

Abstract
The clinical impact of any therapy requires the product be safe and effective. Gammaretroviral vectors pose several unique risks, including inadvertent exposure to replication competent retrovirus (RCR) that can arise during vector manufacture. The US FDA has required patient monitoring for RCR, and the National Gene Vector Biorepository is an NIH resource that has assisted eligible investigators in meeting this requirement. To date, we have found no evidence of RCR in 338 pre-treatment and 1,595 post-treatment blood samples from 737 patients associated with 60 clinical trials. Most samples (75%) were obtained within 1 year of treatment, and samples as far out as 9 years after treatment were analyzed. The majority of trials (93%) were cancer immunotherapy, and 90% of the trials used vector products produced with the PG13 packaging cell line. The data presented here provide further evidence that current manufacturing methods generate RCR-free products and support the overall safety profile of retroviral gene therapy.
Keywords: replication competent retrovirus, gene therapy, safety, patient monitoring, cancer immunotherapy
Graphical abstract
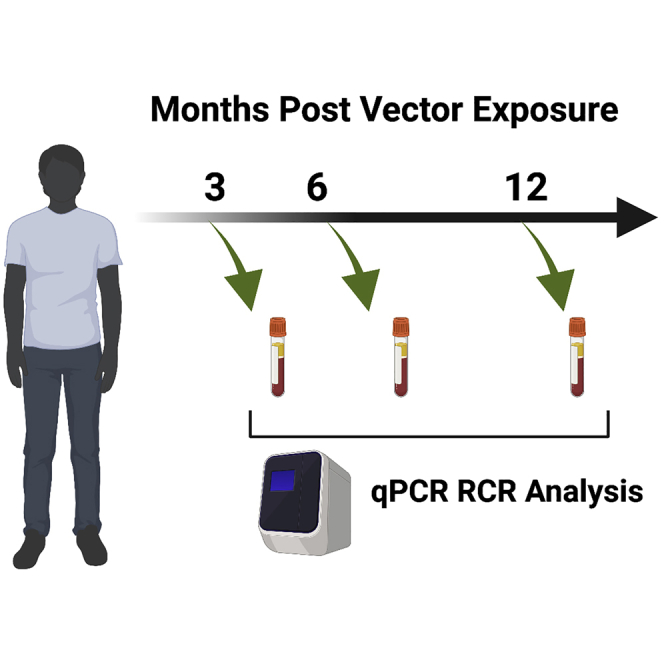
Replication competent virus (RCR) arising during vector manufacturing would significantly increase the risk of insertional oncogenesis prompting the US FDA to RCR test at multiple time points after treatment. This manuscript documents a lack of detectable RCR in 60 clinical trials, attesting to the safety of current manufacturing and screening procedures.
Introduction
Retroviral vectors derived from murine gammaretroviruses have been an important tool for advancing gene therapy, particularly in the setting of cancer immunotherapy. Most retroviral vectors are engineered to be replication defective by separating the transgene of interest from the viral structural genes (gag and pol) and glycoprotein envelope required for vector particle formation. Early packaging systems can recombine and restore replicative function,1,2 and vector-associated replication competent retrovirus (RCR) may cause malignancy in mice and non-human primates.3,4 Therefore, inadvertent exposure to RCR is considered a serious risk factor when using this class of vectors (see Cornetta et al. for a review of RCR risk as it relates to insertional oncogenesis).5 While RCR exposure in humans has not been reported to date, the finding of vector-related malignancy in a subset of patients treated with replication defective vectors targeting hematopoietic stem cells6,7,8,9 has further prompted regulatory agencies to continue to recommend post-trial monitoring for RCR exposure.10
The National Gene Vector Biorepository (NGVB, www.NGVBCC.org),11 a program funded by the US National Heart, Lung and Blood Institute (NHLBI), has assisted in monitoring for RCR using biologic assays12 and quantitative PCR (qPCR). In this paper, we report no evidence of RCR in peripheral blood from patients tested at a variety of time points post-infusion of vector-transduced cells. The predominant method of manufacture has been packaging cell lines using the Gibbon Ape Leukemia Virus (GALV) envelope. The data provide further evidence of the safety of gene therapy using current retroviral vectors and packaging systems.
Results
The NGVB uses qPCR to detect the envelope used in pseudotyping viral vectors as a surrogate test for RCR. This is in line with US Food and Drug Administration (FDA) guidelines for post-treatment RCR monitoring.10 In 2018, investigators who submitted samples to the NGVB received a questionnaire requesting information about the clinical trial, vector manufacture, ex vivo cell target, and whether the vector lot used in generating the ex vivo cell product was screened for RCR. All trials in this study utilized gammaretroviral vectors to transduce ex vivo cell targets. As shown in Table 1, most studies employed the GALV envelope, of which 87% utilized the PG13 packaging cell line in vector production.13 Two studies used vector pseudotyped with the amphotropic retrovirus AM-MLV using PA317 cells.14 Only a minority employed transient transfection methods. The majority of trials prepared genetically modified T cells for use in cancer immunotherapy trials. In two trials, vector was used to modify neuroblastoma cell lines. Three trials, at different sites, transduced CD34+ hematopoietic stem cells (HSCs) using the same vector construct that contained a self-inactivating LTR (Long Terminal Repeat). The disease target was X-linked severe combined immunodeficiency (SCID). Two additional trials used CD34+ HSC, one for treatment of adenosine deaminase deficiency and another conferred a drug resistance gene.
Table 1.
Studies evaluating replication competent retrovirus (RCR) exposure after infusion of gene modified cells
| IU master agreement number | Investigator | Institution | NCT number | Indication | Method | Pseudotype | Target cell | Vector product negative for RCR | SIN LTR |
|---|---|---|---|---|---|---|---|---|---|
| M2 | Ramos | Baylor College of Medicine | NCT00881920 | cancer | PG13 | GALV | T cell | yes | |
| M4 | Williams | Boston Children’s Hospital | NCT01129544 | X-linked SCID | transient | GALV | CD34+ HSC | yes | X |
| M5 | Ramos | Baylor College of Medicine | NCT00709033 | cancer | PG13 | GALV | T cell | yes | |
| M6 | Brenner | Baylor College of Medicine | NCT01192555 | cancer | PA317 | Ampho | neuroblastoma cell line | yes | |
| M7 | Brenner | Baylor College of Medicine | NCT00710892 | cancer | PG13 | GALV | T cell | yes | |
| M8 | Brenner | Baylor College of Medicine | NCT00703222 | cancer | PA317 | Ampho | neuroblastoma cell line | yes | |
| M9 | Ramos | Baylor College of Medicine | NCT00586391 | cancer | PG13 | GALV | T cell | yes | |
| M10 | Heslop | Baylor College of Medicine | NCT00889954 | cancer | PG13 | GALV | T cell | yes | |
| M11 | Brenner | Baylor College of Medicine | NCT00902044 | cancer | PG13 | GALV | T cell | yes | |
| M12 | Kochenderfer | NIH Surgery Branch | NCT01087294 | cancer | PG13 | GALV | T cell | yes | |
| M16 | Heslop | Baylor College of Medicine | NCT00368082 | cancer | PG13 | GALV | T cell | yes | |
| M18 | Ramos | Baylor College of Medicine | NCT00840853 | cancer | PG13 | GALV | T cell | yes | |
| M20 | Brenner | Baylor College of Medicine | NCT01109095 | cancer | PG13 | GALV | T cell | yes | |
| M22 | Heslop | Baylor College of Medicine | NCT01192464 | cancer | PG13 | GALV | T cell | yes | |
| M23 | Shah | NIH Pediatric Oncology Branch | NCT01593696 | cancer | PG13 | GALV | T cell | yes | |
| M27 | Brenner | Baylor College of Medicine | NCT01494103 | cancer | PG13 | GALV | T cell | yes | |
| M31 | Kohn | University of CA Los Angeles | NCT01129544 | X-linked SCID | transient | GALV | CD34+ HSC | yes | X |
| M40 | Rosenberg | NIH Surgery Branch | NCT01273181 | cancer | PG13 | GALV | T cell | yes | |
| M41 | Rosenberg | NIH Surgery Branch | NCT00670748 | cancer | PG13 | GALV | T cell | yes | |
| M42 | Rosenberg | NIH Surgery Branch | NCT01236573 | cancer | PG13 | GALV | T cell | yes | |
| M43 | Rosenberg | NIH Surgery Branch | NCT01218867 | cancer | PG13 | GALV | T cell | yes | |
| M44 | Rosenberg | NIH Surgery Branch | NCT01454596 | cancer | PG13 | GALV | T cell | yes | |
| M45 | Rosenberg | NIH Surgery Branch | NCT01583686 | cancer | PG13 | GALV | T cell | yes | |
| M46 | Heslop | Baylor College of Medicine | NCT01316146 | cancer | PG13 | GALV | T cell | yes | |
| M48 | Brenner | Baylor College of Medicine | NCT01460901 | cancer | PG13 | GALV | T cell | yes | |
| M49 | Kohn | University of CA Los Angeles | NCT00794508 | ADA SCID | PG13 | GALV | CD34+ HSC | yes | |
| M56 | Sauter | Memorial Sloan Kettering | NCT01840566 | cancer | PG13 | GALV | T cell | yes | |
| M57 | J. Park | Memorial Sloan Kettering | NCT03085173 | cancer | PG13 | GALV | T cell | yes | |
| M58 | J. Park | Memorial Sloan Kettering | NCT01416974 | cancer | PG13 | GALV | T cell | yes | |
| M59 | Curran | Memorial Sloan Kettering | NCT01860937 | cancer | PG13 | GALV | T cell | yes | |
| M60 | Rosenberg | NIH Surgery Branch | NCT00509288 | cancer | PG13 | GALV | T cell | yes | |
| M61 | Rosenberg | NIH Surgery Branch | NCT00273910 | cancer | PG13 | GALV | T cell | yes | |
| M62 | Rosenberg | NIH Surgery Branch | NCT00706992 | cancer | PG13 | GALV | T cell | yes | |
| M66 | Kaplan | NIH Pediatric Oncology Branch | NCT02107963 | cancer | PG13 | GALV | T cell | yes | |
| M70 | Kiem | Fred Hutchison Cancer Center | NCT00669669 | cancer | transient | GALV | CD34+ HSC | yes | |
| M76 | Booth/Thrasher | Great Ormond Street Hospital | NCT01175239 | X-linked SCID | transient | GALV | CD34+ HSC | yes | X |
| M77 | Brenner | Baylor College of Medicine | NCT01822652 | cancer | PG13 | GALV | T cell | yes | |
| M83 | Rosenberg | NIH Surgery Branch | NCT02111850 | cancer | PG13 | GALV | T cell | yes | |
| M85 | Rosenberg | NIH Surgery Branch | NCT01967823 | cancer | PG13 | GALV | T cell | yes | |
| M95 | Ramos | Baylor College of Medicine | NCT02050347 | cancer | PG13 | GALV | T cell | yes | |
| M96 | Ramos | Baylor College of Medicine | NCT01853631 | cancer | PG13 | GALV | T cell | yes | |
| M102 | Heslop | Baylor College of Medicine | NCT01953900 | cancer | PG13 | GALV | T cell | yes | |
| M105 | Heslop | Baylor College of Medicine | NCT02065362 | cancer | PG13 | GALV | T cell | yes | |
| M106 | Norberg/Hinrichs | NIH Center for Cancer Research | NCT02280811 | cancer | PG13 | GALV | T cell | yes | |
| M107 | J. Park | Memorial Sloan Kettering | NCT00466351 | cancer | PG13 | GALV | T cell | yes | |
| M112 | Kochenderfer | NIH Surgery Branch | NCT02215967 | cancer | PG13 | GALV | T cell | yes | |
| M113 | Slovin | Memorial Sloan Kettering | NCT01140373 | cancer | PG13 | GALV | T cell | yes | |
| M116 | Curran | Memorial Sloan Kettering | NCT01430390 | cancer | PG13 | GALV | T cell | yes | |
| M122 | Ribas | University of CA Los Angeles | NCT01697527 | cancer | PG13 | GALV | T cell | yes | |
| M123 | Adusumilli | Memorial Sloan Kettering | NCT02414269 | cancer | PG13 | GALV | T cell | yes | |
| M141 | Ribas | University of CA Los Angeles | NCT02070406 | cancer | PG13 | GALV | T cell | yes | |
| M142 | O'Cearbhaill | Memorial Sloan Kettering | NCT02498912 | cancer | PG13 | GALV | T cell | yes | |
| M143 | Rosenberg | NIH Surgery Branch | NCT02153905 | cancer | PG13 | GALV | T cell | yes | |
| M144 | Rosenberg | NIH Surgery Branch | NCT02062359 | cancer | PG13 | GALV | T cell | yes | |
| M152 | J. Park | Memorial Sloan Kettering | NCT02792114 | cancer | PG13 | GALV | T cell | yes | |
| M161 | Mailankody | Memorial Sloan Kettering | NCT03070327 | cancer | PG13 | GALV | T cell | yes | |
| M168 | J. Park | Memorial Sloan Kettering | NCT03085173 | cancer | PG13 | GALV | T cell | yes | |
| M178 | Ramos | Baylor College of Medicine | NCT02379520 | cancer | PG13 | GALV | T cell | yes | |
| M226 | Ribas | University of CA Los Angeles | NCT03240861 | cancer | PG13 | GALV | T cell | yes | |
| M228 | Ribas | University of CA Los Angeles | NCT02775292 | cancer | PG13 | GALV | T cell | yes |
ADA = adenosine deaminase deficiency, SCID = severe combined immunodeficiency, PG13 = PG13 packaging cell line, GALV = Gibbon ape leukemia virus, SIN = self-inactivating LTR.
Investigators were asked to provide the time from cell product transfusion to sample collection (Table 2). The earliest samples described in this report were evaluated on March 9, 2011. For patients who received multiple transfusions (16.7%), the time of RCR testing was calculated from the first administration of a transduced cell product. A total of 737 patients had at least one sample tested. The distribution of timing is shown in Figure 1. Pre-treatment samples accounted for approximately 17.5% of all the samples tested. Not all patients had pre-treatment samples submitted, and a few patients screened did not go on to get transduced cells or received them after the data collection time points in this study. Most samples were collected between 90 and 364 days, and approximately 25% post-treatment samples were collected 1 or more years after treatment. All samples analyzed were negative for RCR.
Table 2.
Timing of replication competent retrovirus assessment by study
| IU master agreement number | Number of unique patients | Number of patients with multiple infusions | Pre-treatment | <90 days | 90–179 days | 180–364 days | year 1 | year 2 | year 3 | year 4 | year 5 or greater |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M2 | 14 | 7 | 2 | 4 | 16 | 17 | 15 | 4 | 0 | 0 | 0 |
| M4 | 4 | 0 | 4 | 0 | 4 | 4 | 7 | 3 | 4 | 3 | 2 |
| M5 | 2 | 0 | 1 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 0 |
| M6 | 9 | 0 | 0 | 0 | 4 | 6 | 3 | 0 | 0 | 0 | 0 |
| M7 | 10 | 4 | 10 | 0 | 11 | 10 | 10 | 0 | 0 | 0 | 0 |
| M8 | 5 | 0 | 0 | 0 | 3 | 4 | 4 | 1 | 0 | 0 | 0 |
| M9 | 12 | 3 | 9 | 3 | 12 | 13 | 9 | 0 | 0 | 0 | 0 |
| M10 | 10 | 7 | 3 | 8 | 12 | 10 | 7 | 2 | 1 | 0 | 0 |
| M11 | 21 | 9 | 9 | 8 | 18 | 23 | 17 | 2 | 0 | 0 | 0 |
| M12 | 28 | 4 | 32 | 2 | 23 | 23 | 15 | 0 | 0 | 0 | 0 |
| M16 | 8 | 6 | 6 | 9 | 12 | 12 | 17 | 2 | 0 | 0 | 0 |
| M18 | 17 | 5 | 6 | 4 | 16 | 13 | 15 | 2 | 0 | 0 | 0 |
| M20 | 10 | 6 | 1 | 5 | 9 | 12 | 8 | 2 | 0 | 0 | 0 |
| M22 | 3 | 1 | 1 | 0 | 3 | 3 | 4 | 0 | 0 | 0 | 0 |
| M23 | 51 | 2 | 47 | 0 | 38 | 25 | 19 | 3 | 0 | 0 | 0 |
| M27 | 8 | 3 | 0 | 0 | 8 | 8 | 9 | 0 | 0 | 0 | 0 |
| M31 | 3 | 0 | 1 | 1 | 2 | 3 | 1 | 1 | 0 | 1 | 0 |
| M40 | 8 | 0 | 0 | 2 | 6 | 5 | 0 | 0 | 0 | 0 | 0 |
| M41 | 39 | 8 | 0 | 18 | 26 | 25 | 6 | 1 | 0 | 1 | 2 |
| M42 | 31 | 0 | 0 | 8 | 33 | 11 | 9 | 1 | 2 | 3 | 3 |
| M43 | 15 | 0 | 0 | 8 | 10 | 1 | 4 | 0 | 0 | 0 | 0 |
| M44 | 8 | 0 | 0 | 4 | 6 | 2 | 1 | 0 | 0 | 0 | 0 |
| M45 | 9 | 0 | 0 | 7 | 4 | 2 | 1 | 0 | 0 | 0 | 0 |
| M46 | 9 | 5 | 0 | 4 | 9 | 11 | 9 | 2 | 0 | 0 | 0 |
| M48 | 2 | 2 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
| M49 | 9 | 0 | 1 | 0 | 2 | 4 | 3 | 0 | 0 | 0 | 0 |
| M56 | 11 | 0 | 13 | 0 | 11 | 4 | 6 | 3 | 0 | 0 | 0 |
| M57 | 56 | 14 | 49 | 10 | 30 | 20 | 9 | 2 | 0 | 0 | 0 |
| M58 | 8 | 0 | 5 | 0 | 6 | 6 | 3 | 1 | 1 | 1 | 0 |
| M59 | 21 | 2 | 21 | 1 | 13 | 10 | 8 | 0 | 0 | 0 | 0 |
| M60 | 18 | 0 | 0 | 7 | 15 | 9 | 6 | 0 | 0 | 0 | 0 |
| M61 | 13 | 0 | 0 | 5 | 11 | 6 | 2 | 1 | 1 | 0 | 0 |
| M62 | 42 | 0 | 0 | 15 | 22 | 37 | 18 | 2 | 3 | 3 | 5 |
| M66 | 14 | 0 | 14 | 0 | 10 | 6 | 2 | 0 | 0 | 0 | 0 |
| M70 | 5 | 0 | 0 | 0 | 4 | 3 | 2 | 0 | 0 | 0 | 0 |
| M76 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 |
| M77 | 11 | 5 | 0 | 3 | 9 | 10 | 6 | 0 | 0 | 0 | 0 |
| M83 | 17 | 0 | 0 | 13 | 6 | 9 | 2 | 2 | 1 | 0 | 0 |
| M85 | 10 | 0 | 0 | 6 | 7 | 9 | 3 | 0 | 0 | 0 | 0 |
| M95 | 5 | 0 | 0 | 1 | 4 | 3 | 2 | 0 | 0 | 0 | 0 |
| M96 | 12 | 4 | 0 | 1 | 12 | 14 | 7 | 0 | 0 | 0 | 0 |
| M102 | 6 | 3 | 0 | 0 | 6 | 8 | 3 | 0 | 0 | 0 | 0 |
| M105 | 8 | 1 | 0 | 1 | 6 | 6 | 5 | 0 | 0 | 0 | 0 |
| M106 | 9 | 2 | 0 | 6 | 2 | 7 | 5 | 0 | 0 | 0 | 0 |
| M107 | 24 | 0 | 21 | 0 | 21 | 13 | 11 | 0 | 0 | 2 | 1 |
| M112 | 20 | 3 | 17 | 0 | 18 | 14 | 10 | 0 | 0 | 0 | 0 |
| M113 | 7 | 0 | 3 | 0 | 4 | 4 | 2 | 0 | 0 | 0 | 0 |
| M116 | 6 | 3 | 4 | 4 | 2 | 4 | 2 | 0 | 0 | 0 | 0 |
| M122 | 7 | 0 | 0 | 1 | 1 | 4 | 0 | 1 | 0 | 0 | 0 |
| M123 | 12 | 0 | 12 | 0 | 3 | 2 | 0 | 0 | 0 | 0 | 0 |
| M141 | 6 | 0 | 0 | 0 | 3 | 2 | 1 | 0 | 0 | 0 | 0 |
| M142 | 13 | 0 | 14 | 0 | 10 | 7 | 2 | 0 | 0 | 0 | 0 |
| M143 | 2 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| M144 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| M152 | 5 | 0 | 5 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| M161 | 11 | 6 | 11 | 3 | 6 | 2 | 0 | 0 | 0 | 0 | 0 |
| M168 | 5 | 2 | 6 | 0 | 5 | 3 | 0 | 0 | 0 | 0 | 0 |
| M178 | 3 | 3 | 0 | 0 | 2 | 5 | 0 | 0 | 0 | 0 | 0 |
| M226 | 2 | 2 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
| M228 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Totals | 737 | 123 | 329 | 174 | 546 | 482 | 312 | 38 | 14 | 15 | 14 |
Figure 1.

Number of samples analyzed using qPCR targeting vector envelope pseudotype
X axis is the time from first exposure to vector-transduced cells. Pre = pre-treatment sample.
We also reviewed qPCR assay performance. All testing was performed under current good manufacturing practice (cGMP) guidelines. In addition to the samples reported in Table 2, we received 15 samples that had insufficient DNA and were not analyzed. An additional 11 samples were tested and were negative for envelope but had insufficient DNA to meet the assay requirements of 0.2 ug of DNA per reaction, so these results are reported as inconclusive. In four samples, the assay detected GALV sequence, and an out-of-specification investigation was initiated. Working with the investigator, it was determined the samples were contaminated with GALV-containing plasmid at the submitting investigator’s site. Additional sample material for the subjects tested negative for RCR, and the initial samples were deemed false positives.
Discussion
Retroviral vectors have been in use for the past 30 years, and the FDA requirements for RCR testing have changed over time. In 1998, the “Guidance for Human Somatic Cell Therapy and Gene Therapy”15 was published and stated, “Patients given retrovirus-related products should be monitored for RCR exposure. Please consult CBER for guidance.” At that time, some investigators were asked to perform monitoring annually. In 2006, the FDA published a specific guidance for RCR testing that recommended “pre-treatment, 3 months, 6 months, 1 year after treatment, and yearly thereafter. If all post-treatment assays are negative during the first year, the yearly samples should be archived.”16 In 2020, this guidance was replaced by “Testing of Retroviral Vector-Based Human Gene Therapy Products for Replication Competent Retrovirus During Product Manufacture and Patient Follow-up. Guidance for Industry,” which kept the testing schedule the same except archiving is not required if all samples in year 1 were negative.10 Given that the first sample in our study was analyzed in 2011, the changing expectations are reflected in a variation in the length of time over which samples were collected.
In this study we compiled results of blood monitoring for RCR in 60 clinical trials. Cell lines of both human and murine origin were utilized in generating vector products, and the majority of studies utilized vectors with intact LTRs, a vector component with a higher risk of RCR than self-inactivating LTRs. While we have some samples out as far as 9 years, the majority were collected within the first 2 years after exposure to retroviral vector. All 338 pre-treatment samples were negative for RCR sequences, suggesting humans have a low incidence of sequences with homology to GALV envelope. All 1,595 samples collected post-infusion of genetically modified cells were negative for RCR using qPCR testing. These findings are consistent with NGVB analysis of gene therapy products using a sensitive RCR biological assay.12 In that study, RCR was undetectable in 282 ex vivo products from 14 clinical trials. This outcome is in line with earlier results noting a lack of RCR in ex vivo transduced products.17,18
Limitations of the study include the small number of vector products generated by transient transfection, as most studies utilized the stable PG13 cell line for vector manufacture. The FDA has stated a preference for stable packaging cell lines given their potential to limit lot-to-lot variability that can be seen with transient transfection methods.10 Nevertheless, transient transfection methods are being increasingly employed, as they do not require the extended time for clone selection, generation of master cell banks, and expansion for vector production. The extensive cell expansion associated with packaging cell lines is also predicted to increase likelihood of recombination and RCR generation. From a safety perspective, our findings add to data from prior studies documenting the generation of RCR-free vector by PG13 cells.12,17,18 It is unclear whether this finding can be extrapolated to other packaging cell lines. Part of the success of PG13 may be in the use of the xenotropic GALV envelope in a murine cell line (NIH3T3). Murine cells lack the receptor for the GALV envelope, so if a GALV pseudotype RCR developed, it would not be able to propagate in culture.
Another limitation is the relatively small number of patients in several of the studies. This issue arises because our study design looks at the time frame when samples were tested. Nonetheless, the inclusion of studies with smaller number of accrued patients allows us to survey more vector products, as the majority of studies used distinct vector products specific to each clinical trial.
The majority of clinical trials in our study were related to cancer immunotherapy. This finding is expected given the time frame of samples collection. The first report of malignancy related to retroviral vectors occurred in 2003 in a trial aimed at treating patients with X-linked SCID.19 This was followed by reports of similar adverse events in trials for chronic granulomatous disease,8 Wiskott-Aldrich syndrome,9 and most recently adenosine deaminase deficiency.20 All these trials targeted HSCs, and our study received samples when most investigators targeting HSCs had moved away from gammaretroviral vectors. To date, malignancy has not been reported when retroviral vectors are used to transduce in T cells, a practice that continues in many cancer trials.
In conclusion, the qPCR testing presented here provides further evidence that patients treated with retroviral vectors have a low risk of RCR exposure. The data also support the safety of current manufacturing methods and release testing requirements for retroviral vector products. This indicates that limited testing of ex vivo products and patient monitoring are sufficient when using established manufacturing systems, although the risk of RCR development will need to be assessed experimentally for new vector production methods, new pseudotypes, and novel packaging cell lines.
Materials and methods
Collection of study data
For this survey, investigators were provided an Excel spreadsheet that contained the NGVB ID number of the samples tested, the lab completion date, and the results. Investigators were requested to provide the following data: (1) ClinicalTrials.gov number, (2) the general indication of the trial (e.g., CAR T cell immunotherapy, genetic disease, etc.), (3) the cell type transduced (e.g., T cell, CD4, CD8, hematopoietic stem cell), (4) production method (transient transfection or packaging cell line), (5) the specific cell line used, if a packaging cell line was used, (6) were the vector supernatant and end of production cells tested and found to be negative for RCR? (Yes/No), and (7) the time from infusion of gene transduced cells to sample collection and whether patients received multiple infusions of transduced cells.
Sample processing
Investigators are requested to submit de-identified samples to the NGVB as DNA, buffy coat cells, isolated cells, or blood. Samples are isolated using either the Qiagen Puregene DNA Isolation Kit or the QIAamp DNA Blood Mini Kit (both available Qiagen USA, Germantown, Maryland). Isolations are performed under cGMP guidelines. DNA is evaluated for purity (OD 260/280), and concentration to ensure sufficient material is present to perform triplicate reactions with 0.2 ug of DNA per reaction.
qPCR reaction
Detection of target sequences was performed with the ABI 7500 (Applied Biosystems, Foster City, CA) using multiplex reactions with primers and probe for the target sequence and a second set of primers and probe for human ApoB. The ApoB determination was used to confirm that a reaction contained a minimum of 0.2 ug of DNA. Negative controls include water (NTC) and target-negative human DNA. Positive control samples were generated from plasmid DNA containing the target sequence diluted in the target-negative control DNA. Positive controls over a 5-log range (from 10 to 105 copies per 0.2 ug DNA) were included in each run to determine the standard curve. Probe and primer sets were qualified by evaluating slope, intercept, and R2 for the standard curve. The NGVB has set the range of amplification efficiency of 85%–105%, which corresponds to a slope of −3.74 to −3.21. R2 limits were values between 0.981 and 1. The Intercept was determined for each probe and primer set, and the acceptable criterion is +2 standard deviations from nine or more runs. The intercept for GALV is 37.74–41.05 and for amphotropic envelope is 38.34–39.13. For the assays to be acceptable, (1) the amplification plots of the NTC must not cross the threshold, (2) the amplification plots of the negative-amplification control must not cross the threshold, and (3) the acceptance criteria for the specific probe and primer set must be met. The ApoB acceptability criteria are determined for each run by calculating the mean and standard deviation Ct value for all samples in the assay. The acceptable range for test articles is less than the mean + 2 standard deviations.
A sample is considered positive if any of the triplicate reactions are above the limits of detection for the envelope (10 copies per 0.2 μg of DNA), regardless of the total DNA content in the reaction. For a sample to be considered negative, all replicates must be less than 10 copies per reaction, and at least two of the three reactions must meet the criteria of >0.2 ug of DNA. If only one or none of the reactions meet the 0.2 ug of DNA requirement, the sample is retested (if sufficient DNA is available). If the repeat remains negative for envelope sequence and fails to show sufficient DNA to meet acceptance criteria, the samples are reported as inconclusive.
Acknowledgments
The NGVB (PI: K.C.) is funded by the National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (NHLBI). Initial funding was provided in grant P40HL116212, and since 2019 funding has been via Contract 75N92019D00018, with additional support from the National Cancer Institute (NCI) under Contract 75N92020F00002. The views expressed in this article are those of the authors and do not necessarily represent the views of the NIH, NHLBI, NCI, or the United States Department of Health and Human Services. The graphic abstract was created with BioRender.
P.S.A. acknowledges grants from the NIH (P30 CA008748, R01 CA236615-01, and R01 CA235667), the U.S. Department of Defense (CA180889,and CA200437), the Batishtia Fellowship, the Comedy vs Cancer Award, the Dalle Pezze Foundation, the Derfner Foundation, the Esophageal Cancer Education Fund, the Geoffrey Beene Foundation, the Memorial Sloan Kettering Technology Development Fund, the Miner Fund for Mesothelioma Research, the Mr. William H. Goodwin and Alice Goodwin, the Commonwealth Foundation for Cancer Research, and the Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center. M.B., B.G., H.H., and C.R. acknowledge the NIH, NCI P50CA126752, 1U54CA232568-01, P01CA094237, P30CA125123, NHLBI U54HL081007, and Stand Up To Cancer (SU2C)/St Baldrick’s Pediatric Cancer Dream Team Translational Research Grant SU2C-AACR-DT1113, SU2C/American Association for Cancer Research (AACR) 604817, Meg Vosburg T-Cell Lymphoma Dream Team, and the Leukemia and Lymphoma Society. SU2C is a program of the Entertainment Industry Foundation administered by the AACR. H-P.K. acknowledges the Indiana University Virus Production Facility for production of the clinical-grade virus for this study R01CA114218, K01DK076973. J.K acknowledges the work of Danielle Natrakul for laboratory contributions. S.M. acknowledges NCI Memorial Sloan Kettering Core Grant P30 CA008748 and Juno/Bristol-Myers Squibb. N.N.S., Intramural Research Program, NCI, NIH (ZIA BC 011823). R.O. acknowledges NIH/NCI Cancer Center Support Grant P3CA008748 and P01CA190174-05. A.R. acknowledges the Parker Institute for Cancer Immunotherapy, NIH grants R35 CA197633 and P01 CA168585. I.R. acknowledges NCI P30 CA08748 and PO1 CA008748-T Cell Therapies. D.A.W. acknowledges NIH U01 AI087628 and GMP vector was provided by Orchard Therapeutics.
Author contributions
K.C., J.Y., K.H., L.D., and T-Y.L. designed the methods, qualified assays, and wrote the article; the remaining authors provided data regarding the manufacturing method and timing of sample collection. All authors contributed to review and editing of the manuscript.
Declaration of interests
Indiana University has licensed technology to Charles River Laboratories and Genezen Inc. based on unrelated work developed by K.Co., L.D., and T-Y. L., who each receive royalties. P.A.S receives research funding from ATARA Biotherapeutics; Scientific Advisory Board and Consultant: ATARA Biotherapeuticcs, Bayer, Carisma Therapeutics, Imugene, ImmPactBio, Johnston & Johnston, Orion, Outpace Bio; research funding and intellectual property licensed to ATARA Biotherapeutics. M.B. has equity in AlloVir, Marker Therapeutics, and Tessa Therapeutics; serves on the Scientific Advisory Board for Tessa Therapeutics, Marker Therapeutics, Allogene, Walking Fish, Cell Genix, Kuur, Turnstone Biologics, Posedia, Tscan, and Bluebird Bio; and receives royalities from Takeda and Bellicum. K.Cu. is a consultant to Novartis and receives research support from Novartis, Cellectis, and Celgene. B.G. has equity in AlloVir, QBRegulatory LLC, and QBRegulatory, and provides consulting services to AlloVir, Marker Therapeutics, Tessa Therapeutics, Lokon Pharma and Proxima Clinical Research. H.H. has equity in AlloVir and Marker Therapeutics, and serves on the Scientific Advisory Board for Gilead Biosciences, Novartis, Tessa Therapeutics, Marker Therapeutics, Kiadis, PACT Pharma, Mesoblast, and receives research support from Tessa Therapeutics and Kuur Therapeutics. C.S.H. performs consulting and advisory board services for Neogene Therapeutics, Capstan Therapeutics, GlaxoSmithKline, and PACT Pharma; patents and royalties for NIH patents in cell and gene therapy and immunotherapy; research funding from Neogene Therapeutics and T-Cure Biosciences. J.K. has research support and royalty from Kite, a Gilead Company; and receives research funding from Bristol-Myers Squibb, Royalties: Kyverna. D.B.K. is a paid Scientific Advisory Board member for Allogene Therapeutics, ImmunoVec, Pluto Therapeutics, MyoGene Bio, Innoskel and an ad hoc consultant for Cimeio Therapeutics, TransformaTx, and Bluebird Bio. S.M. receives research funding from Allogene Therapeutics, Takeda Oncology, Juno Therapeutics, Bristol-Myers Squibb, Janssen Oncology, Fate Therapeutics and serves on the advisory panel for Legend Biotech, Evicore, Janssen Oncology, BioAscend, Optum Oncology, and EcoR1; and receives honoraria from Plexus Communication, OncLive, Physician Education Resource. R.O. receives compensation from Tesaro/GSK, Regeneron, Seattle Genetics, Fresenius Kabi, Gynecologic Oncology Foundation, Bayer, Curio/Onclive, R-Pharm, Immunogen, Hitech Health; non-compensated steering committee member for the PRIMA, Moonstone (Tesaro/GSK) and DUO-O (AstraZeneca) studies; non-compensated advisor for Carina Biotech. A.R. has received honoraria from consulting with Amgen, Bristol-Myers Squibb and Merck, is or has been a member of the scientific advisory board and holds stock in Advaxis, Appia, Apricity, Arcus, Compugen, CytomX, Highlight, ImaginAb, ImmPact, ImmuneSensor, Inspirna, Isoplexis, Kite-Gilead, Lutris, MapKure, Merus, PACT, Pluto, RAPT, Synthekine and Tango; and has received research funding from Agilent and from Bristol-Myers Squibb through Stand Up to Cancer (SU2C), and patent royalties from Arsenal Bio. C.R. is Scientific Board Member for Novartis; research support from Tessa Therapeutics and Kuur Therapeutics. I.R. has equity or property rights with FloDesign Sonics, Takeda Pharmaceuticals, Fate Therapeutics, Mnemo Therapeutics, Juno Therapeutics; Services and Travel: Center for Commercialization of Cancer, Akron. C.S. is a consultant for Juno Therapeutics, Sanofi-Genzyme, Spectrum Pharmaceuticals, Novartis, Genmab, Precision Biosciences, Kite/a Gilead Company, Celgene/BMS, Gamida Cell, Karyopharm Therapeutics, Ono Pharmaceuticals, MorphoSys, CSL Behring, Syncopation Life Sciences, CRISPR Therapeutics, and GSK; research funds: Juno Therapeutics, Celgene/BMS, Bristol-Myers Squibb, Precision Biosciences, Actinium Pharmaceuticals and Sanofi-Genzyme. S.S.: research funding: Sanofi-Aventis, Poseida Pharmaceuticals, Gilead Sciences, Inc, Prostate Cancer Foundation; Honoraria: Physician Education Resources, Janssen, Pfizer, Tolmar. D.A.W. holds intellectual property rights to the vector utilized in the trial reported in this manuscript.
References
- 1.Muenchau D.D., Freeman S.M., Cornetta K., Zwiebel J.A., Anderson W.F. Analysis of retroviral packaging lines for generationof replication-competent virus. Virology. 1990;176:262–265. doi: 10.1016/0042-6822(90)90251-l. [DOI] [PubMed] [Google Scholar]
- 2.Bosselman R.A., Hsu R.-Y., Bruszewski J., Hu S., Martin F., Nicolson M. Replication-defective chimeric helper proviruses and factors affecting generation of competent virus: expression of Moloney Leukemia Virus structural genes via the metallothionein promoter. Mol. Cell. Biol. 1987;7:1797–1806. doi: 10.1128/mcb.7.5.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cornetta K., Nguyen N., Morgan R.A., Muenchau D.D., Hartley J.W., Blaese R.M., Anderson W.F. Infection of human cells with murine amphotropic replication-competent retroviruses. Hum. Gene Ther. 1993;4:579–588. doi: 10.1089/hum.1993.4.5-579. [DOI] [PubMed] [Google Scholar]
- 4.Donahue R.E., Kessler S.W., Bodine D., McDonagh K., Dunbar C., Goodman S., Agricola B., Byrne E., Raffeld M., Moen R., et al. Helper virus induction T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. J. Exp. Med. 1992;176:1125–1135. doi: 10.1084/jem.176.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornetta K., Lin T.-Y., Pellin D., Kohn D.B. Meeting FDA guidance recommendations for replication competent virus and insertional oncogenesis testing. Mol. Ther. Methods Clin. Dev. 2022 doi: 10.1016/j.omtm.2022.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hacein-Bey-Abina S., Garrigue A., Wang G.P., Soulier J., Lim A., Morillon E., Clappier E., Caccavelli L., Delabesse E., Beldjord K., et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howe S.J., Mansour M.R., Schwarzwaelder K., Bartholomae C., Hubank M., Kempski H., Brugman M.H., Pike-Overzet K., Chatters S.J., de Ridder D., et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stein S., Ott M.G., Schultze-Strasser S., Jauch A., Burwinkel B., Kinner A., Schmidt M., Krämer A., Schwäble J., Glimm H., et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 9.Braun C.J., Boztug K., Paruzynski A., Witzel M., Schwarzer A., Rothe M., Modlich U., Beier R., Göhring G., Steinemann D., et al. Gene therapy for Wiskott-Aldrich syndrome--long-term efficacy and genotoxicity. Sci. Transl. Med. 2014;6:227ra33. doi: 10.1126/scitranslmed.3007280. [DOI] [PubMed] [Google Scholar]
- 10.U.S Food and Drug Administration . Guidance for Industry; 2020. Testing of Retroviral Vector-Based Human Gene Therapy Products for Replication Competent Retrovirus during Product Manufacture and Patient Follow-Up. [DOI] [PubMed] [Google Scholar]
- 11.Cornetta K., Matheson L., Long R., Duffy L. The national gene vector biorepository: eleven years of providing resources to the gene therapy community. Hum. Gene Ther. 2020;31:145–150. doi: 10.1089/hum.2019.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cornetta K., Duffy L., Feldman S.A., Mackall C.L., Davila M.L., Curran K.J., Junghans R.P., Tang J.Y., Kochenderfer J.N., O'Cearbhaill R., et al. Screening clinical cell products for replication competent retrovirus: the national gene vector biorepository experience. Mol. Ther. Methods Clin. Dev. 2018;10:371–378. doi: 10.1016/j.omtm.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller A.D., Garcia J.V., Von Suhr N., Lynch C.M., Wilson C., Eiden M.V. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J. Virol. 1991;65:2220–2224. doi: 10.1128/jvi.65.5.2220-2224.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller A.D., Buttimore C. Redesign of retrovirus packaging cell lines to avoid recombination leading to helper virus production. Mol. Cell. Biol. 1986;6:2895–2902. doi: 10.1128/mcb.6.8.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.U.S Food and Drug Administration . 1998. Guidance for Human Somatic Cell Therapy and Gene Therapy. [Google Scholar]
- 16.U.S Food and Drug Administration . 2006. Guidance for Industry - Supplemental Guidance on Testing for Replication Competent Retrovirus in Retroviral Vector Based Gene Therapy Products and during Follow-Up of Patients in Clinical Trials Using Retroviral Vectors. [DOI] [PubMed] [Google Scholar]
- 17.Bear A.S., Morgan R.A., Cornetta K., June C.H., Binder-Scholl G., Dudley M.E., Feldman S.A., Rosenberg S.A., Shurtleff S.A., Rooney C.M., et al. Replication-competent retroviruses in gene-modified T cells used in clinical trials: is it time to revise the testing requirements? Mol. Ther. 2012;20:246–249. doi: 10.1038/mt.2011.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyon D., Lapteva N., Gee A.P. Absence of replication-competent retrovirus in vectors, T cell products, and patient follow-up samples. Mol. Ther. 2018;26:6–7. doi: 10.1016/j.ymthe.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hacein-Bey-Abina S., von Kalle C., Schmidt M., Le Deist F., Wulffraat N., McIntyre E., Radford I., Villeval J.L., Fraser C.C., Cavazzana-Calvo M., et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 20.Orchad Therapeutics . 2020. Orchard Statement on Strimvelis®, a Gammaretroviral Vector-Based Gene Therapy for ADA-SCID. [Google Scholar]