

Abstract
Aspergillus fumigatus and Candida auris are historically problematic fungal pathogens responsible for systemic infections and high mortality rates, especially in immunocompromised populations. The three antifungal classes that comprise our present day armamentarium have facilitated efficacious treatment of these fungal infections in past decades, but their potency has steadily declined over the years as resistance to these compounds has accumulated. Importantly, pan-resistant strains of Candida auris have been observed in clinical settings, leaving affected patients with no treatment options and a death sentence. Many compounds in the ongoing antifungal drug discovery pipeline, similar to those within our aforementioned trinity, are predicated on the binding or inhibition of ergosterol. Recurring accounts of resistance to antifungals targeting this pathway suggest optimization of ergosterol-dependent antifungals is likely not the best solution for the long-term. This review aims to present several natural products with novel or underexplored biological targets, as well as similarly underutilized drug discovery strategies to inspire future biological investigations and medicinal chemistry campaigns.
Keywords: Antifungals, Antimicrobial Resistance, Natural Products, Total Synthesis
1. Introduction
The golden age of antimicrobials has reaped a bleak present and future as antimicrobial resistance (AMR) continues to challenge treatment options in all settings. In 2014, AMR was linked to at least 700,000 deaths worldwide per year with a strong likelihood of reaching well over 10 million by 2050 if current trends persist.1 As of 2019, the Centers for Disease Control and Prevention (CDC) reports nearly 3 million infection events and 36,000 resultant deaths in the United States attributable to AMR in bacteria and fungi, suggesting our current efforts in antimicrobial development are insufficient.2 Although bacterial infections contribute to the bulk of these figures, it is paramount to consider the situation beyond the United States as, worldwide, over 150 million fungal infections and 1.7 million annual deaths were reported in 2017 alone.3
The development of AMR is one of the greatest challenges in the drug discovery effort. Pathogens employ a variety of mechanisms that ultimately increase the minimum inhibitory concentration (MIC) of a given antimicrobial. Following entry into the cell, the drug can be modified or degraded by enzymatic action, as seen in the action of β-lactamases against penicillin.4 Alternatively, rather than targeting the drug, modification of the drug target through mutation can lead to decreased drug affinity and consequent loss of activity. Overexpression of the drug target demands higher drug quantities to achieve the same inhibitory effect, and overexpression of efflux pumps shuttle drugs out of the cell. Another key defensive measure used by microbes is the formation of biofilm – an amalgamation of extracellular polymeric substances such as polysaccharides, proteins, and nucleic acids.5 Biofilms aid in cellular adhesion and denying intracellular access, significantly increasing pathogenicity.
Both the CDC and the World Health Organization (WHO) have recently identified the human fungal pathogens Aspergillus fumigatus and Candida auris as high priority health threats based on several key variables, such as global mortality rates, diagnostic availability, and reports of antifungal resistance.6 To demonstrate the ubiquity of A. fumigatus, it is estimated that the average person inhales several hundred of this species’ conidial spores daily.7 Fortunately, the human immune system can efficiently clear these antigens, with symptoms manifesting as mild allergies.8 Previously, the immunocompromised were most at-risk for developing invasive aspergillosis, where infection was accompanied with an alarmingly high mortality rate of 80–90% in leukemia patients.9 However, the recent increase in immunosuppressive therapies has left a larger portion of the population vulnerable to infection, making A. fumigatus the most common airborne fungal pathogen.10 Agricultural overuse and environmental spillage of antifungal agents has caused widespread resistance to common antifungals to develop across the majority of strains, making treatment of A. fumigatus infections more difficult relative to other fungal pathogens.11
The fungal yeasts Candida albicans and Candida glabrata are the most common cause of vaginal candidiasis, otherwise known as yeast infection. Candidiasis rarely progresses in severity as commercially available treatments remain efficacious against the aforementioned major causes of candidiasis, with few reports of significant resistance development. Although relatively much rarer, candidiasis brought on by C. auris has the potential to be insurmountably problematic. Despite being a rarity in the time following its initial identification in Japan in 2009, C. auris has become far more prevalent following a 318% increase in cases reported in 2018 alone.12 With some isolated strains of C. auris having been found to be resistant to all classes of available commercial antifungals, the necessity for new antifungals has never been clearer.13
2. Current Antifungals and Resistance Mechanisms
Compared to antibacterial drug development, antifungal development is accompanied by some additional challenges. As fungi are eukaryotic, proposed compounds must be capable of selectively inhibiting fungal machinery over those of humans, a consideration that does not have to be made in drug design targeting prokaryotes. For the past several decades, the fungal sterol ergosterol has been leveraged to access this desired selectivity. Ergosterol is important for cellular membrane structure and causes defects in membrane integrity in its absence, leading to eventual cell death.14
There are currently only three major antifungal compound classes for invasive fungal infections, and two of them are dependent on ergosterol in some manner for their desired activity (Figure 1). Polyenes were the first of these major classes to be developed and are large, glycosylated macrocycles with highly unsaturated alkyl regions. The first polyene, nystatin (1), was isolated from Streptomyces noursei in 1950 and briefly used in clinics prior to the adoption of amphotericin B (AmB, 2) by 1958.15 AmB and other polyenes cause fungal cell death by binding to ergosterol and subsequently disrupting the cellular membrane through intercalation of the polyene region.16 Expectedly, mutations in ERG3 that slightly alter sterol structure confers resistance to AmB by reducing membrane binding affinity.17 Unfortunately, approximately 66% of C. auris isolates collected in the United States were found to be resistant to AmB treatment.18 On the other hand, AmB resistance in Aspergillus species outside of A. terreus is exceedingly rare, thus AmB is the first-line treatment option for patients suffering from aspergillosis.19 In spite of this, widespread use of AmB is precluded by its low oral bioavailability restricting administration to intravenous delivery, as well as severe dose-dependent toxicities.20 Recent efforts to modify AmB formulations to minimize these caveats have been met with preliminary success, suggesting that there is still room for optimization of this decades-old drug.21
Figure 1.
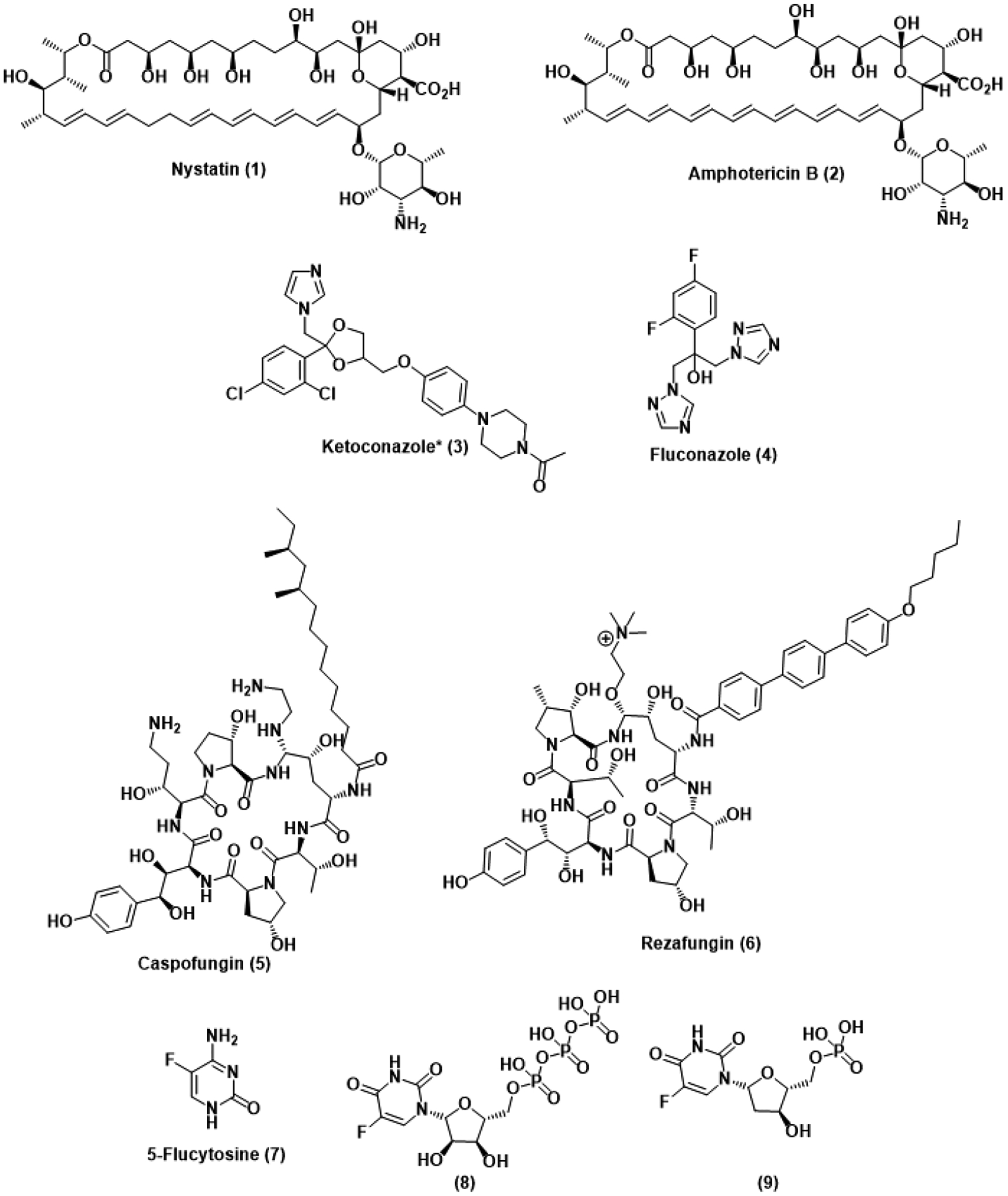
Representative compounds from our current armamentarium. *Ketoconazole is typically prepared and administered as a racemic mixture.
The next antifungal class to be developed are the azoles, beginning with the synthetic lead compound ketoconazole (3) in 1981.22 Celebrated as a broad-spectrum antifungal, the imidazole and piperazine moieties of ketoconazole were soon simplified to the achiral triazole fluconazole (4), the current azole antifungal standard, in 1985.23 Several fluconazole derivatives and optimizations would be developed over the following years, but their mechanism of action remained consistent – inhibition of cytochrome P450 enzyme 51 (CYP51), the enzyme responsible for demethylation of lanosterol in the synthesis towards ergosterol, leads to cytotoxic depletion of ergosterol.24 Resistance to azoles was observed as early as 1989 in Candida krusei, where overexpression and mutation of ERG11 decreases azole binding efficacy.25 Overexpression of the efflux-related genes MDR1, FLU1, CDR1, and CDR2 has also been found to be a major contributor to azole resistance.26 Due to environmental and clinical overuse, fluconazole is almost completely ineffective against a number of fungal pathogens. Approximately 93% of C. auris isolates studied are resistant to fluconazole, while the figure for A. fumigatus approaches 100%.11 Although there have been recent attempts at developing novel azole antifungals to rescue their bioactivity against these resistant pathogens, the consequences of our misuse will likely forever limit azole efficacy and force us to explore other pharmacophores.
The echinocandins ended a 15-year drought in antifungal development following the introduction of caspofungin (5) in the early 2000s, previously isolated from the fungus Glarea lozoyensis in the late 1980s.27 Making a notable departure from the dependence on previously discussed for polyenes and azoles, echinocandins act as non-competitive inhibitors of β-1,3-glucan synthase. This enzyme is responsible for the production of β-1,3-glucan, an integral component of the fungal cellular wall. Resistance to the echinocandins in C. auris has been found to originate from mutations in FKS1, altering the drug binding site at a small fitness cost to the pathogen.28 Interestingly, benzylic dehydroxylation has been found to restore echinocandin activity against mutant glucan synthases.29 Nevertheless, only about 7% of C. auris isolates studied were found to carry this resistance, and thus the echinocandins are the first-line treatment option against C. auris infection.30 However, approximately 1% of C. auris isolates are resistant to echinocandins, AmB, and fluconazole, leaving us with no treatment options for a small percentage of pan-resistant C. auris that will likely to increase over time as more echinocandin regimens are made necessary. The echinocandins have some notable advantages over other antifungal classes – low toxicity and few drug-drug interactions make the very act of treatment less problematic, as seen in the recently developed echinocandin rezafungin (6).31 One major limitation of the echinocandins is poor oral bioavailability, making intravenous administration necessary – similar to AmB, recent efforts at improving echinocandin bioavailability stand to make significant improvements to these decades-old compounds.
Although rarely administered as a monotherapy for invasive fungal infections, 5-fluorocytosine (5-FC, 7) is sometimes employed in combination with the above antifungal classes. Introduced in 1968, 5-FC is an orally available prodrug that is metabolized in vivo to both fluorouridine triphosphate (8), a protein synthesis inhibitor mimicking uridylic acid, and 5-fluorodeoxyuridine monophosphate (9), a strong inhibitor of thymidylate synthase.32 5-FC is one of the few antifungals whose efficacy does not entirely depend on ergosterol, but in order to compensate for the poor selectivity it offers, it is usually co-administered with AmB to ease entry into fungal cells over human cells. Mutations in metabolic and transporter enzymes, as well as upregulation of native pyrimidines, confers resistance to 5-FC in Candida species.33 Furthermore, the prohibitive cost of 5-FC treatments has also contributed to the lack of widespread adoption of this particular therapeutic.34
3. Recent Efforts
Although our current antifungal armament is largely based on the foundations laid several decades ago, the optimization of these foundations has continued well into the present (Figure 2). Treatment of recurrent candidiasis with fluconazole has been met with widespread resistance development and off-target toxicities exacerbated by repeated use. Oteseconazole (10), sold under the brand name Vivjoa, is a tetrazole-containing optimization of the azole scaffold that has been developed to address these concerns.35 Although the mechanism of action remains consistent with other azoles, up to a 40-fold increase in potency has been observed against Candida species, including C. auris, relative to fluconazole, suggesting tighter binding or the presence of a distinct binding site. This is further supported by the 2200-fold selectivity observed for fungal CYP51 inhibition over the human homolog.36 The related triazole opelconazole (11) was similarly developed as a solution for recurrent pulmonary aspergillosis.37 Delivery through inhalation of aerosolized opelconazole yields high concentrations of the therapeutic in the lungs while maintaining low systemic concentrations, facilitating inhibition of azole-resistant Aspergillus residing in pulmonary cavities. Although these new additions to the azole antifungal family fill important clinical niches, continuing to target CYP51, a known locus for resistance development, stands to limit the long-term efficacy of these new drugs.
Figure 2.
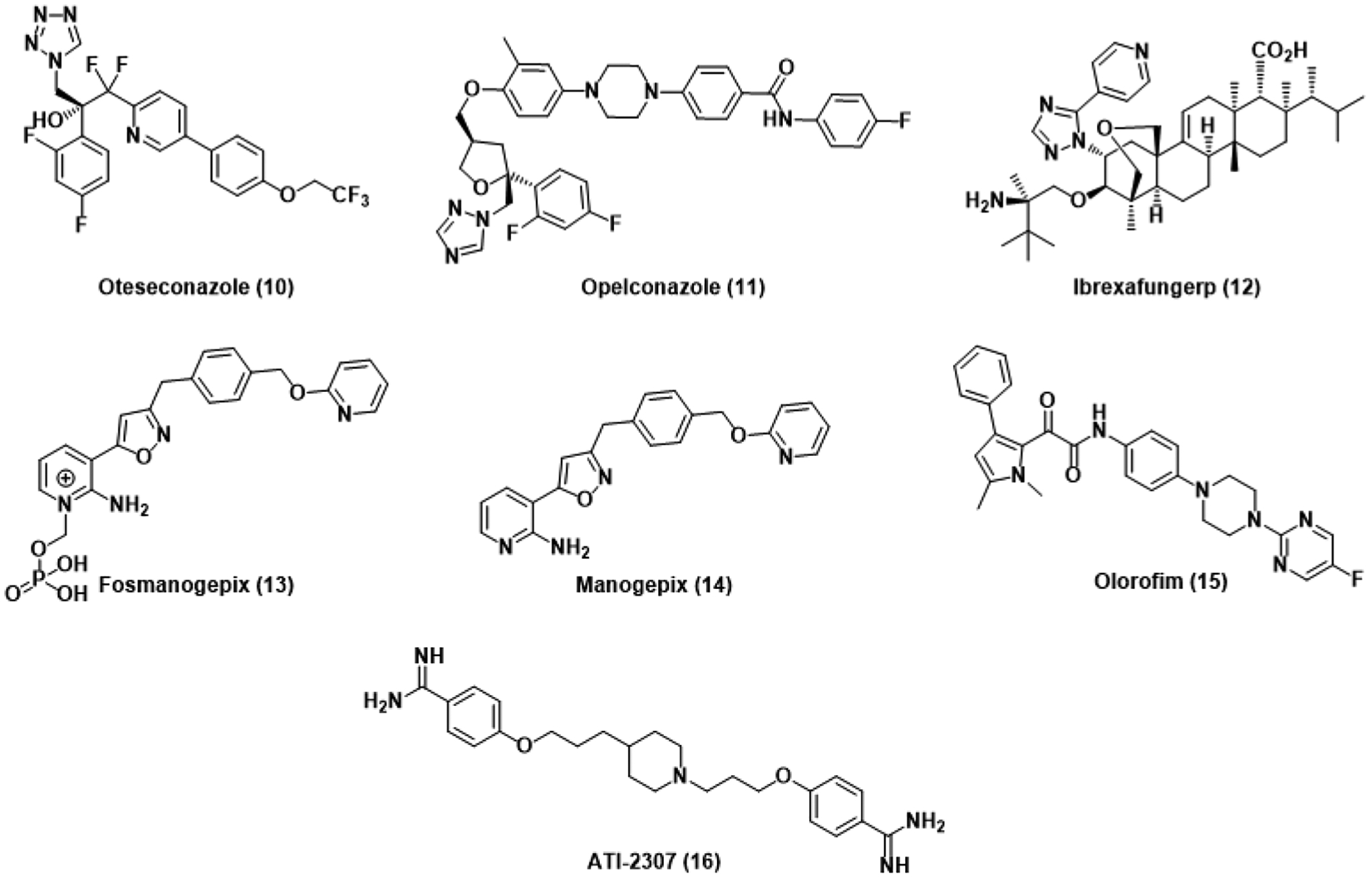
Antifungals recently approved for use or currently in clinical trials.
Similar to other echinocandins, ibrexafungerp (12), sold under the name Brexafemme, affects fungal cell death through the inhibition of β-1,3-glucan synthase.38 In contrast to previous echinocandins, however, optimization of the echinocandin scaffold significantly increases oral bioavailability and affords good activity against azole-resistant Candida and Aspergillus species.39 Interestingly, this activity was conserved against echinocandin-resistant strains as ibrexafungerp interacts differently than other echinocandins.
Rather than investing resources into continually soon-to-be-obsolete antifungals from the 1980s, the development of new drugs with novel targets, good fungal selectivity, and potent activity against clinically relevant Candida and Aspergillus species is more likely to overcome antifungal resistance. Fortunately, several such antifungals have recently entered clinical studies. Similar to 5-FC, the prodrug fosmanogepix (13) is metabolized to the active antifungal manogepix (14) that then inhibits Gwt1, a vital protein involved in the transportation and anchoring of mannoproteins.40 The resulting inhibition of several virulence factors, such as biofilm formation and adhesion, was observed in C. auris.41 As expected, point mutation of the enzymatic target and overexpression of the MDR1 superfamily transporter gene conferred resistance in Candida by decreasing binding affinity and increasing efflux, respectively.42,43
Olorofim (15) similarly evades ergosterol dependence and represents the first advancement towards the development of a new antifungal class, the orotomides. The orotomides inhibit dihydroorotate dehydrogenase, a vital enzyme implicated in pyrimidine synthesis, preventing DNA and glucan synthesis.44 Olorofim is ineffective against the Class 2 DHODH isoforms observed in Candida, Cryptococcus, and humans, limiting the scope of activity to Aspergillus species for now. Otherwise, mutations within DHODH confer resistance to olorofim in A. fumigatus at a significant fitness cost to the pathogen.45 The diamidine ATI-2307 (16) selectively inhibits fungal mitochondrial complexes III and IV, leading to membrane potential collapse.46 Low ng/mL MICs were reported against several Candida and Cryptococcus yeasts, while activity was significantly lower yet comparable to azole and echinocandin controls in filamentous A. fumigatus.47
4. Antifungal Natural Products
Nature has long been an abundant source of bioactive compounds and has provided us with the inspiration and innovation needed to engineer new antifungals. As stated previously, these new antifungals should ideally be both potent and selective inhibitors of fungal machinery without depending on ergosterol. There are several strategies antifungal candidates can employ to achieve this – for example, inhibition of novel enzymatic targets, as seen with the orotomides, stands to introduce wholly new drug classes and significantly bolster current treatment options (Figure 3). Another potential antifungal development strategy involves abandoning the traditional desire for fungicidality and instead opting for an anti-virulence approach (Figure 4). Several Candida species, including C. auris, have been observed to be dimorphic and can alternate between a commensal yeast form and a pathogenic hyphal form in response to environmental stimuli. Although discovered compounds are rarely tested against C. auris, results attained by testing against C. albicans will be cautiously employed as a proxy on account of their homology.48 The absence of biological data against C. auris is another major contributor to the current fungal crisis, but inhibitors of other Candida species stand to be effective against this deadly pathogen.
Figure 3.
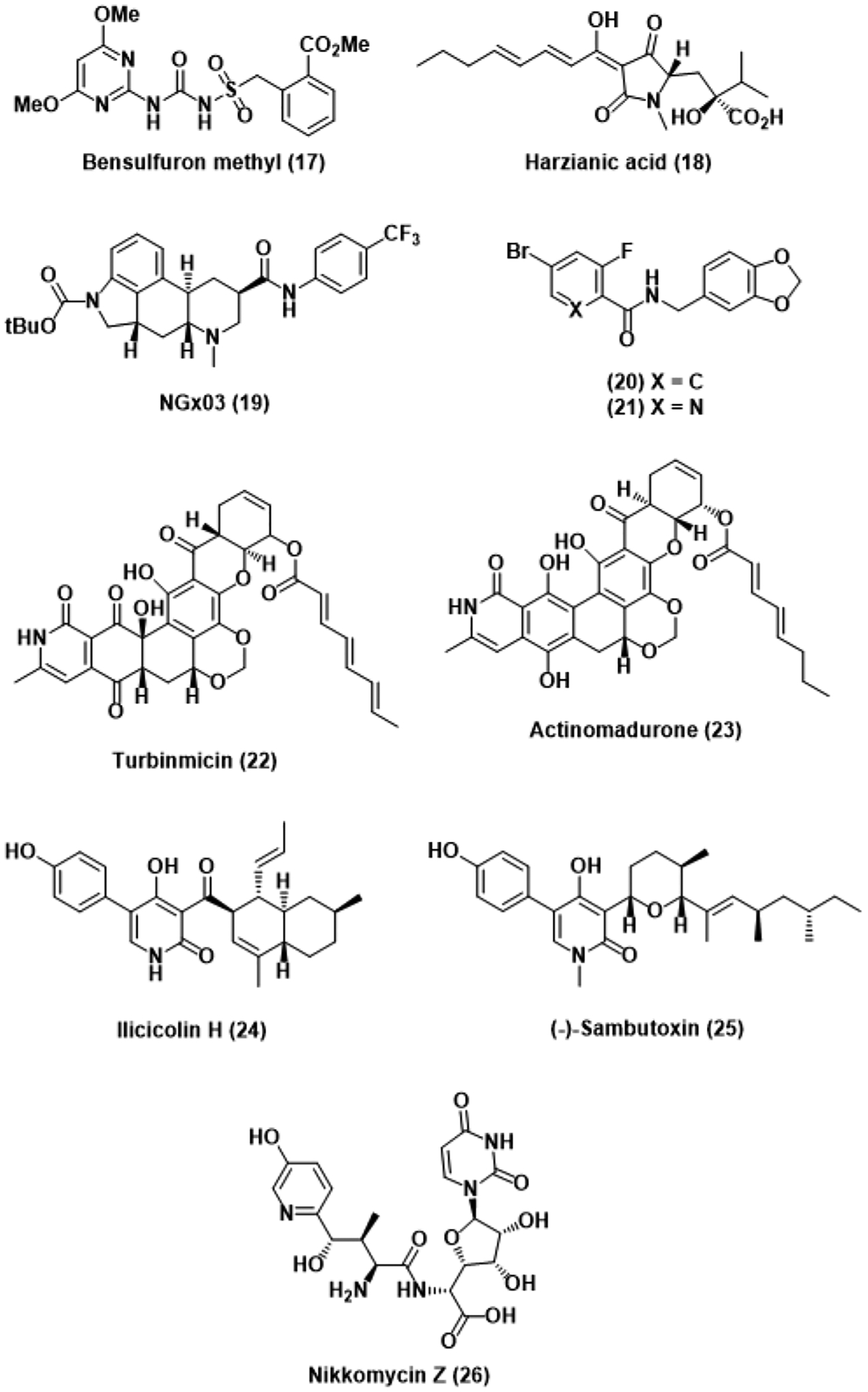
Compounds targeting novel or understudied fungal machinery.
Figure 4.
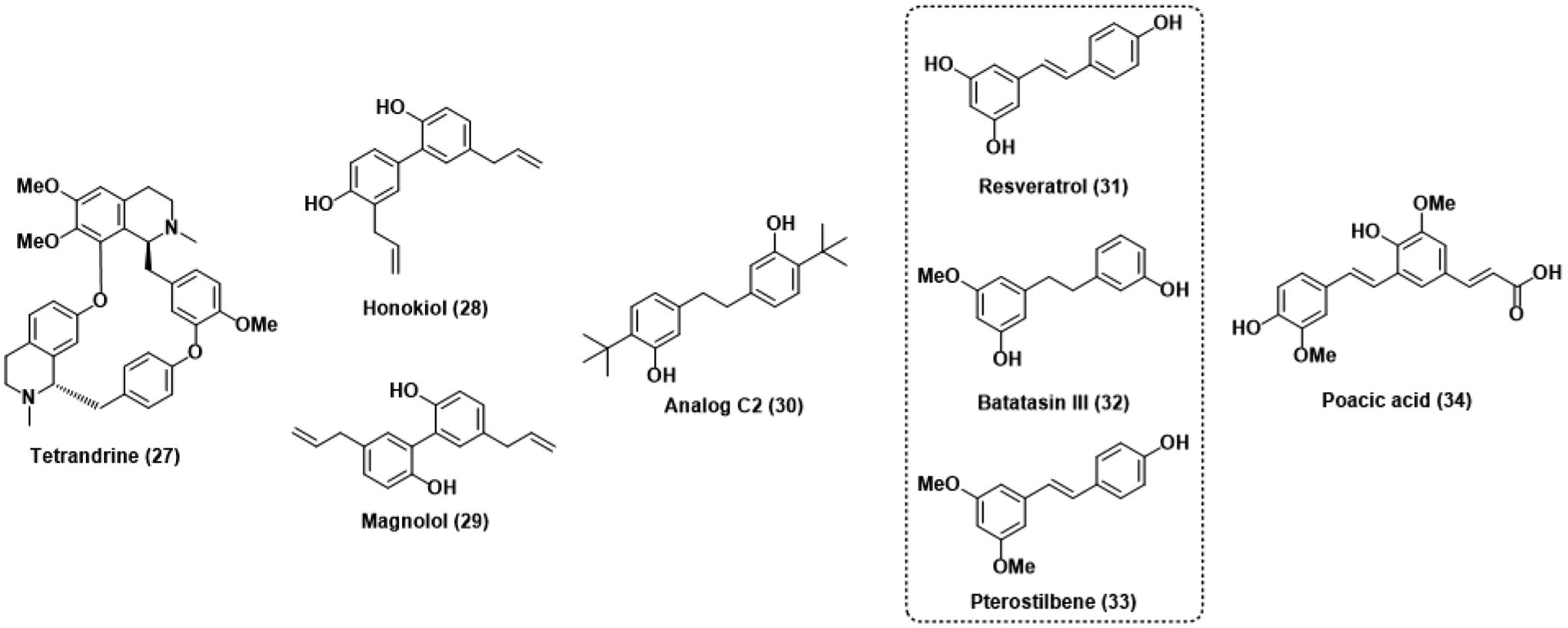
Anti-virulence antifungals and related stilbenoid natural products.
Acetohydroxyacid synthase (AHAS) is the first enzyme within branched chain amino acid synthesis.49 The biological importance of this enzyme, as well as its absence in animals, has previously been leveraged in the design of AHAS-inhibiting herbicides developed in the late 1980s.50 In addition to their original indication, several of these compounds were recently found to be similarly active against fungal AHAS, inhibiting the growth of several Candida species.51 A follow-up study then investigated these herbicides for activity against C. auris in particular, and the sulfonylurea herbicide bensulfuron methyl (17) was found to potently inhibit wild-type C. auris with an MIC of 0.391 μM.52 The herbicide also retained good activity when tested against an azole- and AmB-resistant strain while maintaining a therapeutic index greater than 100. Point mutations of a key proline residue within AHAS was found to confer resistance to these commercial herbicides. Produced by Trichoderma afroharzianum, the known siderophore harzianic acid (HA, 18) was later found to similarly inhibit wild-type AHAS while also retaining activity against AHAS mutants.53 Structural analysis of an AHAS-HA complex revealed that HA binds differently to AHAS than any other commercial herbicide, explaining the retention of activity. Therapeutic parameters of HA beyond fungal activity remain to be investigated, but these preliminary results suggest HA has the potential to develop into a promising candidate.
The phosphatidylinositol and phosphatidylcholine transfer protein Sec14p is exclusive to yeasts and implicated in several cellular processes critical to growth and virulence, such as fatty acid synthesis and hyphal transition, respectively.54 A high-throughput screen searching for activity against Sec14p using chemogenomic profiling found that the ergoline NGx03 (19) inhibited Sec14p in fluconazole-resistant Cryptococcus neoformans with an IC50 of 16 μg/mL through binding competition with native substrates.55 A subsequent study similarly investigated a library of picolinamide and benzamide compounds using the same profiling methodology and ultimately identified the benzamide 20, as well as its picolinamide analog 21, as inhibitors of Sec14p.56 Unfortunately, potent inhibition of Sec14p did not translate to potent activity against several fluconazole-resistant fungal strains as the alternative azole posaconazole vastly outclassed compounds 20 and 21. Although resistance to Sec14p inhibitors can be conferred by Sec14p mutation and one of seven possible genetic loss-of-function events, these changes have nevertheless been found to negatively impact lipid metabolism and generally decrease cellular fitness.
Recently, the marine natural product turbinmicin (22) was isolated from Micromonospora bacteria contributing to a sea squirt microbiome.57 Structurally similar polyketides such as actinomadurone (23) have previously been shown to have fungal activity but no mechanism of action had been elucidated.58 In accordance with these findings, turbinmicin notably possessed an MIC of 0.25 μg/mL against pan-resistant C. auris, suggesting a mechanism of action unique from current antifungals. Indeed, treatment of a knockout library with turbinmicin revealed the selective inhibition of Sec14p as the cause of fungicidal activity. A subsequent investigation studied the effects of turbinmicin on Candida biofilm and found that in vitro biofilm inhibition was observed at concentrations as low as 0.125 μg/mL in C. auris.59 Synergy between fluconazole and turbinmicin was also observed in murine models, further supporting the claim of biofilm inhibition. These findings are consistent with established outcomes for Sec14p inhibition as hyphal transition is a prerequisite for biofilm formation in Candida. Although Sec14p has been validated time and time again as a rational target for antifungal drug design, no such inhibitors have been approved for clinical use nor extensively studied in biological systems. Fortunately, ongoing studies with turbinmicin may eventually culminate in its introduction to clinics if fungal activity and human cell toxicity continues to impress.
Although the pyridone scaffold has been extensively explored across several different medical indications, the 4-hydroxy-2-pyridone subfamily has remained largely biologically unexplored.60 Ilicicolin H (24) was originally isolated from the plant pathogen Cylindrocladium ilicicola and found to be the least cytotoxic towards mammalian cells among the ilicicolin co-isolates.61 The antifungal efficacy of ilicicolin H was soon investigated and found to have potent activity against both C. albicans (MIC = 0.04–0.31 μg/mL) and A. fumigatus (MIC = 0.08 μg/mL). Another similarly cytotoxic and structurally related Fusarium isolate (−)-sambutoxin (25) was also found to have potent antifungal activity.62 Studies investigating the mechanism of action of these pyridones determined the target to be the Qn site of mitochondrial cytochrome bc1 reductase, compromising electron transport and rationalizing the observed potent cytotoxicity.63 A subsequent structure-activity relationship (SAR) campaign of ilicicolin H corroborated these findings and concluded that the 5-aryl-4-hydroxy-2-pyridone and β-diketone moieties are responsible for the observed selective inhibition of fungal cytochrome at sub-μg/mL quantities.64 Unfortunately, these essential moieties also conferred high plasma protein binding that significantly decreased the potency and bioavailability of ilicicolin H in vivo, blockading further progression into clinical trials. Additional research towards the minimization of these compounds’ plasma protein affinity could promote them to drug candidacy, especially as several other 4-hydroxy-2-pyridones remain to be extensively studied in biological systems.
The polyoxin nucleoside antibiotic nikkomycin Z (26) was originally isolated from the soil bacterium Streptomyces tendae in the late 1970s, and later found to inhibit growth of clinically relevant Coccidioides fungal species.65 The mechanism of action of nikkomycin Z against fungi was determined to be competitive inhibition of chitin synthase through mimicking of the native substrate, uridine diphosphate N-acetylglucosamine. Chitin is a polymeric accumulation of N-acetylglucosamine present in the vast majority of fungi and is integral to the stability of the fungal cell wall.66 As a result, inhibition of chitin synthase by nikkomycin Z directly affects cellular integrity. The mechanistic novelty and fungal selectivity of nikkomycin Z have inspired further studies into its activity beyond Coccidioides. Synergy with the azole antifungals fluconazole and itraconazole was observed across a panel of fungal strains, particularly against A. fumigatus.67 Recently, nikkomycin Z activity has also been studied against C. auris, yielding promising results as inhibitory concentrations are similarly low to those necessary to affect inhibition in C. albicans.68 Notably, some of the C. auris strains were particularly resistant to treatment with nikkomycin Z, but the underlying resistance mechanism responsible has yet to be determined.
Although the discovery and development of fungicidal compounds dominate antifungal drug discovery efforts, resistance selection is greatest when the survival of the pathogen is at stake. Recently, a greater abundance of therapeutic strategies seeking to inhibit virulence factors like hyphal transition and biofilm formation have emerged (Figure 4).69 Since inhibition of virulence factors tends to not directly endanger the fungus itself, there is less selective pressure driving resistance development, significantly boosting the longevity of these proposed compounds. Anti-virulence treatments also avoid the adverse effects of disturbing the commensal microbiome, yet another advantage against traditional growth inhibition or cytotoxic approaches.
The traditional Chinese medicine tetrandrine (TET, 27) was originally isolated from the roots of Stephania tetrandra and used to treat inflammation.70 TET was found to synergize with azole antifungals like fluconazole and ketoconazole against azole-resistant C. albicans, suggesting that it is interfering with efflux-dependent resistance mechanisms and restoring the efficacy of the azoles.71,72 Indeed, RT-PCR analysis confirmed that TET affects genes coding for vital efflux machinery, including ATP-binding cassette (ABC) and major facilitator superfamily (MFS) transporters.73 A subsequent study found that TET also inhibits C. albicans hyphal transition through the downregulation of several hyphal-specific genes vital to the Ras-cAMP-PKA pathway, a biochemical signaling pathway necessary for several key cellular processes, including morphological transition from yeast to hyphae.74 Inhibition of hyphal transition also rationalizes the C. albicans biofilm inhibition that has also been observed following treatment with TET.
The phenolic natural products honokiol (28) and magnolol (29) have been isolated from the barks of several Magnolia species and found to have broad antibacterial and antifungal properties, most notably against biofilm formation in Streptococcus mutans and C. albicans.75–77 The promising native activities of these compounds against the bacterium S. mutans originally inspired our group to develop honokiol analogs in an attempt to optimize the scaffold for the treatment of dental caries.78,79 Our SAR campaign ultimately led us to Analog C2 (30) as the most potent S. mutans biofilm inhibitor within the series. The modified dihydrostilbene core of Analog C2 makes it more similar in structure to other stilbenoid natural products such as batatasin III (31), resveratrol (32), and pterostilbene (33). Pterostilbene is a known inhibitor of biofilm formation in C. albicans, and a recent SAR study found that reduction to the dihydrostilbene analog of pterostilbene mostly retained its potency against nascent biofilm, only suffering a 2-fold decrease in activity.80,81 These findings and the structural relationships between Analog C2 and the hydrogenated analog of pterostilbene suggest further structural optimization of a dihydrostilbene scaffold could pivot from known antibacterial activity and towards potent biofilm inhibition in fungal pathogens.
The stilbenoid poacic acid (34), a dehydrodiferulate-derived natural product isolated from the hydrolysate of Poaceae grasses, has also recently been found to have antifungal activity.82 Rather than exhibiting anti-virulence activity, however, chemical genomics and morphological analysis showed that poacic acid damaged fungal cell walls and caused detrimental cellular leakage, similar to echinocandins. By leveraging the intrinsic fluorescent capabilities of the natural product it was then determined that poacic acid causes cell wall damage by directly binding to surface β-1,3-glucan rather than inhibiting β-1,3-glucan synthase. Promising activity was reported against several agriculturally relevant fungal plant pathogens, and efficacy against several Candida species was later determined in a follow-up study.83 This study interestingly found that wild-type C. albicans was significantly more resistant to poacic acid treatment compared to S. cerevisiae, yet caspofungin-resistant mutant strains of C. albicans were more sensitive. This further supported the mechanistic differentiation between poacic acid and echinocandins and demonstrated the lack of overlap between the two compound classes despite both leveraging the biological importance of β-1,3-glucan. Unfortunately, wide variability in fungicidality against a panel of clinically relevant fungal pathogens, including C. auris, have precluded further development of poacic acid into a next-generation commercial antifungal.
Although traditional natural product isolation, characterization, and biological investigation efforts have forged the foundation of modern antimicrobial therapies, debilitating limitations to the methodology have significantly slowed drug discovery efforts in recent decades. As more and more natural reservoirs and their bioactive metabolites have been investigated and archived in the literature, discovery of novel natural products with relevant antimicrobial activity has dramatically increased in difficulty since unproductive re-isolation of known compounds is far more likely. The development and implementation of new methodologies to expedite drug discovery and circumvent re-isolation is paramount if we wish to abolish the current stagnation.
5. Candida albicans Fitness Test
The concept of leveraging the eukaryotic phenomenon of haploinsufficiency in drug discovery was first introduced over two decades ago yet remains thoroughly underutilized in the search for novel antifungals.84 This genomic profiling methodology is based upon the treatment of a library of heterozygous deletion strains with a choice compound. Retention of one functional gene is most often sufficient for the organism to develop without phenotypic abnormalities. However, these deletion mutants are consequently hypersensitive to stressors targeting the deleted locus as there is no compensatory duplicate to aid in resistance efforts. Screening bioactive extracts or pure compound against these deletion libraries quickly elucidates the mechanism of action based on these observed hypersensitivities and enables identification of bioactive extract components with novel chemical structures based on similarly novel bioactivity, doing away with re-isolation complications. This system was originally developed in the model yeast S. cerevisiae but has recently been adapted to the more clinically relevant fungal pathogen Candida albicans.85,86 The aforementioned Candida albicans fitness test (CaFT) has been used to simultaneously identify bioactive compounds in crude extracts and their biological targets in a handful of examples within recent literature (Figure 5).
Figure 5.
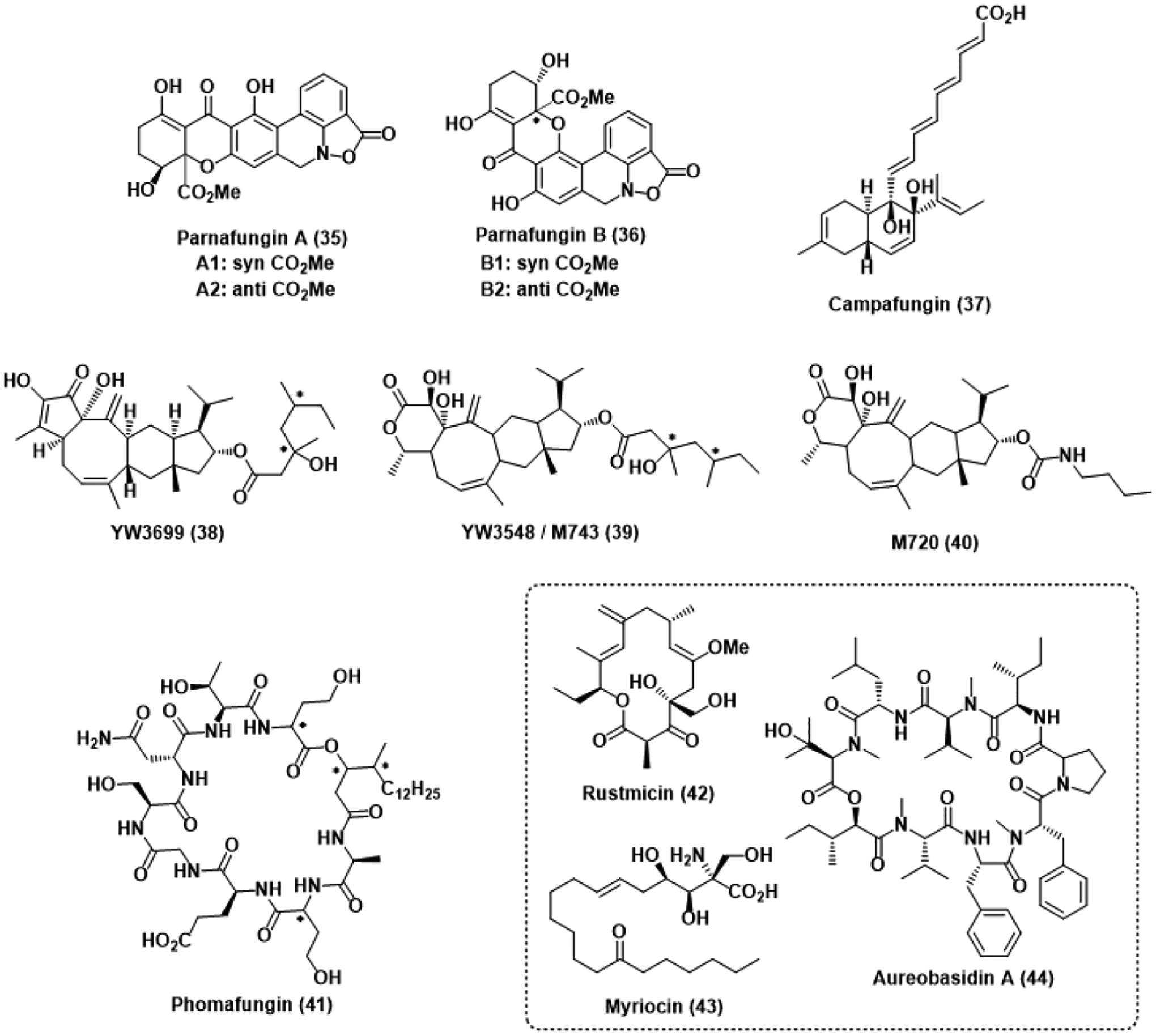
Antifungal natural products identified by the CaFT. * represents an undefined stereocenter.
The isoxazolidinones parnafungin A (35) and B (36) were isolated from a fermentation extract of the insect fungal pathogen Fusarium larvarum as a mixture of syn (A1, B1) and anti (A2, B2) diastereomers. Interestingly, these diastereomers were observed to freely interconvert between one another in DMSO through a proposed retro-Michael ring opening followed by rotation and ring closing.87 Potent activity against C. albicans and A. fumigatus was observed with MICs of 0.014 μg/mL and 8–16 μg/mL, respectively. Notably, this activity against C. albicans was comparable to that of the caspofungin control and retained potency after addition of 50% mouse serum. Evaluation of these compounds using the CaFT identified several heterozygous deletion mutants deficient in genes implicated in mRNA processing as hypersensitive to parnafungin treatment.88 Indeed, the parnafungins were ultimately determined to inhibit the action of polyadenosine polymerase, preventing the polyadenylation of mRNA substrates. This polyadenylation event is crucial to the proper translation, trafficking, and degradation of mRNA. As a result, its absence is significantly deleterious to the fungal cell. In vivo efficacy showcased in a murine model suggests the parnafungins may be viable drug candidates in the future should mammalian cytotoxicity remain minimal.
The decalin polyketide campafungin (37) was isolated from the ascomycete Plenodomus enteroleucus and similarly passaged through the CaFT.89 MIC values against C. albicans and A. fumigatus were greater than desired, but the CaFT profile suggested the compound was targeting some aspect of the cAMP-PKA pathway based on the observed hypersensitivity of heterozygous mutants deficient in genes coding for vital components of the pathway. Although campafungin was limited to fungistatic activity at high concentrations, it was also found to compromise hyphal transition at concentrations as low as 10 μg/mL.
Fermentation of the fungus Codinaea simplex yielded the structurally unique sesterterpene natural product YW3699 (38), which was found to inhibit the biosynthesis of yeast glycosylphosphatidylinositol (GPI) at a MIC of 3.5 μM.90 Modification of proteins with the phosphoglyceride GPI is necessary for proper transportation to the endoplasmic reticulum, and complications in fungal cell wall synthesis arise in its absence.91 Although YW3699 was moderately active against yeast, the related terpene YW3548 (39) was found to have significantly more potent activity with a MIC of 3.4 nM. In order to better understand the mechanisms by which YW3548 (also known as M743) inhibits GPI synthesis, genomic profiling was done using the CaFT approach.90 The resulting CaFT profile revealed that M743 prevents GPI synthesis by inhibiting ethanolaminephosphate transferase activity, stalling the biosynthetic pathway on a mannose precursor. Additionally, the ester moiety of M743 was found to be particularly unstable in a murine candidiasis model and thus, semisynthetically exchanged for a carbamate linkage, giving M720 (40), which was significantly more stable in vivo and retained its target and efficacy based on confirmatory CaFT profiling.
The cyclic lipodepsipeptide phomafungin (41) was isolated from a plant pathogenic Phoma species and passaged through the CaFT.92 Observed hypersensitivities suggested phomafungin was targeting sphingolipid biosynthesis. However, comparison of the phomafungin CaFT profile against the known natural product sphingolipid inhibitors rustmicin (42), myriocin (43), and aureobasidin A (44) implied a novel mechanism of inhibition. Further investigation and development of phomafungin as an antifungal was halted by the observation of mammalian toxicity in a murine candidiasis model. Nevertheless, the CaFT has enabled identification of several antifungal natural products with novel or underrepresented biological targets. Widespread incorporation of the CaFT into antifungal drug discovery efforts is likely to significantly streamline the ongoing search for new options against resistant pathogens.
6. Biosynthetic Gene Cluster Mining
Although microbial genomes encode an enormous number of proteins and metabolites, only a fraction of these compounds are expressed in any given environment. This limitation on isolable natural products has also significantly contributed to unproductive re-isolation and the current stagnation in natural product drug discovery.93 Recently, genome mining and other genetic technologies have been increasingly leveraged to identify biosynthetic gene clusters within microbial genomes to elucidate previously undiscovered natural products and, potentially, natural analogs thereof (Figure 6).94,95
Figure 6.
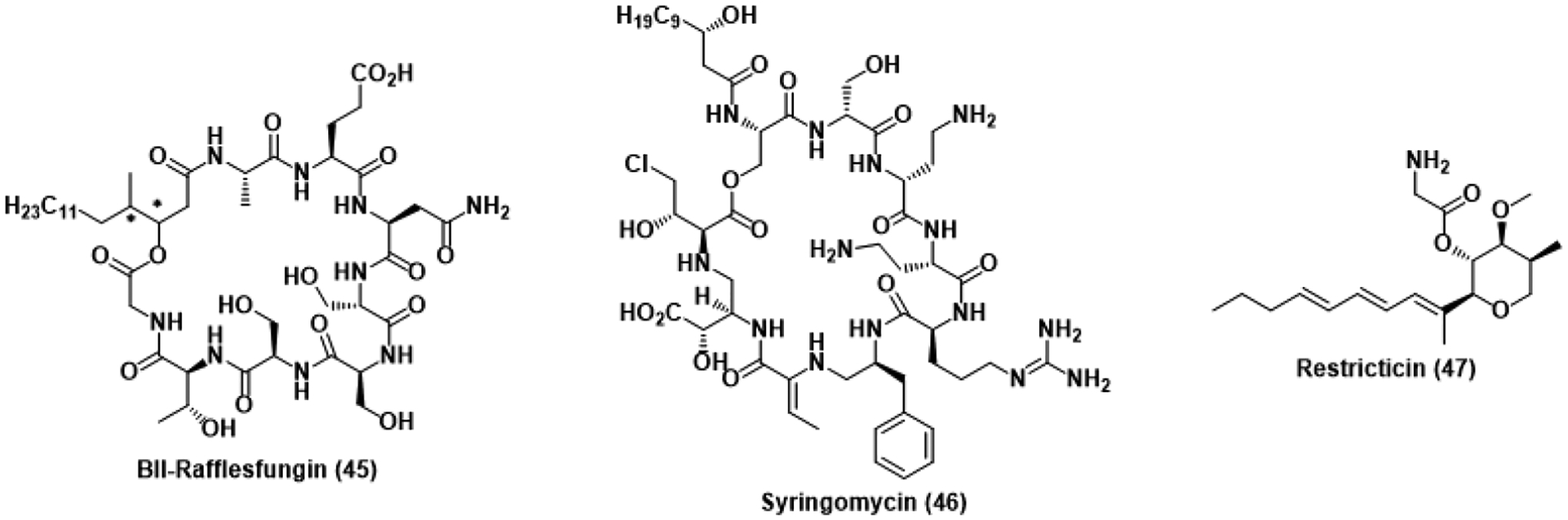
Antifungal natural products studied using genomics-based approaches. * represents an undefined stereocenter.
To decipher the biosynthetic origins of the aforementioned natural product phomafungin, genome mining of an antifungal-producing Phoma species revealed the production of BII-rafflesfungin (45), a structurally similar cyclic lipodepsipeptide.96 BII-Rafflesfungin was found to inhibit the growth of both A. fumigatus and azole-resistant C. albicans strains at MICs of 1.2 and 2.4 μM, respectively. Although a putative mechanism of action has yet to be determined for phomafungin and BII-rafflesfungin, they are likely to act similar to other antifungal cyclic depsipeptides. Syringomycin (46) binds to fungal membranes, inhibiting growth, while aureobasidin inhibits inositol phosphorylceramide, compromising sphingolipid biosynthesis.97,98 Although a distinct mechanism of action has yet to be validated for both phomafungin and BII-rafflesfungin, the ability of genome mining to reveal previously undiscovered natural products with such antifungal potency makes the technique formidable in drug discovery.
This methodology has also recently been employed to study the biosynthesis of restricticin (47) and related molecules.99 Produced by Penicillium restrictum, restricticin is thought to inhibit CYP51 through coordination of the terminal amine to heme, rationalizing the observed similarities in bioactivity and antifungal spectrum with ketoconazole.100 Production of an antifungal CYP51 inhibitor by a fungus dependent on CYP51 suggests the presence of an innate resistance mechanism. The restricticin biosynthetic gene cluster was found to be proximal to self-resistance genes encoding additional CYP51. Searching for similar gene clusters outside of P. restrictum found several fungal species also harbored the necessary genetic information to produce restricticin, previously unreported in these species. Application of this self-resistance-based approach to genome mining stands to surpass the limitations of traditional isolation and identify conserved biosynthetic clusters for novel antifungal natural products.
7. Synthetic Efforts Towards Novel Antifungals
The promising antifungal activity and structural complexity of many of the aforementioned natural products have inspired several total syntheses. Limited by the titer concentrations of these compounds in fermentation extracts, synthetic access to these natural products facilitates SAR studies and analog design that may otherwise be significantly more challenging or impossible through semisynthetic methodologies.
Work towards the construction of natural HA and its synthetic stereoisomers was completed by Westwood et al. in 2015 (Figure 7A).101 Starting from ethyl pyruvate, condensation of the chiral sulfinamide 48 facilitated enantioselective aldol addition to ethyl dimethylpyruvate, yielding the key lactone intermediate 49 after lactonization and deprotection.102 Treatment of this lactone with silver trifluoroacetate and thioester 50 gave HA following basic hydrolysis. Inverting the chirality of the sulfinamide reagent used afforded access to the other pair of diastereomeric lactones. Investigation of the activity of the four stereoisomers prepared against the fungal plant pathogens Sclerotium rolfsii and Pythium ultimum revealed that the synthetic analogs were more potent than natural harzianic acid yet remain to be evaluated against clinically relevant pathogens like C. auris or A. fumigatus.
Figure 7.
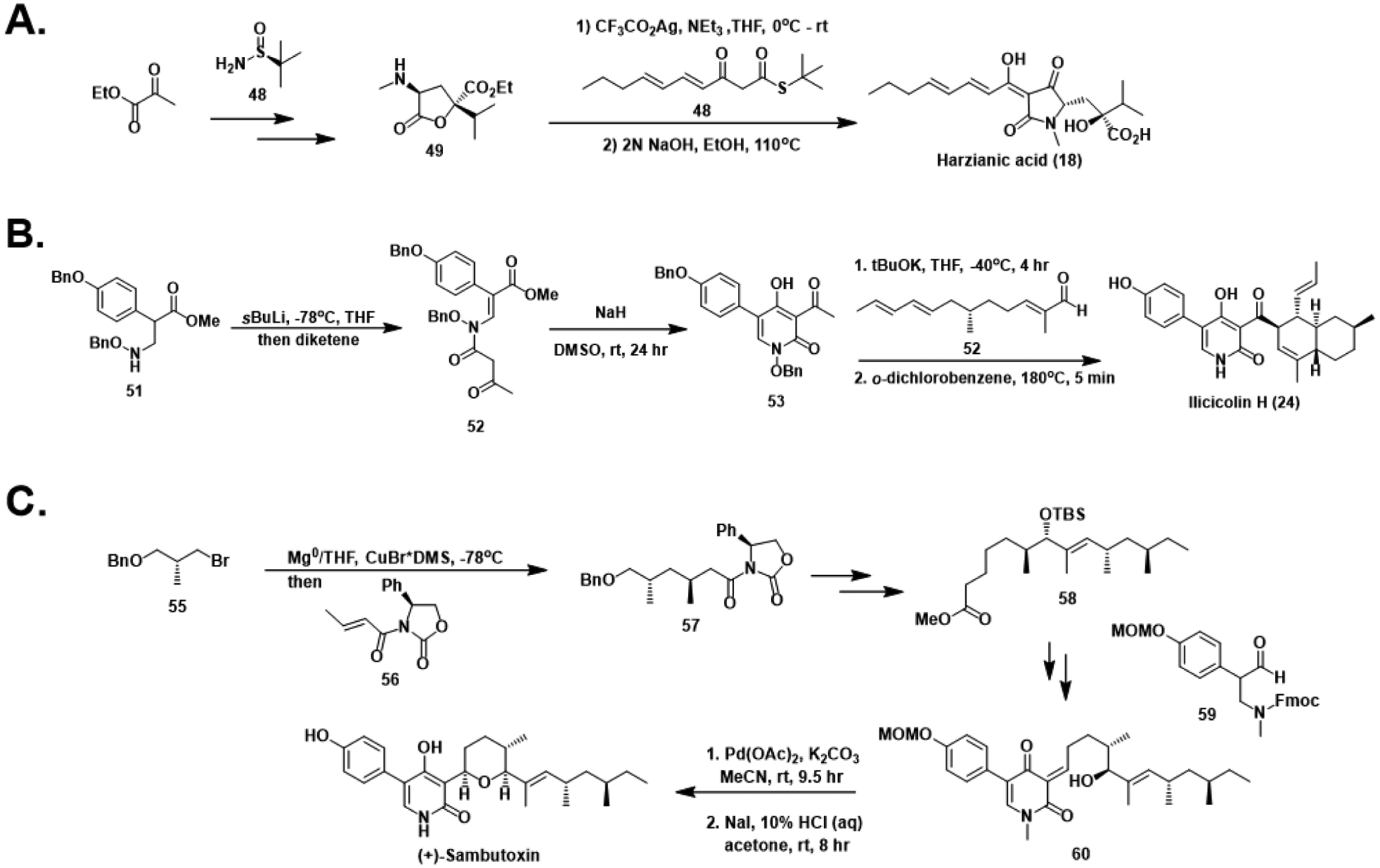
Total syntheses of harzianic acid (A), (±)-ilicicolin H (B), and (+)-sambutoxin (C).
Although the aforementioned SAR studies on ilicicolin H were recently conducted using material fermented from Gliocladium roseum, early synthetic efforts towards 4-hydroxy-2-pyridones resulted in a racemic synthesis as early as 1985 (Figure 7B).103 The methodology for the construction of the pyridone core was reused from previous works targeting the structurally similar natural product tenellin.104 Addition of aryl ester 51 to diketene generated acetoacetamide 52, primed for intramolecular cyclization to the acylated pyridone 53 in basic conditions. Aldol condensation-dehydration with unsaturated aldehyde 54 followed by Diels-Alder cycloaddition established the eastern decalin scaffold and ultimately gave (±)-ilicicolin H after deprotection.
The landmark total synthesis of (+)-sambutoxin by Williams and Turske elucidated the previously unassigned stereochemical configuration of the structure (Figure 7C).105 Addition of the organocuprate generated from chiral bromide 55 to Michael acceptor 56 gave 57, establishing the desired anti-1,3-dimethyl configuration. Conversion of 57 to methyl ester 58 over several steps enabled Claisen condensation with aryl aldehyde 59, yielding the 5-aryl-4-hydroxy-2-pyridone 60 after re-oxidation and deprotection at the nitrogen. Subsequent deprotection of the alcohol facilitated a 6-exo-trig cyclization to construct the pyran. Deprotection of the phenol gave the final compound, which was determined to be the antipode of the naturally occurring (−)-sambutoxin based on the observed optical rotation relative to the natural isolate.
Although semisynthetic efforts towards TET analogs for anticancer evaluation have previously been reported in the literature, not much attention has been placed on TET itself.106,107 A recently published racemic synthesis of tetrandrine described a synthetic strategy allowing for rapid construction of the macrocyclic skeleton (Figure 8A).108 Beginning from tetrasubstituted arene 61, a TFA-catalyzed Pictet-Spengler condensation with enol ether 62 gave isoquinoline 63. A subsequent Ullman coupling with N-acylated arene 64 followed by reductive debenzylation afforded 65. An additional iteration of the Pictet-Spengler / Ullman coupling sequence using enol ether 66 established the complete macrocycle 67, and LAH-mediated reduction of the carbamates yielded a mixture of the four possible stereoisomers of tetrandrine (68).
Figure 8.
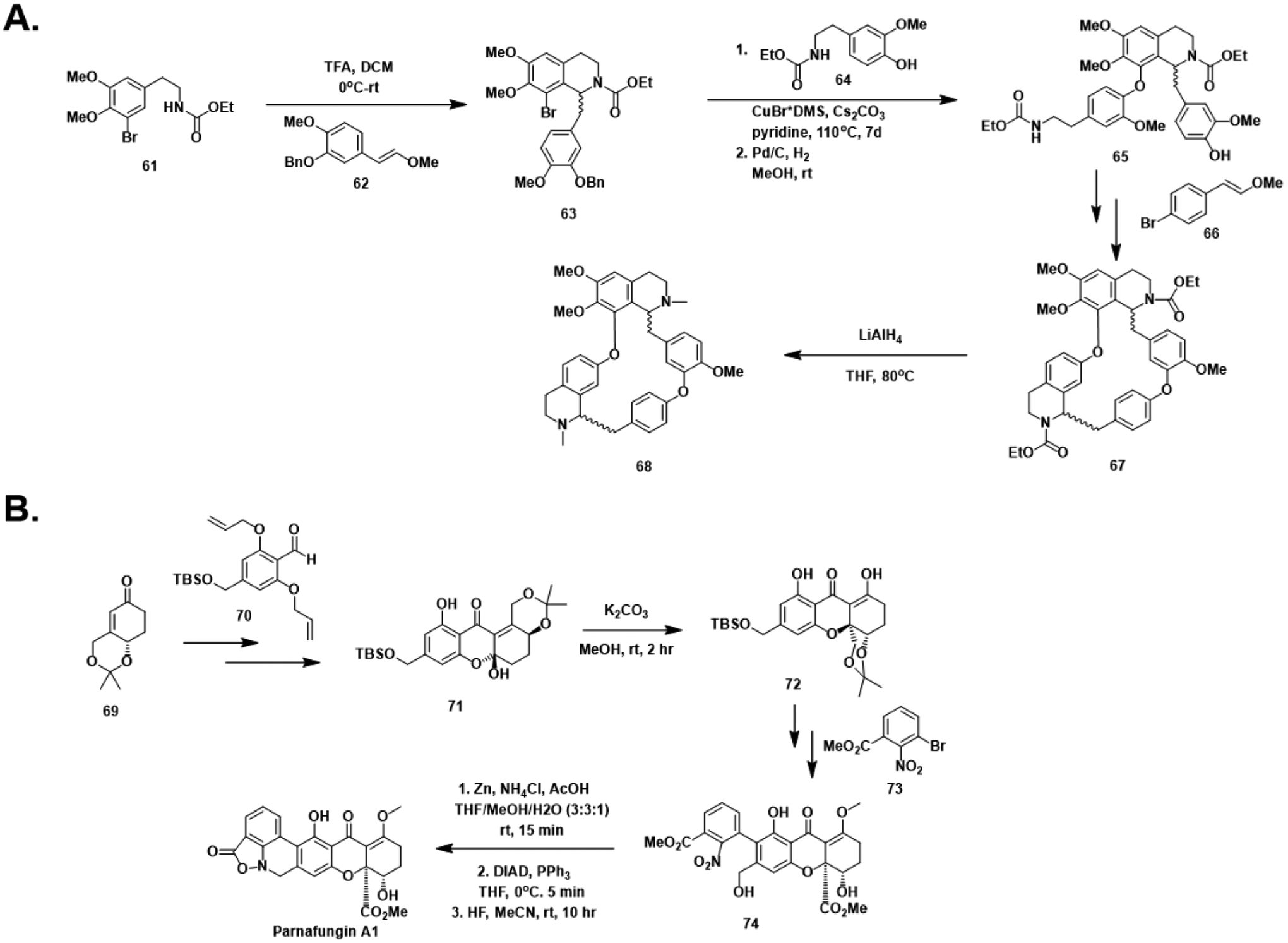
Total synthesis efforts towards the tetrandrines (A) and parnafungins (B).
Synthetic routes towards the syn-epimer of parnafungin A have recently been reported (Figure 8B).109 Starting from known α,β-unsaturated cyclohexanone 69, addition into aryl aldehyde 70 and re-oxidation which, after allyl deprotection, revealed the phenolic oxygen necessary for formation of the desired acetal 72. However, only addition into the ketone was observed, generating the undesired hemiacetal 71. Fortunately, treatment of 66 with base promoted ring-opening and Michael addition to afford acetal 72. An iodination-borylation sequence facilitated aryl coupling with nitroarene 73 giving intermediate 74. Reductive cyclization conditions constructed the unusual benzisoxazolinone moiety following conditions previously reported by Wierenga et al.110 A Mitsunobu reaction promoted the final cyclization and gave parnafungin A1 (70) after deprotection. Dissolution of synthetic parnafungin A1 in DMSO led to conversion of material to the three other possible stereoisomers in ratios closely matching those of the original isolation literature.
Unfortunately, the synthetic derivation of these antifungal natural products has not borne much fruit in regard to the advancement of our biological understanding. Ideally, initial triumphs in total synthesis ought to be succeeded by the development of methodology to facilitate analog design. The construction of analog libraries for these putative antifungals stands to greatly contribute to our knowledge of their biological targets and potency. Synthetic transformation or simplification of molecular scaffolds may serendipitously yield more potent analogs with less structural complexity. Guided by initial target identification results elucidated by technologies like the CaFT, rational analog development for SAR studies is a powerful methodology for natureinspired drug discovery efforts.
8. Conclusion and Future Outlook
The development of resistance against our limited antifungal options in several high-priority pathogens has significantly accelerated in recent years. Unfortunately, antifungal drug discovery efforts have not sped up to match this demand due to the inherent challenges associated with antifungal drug design. Traditional natural product isolation methodologies, such as bioactivity-guided fractionation, often lead to unproductive re-isolation of known compounds. The widespread adoption of newer approaches like the Candida albicans fitness test and genome mining for underexplored biosynthetic gene clusters forego these limitations and instead celebrate efficient identification of new natural products with novel and potent bioactivities. Several literature accounts of such compounds have been presented in hopes to inspire similar creativity in the next generation of antifungals. Pivoting modern antifungal development away from ergosterol dependence and towards novel, underexplored targets is our best hope in this crisis.
Antifungal drug discovery would also greatly benefit in more thorough biological evaluation of compounds of interest. Biological studies of antifungals typically involve testing against the flagship fungal pathogens Candida albicans, Aspergillus fumigatus, and Cryptococcus neoformans. Although Candida auris has been considered a high-priority fungal pathogen for several years, it has still not yet been incorporated into initial inhibition reports, and activity reported against standard C. albicans strains cannot be expected to match that of C. auris on account of their vastly different antifungal resistance profiles. Beyond the void in biological investigation of C. auris, additional synthetic work towards the development of novel C. auris inhibitors is imperative. At the rate that C. auris infection rates have dramatically increased in recent years, it is very much so in our best interest to invest in solutions before a calamitous outbreak takes place.
Acknowledgements
We would like to thank Adrian Demeritte for feedback and helpful suggestions. This work was funded by the National Institute of General Medical Sciences (R35 GM119426) and the National Science Foundation (CHE2003692).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Dedicated to Prof. John Wood for his steadfast dedication to the art of natural product total synthesis and the mentorship of the next generation organic chemists
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- (1).Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations.
- (2).Centers for Disease Control, U. Antibiotic Resistance Threats in the United States, 2019. 10.15620/cdc:82532. [DOI]
- (3).Bongomin F; Gago S; Oladele RO; Denning DW Global and Multi-National Prevalence of Fungal Diseases—Estimate Precision. Journal of Fungi 2017, Vol. 3, Page 57 2017, 3 (4), 57. 10.3390/JOF3040057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bush K; Bradford PA Epidemiology of β-Lactamase-Producing Pathogens. Clinical Microbiology Reviews. American Society for Microbiology April 1, 2020. 10.1128/CMR.00047-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Finkel JS; Mitchell AP Genetic Control of Candida Albicans Biofilm Development. Nature Reviews Microbiology. February 2011, pp 109–118. 10.1038/nrmicro2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).WHO fungal priority pathogens list to guide research, development and public health action. Geneva: World Health Organization; 2022. License: CC BY-NC-SA 3.0 IGO. [Google Scholar]
- (7).Mullins J; Seaton A Fungal Spores in Lung and Sputum. Clin Allergy 1978, 8 (5), 525–533. 10.1111/J.1365-2222.1978.TB01506.X. [DOI] [PubMed] [Google Scholar]
- (8).Lee SH; Lee BJ; Jung DY; Kim JH; Sohn DS; Shin JW; Kim JY; Park IW; Choi BW Clinical Manifestations and Treatment Outcomes of Pulmonary Aspergilloma. Korean J Intern Med 2004, 19 (1), 38. 10.3904/KJIM.2004.19.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lin SJ; Schranz J; Teutsch SM Aspergillosis Case-Fatality Rate: Systematic Review of the Literature. Clin Infect Dis 2001, 32 (3), 358–366. 10.1086/318483. [DOI] [PubMed] [Google Scholar]
- (10).Mousavi B; Hedayati MT; Hedayati N; Ilkit M; Syedmousavi S Aspergillus Species in Indoor Environments and Their Possible Occupational and Public Health Hazards. Curr Med Mycol 2016, 2 (1), 36–42. 10.18869/acadpub.cmm.2.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jeanvoine A; Rocchi S; Bellanger AP; Reboux G; Millon L Azole-Resistant Aspergillus Fumigatus: A Global Phenomenon Originating in the Environment? Medecine et Maladies Infectieuses. Elsevier Masson SAS August 1, 2020, pp 389–395. 10.1016/j.medmal.2019.07.014. [DOI] [PubMed] [Google Scholar]
- (12).Antibiotic Resistance Threats in the United States, 2019; Atlanta, Georgia, 2019. 10.15620/cdc:82532. [DOI] [Google Scholar]
- (13).Forsberg K; Woodworth K; Walters M; Berkow EL; Jackson B; Chiller T; Vallabhaneni S Candida Auris: The Recent Emergence of a Multidrug-Resistant Fungal Pathogen. Med Mycol 2019, 57 (1), 1–12. 10.1093/mmy/myy054. [DOI] [PubMed] [Google Scholar]
- (14).Alcazar-Fuoli L; Mellado E Ergosterol Biosynthesis in Aspergillus Fumigatus: Its Relevance as an Antifungal Target and Role in Antifungal Drug Resistance. Front Microbiol 2012, 3 (JAN), 439. 10.3389/FMICB.2012.00439/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Dismukes WE Introduction to Antifungal Drugs. Clinical Infectious Diseases 2000, 30 (4), 653–657. 10.1086/313748/2/30-4-653-TBL003.GIF. [DOI] [PubMed] [Google Scholar]
- (16).Baginski M; Czub J; Sternal K Interaction of Amphotericin B and Its Selected Derivatives with Membranes: Molecular Modeling Studies. Chemical Record 2006, 6 (6), 320–332. 10.1002/tcr.20096. [DOI] [PubMed] [Google Scholar]
- (17).Sanglard D; Ischer F; Parkinson T; Falconer D; Bille J Candida Albicans Mutations in the Ergosterol Biosynthetic Pathway and Resistance to Several Antifungal Agents. Antimicrob Agents Chemother 2003, 47 (8), 2404–2412. 10.1128/AAC.47.8.2404-2412.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ostrowsky B; Greenko J; Adams E; Quinn M; O’Brien B; Chaturvedi V; Berkow E; Vallabhaneni S; Forsberg K; Chaturvedi S; Lutterloh E; Blog D; Bucher C; Denis RJ; Erazo R; Fernandez R; Southwick K; Zhu YC Candida Auris Isolates Resistant to Three Classes of Antifungal Medications — New York, 2019. MMWR Morb Mortal Wkly Rep 2020, 69 (1), 6–9. 10.15585/MMWR.MM6901A2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Blum G; Perkhofer S; Haas H; Schrettl M; Würzner R; Dierich MP; Lass-Flörl C Potential Basis for Amphotericin B Resistance in Aspergillus Terreus. Antimicrob Agents Chemother 2008, 52 (4), 1553–1555. 10.1128/AAC.01280-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Laniado-Laborín R; Cabrales-Vargas MN Amphotericin B: Side Effects and Toxicity. Revista Iberoamericana de Micologia. Asociacion Espanola de Micologia; 2009, pp 223–227. 10.1016/j.riam.2009.06.003. [DOI] [PubMed] [Google Scholar]
- (21).Dupont B Overview of the Lipid Formulations of Amphotericin B. J Antimicrob Chemother 2002, 49 Suppl 1 (SUPL. S1), 31–36. 10.1093/JAC/49.SUPPL_1.31. [DOI] [PubMed] [Google Scholar]
- (22).Heeres J; Backx LJJ; Mostmans JH; van Cutsem J Antimycotic Imidazoles. Part 4. Synthesis and Antifungal Activity of Ketoconazole, a New Potent Orally Active Broad-Spectrum Antifungal Agent. J Med Chem 1979, 22 (8), 1003–1005. 10.1021/JM00194A023. [DOI] [PubMed] [Google Scholar]
- (23).Maertens JA History of the Development of Azole Derivatives. Clinical Microbiology and Infection 2004, 10 (SUPPL. 1), 1–10. 10.1111/J.14709465.2004.00841.X. [DOI] [PubMed] [Google Scholar]
- (24).Sanati H; Belanger P; Fratti R; Ghannoum M A New Triazole, Voriconazole (UK-109,496), Blocks Sterol Biosynthesis in Candida Albicans and Candida Krusei. Antimicrob Agents Chemother 1997, 41 (11), 2492. 10.1128/AAC.41.11.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mann PA; Parmegiani RM; Wei SQ; Mendrick CA; Li X; Loebenberg D; DiDomenico B; Hare RS; Walker SS; McNicholas PM Mutations in Aspergillus Fumigatus Resulting in Reduced Susceptibility to Posaconazole Appear to Be Restricted to a Single Amino Acid in the Cytochrome P450 14α-Demethylase. Antimicrob Agents Chemother 2003, 47 (2), 577–581. 10.1128/AAC.47.2.577-581.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hagiwara D; Watanabe A; Kamei K; Goldman GH Epidemiological and Genomic Landscape of Azole Resistance Mechanisms in Aspergillus Fungi. Frontiers in Microbiology. Frontiers Media S.A. September 21, 2016. 10.3389/fmicb.2016.01382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Balkovec JM; Hughes DL; Masurekar PS; Sable CA; Schwartz RE; Singh SB Discovery and Development of First in Class Antifungal Caspofungin (CANCIDAS®) - A Case Study. Natural Product Reports. January 2014, pp 15–34. 10.1039/c3np70070d. [DOI] [PubMed] [Google Scholar]
- (28).Denning DW Echinocandins: A New Class of Antifungal. Journal of Antimicrobial Chemotherapy 2002, 49 (6), 889–891. 10.1093/jac/dkf045. [DOI] [PubMed] [Google Scholar]
- (29).Logviniuk D; Jaber QZ; Dobrovetsky R; Kozer N; Ksiezopolska E; Gabaldón T; Carmeli S; Fridman M Benzylic Dehydroxylation of Echinocandin Antifungal Drugs Restores Efficacy against Resistance Conferred by Mutated Glucan Synthase. J Am Chem Soc 2022, 144 (13), 5965–5975. 10.1021/jacs.2c00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ademe M; Girma F Candida Auris: From Multidrug Resistance to Pan-Resistant Strains. Infection and Drug Resistance. Dove Medical Press Ltd. 2020, pp 1287–1294. 10.2147/IDR.S249864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pfaller MA; Carvalhaes C; Messer SA; Rhomberg PR; Castanheira M Activity of a Long-Acting Echinocandin, Rezafungin, and Comparator Antifungal Agents Tested against Contemporary Invasive Fungal Isolates (SENTRY Program, 2016 to 2018). Antimicrob Agents Chemother 2020, 64 (4). 10.1128/AAC.00099-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Waldorf AR; Polak A Mechanisms of Action of 5-Fluorocytosine. Antimicrob Agents Chemother 1983, 23 (1), 79–85. 10.1128/AAC.23.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Delma FZ; Al-Hatmi AMS; Brüggemann RJM; Melchers WJG; de Hoog S; Verweij PE; Buil JB Molecular Mechanisms of 5-Fluorocytosine Resistance in Yeasts and Filamentous Fungi. Journal of Fungi. MDPI November 1, 2021. 10.3390/jof7110909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Merry M; Boulware DR Cryptococcal Meningitis Treatment Strategies Affected by the Explosive Cost of Flucytosine in the United States: A Cost-Effectiveness Analysis. Clinical Infectious Diseases 2016, 62 (12), 1564–1568. 10.1093/cid/ciw151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sobel JD; Nyirjesy P Oteseconazole: An Advance in Treatment of Recurrent Vulvovaginal Candidiasis. Future Microbiology. Future Medicine Ltd. December 1, 2021, pp 1453–1461. 10.2217/fmb-2021-0173. [DOI] [PubMed] [Google Scholar]
- (36).Warrilow AGS; Hull CM; Parker JE; Garvey EP; Hoekstra WJ; Moore WR; Schotzinger RJ; Kelly DE; Kellya SL The Clinical Candidate VT-1161 Is a Highly Potent Inhibitor of Candida Albicans CYP51 but Fails to Bind the Human Enzyme. Antimicrob Agents Chemother 2014, 58 (12), 7121–7127. 10.1128/AAC.03707-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Cass L; Murray A; Davis A; Woodward K; Albayaty M; Ito K; Strong P; Ayrton J; Brindley C; Prosser J; Murray J; French E; Haywood P; Wallis C; Rapeport G Safety and Nonclinical and Clinical Pharmacokinetics of PC945, a Novel Inhaled Triazole Antifungal Agent. Pharmacol Res Perspect 2021, 9 (1). 10.1002/prp2.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Jallow S; Govender NP Ibrexafungerp: A First-in-Class Oral Triterpenoid Glucan Synthase Inhibitor. Journal of Fungi. MDPI AG March 1, 2021, pp 1–19. 10.3390/jof7030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gamal A; Chu S; McCormick TS; Borroto-Esoda K; Angulo D; Ghannoum MA Ibrexafungerp, a Novel Oral Triterpenoid Antifungal in Development: Overview of Antifungal Activity Against Candida Glabrata. Frontiers in Cellular and Infection Microbiology. Frontiers Media S.A. March 11, 2021. 10.3389/fcimb.2021.642358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Miyazaki M; Horii T; Hata K; Watanabe NA; Nakamoto K; Tanaka K; Shirotori; Murai N; Inoue S; Matsukura M; Abe S; Yoshimatsu K; Asada M In Vitro Activity of E1210, a Novel Antifungal, against Clinically Important Yeasts and Molds. Antimicrob Agents Chemother 2011, 55 (10), 4652–4658. 10.1128/AAC.00291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Watanabe NA; Miyazaki M; Horii T; Sagane K; Tsukahara K; Hata K E1210, a New Broad-Spectrum Antifungal, Suppresses Candida Albicans Hyphal Growth through Inhibition of Glycosylphosphatidylinositol Biosynthesis. Antimicrob Agents Chemother 2012, 56 (2), 960–971. 10.1128/AAC.00731-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kapoor M; Moloney M; Soltow QA; Pillar CM; Shaw KJ Evaluation of Resistance Development to the GWT1 Inhibitor Manogepix (APX001A) in Candida Species. Antimicrob Agents Chemother 2020, 64 (1). 10.1128/AAC.01387-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Locke JB; Almaguer AL; Zuill DE; Bartizal K Characterization of In Vitro Resistance Development to the Novel Echinocandin CD101 in Candida Species. 2016. 10.1128/AAC.00620-16. [DOI] [PMC free article] [PubMed]
- (44).Oliver JD; Sibley GEM; Beckmann N; Dobb KS; Slater MJ; McEntee L; du Pré S; Livermore J; Bromley MJ; Wiederhold NP; Hope WW; Kennedy AJ; Law D; Birch M F901318 Represents a Novel Class of Antifungal Drug That Inhibits Dihydroorotate Dehydrogenase. Proc Natl Acad Sci U S A 2016, 113 (45), 12809–12814. 10.1073/pnas.1608304113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Buil JB; Oliver JD; Law D; Baltussen T; Zoll J; J Hokken MW; Tehupeiory-Kooreman M; G Melchers WJ; Birch M; Verweij PE Resistance Profiling of Aspergillus Fumigatus to Olorofim Indicates Absence of Intrinsic Resistance and Unveils the Molecular Mechanisms of Acquired Olorofim Resistance. 2022. 10.1080/22221751.2022.2034485. [DOI] [PMC free article] [PubMed]
- (46).Shibata T; Takahashi T; Yamada E; Kimura A; Nishikawa H; Hayakawa H; Nomura N; Mitsuyama J T-2307 Causes Collapse of Mitochondrial Membrane Potential in Yeast. Antimicrob Agents Chemother 2012, 56 (11), 5892–5897. 10.1128/AAC.05954-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mitsuyama J; Nomura N; Hashimoto K; Yamada E; Nishikawa H; Kaeriyama M; Kimura A; Todo Y; Narita H In Vitro and in Vivo Antifungal Activities of T-2307, a Novel Arylamidine. Antimicrob Agents Chemother 2008, 52 (4), 1318–1324. 10.1128/AAC.01159-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Vila T; Montelongo-Jauregui D; Ahmed H; Puthran T; Sultan AS; Jabra-Rizk MA Comparative Evaluations of the Pathogenesis of Candida Auris Phenotypes and Candida Albicans Using Clinically Relevant Murine Models of Infections. mSphere 2020, 5 (4). 10.1128/MSPHERE.00760-20/FORMAT/EPUB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pue N; Guddat L Acetohydroxyacid Synthase: A Target for Antimicrobial Drug Discovery. Curr Pharm Des 2014, 20 (5), 740–753. 10.2174/13816128113199990009. [DOI] [PubMed] [Google Scholar]
- (50).Garcia MD; Nouwens A; Lonhienne TG; Guddat LW Comprehensive Understanding of Acetohydroxyacid Synthase Inhibition by Different Herbicide Families. Proc Natl Acad Sci U S A 2017, 114 (7), E1091–E1100. 10.1073/PNAS.1616142114/-/DCSUPPLEMENTAL/PNAS.201616142SI.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Garcia MD; Chua SMH; Low YS; Lee YT; Agnew-Francis K; Wang JG; Nouwens A; Lonhienne T; Williams CM; Fraser JA; Guddat LW Commercial AHAS-Inhibiting Herbicides Are Promising Drug Leads for the Treatment of Human Fungal Pathogenic Infections. Proc Natl Acad Sci U S A 2018, 115 (41), E9649–E9658. 10.1073/PNAS.1809422115/-/DCSUPPLEMENTAL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Agnew-Francis KA; Tang Y; Lin X; Low YS; Wun SJ; Kuo A; Elias SMAI; Lonhienne T; Condon ND; Pimentel BNAS; Vergani CE; Smith MT; Fraser JA; Williams CM; Guddat LW Herbicides That Target Acetohydroxyacid Synthase Are Potent Inhibitors of the Growth of Drug-Resistant Candida Auris. ACS Infect Dis 2020, 6 (11), 2901–2912. 10.1021/ACSINFECDIS.0C00229/SUPPL_FILE/ID0C00229_SI_001.PDF. [DOI] [PubMed] [Google Scholar]
- (53).Xie L; Zang X; Cheng W; Zhang Z; Zhou J; Chen M; Tang Y Harzianic Acid from Trichoderma Afroharzianum Is a Natural Product Inhibitor of Acetohydroxyacid Synthase. J Am Chem Soc 2021, 143 (25), 9575–9584. 10.1021/JACS.1C03988/SUPPL_FILE/JA1C03988_SI_001.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Nile AH; Tripathi A; Yuan P; Mousley CJ; Suresh S; Wallace IM; Shah SD; Pohlhaus DT; Temple B; Nislow C; Giaever G; Tropsha A; Davis RW; St Onge RP; Bankaitis VA; Chem N; Author B PITPs as Targets for Selectively Interfering With Phosphoinositide Signaling in Cells HHS Public Access Author Manuscript. Nat Chem Biol 2014, 10 (1), 76–84. 10.1038/nchembio.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Filipuzzi I; Cotesta S; Perruccio F; Knapp B; Fu Y; Studer C; Pries V; Riedl R; Helliwell SB; Petrovic KT; Rao Movva N; Sanglard D; Tao J; Hoepfner D High-Resolution Genetics Identifies the Lipid Transfer Protein Sec14p as Target for Antifungal Ergolines. 2016. 10.1371/journal.pgen.1006374. [DOI] [PMC free article] [PubMed]
- (56).Pries V; Nöcker C; Khan D; Johnen P; Hong Z; Tripathi A; Keller A-L; Fitz M; Perruccio F; Filipuzzi I; Thavam S; Aust T; Riedl R; Ziegler S; Bono F; Schaaf G; Bankaitis V; Waldmann H; Hoepfner D Target Identification and Mechanism of Action of Picolinamide and Benzamide Chemotypes with Antifungal Properties. 10.1016/j.chembiol.2017.12.007. [DOI] [PMC free article] [PubMed]
- (57).Zhang F; Zhao M; Braun DR; Ericksen SS; Piotrowski JS; Nelson J; Peng J; Ananiev GE; Chanana S; Barns K; Fossen J; Sanchez H; Chevrette MG; Guzei A; Zhao C; Guo L; Tang W; Currie CR; Rajski SR; Audhya A; Andes DR; Bugni TS A Marine Microbiome Antifungal Targets Urgent-Threat Drug-Resistant Fungi. https://www.science.org. [DOI] [PMC free article] [PubMed]
- (58).Bunyapaiboonsri T; Yoiprommarat S; Suriyachadkun C; Supothina S; Chanthaket R; Chutrakul C; Vichai V Actinomadurone, a Polycyclic Tetrahydroxanthone from Actinomadura Sp. BCC 35430. 2017. 10.1016/j.tetlet.2017.07.008. [DOI]
- (59).Zhao M; Zhang F; Zarnowski R; Barns K; Jones R; Fossen J; Sanchez H; Rajski R; Audhya A; Bugni TS; Andes DR Turbinmicin Inhibits Candida Biofilm Growth by Disrupting Fungal Vesicle-Mediated Trafficking. J Clin Invest 2021, 131 (5). 10.1172/JCI145123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Zhang Y; Pike A Pyridones in Drug Discovery: Recent Advances. 2021. 10.1016/j.bmcl.2021.127849. [DOI] [PubMed]
- (61).Hayakawa S; Mlnato H; Katagiri K The Ilicicolins, Antibiotics from Cylindrocladium Ilicicola. J Antibiot (Tokyo) 1971, 24 (9), 653–654. 10.7164/ANTIBIOTICS.24.653. [DOI] [PubMed] [Google Scholar]
- (62).Kawai K; Suzuki T; Kitagawa A; Kim JC; Lee YW A Novel Respiratory Chain Inhibitor, Sambutoxin from Fusarium Sambucinum. Cereal Research Communications 1997 25:3 1997, 25 (3), 325–326. 10.1007/BF03543718. [DOI] [Google Scholar]
- (63).Gutierrez-Cirlos EB; Merbitz-Zahradnik T; Trumpower BL Inhibition of the Yeast Cytochrome Bc1 Complex by Ilicicolin H, a Novel Inhibitor That Acts at the Qn Site of the Bc1 Complex. Journal of Biological Chemistry 2004, 279 (10), 8708–8714. 10.1074/jbc.M311805200. [DOI] [PubMed] [Google Scholar]
- (64).Singh SB; Liu W; Li X; Chen T; Shafiee A; Card D; Abruzzo G; Flattery A; Gill C; Thompson JR; Rosenbach M; Dreikorn S; Hornak V; Meinz M; Kurtz M; Kelly R; Onishi JC Antifungal Spectrum, In Vivo Efficacy, and Structure–Activity of Ilicicolin H. ACS Med Chem Lett 2012, 3 (10), 814. 10.1021/ML300173E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Hector RF; Pappagianis D Inhibition of Chitin Synthesis in the Cell Wall of Coccidioides Immitis by Polyoxin D. J Bacteriol 1983, 154 (1), 488. 10.1128/JB.154.1.488-498.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Lenardon MD; Munro CA; Gow NAR Chitin Synthesis and Fungal Pathogenesis. Curr Opin Microbiol 2010, 13 (4), 416. 10.1016/J.MIB.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Li RK; Rinaldi MG In Vitro Antifungal Activity of Nikkomycin Z in Combination with Fluconazole or Itraconazole. Antimicrob Agents Chemother 1999, 43 (6), 1401. 10.1128/AAC.43.6.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Bentz ML; Nunnally N; Lockhart SR; Sexton DJ; Berkow EL Antifungal Activity of Nikkomycin Z against Candida Auris. Journal of Antimicrobial Chemotherapy 2021, 76 (6), 1495–1497. 10.1093/JAC/DKAB052. [DOI] [PubMed] [Google Scholar]
- (69).Dickey SW; Cheung GYC; Otto M Different Drugs for Bad Bugs: Antivirulence Strategies in the Age of Antibiotic Resistance. Nature Reviews Drug Discovery 2017 16:7 2017, 16 (7), 457–471. 10.1038/nrd.2017.23. [DOI] [PubMed] [Google Scholar]
- (70).Liu T; Liu X; Li W; Liu T; Liu X; Li W Tetrandrine, a Chinese Plant-Derived Alkaloid, Is a Potential Candidate for Cancer Chemotherapy. Oncotarget 2016, 7 (26), 40800–40815. 10.18632/ONCOTARGET.8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Zhang H; Wang K; Zhang G; Ho HI; Gao A Synergistic Anti-Candidal Activity of Tetrandrine on Ketoconazole: An Experimental Study. Planta Med 2010, 76 (01), 53–61. 10.1055/S-0029-1185973. [DOI] [PubMed] [Google Scholar]
- (72).Shi J; Li S; Gao A; Zhu K; Zhang H Tetrandrine Enhances the Antifungal Activity of Fluconazole in a Murine Model of Disseminated Candidiasis. Phytomedicine 2018, 46, 21–31. 10.1016/J.PHYMED.2018.06.003. [DOI] [PubMed] [Google Scholar]
- (73).Zhang H; Gao A; Li F; Zhang G; Ho HI; Liao W Mechanism of Action of Tetrandrine, a Natural Inhibitor of Candida Albicans Drug EOEux Pumps. YAKUGAKU ZASSHI 2009, 129 (5), 623–630. [DOI] [PubMed] [Google Scholar]
- (74).Zhao L-X; Li D-D; Hu D-D; Hu G-H; Yan L; Wang Y; Jiang Y-Y Effect of Tetrandrine against Candida Albicans Biofilms. PLoS One 2013, 8 (11), 79671. 10.1371/journal.pone.0079671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Sakaue Y; Domon H; Oda M; Takenaka S; Kubo M; Fukuyama Y; Okiji T; Terao Y Anti-Biofilm and Bactericidal Effects of Magnolia Bark-Derived Magnolol and Honokiol on Streptococcus Mutans. Microbiol Immunol 2016, 60 (1), 10–16. 10.1111/1348-0421.12343. [DOI] [PubMed] [Google Scholar]
- (76).Ho Bang K; Kwan Kim Y; Sun Min B; Kyun Na M; Ha Rhee Y; Pill lee J; Bae H Antifungal Activity of Magnolol and Honokiol. Korea Food & Drug Administration 2000, 23 (1), 122–704. [DOI] [PubMed] [Google Scholar]
- (77).Sun L; Liao K; Wang D Effects of Magnolol and Honokiol on Adhesion, Yeast-Hyphal Transition, and Formation of Biofilm by Candida Albicans. 2015. 10.1371/journal.pone.0117695. [DOI] [PMC free article] [PubMed]
- (78).Solinski AE; Ochoa C; Lee YE; Paniak T; Kozlowski MC; Wuest WM Honokiol-Inspired Analogs as Inhibitors of Oral Bacteria. ACS Infect Dis 2018, 4 (2), 118–122. 10.1021/ACSINFECDIS.7B00178/SUPPL_FILE/ID7B00178_SI_001.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Ochoa C; Solinski AE; Nowlan M; Dekarske MM; Wuest WM; Kozlowski MC A Bisphenolic Honokiol Analog Outcompetes Oral Antimicrobial Agent Cetylpyridinium Chloride via a Membrane-Associated Mechanism. ACS Infect Dis 2020, 6 (1), 74–79. 10.1021/ACSINFECDIS.9B00190/SUPPL_FILE/ID9B00190_SI_001.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Li D-D; Zhao L-X; Mylonakis E; Hu G-H; Zou Y; Huang T-K; Yan L; Wang Y; Jiang Y-Y In Vitro and In Vivo Activities of Pterostilbene against Candida Albicans Biofilms. 2014. 10.1128/AAC.01583-13. [DOI] [PMC free article] [PubMed]
- (81).Hu DD; Zhang RL; Zou Y; Zhong H; Zhang ES; Luo X; Wang Y; Jiang YY The Structure-Activity Relationship of Pterostilbene Against Candida Albicans Biofilms. Molecules : A Journal of Synthetic Chemistry and Natural Product Chemistry 2017, 22 (3). 10.3390/MOLECULES22030360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Piotrowski JS; Okada H; Lu F; Li SC; Hinchman L; Ranjan A; Smith DL; Higbee AJ; Ulbrich A; Coon JJ; Deshpande R; Bukhman Y. v; Mcilwain S; Ong IM; Myers CL; Boone C; Landick R; Ralph J; Kabbage M; Ohya Y Plant-Derived Antifungal Agent Poacic Acid Targets β-1,3-Glucan. 10.1073/pnas.1410400112. [DOI] [PMC free article] [PubMed]
- (83).Lee KK; Kubo K; Abdelaziz JA; Cunningham I; de Silva Dantas A; Chen X; Okada H; Ohya Y; Gow NAR Yeast Species-Specific, Differential Inhibition of β-1,3-Glucan Synthesis by Poacic Acid and Caspofungin. The Cell Surface 2018, 3, 12–25. 10.1016/J.TCSW.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Giaever G; Shoemaker DD; Jones TW; Liang H; Winzeler EA; Astromoff A; Davis RW Genomic Profiling of Drug Sensitivities via Induced Haploinsufficiency. Nature Genetics 1999 21:3 1999, 21 (3), 278–283. 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- (85).Baetz K; McHardy L; Gable K; Tarling T; Rebérioux D; Bryan J; Andersen RJ; Dunn T; Hieter P; Roberge M Yeast Genome-Wide Drug-Induced Haploinsufficiency Screen to Determine Drug Mode of Action. Proc Natl Acad Sci U S A 2004, 101 (13), 4525. 10.1073/PNAS.0307122101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Xu D; Jiang B; Ketela T; Lemieux S; Veillette K; Martel N; Davison J; Sillaots S; Trosok S; Bachewich C; Bussey H; Youngman P; Roemer T Genome-Wide Fitness Test and Mechanism-of-Action Studies of Inhibitory Compounds in Candida Albicans. 10.1371/journal.ppat.0030092. [DOI] [PMC free article] [PubMed]
- (87).Parish CA; Smith SK; Calati K; Zink D; Wilson K; Roemer T; Jiang B; Xu D; Bills G; Platas G; Pelá Ez F; Díez MT; Tsou N; Mckeown AE; Ball RG; Powles MA; Yeung L; Liberator P; Harris G Isolation and Structure Elucidation of Parnafungins, Antifungal Natural Products That Inhibit MRNA Polyadenylation. 10.1021/ja711209p. [DOI] [PubMed]
- (88).Jiang B; Xu D; Allocco J; Parish C; Davison J; Veillette K; Sillaots S; Hu W; Rodriguez-Suarez R; Trosok S; Zhang L; Li Y; Rahkhoodaee F; Ransom T; Martel N; Wang H; Gauvin D; Wiltsie J; Wisniewski D; Salowe S; Kahn JN; Hsu MJ; Giacobbe R; Abruzzo G; Flattery A; Gill C; Youngman P; Wilson K; Bills G; Platas G; Pelaez F; Diez MT; Kauffman S; Becker J; Harris G; Liberator P; Roemer T PAP Inhibitor with In Vivo Efficacy Identified by Candida Albicans Genetic Profiling of Natural Products. Chem Biol 2008, 15 (4), 363–374. 10.1016/J.CHEMBIOL.2008.02.016. [DOI] [PubMed] [Google Scholar]
- (89).Perlatti B; Harris G; Nichols CB; Ekanayake DI; Alspaugh JA; Gloer JB; Bills GF Campafungins: Inhibitors of Candida Albicans and Cryptococcus Neoformans Hyphal Growth. Cite This: J. Nat. Prod 2718, 2020. 10.1021/acs.jnatprod.0c00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Sütterlin C; Horvath A; Gerold P; Schwarz RT; Wang Y; Dreyfuss M; Riezman H Identification of a Species-Specific Inhibitor of Glycosylphosphatidylinositol Synthesis. EMBO J 1997, 16 (21), 6374–6383. 10.1093/EMBOJ/16.21.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Mann PA; McLellan CA; Koseoglu S; Si Q; Kuzmin E; Flattery A; Harris G; Sher X; Murgolo N; Wang H; Devito K; de Pedro N; Genilloud O; Kahn JN; Jiang B; Costanzo M; Boone C; Garlisi CG; Lindquist S; Roemer T Chemical Genomics-Based Antifungal Drug Discovery: Targeting Glycosylphosphatidylinositol (GPI) Precursor Biosynthesis. ACS Infect Dis 2015, 1 (1), 59–72. 10.1021/ID5000212/ASSET/IMAGES/LARGE/ID-2014000212_0007.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Herath K; Harris G; Jayasuriya H; Zink D; Smith S; Vicente F; Bills G; Collado J; González A; Jiang B; Kahn JN; Galuska S; Giacobbe R; Abruzzo G; Hickey E; Liberator P; Xu D; Roemer T; Singh SB Isolation, Structure and Biological Activity of Phomafungin, a Cyclic Lipodepsipeptide from a Widespread Tropical Phoma Sp. Bioorg Med Chem 2009, 17 (3), 1361–1369. 10.1016/J.BMC.2008.12.009. [DOI] [PubMed] [Google Scholar]
- (93).Roemer T; Xu D; Singh SB; Parish CA; Harris G; Wang H; Davies JE; Bills GF Confronting the Challenges of Natural Product-Based Antifungal Discovery. Chem Biol 2011, 18 (2), 148–164. 10.1016/J.CHEMBIOL.2011.01.009. [DOI] [PubMed] [Google Scholar]
- (94).Scherlach K; Hertweck C Mining and Unearthing Hidden Biosynthetic Potential. Nature Communications 2021 12:1 2021, 12 (1), 1–12. 10.1038/s41467-021-24133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Baltz RH Genome Mining for Drug Discovery: Progress at the Front End. J Ind Microbiol Biotechnol 2021, 48 (9–10), 44. 10.1093/JIMB/KUAB044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Sinha S; Nge CE; Leong CY; Ng V; Crasta S; Alfatah M; Goh F; Low KN; Zhang H; Arumugam P; Lezhava A; Chen SL; Kanagasundaram Y; Ng SB; Eisenhaber F; Eisenhaber B Genomics-Driven Discovery of a Biosynthetic Gene Cluster Required for the Synthesis of BII-Rafflesfungin from the Fungus Phoma Sp. F3723. BMC Genomics 2019 20:1 2019, 20 (1), 1–18. 10.1186/S12864-019-5762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Takemoto JY; Zhang L; Taguchi N; Tachikawa T; Miyakawa T Mechanism of Action of the Phytotoxin Syringomycin: A Resistant Mutant of Saccharomyces Cerevisiae Reveals an Involvement of Ca2+ Transport. J Gen Microbiol 1991, 137 (3), 653–659. 10.1099/00221287-137-3-653/CITE/REFWORKS. [DOI] [Google Scholar]
- (98).Aeed PA; Young CL; Nagiec MM; Elhammer ÅP Inhibition of Inositol Phosphorylceramide Synthase by the Cyclic Peptide Aureobasidin A. Antimicrob Agents Chemother 2009, 53 (2), 496–504. 10.1128/AAC.00633-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Liu N; Abramyan ED; Cheng W; Perlatti B; Harvey CJB; Bills GF; Tang Y Targeted Genome Mining Reveals the Biosynthetic Gene Clusters of Natural Product CYP51 Inhibitors. J Am Chem Soc 2021, 143 (16), 6043–6047. 10.1021/JACS.1C01516/SUPPL_FILE/JA1C01516_SI_001.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Schwartz RE; Dufresne C; Flor JE; Kempf AJ; Wilson KE; Lam T; Onishi J; Milligan J; Fromtling RA; Abruzzo GK; Jenkins R; Glazomitsky K; Bills G; Zitano L; Mochales Del Val S; Omstead MN RESTRICTION, A NOVEL GLYCINE-CONTAINING ANTIFUNGAL AGENT. J Antibiot (Tokyo) 44 (5). [DOI] [PubMed] [Google Scholar]
- (101).Healy AR; Vinale F; Lorito M; Westwood NJ Total Synthesis and Biological Evaluation of the Tetramic Acid Based Natural Product Harzianic Acid and Its Stereoisomers. 2015. 10.1021/ol503717r. [DOI] [PMC free article] [PubMed]
- (102).Healy AR; Izumikawa M; Slawin AMZ; Shin-Ya K; Westwood NJ Stereochemical Assignment of the Protein-Protein Interaction Inhibitor JBIR-22 by Total Synthesis. Angewandte Chemie - International Edition 2015, 54 (13), 4046–4050. 10.1002/ANIE.201411141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Williams DR; Bremmer ML; Brown DL; D’Antuono J Total Synthesis of (±)-Ilicicolin H. Journal of Organic Chemistry 1985, 50 (15), 2807–2809. 10.1021/JO00215A053/ASSET/JO00215A053.FP.PNG_V03. [DOI] [Google Scholar]
- (104).Williams DR; Sit S-Y Synthesis of Racemic Tenellin. J. Org. Chem 1982, 47, 2846–2851. [Google Scholar]
- (105).Williams DR; Turske RA Construction of 4-Hydroxy-2-Pyridinones. Total Synthesis of (+)-Sambutoxin. Org Lett 2001, 3 (16), 2619–2619. 10.1021/OL0163070. [DOI] [PubMed] [Google Scholar]
- (106).Hu SC; Yang J; Chen C; Song JR; Pan WD Design, Synthesis of Novel Tetrandrine-14-l-Amino Acid and Tetrandrine-14-l-Amino Acid-Urea Derivatives as Potential Anti-Cancer Agents. Molecules 2020, 25 (7). 10.3390/MOLECULES25071738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Lan J; Wang N; Huang L; Liu Y; Ma X; Lou H; Chen C; Feng Y; Pan W Design and Synthesis of Novel Tetrandrine Derivatives as Potential Anti-Tumor Agents against Human Hepatocellular Carcinoma. Eur J Med Chem 2017, 127, 554–566. 10.1016/J.EJMECH.2017.01.008. [DOI] [PubMed] [Google Scholar]
- (108).Schütz R; Meixner M; Antes I; Bracher F A Modular Approach to the Bisbenzylisoquinoline Alkaloids Tetrandrine and Isotetrandrine. Org Biomol Chem 2020, 18 (16), 3047–3068. 10.1039/D0OB00078G. [DOI] [PubMed] [Google Scholar]
- (109).Sun J; Gu W; Yang H; Tang W Enantioselective Total Synthesis of Parnafungin A1 and 10a-Epi-Hirtusneanine †. 2021. 10.1039/d1sc02919c. [DOI] [PMC free article] [PubMed]
- (110).Wierenga W; Harrison AW; Evans BR; Chidester CG Antibacterial Benzisoxazolones. an Unusual Rearrangement Product From O-Nitrostyrene Oxide En Route to the Photolabile Carbonyl Protecting Group (O-Nitrophenyl)Ethylene Glycol. Journal of Organic Chemistry 1984, 49 (3), 438–442. 10.1021/JO00177A010/SUPPL_FILE/JO00177A010_SI_001.PDF. [DOI] [Google Scholar]