

Abstract
Glycogen synthase kinase 3 (GSK3) remains a therapeutic target of interest for diverse clinical indications. However, one hurdle in the development of small molecule GSK3 inhibitors has been safety concerns related to pan-inhibition of both GSK3 paralogs, leading to activation of the Wnt/β-catenin pathway and potential for aberrant cell proliferation. Development of GSK3α or GSK3β paralog-selective inhibitors that could offer an improved safety profile has been reported but further advancement has been hampered by the lack of structural information for GSK3α. Here we report for the first time the crystal structure for GSK3α, both in apo form and bound to a paralog-selective inhibitor. Taking advantage of this new structural information, we describe the design and in vitro testing of novel compounds with up to ∼37-fold selectivity for GSK3α over GSK3β with favorable drug-like properties. Furthermore, using chemoproteomics, we confirm that acute inhibition of GSK3α can lower tau phosphorylation at disease-relevant sites in vivo, with a high degree of selectivity over GSK3β and other kinases. Altogether, our studies advance prior efforts to develop GSK3 inhibitors by describing GSK3α structure and novel GSK3α inhibitors with improved selectivity, potency, and activity in disease-relevant systems.
Keywords: kinase inhibitor, GSK3α, GSK3β, Alzheimer’s, tau, β-catenin
Introduction
Originally identified as a protein kinase involved in the regulation of glycogen metabolism, glycogen synthase kinase-3 (GSK-3) is now known to be a multi-functional protein with key roles in diverse biological processes including cell proliferation, apoptosis, embryonic development, and insulin response.1−3 In addition, GSK3 is also of considerable interest as a therapeutic target because of its involvement in the core pathophysiologies underlying numerous conditions including Alzheimer’s disease (AD), Fragile X syndrome, diabetes, and several types of cancer.4−7 Therefore, the development of potent and selective GSK3 inhibitors has been of significant interest to the drug discovery field for over 3 decades.
As a therapeutic target, GSK3 has been of particular focus in AD because preclinical data suggest that modulation of this kinase ameliorates both hallmark pathological processes in AD: hyperphosphorylation of tau protein and production of amyloid-β peptides.8−10 Based on the therapeutic potential of targeting GSK3 in AD, several GSK3 inhibitors have previously advanced into clinical trials.11,12 However, these compounds have had limited clinical success due to lack of efficacy and/or significant safety concerns. Safety concerns with targeting GSK3 stem from a key role for this kinase in regulating the Wnt-β-catenin pathway.13 Previous studies have demonstrated that long-term inhibition of GSK3 can induce aberrant proliferation and hyperplasia in several tissues in vivo.14−17 Therefore, there is significant interest in the field to identify potential avenues to mitigate safety concerns while still effectively inhibiting the kinase.
One promising approach to improve the therapeutic index for GSK3 inhibition is the development of paralog-specific GSK3 inhibitors. GSK3 exists as two paralogs, GSK3α and GSK3β. These paralogs are encoded from separate genes and are thought to have arisen evolutionarily through gene duplication. Overall, the GSK3 paralogs are similar in sequence with 95% identity in the ATP binding site (67% amino acid identity overall) and exhibit a high degree of overlap in both tissue expression patterns and in their phosphorylation substrates.18−22 Interestingly, recent studies have also highlighted distinct functional differences for the GSK3 paralogs in several biological processes. Notably, loss of GSK3β causes embryonic lethality in mice whereas loss of GSK3α results in relatively modest defects.23,24 In the context of AD, most of the focus has traditionally been on GSK3β.25 However, in a landmark study using a mouse model that combines amyloid and tau pathologies, Hurtado et al. (2012) reported that knockdown of either GSKα or β ameliorated tau hyper-phosphorylation, yet only knockdown of GSK3α additionally reduced amyloid burden.26 More recently, selective loss or inhibition of GSK3α, but not β, has been shown to rescue deficits in a mouse model of Fragile X27 and suppress tumorigenesis in models of acute myeloid leukemia.28 Importantly, with respect to safety risks, studies have shown that selective reduction or ablation of either GSK3 paralog circumvents β-catenin stabilization29 and that selective genetic suppression of GSK3α impairs leukemia progression in mouse models of AML without increasing β-catenin levels.30 Together, these data suggest that paralog-selective inhibitors could exhibit an improved safety profile and particularly that the selective inhibition of GSK3α could be attractive therapeutically.
Challenges in the discovery of paralog-selective GSK3 inhibitors are predominantly due to the high degree of homology in the ATP binding site in which the primary difference is an aspartate (Asp) to glutamine (Glu) switch located in the hinge region.31 This difference is further complicated by the positioning of corresponding amino acid side chains located outside of the ATP binding site and therefore directed away from potential interactions with ATP competitive inhibitors. Previously, a series of oxadiazole inhibitors were identified that were able to achieve ∼3-fold selectivity for the GSK3α paralog32 and there have also been reports of imide-based33 and thioxoimidazolidine kinase inhibitors that exhibit paralog selectivity up to ∼7-fold.28 Additionally, a series of aminopyrazole inhibitors with up to 8-fold selectivity were reported by Wagner et al. (2018) and their development was based on the crystal structure of a GSK3β D133E mutant thought to mimic key differences between the two paralogs within the ATP binding site. This study also identified differential hydrogen bonding networks outside of the hinge Asp/Glu switch that could potentially be exploited to increase paralog selectivity and cellular potency.31
In the present study, we describe the development of several novel GSK3α-selective inhibitors and evaluate their activity across several in vitro and in vivo models of tau phosphorylation. Importantly, for the first time, we show the crystal structure for GSK3α both bound to a selective inhibitor and in apo form. Taking advantage of this structure, we were able to design compounds with up to ∼37-fold selectivity for GSK3α over β with favorable drug-like properties. Using a rat model of tau phosphorylation and chemoproteomics, we further confirm that acute GSK3α inhibition can lower tau phosphorylation at disease-relevant sites in vivo with a high degree of selectivity over GSK3β and other kinases. Altogether, our studies advance efforts to develop GSK3 paralog selective inhibitors by describing novel GSK3α inhibitors with improved selectivity and potency.
Results
Acute and Selective Inhibition of GSK3α can Reduce Tau Phosphorylation at Disease Relevant Sites
The development of GSK paralog selective compounds is important for the field to investigate the relative contributions of GSK3α or GSK3β to different disease pathologies. To demonstrate the specific effects of each paralog on tau phosphorylation, HEK293 cells stably expressing human 2N4R tau (HEK-huTau) were transfected with plasmids expressing human GSK3α, human GSK3β, or kinase dead mutants in which the catalytic lysine was mutated to an alanine. 24 h after transfection, cells were lysed and run by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE) to probe for phosphorylation of tau at different epitopes. Our results demonstrate that both GSK3 isoforms phosphorylate tau at multiple epitopes including the disease enriched epitopes Thr231 and S202/Thr205 (AT8; Figure 1A). Interestingly, when these kinases were expressed at approximately equal levels, we did not detect selectivity of the GSK3 paralogs for specific phosphorylation epitopes on tau or a difference in total phosphorylation of tau (Figures 1A and S1). These data indicate that in our current HEK-huTau cell assay, both GSK3α and GSK3β have comparable substrate affinity for several disease-enriched phospho-epitopes on tau.
Figure 1.
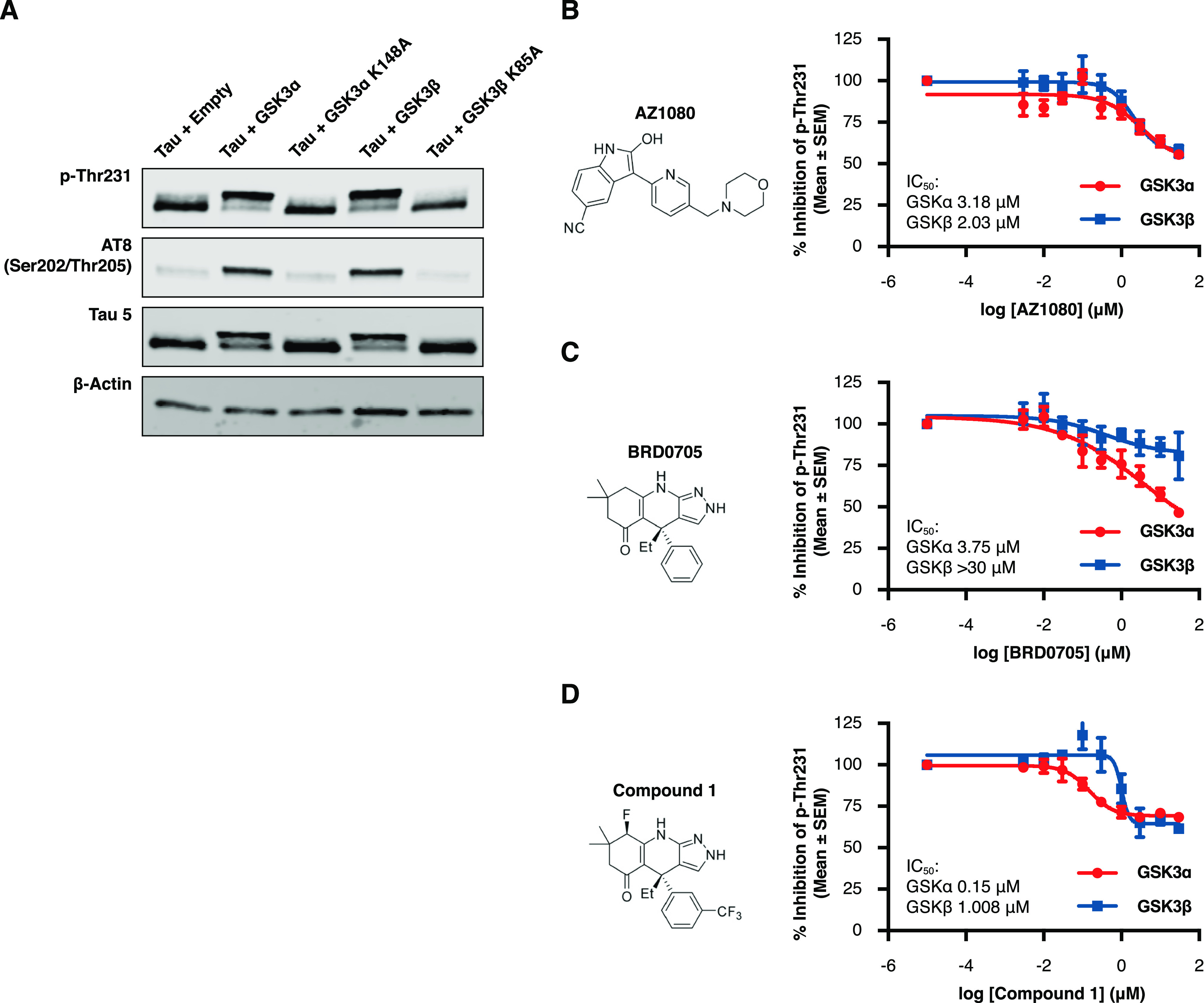
Acute and selective inhibition of GSK3α can reduce tau phosphorylation at disease relevant phospho-sites. HEK293 cells stably expressing human 2N4R tau (HEK-huTau) were transfected with plasmids expressing either human GSK3α, human GSK3β, or kinase dead mutants in which the catalytic lysine was mutated to alanine. 24 h after transfection, cells were lysed. (A) HEK-huTau lysates were run by SDS PAGE to probe for phosphorylation of tau at different epitopes. Both GSK3 isoforms phosphorylate tau at multiple epitopes including the disease enriched epitopes Thr231 and S202/Thr205 (AT8). (B–D) 24 h after transfection, compounds were added at a 10-point dose response (range: 3 nM–30 μM) for 2 h and cell lysates were probed for total tau (Tau5 antibody) and pThr231 using a plate-based assay. The figures represent IC50 curves demonstrating cellular potency and GSK3 isoform selectivity of the non-paralog-selective compound AZ1080 (B), a previously reported GSK3α selective compound BRD0705 (C), and a novel GSK3a selective small molecule, compound 1 (D). Each data point represents the mean ± the standard error of the mean (SEM) from four biological replicates within single run.
To test the potency and isoform selectivity of GSK3 small molecule inhibitors, we transfected HEK-huTau cells with plasmids containing either human GSK3α or GSK3β. 24 h after transfection, compounds were added at a 10-point dose response curve (at a range of 30 μM–3 nM) for a total of 2 h, and cell lysates were probed for total tau and pThr231 using a plate-based assay. Our results demonstrate that this assay is sensitive to GSK3 activity and that the capture antibody is specific to pThr231 as mutating this threonine residue to an alanine abolished assay signal (Figure S2). As a benchmark inhibitor, we used the AstraZeneca compound AZ1080 which was chosen because it is commercially available and has previously shown GSK3 target engagement in clinical trials.11 Our results show similar potencies for AZ1080 to both GSK3 paralogs (Figure 1B; GSK3α IC50 = 3.18 μM; GSK3β IC50 = 2.03 μM), and these results verify previous reports that this compound is not paralog selective. In addition, we also profiled the reported GSK3α selective compound BRD070531 and confirmed selectivity of this compound for GSK3α (Figure 1C; GSK3α IC50 = 3.75 μM; GSK3β IC50 > 30 μM). However, it must be noted that for BRD0705, we were unable to measure the potency of inhibition for GSK3β accurately due to toxicity of this compound at doses >30 μM.
In an effort to further optimize GSK3α selective inhibitors, we performed structural analysis of the previously reported GSK3β D133E mutant31 and screened functional group substitutions at both the phenyl and cyclohexyl ring of BRD0705. Here, we report the characterization of a novel GSK3α selective compound (compound 1) which retains GSK3α paralog selectivity (Figure 1D; GSK3α IC50 = 0.15 μM; GSK3β IC50 = 1 μM) while dramatically increasing cellular potency (GSK3α; BRD0705 IC50 = 3 μM vs compound 1 IC50 = 0.15 μM). Importantly, the relative cellular potencies were similar when these compounds were tested for activity at an additional tau phosphorylation site pS202/pThr205 (AT8; Figure S3), further validating our results and demonstrating the utility of these cell-based assays for further compound screening.
Reported GSK3 Small Molecule Inhibitors Bind at the ATP Binding Pocket and Have Similar Residence Times
To further characterize the novel GSK3 inhibitor compound 1, we also tested this compound in a live-cell target engagement assay to investigate binding mode and residence time. To assess binding at the ATP pocket, HEK293T cells were transiently transfected with NanoLuc-GSK3α or NanoLuc-GSK3β plasmids and the following day treated for 2 h with both inhibitors and the NanoBRET Target Engagement Kinase Tracer-8 (Promega; EC50 concentration GSK3α = 87 nM; GSK3β = 120 nM; Figure S4). In this assay, compound binding was quantified as loss of the BRET signal from baseline due to competitive displacement of the tracer. Our results show competitive displacement of the tracer with each of the inhibitors indicating that all compounds tested bind in the ATP pocket (AZ1080 Figure 2A; BRD0705 Figure 2B; compound 1 Figure 2C). Importantly, the rank order for compound potency and isoform selectivity was similar between the NanoBret assay and HEK293 tau phosphorylation assay providing additional confirmation of our previous results.
Figure 2.
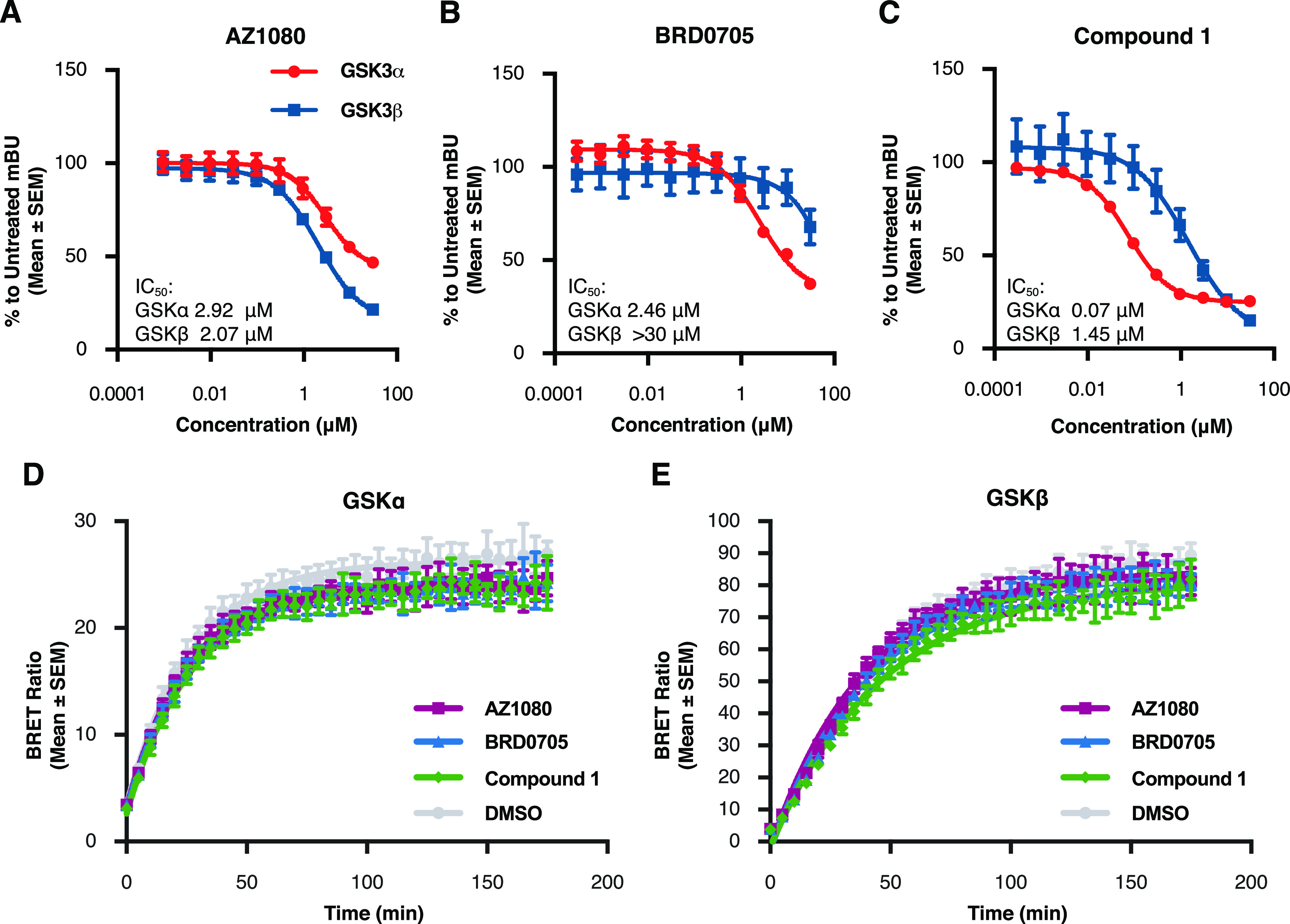
GSK3 small molecule inhibitors bind at the ATP binding pocket and have similar residence times. To test for binding at the ATP pocket, HEK293T cells were transiently transfected with either NanoLuc-GSK3α or NanoLuc-GSK3β plasmids and the following day treated for 2 h with the inhibitor compounds in a 10-point dose response curve (range: 3 nM–30 μM) and the NanoBRET Target Engagement Kinase Tracer-8. (A–C) Our results show competitive binding of inhibitors AZ1080 (A), BRD0705 (B), and compound 1 (C) with the kinase tracer indicating displacement at the kinase ATP pocket. Each data point represents the mean ± SEM from four biological replicates within a single run of the assay. mBU: milliBRET Units (please see Methods). To investigate compound residence time, cells were transiently transfected with NanoLuc plasmids and the following day kinase inhibitors were added at a concentration of 10× the previously identified IC50. After 2 h of treatment, media with compound was removed and Tracer-6908 (Promega) was added. The BRET signal was measured every 5 min for a total of 3 h after tracer addition. (D,E) There was no difference in the residence times between any of the inhibitors and either GSK3α (D) or GSK3β (E). Each data point represents the mean ± SEM from eight biological replicates within a single run of the assay.
To investigate compound residence time, cells were transfected with GSK3 NanoLuc plasmids and treated with each inhibitor at 10× the previously identified IC50. Following the addition of tracer, BRET signal was measured every 5 min for a total of 3 h. Overall, there was no difference in the residence time of the novel compound 1 at either GSK3α or GSK3β when compared to the previously profiled compounds AZ1080 or BRD0705 (GSK3α Figure 2D; GSK3β Figure 2E; Table 1). Together, these data demonstrate that the increase in potency observed with compound 1 was not due to differences in compound binding mode or on/off kinetics.
Table 1. Residence Time of Small Molecule Compound 1, AZ1080, and BRD0705 for GSK3α and GSK3βa.
| Kobserved(normalized to DMSO) | ||
|---|---|---|
| compound | GSK3α | GSK3β |
| AZ1080 | 1.08 | 1.13 |
| BRD0705 | 1.05 | 1.04 |
| compound 1 | 1 | 0.91 |
There was no difference in the residence time of the novel compound 1 at either GSK3α or GSK3β when compared to the previously profiled compounds AZ1080 or BRD0705.
Combined Inhibition of Both GSK3 Paralogs is Necessary for Nuclear Translocation of β-Catenin in SHSY-5Y Cells
One concern in the clinical development of GSK3 kinase inhibitors is the predicted stabilization and nuclear translocation of β-catenin, a substrate whose mutation or overexpression is associated with aberrant cell proliferation and cancer.35,36 Importantly, previous experiments have shown that selective genetic knockdown of either GSK3α or GSK3β does not increase β-catenin levels.29 To determine whether our novel GSK3α inhibitor would show a similar profile, we measured β-catenin activation by looking at its nuclear translocation following compound addition. As expected, the non-selective compound AZ1080 lead to nuclear translation of β-catenin at a potency similar to what we previously reported in the tau phosphorylation assay (Figure 3A). Notably, our results show that compounds which had selectivity toward GSK3α (BRD0705 and compound 1) had a significant window between their reported potencies for GSK3α mediated tau phosphorylation compared to those which resulted in β-catenin stabilization (BRD0705 Figure 3B; compound 1 Figure 3C; Table 2). For example, the novel compound 1 had an IC50 for GSK3α of 0.15 μM and an EC50 in the β-catenin assay of 3.02 μM. These data suggest a window of approximately 20-fold between GSK3α inhibition and potential safety concerns due to β-catenin stabilization (Table 2). Our data confirm that inhibition of the GSK3α paralog alone was not sufficient to cause β-catenin activation because across all three compounds, we only see translocation to the nucleus with compound concentrations high enough to inhibit both paralogs. Altogether, these results demonstrate the identification of a novel GSK3α inhibitor with improved cellular potency and window between inhibition of tau phosphorylation and β-catenin activation.
Figure 3.

Combined inhibition of both GSK3 paralogs is necessary for nuclear translocation of β-catenin in SH-SY5Y cells. To examine β-catenin activation, we quantified translocation of this protein to the nucleus in SH-SY5Y cells 6 h after compound addition at a 9-point dose response curve (range: 20 nM–20 μM). (A–C) EC50 curves demonstrating concentration of GSK3 inhibitors AZ1080 (A), BRD0705 (B) and compound 1 (C) necessary to trigger β-catenin translocation to the nucleus. Each data point represents the mean ± SEM from four biological replicates within a single run of the assay. (D) Representative images showing DAPI nuclear stain in blue and β-catenin in green for each GSK3 inhibitor compound at the highest dose tested (20 μM).
Table 2. Window between Inhibition of GSK3α Mediated Tau Phosphorylation at Thr213 and β-Catenin Translocation for AZ1080, BRD0705, and Compound 1.
| compound | GSK3α p-Thr231 IC50 | β-catenin nuclear translocation EC50 | potency ratio |
|---|---|---|---|
| AZ1080 | 3.18 μM | 3.27 μM | 1.03× |
| BRD0705 | 3.75 μM | >20 μM | >5× |
| compound 1 | 0.15 μM | 3.02 μM | 20× |
Acute GSK3α Inhibition Reduces Tau Phosphorylation at Disease Relevant Sites In Vivo
To determine whether a selective GSK3α inhibitor could also lower tau phosphorylation in a more physiological setting, we administered compound 1 to rats at post-natal day 10. Previous experiments testing tau kinase inhibitors have demonstrated several advantages of using neonatal rats for in vivo PK/PD studies including the observation that these animals have a high level of basal tau phosphorylation37 and that compounds with less-than-ideal physical characteristics are able to cross the blood brain barrier due to the absence of Pgp transporters at this age.38 In the current experiment, we administered compound 1 at 60, 20, and 6 mg/kg in P10 rats and sacrificed the animals at several time points. Our results demonstrate a significant lowering of tau phosphorylation at pThr231 beginning 3 h after drug administration when compared to vehicle treated controls (Figure 4A; 60 mpk p = 0.004; 20 mpk p = 0.0227). Interestingly, over a 24 h period, tau phosphorylation at pThr231 increased by 58% in the vehicle treated animals when compared to the previous baseline level (normalized tau pThr231 at 1 and 24 h in vehicle treated animals was 1 and 1.58 respectively). Animals injected with compound 1 at either 20 or 60 mg/kg did not see a similar increase in tau phosphorylation over this time and were significantly different from the vehicle group at the 24 h timepoint (60 mpk p = 0.005; 20 mpk p = 0.0139).
Figure 4.
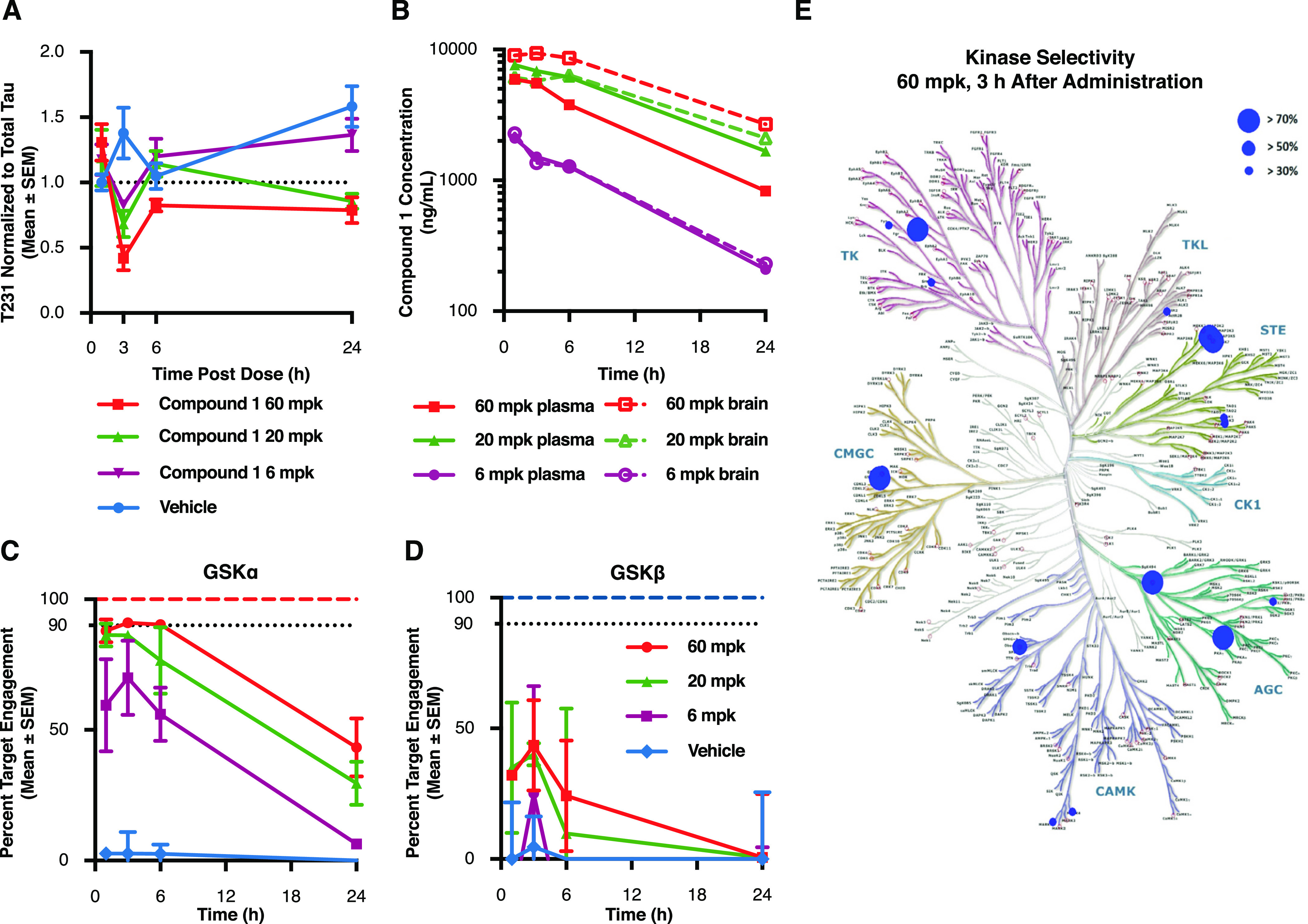
Acute GSK3α inhibition reduces tau phosphorylation at disease relevant sites in vivo. P10 rats were injected with compound 1 at 60, 20, and 6 mg/kg and sacrificed at several time points following treatment. (A) Cortical lysates were run in a plate-based assay to quantify levels of phosphorylated Thr231 normalized to total tau. Results demonstrate a significant lowering of T231 phosphorylation beginning 3 h after drug administration when compared to vehicle treated controls (60 mpk p = 0.004; 20 mpk p = 0.0227). Each data point represents the mean ± SEM from three animals. (B) To determine drug exposure, blood and cortical tissue were analyzed at each time point following dosing. Pharmacokinetic data demonstrate dose responsive compound concentrations with a steady Cmax extending to ∼6 h post injection at the highest dose and comparable exposure between blood and brain at each of the doses tested. (C,D) Target engagement at the GSK3 isoforms was measured using competitive chemoproteomics with Sepharose “kinobeads”. Results demonstrate that compound selectivity for the GSK3α paralog was retained in vivo. Each data point represents the mean ± SEM from three animals. (E) Competitive chemoproteomics was also used to monitor other kinases inhibited by compound 1. Our results show that at the highest dose tested (60 mg/kg), there were only 7 out of 251 unique kinases detected (including GSK3α) that had a target engagement of >70%.
To determine drug exposure, blood and cortical tissue were analyzed at each time point following dosing. Overall, our pharmacokinetic data demonstrate dose responsive compound concentrations with a steady Cmax extending 6 h post injection (Figure 4B). In addition, we noted similar levels of the compound in both the blood and brain (Figure 4B) indicating that in neonatal rats, the compound had no issue crossing the blood brain barrier after peripheral injection. Free drug concentration in the brain was estimated based on plasma protein binding data (Table S1) and demonstrated a Cmax of 715 nM for the 60 mpk dose, 3 h after injection (Figure S5). Together, this data shows a correlation between drug exposure and effects on tau phosphorylation levels.
To measure direct target engagement of the compound for both GSK3α and GSK3β, we used competitive chemoproteomics. Briefly, brain lysates were incubated with Sepharose beads coupled to non-selective kinase inhibitors (kinobeads39). Enriched proteins were analyzed by liquid chromatography coupled with tandem mass spectrometry. Target engagement was measured as competitive displacement of compound 1 by the non-selective beads. Our results demonstrate that compound 1 retained its selectivity for GSK3α over GSK3β in the rat brain (GSK3α Figure 4C; GSK3β Figure 4D). In particular, 3 h after dosing, a timepoint for which we previously demonstrated a significant decrease in tau phosphorylation at Thr231, compound 1 achieved over 90% target engagement for GSK3α (Figure 4C) compared to 40% for GSK3β (Figure 4D). At 24 h, target engagement of compound 1 at GSK3α was still close to 50% at both the 60 and 20 mg/kg dose while there was no significant binding detected at this time point to the GSK3β paralog. This data suggests that the extended effect of this compound preventing tau phosphorylation at Thr231 was likely driven by GSK3α inhibition.
To investigate whether the effects of compound 1 were solely the result of GSK3 inhibition, we also profiled all additional kinases detected by mass spectrometry for compound engagement in the same brain lysates. Our results show that at the highest dose tested (60 mg/kg), there were only 7 out of 251 unique kinases (including GSK3α) that exhibited target engagement greater than 70% at Cmax (Figures 4E and S6). Altogether, these results represent the first demonstration that acute GSK3α inhibition can lead to significant reductions in tau phosphorylation in vivo.
Elucidation of GSK3α Crystal Structure
The development of GSK3 paralog-selective inhibitors has been hindered by the absence of crystal structures for GSK3α, unlike GSK3β, for which structural information has previously been described.40 Wagner et al. (2018) reported challenges associated with directly obtaining a crystal structure for GSK3α and instead utilized a GSK3β mutant (D133E) to mimic key differences between the two paralogs within the ATP binding site. From this structure, it was speculated that a differential hydrogen-bonding outside of the ATP binding site may lead to alternative pockets allowing for paralog selective inhibitors. To confirm this hypothesis, we first sought to obtain a co-crystal structure of BRD0705 with GSK3α. A previous report on the crystallization of GSK3β41 described increased diffraction in the presence of its physiological binding partner axin. Therefore, we hypothesized that this physiologically relevant binding event may also assist in crystallization of the α-paralog. Our results confirmed this hypothesis, and we were able to generate the first crystal structure of GSK3α with an inhibitor (BRD0705) bound to the ATP active site (Figure 5A). We found that the crystallization of both GSK3-α and GSK3-β with axin and BRD0705 led to expected differences in the hydrogen bonding network surrounding Glu196/Asp133 for each paralog (Figure 5B). In particular, Glu196 has an interaction with Lys260 in GSK3α, whereas the corresponding interaction is not observed for GSK3β. Additionally, the two crystal structures contain subtle differences in the sidechain rotamers located within the ATP binding site surrounding BRD0705 which are represented in Figure 5B,C.
Figure 5.

Elucidation of novel GSK3α crystal structures; (A) crystal structure of GSK3α with the small molecule inhibitor BRD0705. The compound interacts with the hinge through three hydrogen bonds (indicated with red dashed lines). (B) Overlay of BRD0705 bound with GSK3α and GSK3β shows similar hinge interactions; however, there are sidechain rotamer differences that lead to subtle differences in the binding pocket in Figure 5B vs Figure 5A. (C) Asp vs Glu difference between GSK3α and GSK3β leads to differences in hydrogen bonding interactions behind the binding pocket. (D) Addition of a trifluoromethyl group to the phenyl ring may lead to increased affinity of these compounds for either GSK3 paralog by enhancing hydrogen bonding interactions between the Fluoro group and Lys 85/148.
Since the primary difference in the two GSK3 active sites is situated at the hinge, for the development of GSK3α selective inhibitors, we first examined substitutions adjacent to the aminopyrazole hinge-binding motif. From the crystal structure of BRD0705, we also noted that the binding pocket would likely tolerate substitution of the 3-position of the aryl ring. Indeed, as previously shown with our novel compound 1, fluorine substitution adjacent to the hinge and an aryl trifluoromethyl substitution led to improved potency while maintaining GSK3α paralog selectivity. Next, using this crystal structure, we sought to further increase both the potency and selectivity of compound 1. First, we wanted to examine whether differential substitution of the phenyl ring could provide improved paralog selectivity despite its position within the binding pocket. We found that the trifluoromethyl group found in compound 1 could be replaced with pyrazole or pyridyl functional groups to direct a hydrogen bond acceptor toward the catalytic lysine residue (Figure 5D). This additional hydrogen bond interaction may lead to increased affinity of these compounds for either GSK3 paralog, and our hypothesis was that additional hydrogen bonds formed from these two heterocycles could lead to subtle differences in the trajectory of the biaryl motif and reinforce GSK3α paralog selectivity.
Structure Based Design Using the GSK3α Structure Leads to Novel, Potent Inhibitors
To determine whether our structure-based designs led to improvements in potency and/or selectivity, we tested the two novel compounds, compound 2 and compound 3 in the previously described series of assays. In our huTau-HEK assay which measures tau phosphorylation at pThr231, each of these compounds demonstrated a significant improvement in both potency and GSK3α selectivity when compared to the parent compound 1 (Figure 6A compound 2, 37-fold selective; Figure 6B compound 3, 11-fold selective). To measure more proximal target engagement, we also verified that compound 2 and compound 3 were ATP-competitive and direct GSK3 binders using the nanobret tracer assay (Figure 6C compound 2; Figure 6D compound 3). Interestingly, when looking at live-cell binding kinetics, both of these compounds demonstrated significantly longer residence times for GSK3α in comparison to the previously characterized GSK3 inhibitors (Figure 6E). This significant change in dissociation speed was only seen for the GSK3α paralog (Figure 6F and Table 3) suggesting that by increasing ligand–protein interactions, we created a tighter interaction in the ATP pocket subsequently leading to slower dissociation. In addition, we also profiled the three novel compounds identified here, along with the parent compound BRD0705, in the Eurofins Kinomescan panel to determine if the increase in GSK3 potency was also able to maintain selectivity across the kinome. Our results show that these compounds are very selective for the GSK3 paralogs across a total of 468 kinase targets tested (Table S4). For example, the only kinases demonstrating >65% competitive binding with compound 2 at 1 μM were GSK3α and GSK3β. For compound 3, 10 out of 468 kinases showed >65% competitive binding at 1 μM. Finally, to verify that selective GSK3α inhibition with these compounds does not lead to β-catenin stabilization, we measured the potency of these compounds in causing translocation of β-catenin to the nucleus in SY5Y cells. For both of these compounds, nuclear translocation of β-catenin occurred only at concentrations in which both GSK3 paralogs were being inhibited (Figure S7). Together, these data represent the characterization of two novel, GSK3α selective compounds with significant improvements in cellular potency compared to those previously reported in the literature.
Figure 6.
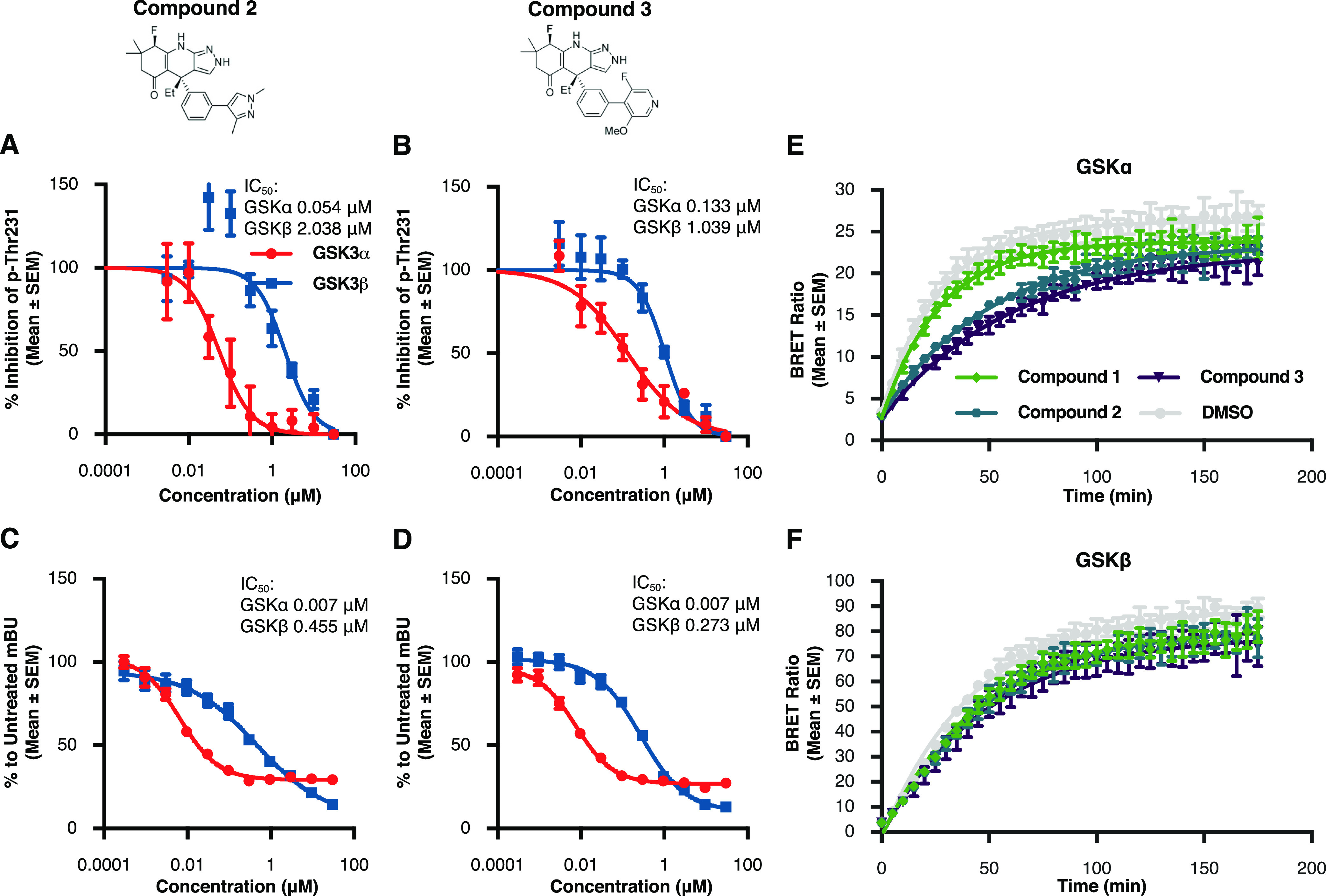
Structure based design leads to novel, potent GSK3α inhibitors (A,B) results from p-Thr231 cell-based assay illustrate improvements in potency of both compound 2 and compound 3 when compared to previously profiled inhibitors. Each data point represents the mean ± SEM from four biological replicates within a single run of the assay. (C,D) Nanobret assay was used to further verify the potency and selectivity of the novel inhibitors compound 2 and compound 3. Each data point represents the mean ± SEM from four biological replicates within a single run of the assay. (E,F) Both compound 2 and compound 3 show significantly longer residence times for GSK3α in comparison to the parent molecule compound 1. These compounds did not differ in their residence times for GSK3β. Each data point represents the mean ± SEM from eight biological replicates within single run of the assay.
Table 3. Residence Time of Small Molecules Compound 1, Compound 2, and Compound 3 for GSK3α and GSK3βa.
| Kobserved(normalized to DMSO) | ||
|---|---|---|
| compound | GSK3α | GSK3β |
| compound 1 | 1 | 0.91 |
| compound 2 | 0.54 | 0.91 |
| compound 3 | 0.46 | 0.87 |
The novel compound 2 and compound 3 demonstrate longer residence times specially at the GSK3α paralog when compared to previously profiled compounds.
Crystal Structure of GSK3α and GSK3β with Novel Compound 3
The in vitro characterization of compound 3 demonstrates a dramatic increase in compound potency and improvements in GSK3α paralog selectivity. To investigate whether this was due to increased interactions of this compound with the GSK3α ATP pocket, we performed crystallization of GSK3α with axin and compound 3. Our results show that compound 3 interacts with Lys148/85 in both GSK3 paralogs, which could account for some of the increased binding affinity (Figure 7A). In addition, the crystal structure also demonstrates minor differences in the orientation of the biaryl motif via the lysine interaction between GSK3α and GSK3β. The orientation of this biaryl motif relative to Lys148/85, and subsequent difference in hydrogen bonding enthalpy, may contribute to the observed paralog selectivity but more importantly highlights the binding differences of compound 3 relative to BRD0705 and potential secondary effects on affinity. Interestingly, both the GSK3α and GSK3β structures with compound 3 show that the central phenyl ring is rotated relative to BRD0705 and that this rotation is required for the biaryl ring to interact with Lys148/85. Additionally, the gem-dimethyl substituents on the solvent exposed ring adjacent to the hinge have undergone a pseudo chair-flip between the BRD0705 and compound 3 structures and the axial methyl group is directed to the N-lobe rather than the C-lobe. Taken together, these different conformational biases captured in the X-ray structures of compound 3 may further contribute to selectivity by reinforcing the interaction of the ethyl moiety with the corresponding lipophilic pocket which was originally identified for BRD0705. Importantly, the structures of compound 3 indicate that the ILE125(/62) residue in this lipophilic pocket is shifted relative to the compound 3 GSK3β structure as well as both GSK3 structures of BRD0705 and provides an expanded binding site (Figure 7A,B). Interestingly, the binding modes of compound 3 do not provide any obvious indications that the alkyl fluorine substitution differs sterically between the α and β structures and could possibly impart additional selectivity by altering the binding affinity of the aminopyrazole hinge binder through inductive electronic effects. Overall, the crystal structure of compound 3 in both GSK3α and GSK3β highlights how subtle differences in binding are able to impart increases in potency and paralog selectivity.
Figure 7.

Crystal structure of GSK3α and GSK3β with the novel small molecule inhibitor compound 3 (A) overlay of compound 3 bound to GSK3α (red), and to GSK3β (slate). (B) GSK3α/β pocket surface overlay shows slight differences due to the rotamers of ILE125/62.
Discussion
In the present study, we describe the development of several novel GSK3α-selective inhibitors and evaluate their activity in both cellular and animal models of tau phosphorylation. The development of these compounds was aided by the first elucidation of a GSK3α crystal structure, and our results describe how increasing ligand interactions with the GSK3α binding pocket could impart improvements in compound potency and target residence time. Using a postnatal rat model and chemoproteomics, we confirm that acute inhibition of GSK3α can suppress tau phosphorylation at epitopes enriched in disease and further demonstrate the selectivity of our inhibitors across the kinome. These results advance prior efforts to develop GSK3 paralog selective inhibitors and demonstrate therapeutically relevant activity in vitro and in vivo.
The hyper-phosphorylation of tau has been shown to negatively affect a wide range of cellular processes including microtubule polymerization,42 axonal transport,43 RNA translational selectivity,44 nuclear import/export,45 pre-synaptic vesicle motility46 and post-synaptic excitotoxicity.47 However, the longest human tau isoform contains a total of 85 possible phosphorylation sites48 making the identification of specific phosphorylation sites as drivers of disease difficult. GSK3 has been shown to phosphorylate tau at 42 different epitopes,49 and the activity of GSK3 has been shown to correlate with the level of neurofibrillary tangles found in AD brains.50 Here we demonstrate in a HEK293 cell system that both GSK3 paralogs phosphorylate tau at several epitopes (T181, S262, S396, Thr231, and AT8) with equal affinity and strength. To test whether acute GSK3α inhibition could also modulate tau phosphorylation in the rat brain, we tested a novel paralog selective inhibitor compound 1 at doses up to 60 mg/kg, which at Cmax engaged GSK3α over 90% while only occupying GSK3β at 40%. Our results demonstrate a reduction in tau phosphorylation at Thr231 at all doses tested 3 h after compound administration. We choose to specifically examine phosphorylation at Thr231 because several lines of evidence indicate that phosphorylation at this epitope is an early event in Alzheimer’s disease progression.51,52 In addition, phosphorylated tau at Thr231 is readily detectable in CSF and was found to be significantly higher in patients with dementia due to AD.53 Together, this data suggest the possibility that Thr231 phosphorylation could serve as a distal biomarker of GSK3 target engagement in the clinical development of similar compounds.
Chemoproteomics-based profiling allows for the simultaneous measurement of both target engagement and selectivity of kinase inhibitors.34 In the current study, following administration of compound 1 we used kinobeads to enrich for endogenous kinases and then measured the differential enrichment of kinases over time at multiple doses using quantitative mass spectrometry. This approach allowed us to determine a direct correlation between free drug levels of our small molecule and target engagement at either the GSK3α or GSK3β active site. These data demonstrate that even at drug Cmax (3 h after injection of 60 mg/kg) we have selectivity of our compound to the GSK3α paralog. In addition, in the current experiment the kinobeads were able to label around 251 unique kinases in the rat brain at post-natal day 10. Of the 251 unique kinases identified, compound 1 at Cmax demonstrated greater than 70% target engagement at only 6 kinases apart from GSK3α/GSK3β. Of the off-target kinases identified, cAMP-dependent Protein Kinase catalytic subunit β (Prkcab) was particularly interesting because previous studies have shown the ability of PKA to phosphorylate tau at Thr23154 and an additional epitope S214, phosphorylation of which is upregulated in the aging primate cortex.55 Therefore, dual inhibition of GSK3α and PKA subunits could constitute an attractive therapeutic strategy. Together, the current chemoproteomics results demonstrate the selectivity of compound 1 between GSK3 paralogs as well as across the kinome, while at the same time establishing an expected range of target engagement in the tissue of interest.
Previous attempts to optimize GSK3 paralog selective compounds have been impeded by the lack of structural information for GSK3α. In this study, we report the first crystal structures for GSK3α and use structural information to design and characterize two novel GSK3α selective inhibitors (compound 2 and compound 3). Our results demonstrate that these compounds have significant improvements in cellular potency compared to previously described GSK3α inhibitors28,31,33 and significantly slower off rates when compared to their parent compound (normalized Koff of parent compound 1 = 1 vs compound 2 = 0.54; compound 3 = 0.46). These data suggest that designing compounds with increased binding interactions within the GSK3α ATP pocket resulted in a tighter interaction with the target and therefore slower dissociation. Further experiments will be necessary to see if this increase in target residence time translates to improved potency in vivo where a slow off-rate may lead to sustained target inhibition. Clinically, this could enable less frequent dosing to maintain efficacious target engagement.
The identification of new GSK3α selective compounds demonstrates that selective inhibition of GSK3 paralogs can be achieved despite high binding site homology. Originally it was thought that selective inhibition would necessitate interaction with the one residue difference between GSK3α and GSK3β in the hinge region (Asp/Glu) due to all additional paralog specific side chains directed outside of the ATP pocket and likely inaccessible to small molecules. Here, we show that modification of the previously identified GSK3α inhibitor BRD0705 on the cyclohexyl ring and aryl substitution led to compound 1, which exhibited a significant improvement in cellular potency, validated in multiple assay systems as well as through determination of binding kinetics. This improvement was most likely due to substitutions adjacent and distal to the hinge binding region. These results confirmed that despite highly homologous primary sequences for the active sites of the GSK3 paralogs, the projection and interactions of these residues occurs in a differential manner and provides avenues to obtain selective binding with small molecule inhibitors. We then used this information to further optimize the aryl functionality and found that the biaryl moieties in compound 2 and compound 3 resulted in further improved in vitro potency and selectivity. Importantly, we found that compound 3 interacted with Lys148/85 after obtaining a crystal structure of this inhibitor with both GSK3 paralogs. The observation that interaction of the inhibitors with a highly conserved residue led to an increase in GSK3 isoform selectivity highlights that further increases in selectivity may be possible through careful inhibitor design.
Previous attempts to use GSK3 inhibitors in the clinic have been accompanied by concerns about toxicity, driven in part by the simultaneous inhibition of both GSK3 paralogs resulting in β-catenin stabilization and potential for aberrant cell proliferation. Here, we investigated GSK3α selective inhibitors in the context of tau phosphorylation; however, GSK3 paralog-selective inhibitors have broader development potential across a multitude of disease targets and indications. In the current experiments, we exploited differences in the crystal structures of GSK paralogs to drive potency and selectivity to GSK3α; however, these data could be also used to gain GSK3β selectivity if preferred. Paralog-selective biology has been implicated in various disorders. For example, aberrant GSK3β activity has been implicated in Type 2 Diabetes56 and in the survival and invasion of tumor cells in several types of cancer.57 Recently, GSK3α has been implicated in the pathology of Fragile X syndrome. Two studies demonstrated that GSK3α, but not GSK3β, is overactive in the Fmr1–/y mouse hippocampus27,58 and McCamphill et al., demonstrated that the GSK3α selective inhibitor BRD0705 corrected susceptibility to audiogenic seizures and reversed learning and memory deficits in a Fragile X mouse model.27 In the current study, we demonstrate improvements in potency and α selectivity of novel compounds when compared to BRD0705. These advancements are likely to increase the therapeutic window with which to test efficacy of these compounds in different disease models.
Methods
Cell Culture and Transfection
HEK293 or SH-SY5Y cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 units/mL penicillin, and 10 μg/mL streptomycin (all reagents were purchased from Gibco, Waltham, MA). Cell cultures were maintained in a humidified 5% (v/v) CO2/air environment at 37 °C. When the cells reached 50–80% confluence, they were transfected with a complex consisting of a 3:1 ratio of FuGENE 6 Transfection Reagent (Promega, Madison, WI) and plasmid DNA. Plasmid DNA was balanced out across reactions using an empty plasmid so that the same amount of DNA was added per well. This transfection complex was prepared following the manufacturer’s protocol and added directly to the cell media for 24 or 48 h prior to lysis.
SDS–PAGE and Western Blot
Samples were lysed in Pierce RIPA lysis and extraction buffer (Thermo Fisher Scientific, Waltham, MA) supplemented with 1% Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA) and centrifuged at 14,000g for 20 min at 4 °C to clear the lysate. Protein concentrations were determined using the Direct Detect Infrared Spectrometer (EMD Millipore Corp., Burlington, MA), and 10 μg of each sample was denatured in 6× SDS sample buffer (Boston Bioproducts, Ashland, MA) for 7 min at 90 °C. Proteins were loaded into a criterion 4–15% tris-glycine gel (Bio-Rad, Hercules, CA) and separated by SDS–PAGE at 120 V for 135 min. The gel was transferred to an IBlot2 nitrocellulose membrane (Invitrogen, Carlsbad, CA), blocked with TBST blocking buffer (Li-cor Biosciences, Lincoln, NE) for 1 h, and washed three times with TBST. The membrane was probed with primary antibodies (1:1000 dilution) in antibody dilution buffer (1:1 TBST blocking buffer and 1× TBST) overnight at 4 °C. The blot was then washed in triplicate with TBST and incubated for 1 h with secondary antibody (1:10,000 dilution of IRDye 800 anti-mouse IgG and IRDye 680 anti-rabbit IgG, Li-cor Biosciences, Lincoln, NE) in antibody dilution buffer. After a final triplicate wash with TBST, the blot was visualized using the Odyssey CLx imaging system (Li-cor Biosciences, Lincoln, NE).
Phospho-Tau: Plate-Based Assays for Epitopes Thr231 and AT8
293T cells stably expressing human 2N4R Tau were transfected with appropriate cDNA plasmids. 24 h following transfection, cells were treated with compound in a 10-point dose-response (from 1000× stocks of compound) and lysates were harvested 2 h later with cell extraction buffer (Life Technologies Cat no. BN0001) or RIPA buffer with Halt protease/phosphatase inhibitors added.
For pThr231 assay: lysate was diluted 1:100 in assay buffer and total Tau, and pTau T231 was assessed by plate-based ELISA as per manufacturer’s instructions (MSD Cat no. K15121D-1). Total fluorescence was measured using an MSD plate reader.
For AT8 assay: FRET antibodies (custom designed Cisbio assay kit; S202/T205 Tb and total Tau D2 FRET antibodies) were added to lysate in a 384 well assay plate at 1:4 ratio (antibody/lysate). After antibody addition, the plate was left at room temperature for 60 min incubation time. Plate was read with appropriate wavelength settings using an Envision plate reader (ex320 and em620 and em665).
NanoBRET
HEK293T cells were transiently transfected with NanoLuc-GSK3-α or NanoLuc-GSK3-β constructs using FuGene-HD (Promega). Next day, the live cell NanoBRET target engagement assay was performed. Cells were treated with serially diluted compound in addition to NanoBRET In-cell Kinase Tracer-8 at EC50 concentration (87 nM for GSK3-α and at 120 nM for GSK3-β) for 2 h at 37 °C + 5% CO2 incubator. Immediately prior to BRET measurements, NanoBRET Nano-Glo Substrate and Extracellular NanoLuc Inhibitor were added on cells and incubated for 2–3 min at room temperature. Donor emission at 450 nm and acceptor emission at 610 nm were measured using a EnVision plate reader. Raw BRET ratio values were calculated by dividing the acceptor emission value by the donor emission value for each sample. Raw BRET units were converted to milliBRET units (mBU) by multiplying each raw BRET value by 1000. mBU values for compound treatments were normalized to DMSO treatment. Ki values are calculated using the formula: Ki = (IC50)/(1 + ([Tracer]/EC50)). All experiments were performed as biological triplicates with three technical replications.
β-Catenin Translocation Assay
SH-SY5Y cells were seeded into a 96-well CellCarrier Ultra (PerkinElmer) plate at a density of 1 × 105 cells/mL in complete DMEM. 16 h following plating, media was changed to DMEM with 2% serum, and compound was added with a D300e Digital Dispenser (Tecan) for an incubation time of 6 h. After compound incubation, media was removed, and cells were fixed with 4% PFA for 10 min at room temperature. Each well was washed three times with PBS-T and then blocked with buffer (1× PBS with 5% normal goat serum and 0.3% Triton X-100) for 30 min at room temperature. The β-Catenin primary antibody (Cell Signaling no. 8480) was added at 1:500 dilution in blocking buffer, and the cells were allowed to incubate in primary antibody overnight at 4 °C. The following day, the primary antibody was removed, each well was washed three times with PBS-T, and the secondary antibody (1:2000 anti-rabbit AlexaFluor Plus 488 and 1:10,000 Hoechst 33342) was added and allowed to incubate for 60 min at room temperature protected from light. After 1 h, the secondary antibody was removed and wells were washed four times with PBS-T before imaging on the Opera Phenix. Sixteen fields per well were imaged with a 20× water lens. Images were quantified using Columbus Image Data Storage and Analysis software. Cells were identified using the Hoechst nuclear stain to identify individual nuclei. Intensity of the nuclear stain was also used to determine cell viability (viable cells < 30% CV of Hoechst intensity).
Animal Use
All experiments were conducted in compliance with the rules set forth by the Biogen Institutional Animal Use and Care Committee in accordance with the guidelines established in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. These protocols were approved by the Biogen IACUC committee.
Rat Developmental Model of Tau Phosphorylation
Sprague Dawley rats were bred in-house, and litters were injected with the compound, I.P. at post-natal day 10. Rats were then euthanized at indicated times following drug administration. For tissue collection, animals were euthanized via decapitation, brains were quickly removed, and bisected sagittally. The right cortex was isolated, placed into 2 mL microtubes along with a single 5 mm stainless steel bead, and the left cortex was placed into 2 mL lysing matrix A tubes (MP Biomedicals, Santa Ana, CA). Both tubes were immediately snap frozen in liquid nitrogen.
Determination of Free Drug Concentration in the Brain
Aqueous buffered homogenate was created from cortical tissue via bead beater, at a consistent homogenization factor, which was normalized by tissue weight. Total drug in brain was determined by measuring total exposure in brain in ng/mL (cortical tissue) via LC–MS/MS. The LC/MS/MS system consisted of a Thermo LX4 Multiplexing Agilent HPLC system (Agilent Technologies, Santa Clara, CA) coupled to an API-5500 mass spectrometer (Applied Biosystems, Foster City, CA). Separation was performed on a Phenomenex Kinetix C18 column (50 × 3.0 mm, 2.62.6 μm; Phenomenex, Torrance, CA). This was then corrected to free concentration via an experimentally derived fu, br using rapid equilibrium dialysis with brain homogenate. A detailed protocol describing the LC–MS/MS conditions used for the analysis is provided in Table S3.
In Vivo Target Engagement Assay
Chemoproteomics competition was used to evaluate inhibitor selectivity and target engagement in vivo. Frozen cortexes were prepared for homogenization with 1.2 mL of lysis buffer [50 mM Tris-HCl, 0.8% NP-40 ((octylphenoxy poly(ethyleneoxy)ethanol)), 5% glycerol, 150 mM NaCl, 1.5 mM MgCl2, 25 mM NaF, 1 mM sodium vanadate, 1 mM DTT, pH 7.5, and supplemented with protease inhibitors]. Tissues were homogenized in a FastPrep-24 5G instrument at maximum oscillations for 20 s. The resulting brain homogenate was clarified by centrifugation for 1 h at 145,000g, 4 °C. The protein concentration of the supernatant was determined by BCA assay and each sample was diluted to final concentration of 5 mg/mL with lysis buffer. Kinome enrichment was performed similarly as described previously34 with minor modifications. In short, in a 96 deep-well filter plate, 1 mL of the brain lysate (5 mg/mL protein concentration) was incubated with 200 μL (50% slurry) of probe functionalized beads for 30 min on an end-over-end rotator at 4 °C. Beads were thoroughly washed in two steps with lysis buffer containing 0.4 and 0.2% NP-40, respectively, and eluted with 100 μL of a 2 × LDS sample buffer supplemented with 20 mM DTT. Eluates containing the enriched kinome were alkylated with 40 mM iodoacetamide for 30 min in the dark, then separated on 4–12% NuPAGE for approximately 1 cm, and stained with colloidal coomassie. Gel bands were first cut and diced into ∼1 mm cubes and then de-stained with 50/50 acetonitrile/50 mM NH4HCO3 solution, followed by tryptic digestion overnight at 37 °C. The tryptic peptides were extracted into 40/60 acetonitrile/0.1% formic acid solution and dried in speed-vac. The dry peptide mixture was reconstituted in 15 μL of 0.1% trifluoro acetic acid and analyzed on 1D nanoLC-MS/MS platform using a standardized 140 min method. Peptides were separated on an Easy-Spray column (75 μm × 50 cm, PepMAP C18, 1.9 μm) at 250 nL/min and analyzed on a QExactive HF mass spectrometer at data dependent acquisition (DDA) mode with MS1 at 60,000 and MS2 at 15,000 rpm, respectively. Raw data were first QC checked using in-house developed software and were subsequently searched against the Swissprot human database using Andromeda integrated in Maxquant (V 1.6.38) with mass tolerance of 20 ppm (MS1) and 4.5 ppm (MS2). Carbamidomethylation of cysteine residues was set as fixed modifications, and (ST) phosphorylation, methionine oxidation, and (NQ) deamidation were set as variable modifications. LFQ values generated in the protein groups file were used to create the selectivity and target engagement graphs. Both selectivity and target engagement data at different doses and multiple time points are reported percentage inhibition compared to the vehicle groups.
Protein Expression, Purification, and X-ray Crystallography
GSK3α residues 98–446 were cloned into a his-tag-MBP-TEV-vector and grown in T.ni cells. 6 L of cell pellets were lysed in 50 mM Hepes 7.4, 150 mM NaCl, and 1 mM TCEP with protease inhibitors. Cells were lysed using a microfluidizer at 15,000 PSI and centrifuged at 18,000 rcf for 30 min. The resulting supernatant was run over a 3 × 5 mL MBPTrap HP column and washed with the lysis buffer. The protein was eluted in the lysis buffer containing 20 mM maltose. The eluted protein was treated with 200 μL of TEC (20,000 units) at 4 °C and dialyzed overnight into 50 mM Hepes 7.4, 150 mM NaCl, and 1 mM TECP. The cleaved protein was put over an MBPTrap column, and the eluant was collected. This was then put over a 16/60 Superdex200 column in 50 mM Hepes 7.4, 150 mM NaCl, 1 mM TCEP. Finally, the protein was then concentrated to 6 mg/mL for crystallography.
Crystallization and Data Collection and Refinement
Sitting drop plates were setup with GSK3α at 6 mg/mL complexed with 0.5 mM of the small molecule inhibitor and 0.5 mM of synthetic axin peptide. The complex crystallized in 0.1 M Bis-TRIS pH 5.0, 0.2 M CaCl2, 5% glycerol, and 30% PEG3350. The crystals were cryoprotected in 0.1 M Bis-TRIS pH 5.0, 0.2 M CaCl2, 25% glycerol, and 30% PEG3350. Data collection was performed at the LRL-CAT beamline at APS on a MAR-CCD detector at 0.97 Å. The data merged and scaled using Aimless. A structure of GSK3β was used as a search model to do molecular refinement using phaser in Phenix. Refinement was also done using Phenix. The statistics for the quality of the data is shown in Table S2.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.2c00476.
GSK3 phosphorylation of tau at additional epitopes, validation of Thr231 plate-based assay, effect of GSK3 inhibitors on tau phosphorylation at AT8 site, EC50 for Nanobret Tracer 8, kinetics of free drug concentration for compound 1 in vivo, off-target kinases demonstrating greater than 70% occupancy by compound 1 3 h post-dosing, potency of compound 2 and compound 3 to activate β-catenin nuclear translocation, cellular toxicity of all compounds evaluated, high resolution image, ADME properties of compound 1, data collection and refinement statistics for crystallography, LC–MS/MS conditions for the analysis of free drugs in the brain tissues, kinase selectivity data of BRD0705, compound 1, compound 2, and compound 3 at 1 μM, and methods describing synthesis and characterization of compounds (PDF)
Author Contributions
B.A. and A.C. contributed equally to the work described in this study. G.M.D. and S.K. contributed equally to design, execution, and publication of this study. B.A., T.A., C.T., P.M., T.W., and G.M.D. performed the in vitro assays and analyzed the data. M.C., O.G., and G.M.D. performed the in vivo assays and analyzed the data. A.C., B.L., T.P., I.E., B.M., Y.C., and Z.Y. were responsible for the design and synthesis of novel chemical compounds. J.V.C. and D.M. were responsible for generating the described crystal structures. R.K., K.S., and M.C. were responsible for protein production. B.B. performed the chemical proteomics competition assay. O.G., S.K., and A.C. conceived the study. S.K., B.A., and G.D. drafted the manuscript. All authors contributed to the final submission.
The authors declare no competing financial interest.
Supplementary Material
References
- Cole A. R. GSK3 as a Sensor Determining Cell Fate in the Brain. Front. Mol. Neurosci. 2012, 5, 4. 10.3389/fnmol.2012.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racaud-Sultan C.; Vergnolle N. GSK3β, a Master Kinase in the Regulation of Adult Stem Cell Behavior. Cells 2021, 10, 225. 10.3390/cells10020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen E. J.; Dokken B. B. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets 2006, 7, 1435–1441. 10.2174/1389450110607011435. [DOI] [PubMed] [Google Scholar]
- Bhat R.; Xue Y.; Berg S.; Hellberg S.; Ormö M.; Nilsson Y.; Radesäter A. C.; Jerning E.; Markgren P. O.; Borgegård T.; Nylöf M.; Giménez-Cassina A.; Hernández F.; Lucas J. J.; Díaz-Nido J.; Avila J. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- Beurel E.; Grieco S. F.; Jope R. S. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary O.; Nolan Y. Glycogen synthase kinase-3 as a therapeutic target for cognitive dysfunction in neuropsychiatric disorders. CNS Drugs 2015, 29, 1–15. 10.1007/s40263-014-0213-z. [DOI] [PubMed] [Google Scholar]
- McCubrey J. A.; Steelman L. S.; Bertrand F. E.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. 10.18632/oncotarget.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel C. J.; Wilson C. A.; Lee V. M.; Klein P. S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Plattner F.; Angelo M.; Giese K. P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006, 281, 25457–25465. 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- Ly P. T.; Wu Y.; Zou H.; Wang R.; Zhou W.; Kinoshita A.; et al. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 2013, 123, 224–235. 10.1172/JCI64516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgievska B.; Sandin J.; Doherty J.; Mörtberg A.; Neelissen J.; Andersson A.; Gruber S.; Nilsson Y.; Schött P.; Arvidsson P. I.; Hellberg S.; Osswald G.; Berg S.; Fälting J.; Bhat R. V. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J. Neurochem. 2013, 125, 446–456. 10.1111/jnc.12203. [DOI] [PubMed] [Google Scholar]
- del Ser T.; Steinwachs K. C.; Gertz H. J.; Andrés M. V.; Gómez-Carrillo B.; Medina M.; Vericat J. A.; Redondo P.; Fleet D.; León T. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J. Alzheimer’s Dis. 2012, 33, 205–215. 10.3233/JAD-2012-120805. [DOI] [PubMed] [Google Scholar]
- Behrens J.; Jerchow B. A.; Würtele M.; Grimm J.; Asbrand C.; Wirtz R.; Kühl M.; Wedlich D.; Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science 1998, 280, 596–599. 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- Meijer L.; Flajolet M.; Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 2004, 25, 471–480. 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Sato N.; Meijer L.; Skaltsounis L.; Greengard P.; Brivanlou A. H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 2004, 10, 55–63. 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- Coghlan M. P.; Culbert A. A.; Cross D. A.; Corcoran S. L.; Yates J. W.; Pearce N. J.; Rausch O. L.; Murphy G. J.; Carter P. S.; Roxbee Cox L.; Mills D.; Brown M. J.; Haigh D.; Ward R. W.; Smith D. G.; Murray K. J.; Reith A. D.; Holder J. C. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Stump B.; Shrestha S.; Lamattina A. M.; Louis P. H.; Cho W.; Perrella M. A.; Ai X.; Rosas I. O.; Wagner F. F.; Priolo C.; Astin J.; El-Chemaly S. Glycogen synthase kinase 3-β inhibition induces lymphangiogenesis through β-catenin-dependent and mTOR-independent pathways. PLoS One 2019, 14, e0213831 10.1371/journal.pone.0213831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett J. R. cDNA cloning and properties of glycogen synthase kinase-3. Methods Enzymol. 1991, 200, 564–577. 10.1016/0076-6879(91)00172-s. [DOI] [PubMed] [Google Scholar]
- Yao H. B.; Shaw P. C.; Wong C. C.; Wan D. C. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J. Chem. Neuroanat. 2002, 23, 291–297. 10.1016/s0891-0618(02)00014-5. [DOI] [PubMed] [Google Scholar]
- Soutar K.; Kim W.-Y.; et al. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 2010, 115, 974–983. 10.1111/j.1471-4159.2010.06988.x. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O.; Woodgett J. R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40. 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski T.; Dewachter I.; Lechat B.; Gees M.; Kremer A.; Demedts D.; Borghgraef P.; Devijver H.; Kügler S.; Patel S.; Woodgett J. R.; Van Leuven F. GSK-3α/β kinases and amyloid production in vivo. Nature 2011, 480, E4–E5. 10.1038/nature10615. [DOI] [PubMed] [Google Scholar]
- Kim W. Y.; Wang X.; Wu Y.; Doble B. W.; Patel S.; Woodgett J. R.; et al. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. 10.1038/nn.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Smith M.; Wu Y.; Zhu X.; Pringle J.; Snider W. D. GSK-3 signaling in developing cortical neurons is essential for radial migration and dendritic orientation. Elife 2014, 3, 1057–1066. 10.7554/eLife.02663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Martín M.; Jurado J.; Hernández F.; Avila J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 20142014, 7, 46. 10.3389/fnmol.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado D. E.; Molina-Porcel L.; Carroll J. C.; MacDonald C.; Aboagye A. K.; Trojanowski J. Q.; Lee V. M. Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of Alzheimer’s disease. J. Neurosci. 2012, 32, 7392–7402. 10.1523/JNEUROSCI.0889-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCamphill P. K.; Stoppel L. J.; Senter R. K.; Lewis M. C.; Heynen A. J.; Stoppel D. C.; Sridhar V.; Collins K. A.; Shi X.; Pan J. Q.; Madison J.; Cottrell J. R.; Huber K. M.; Scolnick E. M.; Holson E. B.; Wagner F. F.; Bear M. F. Selective inhibition of glycogen synthase kinase 3α corrects pathophysiology in a mouse model of fragile X syndrome. Sci. Transl. Med. 2020, 12, eaam8572 10.1126/scitranslmed.aam8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Dou X.; Jiang L.; Jin H.; Zhang L.; Zhang L.; Liu Z. Discovery of novel glycogen synthase kinase-3α inhibitors: Structure-based virtual screening, preliminary SAR and biological evaluation for treatment of acute myeloid leukemia. Eur. J. Med. Chem. 2019, 171, 221–234. 10.1016/j.ejmech.2019.03.039. [DOI] [PubMed] [Google Scholar]
- Doble B. W.; Patel S.; Wood G. A.; Kockeritz L. K.; Woodgett J. R. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 2007, 12, 957–971. 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji V.; Frumm S. M.; Ross K. N.; Li L. S.; Schinzel A. C.; Hahn C. K.; Kakoza R. M.; Chow K. T.; Ross L.; Alexe G.; Tolliday N.; Inguilizian H.; Galinsky I.; Stone R. M.; DeAngelo D. J.; Roti G.; Aster J. C.; Hahn W. C.; Kung A. L.; Stegmaier K. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J. Clin. Invest. 2012, 122, 935–947. 10.1172/JCI46465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner F. F.; Benajiba L.; Campbell A. J.; Weïwer M.; Sacher J. R.; Gale J. P.; Ross L.; Puissant A.; Alexe G.; Conway A.; Back M.; Pikman Y.; Galinsky I.; DeAngelo D. J.; Stone R. M.; Kaya T.; Shi X.; Robers M. B.; Machleidt T.; Wilkinson J.; Hermine O.; Kung A.; Stein A. J.; Lakshminarasimhan D.; Hemann M. T.; Scolnick E.; Zhang Y. L.; Pan J. Q.; Stegmaier K.; Holson E. B. Exploiting an Asp-Glu “switch” in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia. Sci. Transl. Med. 2018, 10, eaam8460 10.1126/scitranslmed.aam8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Monte F.; Kramer T.; Gu J.; Brodrecht M.; Pilakowski J.; Fuertes A.; Dominguez J. M.; Plotkin B.; Eldar-Finkelman H.; Schmidt B. Structure-based optimization of oxadiazole-based GSK-3 inhibitors. Eur. J. Med. Chem. 2013, 61, 26–40. 10.1016/j.ejmech.2012.06.006. [DOI] [PubMed] [Google Scholar]
- Palomo V.; Perez D. I.; Perez C.; Morales-Garcia J. A.; Soteras I.; Alonso-Gil S.; Encinas A.; Castro A.; Campillo N. E.; Perez-Castillo A.; Gil C.; Martinez A. 5-imino-1,2,4-thiadiazoles: first small molecules as substrate competitive inhibitors of glycogen synthase kinase 3. J. Med. Chem. 2012, 55, 1645–1661. 10.1021/jm201463v. [DOI] [PubMed] [Google Scholar]
- Reinecke M., Heinzlmeir S., Wilhelm M., Médard G., Klaeger S., Kuster K. B.. Kinobeads: A Chemical Proteomic Approach for Kinase Inhibitor Selectivity Profiling and Target Discovery; Wiley-VCH Verlag GmbH & Co. KGaA, 2019; Book Chapter, pp.97–130 [Google Scholar]
- Miyoshi Y.; Iwao K.; Nagasawa Y.; Aihara T.; Sasaki Y.; Imaoka S.; Murata M.; Shimano T.; Nakamura Y. Activation of the beta-catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res. 1998, 58, 2524–2527. [PubMed] [Google Scholar]
- Kurnit K.; Kim G.; Fellman B.; et al. CTNNB1 (beta-catenin) mutation identifies low grade, early stage endometrial cancer patients at increased risk of recurrence. Mod. Pathol. 2017, 30, 1032–1041. 10.1038/modpathol.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halkina T.; Henderson J. L.; Lin E. Y.; Himmelbauer M. K.; Jones J. H.; Nevalainen M.; Feng J.; King K.; Rooney M.; Johnson J. L.; Marcotte D. J.; Chodaparambil J. V.; Kumar P. R.; Patterson T. A.; Murugan P.; Schuman E.; Wong L.; Hesson T.; Lamore S.; Bao C.; Calhoun M.; Certo H.; Amaral B.; Dillon G. M.; Gilfillan R.; de Turiso F. G. Discovery of Potent and Brain-Penetrant Tau Tubulin Kinase 1 (TTBK1) Inhibitors that Lower Tau Phosphorylation In Vivo. J. Med. Chem. 2021, 64, 6358–6380. 10.1021/acs.jmedchem.1c00382. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y.; Okazaki M.; Kitamura Y.; Taniguchi T. Developmental expression of P-glycoprotein (multidrug resistance gene product) in the rat brain. J. Neurobiol. 1999, 39, 383–392. . [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C.; Reader V.; Sweetman G.; Bauer A.; Bouwmeester T.; Hopf C.; Kruse U.; Neubauer G.; Ramsden N.; Rick J.; Kuster B.; Drewes G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044. 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- Dajani R.; Fraser E.; Roe S. M.; Young N.; Good V.; Dale T. C.; Pearl L. H. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 2001, 105, 721–732. 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Dajani R.; Fraser E.; Roe S. M.; Yeo M.; Good V. M.; Thompson V.; Dale T. C.; Pearl L. H. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J. 2003, 22, 494–501. 10.1093/emboj/cdg068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon G. M.; Henderson J. L.; Bao C.; Joyce J. A.; Calhoun M.; Amaral B.; King K. W.; Bajrami B.; Rabah D. Acute inhibition of the CNS-specific kinase TTBK1 significantly lowers tau phosphorylation at several disease relevant sites. PLoS One 2020, 15, e0228771 10.1371/journal.pone.0228771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Martín T.; Cuchillo-Ibáñez I.; Noble W.; Nyenya F.; Anderton B. H.; Hanger D. P. Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 2013, 34, 2146–2157. 10.1016/j.neurobiolaging.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S. A.; Hamm M. J.; Meier S. E.; Weiss B. E.; Nation G. K.; Chishti E. A.; Arango J. P.; Chen J.; Zhu H.; Blalock E. M.; Abisambra J. F. Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 2019, 137, 571–583. 10.1007/s00401-019-01970-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftekharzadeh B.; Daigle J. G.; Kapinos L. E.; Coyne A.; Schiantarelli J.; Carlomagno Y.; Cook C.; Miller S. J.; Dujardin S.; Amaral A. S.; Grima J. C.; Bennett R. E.; Tepper K.; DeTure M.; Vanderburg C. R.; Corjuc B. T.; DeVos S. L.; Gonzalez J. A.; Chew J.; Vidensky S.; Gage F. H.; Mertens J.; Troncoso J.; Mandelkow E.; Salvatella X.; Lim R. Y. H.; Petrucelli L.; Wegmann S.; Rothstein J. D.; Hyman B. T. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940. 10.1016/j.neuron.2018.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F. Q.; Snider W. D. GSK-3β and microtubule assembly in axons. Science 2005, 308, 211–214. 10.1126/science.1110301. [DOI] [PubMed] [Google Scholar]
- Ittner L. M.; Ke Y. D.; Delerue F.; Bi M.; Gladbach A.; van Eersel J.; Wölfing H.; Chieng B. C.; Christie M. J.; Napier I. A.; Eckert A.; Staufenbiel M.; Hardeman E.; Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Goedert M.; Spillantini M. G.; Jakes R.; Rutherford D.; Crowther R. A. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Hanger D. P.; Noble W. Functional implications of glycogen synthase kinase-3-mediated tau phosphorylation. Int J Alzheimers Dis 2011, 2011, 1. 10.4061/2011/352805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutajangout K.; Leroy A. M.; Anderton J. R.; Brion B. H.; Woodgett J. P. The active form of glycogen synthase kinase-3beta is associated with granulovacuolar degeneration in neurons in Alzheimer’s disease. Acta Neuropathol. 2002, 103, 91–99. 10.1007/s004010100435. [DOI] [PubMed] [Google Scholar]
- Luna-Muñoz J.; García-Sierra F.; Falcón V.; Menéndez I.; Chávez-Macías L.; Mena R. Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz-50 antibody in Alzheimer’s disease. J. Alzheimer’s Dis. 2005, 8, 29–41. 10.3233/jad-2005-8104. [DOI] [PubMed] [Google Scholar]
- Neddens J.; Temmel M.; Flunkert S.; et al. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 52. 10.1186/s40478-018-0557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos J. R. F.; Bauer C.; Schuchhardt J.; Wedekind D.; Waniek K.; Lachmann I.; Wiltfang J.; Vogelgsang J. Validation of a prototype tau Thr231 phosphorylation CSF ELISA as a potential biomarker for Alzheimer’s disease. J. Neural Transm. 2019, 126, 339–348. 10.1007/s00702-019-01982-5. [DOI] [PubMed] [Google Scholar]
- Carlyle B. C.; Nairn A. C.; Wang M.; Yang Y.; Jin L. E.; Simen A. A.; Ramos B. P.; Bordner K. A.; Craft G. E.; Davies P.; Pletikos M.; Šestan N.; Arnsten A. F.; Paspalas C. D. cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 5036–5041. 10.1073/pnas.1322360111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benítez M. J.; Cuadros R.; Jiménez J. S. Phosphorylation and Dephosphorylation of Tau Protein by the Catalytic Subunit of PKA, as Probed by Electrophoretic Mobility Retard. J. Alzheimer’s Dis. 2021, 79, 1143–1156. 10.3233/JAD-201077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H.; Schreyer S. A.; Shinohara M. M.; LeBoeuf R. C.; Krebs E. G. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes 1999, 48, 1662–1666. 10.2337/diabetes.48.8.1662. [DOI] [PubMed] [Google Scholar]
- Domoto T.; Pyko I. V.; Furuta T.; et al. Glycogen synthase kinase-3β is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci. 2016, 107, 1363–1372. 10.1111/cas.13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min W. W.; Yuskaitis C. J.; Yan Q.; Sikorski C.; Chen S.; Jope R. S.; Bauchwitz R. P. Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 2009, 56, 463–472. 10.1016/j.neuropharm.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.