bezerra MRL, Pontes LQ, Fernandes CFC. Boosting half-life and effector functions of therapeutic antibodies by Fc-engineering: an interaction-function review. Int J Biol Macromol. 2018;119:306–11. doi: 10.1016/j.ijbiomac.2018.07.141. PMID: 30041038. [DOI] [PubMed] [Google Scholar]
- 9.Kim SJ, Park Y, Hong HJ. Antibody engineering for the development of therapeutic antibodies. Mol Cells PMID: 16258237. 2005;20:17–29. https://www.ncbi.nlm.nih.gov/pubmed/16258237. [PubMed] [Google Scholar]
- 10.Maynard J, Georgiou G. Antibody engineering. Annu Rev Biomed Eng. 2000;2:339–76. doi: 10.1146/annurev.bioeng.2.1.339. PMID: 11701516. [DOI] [PubMed] [Google Scholar]
- 11.Yang X, Xu W, Dukleska S, Benchaar S, Mengisen S, Antochshuk V, Cheung J, Mann L, Babadjanova Z, Rowand J, et al. Developability studies before initiation of process development: improving manufacturability of monoclonal antibodies. MAbs. 2013;5:787–94. PMID: 23883920. doi: 10.4161/mabs.25269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Wang D, Mason B, Rossomando T, Li N, Liu D, Cheung JK, Xu W, Raghava S, Katiyar A, et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11:239–64. PMID: 30543482. doi: 10.1080/19420862.2018.1553476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garripelli VK, Wu Z, Gupta S. Developability assessment for monoclonal antibody drug candidates: a case study. Pharm Dev Technol. 2021;26:11–20. doi: 10.1080/10837450.2020.1829641. PMID: 32986499. [DOI] [PubMed] [Google Scholar]
- 14.Kaur H. Stability testing in monoclonal antibodies. Crit Rev Biotechnol. 2021;41:692–714. doi: 10.1080/07388551.2021.1874281. PMID: 33596751. [DOI] [PubMed] [Google Scholar]
- 15.Wang SS, Yan YS, Ho K. US FDA-approved therapeutic antibodies with high-concentration formulation: summaries and perspectives. Antib Ther. 2021;4:262–72. doi: 10.1093/abt/tbab027. PMID: 34909579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kannan A, Shieh IC, Hristov P, Fuller GG. In-Use Interfacial Stability of Monoclonal Antibody Formulations Diluted in Saline i.V. Bags J Pharm Sci. 2021;110:1687–92. doi: 10.1016/j.xphs.2020.10.036. PMID: 33141046. [DOI] [PubMed] [Google Scholar]
- 17.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel. 2010;23:385–92. PMID: 20159773. doi: 10.1093/protein/gzq009. [DOI] [PubMed] [Google Scholar]
- 18.Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJ, Middaugh CR, Winter G, Fan YX, Kirshner S, Verthelyi D, Kozlowski S, et al. Overlooking subvisible particles in therapeutic protein products: gaps that may compromise product quality. J Pharm Sci. 2009;98:1201–05. PMID: 18704929. doi: 10.1002/jps.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114:944–49. PMID: 28096333. doi: 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, Kettenberger H. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci. 2015;104:1885–98. doi: 10.1002/jps.24430. PMID: 25821140. [DOI] [PubMed] [Google Scholar]
- 21.Vi R, Mj T, Gw B. Accelerated formulation development of monoclonal antibodies (mAbs) and mAb-based modalities: review of methods and tools. J Biomol Screen. 2015;20:468–83. doi: 10.1177/1087057114565593. PMID: 25576149. [DOI] [PubMed] [Google Scholar]
- 22.Svilenov HL, Arosio P, Menzen T, Tessier P, Sormanni P. Approaches to expand the conventional toolbox for discovery and selection of antibodies with drug-like physicochemical properties. MAbs. 2023;15:2164459. doi: 10.1080/19420862.2022.2164459. PMID: 36629855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spencer S, Bethea D, Raju TS, Giles-Komar J, Feng Y. Solubility evaluation of murine hybridoma antibodies. MAbs. 2012;4:319–25. doi: 10.4161/mabs.19869. PMID: 22531448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu SJ, Luo J, O’neil KT, Kang J, Lacy ER, Canziani G, Baker A, Huang M, Tang QM, Raju TS, et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng Des Sel. 2010;23:643–51. PMID: 20543007. doi: 10.1093/protein/gzq037. [DOI] [PubMed] [Google Scholar]
- 25.Pepinsky RB, Silvian L, Berkowitz SA, Farrington G, Lugovskoy A, Walus L, Eldredge J, Capili A, Mi S, Graff C, et al. Improving the solubility of anti-LINGO-1 monoclonal antibody Li33 by isotype switching and targeted mutagenesis. Protein Sci. 2010;19:954–66. PMID: 20198683. doi: 10.1002/pro.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cunningham O, Scott M, Zhou ZS, Finlay WJJ. Polyreactivity and polyspecificity in therapeutic antibody development: risk factors for failure in preclinical and clinical development campaigns. MAbs. 2021;13:1999195. doi: 10.1080/19420862.2021.1999195. PMID: 34780320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sidhu SS, Li B, Chen Y, Fellouse FA, Eigenbrot C, Fuh G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol. 2004;338:299–310. doi: 10.1016/j.jmb.2004.02.050. PMID: 15066433. [DOI] [PubMed] [Google Scholar]
- 28.Jespers L, Schon O, Famm K, Winter G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol. 2004;22:1161–65. doi: 10.1038/nbt1000. PMID: 15300256. [DOI] [PubMed] [Google Scholar]
- 29.Traxlmayr MW, Obinger C. Directed evolution of proteins for increased stability and expression using yeast display. Arch Biochem Biophys. 2012;526:174–80. doi: 10.1016/j.abb.2012.04.022. PMID: 22575387. [DOI] [PubMed] [Google Scholar]
- 30.Cherf GM, Cochran JR. Applications of Yeast Surface Display for Protein Engineering. Methods Mol Biol. 2015;1319:155–75. doi: 10.1007/978-1-4939-2748-7_8. PMID: 26060074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rossant CJ, Carroll D, Huang L, Elvin J, Neal F, Walker E, Benschop JJ, Kim EE, Barry ST, Vaughan TJ. Phage display and hybridoma generation of antibodies to human CXCR2 yields antibodies with distinct mechanisms and epitopes. MAbs. 2014;6:1425–38. doi: 10.4161/mabs.34376. PMID: 25484064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta N, Maddineni S, Kelly RL, Lee RB, Hunter SA, Silberstein JL, Parra Sperberg RA, Miller CL, Rabe A, Labanieh L, et al. An engineered antibody binds a distinct epitope and is a potent inhibitor of murine and human VISTA. Sci Rep. 2020;10:15171. PMID: 32938950. doi: 10.1038/s41598-020-71519-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Safdari Y, Farajnia S, Asgharzadeh M, Khalili M. Antibody humanization methods - a review and update. Biotechnol Genet Eng Rev. 2013;29:175–86. doi: 10.1080/02648725.2013.801235. PMID: 24568279. [DOI] [PubMed] [Google Scholar]
- 34.Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S, et al. Predicting Antibody Developability Profiles Through Early Stage Discovery Screening. MAbs. 2020;12:1743053. PMID: 32249670. doi: 10.1080/19420862.2020.1743053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu A, Kim HS, Estee S, ViaJar S, Galush WJ, Gill A, Hotzel I, Lazar GA, McDonald P, Andersen N, et al. Susceptibility of Antibody CDR Residues to Chemical Modifications Can Be Revealed Prior to Antibody Humanization and Aid in the Lead Selection Process. Mol Pharm. 2018;15:4529–37. PMID: 30118239. doi: 10.1021/acs.molpharmaceut.8b00536. [DOI] [PubMed] [Google Scholar]
- 36.Sawant MS, Streu CN, Wu L, Tessier PM. Toward Drug-Like Multispecific Antibodies by Design. Int J Mol Sci. 2020;21 PMID: 33053650. doi: 10.3390/ijms21207496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trianni Inc . Whitepaper: transgenic Mice: transforming Targeted Monoclonal Antibody (mAb) Therapeutics (2017).
- 38.Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, Wu HC. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27:1. doi: 10.1186/s12929-019-0592-z. PMID: 31894001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. doi: 10.3389/fimmu.2014.00520. PMID: 25368619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phakham T, Boonkrai C, Wongtangprasert T, Audomsun T, Attakitbancha C, Saelao P, Muanwien P, Sooksai S, Hirankarn N, Pisitkun T. Highly efficient hybridoma generation and screening strategy for anti-PD-1 monoclonal antibody development. Sci Rep. 2022;12:17792. doi: 10.1038/s41598-022-20560-6. PMID: 36273231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Estes B, Hsu YR, Tam LT, Sheng J, Stevens J, Haldankar R. Uncovering methods for the prevention of protein aggregation and improvement of product quality in a transient expression system. Biotechnol Prog. 2015;31:258–67. doi: 10.1002/btpr.2021. PMID: 25395220. [DOI] [PubMed] [Google Scholar]
- 42.Ma H, O’fagain C, O’kennedy R. Antibody stability: a key to performance - Analysis, influences and improvement. Biochimie. 2020;177:213–25. PMID: 32891698. doi: 10.1016/j.biochi.2020.08.019. [DOI] [PubMed] [Google Scholar]
- 43.Cromwell ME, Hilario E, Jacobson F. Protein aggregation and bioprocessing. Aaps J. 2006;8:E572–579. doi: 10.1208/aapsj080366. PMID: 17025275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Some D, Amartely H, Tsadok A, Lebendiker M. Characterization of Proteins by Size-Exclusion Chromatography Coupled to Multi-Angle Light Scattering (SEC-MALS). J Vis Exp [PMID: 31282880]. 2019. doi: 10.3791/59615-v. [DOI] [PubMed] [Google Scholar]
- 45.Vazquez-Rey M, Lang DA. Aggregates in monoclonal antibody manufacturing processes. Biotechnol Bioeng. 2011;108:1494–508. doi: 10.1002/bit.23155. PMID: 21480193. [DOI] [PubMed] [Google Scholar]
- 46.Lechner A, Giorgetti J, Gahoual R, Beck A, Leize-Wagner E, Francois YN. Insights from capillary electrophoresis approaches for characterization of monoclonal antibodies and antibody drug conjugates in the period 2016-2018. J Chromatogr B Analyt Technol Biomed Life Sci. 2019;1122-1123:1–17. doi: 10.1016/j.jchromb.2019.05.014. PMID: 31128359. [DOI] [PubMed] [Google Scholar]
- 47.Vlasak J, Ionescu R. Fragmentation of monoclonal antibodies. MAbs. 2011;3:253–63. doi: 10.4161/mabs.3.3.15608. PMID: 21487244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le Basle Y, Chennell P, Tokhadze N, Astier A, Sautou V. Physicochemical Stability of Monoclonal Antibodies: a Review. J Pharm Sci. 2020;109:169–90. doi: 10.1016/j.xphs.2019.08.009. PMID: 31465737. [DOI] [PubMed] [Google Scholar]
- 49.Wang W. Protein aggregation and its inhibition in biopharmaceutics. Int J Pharm. 2005;289:1–30. doi: 10.1016/j.ijpharm.2004.11.014. PMID: 15652195. [DOI] [PubMed] [Google Scholar]
- 50.Philo JS, Arakawa T. Mechanisms of protein aggregation. Curr Pharm Biotechnol. 2009;10:348–51. doi: 10.2174/138920109788488932. PMID: 19519409. [DOI] [PubMed] [Google Scholar]
- 51.Mieczkowski C, Bahmanjah S, Yu Y, Baker J, Raghunathan G, Tomazela D, Hsieh M, McCoy M, Strickland C, Fayadat-Dilman L. Crystal Structure and Characterization of Human Heavy-Chain Only Antibodies Reveals a Novel, Stable Dimeric Structure Similar to Monoclonal Antibodies. Antibodies (Basel). 2020;9 PMID: 33266498. doi: 10.3390/antib9040066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stoyle CL, Stephens PE, Humphreys DP, Heywood S, Cain K, Bulleid NJ. IgG light chain-independent secretion of heavy chain dimers: consequence for therapeutic antibody production and design. Biochem J. 2017;474:3179–88. doi: 10.1042/BCJ20170342. PMID: 28784690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rajendra Y, Peery RB, Hougland MD, Barnard GC, Wu X, Fitchett JR, Bacica M, Demarest SJ. Transient and stable CHO expression, purification and characterization of novel hetero-dimeric bispecific IgG antibodies. Biotechnol Prog. 2017;33:469–77. doi: 10.1002/btpr.2414. PMID: 27977915. [DOI] [PubMed] [Google Scholar]
- 54.Tam SH, McCarthy SG, Armstrong AA, Somani S, Wu SJ, Liu X, Gervais A, Ernst R, Saro D, Decker R, et al. Functional, Biophysical, and Structural Characterization of Human IgG1 and IgG4 Fc Variants with Ablated Immune Functionality. Antibodies (Basel). 2017;6: PMID: 31548527. doi: 10.3390/antib6030012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thiagarajan G, Semple A, James JK, Cheung JK, Shameem M. A comparison of biophysical characterization techniques in predicting monoclonal antibody stability. MAbs. 2016;8:1088–97. doi: 10.1080/19420862.2016.1189048. PMID: 27210456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci. 2008;97:1414–26. doi: 10.1002/jps.21104. PMID: 17721938. [DOI] [PubMed] [Google Scholar]
- 57.Rabia LA, Desai AA, Jhajj HS, Tessier PM. Understanding and overcoming trade-offs between antibody affinity, specificity, stability and solubility. Biochem Eng J. 2018;137:365–74. doi: 10.1016/j.bej.2018.06.003. PMID: 30666176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vermeer AW, Norde W. The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys J. 2000;78:394–404. doi: 10.1016/S0006-3495(00)76602-1. PMID: 10620303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brader ML, Estey T, Bai S, Alston RW, Lucas KK, Lantz S, Landsman P, Maloney KM. Examination of thermal unfolding and aggregation profiles of a series of developable therapeutic monoclonal antibodies. Mol Pharm. 2015;12:1005–17. doi: 10.1021/mp400666b. PMID: 25687223. [DOI] [PubMed] [Google Scholar]
- 60.Lehermayr C, Mahler HC, Mader K, Fischer S. Assessment of net charge and protein-protein interactions of different monoclonal antibodies. J Pharm Sci. 2011;100:2551–62. doi: 10.1002/jps.22506. PMID: 21294130. [DOI] [PubMed] [Google Scholar]
- 61.Li Y. Effective strategies for host cell protein clearance in downstream processing of monoclonal antibodies and Fc-fusion proteins. Protein Expr Purif. 2017;134:96–103. doi: 10.1016/j.pep.2017.04.006. PMID: 28414067. [DOI] [PubMed] [Google Scholar]
- 62.Cui Y, Cui P, Chen B, Li S, Guan H. Monoclonal antibodies: formulations of marketed products and recent advances in novel delivery system. Drug Dev Ind Pharm. 2017;43:519–30. doi: 10.1080/03639045.2017.1278768. PMID: 28049357. [DOI] [PubMed] [Google Scholar]
- 63.Strickley RG, Lambert WJ. A review of Formulations of Commercially Available Antibodies. J Pharm Sci. 2021;110:2590–608 e2556. doi: 10.1016/j.xphs.2021.03.017. PMID: 33789155. [DOI] [PubMed] [Google Scholar]
- 64.Singh SK, Kumar D, Nagpal S, Dubey SK, Rathore AS. A Charge Variant of Bevacizumab Offers Enhanced FcRn-Dependent Pharmacokinetic Half-Life and Efficacy. Pharm Res. 2022;39:851–65. doi: 10.1007/s11095-022-03236-8. PMID: 35355206. [DOI] [PubMed] [Google Scholar]
- 65.Li B, Tesar D, Boswell CA, Cahaya HS, Wong A, Zhang J, Meng YG, Eigenbrot C, Pantua H, Diao J, et al. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. MAbs. 2014;6:1255–64. PMID: 25517310. doi: 10.4161/mabs.29809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem. 2010;21:2153–63. doi: 10.1021/bc100261d. PMID: 21053952. [DOI] [PubMed] [Google Scholar]
- 67.Bumbaca Yadav D, Sharma VK, Boswell CA, Hotzel I, Tesar D, Shang Y, Ying Y, Fischer SK, Grogan JL, Chiang EY, et al. Evaluating the Use of Antibody Variable Region (Fv) Charge as a Risk Assessment Tool for Predicting Typical Cynomolgus Monkey Pharmacokinetics. J Biol Chem. 2015;290:29732–41. PMID: 26491012. doi: 10.1074/jbc.M115.692434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci U S A. 2015;112:5997–6002. doi: 10.1073/pnas.1408766112. PMID: 25918417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gupta S, Jiskoot W, Schoneich C, Rathore AS. Oxidation and Deamidation of Monoclonal Antibody Products: potential Impact on Stability, Biological Activity, and Efficacy. J Pharm Sci. 2022;111:903–18. doi: 10.1016/j.xphs.2021.11.024. PMID: 34890632. [DOI] [PubMed] [Google Scholar]
- 70.Voynov V, Chennamsetty N, Kayser V, Helk B, Trout BL. Predictive tools for stabilization of therapeutic proteins. MAbs. 2009;1:580–82. doi: 10.4161/mabs.1.6.9773. PMID: 20068399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu H, Randolph TW. Aggregation and Particle Formation During Pumping of an Antibody Formulation are Controlled by Electrostatic Interactions Between Pump Surfaces and Protein Molecules. J Pharm Sci. 2020;109:1473–82. doi: 10.1016/j.xphs.2020.01.023. PMID: 32004539. [DOI] [PubMed] [Google Scholar]
- 72.Tomar DS, Kumar S, Singh SK, Goswami S, Li L. Molecular basis of high viscosity in concentrated antibody solutions: strategies for high concentration drug product development. MAbs. 2016;8:216–28. doi: 10.1080/19420862.2015.1128606. PMID: 26736022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shire SJ. Formulation and manufacturability of biologics. Curr Opin Biotechnol. 2009;20:708–14. doi: 10.1016/j.copbio.2009.10.006. PMID: 19880308. [DOI] [PubMed] [Google Scholar]
- 74.Yadav S, Shire SJ, Kalonia DS. Factors affecting the viscosity in high concentration solutions of different monoclonal antibodies. J Pharm Sci. 2010;99:4812–29. doi: 10.1002/jps.22190. PMID: 20821382. [DOI] [PubMed] [Google Scholar]
- 75.Quigley A, Williams DR. The second virial coefficient as a predictor of protein aggregation propensity: a self-interaction chromatography study. Eur J Pharm Biopharm. 2015;96:282–90. doi: 10.1016/j.ejpb.2015.07.025. PMID: 26259782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sule SV, Sukumar M, WFt W, Marcelino-Cruz AM, Sample T, Tessier PM. High-throughput analysis of concentration-dependent antibody self-association. Biophys J. 2011;101:1749–57. doi: 10.1016/j.bpj.2011.08.036. PMID: 21961601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu J, Schultz JS, Weldon CL, Sule SV, Chai Q, Geng SB, Dickinson CD, Tessier PM. Discovery of highly soluble antibodies prior to purification using affinity-capture self-interaction nanoparticle spectroscopy. Protein Eng Des Sel. 2015;28:403–14. doi: 10.1093/protein/gzv045. PMID: 26363633. [DOI] [PubMed] [Google Scholar]
- 78.Phan S, Walmer A, Shaw EW, Chai Q. High-throughput profiling of antibody self-association in multiple formulation conditions by PEG stabilized self-interaction nanoparticle spectroscopy. MAbs. 2022;14:2094750. doi: 10.1080/19420862.2022.2094750. PMID: 35830420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu X, Nobrega RP, Lynaugh H, Jain T, Barlow K, Boland T, Sivasubramanian A, Vasquez M, Xu Y. Deamidation and isomerization liability analysis of 131 clinical-stage antibodies. MAbs. 2019;11:45–57. doi: 10.1080/19420862.2018.1548233. PMID: 30526254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yuk IH, Zhang B, Yang Y, Dutina G, Leach KD, Vijayasankaran N, Shen AY, Andersen DC, Snedecor BR, Joly JC. Controlling glycation of recombinant antibody in fed-batch cell cultures. Biotechnol Bioeng. 2011;108:2600–10. doi: 10.1002/bit.23218. PMID: 21618472. [DOI] [PubMed] [Google Scholar]
- 81.Bee JS, Goletz TJ, Ragheb JA. The future of protein particle characterization and understanding its potential to diminish the immunogenicity of biopharmaceuticals: a shared perspective. J Pharm Sci. 2012;101:3580–85. doi: 10.1002/jps.23247. PMID: 22736570. [DOI] [PubMed] [Google Scholar]
- 82.Narhi LO, Corvari V, Ripple DC, Afonina N, Cecchini I, Defelippis MR, Garidel P, Herre A, Koulov AV, Lubiniecki T, et al. Subvisible (2-100 μm) Particle Analysis During Biotherapeutic Drug Product Development: part 1, Considerations and Strategy. J Pharm Sci. 2015;104:1899–908. PMID: 25832583. doi: 10.1002/jps.24437. [DOI] [PubMed] [Google Scholar]
- 83.Vargas SK, Eskafi A, Carter E, Ciaccio N. A comparison of background membrane imaging versus flow technologies for subvisible particle analysis of biologics. Int J Pharm. 2020;578:119072. doi: 10.1016/j.ijpharm.2020.119072. PMID: 32001293. [DOI] [PubMed] [Google Scholar]
- 84.Singh SK, Afonina N, Awwad M, Bechtold-Peters K, Blue JT, Chou D, Cromwell M, Krause HJ, Mahler HC, Meyer BK, et al. An industry perspective on the monitoring of subvisible particles as a quality attribute for protein therapeutics. J Pharm Sci. 2010;99:3302–21. PMID: 20310025. doi: 10.1002/jps.22097. [DOI] [PubMed] [Google Scholar]
- 85.Grabarek AD, Bozic U, Rousel J, Menzen T, Kranz W, Wuchner K, Jiskoot W, Hawe A. What Makes Polysorbate Functional? Impact of Polysorbate 80 Grade and Quality on IgG Stability During Mechanical Stress. J Pharm Sci. 2020;109:871–80. doi: 10.1016/j.xphs.2019.10.015. PMID: 31614127. [DOI] [PubMed] [Google Scholar]
- 86.Morar-Mitrica S, Brisbane C, Nesta D, Ketkar A. Biophysical approaches to the detection and characterization of particles in an antibody solution. American Pharma Rev. 2009;12:34–40. [Google Scholar]
- 87.Durbin KR, Nottoli MS, Catron ND, Richwine N, Gj J. High-Throughput, Multispecies, Parallelized Plasma Stability Assay for the Determination and Characterization of Antibody-Drug Conjugate Aggregation and Drug Release. ACS Omega. 2017;2:4207–15. doi: 10.1021/acsomega.7b00452. PMID: 30023717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rodrigues D, Tanenbaum LM, Thirumangalathu R, Somani S, Zhang K, Kumar V, Amin K, Thakkar SV. Product-Specific Impact of Viscosity Modulating Formulation Excipients During Ultra-High Concentration Biotherapeutics Drug Product Development. J Pharm Sci. 2021;110:1077–82. doi: 10.1016/j.xphs.2020.12.016. PMID: 33340533. [DOI] [PubMed] [Google Scholar]
- 89.Bramham JE, Davies SA, Podmore A, Golovanov AP. Stability of a high-concentration monoclonal antibody solution produced by liquid-liquid phase separation. MAbs. 2021;13:1940666. doi: 10.1080/19420862.2021.1940666. PMID: 34225583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang T, Zhang J, Hewitt D, Tran B, Gao X, Qiu ZJ, Tejada M, Gazzano-Santoro H, Kao YH. Identification and characterization of buried unpaired cysteines in a recombinant monoclonal IgG1 antibody. Anal Chem. 2012;84:7112–23. doi: 10.1021/ac301426h. PMID: 22794164. [DOI] [PubMed] [Google Scholar]
- 91.Robotham AC, Kelly JF. Detection and quantification of free sulfhydryls in monoclonal antibodies using maleimide labeling and mass spectrometry. MAbs. 2019;11:757–66. doi: 10.1080/19420862.2019.1595307. PMID: 30894096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shukla AA, Hubbard B, Tressel T, Guhan S, Low D. Downstream processing of monoclonal antibodies–application of platform approaches. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;848:28–39. doi: 10.1016/j.jchromb.2006.09.026. PMID: 17046339. [DOI] [PubMed] [Google Scholar]
- 93.Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat Biotechnol. 2004;22:1393–98. doi: 10.1038/nbt1026. PMID: 15529164. [DOI] [PubMed] [Google Scholar]
- 94.Liu HF, Ma J, Winter C, Bayer R. Recovery and purification process development for monoclonal antibody production. MAbs. 2010;2:480–99. doi: 10.4161/mabs.2.5.12645. PMID: 20647768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Falconer RJ, Chan C, Hughes K, Munro TP. Stabilization of a monoclonal antibody during purification and formulation by addition of basic amino acid excipients. J Chem Tech Biotech. 2011;86:942–48. doi: 10.1002/jctb.2657. [DOI] [Google Scholar]
- 96.ICH Harmonised Tripartite Guideline . Evaluation for Stability Data Q1E Step 4. Geneva, Switzerland: The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; 2003. [accessed 2022 Dec 1]. https://www.ich.org/page/quality-guidelines [Google Scholar]
- 97.Kuzman D, Bunc M, Ravnik M, Reiter F, Zagar L, Boncina M. Long-term stability predictions of therapeutic monoclonal antibodies in solution using Arrhenius-based kinetics. Sci Rep. 2021;11:20534. doi: 10.1038/s41598-021-99875-9. PMID: 34654882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang W, Roberts CJ. Non-Arrhenius protein aggregation. Aaps J. 2013;15:840–51. doi: 10.1208/s12248-013-9485-3. PMID: 23615748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hauptmann A, Podgorsek K, Kuzman D, Srcic S, Hoelzl G, Loerting T. Impact of Buffer, Protein Concentration and Sucrose Addition on the Aggregation and Particle Formation during Freezing and Thawing. Pharm Res. 2018;35:101. doi: 10.1007/s11095-018-2378-5. PMID: 29556730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haeuser C, Goldbach P, Huwyler J, Friess W, Allmendinger A. Excipients for Room Temperature Stable Freeze-Dried Monoclonal Antibody Formulations. J Pharm Sci. 2020;109:807–17. doi: 10.1016/j.xphs.2019.10.016. PMID: 31622600. [DOI] [PubMed] [Google Scholar]
- 101.Halley J, Chou YR, Cicchino C, Huang M, Sharma V, Tan NC, Thakkar S, Zhou LL, Al-Azzam W, Cornen S, et al. An Industry Perspective on Forced Degradation Studies of Biopharmaceuticals: survey Outcome and Recommendations. J Pharm Sci. 2020;109:6–21. PMID: 31563512. doi: 10.1016/j.xphs.2019.09.018. [DOI] [PubMed] [Google Scholar]
- 102.Sule SV, Cheung JK, Antochshuk V, Bhalla AS, Narasimhan C, Blaisdell S, Shameem M, Tessier PM. Solution pH that minimizes self-association of three monoclonal antibodies is strongly dependent on ionic strength. Mol Pharm. 2012;9:744–51. doi: 10.1021/mp200448j. PMID: 22221144. [DOI] [PubMed] [Google Scholar]
- 103.Hong T, Iwashita K, Shiraki K. Viscosity Control of Protein Solution by Small Solutes: a Review. Curr Protein Pept Sci. 2018;19:746–58. doi: 10.2174/1389203719666171213114919. PMID: 29237380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang S, Zhang N, Hu T, Dai W, Feng X, Zhang X, Qian F. Viscosity-Lowering Effect of Amino Acids and Salts on Highly Concentrated Solutions of Two IgG1 Monoclonal Antibodies. Mol Pharm. 2015;12:4478–87. doi: 10.1021/acs.molpharmaceut.5b00643. PMID: 26528726. [DOI] [PubMed] [Google Scholar]
- 105.Zeng Y, Tran T, Wuthrich P, Naik S, Davagnino J, Greene DG, Mahoney RP, Soane DS. Caffeine as a Viscosity Reducer for Highly Concentrated Monoclonal Antibody Solutions. J Pharm Sci. 2021;110:3594–604. doi: 10.1016/j.xphs.2021.06.030. PMID: 34181992. [DOI] [PubMed] [Google Scholar]
- 106.Nowak C, Kc J, Md S, Katiyar A, Bhat R, Sun J, Ponniah G, Neill A, Mason B, Beck A, et al. Forced degradation of recombinant monoclonal antibodies: a practical guide. MAbs. 2017;9:1217–30. doi: 10.1080/19420862.2017.1368602. PMID: 28853987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci. 2007;96:1–26. doi: 10.1002/jps.20727. PMID: 16998873. [DOI] [PubMed] [Google Scholar]
- 108.Chelius D, Rehder DS, Bondarenko PV. Identification and characterization of deamidation sites in the conserved regions of human immunoglobulin gamma antibodies. Anal Chem. 2005;77:6004–11. doi: 10.1021/ac050672d. PMID: 16159134. [DOI] [PubMed] [Google Scholar]
- 109.Peters B, Trout BL. Asparagine deamidation: pH-dependent mechanism from density functional theory. Biochemistry. 2006;45:5384–92. doi: 10.1021/bi052438n. PMID: 16618128. [DOI] [PubMed] [Google Scholar]
- 110.Svilenov HL, Kulakova A, Zalar M, Golovanov AP, Harris P, Winter G. Orthogonal Techniques to Study the Effect of pH, Sucrose, and Arginine Salts on Monoclonal Antibody Physical Stability and Aggregation During Long-Term Storage. J Pharm Sci. 2020;109:584–94. doi: 10.1016/j.xphs.2019.10.065. PMID: 31689429. [DOI] [PubMed] [Google Scholar]
- 111.Robinson NE, Robinson AB. Prediction of protein deamidation rates from primary and three-dimensional structure. Proc Natl Acad Sci U S A. 2001;98:4367–72. doi: 10.1073/pnas.071066498. PMID: 11296285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sydow JF, Lipsmeier F, Larraillet V, Hilger M, Mautz B, Molhoj M, Kuentzer J, Klostermann S, Schoch J, Voelger HR, et al. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS One. 2014;9:e100736. PMID: 24959685. doi: 10.1371/journal.pone.0100736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Qiu H, Wei R, Jaworski J, Boudanova E, Hughes H, VanPatten S, Lund A, Day J, Zhou Y, McSherry T, et al. Engineering an anti-CD52 antibody for enhanced deamidation stability. MAbs. 2019;11:1266–75. PMID: 31199181. doi: 10.1080/19420862.2019.1631117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang W, Meeler AR, Bergerud LT, Hesselberg M, Byrne M, Wu Z. Quantification and characterization of antibody deamidation by peptide mapping with mass spectrometry. Int J Mass Spectrom. 2012;312:107–13. doi: 10.1016/j.ijms.2011.06.006. [DOI] [Google Scholar]
- 115.Dashivets T, Stracke J, Dengl S, Knaupp A, Pollmann J, Buchner J, Schlothauer T. Oxidation in the complementarity-determining regions differentially influences the properties of therapeutic antibodies. MAbs. 2016;8:1525–35. doi: 10.1080/19420862.2016.1231277. PMID: 27612038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Folzer E, Diepold K, Bomans K, Finkler C, Schmidt R, Bulau P, Huwyler J, Mahler HC, Koulov AV. Selective Oxidation of Methionine and Tryptophan Residues in a Therapeutic IgG1 Molecule. J Pharm Sci. 2015;104:2824–31. doi: 10.1002/jps.24509. PMID: 26010344. [DOI] [PubMed] [Google Scholar]
- 117.Mieczkowski C, Cheng A, Fischmann T, Hsieh M, Baker J, Uchida M, Raghunathan G, Strickland C, Fayadat-Dilman L. Characterization and Modeling of Reversible Antibody Self-Association Provide Insights into Behavior, Prediction, and Correction. Antibodies (Basel). 2021;10 PMID: 33671864. doi: 10.3390/antib10010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ji JA, Zhang B, Cheng W, Wang YJ. Methionine, tryptophan, and histidine oxidation in a model protein, PTH: mechanisms and stabilization. J Pharm Sci. 2009;98:4485–500. doi: 10.1002/jps.21746. PMID: 19455640. [DOI] [PubMed] [Google Scholar]
- 119.U.S. Department of Health and Human Services, Food and Drug Administration . Guidance for Industry: Container Closure Systems for Packaging Human Drugs and Biologics. Chemistry, Manufacturing, and Controls Documentation. Rockville, Maryland: Office of Training and Communications; 1999. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/container-closure-systems-packaging-human-drugs-and-biologics [Google Scholar]
- 120.Bhattacharya S, De A, Narasimhan CN, Sharma MK, Yang X, Sharpe M, Dohme A. Stable formulations of anti-ctla4 antibodies alone and in combination with programmed death receptor 1 (pd-1) antibodies and methods of use thereof. WIPO (PCT) Patent WO2018204343A1. 2018 May 1. [Google Scholar]
- 121.Chu GC, Chelius D, Xiao G, Khor HK, Coulibaly S, Bondarenko PV. Accumulation of succinimide in a recombinant monoclonal antibody in mildly acidic buffers under elevated temperatures. Pharm Res. 2007;24:1145–56. doi: 10.1007/s11095-007-9241-4. PMID: 17385019. [DOI] [PubMed] [Google Scholar]
- 122.Carpenter JF, Arakawa T, Crowe JH. Interactions of stabilizing additives with proteins during freeze-thawing and freeze-drying. Dev Biol Stand. 1992;74:225–38. discussion 238-229. PMID: 1592173. [PubMed] [Google Scholar]
- 123.Kueltzo LA, Wang W, Randolph TW, Carpenter JF. Effects of solution conditions, processing parameters, and container materials on aggregation of a monoclonal antibody during freeze-thawing. J Pharm Sci. 2008;97:1801–12. doi: 10.1002/jps.21110. PMID: 17823949. [DOI] [PubMed] [Google Scholar]
- 124.Kolhe P, Amend E, Singh SK. Impact of freezing on pH of buffered solutions and consequences for monoclonal antibody aggregation. Biotechnol Prog. 2010;26:727–33. doi: 10.1002/btpr.377. PMID: 20039442. [DOI] [PubMed] [Google Scholar]
- 125.Wang X, An Z, Luo W, Xia N, Zhao Q. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell. 2018;9:74–85. doi: 10.1007/s13238-017-0447-x. PMID: 28733914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ding Y, Marino M, Zen K, Sheffer J, Almaguer N, Caddy K, Praseuth A. Considerations for Monoclonal Antibody Bioprocess and Manufacturing Validation. Pharma Tech. 2022;44:31–36. https://www.pharmtech.com/view/considerations-for-monoclonal-antibody-bioprocess-and-manufacturing-validation. [Google Scholar]
- 127.Reinhart D, Damjanovic L, Kaisermayer C, Sommeregger W, Gili A, Gasselhuber B, Castan A, Mayrhofer P, Grunwald-Gruber C, Kunert R. Bioprocessing of Recombinant CHO-K1, CHO-DG44, and CHO-S: cHO Expression Hosts Favor Either mAb Production or Biomass Synthesis. Biotechnol J. 2019;14:e1700686. doi: 10.1002/biot.201700686. PMID: 29701329. [DOI] [PubMed] [Google Scholar]
- 128.Schmieder V, Fieder J, Drerup R, Gutierrez EA, Guelch C, Stolzenberger J, Stumbaum M, Mueller VS, Higel F, Bergbauer M, et al. Towards maximum acceleration of monoclonal antibody development: leveraging transposase-mediated cell line generation to enable GMP manufacturing within 3 months using a stable pool. J Biotechnol. 2022;349:53–64. PMID: 35341894. doi: 10.1016/j.jbiotec.2022.03.010. [DOI] [PubMed] [Google Scholar]
- 129.Wang Y, Qiu H, Minshull J, Tam W, Hu X, Mieczkowski C, Zheng W, Chu C, Liu W, Boldog F, et al. An innovative platform to improve asymmetric bispecific antibody assembly, purity, and expression level in stable pool and cell line development. Biochem Eng J. pp.188. 2022. doi: 10.1016/j.bej.2022.108683 [DOI] [Google Scholar]
- 130.Hunter M, Yuan P, Vavilala D, Fox M. Optimization of Protein Expression in Mammalian Cells. Curr Protoc Protein Sci. 2019;95:e77. doi: 10.1002/cpps.77. PMID: 30265450. [DOI] [PubMed] [Google Scholar]
- 131.Mimura Y, Katoh T, Saldova R, O’flaherty R, Izumi T, Mimura-Kimura Y, Utsunomiya T, Mizukami Y, Yamamoto K, Matsumoto T, et al. Glycosylation engineering of therapeutic IgG antibodies: challenges for the safety, functionality and efficacy. Protein Cell. 2018;9:47–62. PMID: 28597152. doi: 10.1007/s13238-017-0433-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aldington S, Bonnerjea J. Scale-up of monoclonal antibody purification processes. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;848:64–78. doi: 10.1016/j.jchromb.2006.11.032. PMID: 17224311. [DOI] [PubMed] [Google Scholar]
- 133.Jin W, Xing Z, Song Y, Huang C, Xu X, Ghose S, Li ZJ. Protein aggregation and mitigation strategy in low pH viral inactivation for monoclonal antibody purification. MAbs. 2019;11:1479–91. doi: 10.1080/19420862.2019.1658493. PMID: 31441367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Feroz H, Chennamsetty N, Byers S, Holstein M, Li ZJ, Ghose S. Assessing detergent-mediated virus inactivation, protein stability, and impurity clearance in biologics downstream processes. Biotechnol Bioeng. 2022;119:1091–104. doi: 10.1002/bit.28034. PMID: 35023152. [DOI] [PubMed] [Google Scholar]
- 135.Low D, O’leary R, Pujar NS. Future of antibody purification. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;848:48–63. doi: 10.1016/j.jchromb.2006.10.033. PMID: 17134947. [DOI] [PubMed] [Google Scholar]
- 136.Kelley B. Very large scale monoclonal antibody purification: the case for conventional unit operations. Biotechnol Prog. 2007;23:995–1008. doi: 10.1021/bp070117s. PMID: 17887772. [DOI] [PubMed] [Google Scholar]
- 137.Teeters M, Bezila D, Benner T, Alfonso P, Alred P. Predicting diafiltration solution compositions for final ultrafiltration/diafiltration steps of monoclonal antibodies. Biotechnol Bioeng. 2011;108:1338–46. doi: 10.1002/bit.23067. PMID: 21328314. [DOI] [PubMed] [Google Scholar]
- 138.Lolas A, Hughes P, Suvarna K, Chi B. CMC Microbiology Review of Biologics License Applications and Pre-License/Pre-Approval Inspections: therapeutic Biological Proteins. American Pharma Rev. 2010. https://www.americanpharmaceuticalreview.com/Featured-Articles/117310-CMC-Microbiology-Review-of-Biologics-License-Applications-and-Pre-License-Pre-Approval-Inspections-Therapeutic-Biological-Proteins/ [Google Scholar]
- 139.Ausserwöger H, Schneider MM, Herling TW, Arosio P, Invernizzi G, Knowles TPJ, Lorenzen N. Non-specificity as the sticky problem in therapeutic antibody development. Nat Rev Chem. 2022;6:844–61. doi: 10.1038/s41570-022-00438-x. [DOI] [PubMed] [Google Scholar]
- 140.Alam ME, Slaney TR, Wu L, Das TK, Kar S, Barnett GV, Leone A, Tessier PM. Unique Impacts of Methionine Oxidation, Tryptophan Oxidation, and Asparagine Deamidation on Antibody Stability and Aggregation. J Pharm Sci. 2020;109:656–69. doi: 10.1016/j.xphs.2019.10.051. PMID: 31678251. [DOI] [PubMed] [Google Scholar]
- 141.Li W, Prabakaran P, Chen W, Zhu Z, Feng Y, Dimitrov DS. Antibody Aggregation: insights from Sequence and Structure. Antibodies (Basel). 2016;5 PMID: 31558000. doi: 10.3390/antib5030019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chi EY, Krishnan S, Randolph TW, Carpenter JF. Physical stability of proteins in aqueous solution: mechanism and driving forces in nonnative protein aggregation. Pharm Res. 2003;20:1325–36. doi: 10.1023/a:1025771421906. PMID: 14567625. [DOI] [PubMed] [Google Scholar]
- 143.Brummitt RK, Nesta DP, Chang L, Chase SF, Laue TM, Roberts CJ. Nonnative aggregation of an IgG1 antibody in acidic conditions: part 1. Unfolding, colloidal interactions, and formation of high-molecular-weight aggregates. J Pharm Sci. 2011;100:2087–103. doi: 10.1002/jps.22448. PMID: 21213308. [DOI] [PubMed] [Google Scholar]
- 144.Kim N, Remmele RL Jr., Liu D, Vi R, Ej F, Cj R. Aggregation of anti-streptavidin immunoglobulin gamma-1 involves Fab unfolding and competing growth pathways mediated by pH and salt concentration. Biophys Chem. 2013;172:26–36. doi: 10.1016/j.bpc.2012.12.004. PMID: 23334430. [DOI] [PubMed] [Google Scholar]
- 145.Mahler HC, Friess W, Grauschopf U, Kiese S. Protein aggregation: pathways, induction factors and analysis. J Pharm Sci. 2009;98:2909–34. doi: 10.1002/jps.21566. PMID: 18823031. [DOI] [PubMed] [Google Scholar]
- 146.Knight MJ, Floret L, Patel N, O’hara J, Rodriguez E. The impact of forced degradation conditions on mAb dimer formation and subsequent influence on aggregation propensity. MAbs. 2022;14:2127172. doi: 10.1080/19420862.2022.2127172. PMID: 36198003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schon A, Freire E. Reversibility and irreversibility in the temperature denaturation of monoclonal antibodies. Anal Biochem. 2021;626:114240. doi: 10.1016/j.ab.2021.114240. PMID: 33964250. [DOI] [PubMed] [Google Scholar]
- 148.Sahin E, Grillo AO, Perkins MD, Roberts CJ. Comparative effects of pH and ionic strength on protein-protein interactions, unfolding, and aggregation for IgG1 antibodies. J Pharm Sci. 2010;99:4830–48. doi: 10.1002/jps.22198. PMID: 20821389. [DOI] [PubMed] [Google Scholar]
- 149.Bauer J, Mathias S, Kube S, Otte K, Garidel P, Gamer M, Blech M, Fischer S, Karow-Zwick AR. Rational optimization of a monoclonal antibody improves the aggregation propensity and enhances the CMC properties along the entire pharmaceutical process chain. MAbs. 2020;12:1787121. doi: 10.1080/19420862.2020.1787121. PMID: 32658605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Haverick M, Mengisen S, Shameem M, Ambrogelly A. Separation of mAbs molecular variants by analytical hydrophobic interaction chromatography HPLC: overview and applications. MAbs. 2014;6:852–58. doi: 10.4161/mabs.28693. PMID: 24751784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Demeule B, Messick S, Shire SJ, Liu J. Characterization of particles in protein solutions: reaching the limits of current technologies. Aaps J. 2010;12:708–15. doi: 10.1208/s12248-010-9233-x. PMID: 20953747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wakankar AA, Borchardt RT, Eigenbrot C, Shia S, Wang YJ, Shire SJ, Liu JL. Aspartate Isomerization in the Complementarity-Determining Regions of Two Closely Related Monoclonal Antibodies. Biochemistry. 2007;46:1534–44. doi: 10.1021/bi061500t. [DOI] [PubMed] [Google Scholar]
- 153.Dillon TM, Bondarenko PV, Speed Ricci M. Development of an analytical reversed-phase high-performance liquid chromatography-electrospray ionization mass spectrometry method for characterization of recombinant antibodies. J Chromatogr A. 2004;1053:299–305. doi: 10.1016/S0021-9673(04)01410-4. PMID: 15543996. [DOI] [PubMed] [Google Scholar]
- 154.Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog. 2005;21:11–16. doi: 10.1021/bp040016j. PMID: 15903235. [DOI] [PubMed] [Google Scholar]
- 155.Grassi L, Cabrele C. Susceptibility of protein therapeutics to spontaneous chemical modifications by oxidation, cyclization, and elimination reactions. Amino Acids. 2019;51:1409–31. doi: 10.1007/s00726-019-02787-2. PMID: 31576455. [DOI] [PubMed] [Google Scholar]
- 156.Zeunik R, Ryuzoji AF, Peariso A, Wang X, Lannan M, Spindler LJ, Knierman M, Copeland V, Patel C, Wen Y. Investigation of Immune Responses to Oxidation, Deamidation, and Isomerization in Therapeutic Antibodies using Preclinical Immunogenicity Risk Assessment Assays. J Pharm Sci. 2022;111:2217–29. doi: 10.1016/j.xphs.2022.05.005. PMID: 35577116. [DOI] [PubMed] [Google Scholar]
- 157.Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Prueksaritanont T, Ionescu R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol. 2011;48:860–66. doi: 10.1016/j.molimm.2010.12.009. PMID: 21256596. [DOI] [PubMed] [Google Scholar]
- 158.Laue TM, Shire SJ. The Molecular Interaction Process. J Pharm Sci. 2020;109:154–60. doi: 10.1016/j.xphs.2019.10.045. PMID: 31676268. [DOI] [PubMed] [Google Scholar]
- 159.Liu L, Randolph TW, Carpenter JF. Particles shed from syringe filters and their effects on agitation-induced protein aggregation. J Pharm Sci. 2012;101:2952–59. doi: 10.1002/jps.23225. PMID: 22674153. [DOI] [PubMed] [Google Scholar]
- 160.Liu J, Yadav S, Andya J, Demeule B, Shire SJ. Analytical Ultracentrifugation and Its Role in Development and Research of Therapeutical Proteins. Methods Enzymol. 2015;562:441–76. doi: 10.1016/bs.mie.2015.04.008. PMID: 26412663. [DOI] [PubMed] [Google Scholar]
- 161.Shire SJ. The molecular basis of high viscosity of monoclonal antibodies (mAbs) at high concentration. Monoclonal Antibodies. Cambridge, UK: Woodhead Publishing; 2015. p. 163–192. doi: 10.1016/C2014-0-04095-6. [DOI] [Google Scholar]
- 162.Liu J, Nguyen MD, Andya JD, Shire SJ. Reversible self-association increases the viscosity of a concentrated monoclonal antibody in aqueous solution. J Pharm Sci. 2005;94:1928–40. doi: 10.1002/jps.20347. PMID: 16052543. [DOI] [PubMed] [Google Scholar]
- 163.Bethea D, Wu SJ, Luo J, Hyun L, Lacy ER, Teplyakov A, Jacobs SA, O’neil KT, Gilliland GL, Feng Y. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng Des Sel. 2012;25:531–37. doi: 10.1093/protein/gzs047. PMID: 22915597. [DOI] [PubMed] [Google Scholar]
- 164.Salinas BA, Sathish HA, Bishop SM, Harn N, Carpenter JF, Randolph TW. Understanding and modulating opalescence and viscosity in a monoclonal antibody formulation. J Pharm Sci. 2010;99:82–93. doi: 10.1002/jps.21797. PMID: 19475558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Casaz P, Boucher E, Wollacott R, Pierce BG, Rivera R, Sedic M, Ozturk S, Thomas WD Jr., Wang Y. Resolving self-association of a therapeutic antibody by formulation optimization and molecular approaches. MAbs. 2014;6:1533–39. doi: 10.4161/19420862.2014.975658. PMID: 25484044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dobson CL, Devine PW, Phillips JJ, Higazi DR, Lloyd C, Popovic B, Arnold J, Buchanan A, Lewis A, Goodman J, et al. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci Rep. 2016;6:38644. PMID: 27995962. doi: 10.1038/srep38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Schrag JD, Picard ME, Gaudreault F, Gagnon LP, Baardsnes J, Manenda MS, Sheff J, Deprez C, Baptista C, Hogues H, et al. Binding symmetry and surface flexibility mediate antibody self-association. MAbs. 2019;11:1300–18. PMID: 31318308. doi: 10.1080/19420862.2019.1632114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nishi H, Miyajima M, Wakiyama N, Kubota K, Hasegawa J, Uchiyama S, Fukui K. Fc domain mediated self-association of an IgG1 monoclonal antibody under a low ionic strength condition. J Biosci Bioeng. 2011;112:326–32. doi: 10.1016/j.jbiosc.2011.06.017. PMID: 21783411. [DOI] [PubMed] [Google Scholar]
- 169.Scherer TM, Liu J, Shire SJ, Minton AP. Intermolecular interactions of IgG1 monoclonal antibodies at high concentrations characterized by light scattering. J Phys Chem B. 2010;114:12948–57. doi: 10.1021/jp1028646. PMID: 20849134. [DOI] [PubMed] [Google Scholar]
- 170.Wang W, Ignatius AA, Thakkar SV. Impact of residual impurities and contaminants on protein stability. J Pharm Sci. 2014;103:1315–30. doi: 10.1002/jps.23931. PMID: 24623189. [DOI] [PubMed] [Google Scholar]
- 171.Bee JS, Chiu D, Sawicki S, Stevenson JL, Chatterjee K, Freund E, Carpenter JF, Randolph TW. Monoclonal antibody interactions with micro- and nanoparticles: adsorption, aggregation, and accelerated stress studies. J Pharm Sci. 2009;98:3218–38. doi: 10.1002/jps.21768. PMID: 19492408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Huang M, Horwitz TS, Zweiben C, Singh SK. Impact of extractables/leachables from filters on stability of protein formulations. J Pharm Sci. 2011;100:4617–30. doi: 10.1002/jps.22670. PMID: 21695696. [DOI] [PubMed] [Google Scholar]
- 173.Bee JS, Davis M, Freund E, Carpenter JF, Randolph TW. Aggregation of a monoclonal antibody induced by adsorption to stainless steel. Biotechnol Bioeng. 2010;105:121–29. doi: 10.1002/bit.22525. PMID: 19725039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Brusotti G, Calleri E, Colombo R, Massolini G, Rinaldi F, Temporini C. Advances on Size Exclusion Chromatography and Applications on the Analysis of Protein Biopharmaceuticals and Protein Aggregates: a Mini Review. Chromatographia. 2017;81:3–23. doi: 10.1007/s10337-017-3380-5. [DOI] [Google Scholar]
- 175.Khan TA, Mahler HC, Kishore RS. Key interactions of surfactants in therapeutic protein formulations: a review. Eur J Pharm Biopharm. 2015;97:60–67. doi: 10.1016/j.ejpb.2015.09.016. PMID: 26435336. [DOI] [PubMed] [Google Scholar]
- 176.Roy I, Patel A, Kumar V, Nanda T, Assenberg R, Wuchner K, Amin K. Polysorbate Degradation and Particle Formation in a High Concentration mAb: formulation Strategies to Minimize Effect of Enzymatic Polysorbate Degradation. J Pharm Sci. 2021;110:3313–23. doi: 10.1016/j.xphs.2021.05.012. PMID: 34077768. [DOI] [PubMed] [Google Scholar]
- 177.Molden R, Hu M, Yen ES, Saggese D, Reilly J, Mattila J, Qiu H, Chen G, Bak H, Li N. Host cell protein profiling of commercial therapeutic protein drugs as a benchmark for monoclonal antibody-based therapeutic protein development. MAbs. 2021;13:1955811. doi: 10.1080/19420862.2021.1955811. PMID: 34365906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Magalhaes PO, Lopes AM, Mazzola PG, Rangel-Yagui C, Penna TC, Pessoa A Jr.. Methods of endotoxin removal from biological preparations: a review. J Pharm Pharm Sci. 2007;10:388–404. PMID: 17727802. [PubMed] [Google Scholar]
- 179.Basu P, Krishnan S, Thirumangalathu R, Randolph TW, Carpenter JF. IgG1 aggregation and particle formation induced by silicone-water interfaces on siliconized borosilicate glass beads: a model for siliconized primary containers. J Pharm Sci. 2013;102:852–65. doi: 10.1002/jps.23434. PMID: 23280943. [DOI] [PubMed] [Google Scholar]
- 180.Thirumangalathu R, Krishnan S, Ricci MS, Brems DN, Randolph TW, Carpenter JF. Silicone oil- and agitation-induced aggregation of a monoclonal antibody in aqueous solution. J Pharm Sci. 2009;98:3167–81. doi: 10.1002/jps.21719. PMID: 19360857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hollowell P, Li Z, Hu X, Ruane S, Kalonia C, Cf VDW, Lu JR. Recent Advances in Studying Interfacial Adsorption of Bioengineered Monoclonal Antibodies. Molecules. 2020;25 PMID: 32353995. doi: 10.3390/molecules25092047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. MAbs. 2015;7:483–93. doi: 10.1080/19420862.2015.1016696. PMID: 25695748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hotzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. A strategy for risk mitigation of antibodies with fast clearance. MAbs. 2012;4:753–60. PMID: 23778268. doi: 10.4161/mabs.22189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu TY, Torrey J, Thomas J, Bobrowicz P, Vasquez M, et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng Des Sel. 2013;26:663–70. PMID: 24046438. doi: 10.1093/protein/gzt047. [DOI] [PubMed] [Google Scholar]
- 185.Kraft TE, Richter WF, Emrich T, Knaupp A, Schuster M, Wolfert A, Kettenberger H. Heparin chromatography as an in vitro predictor for antibody clearance rate through pinocytosis. MAbs. 2020;12:1683432. doi: 10.1080/19420862.2019.1683432. PMID: 31769731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hu S, Datta-Mannan A, D’argenio DZ. Physiologically Based Modeling to Predict Monoclonal Antibody Pharmacokinetics in Humans from in vitro Physiochemical Properties. MAbs. 2022;14:2056944. doi: 10.1080/19420862.2022.2056944. PMID: 35491902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jacobs SA, Wu SJ, Feng Y, Bethea D, O’neil KT. Cross-interaction chromatography: a rapid method to identify highly soluble monoclonal antibody candidates. Pharm Res. 2010;27:65–71. doi: 10.1007/s11095-009-0007-z. PMID: 19911257. [DOI] [PubMed] [Google Scholar]
- 188.Kohli N, Jain N, Geddie ML, Razlog M, Xu L, Lugovskoy AA. A novel screening method to assess developability of antibody-like molecules. MAbs. 2015;7:752–58. doi: 10.1080/19420862.2015.1048410. PMID: 25961854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kuroda D, Tsumoto K. Engineering Stability, Viscosity, and Immunogenicity of Antibodies by Computational Design. J Pharm Sci. 2020;109:1631–51. doi: 10.1016/j.xphs.2020.01.011. PMID: 31958430. [DOI] [PubMed] [Google Scholar]
- 190.Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302–06. doi: 10.1038/nbt1012. PMID: 15361882. [DOI] [PubMed] [Google Scholar]
- 191.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci U S A. 2009;106:11937–42. doi: 10.1073/pnas.0904191106. PMID: 19571001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharm Sci. 2012;101:102–15. doi: 10.1002/jps.22758. PMID: 21935950. [DOI] [PubMed] [Google Scholar]
- 193.Vatsa S. In silico prediction of post-translational modifications in therapeutic antibodies. MAbs. 2022;14:2023938. doi: 10.1080/19420862.2021.2023938. PMID: 35040751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Thorsteinson N, Gunn JR, Kelly K, Long W, Labute P. Structure-based charge calculations for predicting isoelectric point, viscosity, clearance, and profiling antibody therapeutics. MAbs. 2021;13:1981805. doi: 10.1080/19420862.2021.1981805. PMID: 34632944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Agrawal NJ, Helk B, Kumar S, Mody N, Sathish HA, Samra HS, Buck PM, Li L, Trout BL. Computational tool for the early screening of monoclonal antibodies for their viscosities. MAbs. 2016;8:43–48. doi: 10.1080/19420862.2015.1099773. PMID: 26399600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tomar DS, Li L, Broulidakis MP, Luksha NG, Burns CT, Singh SK, Kumar S. In-silico prediction of concentration-dependent viscosity curves for monoclonal antibody solutions. MAbs. 2017;9:476–89. doi: 10.1080/19420862.2017.1285479. PMID: 28125318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A. 2014;111:18601–06. PMID: 25512516. doi: 10.1073/pnas.1421779112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, Deane CM. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci U S A. 2019;116:4025–30. doi: 10.1073/pnas.1810576116. PMID: 30765520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hebditch M, Warwicker J. Charge and hydrophobicity are key features in sequence-trained machine learning models for predicting the biophysical properties of clinical-stage antibodies. PeerJ. 2019;7:e8199. PMID: 31976163. doi: 10.7717/peerj.8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Dunbar J, Krawczyk K, Leem J, Marks C, Nowak J, Regep C, Georges G, Kelm S, Popovic B, Deane CM. SAbPred: a structure-based antibody prediction server. Nucleic Acids Res. 2016;44:W474–478. doi: 10.1093/nar/gkw361. PMID: 27131379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chen X, Dougherty T, Hong C, Schibler R, Zhao YC, Sadeghi R, Matasci N, Wu Y-C, Kerman I. bioRxiv. 2020. Predicting Antibody Developability from Sequence using Machine Learning. [DOI] [Google Scholar]
- 202.Wilman W, Wrobel S, Bielska W, Deszynski P, Dudzic P, Jaszczyszyn I, Kaniewski J, Mlokosiewicz J, Rouyan A, Satlawa T, et al. Machine-designed biotherapeutics: opportunities, feasibility and advantages of deep learning in computational antibody discovery. Brief Bioinform. 2022;23: PMID: 35830864. doi: 10.1093/bib/bbac267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Akbar R, Bashour H, Rawat P, Robert PA, Smorodina E, Cotet TS, Flem-Karlsen K, Frank R, Mehta BB, Vu MH, et al. Progress and challenges for the machine learning-based design of fit-for-purpose monoclonal antibodies. MAbs. 2022;14:2008790. PMID: 35293269. doi: 10.1080/19420862.2021.2008790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Makowski EK, Kinnunen PC, Huang J, Wu L, Smith MD, Wang T, Desai AA, Streu CN, Zhang Y, Zupancic JM, et al. Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space. Nat Commun. 2022;13:3788. PMID: 35778381. doi: 10.1038/s41467-022-31457-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jia L, Sun Y, de Brevern AG. Protein asparagine deamidation prediction based on structures with machine learning methods. PLoS One. 2017;12:e0181347. PMID: 28732052. doi: 10.1371/journal.pone.0181347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sankar K, Hoi KH, Yin Y, Ramachandran P, Andersen N, Hilderbrand A, McDonald P, Spiess C, Zhang Q. Prediction of methionine oxidation risk in monoclonal antibodies using a machine learning method. MAbs. 2018;10:1281–90. doi: 10.1080/19420862.2018.1518887. PMID: 30252602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lai PK, Gallegos A, Mody N, Sathish HA, Trout BL. Machine learning prediction of antibody aggregation and viscosity for high concentration formulation development of protein therapeutics. MAbs. 2022;14:2026208. doi: 10.1080/19420862.2022.2026208. PMID: 35075980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Narayanan H, Dingfelder F, Butte A, Lorenzen N, Sokolov M, Arosio P. Machine Learning for Biologics: opportunities for Protein Engineering, Developability, and Formulation. Trends Pharmacol Sci. 2021;42:151–65. doi: 10.1016/j.tips.2020.12.004. PMID: 33500170. [DOI] [PubMed] [Google Scholar]
- 209.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–89. PMID: 34265844. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bryant P, Pozzati G, Elofsson A. Improved prediction of protein-protein interactions using AlphaFold2. Nat Commun. 2022;13:1265. doi: 10.1038/s41467-022-28865-w. PMID: 35273146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Abanades B, Georges G, Bujotzek A, Deane CM, Xu J. Ablooper: fast accurate antibody CDR loop structure prediction with accuracy estimation. Bioinformatics. 2022;38:1877–80. PMID: 35099535. doi: 10.1093/bioinformatics/btac016. [DOI] [PMC free article] [PubMed] [Google Scholar]