

Abstract
RNA structures regulate a wide range of processes in biology and disease, yet small molecule chemical probes or drugs that can modulate these functions are rare. Machine learning and other computational methods are well poised to fill gaps in knowledge and overcome the inherent challenges in RNA targeting, such as the dynamic nature of RNA and the difficulty of obtaining RNA high-resolution structures. Successful tools to date include principal component analysis, linear discriminate analysis, k-nearest neighbor, artificial neural networks, multiple linear regression, and many others. Employment of these tools has revealed critical factors for selective recognition in RNA:small molecule complexes, predictable differences in RNA- and protein-binding ligands, and quantitative structure activity relationships that allow the rational design of small molecules for a given RNA target. Herein we present our perspective on the value of using machine learning and other computation methods to advance RNA:small molecule targeting, including select examples and their validation as well as necessary and promising future directions that will be key to accelerate discoveries in this important field.
Keywords: machine learning, small molecule, RNA, cheminformatics, pattern recognition, quantitative structure activity relationships
INTRODUCTION
Modulating RNA biological function by RNA-targeting small molecules (SMs) can be extremely challenging yet the potential is immeasurable. The ability to control RNA-dependent processes with drug-like molecules will not only reveal novel fundamental biology but create pathways to orally available therapeutics for a wide range of human diseases (McKnight and Heinz 2003; Hong et al. 2014; Bernat and Disney 2015; Slaby et al. 2017; Fedorova et al. 2018; Lekka and Hall 2018; Tang et al. 2020; Zamani and Suzuki 2021). The medicinal chemistry community has been exhibiting a growing interest in targeting nonribosomal RNAs (Warner et al. 2018; Costales et al. 2020), and venture capital and pharmaceutical companies are making significant investments in this space. The first and only SM FDA approved drug targeting nonribosomal RNA, risdiplam (Evrysdi), was approved for treatment of spinal muscular atrophy in August of 2020 (Ratni et al. 2018; Markati et al. 2022). However, this gold rush has revealed its Achilles’ heel in the form of our still incomplete knowledge of the molecular recognition underlying RNA–SM interactions.
Our knowledge of SM recognition of biomacromolecules comes almost exclusively from protein targeting, but the chemical and physical properties of RNA are distinct (Falese et al. 2021). For example, RNA has four nucleobase monomers that have similar chemical functionality, namely one to two heteroaromatic rings substituted with amine and carbonyl functional groups (Fig. 1). Protein amino acid side chains run the gamut of chemical functionality, including a wide range of pKa's (∼3–13) leading to both positive and negative charges, alkyl and aromatic groups, a range of hydrogen bonding acceptors and donors, and thiols capable of disulfide interactions. This diversity combined with the neutral amide backbone of proteins allows close packing and the formation of hydrophobic pockets, both of which are more difficult with the densely charged backbone of RNA (Fig. 1). In addition, specific drivers of molecular recognition are often inferred from high-resolution 3D structures, yet there are fewer than 50 high resolution (<2.7 Å) unique RNA:SM complexes, which is less than 0.5% of the respective number for proteins (Padroni et al. 2020). This paucity is in part due to challenges in RNA structure determination techniques, such as X-ray crystallography, as a result of the ability of RNA to adopt multiple conformational states and its highly charged backbone. These challenges hinder both a fundamental understanding of RNA:SM recognition and the feasibility of structure-guided drug design. Therefore, alternative approaches are required to advance the field of SM RNA targeting.
FIGURE 1.

Schematic representation of the four RNA nucleosides and negatively charged backbone. Curved dashed lines highlight the amine and carbonyl functional groups as sites of hydrogen bonding on heteroaromatic rings.
Pattern recognition and other machine-learning methods have proven to be a valuable alternative to elucidate the drivers of RNA:SM recognition and SM functional impact without a priori knowledge of RNA 3D structure. Methods such as linear discriminate analysis (LDA), principal component analysis (PCA), and artificial neural networks (ANN) have been used to identify patterns of molecular recognition in a wide variety of analytes, from ions to whole cells, and more recently RNA (Eubanks and Hargrove 2019; Yazdani et al. 2022). At a high level, both LDA and PCA produce a set of linear combinations of the input variables that are plotted orthogonally for data visualization and provide insight into the properties that drive clustering. In LDA, the goal is to increase clustering within and maximize differences between predefined classes. PCA does not use predefined classes but instead maximizes variance and covariance within the entire data set. The most common ANNs are feedforward neural networks (NNs) in which input data is weighted and transformed in a hidden layer to give the desired output, often classifications. While powerful, ANNs are often a “black box” in terms of what factors lead to classification.
In the case of RNA:SM recognition, clustering in LDA and PCA can be driven by the SMs or by the RNA. In the former case, SM physicochemical and other quantifiable properties have been combined with a behavior, that is, RNA binding or protein binding, to generate insight into the most critical SM properties for achieving those behaviors (Morgan et al. 2017). Conversely, patterns in RNA structures have been elucidated via differential SM binding to better understand the RNA features that allow for specific RNA:SM interactions (Eubanks et al. 2017; Eubanks and Hargrove 2017). More complex machine-learning methods, such as the tree-like method Tree MAP, have allowed for prediction of RNA-binding behavior versus protein-binding behavior (Yazdani et al. 2022). For a specific RNA target, quantitative structure–activity relationship (QSAR) studies can substitute for structure-based drug design (Cai et al. 2022). In this method, hundreds of SM physicochemical and other properties are calculated, and the descriptors most important for differentiation in the studied behavior (e.g., binding affinity or kinetic rate constants) are selected. These select descriptors can be used to build predictive models using a variety of machine-learning approaches, including multiple linear regression (MLR) and ensemble tree methods. Herein, we overview representative examples from our laboratory and others to leverage machine-learning methods to elucidate the drivers of RNA:SM recognition as well as our perspective on the needs and opportunities moving forward. We hope to inspire others interested in RNA:SM targeting to utilize these and other machine-learning methods, as we collectively build tools that will allow SM modulators to be identified for the full range of biologically functional RNA structures.
REVEALING PATTERNS IN SELECTIVE RNA:SMALL MOLECULE INTERACTIONS
Cheminformatic properties of bioactive RNA ligands
The recent expansion of the number of reported bioactive RNA ligands opens the opportunity to use cheminformatics tools and machine learning to better understand RNA:SM recognition. Based on the differences in chemical functionalities of RNA and protein, our initial hypothesis was that bioactive RNA ligands have specific molecular properties that drive selectivity in RNA targeting and, at the same time, are distinct from protein ligands. Indeed, structural and chemical differences between protein-binding pockets and RNA motifs supported the existence of specific molecular recognition patterns in RNA-targeting molecules versus protein ligands (Hewitt et al. 2019; Padroni et al. 2020).
To test this hypothesis, we built the RNA-targeted BIoactive ligaNd Database (R-BIND) (Morgan et al. 2017). Created in 2017, R-BIND incorporated molecules reported in the literature for in vitro binding to nonribosomal RNA through noncovalent interactions and was the first database to include biological activity in cellular and/or animal models as an essential criterion (Morgan et al. 2017, 2019; Donlic et al. 2022). Aminoglycosides (Ags), covalent ligands, peptides, and oligonucleotides were excluded in this analysis due to dramatic differences in molecular properties relative to traditional “drug-like” small molecules. R-BIND characterized ligands with 20 physicochemical and structural descriptors as well as three-dimensional shapes, that is, disk-, rod-, and sphere-like, using principal moments of inertia (PMI) (Morgan et al. 2017). Comparison of the properties found in FDA-approved SM drugs, as a source of bioactive protein ligands, revealed that R-BIND occupied a specific subregion of the space occupied by the FDA library, considered as “drug-like” space (Fig. 2A; Donlic et al. 2022). Here, k-NN clustering analysis determined the quantitative overlap of the two libraries, revealing that 31% of R-BIND SM and 9% of FDA library overlap with the other library's cluster (Morgan et al. 2017). The differentiation between R-BIND and FDA libraries was described in terms of physicochemical, structural and spatial descriptors (Donlic et al. 2022). Several physicochemical and structural descriptors were significantly different between the two libraries. Cell-based partitioning assisted the quantitative comparison of SM distribution in the PMI triangle (Fig. 2B), and cumulative frequency distribution found that while both are enriched with rod-like SM, R-BIND is significantly more enriched than the FDA library (Morgan et al. 2017).
FIGURE 2.

(A) 3D representation of principal components (PCs) 1, 2, and 3 that plots R-BIND SMs and FDA-approved SMs (adapted with permission from Donlic et al. 2022, © American Chemical Society; (B) Principal moments of inertia (PMI) triangle partitions in four subtriangles, representing rod-like (1), sphere-like (2), disc-like (3) and hybrid shapes (4); example molecules are provided for each shape, (1) NVS-SM1 (Palacino et al. 2015), (2) compound 139 from FDA curated library (Donlic et al. 2022), (3) roseoflavin (Lee et al. 2009), and (4) CP6 (Khan et al. 2019) (adapted with permission from Morgan et al. 2017, © Wiley VCH); the dashed triangle outlines the subtriangle 1 where library averages are located. (C) Schematic representation of the k-nearest neighbor (k-NN) algorithm. The R-BIND SMs, plotted in the chemical space, are used to define nearest neighbors, averaging the smallest distance for each new molecule (adapted with permission from Morgan et al. 2019, © American Chemical Society).
Through PCA, we mapped the physicochemical and structural descriptors of R-BIND SMs to define an RNA-privileged chemical space. R-BIND also led to the identification of RNA-privileged scaffolds and subunits, and “R-BIND-likeness” became a parameter to be leveraged in the design of selective ligands for RNA via k-NN (Fig. 2C; Hargrove 2020). The database was made accessible to the community as a website platform (https://rbind.chem.duke.edu/) (Morgan et al. 2019). Here, the user can explore the latest R-BIND version, using “Parameter Search,” “Structure Search,” and “Advanced Search,” or use the “Nearest Neighbor Search” function to assess the proximity in the chemical space between R-BIND members and user-input molecules (Fig. 2C; Morgan et al. 2019). As R-BIND undergoes biennial updates, becoming more and more populated, the descriptor averages and distributions have proven consistent (Morgan et al. 2019; Donlic et al. 2022).
Leveraging the increased size of the most recent R-BIND update, we were able to use pattern recognition to further differentiate this space into RNA substructures and identify privileged SM properties for binding to specific structural motifs. Using LDA, R-BIND SMs could successfully classify five selected RNA structures, namely bulges, G-quadruplexes, double-stranded RNA, internal loops, and stem–loops (Donlic et al. 2022). This analysis also revealed several physicochemical properties that contributed to RNA structure classification. For instance, G-quadruplex-binding ligands had the highest average number of rings and aromatic rings while double-stranded RNA-binding ligands had an enriched lipophilic character, and ligands targeting more flexible and solvent-exposed bulges had a higher MW, number of rotatable bonds, and surface area parameters (Donlic et al. 2022).
Other studies have found consistency in RNA-binding SM properties, including collaborative work between the Disney laboratory and the medicinal chemistry group at AstraZeneca in which RNA binders were identified from an AstraZeneca corporate collection of two million compounds (Haniff et al. 2020). The collection was first filtered in silico based on physicochemical properties of varied RNA binders from the Inforna database (Disney et al. 2016), and the remaining 1967 compounds were screened via high-throughput structure–activity relationships through sequencing (HiT-StARTS), which identified 27 hits (Haniff et al. 2020). The comparison between RNA binders and nonbinders revealed features suggested to define RNA binding, including increased lipophilicity, reduced flexibility, increased polar surface area, and an enrichment in nitrogen atoms, aromatic rings and hydrogen-bond donors, largely in agreement with the features of R-BIND members. Scaffold-based comparison with R-BIND (Morgan et al. 2019) unveiled dissimilarities between the reported new hits and R-BIND ligands (Haniff et al. 2020), which is consistent with R-BIND molecular properties accommodating many scaffolds. These results support the use of R-BIND for the discovery of novel privileged scaffolds and chemotypes to modulate RNAs.
R-BIND established the power of cheminformatics and machine learning in the RNA-targeting field, quantitatively defining features that drive RNA:SM recognition and identifying both RNA-privileged molecular properties and targetable RNAs without requiring a priori knowledge of the RNA structure. As the R-BIND platform grows, we expect to generate a consolidated toolbox of machine-learning methods to access and optimize RNA-focused libraries in structure- and ligand-based design.
Classifying RNA binders versus protein binders
As the number of RNA-privileged chemotypes and scaffolds is increasing, the field is identifying the properties that favor RNA binding over protein binding and consolidating the boundaries of RNA-targeting chemical space. A recent collaborative study between the Schneekloth laboratory and Ladder Therapeutics used machine learning to develop predictive models that identify RNA-targeting ligands from drug-like libraries (Yazdani et al. 2022). Here, a library of more than 24,000 compounds was selected on the basis of commercial availability, synthetic feasibility, and drug-like parameters, and screened in a small molecule microarray (SMM) against 36 different nucleic acids with varied structures. From this, a Repository Of BInders to Nucleic acids (ROBIN) library was derived, containing 2003 RNA binders (Yazdani et al. 2022). A comparative analysis of ROBIN with FDA-approved drugs used least absolute shrinkage and selection operator (LASSO) logistic regression to achieve a binary classification and identified 41/1664 descriptors, generated by the Mordred software package (Moriwaki et al. 2018), as the most important to differentiate RNA and protein binding within a set of drug-like molecules (Yazdani et al. 2022). Further, tree-MAP (TMAP) was used to map ROBIN RNA binders, FDA-approved drugs, and 10,000 protein binders from BindingDB in chemical space, clustering them into branches. Ligands were encoded by extended connectivity fingerprint up to four bonds (ECFP4 fingerprints). Additionally, this clustering may identify RNA-off targets among current drugs and inform the design of bioactive RNA-targeting molecules. Further, LASSO logistic regression and multilayer perceptron (MLP), a class of neural networks, were compared for their performance in the classification of ROBIN RNA binders and the entire BindingDB set of 77,678 protein ligands, using an oversampling strategy to augment the size of the ROBIN RNA binders set (Yazdani et al. 2022). According to mean area under the receiver operating characteristic (AUROC), the nonlinear model MLP performed better than LASSO in the classification and identified properties such as van der Waals surface, aromaticity, topological charge, hydrogen-bond acceptors, nitrogen number, and fraction of sp3 hybridized carbons as strongly predictive for RNA recognition in drug-like libraries, many in line with R-BIND findings (Yazdani et al. 2022). This represents additional evidence that machine-learning algorithms can successfully address the complexity of molecular properties driving RNA recognition.
An experimental approach to identify off-target RNA binding of approved drugs was developed in the Kool laboratory (Fang et al. 2022). Reactivity-based RNA profiling (RBRP) tested transcriptome-wide targeting of protein-targeting molecules selected from preclinical studies, Phase 3, and FDA-approved drugs, and many identified binding events were proposed as responsible for known biological side effects. The study used R-BIND as a source of RNA-binding SMs to perform a structural comparative analysis with their tested set of protein-targeting molecules and found significant chemical similarity between their newly identified RNA binders and R-BIND.
These studies reveal the power of machine learning to identify molecules with both RNA-binding and drug-like properties and to unveil potential off-target effects of existing ligands.
RNA structures differentiated by small molecules
Given the paucity of high-resolution RNA:SM structures, alternative methods are needed to understand the RNA properties that allow selective recognition by SMs. Toward this end, we developed pattern recognition of RNA by small molecules (PRRSM) based on a training set of RNA sequences with a range of simple, well-predicted secondary structure motifs and incorporated benzofuranyluridine (BFU) (Eubanks et al. 2017) as a solvatochromic dye within each secondary structure (Fig. 3A). Each construct was exposed to eleven AGs at various concentrations, and emission intensity was used as input for PCA. Unbiased clustering of each secondary structure class, as well as unique structures within each class, provided evidence for structure-specific recognition properties (Fig. 3B). Additionally, the results in different buffer conditions suggested that RNA secondary structures are best differentiated under conditions that favor dynamic motion (Eubanks and Hargrove 2017). These results underscored the importance of shape complementarity and provided a better understanding of the role of RNA conformational dynamics in SM recognition, with bulge structures as particularly promising targets among the secondary structures studied. The classification of targets using R-BIND ligands mentioned above (Donlic et al. 2022) supported the value and feasibility of using pattern recognition to understand more complex structures in the future. The initial classification was validated using differential labeling of human immunodeficiency virus-1 trans-activation response (HIV-1-TAR) element where PRRSM could predict bulge-labeled or apical-loop labeled RNA. Using this predictive power, we were able to distinguish and predict specific RNA-folded states as represented by the PreQ1 and fluoride riboswitches (Fig. 3C; Eubanks et al. 2019).
FIGURE 3.

(A) The 16 RNA training set sequences, including stems, bulges (Blg), internal loops (IL), asymmetrical internal loops (AIL), and hairpins (HP) used in PRRSM. BFU-labeled position shown with blue star. (B) Differentiation of the five structural classes of the training set using PCA. (C) Differentiation of the individual training set sequences. PC1 correlated to the increasing motif size (from stem to AIL), while PC2 correlated to the purine: pyrimidine ratio, which is dependent on the sequence of the RNA (HP to IL); (D) PRRSM classification of Pre-Queuosine1 (PreQ1) and fluoride riboswitch conformational changes. Each construct was labeled with BFU in three positions and subjected to the assay. PRRSM was able to classify these RNA structures, including folded and unfolded states, and provide insight into sites that are critical for these structural changes. All PRRSM-based observations of unfolded and folded riboswitch states were confirmed via NMR (adapted with permission from Eubanks et al. 2017, 2019, © American Chemical Society).
Other tools also allow for complementary methods of elucidating binding patterns between SMs and RNA secondary structural motifs, which emerge from several RNA-targeting curated platforms, such as Inforna by Disney (Disney et al. 2016), RNALigands by Zhang (Sun et al. 2022), and RNAmigos by Waldispühl (Oliver et al. 2020). The last platform tries to bridge RNA targeting and machine learning and gives more contribution to noncanonical base pairs to explain higher-order RNA structures and ligand modulation, in agreement with the most recent molecular dynamic (MD) simulation models.
Exploration of RNA-privileged small molecule space via scaffold-based synthetic libraries
One strategy to continue the exploration, validation, and expansion of RNA-privileged chemical space is to generate synthetic SM libraries, and we chose to use scaffolds with a known propensity to bind nucleic acids and demonstrated clinical utility (Patwardhan et al. 2017; Donlic et al. 2018; Zafferani et al. 2022). Compared to current commercially available libraries, which generally occupy a similar subsection of R-BIND chemical space, we can generate and analyze synthetic libraries that complete coverage and push the boundaries of R-BIND space. Continuous synthetic tuning of these scaffolds has led to the discovery of lead molecules for chemical probe development of medicinally relevant RNA targets, including viral and long noncoding RNA structures (Patwardhan et al. 2019b). In subsequent analysis, machine-learning techniques have allowed insight into drivers of selectivity and informed novel SM design based on the analyzed physicochemical and spatial properties.
Our first efforts explored amiloride, an FDA-approved diuretic, as an RNA-binding scaffold. Dimethyl amiloride (DMA) was previously identified by the Al-Hashimi group as a weak but selective binder to HIV-1 TAR RNA (Stelzer et al. 2011). We synthesized a library of 28 amilorides and evolved the lead molecule (DMA-1) into a strong, selective TAR ligand (DMA-169) through modifications at the C5 and C6 positions (Fig. 4A). Each member was assessed for its affinity and selectivity for TAR with addition of excess tRNA and DNA. This screening data allowed for cheminformatic analysis by LDA to determine whether a combination of the 20 cheminformatic parameters used to analyze R-BIND could predict binding and/or selectivity (Fig. 4B; Patwardhan et al. 2017). Predictive power was achieved for selective binders, but separation was not seen for promiscuous or nonbinders. Cheminformatics with LDA provided specific insights into parameters critical for the selective molecular recognition of amilorides to TAR RNA, specifically those related to stacking and hydrogen bonding interactions.
FIGURE 4.
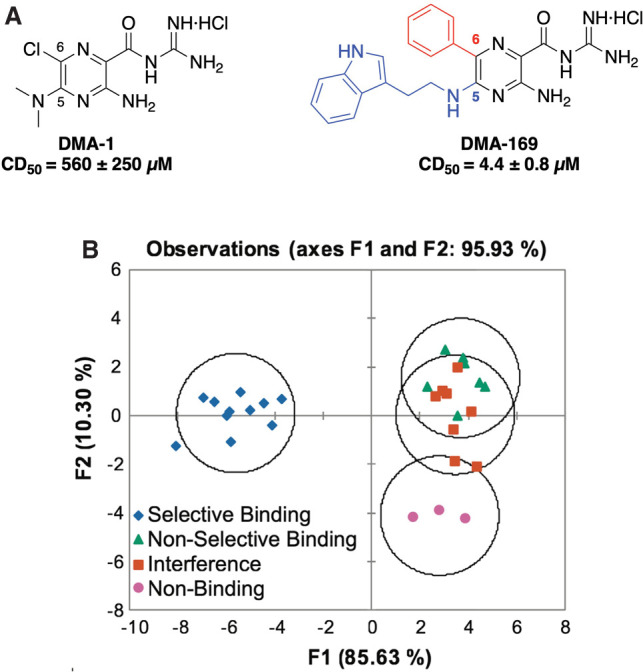
(A) Combinatorial modifications at the C5 (blue) and C6 (red) positions of the amiloride scaffold optimized affinity of DMA-1 to give lead DMA-169. Selective ligands showed competitive displacement doses (CD50) of ∼4–200 µM. (B) Linear discriminate analysis (LDA) plot based on 20 cheminformatic parameters clusters selective amiloride derivative ligands from nonbinding and nonselective ligands (panels A and B adapted with permission from Patwardhan et al. 2017, © Royal Society of Chemistry).
To achieve better insights into differential drivers of selectivity, promiscuity, and nonbinding SMs, we leveraged the tunability of the amiloride scaffold, synthesizing new derivatives and investigating their selectivity profile against five HIV RNAs (Fig. 5A; Le Grice 2015). Some trends could be observed directly from the screening results, particularly for ligands demonstrating differential binding between TAR and ESSV, but important insights into selective RNA recognition were gained from pattern recognition. We used hierarchical clustering of the screening data to define classes and then combined cheminformatic analysis with LDA. In this case, the LDA loading plots revealed several qualitative trends for promiscuous and nonbinding ligands. For example, nonbinding ligands tended to have more oxygens, more sp3 centers, and higher relative polar surface area (Fig. 5B,C), which correlated to the descriptors defining FDA chemical space relative to R-BIND, validating our previous R-BIND analysis (Patwardhan et al. 2019b).
FIGURE 5.

(A) Secondary structures of HIV RNAs screened with DMA library. (TAR) Trans-activation response element, (RRE) rev response element, (FSS) frameshift-stimulating, (ESSV) exonic splicing silencer of Vpr. (B) Linear discriminate analysis (LDA) plot based on 20 cheminformatic parameters clusters to differentiate five groups of ligands for HIV RNA targets. (C) LDA loading plot for the qualitative analysis of the contribution of each cheminformatic parameter contributing on F1 versus F2. (MW) Molecular weight, (HBA) number of hydrogen bond acceptors, (HBD) number of hydrogen bond donors, (LogP) n-octanol/water partition coefficient, (RotB) number of rotatable bonds, (tPSA) topological polar surface area, (LogD) n-octanol/water distribution coefficient, (N) number of nitrogen atoms, (O) number of oxygen atoms, (Rings) number of rings, (ArRings) number of aromatic rings, (HetRings) number of heteroatom-containing rings, (SysRings) number of ring systems, (SysRR) ring complexity, (Fsp3) fraction of sp3 hybridized carbons, (ASA) accessible surface area, (relPSA) relative polar surface area, (VWSA) van der Waals surface area (panels A–C adapted with permission from Patwardhan et al. 2019b, © Royal Society of Chemistry).
Diphenylfuran (DPF) and diminazine (DMZ) are other promising scaffolds with synthetic versatility and known nucleic acid binding properties (Fig. 6A; Pilch et al. 1995; Zhao et al. 1995; Gelus et al. 1999; Chaires et al. 2004; Nguyen et al. 2009). We first explored targeting of the 3′-triple helix of the long noncoding RNA metastasis associated lung adenocarcinoma transcript 1 (MALAT-1; Donlic et al. 2020), which has been observed to accumulate at high levels in many cancer types and where the 3′-triple helix acts as a stabilizing structural motif to prevent the degradation of the MALAT1 transcript. We synthesized derivatives with varying subunits at the ortho-, para-, and meta-positions of the phenyl rings, which led to diversity of 3D shapes as determined by PMI (Donlic et al. 2020). Combining screening with PMI analysis of both libraries revealed a general trend that more rod-like shapes, that is, para-substituted derivatives, exhibited the strongest binding to the MALAT1 triple helix (Fig. 6C). Collectively, these studies established the importance of shape-based recognition in high affinity triple helix binding, particularly validating the R-BIND analysis of biased rod-like shapes. In addition to PMI, other methods such as QSAR, which is discussed in more detail below, can be utilized to allow identification of other 3D parameters that directly contribute to SM binding events.
FIGURE 6.
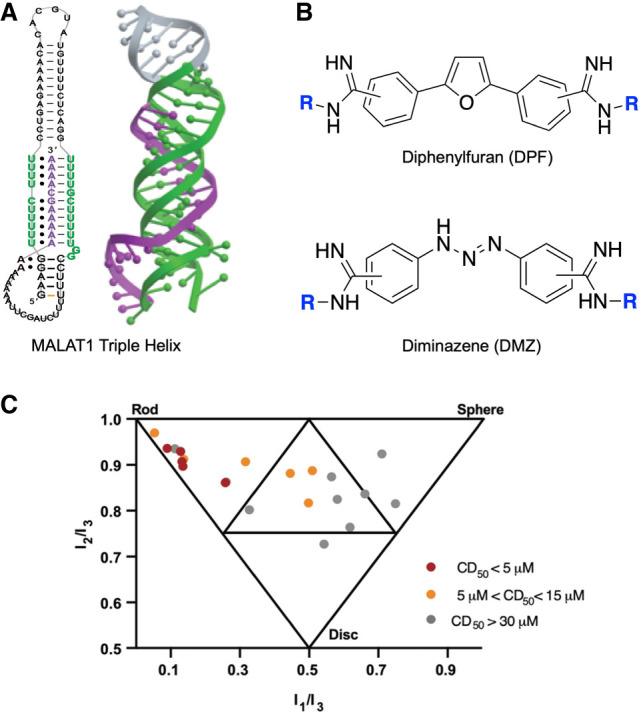
(A) Schematic diagram of MALAT1 triple helix base-pairing and crystal structure. Protein Data Bank entry 4PLX. (B) Diphenylfuran (DPF) and diminazene (DMZ) core scaffold structures. (C) Envelop diagram of the principal moments of inertia (PMI) calculations of the 21-member DMZ-based focus library (panels A and C adapted with permission from Zafferani et al. 2022, © American Chemical Society).
Together, the studies of the DMA, DPF, and DMZ scaffold-based libraries have showcased the power of computational analysis to reveal important properties of tunable and diverse RNA-privileged scaffolds and build diverse libraries that reveal important RNA-targeting properties for additional investigation. Efforts to discover additional novel RNA-binding scaffolds will lead to further insights into the unique RNA features and SM properties critical for binding. By leveraging this scaffold-based library strategy in combination with cheminformatic and machine-learning–based tools, we have revealed preliminary guidelines to understand the chemotypes and chemical properties that drive affinity and selectivity in RNA:SM interactions and unveil which factors are critical for development of future novel chemical probes and therapeutics.
APPLICATIONS TO RNA-TARGETED CHEMICAL PROBE DISCOVERY
Design of diverse RNA-privileged libraries
One promising application of these insights in RNA:SM recognition is the design of RNA-specific screening libraries. Such libraries will be valuable given that RNA ligands have distinct features from the protein ligands or FDA-approved SMs around which most libraries are constructed. For example, Nickbarg and coworkers used affinity mass spectrometry to screen 42 RNA ligands against ∼55,000 SMs that had also been screened against protein targets. Naïve Bayesian models were constructed with this data to identify cheminformatic properties biased toward RNA targeting. These features were used to build an ∼3800-member RNA-specific library that showed increased hit rates for RNA (Rizvi et al. 2020).
Recently, we chose to use the k-NN method using 20 cheminformatic parameters to design a screening set based on the R-BIND library (SL Wicks, BS Morgan, A Wilson, AE Hargrove, in prep.). k-NN effectively created a 20-dimensional space in which distances between each molecule were measured. We could then evaluate commercial libraries using this same space to identify molecules close in properties to R-BIND molecules. One advantage of this method is that it avoids the use of scaffold hopping or fingerprints, computational techniques that rely on chemical substructures, which can limit the structural diversity of the generated library. From more than 2,500,000 commercially available ligands we selected a diverse set of 804 molecules. Screens of this library have identified ligands for all unique RNA targets tested to date (n > 10), validating this strategy (SL Wicks, BS Morgan, A Wilson, AE Hargrove, in prep.). At the same time, we discovered that specific regions of R-BIND chemical space were either inaccessible or sparsely occupied by commercially available molecules. Using methods such as k-NN and QSAR, we are generating synthetic molecules that cover this space and will allow for a comprehensive RNA-targeting library.
Quantitative structure activity relationship (QSAR) studies for RNA
The rational design of SMs for a specific RNA target is still a hindered milestone, due to the difficulties in RNA structure characterization and our incomplete understanding of RNA:SM binding at the molecular level (Morgan et al. 2018; Cai et al. 2022). In this scenario, machine-learning–aided tools have the potential to take up the challenge (Dara et al. 2022). Recently, QSAR models have landed in the RNA-targeting field as predictive tools to assist hit-to-lead optimization against specific RNAs (Patwardhan et al. 2019b; Cai et al. 2022). The QSAR model defines a quantitative correlation between the experimental binding profile and the molecular descriptors of ligands against a target. A training set is used to create the model, and a test data set evaluates the predictive power of the model. The amount and quality of data in the training set are generally the limiting factors, and the data is usually processed and refined prior to model construction (Cai et al. 2022).
We first pioneered RNA-targeting QSAR modeling with a “one-library-one-target” approach using the data from the amiloride screen mentioned above, which was trained with data from DMA titrations via a Tat peptide displacement assay (Patwardhan et al. 2019a, b) as a proxy for binding affinity, and molecular descriptor values from cheminformatic analysis. Here HIV-1 TAR and ESSV were used, while the other three RNAs were discarded for lacking a sufficient number of binders for analysis (Patwardhan et al. 2019b). Linear regression, exhaustive search and leave-one-out cross validation (LOOCV) identified the best two-parameter linear models for each RNA target, with R2 used to evaluate fit and Q2 used to measure the predictive power (Patwardhan et al. 2019b). The use of different RNA constructs allowed us to test whether a predictive QSAR model could be applied to any RNA construct and to evaluate differences in molecular properties contributing to binding of different targets, paving the way to design selective ligands.
To expand beyond a single scaffold, we challenged the QSAR predictive power in a “multiple-libraries-one-target” model using HIV-1 TAR, for which ligands from several SM classes have been published. Here, we combined our three RNA-binding scaffold-based libraries, DMAs (Patwardhan et al. 2017, 2019b), DPFs (Donlic et al. 2018; Donlic et al. 2020), DMZs (Zhou et al. 2014) with AGs, and nucleic acid dyes (Fig. 7A). We calculated nearly 400 molecular descriptors using a molecular operating environment (MOE) and, due to the increased complexity, optimized data refinement by Pearson correlation coefficient to remove descriptors with redundancy and/or multicollinearity. The optimized data was split using Kennard-Stone subsampling (Kennard and Stone 1969), and guaranteed that the training and test set were obtained from uniform regions of the descriptor space (Cai et al. 2022). This study used surface plasmon resonance (SPR) data, which generates KD, koff, and kon values, allowing QSAR models to be trained for thermodynamic and kinetics parameters. Kinetic insight is particularly valuable as rates are rarely measured for RNA:SM binding, a complex event that must be properly considered to better predict biological activity and eventual progression to in vivo studies (Patwardhan et al. 2019b; Cai et al. 2022). SMs generally have slower binding to RNA compared to protein–ligand interactions, suggesting that taking kinetics into account can be crucial to build reliable and robust predictive models (Cai et al. 2022). To control model complexity, LASSO was used for descriptor selection to afford the final model, which was derived from MLR after exhaustive search and reported in Figure 7B (Cai et al. 2022). The predictive performance of MLR was compared to ensemble tree methods, such as random forest and gradient boosting machine (Cai et al. 2022). As shown in Figure 7C,D, ensemble tree methods improved the R2, while Q2 values were unchanged compared to MLR. While MLR allows increased interpretability of the trends, successful model-building with the other methods confirmed that the selected descriptors are appropriate to interpret RNA:SM interactions (Cai et al. 2022). This work established the predictive power of QSAR models and the value of multiple machine-learning methods. Moving forward, the use of additional machine-learning algorithms, such as neural networks, will improve the versatility of the QSAR predictive models and make it possible to couple QSAR predictions with generative models to increase the chemical diversity of RNA-binding molecules (Dara et al. 2022).
FIGURE 7.

(A) Representative structure of the five scaffolds used in the QSAR model (Cai et al. 2022); graph plotting of observed and predicted ln KD, training set in red, test set in blue, comparing (B) multiple linear regression (MLR), (C) random forest, and (D) gradient boosting machine (adapted with permission from Cai et al. 2022, © American Chemical Society).
EMERGING OPPORTUNITIES
The application of machine learning to the development of RNA-targeted chemical probes has led to growing and convergent understanding of the ideal properties of RNA ligands and initial insights into ideal RNA targets, allowing applications such as RNA-specific library design, quantitative methods for RNA-binding SM optimization, and even predicted off-target effects of approved drugs. Expansion in these areas will require a significant increase in data, including new chemical matter to explore the fullness of RNA-targeted chemical space, the creation of additional methods to evaluate not only binding and kinetics but also conformation and function-related behaviors, and the incorporation of more complex RNA targets. These advances will also allow the coupling of SM generation methods (Popova et al. 2018; Brown et al. 2019; Polykovskiy et al. 2020) and established machine-learning methods for RNA, such as nearest neighbor searches or neural networks to allow for the generation of entirely novel RNA–SM binders.
In an ideal scenario, there would be an increase in our understanding of individual noncovalent interactions. The analysis of the limited structures available has revealed rod-like binding pockets (Hewitt et al. 2019) and a prevalence of hydrogen bonding and stacking interactions (Padroni et al. 2020), in line with RNA-targeting SM properties. Cryogenic electron microscopy (Cryo-EM) may offer additional high-resolution structures, and the establishment of general methods for the smaller sizes of RNA often used to study binding is ongoing (Kappel et al. 2020). Another opportunity may lie in docking, though this requires an ensemble of structures most often identified through in-depth nuclear magnetic resonance (NMR) experiments combined with molecular dynamics (MD) (Ganser et al. 2018). RNA 3D structure prediction methods such as Rosetta's Fragment Assembly of RNA with Full Atom Refinement (FARFAR; Watkins et al. 2020) may also facilitate progress, though these algorithms have been largely trained on well-structured RNAs, such as those amenable to crystallography, and it is not clear how representative the output ensembles are for more dynamic RNAs. Finally, the current force-fields (FFs) used in MD and docking are based on protein training sets, and the development of FFs specific to RNA will greatly facilitate progress (Sponer et al. 2018; Manigrasso et al. 2021). All of these advances will rely on machine learning.
Finally, progression into functional and biological assays is required to fully understand how to design bioactive RNA ligands, the ultimate goal of most in the field. Below we discuss outlooks for the use of machine learning in improved molecular dynamics and in understanding how to modulate RNA biological functions.
Machine learning and molecular dynamics to assess RNA druggability
RNA druggability is no longer the myth that it used to be in the past, and RNA ordered architectures can become suitable sites for structure-based SM design, leading to drug discovery. Given challenges and limitations of RNA X-ray crystallography, the characterization of structured RNAs to examine potential binding pockets and SM interactions is generally the result of a joint effort of computational, biophysical and structural biology techniques (Ganser et al. 2018; Zhang et al. 2022). These methods include Cryo-EM (Ma et al. 2022), NMR spectroscopy (Scott and Hennig 2008; Barnwal et al. 2017), small-angle X-ray scattering (SAXS) (Chen and Pollack 2016; He et al. 2022), chemical probing, such as selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE; Deigan et al. 2009; Mlýnský and Bussi 2018; Busan et al. 2019) and dimethyl sulfate (DMS; Tijerina et al. 2007), computational methods for de novo prediction (Manigrasso et al. 2021), homology-based models (Flores et al. 2010; Rother et al. 2011) and covariance models (Fig. 8; Tourasse and Darfeuille 2020). This combination allows for ever more detailed descriptions of RNA as either conformational ensembles or single energy-minimized structure, including studying solvent effect and ion-dependency on RNA molecular dynamics, as reported by Bernetti et al. (2021). The combination of computation prediction and Cryo-EM is particularly promising (Kappel et al. 2020). As a de novo prediction model, FARFAR2 leverages a combination of score filters for a library of RNA fragments, Monte Carlo minimization to describe base-pairing interactions and an all-atom scoring function (Watkins et al. 2018) to predict the conformations of complex folded ncRNAs, which has been successful for structures such as riboswitches, T-box riboswitches and the adenovirus viral-associated (VA)-I noncoding RNA (Watkins et al. 2020). FARFAR was combined with Cryo-EM in DRRAFTER, which leveraged Cryo-EM maps of ribonucleoprotein complexes to inform the de novo modeling of RNA structures (Kappel et al. 2018).
FIGURE 8.

Potential workflows to allow the computational prediction of RNA:SM structures and used methods.
Once a structural ensemble or single conformation is determined, the target druggability and the design of ligands can be informed by computational tools able to generate predictions of the RNA:SM complex. These techniques are mainly molecular docking model and MD simulation (Fig. 8).
Similar to protein targeting, docking studies search for putative SM binding pockets within structures, identify hits through virtual screening (VS), and predict RNA:SM complexes. Internal Coordinate Mechanics (ICM, Molsoft; Neves et al. 2012) represents a modeling-docking platform successfully used to search for binding pockets in RNA molecules and study RNA:protein (Arnautova et al. 2018) and RNA:SM (Zafferani et al. 2021) complexes. Validated prospective docking studies have required experimentally informed ensembles but have also been very successful, such as the work by the Al-Hashimi laboratory targeting HIV-1-TAR RNA (Ganser et al. 2018). The analysis of RNA:SM complexes may also serve as a retrospective tool to corroborate binding and activity data. For example, we reported docking studies of DMA-132, DMA-135, and DMA-155 in complex with FARFAR-generated structures of SL 1 and 6 of SARS-CoV-2 (Zafferani et al. 2021) and of DPF-p8 on MALAT1 triple helix, using the available crystallographic structure (PDB ID: 4PLX) (Donlic et al. 2020). Often, however, shallow binding pockets and RNA dynamics and flexibility make it difficult to generate reliable binding poses with conventional docking protocols (Manigrasso et al. 2021). Promisingly, some protein-based scoring functions have been adapted to address RNA molecular properties, for instance considering the water solvation state of bound RNA, the desolvation effect and the high structural flexibility (Manigrasso et al. 2021). These functions include AutoDock (Moitessier et al. 2006), MOlecular Recognition with a Driven dynamics OptimizeR (MORDOR; Guilbert and James 2008), rDock (Ruiz-Carmona et al. 2014), DrugScoreRNA (Pfeffer and Gohlke 2007), and LigandRNA (Philips et al. 2013). Nonetheless, the description of SM binding mode, including intercalation, together with the dynamic conformational states that may be sampled or induced by SMs can be better addressed by the use of methods to study the complex dynamicity in a time-dependent way, such as MD simulation systems assisted by machine learning (Manigrasso et al. 2021; Dara et al. 2022).
Given the dynamic RNA conformational landscape, identifying metastable states is both critical and challenging (Sponer et al. 2018). MD simulations methods predict RNA molecular conformations at the atomistic level and would also increase understanding of RNA:SM binding kinetics (Manigrasso et al. 2021). As extensively reviewed by Ganser et al. (2019), the postulated RNA-ensemble-function paradigm strongly underscores the importance of understanding how RNA conformational fluctuations modulate RNA, how to exploit them as drug targets, and how to consider ligand-induced reorganization of RNA structure (Ganser et al. 2020). Overall, development of performant computer-aided simulations is a compelling goal to expand the window of RNA-targeting drug discovery.
MD simulations require basic approximation combined to specific parametrization to simulate atom positions in a time-dependent manner (Sponer et al. 2018). The approximation is achieved by molecular mechanical (MM) FFs, which establish a correlation between molecular geometry and potential energy (Sponer et al. 2018). Generally employed FFs can be suitable to study nucleic acids. FFs differ in terms of accuracy and performance and can be classified in two categories, namely all-atom FFs and the coarse-grained FFs. All-atom FFs are the most used and include Assisted Model Building with Energy Refinement (AMBER; Cornell et al. 1995; Perez et al. 2007; Yildirim et al. 2010; Zgarbova et al. 2011), Merck molecular force field (MMFF; Tosco et al. 2014), and Chemistry at Harvard Macromolecular Mechanics (CHARMM; Denning et al. 2011). Due to some limitations such as the nucleobase overstacking of AMBER and the nucleobase understacking of CHARMM (Hall 2013), FFs are being reparametrized (Cesari et al. 2016) to become more descriptive of RNA. Refined FFs can incorporate different contributions, including the ribose 2′ OH, noncanonical interactions and charge transfer, as in the polarizable FF Drude-2017 (Lemkul and MacKerell 2018). On the other hand, coarse-grained models have also been successfully applied as demonstrated by RACER (RnA CoarsE gRained), an example of an MD coarse-grained RNA model used for the determination of RNA secondary structures (Bell et al. 2017).
Despite the achievement of FFs predictions, quantum mechanical (QM) calculations would increase the accuracy of the model and give new insights into RNA structural conformations. However, QM is still difficult to be applied on large systems such as biomolecules due to its high time-consumption (Noe et al. 2020). Machine learning can be the keystone to speed up the transferability of QM calculations to more complex systems and depict an ever more reliable RNA conformational landscape (Noe et al. 2020).
In the future, the reparametrization of existing FFs and the support of machine learning in the use of QM calculations will help characterize the complex RNA conformations, unveiling pockets and sites to climb the cliff to RNA druggability. Additionally, we also expect that MD simulations will reveal how SMs can modulate RNA conformations and ultimately correlate these modulations to their bioactivity. For higher ordered architectures such as triple helixes and pseudoknot, for example, it is still an open question whether it would be more beneficial to achieve RNA modulation by the stabilization into a locked conformation or by destabilization followed by degradation or enzymatic read-through, respectively. Such insights, which also depend on machine learning, would revolutionize the process of drug-discovery.
Application in biological systems
Discovering novel SMs with the desired biological activity is an essential step to develop new therapeutics and gather a greater understanding of biological systems. Given that RNA is highly diverse both in structure and function and has pivotal roles that act at many levels of regulation including diseases, a key area of need within the field is the combination of large, complex biological data and machine learning to predict broader ranges of biological activity. Recent advances in machine learning have allowed biological information to be systematically measured and mined at unprecedented levels using the increasing availability of “omics” data and data-driven algorithms (Vamathevan et al. 2019; Xu and Jackson 2019). Current machine-learning approaches within biological systems are being utilized for the challenging prediction of genomic features, such as binding sites of DNA- and RNA-binding proteins, enhancer sites, and other regulatory regions (Libbrecht and Noble 2015). For instance, deep learning methods were used to build models to predict regulatory elements and noncoding variant effects de novo from a DNA sequence that can then be experimentally validated for their contribution to gene regulation (Zou et al. 2019). Other applications have predicted DNA transcript abundance, imputation of missing single-nucleotide polymorphisms, and DNA methylation states (Zhao et al. 2008; Angermueller et al. 2017; Washburn et al. 2019). It is imperative to continue to refine and implement machine-learning–based tools that expand applications to other underexplored areas, such as RNA biology and RNA:SM targeting.
Current machine-learning approaches for protein-based endeavors, particularly the functional prediction derived from protein sequence and 3D structure, have foreseeable applications to RNA:SM targeting (Dara et al. 2022). To narrow the growing gap between the number of proteins being discovered and their functional characterization due to experimental limitations, established methods such as random forests, support vector machines (SVM), and ANN have shown to provide reliable protein function prediction, even when the underlying mechanisms were not well understood, and they have demonstrated effective use in drug discovery (Bernardes and Pedreira 2013). The random forest technique has been used to improve the prediction to select molecular descriptors of ligands of kinases, nuclear hormone receptors, and other enzymes, which is seen as an important step in VS to identify bioactive molecules during the drug development process (Cano et al. 2017). Similarly, the SVM model has the ability to classify different varieties of active or inactive compounds and to predict the biological activity of new molecules from regression models, which has enabled the identification of compounds in VS libraries that are not only active for a target protein, but also selective for a particular target over a closely related member of the same protein family (Maltarollo et al. 2019). Additionally, there has been a rise in the use and rapid evolution of deep learning techniques to extract meaningful features and develop high performing predictors, even of multiple protein functions (Clark and Radivojac 2011; Gligorijevic et al. 2018). For example, autoencoders have been utilized for generating de novo drug design (Gómez-Bombarelli et al. 2018). Collectively, these methods should be explored as potential tools to predict the probability that an RNA, especially as an experimentally informed RNA conformational ensembles, is associated with a particular function to validate as a target for SM probing.
Previous machine-learning applications, including our QSAR studies, were aimed at exploring the chemical and geometrical features important for binding of SMs to a specific RNA target in vitro. To increase the utility of these models, they must also show experimental validation in a biological system. Most likely, activity-based measurements within a biological context will be required to train new, complementary models. Forging into this direction will improve rational chemical optimization, particularly for function-based design of RNA-targeting SMs, and provide essential insight into the future work to evaluate both ligand selectivity and its correlation with biological activity.
CONCLUSION
In this Perspective, we contend that machine learning has made significant contributions in advancing the field of RNA:SM targeting and offers untapped opportunities to bring RNA targeting on par with protein targeting, and even beyond. We have discussed how currently available machine-learning–based approaches, often refined and optimized for protein targeting, can be used in the underexplored context of RNA targeting. Moreover, these models and techniques have been used to characterize RNA-privileged chemical space and reveal pertinent molecular features that can identify putative RNA-binding SMs with drug-like properties. Finally, this work emphasizes several machine-learning approaches that, if developed and optimized, would significantly accelerate the understanding and efficient development of RNA-targeted SMs as novel therapeutics.
ACKNOWLEDGMENTS
The authors would like to thank the members of the Hargrove Laboratory for their invaluable feedback on this manuscript, particularly Daniel Santana Garcia for insight and initial planning and Anita Donlic, Emily Swanson, and Zhengguo (Alex) Cai for input and proofreading. The authors acknowledge financial support from Duke University, the National Science Foundation (CAREER 1750375), the U.S. National Institute of General Medicine (R35 GM124785), the U.S. National Institute of Allergy and Infectious Diseases (U54AI150470) and the Sloan Foundation.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079497.122.
Freely available online through the RNA Open Access option.
REFERENCES
- Angermueller C, Lee HJ, Reik W, Stegle O. 2017. DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol 18: 67. 10.1186/s13059-017-1189-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnautova YA, Abagyan R, Totrov M. 2018. Protein-RNA docking using ICM. J Chem Theory Comput 14: 4971–4984. 10.1021/acs.jctc.8b00293 [DOI] [PubMed] [Google Scholar]
- Barnwal RP, Yang F, Varani G. 2017. Applications of NMR to structure determination of RNAs large and small. Arch Biochem Biophys 628: 42–56. 10.1016/j.abb.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell DR, Cheng SY, Salazar H, Ren P. 2017. Capturing RNA folding free energy with coarse-grained molecular dynamics simulations. Sci Rep 7: 45812. 10.1038/srep45812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes JS, Pedreira CE. 2013. A review of protein function prediction under machine learning perspective. Recent Pat Biotechnol 7: 122–141. 10.2174/18722083113079990006 [DOI] [PubMed] [Google Scholar]
- Bernat V, Disney MD. 2015. RNA structures as mediators of neurological diseases and as drug targets. Neuron 87: 28–46. 10.1016/j.neuron.2015.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernetti M, Hall KB, Bussi G. 2021. Reweighting of molecular simulations with explicit-solvent SAXS restraints elucidates ion-dependent RNA ensembles. Nucleic Acids Res 49: e84. 10.1093/nar/gkab459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N, Fiscato M, Segler MHS, Vaucher AC. 2019. GuacaMol: benchmarking models for de novo molecular design. J Chem Inf Model 59: 1096–1108. 10.1021/acs.jcim.8b00839 [DOI] [PubMed] [Google Scholar]
- Busan S, Weidmann CA, Sengupta A, Weeks KM. 2019. Guidelines for SHAPE reagent choice and detection strategy for RNA structure probing studies. Biochemistry 58: 2655–2664. 10.1021/acs.biochem.8b01218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Zafferani M, Akande OM, Hargrove AE. 2022. Quantitative structure-activity relationship (QSAR) study predicts small-molecule binding to RNA structure. J Med Chem 65: 7262–7277. 10.1021/acs.jmedchem.2c00254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano G, Garcia-Rodriguez J, Garcia-Garcia A, Perez-Sanchez H, Benediktsson JA, Thapa A, Barr A. 2017. Automatic selection of molecular descriptors using random forest: application to drug discovery. Expert Syst Appl 72: 151–159. 10.1016/j.eswa.2016.12.008 [DOI] [Google Scholar]
- Cesari A, Gil-Ley A, Bussi G. 2016. Combining simulations and solution experiments as a paradigm for RNA force field refinement. J Chem Theory Comput 12: 6192–6200. 10.1021/acs.jctc.6b00944 [DOI] [PubMed] [Google Scholar]
- Chaires JB, Ren J, Hamelberg D, Kumar A, Pandya V, Boykin DW, Wilson WD. 2004. Structural selectivity of aromatic diamidines. J Med Chem 47: 5729–5742. 10.1021/jm049491e [DOI] [PubMed] [Google Scholar]
- Chen Y, Pollack L. 2016. SAXS studies of RNA: structures, dynamics, and interactions with partners. Wiley Interdiscip Rev RNA 7: 512–526. 10.1002/wrna.1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark WT, Radivojac P. 2011. Analysis of protein function and its prediction from amino acid sequence. Proteins 79: 2086–2096. 10.1002/prot.23029 [DOI] [PubMed] [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc 117: 5179–5197. 10.1021/ja00124a002 [DOI] [Google Scholar]
- Costales MG, Childs-Disney JL, Haniff HS, Disney MD. 2020. How we think about targeting RNA with small molecules. J Med Chem 63: 8880–8900. 10.1021/acs.jmedchem.9b01927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dara S, Dhamercherla S, Jadav SS, Babu CM, Ahsan MJ. 2022. Machine learning in drug discovery: a review. Artif Intell Rev 55: 1947–1999. 10.1007/s10462-021-10058-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deigan KE, Li TW, Mathews DH, Weeks KM. 2009. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci 106: 97–102. 10.1073/pnas.0806929106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning EJ, Priyakumar UD, Nilsson L, Mackerell AD Jr. 2011. Impact of 2′-hydroxyl sampling on the conformational properties of RNA: update of the CHARMM all-atom additive force field for RNA. J Comput Chem 32: 1929–1943. 10.1002/jcc.21777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney MD, Winkelsas AM, Velagapudi SP, Southern M, Fallahi M, Childs-Disney JL. 2016. Inforna 2.0: a platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem Biol 11: 1720–1728. 10.1021/acschembio.6b00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlic A, Morgan BS, Xu JL, Liu A, Roble C Jr, Hargrove AE. 2018. Discovery of small molecule ligands for MALAT1 by tuning an RNA-binding scaffold. Angew Chem Int Ed Engl 57: 13242–13247. 10.1002/anie.201808823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlic A, Zafferani M, Padroni G, Puri M, Hargrove AE. 2020. Regulation of MALAT1 triple helix stability and in vitro degradation by diphenylfurans. Nucleic Acids Res 48: 7653–7664. 10.1093/nar/gkaa585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlic A, Swanson EG, Chiu LY, Wicks SL, Juru AU, Cai Z, Kassam K, Laudeman C, Sanaba BG, Sugarman A, et al. 2022. R-BIND 2.0: an updated database of bioactive RNA-targeting small molecules and associated RNA secondary structures. ACS Chem Biol 17: 1556–1566. 10.1021/acschembio.2c00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubanks CS, Hargrove AE. 2017. Sensing the impact of environment on small molecule differentiation of RNA sequences. Chem Commun 53: 13363–13366. 10.1039/C7CC07157D [DOI] [PubMed] [Google Scholar]
- Eubanks CS, Hargrove AE. 2019. RNA structural differentiation: opportunities with pattern recognition. Biochemistry 58: 199–213. 10.1021/acs.biochem.8b01090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubanks CS, Forte JE, Kapral GJ, Hargrove AE. 2017. Small molecule-based pattern recognition to classify RNA structure. J Am Chem Soc 139: 409–416. 10.1021/jacs.6b11087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubanks CS, Zhao B, Patwardhan NN, Thompson RD, Zhang Q, Hargrove AE. 2019. Visualizing RNA conformational changes via pattern recognition of RNA by small molecules. J Am Chem Soc 141: 5692–5698. 10.1021/jacs.8b09665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falese JP, Donlic A, Hargrove AE. 2021. Targeting RNA with small molecules: from fundamental principles towards the clinic. Chem Soc Rev 50: 2224–2243. 10.1039/D0CS01261K [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Velema WA, Lee Y, Lu X, Mohsen MG, Kietrys AM, Kool ET. 2022. Pervasive transcriptome interactions of protein-targeted drugs. bioRxiv 10.1101/2022.07.18.500496 [DOI] [PubMed] [Google Scholar]
- Fedorova O, Jagdmann GE Jr, Adams RL, Yuan L, Van Zandt MC, Pyle AM. 2018. Small molecules that target group II introns are potent antifungal agents. Nat Chem Biol 14: 1073–1078. 10.1038/s41589-018-0142-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores SC, Wan Y, Russell R, Altman RB. 2010. Predicting RNA structure by multiple template homology modeling. Pac Symp Biocomput 216–227. 10.1142/9789814295291_0024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser LR, Lee J, Rangadurai A, Merriman DK, Kelly ML, Kansal AD, Sathyamoorthy B, Al-Hashimi HM. 2018. High-performance virtual screening by targeting a high-resolution RNA dynamic ensemble. Nat Struct Mol Biol 25: 425–434. 10.1038/s41594-018-0062-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser LR, Kelly ML, Herschlag D, Al-Hashimi HM. 2019. The roles of structural dynamics in the cellular functions of RNAs. Nat Rev Mol Cell Biol 20: 474–489. 10.1038/s41580-019-0136-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser LR, Kelly ML, Patwardhan NN, Hargrove AE, Al-Hashimi HM. 2020. Demonstration that small molecules can bind and stabilize low-abundance short-lived RNA excited conformational states. J Mol Biol 432: 1297–1304. 10.1016/j.jmb.2019.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelus N, Bailly C, Hamy F, Klimkait T, Wilson W D, Boykin D W. 1999. Inhibition of HIV-1 Tat-TAR interaction by diphenylfuran derivatives: effects of the terminal basic side chains. BioorgMed Chem 7: 1089–1096. 10.1016/S0968-0896(99)00041-3 [DOI] [PubMed] [Google Scholar]
- Gligorijevic V, Barot M, Bonneau R. 2018. deepNF: deep network fusion for protein function prediction. Bioinformatics 34: 3873–3881. 10.1093/bioinformatics/bty440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Bombarelli R, Wei JN, Duvenaud D, Hernández-Lobato JM, Sánchez-Lengeling B, Sheberla D, Aguilera-Iparraguirre J, Hirzel TD, Adams RP, Aspuru-Guzik A. 2018. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent Sci 4: 268–276. 10.1021/acscentsci.7b00572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbert C, James TL. 2008. Docking to RNA via root-mean-square-deviation-driven energy minimization with flexible ligands and flexible targets. J Chem Inf Model 48: 1257–1268. 10.1021/ci8000327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KB. 2013. RNA does the folding dance of twist, turn, stack. Proc Natl Acad Sci 110: 16706–16707. 10.1073/pnas.1316029110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haniff HS, Knerr L, Liu X, Crynen G, Boström J, Abegg D, Adibekian A, Lekah E, Wang KW, Cameron MD, et al. 2020. Design of a small molecule that stimulates vascular endothelial growth factor A enabled by screening RNA fold–small molecule interactions. Nat Chem 12: 952–961. 10.1038/s41557-020-0514-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove AE. 2020. Small molecule–RNA targeting: starting with the fundamentals. Chem Commun 56: 14744–14756. 10.1039/D0CC06796B [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Henning-Knechtel A, Kirmizialtin S. 2022. Visualizing RNA structures by SAXS-driven MD simulations. Front Bioinform 2: 781949. 10.3389/fbinf.2022.781949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt WM, Calabrese DR, Schneekloth JS Jr. 2019. Evidence for ligandable sites in structured RNA throughout the Protein Data Bank. Bioorg Med Chem 27: 2253–2260. 10.1016/j.bmc.2019.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W, Zeng J, Xie J. 2014. Antibiotic drugs targeting bacterial RNAs. Acta Pharm Sin B 4: 258–265. 10.1016/j.apsb.2014.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel K, Liu S, Larsen KP, Skiniotis G, Puglisi EV, Puglisi JD, Zhou ZH, Zhao R, Das R. 2018. De novo computational RNA modeling into cryo-EM maps of large ribonucleoprotein complexes. Nat Methods 15: 947–954. 10.1038/s41592-018-0172-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel K, Zhang K, Su Z, Watkins AM, Kladwang W, Li S, Pintilie G, Topkar VV, Rangan R, Zheludev IN, et al. 2020. Accelerated cryo-EM-guided determination of three-dimensional RNA-only structures. Nat Methods 17: 699–707. 10.1038/s41592-020-0878-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennard RW, Stone LA. 1969. Computer aided design of experiments. Technometrics 11: 137–148. 10.2307/1266770 [DOI] [Google Scholar]
- Khan E, Mishra SK, Mishra R, Mishra A, Kumar A. 2019. Discovery of a potent small molecule inhibiting Huntington's disease (HD) pathogenesis via targeting CAG repeats RNA and Poly Q protein. Sci Rep 9: 16872. 10.1038/s41598-019-53410-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ER, Blount KF, Breaker RR. 2009. Roseoflavin is a natural antibacterial compound that binds to FMN riboswitches and regulates gene expression. RNA Biol 6: 187–194. 10.4161/rna.6.2.7727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grice SF. 2015. Targeting the HIV RNA genome: high-hanging fruit only needs a longer ladder. Curr Top Microbiol Immunol 389: 147–169. 10.1007/82_2015_434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekka E, Hall J. 2018. Noncoding RNAs in disease. FEBS Lett 592: 2884–2900. 10.1002/1873-3468.13182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkul JA, MacKerell AD Jr. 2018. Polarizable force field for RNA based on the classical drude oscillator. J Comput Chem 39: 2624–2646. 10.1002/jcc.25709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbrecht MW, Noble WS. 2015. Machine learning applications in genetics and genomics. Nat Rev Genet 16: 321–332. 10.1038/nrg3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Jia X, Zhang K, Su Z. 2022. Cryo-EM advances in RNA structure determination. Signal Transduct Target Ther 7: 58. 10.1038/s41392-022-00916-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltarollo VG, Kronenberger T, Espinoza G Z, Oliveira P R, Honorio K M. 2019. Advances with support vector machines for novel drug discovery. Expert Opin Drug Discov 14: 23–33. 10.1080/17460441.2019.1549033 [DOI] [PubMed] [Google Scholar]
- Manigrasso J, Marcia M, De Vivo M. 2021. Computer-aided design of RNA-targeted small molecules: a growing need in drug discovery. Chemistry (Easton) 7: 2965–2988. 10.1016/j.chempr.2021.05.021 [DOI] [Google Scholar]
- Markati T, Fisher G, Ramdas S, Servais L. 2022. Risdiplam: an investigational survival motor neuron 2 (SMN2) splicing modifier for spinal muscular atrophy (SMA). Expert Opin Investig Drugs 31: 451–461. 10.1080/13543784.2022.2056836 [DOI] [PubMed] [Google Scholar]
- McKnight KL, Heinz BA. 2003. RNA as a target for developing antivirals. Antivir Chem Chemother 14: 61–73. 10.1177/095632020301400201 [DOI] [PubMed] [Google Scholar]
- Mlýnský V, Bussi G. 2018. Molecular dynamics simulations reveal an interplay between SHAPE reagent binding and RNA flexibility. J Phys Chem Lett 9: 313–318. 10.1021/acs.jpclett.7b02921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitessier N, Westhof E, Hanessian S. 2006. Docking of aminoglycosides to hydrated and flexible RNA. J Med Chem 49: 1023–1033. 10.1021/jm0508437 [DOI] [PubMed] [Google Scholar]
- Morgan BS, Forte JE, Culver RN, Zhang Y, Hargrove AE. 2017. Discovery of key physicochemical, structural, and spatial properties of RNA-targeted bioactive ligands. Angew Chem Int Ed Engl 56: 13498–13502. 10.1002/anie.201707641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BS, Forte JE, Hargrove AE. 2018. Insights into the development of chemical probes for RNA. Nucleic Acids Res 46: 8025–8037. 10.1093/nar/gky718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BS, Sanaba BG, Donlic A, Karloff DB, Forte JE, Zhang Y, Hargrove AE. 2019. R-BIND: an interactive database for exploring and developing RNA-targeted chemical probes. ACS Chem Biol 14: 2691–2700. 10.1021/acschembio.9b00631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki H, Tian YS, Kawashita N, Takagi T. 2018. Mordred: a molecular descriptor calculator. J Cheminform 10: 4. 10.1186/s13321-018-0258-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves MA, Totrov M, Abagyan R. 2012. Docking and scoring with ICM: the benchmarking results and strategies for improvement. J Comput Aided Mol Des 26: 675–686. 10.1007/s10822-012-9547-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B, Neidle S, Wilson WD. 2009. A role for water molecules in DNA−ligand minor groove recognition. Acc Chem Res 42: 11–21. 10.1021/ar800016q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noe F, Tkatchenko A, Muller KR, Clementi C. 2020. Machine learning for molecular simulation. Annu Rev Phys Chem 71: 361–390. 10.1146/annurev-physchem-042018-052331 [DOI] [PubMed] [Google Scholar]
- Oliver C, Mallet V, Gendron RS, Reinharz V, Hamilton WL, Moitessier N, Waldispuhl J. 2020. Augmented base pairing networks encode RNA-small molecule binding preferences. Nucleic Acids Res 48: 7690–7699. 10.1093/nar/gkaa583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padroni G, Patwardhan NN, Schapira M, Hargrove AE. 2020. Systematic analysis of the interactions driving small molecule-RNA recognition. RSC Med Chem 11: 802–813. 10.1039/d0md00167h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, Van Hoosear M, Shin Y, Chin DN, Keller CG, et al. 2015. SMN2 splice modulators enhance U1–pre-mRNA association and rescue SMA mice. Nat Chem Biol 11: 511–517. 10.1038/nchembio.1837 [DOI] [PubMed] [Google Scholar]
- Patwardhan NN, Ganser LR, Kapral GJ, Eubanks CS, Lee J, Sathyamoorthy B, Al-Hashimi HM, Hargrove AE. 2017. Amiloride as a new RNA-binding scaffold with activity against HIV-1 TAR. Medchemcomm 8: 1022–1036. 10.1039/C6MD00729E [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan NN, Cai Z, Newson CN, Hargrove AE. 2019a. Fluorescent peptide displacement as a general assay for screening small molecule libraries against RNA. Org Biomol Chem 17: 1778–1786. 10.1039/c8ob02467g [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan NN, Cai Z, Umuhire Juru A, Hargrove AE. 2019b. Driving factors in amiloride recognition of HIV RNA targets. Org Biomol Chem 17: 9313–9320. 10.1039/c9ob01702j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE III, Laughton CA, Orozco M. 2007. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys J 92: 3817–3829. 10.1529/biophysj.106.097782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer P, Gohlke H. 2007. DrugScoreRNA—knowledge-based scoring function to predict RNA-ligand interactions. J Chem Inf Model 47: 1868–1876. 10.1021/ci700134p [DOI] [PubMed] [Google Scholar]
- Philips A, Milanowska K, Lach G, Bujnicki JM. 2013. LigandRNA: computational predictor of RNA-ligand interactions. RNA 19: 1605–1616. 10.1261/rna.039834.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilch DS, Kirolos MA, Breslauer KJ. 1995. Berenil binding to higher ordered nucleic acid structures: complexation with a DNA and RNA triple helix. Biochemistry 34: 16107–16124. 10.1021/bi00049a026 [DOI] [PubMed] [Google Scholar]
- Polykovskiy D, Zhebrak A, Sanchez-Lengeling B, Golovanov S, Tatanov O, Belyaev S, Kurbanov R, Artamonov A, Aladinskiy V, Veselov M, et al. 2020. Molecular sets (MOSES): a benchmarking platform for molecular generation models. Front Pharmacol 11: 565644. 10.3389/fphar.2020.565644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova M, Isayev O, Tropsha A. 2018. Deep reinforcement learning for de novo drug design. Sci Adv 4: eaap7885. 10.1126/sciadv.aap7885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratni H, Ebeling M, Baird J, Bendels S, Bylund J, Chen KS, Denk N, Feng Z, Green L, Guerard M, et al. 2018. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem 61: 6501–6517. 10.1021/acs.jmedchem.8b00741 [DOI] [PubMed] [Google Scholar]
- Rizvi NF, Santa Maria JP, Nahvi A, Klappenbach J, Klein DJ, Curran PJ, Richards MP, Chamberlin C, Saradjian P, Burchard J, et al. 2020. Targeting RNA with small molecules: identification of selective, RNA-binding small molecules occupying drug-like chemical space. SLaS Discov 25: 384–396. 10.1177/2472555219885373 [DOI] [PubMed] [Google Scholar]
- Rother M, Rother K, Puton T, Bujnicki J M. 2011. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic Acids Res 39: 4007–4022. 10.1093/nar/gkq1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, Garmendia-Doval AB, Juhos S, Schmidtke P, Barril X, Hubbard RE, Morley SD. 2014. rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput Biol 10: e1003571. 10.1371/journal.pcbi.1003571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LG, Hennig M. 2008. RNA structure determination by NMR. Methods Mol Biol 452: 29–61. 10.1007/978-1-60327-159-2_2 [DOI] [PubMed] [Google Scholar]
- Slaby O, Laga R, Sedlacek O. 2017. Therapeutic targeting of non-coding RNAs in cancer. Biochem J 474: 4219–4251. 10.1042/BCJ20170079 [DOI] [PubMed] [Google Scholar]
- Sponer J, Bussi G, Krepl M, Banas P, Bottaro S, Cunha RA, Gil-Ley A, Pinamonti G, Poblete S, Jurecka P, et al. 2018. RNA structural dynamics as captured by molecular simulations: a comprehensive overview. Chem Rev 118: 4177–4338. 10.1021/acs.chemrev.7b00427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM, Al-Hashimi HM. 2011. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat Chem Biol 7: 553–559. 10.1038/nchembio.596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Yang J, Zhang Z. 2022. RNALigands: a database and web server for RNA-ligand interactions. RNA 28: 115–122. 10.1261/rna.078889.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang R, Long T, Lui KO, Chen Y, Huang ZP. 2020. A roadmap for fixing the heart: RNA regulatory networks in cardiac disease. Mol Ther Nucleic Acids 20: 673–686. 10.1016/j.omtn.2020.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijerina P, Mohr S, Russell R. 2007. DMS footprinting of structured RNAs and RNA–protein complexes. Nat Protoc 2: 2608–2623. 10.1038/nprot.2007.380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosco P, Stiefl N, Landrum G. 2014. Bringing the MMFF force field to the RDKit: implementation and validation. J Cheminform 6: 37. 10.1186/s13321-014-0037-3 [DOI] [Google Scholar]
- Tourasse NJ, Darfeuille F. 2020. Structural alignment and covariation analysis of RNA sequences. Bio Protoc 10: e3511. 10.21769/BioProtoc.3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E, Lee G, Li B, Madabhushi A, Shah P, Spitzer M, et al. 2019. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 18: 463–477. 10.1038/s41573-019-0024-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner KD, Hajdin CE, Weeks KM. 2018. Principles for targeting RNA with drug-like small molecules. Nat Rev Drug Discov 17: 547–558. 10.1038/nrd.2018.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn JD, Mejia-Guerra MK, Ramstein G, Kremling KA, Valluru R, Buckler ES, Wang H. 2019. Evolutionarily informed deep learning methods for predicting relative transcript abundance from DNA sequence. Proc Natl Acad Sci 116: 5542–5549. 10.1073/pnas.1814551116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins AM, Geniesse C, Kladwang W, Zakrevsky P, Jaeger L, Das R. 2018. Blind prediction of noncanonical RNA structure at atomic accuracy. Sci Adv 4: eaar5316. 10.1126/sciadv.aar5316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins AM, Rangan R, Das R. 2020. FARFAR2: improved de novo rosetta prediction of complex global RNA folds. Structure 28: 963–976.e6. 10.1016/j.str.2020.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Jackson SA. 2019. Machine learning and complex biological data. Genome Biol 20: 76. 10.1186/s13059-019-1689-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani K, Jordan D, Yang M, Fullenkamp CR, Calabrese DR, Boer R, Hilimire T, Allen TEH, Khan RT, Schneekloth JS Jr. 2022. Machine learning informs RNA-binding chemical space. Angew Chem Int Ed Engl 30: e202211358. 10.1002/anie.202211358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim I, Stern HA, Kennedy SD, Tubbs JD, Turner DH. 2010. Reparameterization of RNA χ torsion parameters for the AMBER force field and comparison to NMR spectra for cytidine and uridine. J Chem Theory Comput 6: 1520–1531. 10.1021/ct900604a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafferani M, Haddad C, Luo L, Davila-Calderon J, Chiu LY, Mugisha CS, Monaghan AG, Kennedy AA, Yesselman JD, Gifford RJ, et al. 2021. Amilorides inhibit SARS-CoV-2 replication in vitro by targeting RNA structures. Sci Adv 7: eabl6096. 10.1126/sciadv.abl6096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafferani M, Martyr JG, Muralidharan D, Montalvan NI, Cai Z, Hargrove AE. 2022. Multiassay profiling of a focused small molecule library reveals predictive bidirectional modulation of the lncRNA MALAT1 triplex stability in vitro. ACS Chem Biol 17: 2437–2447. 10.1021/acschembio.2c00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamani F, Suzuki T. 2021. Synthetic RNA modulators in drug discovery. J Med Chem 64: 7110–7155. 10.1021/acs.jmedchem.1c00154 [DOI] [PubMed] [Google Scholar]
- Zgarbova M, Otyepka M, Sponer J, Mladek A, Banas P, Cheatham TE III, Jurecka P. 2011. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J Chem Theory Comput 7: 2886–2902. 10.1021/ct200162x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fei Y, Sun L, Zhang QC. 2022. Advances and opportunities in RNA structure experimental determination and computational modeling. Nat Methods 19: 1193–1207. 10.1038/s41592-022-01623-y [DOI] [PubMed] [Google Scholar]
- Zhao M, Ratmeyer L, Peloquin RG, Yao S, Kumar A, Spychala J, Boykin DW, David Wilson W. 1995. Small changes in cationic substituents of diphenylfuran derivatives have major effects on the binding affinity and the binding mode with RNA helical duplexes. Bioorg Med Chem 3: 785–794. 10.1016/0968-0896(95)00057-N [DOI] [PubMed] [Google Scholar]
- Zhao Z, Timofeev N, Hartley SW, Chui DH, Fucharoen S, Perls TT, Steinberg MH, Baldwin CT, Sebastiani P. 2008. Imputation of missing genotypes: an empirical evaluation of IMPUTE. BMC Genet 9: 85. 10.1186/1471-2156-9-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Le V, Kalia D, Nakayama S, Mikek C, Lewis EA, Sintim HO. 2014. Diminazene or berenil, a classic duplex minor groove binder, binds to G-quadruplexes with low nanomolar dissociation constants and the amidine groups are also critical for G-quadruplex binding. Mol Biosyst 10: 2724–2734. 10.1039/c4mb00359d [DOI] [PubMed] [Google Scholar]
- Zou J, Huss M, Abid A, Mohammadi P, Torkamani A, Telenti A. 2019. A primer on deep learning in genomics. Nat Genet 51: 12–18. 10.1038/s41588-018-0295-5 [DOI] [PMC free article] [PubMed] [Google Scholar]