

Abstract
In light of the dramatic decline in amphibian biodiversity, new cost-efficient tools to rapidly monitor species abundance and population genetic diversity in space and time are urgently needed. It has been amply demonstrated that the use of environmental DNA (eDNA) for single-species detection and characterization of community composition can increase the precision of amphibian monitoring compared to traditional (observational) approaches. However, it has been suggested that the efficiency and accuracy of the eDNA approach could be further improved by more timely sampling; in addition, the quality of genetic diversity data derived from the same DNA has been confirmed in other vertebrate taxa, but not amphibians. Given the availability of previous tissue-based genetic data, here we use the common frog Rana temporaria Linnaeus, 1758 as our target species and an improved eDNA protocol to: (i) investigate differences in species detection between three developmental stages in various freshwater environments; and (ii) study the diversity of mitochondrial DNA (mtDNA) haplotypes detected in eDNA (water) samples, by amplifying a specific fragment of the COI gene (331 base pairs, bp) commonly used as a barcode. Our protocol proved to be a reliable tool for monitoring population genetic diversity of this species, and could be a valuable addition to amphibian conservation and wetland management.
Subject terms: Biological techniques, Genetics, Molecular biology, Zoology
Introduction
Of all taxa affected by the current biodiversity crisis, amphibians are the most endangered group of vertebrates1, with 41% of globally-evaluated IUCN species included in ‘threatened’ categories (IUCN, 2021). Due to the general elusiveness of amphibian adults and limited detectability of early life stages, which often live in protected, difficult-to-access and/or season-dependent aquatic environments, accurate estimates of the presence and distribution of amphibian species are logistically difficult, costly and time-consuming to obtain2,3.
In addition to species richness, within-species genetic diversity is of crucial importance for the persistence and evolution of natural populations4,5, enabling them to adapt to environmental changes, and to the spread of new pathogens6,7. Moreover, the loss of genetic variability may have detrimental effects on individual health due to inbreeding depression, reducing fitness and ultimately increasing the risk of population and species extinction8. Due to their particular breeding strategy (often r-strategists with small effective population sizes and high clutch mortality), low dispersal rates and high philopatry, all of which limit population connectivity, amphibians seem to be especially prone to genetic erosion9. Therefore, regular, cost-effective genetic monitoring should be considered a fundamental aspect of amphibian conservation strategies. Although the importance of protecting genetic diversity is widely recognized and has been addressed under the Aichi Biodiversity Targets10, the development of standardized monitoring frameworks for an accurate surveillance of genetic diversity trends in natural populations is still limited to few charismatic or economically relevant species5,11.
For the last decade, environmental DNA (i.e. DNA that can be extracted from noninvasive samples such as soil, water, fecal pellets, hair or feathers; eDNA) has been used for accurate, cost-efficient species detection in aquatic environments12,13, including for amphibians (for a recent review, see3). Specifically, single species eDNA surveys (mainly using quantitative PCR; qPCR) have increased the speed and efficiency of amphibian detection, compared to traditional observational monitoring14,15, and have proven to be particularly useful for detecting rare or elusive amphibian species (e.g.,16–18), improving the knowledge of habitat requirements and species distributions (e.g.19–21), and tracking invasive alien amphibians (e.g.22–24.). In addition, the eDNA metabarcoding approach is increasingly used as a cost-effective method for the simultaneous assessment of species composition in amphibian communities3,25. However, to our knowledge, only one study aiming at the development of a long-term eDNA-based monitoring program for an amphibian species has been published thus far26.
Very recently, the possibility of inferring intraspecific genetic diversity from eDNA has also been explored3,27–29; however, only a few pilot studies on amphibians have been published. For example, Gorički et al.30 used two short CytB fragments (about 100–150 bp) for the discrimination of two putative olm (Proteus anguinus) subspecies, but the protocol was not designed to estimate population-level genetic diversity. Wang et al.31 developed an eDNA metabarcoding protocol for the Chinese giant salamander (Andrias davidianus) to allow the detection of seven haplotypes corresponding to distinct evolutionary lineages (reporting the results of laboratory amplicon mixture), but again the study was not aimed at the assessment of within-population genetic variability.
Despite their great potential, eDNA methods are extremely sensitive to sampling design and field and laboratory protocols, as eDNA is not abundant and persists in aquatic environments for a limited time, from a few days to several weeks32–34. Therefore, developing and validating effective field sampling methods is essential to the application of eDNA-based survey methods35–37.
In this study, we chose the common frog (Rana temporaria Linnaeus, 1758), a widespread European pond-breeding amphibian38, showing high genetic diversity39,40, to develop and validate an eDNA metabarcoding protocol allowing rapid and standardized assessments of within-population genetic variability from water samples. Specifically, by selecting 10 wetland sites, for which we had previous information on R. temporaria haplotypes identified using traditional tissue sampling41, we: (a) developed a metabarcoding protocol targeting a 331 bp long fragment of the COI region, which allowed discrimination of previously identified haplotypes; (b) optimized the sampling design, in terms of temporal and spatial replicates, for the collection of water samples in a variety of wetland habitats; (c) computed standard genetic diversity estimates for R. temporaria populations from eDNA metabarcoding, to compare eDNA-based results with previously available genetic data.
Materials and methods
Study species and study area
The common frog has been a model species for previous genetic diversity studies (e.g.,39,40); however, despite being the most widespread amphibian species in Europe42, local population declines are frequent due to climate change and habitat degradation43,44. Our study area covers the Autonomous Province of Trento (Italy), a mountainous region located in the eastern Alps; mtDNA haplotypes data are available for R. temporaria across the study area from a recent genetic survey41.
Sample collection
For the present study, 10 sites were selected from Marchesini et al.41 (Fig. 1 and Supplementary Information, Table S1) that encompassed all three mitochondrial lineages of R. temporaria known to be present in the study area, as well as the majority of the haplotypes (8/12) noted in41.
Figure 1.
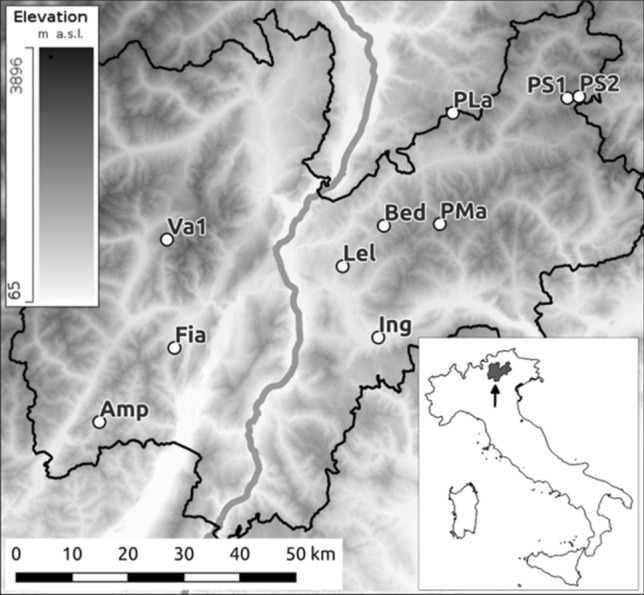
Map of the study region (Province of Trento, Italy) showing the 10 selected wetland sample sites, labeled according to Table 1. Site locations, abbreviations, coordinates, elevation and description are listed in Supplementary Information, Table S1. The map was generated using QGIS version 3.20 (http://www.qgis.org); Digital Terrain Model (DTM) for the study area was extracted from the 20 m-resolution DTM of Italy, publicly available at the National Geoportal of Italy (http://www.pcn.minambiente.it/mattm/servizio-di-scaricamento-wcs/).
Water samples were collected at each of the 10 sites during the reproductive season (March-September depending on altitude) from 2019 to 2021. In order to define a standard protocol, three temporal (T1-T3) and three spatial (S1-S3) replicates were collected, where T1 corresponds to a replicate collected when eggs or early-stage larvae were present at the site (early spring), T2, when late-stage larvae were present (mid-summer), and T3, when both adults and juveniles had abandoned the sites following reproduction (end of summer/ early autumn). Some of the wetland sites became partially desiccated during the study, thus collecting the T3 replicates was not always possible. When possible (i.e. whenever spatially separated, accessible water microhabitats were available at the site), for each temporal replicate, three spatial replicates were sampled (5/10 sites) close to observed life stages. For three of these sites, for one of the three spatial replicates (Amp-T1-S2; Ing-T1-S3; PMa-T1-S1, Table 1 and in Supplementary Information, Table S2), no egg clutches or tadpoles were detected at T1; nonetheless, sampling was performed in any case, and all sampled microhabitats were included in the experimental design. In the remaining five sites, only two spatial replicates were possible. In order to capture as many haplotypes as possible from the same sampling site, the spatial replicates were collected at least 20 m apart (depending on the area of wetland on the sampling site).
Table 1.
Spatial and temporal replicates included in the eDNA metabarcoding study of Rana temporaria in the Province of Trento, Italy.
| Site | T1 | T2 | T3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | Repl. T1 | S1 | S2 | s3 | Repl. T2 | S1 | S2 | S3 | Repl. T3 | Tot Repl | |
| Amp | x | nd | x | 2/3 | nd | x | x | 2/3 | nd | nd | nd | 0/3 | 4/9 |
| Bed | x | x | / | 2/2 | x | x | / | 2/2 | nd | nd | / | 0/2 | 4/6 |
| Fia | x | x | x | 3/3 | x | x | nd | 2/3 | nd | nd | nd | 0/3 | 5/9 |
| Ing | x | x | nd | 2/3 | x | nd | nd | 1/3 | nd | nd | nd | 0/3 | 3/9 |
| Lel | x | nd | / | 1/2 | nd | / | / | 0/1 | / | / | / | 0/0 | 1/3 |
| Pla | x | x | x | 3/3 | nd | nd | nd | 0/3 | nd | nd | nd | 0/3 | 3/9 |
| PMa | nd | x | x | 2/3 | nd | nd | nd | 0/3 | nd | nd | nd | 0/3 | 2/9 |
| PS1 | x | x | / | 2/2 | x | nd | / | 1/2 | nd | nd | / | 0/2 | 3/6 |
| PS2 | x | x | / | 2/2 | nd | nd | / | 0/2 | nd | nd | / | 0/2 | 2/6 |
| Va1 | x | x | / | 2/2 | x | x | / | 2/2 | x | nd | / | 1/2 | 5/6 |
| TOT | 32/72 | ||||||||||||
T1, T2, T3 = temporal replicates 1, 2, and 3; Repl. T1, Repl. T2, Repl. T3: for every temporal point, the sum of spatial replicates per site is reported (positive/total); S1, S2, S3 = spatial replicates 1, 2, and 3; x = sampling was performed and positive results were obtained after sequencing; nd = sampling was performed but amplification was not successful (no DNA bands visible on the Qiaxcel Advanced System (Qiagen) and/or several samples were sequenced for confirmation and no R. temporaria DNA was detected); / = sampling was not performed because the structure of the wetland site was not compatible with three spatial replicates (see text for details) or, (for T3) one of the previously selected water microhabitats was no longer physically accessible or non-existent (due to seasonal changes in wetland extent and hydrology).
Each replicate sample was collected manually from just under the surface of the water body using a sterile plastic canister. Stirring up sediment was carefully avoided. To minimize sample contamination, field workers wore laboratory gloves (changed between each replicate), collected samples from the shore without disturbing the water body, and wiped all equipment with bleach and alcohol between each site. The water collected in the canister was stirred before being drawn up with a 100 ml syringe and filtered through a Sterivex-GP Filter unit (pore size 0.22 µm, Millipore cat. no. SVGPL10RC); this step was repeated until the filter clogged. A second filter was also used for the same spatial replicate. The quantity of water that could be filtered through two Sterivex-GP Filter units varied widely among sites, ranging from about 100 ml to more than one liter, depending on the suspended organic matter. Each filter was drained and capped at both ends with the inlet and outlet caps. Following the manufacturer’s instructions, all filters were kept at ambient temperature out of direct sunlight until arrival in the laboratory later the same day, and archived at − 20 °C until DNA extraction.
Sample processing, including primer validation
All laboratory procedures were carried out at the Platform of Animal, Environmental and Antique DNA of the Conservation Genomics Research Unit, Fondazione Edmund Mach, following recommended guidelines for eDNA analyses, including separate pre- and post-PCR laboratories45,46. All procedures were performed under BSL2 biological hoods. DNA extraction from filters was carried out using the DNeasy PowerWater Sterivex Kit (Qiagen), following the manufacturer’s instructions with these modifications: the heating step and bead beating tubes (PowerBead Tubes) were eliminated to reduce extraction of non-target genetic material from microorganisms. The two filters corresponding to the same spatial replicate S1, S2, or S3 were processed simultaneously and extracts were merged into a single tube at the 14th step of the protocol. DNA extraction was performed in batches of a maximum of 10 water replicate samples, including one negative control (extraction blank) for each extraction batch.
To design a primer pair for amplifying the diagnostic mtDNA region for R. temporaria, Primer3Plus47 was used with the haplotype sequences available from Marchesini et al.41. The resulting primer pair, named Rt-aplo_COI F (GTAATAATTTTCTTTATGGT) and Rt-aplo_COI R (TCAAACAAAGAGGGGTGT), amplifies a fragment 331 bp long distinguishing 11 of the 12 mtDNA haplotypes listed in Marchesini et al.41 for the Province of Trento. Specifically, haplotype pairs CA2 and TN4, and SA1 and PR7 (the latter not being previously found in the study area) were not distinguishable, and therefore, were named CA2_TN4 and SA1_PR7, respectively, in the Results section. To distinguish all known haplotypes for the study area, a 569 bp sequence plus adapters (thus about 690 bp in total) would have been required, but such a long fragment cannot be processed using the Illumina technology commonly adopted for metabarcoding (maximum length 600 bp). The primer pair was tested in vitro on DNA extracted from R. temporaria tissue samples available from previous studies, and amplification success of all samples was confirmed via screening on a Qiaxcel Advanced System (Qiagen). The amplification reaction took place in a final volume of 50µL, containing H2O (22.25µL), Promega Flexi Buffer 5X (10µL), MgCl2 25 mM (4µL), BSA 10 mg/mL (0.5µL), Rt-aplo_COI-F 10 ρmol/μL (1µL) and Rt-aplo_COI-R 10 ρmol/μL (1µL), dNTP’s 10 mM each (1µL total), Promega GoTaq G2 5U/µL (0.25µL) and template DNA (10µL). The PCR mixture was denatured at 95 °C for 2 min, followed by 40 cycles of 30 s at 95 °C, 15 s at 48 °C and 40 s at 72 °C, and a final elongation step at 72 °C for 5 min. Only one PCR was run per replicate, and a second PCR was run only if the first one failed to produce results. One negative control (PCR blank) was included for each PCR reaction, along with all extraction negative controls.
Each amplification product was then purified with the MinElute PCR Purification Kit (Qiagen) following manufacturer’s instructions. 20 µl of each purified product were loaded into a single 96-well plate and sequenced at the FEM Sequencing and Genotyping Platform using paired-end sequencing (2 × 300 bp) on an Illumina Miseq (Illumina, San Diego, CA) with a 30 000 bp coverage.
Bioinformatics and statistical analysis
All bioinformatic analyses were performed with the software MICCA48. Overlapping paired-end sequences were merged to obtain consensus sequences using the command ‘mergepairs’ with a minimum overlap length of 100 bp and maximum of one mismatch in the overlap region. Reads that did not contain the forward or reverse primers were discarded, and primers were trimmed from the remaining fragments with the command ‘trim’. Sequences were quality filtered using the command ‘filter’, assuming a maximum allowed expected error rate of 0.1% and a minimum length of 331 bp (exact length of the target sequence without primers, as is becoming common practice for genetic diversity studies based on eDNA metabarcoding, e.g.,27,49). The reference database for haplotype classification was assembled using published R. temporaria haplotype sequences found in Italy. The method ‘otu open_ref’ was used to cluster our sequences against this database with an identity threshold of 0.99, rejecting a sequence if the fraction of alignment to the reference sequence was lower than one, and discarding sequences with a read abundance value lower than 100 after dereplication. To avoid false positives, either from PCR or sequencing errors, or minor cross-contaminations between the replicate samples, OTUs represented by fewer reads than 5% of the total reads for a specific replicate sample were discarded27.
To calculate the diversity indices, haplotype relative frequencies were obtained across spatial and temporal replicates through simulated datasets created with a Linux shell script. The datasets were built such as that, for each site and each recorded haplotype, the haplotypes frequencies were equal to the haplotype total number of reads (sequence counts) divided by 100. The simulated datasets thus represent hypothetical populations in which absolute abundance (in terms of sequence counts) of each haplotype ideally correspond to the number of individuals carrying the haplotype. As the abundances calculated from eDNA data could be influenced by several limitations (i.e. non-exhaustive sampling in terms of spatial coverage of the site, potential preferential amplification between different haplotypes, potential differences in DNA particles release from different coexisting life stages of the species), these estimates may not be as accurate as more invasive methods. For a more precise estimate of the population genetic diversity, a traditional sampling is still desirable. However, the reproductive behavior of R. temporaria, similar to other pond-breeding amphibians, is characterized by an ‘explosive’ reproduction, producing large numbers of egg masses sometimes crowded together. Furthermore, in our target species, generally one female lies one egg clutch, therefore the number of egg clutches roughly corresponds to the number of females in the population. Therefore, as eDNA samples were collected when larvae/eggs presence was visually confirmed, and by sampling multiple spatial replicates, the chances of obtaining a sufficiently representative sampling of the population’s gene pool were increased.
Standard diversity indices were calculated for each site (number of haplotypes, n; haplotype diversity, h; nucleotide diversity, π) using the software DnaSP v650, and Spearman rank correlations tests were performed using RStudio to compare these estimates with those reported in Marchesini et al.41. Since the estimates of haplotype frequencies and diversity indices from the two methods are not exactly the same (i.e. the tissue based measures were based on the number of individuals in which a haplotype was found, whereas eDNA haplotype frequencies and diversity were based on the number of samples through space in which each haplotype was detected), non-metric multidimensional scaling (NMDS), based on Bray–Curtis dissimilarity matrices and computed using the R package ‘vegan’ 2.6–4, was used to graphically represent differences between the eDNA metabarcoding and the reference datasets; these two datasets were then statistically compared with a Mantel test with 10000 permutations in RStudio. A correlation between the two measures was expected if eDNA was capable of detecting nearly all haplotypes (since common haplotypes would also be more widespread).
Results
A total of 72 water samples were collected and filtered at the 10 sampling sites. Rana temporaria DNA was successfully amplified from 32 of these. T1 replicates were the most successful, with 21/25 eDNA-positive replicate samples, while T2 replicates yielded results in 10/24 replicates (Table 1). T3 replicates did not produce positive results for any sites except Va1 (data not shown). All negative controls from both the extraction and PCR steps were considered not contaminated, as none had more than 10 reads; therefore, these were removed during the OTU clustering step. The spatial replicates showed high variability in terms of amplification success, identified haplotypes and their relative frequencies for each sampling site (Tables 1 and 2).
Table 2.
COI haplotype frequencies for Rana temporaria populations in the Province of Trento detected by eDNA metabarcoding across temporal replicates. For each wetland site, haplotype frequencies (mean of spatial replicates S1-3) detected by eDNA metabarcoding at the different temporal points (T1, T2 and mean T1 + T2) are reported, together with available frequencies derived from traditional tissue-based genetic sampling (reference dataset: 41). nd = no data.
| Site | Dataset | COI haplotypes (frequencies) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CA2_TN4 | VC6 | PR4 | SA1_PR7 | TN2 | TN3 | TN5 | MT5 | ||
| Amp | eDNA (T1) | 0.644 | 0 | 0 | 0 | 0.74 | 0 | 0 | 0 |
| eDNA (T2) | 0.317 | 0 | 0 | 0 | 0.68 | 0 | 0 | 0 | |
| eDNA (T1 + T2) | 0.2907 | 0 | 0 | 0 | 0.71 | 0 | 0 | 0 | |
| reference dataset | 0.4 | 0 | 0 | 0 | 0.6 | 0 | 0 | 0 | |
| Bed | eDNA (T1) | 0.2637 | 0.61 | 0.122 | 0 | 0 | 0 | 0 | 0 |
| eDNA (T2) | 0.2006 | 0.59 | 0.209 | 0 | 0 | 0 | 0 | 0 | |
| eDNA (T1 + T2) | 0.2321 | 0.6 | 0.166 | 0 | 0 | 0 | 0 | 0 | |
| reference dataset | 0.1 | 0.8 | 0.1 | 0 | 0 | 0 | 0 | 0 | |
| Fia | eDNA (T1) | 0.3871 | 0 | 0 | 0 | 0.16 | 0.43 | 0.0196 | 0 |
| eDNA (T2) | 0.4415 | 0 | 0 | 0 | 0.06 | 0.49 | 0 | 0 | |
| eDNA (T1 + T2) | 0.4089 | 0 | 0 | 0 | 0.12 | 0.46 | 0.0118 | 0 | |
| reference dataset | 0.3 | 0 | 0 | 0 | 0.1 | 0.3 | 0.3 | 0 | |
| Ing | eDNA (T1) | 0.9528 | 0.05 | 0 | 0 | 0 | 0 | 0 | 0 |
| eDNA (T2) | 0.2345 | 0.77 | 0 | 0 | 0 | 0 | 0 | 0 | |
| eDNA (T1 + T2) | 0.7133 | 0.29 | 0 | 0 | 0 | 0 | 0 | 0 | |
| reference dataset | 0.8 | 0.2 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Lel | eDNA (T1) | 0.2203 | 0.78 | 0 | 0 | 0 | 0 | 0 | 0 |
| eDNA (T2) | nd | nd | nd | nd | nd | nd | nd | nd | |
| eDNA (T1 + T2) | 0.2203 | 0.78 | 0 | 0 | 0 | 0 | 0 | 0 | |
| reference dataset | 0.5 | 0.4 | 0 | 0.1a | 0 | 0 | 0 | 0 | |
| PLa | eDNA (T1) | 0.8031 | 0 | 0.134b | 0.0628 | 0 | 0 | 0 | 0 |
| eDNA (T2) | nd | nd | nd | nd | nd | nd | nd | nd | |
| eDNA (T1 + T2) | 0.8031 | 0 | 0.134b | 0.0628 | 0 | 0 | 0 | 0 | |
| reference dataset | 0.8 | 0 | 0 | 0.2 | 0 | 0 | 0 | 0 | |
| PMa | eDNA (T1) | 0.1754 | 0.82 | 0 | 0 | 0 | 0 | 0 | 0 |
| eDNA (T2) | nd | nd | nd | nd | nd | nd | nd | nd | |
| eDNA (T1 + T2) | 0.1754 | 0.82 | 0 | 0 | 0 | 0 | 0 | 0 | |
| reference dataset | 0.7 | 0.3 | 0 | 0 | 0 | 0 | 0 | 0 | |
| PS1 | eDNA (T1) | 0.3936 | 0.35 | 0.26 | 0 | 0 | 0 | 0 | 0 |
| eDNA (T2) | 0.7635 | 0 | 0.237 | 0 | 0 | 0 | 0 | 0 | |
| eDNA (T1 + T2) | 0.5169 | 0.23 | 0.252 | 0 | 0 | 0 | 0 | 0 | |
| reference dataset | 0.3 | 0.2 | 0.5 | 0 | 0 | 0 | 0 | 0 | |
| PS2 | eDNA (T1) | 0.6715 | 0.09 | 0.238 | 0 | 0 | 0 | 0 | 0 |
| eDNA (T2) | nd | nd | nd | nd | nd | nd | nd | nd | |
| eDNA (T1 + T2) | 0.6715 | 0.09 | 0.238 | 0 | 0 | 0 | 0 | 0 | |
| reference dataset | 0.5 | 0.1 | 0.4 | 0 | 0 | 0 | 0 | 0 | |
| Va1 | eDNA (T1) | 0.5235 | 0 | 0 | 0 | 0.24 | 0.19 | 0.0373b | 0 |
| eDNA (T2) | 0.4887 | 0 | 0 | 0 | 0.1 | 0.25 | 0.1544b | 0 | |
| eDNA (T1 + T2) | 0.5061 | 0 | 0 | 0 | 0.17 | 0.22 | 0.0959b | 0 | |
| reference dataset | 0.4 | 0 | 0 | 0 | 0.1 | 0.2 | 0 | 0.3a | |
Ten COI haplotypes (including CA2_TN4 and SA1_Pr7) were detected, all belonging to the three Alpine lineages already known to be present in the province of Trento41. The genetic diversity estimates calculated from the eDNA dataset are reported in Table 3. Considering the haplotype detected in all replicates from each site, the number of haplotypes detected with our eDNA protocol showed a strong and statistically significant correlation with the number of haplotypes found in the previous study (R = 0.78, p = 0.008). For eight out of 10 sites, in fact, the number of haplotypes detected by the two approaches was the same. The remaining two sites only differed for one haplotype. Similarly, nucleotide diversity for the eDNA data was strongly correlated with π of the reference dataset (R = 0.88, p = 0.002), as shown in Fig. 2. Haplotype diversity (h) from the two datasets (Fig. 3) showed a more moderate but statistically significant correlation (R = 0.63, p = 0.05). If only T1 (the temporal replicate with the highest number of positive samples) was considered, the correlation between the number of haplotypes calculated with the two datasets remained unchanged (R = 0.78, p = 0.008), while both h and π estimated from eDNA data showed a slightly higher correlation with the same indices from the reference dataset (R = 0.74, p = 0.014 and R = 0.9, p = 0.001, respectively).
Table 3.
Genetic diversity estimates for Rana temporaria populations in the Province of Trento: results of eDNA metabarcoding from the present study compared to the reference dataset41.
| Site | No haplotypes (n) | Haplotype div. (h) | Nucleotide div. (pi) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| eDNA (T1) | eDNA (T1 + T2) | tissue DNA |
eDNA (T1) | eDNA (T1 + T2) | DNA | eDNA (T1) | eDNA (T1 + T2) | tissue DNA |
|
| Amp | 2 | 2 | 2 | 0.368 | 0.428 | 0.533 | 0.001 | 0.001 | 9E-04 |
| Bed | 3 | 3 | 3 | 0.543 | 0.545 | 0.378 | 0.009 | 0.01 | 0.005 |
| Fia | 4 | 4 | 4 | 0.644 | 0.639 | 0.8 | 0.002 | 0.002 | 0.002 |
| Ing | 2 | 2 | 2 | 0.121 | 0.352 | 0.351 | 0.001 | 0.004 | 0.003 |
| Lel | 2 | 2 | 3 | 0.344 | 0.344 | 0.644 | 0.004 | 0.004 | 0.008 |
| PLa | 3 | 3 | 2 | 0.349 | 0.349 | 0.356 | 0.009 | 0.009 | 0.008 |
| PMa | 2 | 2 | 2 | 0.256 | 0.256 | 0.467 | 0.003 | 0.003 | 0.004 |
| PS1 | 3 | 3 | 3 | 0.533 | 0.509 | 0.689 | 0.011 | 0.01 | 0.011 |
| PS2 | 3 | 3 | 3 | 0.575 | 0.575 | 0.644 | 0.013 | 0.013 | 0.011 |
| Va1 | 4 | 4 | 4 | 0.601 | 0.632 | 0.778 | 0.002 | 0.002 | 0.002 |
eDNA (T1): eDNA metabarcoding considering only T1 replicates; eDNA (T1 + T2): eDNA metabarcoding considering T1 + T2 replicates; tissue DNA: estimates from the reference dataset41.
Figure 2.

Scatterplot of the correlation between the nucleotide diversity index values for Rana temporaria in the Province of Trento Italy, for two datasets: x-axis: standard tissue-based genetic survey 41; y-axis: current study (eDNA metabarcoding).
Figure 3.

Scatterplot of the correlation between the haplotype diversity index values for Rana temporaria in the Province of Trento Italy, for two datasets: x-axis: standard tissue-based genetic survey 41; y-axis: current study (eDNA metabarcoding).
The NMDS plot (shown in Fig. 4) showed that, for each sampling site, the eDNA metabarcoding datapoints were closer to the reference datapoints from the same site, than to those of other sites, suggesting a reasonable match between eDNA metabarcoding results and the reference dataset, except for two sites (i.e.: Lel, PMa). Overall, the Mantel test consistently showed a statistically significant correlation between population genetic dissimilarities based on eDNA metabarcoding data and those based on the reference dataset (R = 0.44, p = 0.005). In addition, the NMDS plot showed that three sites (Amp, Fia and Va1) cluster closely together with respect to the remaining seven, confirming the previously known distribution of R. temporaria haplotypes; that is, two clusters, one in western and one in eastern Trentino41.
Figure 4.

Non-metric multidimensional scaling (NMDS) plot, based on two Bray- Curtis dissimilarity matrices for Rana temporaria in the Province of Trento Italy, for two datasets: sites represented by a triangle and labeled with ‘*’ refer to the standard genetic survey dataset 41; sites represented with a circle and without ‘*’ refer to the eDNA metabarcoding dataset.
Discussion
In this study, all statistical tests showed that our eDNA metabarcoding protocol targeting a 331 bp long fragment of the COI region could identify haplotypes of R. temporaria previously identified from tissue samples, most reliably in the spring season. In addition, our protocol allowed us, for the first time, to provide an estimation of standard genetic diversity indices for an amphibian species from eDNA.
Our experimental design allowed us to identify an optimal sampling regime, in terms of temporal and spatial replicates, for the collection of eDNA water samples in a variety of small wetland habitats. Our results are in agreement with the recommendation from previous studies51,52 of sampling an adequate number of spatial replicates per site (up to one per 10–20 m perimeter), as this distribution of replicates allowed us to capture the full set of expected haplotypes from each sampling site. Regarding the temporal replicates, T1 alone allowed us to identify almost all haplotypes present per site in a given monitoring year. In fact, T1 yielded the same results, in terms of number of haplotypes identified, as those attained from the union of temporal replicates 1 and 2 (while no T3 replicates yielded target species DNA). Thus, if a multi-year monitoring program is being considered, we suggest implementing a simplified version of our protocol, with T1 as the optimal time point, as a strategy to maximize the cost/benefit ratio. However, as the combination of T1 and T2 replicates provided the most accurate data on haplotypes frequencies, for a more complete view of R. temporaria genetic diversity we propose that the protocol should be applied at least every three to five years. Replicate T3 did not provide any information on species presence and genetic diversity, and should not be considered for future eDNA protocol applications for amphibians that have a reproductive cycle similar to that of R. temporaria.
All the standard genetic diversity estimates computed here (namely, the number of haplotypes, h and π) proved to be statistically correlated with the same indices computed in the reference dataset from Marchesini et al.41. In agreement with the NMDS plot (Fig. 4), the Mantel test shows a moderate, statistically significant, correlation between the dissimilarities computed based on eDNA metabarcoding data and those computed based on the reference dataset. Moreover, the NMDS plot based on eDNA data further confirms previous genetic distributions of R. temporaria haplotypes41, identifying the same two geographical clusters formed by the three sites Amp, Fia and Va1 (western Trentino) and the remaining seven (eastern Trentino).
Only three of the 10 sampling sites consistently showed different haplotype counts and frequencies between the two approaches, namely sites Lel, PLa and PMa (Laghestel, Passo Lavazé and Passo Manghen; Table 2 and Table S1 in Supplementary Information). Interestingly, in these sites, the target species is known to breed in small temporary ponds near a bigger pond (Lel), or in small alpine lakes (PLa, PMa)53. These habitats are especially prone to seasonal fluctuations in water availability; therefore, it is possible that the differences in the haplotypes detected with the two methods, as well as in their relative frequencies, might be due to the ephemeral nature and repeated recolonization of these three sampling areas. In fact, the influence of short-term climatic fluctuations on demographic and genetic characteristics of wild populations has been demonstrated recently54. Even so, the eDNA protocol was able to detect two haplotypes out of the three found at Lel in the reference dataset. In the case of PLa, the most abundant haplotype shows the same relative frequency according to both datasets, and our protocol was also able to detect the less common haplotype. In addition, our eDNA metabarcoding approach detected another haplotype, not previously reported for this site but known to be present in eastern Trentino (PR4, see Table 2). For PMa, the eDNA protocol identified the same haplotypes as the reference dataset, but frequencies were reversed (see Table 2). Finally, the differences between the two datasets could also be explained by fluctuations of allele frequencies over time (with samples collected in 2017 and 2021, respectively), and considering that the reference dataset did not necessarily represent the entire gene pool of the considered populations, being based on only 10 samples per site.
The protocol developed here, which is completely non-invasive and less time-consuming than traditional observational or tissue-based genetic surveys, could be implemented routinely in amphibian monitoring programs that integrate genetic diversity estimates with confirmation of the target species presence. We are currently improving and extending this holistic approach to all European amphibian species, but also to even rarer and more elusive invertebrates such as the European freshwater crayfish Austropotamobius pallipes. The same approach could also be used to determine the presence of invasive species, such as the American bullfrog Lithobates catesbeianus, which occurs in the regions surrounding our study site, but has not been reported thus far in the Province of Trento. Finally, we advocate the integration of nucleotide diversity estimates in eDNA-based approaches, included in very few studies thus far (e.g.,28,29, in order to obtain a detailed reconstruction of the potential adaptability of populations living in human- and/or climate change-impacted areas.
Supplementary Information
Acknowledgements
This research was co-financed by the project ACQUA E VITA funded by the Ledro Alps and Judicaria MAB UNESCO Biosphere Reserve. LZ was also supported by a PhD scholarship from the University of Ferrara and MUSE - Science Museum Trento. The authors would like to thank the Fondazione E. Mach for the use of laboratory facilities, and the Editor and anonymous reviewers for their comments which improved the manuscript.
Author contributions
Conceived and designed the study: L.Z., G.M., A.M., P.P, H.C.H.; acquired the funding: G.M., P.P, G.B., H.C.H.; collected the samples: L.Z., G.M., S.C., S.E., C.F.; performed laboratory analyses: L.Z., G.M., S.C.; completed bioinformatic and statistical analyses: L.Z., A.M., G.G., D.M; drafted the manuscript: L.Z., A.M., H.C.H; read, commented and approved the manuscript: all authors.
Data availability
Sequences available upon acceptance for publication of the manuscript or upon request. Correspondence and requests for materials should be addressed to H.C.H. (email: heidi.hauffe@fmach.it).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-023-31410-4.
References
- 1.Hoffmann M, et al. The impact of conservation on the status of the world’s vertebrates. Science. 2010;330:1503–1509. doi: 10.1126/science.1194442. [DOI] [PubMed] [Google Scholar]
- 2.Mazerolle MJ, et al. Making great leaps forward: Accounting for detectability in herpetological field studies. J. Herpetol. 2007;41:672–689. doi: 10.1670/07-061.1. [DOI] [Google Scholar]
- 3.Ficetola GF, Manenti R, Taberlet P. Environmental DNA and metabarcoding for the study of amphibians and reptiles: species distribution, the microbiome, and much more. Amphibia-Reptilia. 2019;40(2):129–148. doi: 10.1163/15685381-20191194. [DOI] [Google Scholar]
- 4.Hoban SM, et al. Bringing genetic diversity to the forefront of conservation policy and management. Conserv. Genet. Resour. 2013;5:593–598. doi: 10.1007/s12686-013-9859-y. [DOI] [Google Scholar]
- 5.Pärli R, et al. Developing a monitoring program of genetic diversity: What do stakeholders say? Conserv. Gen. 2021;22(5):673–684. doi: 10.1007/s10592-021-01379-6. [DOI] [Google Scholar]
- 6.Booy G, Hendriks RJJ, Smulders MJM, Van Groenendael JM, Vosman B. Genetic diversity and the survival of populations. Plant Biol. 2000;2:379–395. doi: 10.1055/s-2000-5958. [DOI] [Google Scholar]
- 7.Höglund J. Evolutionary conservation genetics. Oxford University Press; 2009. [Google Scholar]
- 8.Frankham R, et al. Genetic management of fragmented animal and plant populations. Oxford University Press; 2017. [Google Scholar]
- 9.Allentoft ME, O’Brien J. Global amphibian declines, loss of genetic diversity and fitness: A review. Diversity. 2010;2:47–71. doi: 10.3390/d2010047. [DOI] [Google Scholar]
- 10.Secretariat of the Convention on Biological Diversity. COP-10 Decision X/2. (2010). https://www.cbd.int/decisions/cop/10/2
- 11.Hoban SM, et al. Global commitments to conserving and monitoring genetic diversity are now necessary and feasible. Bioscience. 2021;71(9):964–976. doi: 10.1093/biosci/biab054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deiner K, et al. Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Mol. Ecol. 2017;6:5872–5895. doi: 10.1111/mec.14350. [DOI] [PubMed] [Google Scholar]
- 13.Taberlet P, Bonin A, Zinger L, Coissac E. Environmental DNA: For biodiversity research and monitoring. Oxford University Press; 2018. [Google Scholar]
- 14.Eiler A, Löfgren A, Hjerne O, Nordén S, Saetre P. Environmental DNA (eDNA) detects the pool frog (Pelophylax lessonae) at times when traditional monitoring methods are insensitive. Sci. Rep. 2018;8:5452. doi: 10.1038/s41598-018-23740-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fediajevaite J, Priestley V, Arnold R, Savolainen V. Meta-analysis shows that environmental DNA outperforms traditional surveys, but warrants better reporting standards. Ecol. Evol. 2021;1:4803–4815. doi: 10.1002/ece3.7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierson TW, et al. Detection of an enigmatic plethodontid salamander using environmental DNA. Copeia. 2016;104(1):78–82. doi: 10.1643/CH-14-202. [DOI] [Google Scholar]
- 17.Vörös J, Márton O, Schmidt BR, Gál JT, Jelić D. Surveying Europe’s only cave-dwelling chordate species (Proteus anguinus) using environmental DNA. PLoS ONE. 2017;12(1):e0170945. doi: 10.1371/journal.pone.0170945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruppert KM, Davis DR, Rahman MS, Kline RJ. Development and assessment of an environmental DNA (eDNA) assay for a cryptic Siren (Amphibia: Sirenidae) Environ. Adv. 2022;7:100163. doi: 10.1016/j.envadv.2021.100163. [DOI] [Google Scholar]
- 19.Renan S, et al. Living quarters of a living fossil—uncovering the current distribution pattern of the rediscovered Hula painted frog (Latonia nigriventer) using environmental DNA. Mol. Ecol. 2017;26:6801–6812. doi: 10.1111/mec.14420. [DOI] [PubMed] [Google Scholar]
- 20.Franklin TW, et al. Repurposing environmental DNA samples to verify the distribution of rocky mountain tailed frogs in the warm springs creek basin, Montana. Northwest Sci. 2019;93(1):85–92. doi: 10.3955/046.093.0108. [DOI] [Google Scholar]
- 21.Perl RGB, et al. Using eDNA presence/non-detection data to characterize the abiotic and biotic habitat requirements of a rare, elusive amphibian. Environ. DNA. 2022;4:642–653. doi: 10.1002/edn3.276. [DOI] [Google Scholar]
- 22.Ficetola GF, Miaud C, Pompanon F, Taberlet P. Species detection using environmental DNA from water samples. Biol. Lett. 2008;4(4):423–425. doi: 10.1098/rsbl.2008.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dejean T, et al. Improved detection of an alien invasive species through environmental DNA barcoding: The example of the American bullfrog Lithobates catesbeianus. J. Appl. Ecol. 2012;49:953–959. doi: 10.1111/j.1365-2664.2012.02171.x. [DOI] [Google Scholar]
- 24.Secondi J, Dejean T, Valentini A, Audebaud B, Miaud C. Detection of a global aquatic invasive amphibian, Xenopus laevis, using environmental DNA. Amphibia-Reptilia. 2016;37(1):131–136. doi: 10.1163/15685381-00003036. [DOI] [Google Scholar]
- 25.Valentini A, et al. Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol. Ecol. 2016;25:929–942. doi: 10.1111/mec.13428. [DOI] [PubMed] [Google Scholar]
- 26.Biggs J, et al. Using eDNA to develop a national citizen science-based monitoring programme for the great crested newt (Triturus cristatus) Biol. Conserv. 2015;183:19–28. doi: 10.1016/j.biocon.2014.11.029. [DOI] [Google Scholar]
- 27.Elbrecht V, Vamos EE, Steinke D, Leese F. Estimating intraspecific genetic diversity from community DNA metabarcoding data. PeerJ. 2018;6:e4644. doi: 10.7717/peerj.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sigsgaard EE, et al. Population-level inferences from environmental DNA: Current status and future perspectives. Evol. Appl. 2020;3:245–262. doi: 10.1111/eva.12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weitemeier K, et al. Estimating the genetic diversity of Pacific salmon and trout using multigene eDNA metabarcoding. Mol. Ecol. 2021;30:4970–4990. doi: 10.1111/mec.15811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorički Š, et al. Environmental DNA in subterranean biology: Range extension and taxonomic implications for Proteus. Sci. Rep. 2017;7:45054. doi: 10.1038/srep45054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, et al. Development of an eDNA metabarcoding tool for surveying the world’s largest amphibian. Curr. Zool. 2022;68(5):608–614. doi: 10.1093/cz/zoab094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dejean T, et al. Persistence of environmental DNA in freshwater ecosystems. PLoS ONE. 2011;6(8):e23398. doi: 10.1371/journal.pone.0023398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnes MA, Turner CR. The ecology of environmental DNA and implications for conservation genetics. Conserv. Genet. 2016;17:1–17. doi: 10.1007/s10592-015-0775-4. [DOI] [Google Scholar]
- 34.Buxton A, Groombridge J, Zakaria N, Griffiths RA. Seasonal variation in environmental DNA in relation to population size and environmental factors. Sci. Rep. 2017;7:46294. doi: 10.1038/srep46294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eichmiller JJ, Miller LM, Sorensen PW. Optimizing techniques to capture and extract environmental DNA for detection and quantification of fish. Mol. Ecol. Resour. 2016;16:56–68. doi: 10.1111/1755-0998.12421. [DOI] [PubMed] [Google Scholar]
- 36.Carraro L, Stauffer JB, Altermatt F. How to design optimal eDNA sampling strategies for biomonitoring in river networks. Environ. DNA. 2021;3(1):157–172. doi: 10.1002/edn3.137. [DOI] [Google Scholar]
- 37.Buxton A, Matechou E, Griffin J, Diana A, Griffiths RA. Optimising sampling and analysis protocols in environmental DNA studies. Sci. Rep. 2021;11:11637. doi: 10.1038/s41598-021-91166-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sillero N, et al. Updated distribution and biogeography of amphibians and reptiles of Europe. Amphibia-Reptilia. 2014;35:1–31. doi: 10.1163/15685381-00002935. [DOI] [Google Scholar]
- 39.Vences M, et al. Radically different phylogeographies and patterns of genetic variation in two European brown frogs, genus Rana. Mol. Phylogenet. Evol. 2013;68:657–670. doi: 10.1016/j.ympev.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Stefani F, et al. Refugia within refugia as a key to disentangle the genetic pattern of a highly variable species: The case of Rana temporaria Linnaeus, 1758 (Anura, Ranidae) Mol. Phylogenet. Evol. 2012;65:718–726. doi: 10.1016/j.ympev.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 41.Marchesini A, Ficetola GF, Cornetti L, Battisti A, Vernesi C. Fine-scale phylogeography of Rana temporaria (Anura: Ranidae) in a putative secondary contact zone in the southern Alps. Biol. J. Linn. Soc. 2017;122(4):824–837. doi: 10.1093/biolinnean/blx114. [DOI] [Google Scholar]
- 42.Gasc JP, et al. Atlas of amphibians and reptiles in Europe. Societas Europaea Herpetologica & Museum National d’Histoire Naturelle; 1997. [Google Scholar]
- 43.Bartolini S, et al. Late Pleistocene fossils and the future distribution of Rana temporaria (Amphibia, Anura) along the Apennine Peninsula (Italy) Zool. Stud. 2014;53:76. doi: 10.1186/s40555-014-0076-5. [DOI] [Google Scholar]
- 44.Kyek M, Kaufmann PH, Linden R. Differing long term trends for two common amphibian species (Bufo bufo and Rana temporaria) in alpine landscapes of Salzburg. Austria. PLOS ONE. 2017;12(11):e0187148. doi: 10.1371/journal.pone.0187148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldberg CS, Strickler KM, Pilliod DS. Moving environmental DNA methods from concept to practice for monitoring aquatic macroorganisms. Biol. Conserv. 2015;183:1–3. doi: 10.1016/j.biocon.2014.11.040. [DOI] [Google Scholar]
- 46.Harper LR, et al. Prospects and challenges of environmental DNA (eDNA) monitoring in freshwater ponds. Hydrobiologia. 2019;826:25–41. doi: 10.1007/s10750-018-3750-5. [DOI] [Google Scholar]
- 47.Untergasser A, et al. Primer 3: New capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albanese D, Fontana P, De Filippo C, Cavalieri D, Donati C. MICCA: A complete and accurate software for taxonomic profiling of metagenomic data. Sci. Rep. 2015;5:9743. doi: 10.1038/srep09743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Antich A, Palacin C, Wangensteen OS, Turon X. To denoise or to cluster, that is not the question: Optimizing pipelines for COI metabarcoding and metaphylogeography. BMC Bioinform. 2021;22:177. doi: 10.1186/s12859-021-04115-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Librado P, Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 51.Bedwell ME, Goldberg CS. Spatial and temporal patterns of environmental DNA detection to inform sampling protocols in lentic and lotic systems. Ecol. Evol. 2020;10:1602–1612. doi: 10.1002/ece3.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldberg CS, Strickler KM, Fremier AK. Degradation and dispersion limit environmental DNA detection of rare amphibians in wetlands: Increasing efficacy of sampling designs. Sci. Total Environ. 2018;633:695–703. doi: 10.1016/j.scitotenv.2018.02.295. [DOI] [PubMed] [Google Scholar]
- 53.Caldonazzi, M., Pedrini, P., & Zanghellini, S. Atlante degli Anfibi e dei Rettili della Provincia di Trento (Amphibia- Reptilia). 1987- 1996 con aggiornamenti al 2001. Museo Tridentino di Scienze Naturali- Trento (2002).
- 54.Cohen O, Ram Y, Hadany L, Gafny S, Geffen E. Annual climatic fluctuations and short-term genetic variation in the eastern spadefoot toad. Sci. Rep. 2021;11(1):1–10. doi: 10.1038/s41598-021-92696-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequences available upon acceptance for publication of the manuscript or upon request. Correspondence and requests for materials should be addressed to H.C.H. (email: heidi.hauffe@fmach.it).