

Summary
Cell type-specific transcriptional differences between brain tissues from donors with Alzheimer’s disease (AD) and unaffected controls have been well documented, but few studies have rigorously interrogated the regulatory mechanisms responsible for these alterations. We performed single nucleus multiomics (snRNA-seq plus snATAC-seq) on 105,332 nuclei isolated from cortical tissues from 7 AD and 8 unaffected donors to identify candidate cis-regulatory elements (CREs) involved in AD-associated transcriptional changes. We detected 319,861 significant correlations, or links, between gene expression and cell type-specific transposase accessible regions enriched for active CREs. Among these, 40,831 were unique to AD tissues. Validation experiments confirmed the activity of many regions, including several candidate regulators of APP expression. We identified ZEB1 and MAFB as candidate transcription factors playing important roles in AD-specific gene regulation in neurons and microglia, respectively. Microglia links were globally enriched for heritability of AD risk and previously identified active regulatory regions.
Keywords: Alzheimer’s, multiomics, microglia, neuron, single cell, transcription factor, cis-regulatory element, APP, ZEB1, MAFB
Graphical abstract
Highlights
-
•
Single nucleus multiomics to identify cell type-specific regulatory elements in Alzheimer's
-
•
Identified 40,831 candidate regulatory elements specific to Alzheimer’s
-
•
Confirmed enhancer-like activity of candidate regulators of APP expression
-
•
Nominated ZEB1 and MAFB as candidate Alzheimer’s-specific trans-regulators
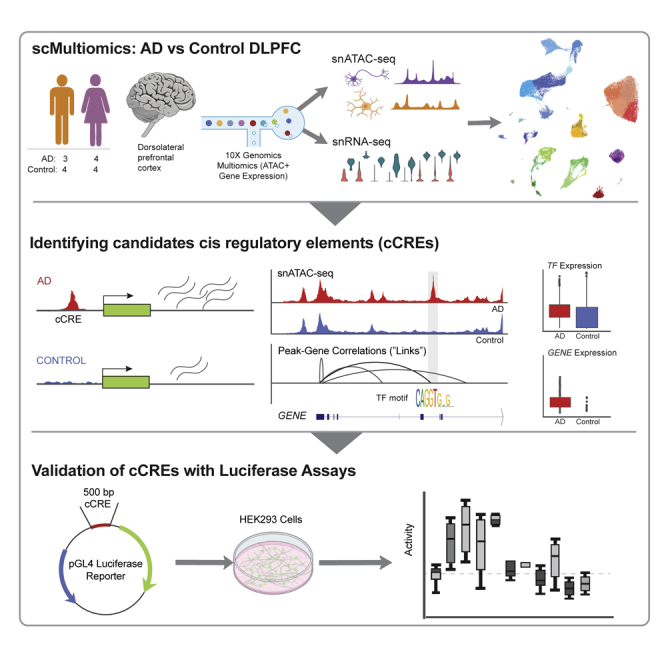
Linking regulatory elements to target genes in specific cell types can reveal regulatory mechanisms disrupted in disease. Here, Anderson and Rogers et al. identify Alzheimer’s-specific elements in cortical tissues and candidate transcription factors regulating their activity. Enhancer-like activity was demonstrated for twelve elements including several regulators of APP.
Introduction
Identification of genetic contributors to Alzheimer’s disease (AD) has provided critical insights into potential disease mechanisms. Rare, protein-altering variants in APP, PSEN1, or PSEN2 cause early-onset, autosomal-dominant AD,1 and genome-wide association studies (GWASs) have identified common variants for late-onset AD that increase disease risk to varying degrees.2,3,4,5,6 However, the majority of GWAS variants are in non-coding regions of the genome and many presumably affect gene regulation. Linkage disequilibrium makes identification of the causal variant difficult, particularly for putative regulatory regions where conservation and deleteriousness estimates may not be as informative. Associating common and rare regulatory variants with affected genes is also challenging.7,8,9 In addition, disease-associated variants often function only in specific cell types, further complicating interpretation of their effects.10,11 Thus, determining which genes are contributing to disease requires assessments in specific cell types.
Recent advances in single cell technologies have allowed profiling of gene expression12,13,14,15,16,17,18 and chromatin accessibility,10 either separately or in parallel from the same samples.19,20 Although studies have examined the cell type-specific transcriptional and epigenetic differences between tissues from brain donors with AD and unaffected controls, few have rigorously interrogated the regulatory mechanisms responsible for these alterations.11,21 Integrating single nucleus RNA-seq (snRNA-seq) and single nucleus assay for transposase-accessible chromatin sequencing (snATAC-seq) data allow the identification of potential cis-regulatory elements (CREs) by correlating chromatin accessibility with nearby gene expression. Here, we simultaneously measure both gene expression and chromatin accessibility in the same nuclei to identify cell type-specific regulatory regions and their target genes in dorsolateral prefrontal cortex (DLPFC) tissues from both AD and unaffected donors. In addition, we assessed regulatory mechanisms unique to nuclei from donors with AD.
Results
Cellular diversity within the human dorsolateral prefrontal cortex
We used the 10x Genomics Multiome technology to perform snATAC-seq and snRNA-seq on nuclei isolated from human postmortem DLPFC tissues from seven individuals diagnosed with AD (mean age 78 years, Braak stages 4–6) and eight sex-matched unaffected control donors (mean age 63 years) (Table S1; Figure 1A). This assay allows direct mapping of both gene expression and chromatin accessibility within the same nuclei without the need to computationally infer cell type identification during cross-modality integration. After removing low quality nuclei and doublets (STAR Methods), we retained a total of 105,332 nuclei with an average of 7,022 nuclei per donor (range 1,410–11,723). We detected a median of 2,659 genes and 11,647 ATAC fragments per cell. We performed normalization and dimensionality reduction for snRNA-seq and snATAC-seq data using Seurat22 and Signac,23 respectively. We used weighted nearest neighbor (WNN) analysis to determine a joint representation of expression and accessibility and identified 36 distinct clusters composed of eight major cell types and their associated subclusters (Figures 1B, S1A, and S1B). Consistent with previous snRNA-seq datasets,12,13,15,19 we identified all expected cell types in the brain with similar relative abundances across AD and control donors (Figures 1B, 1C, and S1C). Pericytes and endothelial cell clusters contained <500 nuclei and were excluded from further analyses. Cluster annotations were supported by both gene expression and promoter accessibility of well-established cell type marker genes (Figures 1D and 1E). There were strong correlations in global gene expression across donors within each cell type and between excitatory/inhibitory neurons (Figure 1F). The only cell type to display variable correlation values across donors was microglia, a cell type known to be dysregulated in AD. In addition, we identified distinct subpopulations within each major cell type with the exception of oligodendrocyte precursor cells (OPCs), pericytes, and endothelial cells (Figure S2). These subtype annotations were consistent with those from prior studies,14,22,24 and distributions were similar across AD and control donors, with the exception of microglia subpopulations and two inhibitory neuron subtypes (Inh_1 and Inh_2; Figure S2).
Figure 1.

Cellular diversity of DLPFC from Alzheimer’s disease and unaffected donors revealed by single cell multiomics
(A) Experimental design.
(B) Uniform manifold approximation and projection (UMAP) visualization of the weighted nearest neighbor (WNN) clustering of single nuclei colored by cell type and cluster assignment.
(C) Total number of cells in each subcluster and the proportion of cells from each individual (red, AD donors; blue, unaffected donors) in the subcluster.
(D) Row-normalized gene expression of scREAD cell type markers.
(E) Chromatin accessibility across cell types for cell type marker genes (indicated below).
(F) Correlation of pseudo-bulked cell type-specific expression profiles between individuals. Colors indicating cell type are consistent throughout the figure.
Cell type-specific transcriptome changes in Alzheimer’s DLPFC
Within each cell type, we identified differentially expressed genes (DEGs) between AD and control tissues. A total of 911 DEGs were identified after considering sex and age as covariates (Figure 2A; Table S3). Although significant sex-specific differences in gene expression between AD and controls have been shown previously,24 because of our smaller sample size we did not detect such changes. Although the majority of DEGs were cell type specific, 141 were identified across multiple cell types (Figure 2B). Of these DEGs, 62 were also identified as differentially up- or downregulated in the same cell type in both Mathys et al.12 and Morabito et al.,19 including PTPRG (up in AD Mic) and GRIA2 (down in AD Ast) (Figures 2C and 2D). PTPRG encodes a protein tyrosine phosphatase associated with inflammation25 and AD disease risk (rs7609954).26 GRIA2 encodes the glutamate receptor 2 (GluR2) subunit that reduces calcium channel permeability and may protect against excitotoxicity.27 A recent meta-analysis28 of three snRNA studies12,13,18 found 41 DEGs that were shared across these three studies, only 7 of which were discordant in direction. We also identified 17 of them, including GRM3 (down in AD Ast) and SLC38A2 (up in AD Oli) involved in glutamate signaling and RNF149 (up in AD Mic) encoding an E3 ubiquitin ligase. Most DEGs were upregulated in AD and were enriched for cell type-specific Gene Ontology (GO) terms related to PDGFR beta signaling in microglia, apoptosis in astrocytes, and Notch and BDNF signaling in oligodendrocytes (Figure 2E; Table S4). In contrast, most DEGs downregulated in AD were in neurons and showed enrichment in GO terms related to regulation of tau activity (HSP90AB1, HSP90AA1) and calcium channel activity (CALM2, CALM3) (Figure 2E; Table S4). Example DEGs include MDGA2 whose overexpression results in reduced excitatory synapse density,29 GPR158 which is an osteocalcin receptor associated with cognition30 and SAMD4A whose expression is correlated with AD-associated neuropathology and cognitive status (https://agora.adknowledgeportal.org). SAMD4A, along with several other DEGs (NRXN1, LUZP2, RBMS3, ARHGAP15, ARHGAP24, LRP1B, and ATP1B3), was also newly identified as AD associated in a recent genome-wide association neural network analysis of AD family history in the UK Biobank.31
Figure 2.

Cell type-specific transcriptome dysregulation in Alzheimer’s DLPFC
(A) MAST log2(fold change [FC]) of all up- and downregulated genes in AD for each cell type.
(B) Number of shared DEGs between cell types in both directions (upper triangle, upregulated in AD; lower triangle, downregulated in AD).
(C) Normalized expression of the top DEG in the indicated cell types (log2[FC] > 1).
(D) Overlap of DEGs with agreement on cell type and direction with Morabito et al.19 and Mathys et al.12
(E) Heatmap showing the odds ratio of the top enrichR GO terms for up and downregulated DEGs within each cell type (∗adjusted p < 0.01).
Identification of candidate CREs
Previous single cell studies have characterized altered gene expression in AD brain tissues and cell types,12,13,14,17,19 and we observed signals consistent with those studies. Additionally, we sought to leverage single cell multiomics data to identify cell type- and disease-specific CREs and their target genes by correlating gene expression with chromatin accessibility across all nuclei in the dataset. The Cell Ranger ARC (v2.0) analysis pipeline produces these correlations as “feature linkages” (STAR Methods). A feature linkage, or link, is defined as a significant correlation between accessibility of an ATAC peak and the expression of a gene32 (Figure 3A). We restricted this correlation analysis to consider only peaks within 500 kb of each transcription start site (TSS), a generous search space, as previous studies have found the majority of enhancers are within 50–100 kb of their target genes,33 but chosen to allow the identification of longer range interactions that can have high impact.34 We first took the union of ATAC peaks identified in each cell type and retained only those present in ≥2% of cells in at least one cell type for a total of 189,925 peaks. Nearly half of all peaks overlapped H3K27ac (46%) from the corresponding cell type, and 43% overlapped ENCODE (Encyclopedia of DNA Elements)35 distal enhancer-like sequences. Using this peak set, links were then calculated independently using gene expression data from either AD or control nuclei allowing classification of links as AD-specific, control-specific, or common (STAR Methods). We consider the linked peaks to be candidate CREs. Given the gene expression changes observed in AD, we hypothesized that there would be differential use of CREs between AD and control samples that would be identified in this analysis as AD- or control-specific links. Cell type specificity of each link was determined by the cell type(s) in which the ATAC peak was identified. A total of 319,905 peak-gene links were found involving 15,471 linked genes and 126,213 linked peaks with a minimum absolute correlation value of 0.2 (Figure 3A; Table S5). The median distance between the linked peak and the TSS of the linked gene was 201,506 bp, and there was an inverse relationship between absolute correlation value and distance to TSS (Figure S3A).
Figure 3.

Identification of candidate CREs
(A) Schematic of gene-peak association (top). Heatmap of row-normalized accessibility and expression for the most correlated peak-gene link for each gene (bottom). Columns are pseudo-bulked on cell type and disease status.
(B) Distributions of the number of linked peaks per gene (left) and the number of linked genes per peak (right) for AD (red) and control (blue) samples.
(C) Total number of links per cell type for AD and control. Cell type of the link is assigned by the cell type in which the peak was called.
(D) ENCODE annotation of linked peaks by cell type.
(E) Shared (across cell types) and cell type-specific linked peaks that overlap H3K27ac of the corresponding cell type.
(F) Normalized expression of KANSL1 from AD and control samples in each cell type. Expression is significantly different in AD versus control for all cell types.
(G) Linkage plot for all links to KANSL1. Top: coverage plot of pseudo-bulked accessibility in excitatory neurons separated by status (red, AD; blue, control). Bottom: significant AD and control peak-gene links. Arc height represents strength and direction of correlation. Arc color indicates if the link was identified in both AD and control (common, gray) or control donors only (blue). A linked peak overlapping a single SNP is highlighted in gray.
For most genes, we identified a similar number of links in both AD (median = 12) and control samples (median = 13). However, we found 1,294 genes that had only AD links and 1,596 that had only control links (Figure S3B). We observed no significant bias when comparing the number of links identified in either AD or control for a given gene (Figure S3B). Most genes were linked to multiple peaks across all cell types, with a median of 14 linked peaks per gene. However, 16% of genes were linked with 40 or more peaks (Figure 3B), and these genes were significantly longer and more highly expressed than those with fewer links (Figure S3C). This finding is likely due to links being called for peaks within the gene body of longer genes as excluding these peaks abolishes the difference in number of links (p = 0.12, t test). Across the entire dataset, 17.8% of linked peaks are present in the promoter or gene body of the target gene. Although positively correlated links in gene bodies may often be merely a consequence of target gene expression, we retained these peaks in our analyses as enhancers are often located within the introns of their target genes.
ATAC peaks often interacted with more than one gene. Nearly 70% (126,213) of the ATAC peaks analyzed were linked to a gene with an average of two genes linked to each peak and a range of 1–21 linked genes (Figure 3B). Almost a third (30.24%) of the links were unique to a single cell type, while 21% were common across all cell types (Figure 3C). We identified 40,831 AD-specific links and 74,028 control-specific links with the majority of links identified in both (205,046). We performed permutation analyses and determined that this fraction of AD- and control-specific links (0.36 of total links) was greater than expected by chance (p = 0.027, Z test; Figure S3D). Target genes of cell type-specific links identified in both AD and control samples were enriched in expected pathways (Figure S3E). To evaluate whether linked peaks associate with regulatory regions, we evaluated their overlap with a curated set of candidate CREs identified by ENCODE.35 We found that linked peaks were significantly enriched for proximal (odds ratio [OR] = 1.24, p = 2.4 × 10−15) and distal (OR = 1.06, p = 3.06 × 10−9) enhancer-like sequences and the proportion of overlap was similar across cell types (Figure 3D). As these annotations were not generated in our particular cell types and tissue, we also intersected these linked peaks with regions of H3K27ac previously identified within cell types isolated from prefrontal cortex tissues.11,36 We found that on average, 57.5% of linked peaks overlap a H3K27ac peak from the corresponding cell type and this increases to 79% for cell type-specific linked peaks (Figure 3E). The majority (76.11%) of linked peaks were positively correlated with gene expression, as is expected given the association between open chromatin and transcriptional activation,37,38,39,40,41 though negative correlations may be indicative of repressor binding.37,40
We then asked if the differential gene expression we observed could be mediated by candidate CREs. Nearly all (94%) the DEGs identified between AD and control nuclei had a linked peak in the same cell type where the gene was differentially expressed and 85% of these linked peaks overlapped H3K27ac in the same cell type. In addition, we observed that some links to differentially expressed genes were uniquely identified in either AD or control datasets. For genes upregulated in AD, 72% of their positively correlated links were AD specific, while for downregulated genes 62% were control specific. For example, BIN1 expression is significantly reduced in AD microglia compared with controls and this reduced expression hampers proinflammatory microglial responses.42 We identified six control-specific links and no AD-specific links for BIN1 in microglia. One of these control-specific links was validated as a microglia-specific BIN1 enhancer in Nott et al.11 and harbors an AD-associated SNP (rs733839). Together, these findings suggest that this CRE may no longer be used in AD microglia, leading to lower BIN1 expression, though it remains possible that these observations could also result from less sensitive detection with lower expression. Another example is KANSL1, located in the MAPT locus, that was downregulated in AD in all cell types (Figure 3F). Twenty-eight of the 37 KANSL1 linked peaks are unique to control samples and the rest are common to both AD and control (Figure 3G). One of these linked peaks found in the promoter overlaps an expression quantitative trait locus (eQTL)43 (rs2532404) associated with progressive supranuclear palsy44 and was recently shown via CRISPRi to regulate KANSL1 expression in induced pluripotent stem cell (iPSC)-derived neurons.21 These results are consistent with the hypothesis that other AD- and control-specific regions contribute to the differential expression observed in AD.
Identification of AD-specific peak-gene-TF trios
We next sought to identify transcription factors (TFs) that could be driving the positive correlations between accessible peaks and target genes. To further investigate the regulatory roles of links, we identified peak-gene-TF “trios” in which (1) there was a correlation between the linked peak and linked gene, (2) the accessibility of a linked peak harboring a specific TF motif was correlated with the expression of that TF, and (3) the expression of the TF was correlated with the expression of the linked gene (Figure 4A; STAR Methods). This approach is conceptually similar to a recently described method called TRIPOD that employs nonparametric models to identify peak-gene-TF associations and from which we adopted the term “trio.”45 To identify trios, we performed these additional correlation analyses (linked peak:TF expression and TF expression:target gene expression) separately using either AD or control datasets to enable identification of TFs whose activities may be associated with disease. AD- or control-specific trios were those uniquely identified in the AD or control dataset, respectively. Cell type specificity was defined on the basis of the cell type in which the linked peak was identified. We restricted these analyses to links with a correlation value >0.3 that were within 100 kb of the linked gene’s TSS (115,107) and identified 60,120 peak-gene-TF trios involving 17,149 unique peaks and 437 TFs (Table S6). Fewer than 20% of the peaks in these trios are found in promoters, with the majority present in intronic regions (Figure 4B). Trio peaks were enriched for ENCODE distal (OR = 1.26, p = 2.2 × 10−16) and proximal (OR = 1.12, p = 5.9 × 10−7) enhancer-like sequences. There was a median of 37 trios per TF. The TF MEF2C was the most common trio participant, appearing in nearly 5% of all trios. Although MEF2C was expressed in most cell types, expression of target genes in MEF2C trios were distinct between cell types (Figures 4C and 4D). In microglia, target genes were enriched in GO terms related to pattern recognition receptor (PRR) signaling, while in neurons they were enriched in synaptic transmission in neurons (Figure 4E; Table S7). PRRs consist of several receptor families including Toll-like receptors that are critical for microglial activation.46 Of the 911 DEGs, we found that 601 participated in a trio in the same cell type in which differential expression was observed. Ninety-six DEGs were in AD-specific trios (including MAPT, APOE, and BIN1), while 89 were in control-specific trios (PADI2/PAD2, PDE10A, and SNAP25). The presence of SNAP-25 in cerebral spinal fluid is associated with amyloid pathology,47 and decreased expression in AD brain tissues has been observed,48 consistent with the decreased expression we find in AD astrocytes and neurons.
Figure 4.

Identification of AD-specific TF regulatory networks
(A) Strategy for defining peak-gene-TF trios. A linked peak containing a TF motif must be correlated with that TF and the expression of that TF must be correlated with the linked gene for that peak to be considered a part of trio.
(B) Genome annotations for location of linked peaks within trios.
(C) Heatmap of column-normalized expression of genes within MEF2C trios by cell type.
(D) Normalized expression of MEF2C by cell type.
(E) Top enriched GO terms for genes within MEF2C trios from excitatory and inhibitory neurons (green, “neuron”) and microglia (purple, “microglia”).
(F) Heatmap of correlation values of AD and control-specific trios identified in microglia (left) and excitatory/inhibitory neurons (right) for TFs involved in at least 3 trios.
(G) Linkage plot for GABRA5. Top: coverage plot of pseudo-bulked accessibility in indicated cell types. Middle: coverage plot of ZEB1 ChIP-seq signal from NeuN+ nuclei isolated from DLPFC tissue from two unaffected donors (1238 and 1242). Bottom: significant peak-gene links; green indicates overlap with ZEB1 motif. Arc height represents strength and direction of correlation. Track of ZEB1 motifs (green) and H3K27ac peaks from neurons (black; Nott et al.11). Linked peak of interest is highlighted in gray.
(H) ZEB1 motif from JASPAR 2022 (top). Normalized expression of ZEB1 and GABRA5 in excitatory/inhibitory neurons and microglia.
Within this set of trios, there was a small subset that were specific to either AD or control groups (n = 2,718). Although many of these were specific to a single cell type, 55% were shared across two or more (Figure S4). All cell type-specific trios overlapped H3K27ac peaks from their respective cell types (Table S6). Within microglia trios, NR4A2 (Nurr1) was identified most frequently in control-specific trios (Figure 4F). NR4A2 can function as both an activator and repressor and has been shown to repress inflammatory responses in microglia through recruitment of the CoREST complex.49 Target genes in NR4A2 trios are enriched in neutrophil degranulation (OR = 9.01, q = 5.3 × 10−6) and include interleukin genes IL1A and IL1B, as well as TGFB1. Similarly, MAFB was involved in 24% of the AD-specific trios (Figure 4F) where it was linked to the microglial marker gene CX3CR1 and genes involved in microglial activation (TLR3, CD84, HAVCR2).50 In healthy microglia, MAFB inhibits inflammatory responses51 consistent with our finding that target genes in AD-specific trios were enriched for negative regulation of myeloid leukocyte mediated immunity (OR = 332, q = 0.0004).
Within neuron-specific trios, we identified KLF10 and ZEB1 most frequently in control- and AD-specific trios, respectively (Figure 4F). In neurons, we identified ZEB1 in half of all AD-specific trios with target genes involved in regulating ion channel signaling (ITPR1, CAMK2A, CACNB3, KCNH3, KCNQ5, KCNT1). We found that one ZEB1 target gene encoding a neuronal Ca2+ sensor, VSNL1, was downregulated in AD inhibitory neurons consistent with previous studies, and this reduced expression has been correlated with amyloid plaques and neurofibrillary tangles.52 ZEB1 was never found in control-specific trios. Given the frequency of ZEB1 participation in neuronal AD-specific trios, we performed ZEB1 chromatin immunoprecipitation sequencing (ChIP-seq) in NeuN+ nuclei isolated from two control donors (1238 and 1242). We found that 33% (55/167) of neuronal ZEB1 trios are bound by ZEB1, and 25 of these are AD-specific trios. The GABAA receptor ɑ5 subunit, encoded by GABRA5, is one gene that we find likely to be regulated by ZEB1 in AD (Figure 4G). α5 GABAA receptors are associated with learning and memory, consistent with highest expression of GABRA5 in hippocampal neurons and association of reduced expression with neurodevelopmental disorders.53 In our data, ZEB1 is expressed in both neurons and microglia; however, GABRA5 is primarily expressed in excitatory neurons (Figure 4G, right). In excitatory neurons, we identified a linked peak correlated with GABRA5 expression that was marked with H3K27ac and contained a ZEB1 motif. ChIP-seq data from two of our unaffected donors confirmed ZEB1 binding at this site providing additional evidence to suggest cis-regulatory activity of this region for GABRA5.
Genetic variation at candidate CREs
We performed stratified linkage disequilibrium score (sLDSC) regression54 to determine if our linked peaks were significantly enriched for SNPs associated with complex brain-related traits (Figure 5A; Table S8). Link categories are defined as “AD” or “control” if the links were only identified in the analysis of AD or control samples, respectively. “Common” links were identified in both analyses, and “all” is the union of all linked peaks. Although a peak with multiple links can be duplicated across categories, fewer than a third of peaks with AD-specific links also have control-specific links, emphasizing the specificity of these linked peaks. Within each link category for each cell type, the union of linked peaks was used for this analysis. Cell type was assigned on the basis of the cell type(s) in which the linked peak was identified. Consistent with previous studies,55,56 linked peaks identified in microglia were significantly enriched for heritability of AD across five different studies2,3,4,5,6; however, this was not true for those microglial linked peaks identified in control samples, suggesting that variants in AD-specific linked peaks could have a greater contribution to AD risk. Specificity of microglial linked peaks for AD heritability is also supported by the lack of significant enrichment of these links with risk variants from other brain-related traits,57,58,59,60,61 or traits where other immune cells play important roles.62,63,64 In contrast, linked peaks identified in other cell types were enriched for heritability of brain-related traits including autism spectrum disorder (ASD), bipolar disorder (BD), and schizophrenia (SZ) with AD-specific linked peaks largely excluded from any significant enrichment in these traits. These findings are consistent with previous studies in which candidate CREs identified in excitatory and inhibitory neurons were significantly associated with neuropsychiatric traits.11 As expected, we did not identify significant enrichments with immune diseases or with other phenotypic traits, such as body mass index (BMI)65 or height.66
Figure 5.

Validation of candidate CREs
(A) sLDSC results using 16 GWAS traits as indicated with our linked peaks stratified by cell type and group (“All” = all links, “Common” = links identified in both AD and control data, “AD” = links specific to AD, “Control” = links specific to control). Heatmap indicates coefficient Z score from running sLDSC with each set of links combined with the 97 baseline features. Feature-trait combinations with a Z score significantly larger than 0 (one-sided Z test with alpha = 0.05, p values corrected within each trait using Benjamini-Hochberg method) are indicated with a numeric value reporting the enrichment score.
(B) Bar plot showing enrichment (±95% confidence interval [CI]) of links for previously nominated regulatory regions: active MPRA elements (blue), eQTLs where target gene is same as linked gene (pink), and HiC loops linking region to same target gene (green). MPRA, massively parallel reporter assay; NPC, neural precursor cells; ESC, embryonic stem cells.
(C) Boxplots showing statistically significant (∗p < 0.05, ANOVA with Fisher’s LSD) elements representing links tested in luciferase assays. Luciferase elements are denoted by the linked gene for the nominated region.
(D) Boxplots showing comparison of rs12445022 with its corresponding reference element linked to JPH3 (∗p < 0.05, ANOVA with Fisher’s LSD).
(E) Top: normalized expression of APP in each cell type. Middle: coverage plot of accessibility in indicated cell types. Bottom: significant control (blue) and common (gray) peak-gene links to APP tested in luciferase assays. Arc height represents strength and direction of correlation. Links that contained CREs that increased expression of the luciferase reporter are highlighted in gray.
(F) Boxplots showing all tested luciferase elements representing APP-peak links. Elements highlighted in gray are located within the APP gene body (∗p < 0.05, ANOVA with Fisher’s LSD).
Validation of candidate CREs
We compared the 319,905 links we identified to pre-existing, large-scale functional genomic datasets and identified 67,541 links representing candidate CREs with orthogonal evidence of regulatory activity. This evidence was provided by three data types: (1) massively parallel reporter assay (MPRA),21,67,68,69 (2) eQTL studies,43,70 and (3) HiC71 datasets. We found significant enrichments of links across each of these datasets despite several MPRAs being performed in cancer cell lines (Figure 5B). The MPRA data provided evidence that linked peaks could stimulate transcription, but this assay is not capable of identifying the target gene. In contrast, HiC data from NeuN+ nuclei provided orthogonal validation of a linked peak’s target gene, but no evidence of promoting transcriptional activity. We intersected the results from these analyses and found that 1,542 of the 60,473 links that displayed regulatory activity in one or more MPRAs also identified the same target gene as the HiC data. In addition, 617 linked peaks overlapped eQTLs and were linked to the same gene providing both evidence of activity and confirming the target gene.
For additional validation, we selected 51 neuronal links for testing in a luciferase reporter assay (Table S9). We performed these assays in the neuroepithelial-derived human embryonic kidney 293 (HEK293 and 293FT) cell lines because of the similar chromatin accessibility landscape to that found in brain tissues.21 These cell lines are also technically tractable as they are highly transfectable and allow efficient screening of regions of interest. We did not select any AD-specific links for validation, as we are using cell lines from a presumably unaffected individual. Thirteen of these 51 links contained SNPs associated with a brain-related trait (e.g. AD, epilepsy, neurodegeneration), and we tested both alleles of these SNPs (Table S10). Twelve of the elements increased activity of the luciferase reporter including regions linked to SNCA (ɑ-synuclein) and APP (amyloid precursor protein) (Figures 5C–5F). Three of these active elements were involved in peak-gene-TF trios (CCSER1-MEF2C, JPH3-RARB, and ADAMTS1-SOX10). ChIP-seq analysis of NeuN+ nuclei confirmed that MEF2C is bound at the peak linked to CCSER1, a gene associated with autism72 (data not shown). Only one of the 15 variants tested abolished activity, rs12445022, a G/A substitution in a peak linked to JPH3 (p = 0.0003 by ANOVA with Fisher’s least significant difference [LSD]) (Figure 5D). JPH3 encodes junctophilin-3, important for regulating neuronal excitability.73 This JPH3 linked peak was highly correlated (r = 0.64, q < 2.2 × 10−16) with JPH3 expression in both AD and control samples in all cell types except microglia. The linked peak is located 45,503 bp upstream of the JPH3 TSS and was also linked to ZCCHC14-DT, although with a much lower correlation (r = 0.36, q < 2.2 × 10−16). Repeat expansions in JPH3 have been associated with a Huntington disease-like phenotype.74,75
Because of its importance in AD pathogenesis, we focused our validation efforts particularly on the APP locus (Figure 5E) where we tested 15 elements and identified three that increased expression in the luciferase reporter assay (Figure 5F). APP is expressed across all cell types (Figure 5E, top panel), consistent with the high promoter accessibility observed (Figure 5E, middle panels). We also found one element negatively correlated with APP expression that significantly reduced reporter activity; however, this assay was not designed to detect repressor activity (as it employs a minimal promoter), thus further experiments would be required to definitively assign a repressive function to this element. Although we have validated several CREs in an unaffected cell line, future work in patient-derived iPSCs would be necessary to evaluate the activity of AD-specific elements identified in this study, with the caveat that this system may still have limitations.
Discussion
Single cell multiomics has allowed the generation of a rich source of disease- and cell type-specific candidate CREs enriched in variants associated with AD. Our study provides tangible advances by employing snRNA-seq and snATAC-seq in the same cells. Other studies have generated snRNA-seq and snATAC-seq separately and integrated them to identify CREs in AD19; however, profiling gene expression and chromatin accessibility simultaneously in the same nuclei allows greater confidence in the correlations linking potential CREs to target genes. As such, we identified five times as many new candidate CREs than previously reported (319,905 links vs 56,552 gene-linked cCREs).19 To our knowledge to date, only one other study of another human neurodegenerative disease, Parkinson’s, used the 10x Genomics Multiomics (ATAC+Gene Expression) technology76 and identified a similarly large number of peak-gene links (193,732 compared with our 319,905). Our approach is unique in that we identified peak-gene correlations independently in control and AD datasets allowing us to identify 40,831 peak-gene links specific to AD.
We identified many DEGs associated with calcium homeostasis consistent with the calcium hypothesis of AD which postulates that a synergistic relationship between Aβ accumulation and Ca2+ levels promotes neurodegeneration.77 In AD neurons, we found decreased expression of ryanodine receptor 3 (RYR3) and inositol 1,4,5-trisphosphate receptor type 2 (ITPR2) that both release internal stores of Ca2+ from the endoplasmic reticulum.78 We also measured decreased expression of the Ca2+ sensors calmodulin (CALM1, CALM2, CALM3) and VILIP-1 (VSNL1), the latter which is associated with neuropathologic lesions.52,79 In contrast, two genes encoding calcium channel subunits (CACNA1C and CACNA1B) were upregulated in AD neurons. In addition, astrocytes also demonstrated decreased expression of calneuron 1 (CALN1, a Ca2+ sensor similar to calmodulin), glutamate receptor 2 subunit (GRIA2, limits Ca2+ permeability of AMPA receptors), and glutamate receptor NMDA 2C (GRIN2C, a subunit of the NMDA receptors). Both AD neurons and astrocytes showed decreased expression of the glutamate transporter GLT-1 (SLC1A2). We identified AD-specific links for all these genes except GRIN2C. Altered expression of these calcium-associated proteins is likely to exhibit complex and cell type-specific effects making the resulting network effect on excitability uncertain. However, one possibility is that this altered expression could lead to increased sensitivity of neurons to glutamate and thus neurotoxicity.78,80 Further study of the candidate regulatory elements we identified for these genes would improve our understanding of how these genes become dysregulated in AD and the emergent resulting effects.
Our study provides two main advances in our understanding of altered gene regulation in AD. First, by leveraging the AD- and control-specific links identified here we constructed peak-gene-TF trios to determine which TFs were particularly involved in regulating AD-specific transcriptional programs. MAFB and ZEB1 were found to be enriched in AD-specific trios in microglia and neurons, respectively. MAFB has previously been implicated in exercise-associated responses in the peripheral immune system in AD81 and in regulation of the receptor VISTA in microglia, which is upregulated in AD.82 In this study, we identify a previously unknown role for ZEB1 in AD-specific transcriptional regulation in neurons. Previously, ZEB1 was shown to play a critical role in epithelial-mesenchymal transition in neural crest migration and glioblastoma83,84 and further investigation is necessary to fully understand its role in AD. Second, we demonstrated enhancer-like activity for 12 candidate CREs linked to neurodegeneration-associated genes APP, SNCA, PHF24, and ADAMTS1. Amyloid precursor protein (APP) is the precursor to the AD hallmark pathology Aβ, and, while characteristic of Lewy body diseases, α-synuclein (SNCA) aggregates are highly prevalent in AD postmortem brains as well.85 PHF24 is a modulator of GABAB receptor activity86 and was recently identified in a study of AD resilience genes.87 ADAMTS1 has been implicated in AD both biochemically88 and genetically.6 Our study lays the groundwork for additional functional validation in future studies to confirm these genes as targets of these CREs. Understanding how these genes are regulated and by which TFs could provide new therapeutic targets. In fact, a recent study89 identified TFs contributing to disruption of gene regulatory networks in AD, demonstrated their ability to predict AD cognitive phenotypes, and used them to prioritize candidate drugs that could be repurposed for AD.
In summary, our study provides important new insights into the contribution of CREs to AD including the roles of TFs ZEB1 and MAFB in neurons and microglia. These findings could provide additional insights for interpreting SNPs associated with AD risk should they disrupt binding motifs for these TFs. Furthermore, these TFs could be therapeutic targets for manipulating aberrant gene regulation in AD. Our study lays the groundwork for future research to expand on the candidate- and literature-based validation approaches taken here. High-throughput CRISPRi screens are well suited to test the necessity and sufficiency of regulatory elements for linked gene expression. Future validation efforts will greatly contribute to advancing our understanding of the effects of non-coding variation on risk for AD.
Limitations of the study
One limitation of this study is that snATAC-seq data can contain spurious signals, as well as bias from transcribed genes. This limitation underscores the importance of evaluation via orthogonal methods, which we have provided using both published and newly generated data. A second limitation is that our sample size is small. This can be addressed in future studies by increasing sample size; however, the shared signals we observed with larger AD snRNA-seq studies emphasizes the representative nature of our sample set, and that our total number of cells per biological sample is adequate. Finally, as with any study from postmortem tissue, we are measuring by definition the material that remains in a neurodegenerative disease, which can confound interpretation. For this reason, we chose to evaluate DLPFC, which is preserved later into the disease course of AD than tissues affected earlier such as entorhinal cortex and hippocampus. Although this study is focused on identifying CREs, there are other non-coding regulatory mechanisms that could alter gene expression in AD including microRNAs (miRNAs),90 long non-coding RNAs (lncRNAs),91 transposable elements,92 etc., that are not assessed.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-ZEB1 antibody | Bethyl | Cat#: A301-921A; RRID: AB_1524109 |
| Rabbit polyclonal MEF2C antibody | proteintech | Cat#: 18290-1-AP; RRID: AB_2142849 |
| Dynabeads™ M−280 Sheep Anti-Rabbit IgG | ThermoFisher Scientific | Cat#: 11203D; RRID: AB_2783009 |
| Anti-NeuN Antibody, clone A60, Alexa Fluor®488 conjugated | Millipore Sigma | Cat#: MAB377X; RRID: AB_2149209 |
| Recombinant Anti-Olig2 antibody [EPR2673] | Abcam | Cat#: ab109186; RRID: AB_10861310 |
| Biological samples | ||
| Human brain tissue | NIH NeuroBioBank | https://neurobiobank.nih.gov |
| Human brain tissue | Pritzker Neuropsychiatric Disorders Research Consortium | https://pritzkerneuropsych.org/www/about-us/scientific-approach/brain-bank |
| Chemicals, peptides, and recombinant proteins | ||
| SPRI Select Reagent | Fisher Scientific | Cat#: NC0406407 |
| Lipofectamine LTX with Plus Reagent | ThermoFisher Scientific | Cat#: 15338100 |
| Protector RNAse inhibitor | Millipore Sigma | Cat#: 3335399001 |
| Poly-L-ornithine solution | Millipore Sigma | Cat#: P4957-50ML |
| UltraPure 1M Tris-HCl, pH 8.0 | Fisher Scientific | Cat#: 15568-025 |
| Calcium Chloride Solution | Millipore Sigma | Cat#: 21115-100ML |
| Magnesium Acetate Solution | Millipore Sigma | Cat#: 63052-100ML |
| DTT, 1M | ThermoFisher Scientific | Cat#: P2325 |
| EDTA, 0.5M | Millipore Sigma | Cat#: 324506-100ML |
| Triton X-100 | Millipore Sigma | Cat#: T8787-100ML |
| OmniPur Sucrose | Millipore Sigma | Cat#: 8510-500GM |
| Bovine Serum Albumin Fraction V, protease-free | Millipore Sigma | Cat#: 3117332001 |
| Tween 20 | Millipore Sigma | Cat#: P9416-50ML |
| NP-40 | Fisher Scientific | Cat#: 28324 |
| Digitonin (5%) | ThermoFisher Scientific | Cat#: BN2006 |
| PMSF | Millipore Sigma | Cat#: 10837091001 |
| cOmplete protease inhibitor cocktail | Millipore Sigma | Cat#: 11697498001 |
| Corning® 10X Phosphate-Buffered Saline (PBS), pH 7.4 ± 0.1, Liquid without calcium and magnesium, RNase-/DNase- and protease-free | Corning | Cat#: 46-013-CM |
| Sodium Deoxycholate 10% | BioWorld | Cat#: 40430018-2 |
| UltraPure SDS Solution, 10% | ThermoFisher Scientific | Cat#: 15553027 |
| Pierce™ 16% Formaldehyde (w/v), Methanol-free | ThermoFisher Scientific | Cat#: 28906 |
| BioUltra Glycine | Millipore Sigma | Cat#: 50046-250G |
| DAPI Solution (1 mg/mL) | ThermoFisher Scientific | Cat#: 62248 |
| Lithium chloride solution | Millipore Sigma | Cat#: L7026-500ML |
| Sodium Bicarbonate | Millipore Sigma | Cat#: S5761-500G |
| Proteinase K | Lucigen | Cat#: MPRK092 |
| RNase A | Qiagen | Cat#: 19101 |
| Critical commercial assays | ||
| Nano-Glo Dual-Luciferase Reporter Assay System | Promega | Cat#: N1630 |
| Global Diversity Array + NeuroBooster | Illumina | Cat#: 20031816 |
| Chromium Next GEM Chip J | 10X Genomics | Cat#: PN-1000230 |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10X Genomics | Cat#: PN-1000283 |
| Dual Index Kit TT Set A | 10X Genomics | Cat#: PN-1000215 |
| DNeasy Blood and Tissue Kit | Qiagen | Cat#: 69506 |
| Qubit dsDNA HS Assay Kit | ThermoFisher Scientific | Cat#: Q32854 |
| Standard Sensitivity NGS Fragment Analysis Kit | Advanced Analytical | Cat#: DNF-473 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE214637 |
| Code | https://doi.org/10.5281/zenodo.7405971 | https://github.com/aanderson54/scMultiomics_AD |
| Experimental models: Cell lines | ||
| HEK293 | ATCC | Cat# CRL-1573, RRID:CVCL_0045 |
| 293FT | ThermoFisher Scientific | Cat#: R70007, RRID:CVCL_6911 |
| Recombinant DNA | ||
| pGL4.23 [luc2/minP] | Promega | Cat#: E8411 |
| pNL1.1.CMV[Nluc/CMV] | Promega | Cat#: N1091 |
| Software and algorithms | ||
| Prism v9 | Graph Pad | https://www.graphpad.com/scientific-software/prism/ |
| BioConductor v1.30.16 | Huber et al.93 | https://www.bioconductor.org |
| scDblFinder v1.9.4 | Germain et al.94 | https://github.com/plger/scDblFinder |
| Seurat v4.1.1 | Hao et al.22 | https://github.com/satijalab/seurat |
| sctransform v0.3.3 | Hafemeister and Satija95 | https://github.com/satijalab/sctransform |
| Harmony v0.1.0 | Korsunsky et al.96 | https://github.com/immunogenomics/harmony |
| MAST v1.20.0 | Finak et al.97 | https://github.com/RGLab/MAST |
| R v4.1.1 | CRAN | https://cran.r-project.org |
| enrichR v3.0.0 | Chen et al.98 | https://github.com/wjawaid/enrichR |
| MACS2 v2.2.71 | Zhang et al.99 | https://github.com/macs3-project/MACS |
| Signac v1.6.0 | Stuart et al.23 | https://github.com/stuart-lab/signac |
| ComplexHeatmap v2.10.0 | Gu et al.100 | https://github.com/jokergoo/ComplexHeatmap |
| GenomicRanges v1.46.1 | Lawrence et al.101 | https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html |
| rtracklayer v1.54.0 | Lawrence et al.102 | https://bioconductor.org/packages/release/bioc/html/rtracklayer.html |
| ChIPseeker v1.30.3 | Yu et al.103 | https://github.com/YuLab-SMU/ChIPseeker |
| cellSNP 1.2.2 | Huang and Huang104 | https://github.com/hxj5/cellsnp-lite |
| vireo v0.5.6 | Huang et al.105 | https://github.com/single-cell-genetics/vireo |
| sLDSC v1.0.1 | Finucane et al.54 | https://github.com/bulik/ldsc |
| ENCODE ChIP-seq pipeline2 | ENCODE DCC | https://github.com/ENCODE-DCC/chip-seq-pipeline2 |
| CellRanger arc 2.0.1 | 10X Genomics | http://software.10xgenomics.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lindsay Rizzardi, Ph.D. (lrizzardi@hudsonalpha.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell lines
HEK293 cells were obtained from ATCC (CRL-1573) and grown in DMEM (high glucose, L-glutamine, no sodium pyruvate) (ThermoFisher Scientific Cat#11955-126), supplemented with 10% fetal bovine serum (FBS). 293FT cells were obtained from ThermoFisher Scientific (Cat#R70007) and maintained in DMEM (high glucose, L-Glutamine, 100 mg/L Sodium Pyruvate) (ThermoFisher Scientific Cat#11995081) supplemented with 10% FBS, 1% Glutamax (ThermoFisher Scientific Cat#35050061, 1% non-essential amino acids (NEAA) (ThermoFisher Scientific Cat#11140050), and 500 mg/mL Geneticin (G418 Sulfate) (ThermoFisher Scientific Cat#10131-035). All cells were cultured at 37°C with 5% CO2.
Human brain tissues
Postmortem human brain biospecimens were obtained from the NIH Neurobiobank at the University of Miami and the Human Brain and Spinal Fluid Resource Center (HBSFRC) and from collaborators from the Pritzker Neuropsychiatric Disorders Research Consortium in the Department of Psychiatry and Human Behavior, University of California Irvine (UCI) as noted in Table S1. Flash-frozen tissues were obtained from the dorsolateral prefrontal cortex (BA9/46) of seven donors diagnosed with Alzheimer’s (Braak stages 4–6) and eight unaffected controls. AD donors were neuropathologically diagnosed according to CERAD criteria and Braak staging. All AD donors had a clinical diagnosis of AD and evidence of both amyloid beta plaques and neurofibrillary tangles. Demographic information for each donor is presented in Table S1. No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those reported in previous publications.18,19 Data collection and analyses were not performed blind to tissue of origin. We did not pre-select samples based on APOE genotype, but genotype information was generated for each sample through TaqMan genotyping assays (see APOE genotyping).
Method details
Nuclei isolation from human brain tissues
Approximately 50–100 mg of frozen tissue per sample was homogenized in 4 mL of nuclei extraction buffer [0.32 M sucrose, 10 mM Tris pH 7.4, 5 mM CaCl2, 3 mM Mg acetate, 1 mM DTT, 0.1 mM EDTA, 0.1% Triton X-100, 0.2U/μL Protector RNAse inhibitor (Sigma cat. 3335399001)] by douncing 30 times in a 40 mL dounce homogenizer. Filter through 70 μm filter and spin at 500 x g, 5 min at 4°C in a swinging bucket centrifuge. Resuspend nuclei in 500 μL nuclei extraction buffer and layer over 750 μL sucrose solution (1.8 M sucrose, 10 mM Tris pH 7.4, 3 mM Mg acetate, 1 mM DTT) in a 1.5 mL tube. The samples were then centrifuged at >16,000 x g for 30 min at 4°C. After centrifugation, the supernatant was removed by aspiration and the nuclear pellet was resuspended in 125 μL PBS with 1% BSA and centrifuged 5 min at 500 x g at 4°C in a swinging bucket centrifuge. Permeabilization was performed according to 10X Genomics protocol CG000375 Rev B: nuclei were resuspended in 100 μL lysis buffer (10 mM Tris-HCl pH 7.4.10 mM NaCl, 3 mM MgCl2, 1% BSA, 0.01% Tween 20, 0.01% NP-40, 0.001% digitonin, 1 mM DTT, 1 U/μL Protector RNase inhibitor) and incubated 2 min on ice. Nuclei were washed once and resuspended in 30 μL of 1X nuclei buffer with 1 mM DTT and 0.5 U/μL of Protector RNAse inhibitor. Nuclei quality and concentrations were determined using the Countess II FL.
Single nucleus multiomics
Transposition, nuclei isolation, barcoding, and library preparation were performed according to the 10X Genomics Chromium Next GEM Single Cell Multiome protocol CG000338 Rev E with the following alterations. The initial set of eight samples were processed as above (noted as “batch 1” in Table S1) and each sample was loaded across two lanes of the Chromium Next GEM Chip J. Nuclei were loaded according to manufacturer’s recommendations to target recovery of 10,000 nuclei per lane. The second batch of ten samples were processed as above, but two samples were pooled per lane of the Chromium Next GEM Chip J (each pool is indicated by sub-batch in Table S1). Each pool consisted of a male and female donor to facilitate assignment of each single cell back to the donor based on genotype and chrY gene expression (see sample demultiplexing). For these samples, we pooled 20,000 nuclei from each sample and the entire pool was processed according to the multiome protocol. Libraries were sequenced by HudsonAlpha Discovery using Illumina NovaSeq S4 flowcells.
Sample demultiplexing
For lanes where a male and female sample were pooled together, reads were assigned to samples by genotyping cells. Variants were called from the cellranger output bam file for each cell using cellsnp-lite.104 High-confidence SNPs from the 1000 Genome Project were used as a reference panel to call variants. Cell genotypes were then split by individual using vireoSNP105 with the number of donors set to two. Cells were labeled as donor_0, donor_1, unassigned, or doublet. Unassigned and doublet cells were removed. Donor ID was assigned to the sample by observing the number of UMIs for genes on chrY. The donor ID with the higher mean counts was assigned to the male sample (Table S11).
Joint snRNA-seq and snATAC-seq workflow
Low-quality cells were filtered on gene expression data (nFeatures >200, nFeatures <10,000, and mitochondrial percent <5) and chromatin accessibility data (nucleosome signal <2 and TSS enrichment >2). PMI-associated genes106 were removed from the RNA counts matrix. Peaks that were present in less than 10 cells were removed from the ATAC matrix. Functions within Seurat22 (v4.1.1, installed via Bioconductor v1.30.1693) were used for analysis. RNA counts were normalized with SCTransform95 with mitochondrial percent per cell regressed out. Principal component analysis (PCA) was performed on RNA, and UMAP was run on the first 30 principal components (PCs). The optimum number of PCs was determined to be 30 PCs using an elbow plot. The ATAC counts were normalized with term-frequency inverse-document-frequency (TFIDF). Dimension reduction was performed with singular value decomposition (SVD) of the normalized ATAC matrix. The ATAC UMAP was created using the second through the 50th LSI components. Doublet density was computed using computeDoubletDensity from scDblFinder94 where doublet score is the ratio of densities of simulated doublets to the density in the data. Cells with a doublet score >3.5 were removed. Normalization and dimension reduction were performed again on the filtered set with the same parameters. Predicted cell types were determined for each cell using Seurat SCT-normalized reference mapping. Gene expression data was mapped to SCT-normalized DLPFC data12 and annotated with the cell types of the reference map. Cells with a predicted cell type score less than 0.95 were removed from the data. Batch effects were corrected in RNA (theta = 1) and ATAC (theta = 2) with Harmony (v1.0.0)96 by removing the effect of sample.
WNN analysis of snRNA-seq and snATAC-seq
The weighted nearest neighbor (wnn) graph was determined with Seurat’s FindMultiModalNeighbors to represent a weighted combination of both modalities. The first 30 dimensions of the harmony-corrected RNA reduction and the second through the 50th dimensions from the harmony-corrected ATAC reduction were used to create the graph. The WNN UMAP was created using the wknn (k = 20) (Figure S5).
Differential expression
Differentially expressed genes (DEGs) were determined for AD versus control for each cell type. Within each cell type, the gene expression data was log-normalized with a scale factor of 1 × 105. Pericytes and Endothelial cells were not included in the analysis because of small cell counts. Differential expression was assessed using MAST97 for genes present in at least 25% of either AD or control cells. Age and sex were included as covariates in the MAST model. Genes with a Bonferroni-adjusted p value <0.01 and an absolute log2(fold change) >0.25 were determined to be significant. DEGs between cell types were determined using MAST with age and sex as covariates for genes present in at least 25% of cells. Genes with a Bonferroni adjusted p value <0.01 an absolute log2(fold change) >0.5 were determined to be significant.
Annotation of cell subpopulations
Cell type subclusters were identified using weighted snRNA and snATAC modalities. Expression data were normalized with SCTransform, and chromatin accessibility data were normalized with TFIDF within each cell type. Normalized values were used to construct a multimodal weighted nearest neighbor graph (k = 20). Clusters were identified using wknn and the SLM algorithm. The resolution (0.3, 0.2, 0.3, 0.3, 0.45) was adjusted for each cell type (Astro, Inh, Exc, Olig, Mic). Any clusters with <100 cells were excluded from DEG analysis. Within each cell type, cluster DEGs were determined for each subcluster versus all other subclusters. DEGs were defined as those with a Bonferroni adjusted p value <0.01 using MAST with age and sex as covariates. Only genes that were detected in at least 25% of cells in a subcluster were considered.
Neuronal subclusters were further annotated with Azimuth22 Human motor cortex107 clusters to identify known neuronal subpopulations. For each neuronal subcluster, a subtype was assigned by the enrichment for upregulated subcluster DEGs in Azimuth gene sets. Enrichment was performed using enrichR98,108,109 and the Azimuth Cell Types 2021 gene sets. The top subtype annotation was assigned to a subcluster if the adjusted p value was <0.01.
AD-specific subclusters and subtypes were determined by observing overrepresentation of cells isolated from AD individuals. Statistically significant overrepresentation was evaluated with a Fisher exact test and adjusted p values.
Gene set enrichment
The R package enrichR98,108,109 was used for all gene set enrichment analyses. Sets of DEGs and peak-linked genes were used as input to look for enrichment in GO Biological Process 2021, GO Molecular Function 2021, GO Cellular Component 2021, and KEGG 2021 databases. Terms with an adjusted p value less than 0.05 were considered to be enriched.
Feature linkage analysis
ATAC peaks were called independently for each cell type using MACS299 and Signac23 CallPeaks and the union of these peaks was used in subsequent analyses retaining the cell type annotations. The peaks were then annotated with ChIPseeker103 and TxDb.Hsapiens.UCSC.hg38.knownGene where promoters were considered to be 1 kb upstream and 100 bp downstream of the TSS. Only ATAC peaks that were present in at least 2% of cells in at least one cell type were included in the analyses. AD and control links were identified separately via the cellranger-arc (v2.0.1) reanalyze function using the filtered cell type ATAC peaks and either AD or control expression and accessibility data as input. The maximum interaction distance was restricted to 500 kb. Peak-peak links were not used for downstream analysis. For feature linkage calculation, ATAC and gene expression counts were normalized independently using depth-adaptive negative binomial normalization. To account for sparsity in the data, the normalized counts were smoothed by taking the weighted sum of the 30 closest neighbors from the KNN graph. The cell weights are determined by using a Gaussian kernel transformation of the euclidean distance. Feature linkage scores were calculated by taking the Pearson correlation between the smoothed counts, while the significance of the correlation was determined using the Hotspot algorithm.32 Links with an absolute correlation score <0.2 and linked to a gene with <200 UMIs were removed. Other packages used in these analyses were GenomicRanges (v1.46.1)101 and ComplexHeatmap (v2.10.0).100
Permutation testing
We performed 100 sample permutations calling links. Sample sets for the permutations were determined by randomly selecting seven or eight individuals regardless of disease status. Links were called with the same parameters as the true data. Links were independently overlapped with links from all other permutations to get the proportion of links that were group-specific for each pair of permutations. Group-specific links were defined as the links that were not shared between any two permutations. Given the distribution of group-specific links across all permutation pairs, we tested if AD and control-specific links were an outlier for group-specificity using a Z-test.
Peak-gene-TF trios
Trios were called for a filtered set of links by removing those >100kb from a TSS and those with an absolute score <0.2. Motifs were then called in each linked-peak using Signac AddMotifs and the JASPAR 2022110 CORE PFM. Peaks with >100 motifs were additionally filtered from the link set. TF expression, linked gene expression, and linked peak accessibility matrices for trio correlation were derived from the average counts within metacells. Metacells were determined using WNN clusters for all AD cells and all control cells separately. TF-peak scores are the Pearson correlation between peak accessibility and the expression of the TF whose motif was called in the peak. TF-gene scores are the Pearson correlation between a gene and the TF whose motif was called in the linked peak. Significant associations were defined as those with a p value <0.001. Significant trios were then defined as those with a significant positive TF-peak correlation and a significant TF-gene correlation.
Partitioned heritability analysis
To evaluate whether linked peaks are enriched for common genetic variants that have been associated with AD or other traits by GWAS, we performed stratified linkage disequilibrium (LD) score regression (sLDSC v1.0.1).60,111 sLDSC estimates the proportion of genome-wide SNP-based heritability that can be attributed to SNPs within a given genomic feature by a regression model that combines GWAS summary statistics with estimates of linkage disequilibrium from an ancestry-matched reference panel. Summary statistics for AD were downloaded from.2,3,4,5,6 To estimate SNP heritability from AD GWAS summary statistics, we excluded the APOE and MHC/HLA genomic regions. Additional GWAS summary statistics were downloaded for brain-related57,58,59,60,61,112 and other traits.62,63,64,65,66 Each category (all, common, AD, control) corresponds to the analysis in which the peak-gene link was identified. Cell type is assigned based on the cell type(s) in which the linked peak was identified. Peaks were resized to 1 kb and each set of unique peaks with these categories was tested individually along with the full baseline model (baseline-LD model v2.2.) that included 97 categories capturing a broad set of genomic annotations. Note that a peak can have multiple links that fall in different categories. GWAS summary statistics are available from the websites listed in Table S8. Additional files needed for the sLDSC analysis were downloaded from https://alkesgroup.broadinstitute.org/LDSCORE/ following instructions at https://github.com/bulik/ldsc/wiki.
APOE genotyping
To determine APOE status, TaqMan genotyping assays (cat#: 4371353) were used to genotype SNPs rs429358 and rs7412 (cat#: 4351379, C___3084793_20 and C___904973_10, respectively) following the manufacturer’s instructions. Genotyping calls were made using QuantStudio software (v1.3) for all individuals in this study. APOE status is reported in Table S1.
Comparisons to external data sources
Cell type-specific H3K27ac peak calls were obtained from11 and converted to hg38 coordinates using the liftOver function from the R package rtracklayer.102 GABA and GLU neuronal subtype H3K27ac fastqs from Kozlenkov et al.36 were downloaded from Synapse (syn12033252) and processed as individual replicates using the AQUAS Transcription Factor and Histone ChIP-Seq processing pipeline113 (https://github.com/kundajelab/chipseq_pipeline). Peaks were called using the IDR naive overlapping method with a threshold of 0.05 and the optimal peak sets were used. For each cell type, only peaks identified in at least 3 individuals were retained for downstream analyses. ATAC-seq peaks from non-neuronal cell types were intersected with H3K27ac data from the corresponding cell type obtained from Nott et al.11 Excitatory and inhibitory neuron ATAC-seq peaks were intersected with H3K27ac peaks identified from GLU (NeuN+/SOX6-) or GABA (NeuN+/SOX6+) neuronal nuclei36 and from neuronal (NeuN+) nuclei.11 MPRA data were obtained from,21,67,68,69 eQTL data were obtained from,43,70 and neuronal HiC loop calls were obtained from.71
Plasmids
The pNL1.1.CMV [Nluc/CMV] (Cat# N1091) and pGL4.23 [luc2/minP] (Cat# E8411) vectors were obtained from Promega. Luciferase elements were generated by selecting 467 bp of the nominated region using hg38 coordinates. Both the forward and reverse complement sequences were ordered as gBlocks from Integrated DNA Technologies (IDT). Gibson assembly was performed by cloning elements into the pGL4.23 [luc2/minP] vector digested with EcoRV. Element insertion was confirmed by Sanger sequencing (MCLAB). Each element was individually prepped 3 times for a total of 6 individual plasmid preparations per nominated region.
Transfection
HEK293 and 293FT cells were plated at 70,000 cells/cm2 in a 24-well format. Before plating 293FT cells, culture plates were pre-coated with poly-L-ornithine solution (Millipore Sigma Cat#P4957-50ML). The next day, cells were transfected with 1 μg of plasmid DNA using Lipofectamine LTX with Plus Reagent (ThermoFisher Scientific Cat#15338-100) following the manufacturer’s recommendations. Per transfection, 900 ng of luciferase element and 100 ng of pNL1.1.CMV [Nluc/CMV] were used. A transfection reaction of 900 ng pGL4.23 [luc2/minP] and 100 ng pNL1.1.CMV [Nluc/CMV] was used as a baseline control. Both vectors were also transfected as background controls (100 ng) with pmaxGFP (900 ng, Lonza). Cell lysates were harvested by freezing at −80°C 48 h post-transfection.
Luciferase assays
Luciferase assays were performed using the Nano-Glo Dual-Luciferase Reporter Assay System (Promega cat#: N1630) following the manufacturer’s protocol. Cell lysis was performed on the 24 well plate and aliquoted across 4 wells of a white 96-well plate for 4 technical replicates per biological replicate. Assays were completed in quadruplicate. Firefly luminescence was first normalized across the average plate luminescence and then normalized to the average control luminescence. For each biological replicate, the median fold luminescence value was determined for the four technical replicates. Four biological replicates were compared to the pGL4.23 [luc2/minP]/pNL1.1.CMV [Nluc/CMV] control using an ordinary one-way ANOVA with Fisher’s LSD.
Chromatin preparation for sorted nuclei
Buffers required: Nuclei Extraction Buffer (NEB): 0.32 M Sucrose, 5 mM CaCl2, 3 mM Mg(Ac)2, 0.1 mM EDTA, 10 mM Tris-HCl, 0.1 mM PMSF, 0.1% Triton X-100, 1 mM DTT. Before use, add Roche cOmplete protease inhibitor cocktail according to manufacturer recommendation (Sigma 11697498001). Sucrose Cushion Buffer (SCB): 1.6 M Sucrose, 3 mM Mg(Ac)2, 10 mM Tris-HCl, 1 mM DTT. Interphase Buffer: 0.8 M Sucrose, 3 mM Mg(Ac)2, 10 mM Tris-HCl. Blocking buffer: 1x PBS, 1% BSA, 1 mM EDTA. Pellet buffer: add up to 200 μL 1 M CaCl2 to 10 mL SCB. RIPA: 1x PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS.
Methods for extracting and sorting nuclei from postmortem brain are similar to previously published methods.114 Here, approximately 500 mg of tissue was placed into a chilled 40 mL Dounce homogenizer containing 5 mL of NEB on ice and allowed to partially thaw to ease douncing (2-3 min). Extract nuclei by douncing with “tight” pestle 30-40 times until the tissue is homogenized. Transfer to 15 mL conical tube on ice, wash glassware with 5 mL NEB and add to 15 mL tube. Fix chromatin by adding 625 μL of 16% formaldehyde (methanol free, Thermo 28,906) to a final concentration of 1% and rotate end-over-end at room temperature for 10 min. Halt fixation by adding 500 μL of 2.5 M Glycine and incubate another 5 min rotating at room temperature then place homogenate back on ice. During fixation, prepare sucrose gradient in 2 ultracentrifuge buckets (Beckman Coulter cat: 344058) by layering 5 mL of Interphase buffer atop 10 mL of SCB in each. Carefully layer nuclei homogenate atop sucrose gradient, balance with NEB, then ultracentrifuge at 24,000 rpm for 2 h using SW28 swinging bucket rotor (Beckman Coulter). Upon completion, inspect tubes for a visible pellet of nuclei at the bottom of tube. Remove debris at interphase first by using a 25 mL graduated pipette, then continue removing the remaining sucrose gradient being careful not to disturb the nuclei pellet. Carefully resuspend the pellet in 1 mL cold PBS and transfer to a 15 mL lo-bind tube containing 2 mL PBS on ice. (Optional: if pellet appears to contain large debris then pass through 70 μm filter). Wash ultracentrifuge tubes with 1 mL cold PBS and combine in 15 mL tube to a final volume of 10 mL, inverting to mix. Centrifuge the nuclei at 1,000 x g for 10 min at 4°C to remove residual sucrose. Label nuclei by resuspending pellet in 5 mL blocking buffer with NeuN-488 antibody (Millipore cat: MAB377X) and OLIG2 antibody (Abcam cat: ab109186) at 1:5,000 each. Incubate nuclei in staining buffer with rotation for at least 1 h at 4°C. Spin nuclei 500 x g for 5 min to pellet, remove supernatant, then resuspend in 5 mL blocking buffer with goat-anti-rabbit-647 (ThermoFisher cat: A-21245) at 1:5,000 and DAPI at 1:100,000. Incubate for at least 1 h at 4°C with rotation. Remove stain by centrifuging 500 x g 5 min at 4°C and resuspending in 3 mL cold PBS. Hold on ice and proceed immediately to sorting.
Nuclei were sorted using Sony MA900 with a 70 μm nozzle and pressure not exceeding pressure setting of 7. Gates were set to capture those populations that were positive for 488 signal (NeuN+), positive for 647 signal (OLIG+), or negative for both (NeuN-;OLIG-). The NeuN+ population was collected into 5 mL tubes held at 4°C and pooled into 15 mL lo-bind tubes on ice. Purity of selected samples were typically >95% based on reanalysis of sorted samples. To concentrate nuclei for downstream analysis, add approximately 2 mL of pellet buffer per 10 mL of sorted nuclei and rotated at 4°C for 15 min. Centrifuge 500 x g for 10 min at 4°C, after which a pellet should be visible. Remove supernatant and carefully resuspend pelleted nuclei in at least 3 mL cold PBS. Centrifuge 500 x g for 5 min at 4°C.
To generate chromatin for ChIP-seq, resuspend pellet in cold RIPA plus protease inhibitor (Roche, cat: 11836153001) at approximately 3 million nuclei per 250 μL. Transfer 250 μL of each sample to the Bioruptor tubes (Diagenode, cat: C30010016) and sonicate tissue using a Bioruptor Pico (8 cycles; 30 s on/30 s off). Pool the sonicated chromatin into a 1.5 mL DNA lo-bind conical tube and centrifuge 12,000 x g for 5 min at 4°C to remove any insoluble debris. Collect supernatant into a separate tube, add RIPA to final volume equivalent to 500,000 nuclei per 100 μL, then dispense working aliquots into 1.5 mL tubes held on dry ice. Store at −80°C.
ChIP-seq protocol
ChIP-seq for ZEB1 was performed using chromatin from NeuN+ nuclei from DLPFC, occipital lobe, and frontal pole from two control donors serving as biological replicates. ChIP-seq for MEF2C was performed on bulk DLPFC tissue from two control donors serving as biological replicates. Protocols for ChIP-seq are similar to those for frozen tissue previously described by our lab115,116 and consistent with techniques recommended by the ENCODE Consortium (www.encodeproject.org/documents). Briefly, 60 μL Dynabeads (ThermoFisher, cat: 11203D) were washed with cold 1x PBS +5 mg/mL BSA then combined with 8 μL antibody targeting ZEB1 (Bethyl, cat: A301-921A) or MEF2C (proteintech, cat: 18290-1-AP) in a final volume of 200 μL and held at 4°C overnight with rotation. Tubes of aliquoted chromatin are thawed on ice and bead/antibody complex is washed with PBS +5 mg/mL BSA solution. Beads are ultimately resuspended in 100 μL RIPA and brought to 200 μL with 100 μL chromatin aliquot. Incubate bead/antibody with chromatin using rotation for 1 h at room temperature then move to 4°C for another hour. After incubation, bead complexes were washed five times with a LiCl wash buffer (100 mM Tris at pH 7.5, 500 mM LiCl, 1% NP-40, 1% sodium deoxycholate) and wash with 1 mL of cold TE (10 mM Tris-HCl at pH 7.5, 0.1 mM Na2EDTA). Chromatin was eluted from beads by incubating with intermittent shaking for 1 h at 65°C in IP elution buffer (1% SDS, 0.1 M NaHCO3), followed by incubating overnight at 65°C to reverse formaldehyde cross-links. DNA was purified using DNeasy Blood and Tissue kit (Qiagen 69,506) and eluted in a final volume of 50 μL EB. Recovered DNA was quantified using Qubit dsDNA HS Assay kit (Thermo Q32854). For input controls, one aliquot of each tissue was brought to 200 μL with RIPA and reverse-crosslinked overnight at 65°C. The following morning, samples were incubated an additional 30 min with 20 μL Proteinase K and 4 μL RNase A (Qiagen 19,101) and subsequently eluted for DNA using DNeasy Blood and Tissue kit. The entirety of the remaining IP DNA (and approximately 250 ng Input control) was used to generate sequencing libraries. Libraries were prepared by blunting and ligating ChIP DNA fragments to sequencing adapters for amplification with barcoded primers (30 s at 98°C; [10 s at 98°C, 30 s at 65°C, 30 s at 72°C] x 15 cycles; 5 min at 72°C). Libraries were quantified with Qubit dsDNA HS Assay kit and visualized with Standard Sensitivity NGS Fragment Analysis Kit (Advanced Analytical DNF-473) and Fragment Analyzer 5200 (Agilent). Libraries were sequenced using Illumina NovaSeq flow cell with 100 bp single-end runs.
ChIP-seq analysis
Prior to analysis, reads were processed to remove optical duplicates with clumpify (BBMap v38.20; https://sourceforge.net/projects/bbmap/) [dedupe = t optical = t dupedist = 2500] and remove adapter reads with Cutadapt (v1.16)117 [-a AGATCGGAAGAGC -m 40]. Input reads were capped at 40 million using Seqtk (v1.2; https://github.com/lh3/seqtk). Individual experiments were constructed following ENCODE guidelines (https://www.encodeproject.org/about/experiment-guidelines/) and analyzed with the chip-seq-pipeline2 processing pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline2). All software within the package was run using the default settings or those recommended by the authors for transcription factors. Final peaks were called using the IDR naive overlapping method with a threshold of 0.05. For ZEB1, IDR optimal peaks from the three cortical brain regions were merged for downstream analyses.
Quantification and statistical analysis
The quantitative and statistical analyses are described in the relevant sections of the method details or in the figure legends.
Acknowledgments
We thank Paige I. Hall for assisting L.W. and J.N.B. and Rebecca M. Hauser for APOE genotyping. This study was supported in part by NIH grants 5R01MH110472 and 3R01MH110472-03S1 awarded to R.M.M. and NIH grant 5R00AG068271 awarded to J.N.C. Additional funding was generously provided by donors to the HudsonAlpha Foundation Memory and Mobility Program and to the Leo Fund. We especially thank the brain donors and their families without whom this research would not have been possible.
Author contributions
Conceptualization, L.F.R., J.N.C., and J.M.L.; formal analysis, A.G.A., L.F.R., and I.R.-N.; investigation, J.N.B., L.W., S.R., L.F.R., J.M.L., and B.B.R.; resources, W.E.B., B.G.B., and S.J.W.; data curation, A.G.A.; writing – original draft, L.F.R., A.G.A., and B.B.R.; writing – review & editing, J.N.C., R.M.M., and J.M.L.; supervision, L.F.R., J.N.C., and R.M.M; funding, J.N.C. and R.M.M.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: February 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100263.
Contributor Information
J. Nicholas Cochran, Email: ncochran@hudsonalpha.org.
Richard M. Myers, Email: rmyers@hudsonalpha.org.
Lindsay F. Rizzardi, Email: lrizzardi@hudsonalpha.org.
Supplemental information
Description of samples and data. All QC stats are mean.
DEGs for one subcluster versus all other subclusters of the same cell type. DEGs were found using MAST with age and sex as covariates. Genes must be expressed in at least 10% of the cells in the subcluster. Significant genes are those with an absolute log2(FC) > 0.5 and an adjusted p value < 0.01.
DEGs for AD versus control within each cell type. MAST was used with age and sex as covariates. Genes must be expressed in at least 25% of the cells in that cell type. Significant genes have an adjusted p value < 0.01 and an absolute log2(FC) > 0.25.
GO Biological Process 2021 enrichment for AD versus control DEGs (worksheet 1) split by cell type and effect direction and GO Biological Process 2021 enrichment for genes linked to cell type-specific peaks (worksheet 2). Significant GO terms have adjusted p value < 0.01.
Merged AD and control feature linkages with correlations between peak accessibility and gene expression within each cell type.
Trios that showed significant correlation between peak accessibility-gene expression, peak accessibility-TF expression, and TF expression-gene expression.
GO Biological Process 2021 enrichments are split into genes in MEF2C trios where the linked peak was called in neurons and genes in MEF2C trios where the linked peak was called in microglia.
Partitioned heritability from LDSC analysis. Linked peaks were split by cell type that the peak was called in and by the dataset that the link was called in (AD, control, common).
Genomic location of elements cloned into luciferase reporter vector, genes linked to peaks that are represented by luciferase elements, and individual replicate data for luciferase assays where reference elements are compared with empty vector control.
Genomic location of elements cloned into luciferase reporter vector, genes linked to peaks that are represented by elements tested, SNPs tested, and individual replicate data for luciferase assays performed in HEK293 cells where alternate elements are compared with corresponding reference elements.
Sample ID assignment for 10X lanes demultiplexed with cellSNP/vireo where 2 samples were pooled on the same lane.
Document S2. Transparent peer review records for Anderson et al.
Data and code availability
-
•
Single nuclei RNA-seq, single nuclei ATAC-seq, and ChIP-seq data have been deposited at GEO and are publicly available under series accession number GSE214637.
-
•
All original code generated during this study is publicly available at https://github.com/aanderson54/scMultiomics_AD and has been deposited at Zenodo. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Neuner S.M., Tcw J., Goate A.M. Genetic architecture of Alzheimer’s disease. Neurobiol. Dis. 2020;143:104976. doi: 10.1016/j.nbd.2020.104976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunkle B.W., Grenier-Boley B., Sims R., Bis J.C., Damotte V., Naj A.C., Boland A., Vronskaya M., van der Lee S.J., Amlie-Wolf A., et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019;51:414–430. doi: 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jansen I.E., Savage J.E., Watanabe K., Bryois J., Williams D.M., Steinberg S., Sealock J., Karlsson I.K., Hägg S., Athanasiu L., et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat. Genet. 2019;9:63. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartzentruber J., Cooper S., Liu J.Z., Barrio-Hernandez I., Bello E., Kumasaka N., Young A.M.H., Franklin R.J.M., Johnson T., Estrada K., et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet. 2021;53:392–402. doi: 10.1038/s41588-020-00776-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wightman D.P., Jansen I.E., Savage J.E., Shadrin A.A., Bahrami S., Holland D., Rongve A., Børte S., Winsvold B.S., Drange O.K., et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 2021;53:1276–1282. doi: 10.1038/s41588-021-00921-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellenguez C., Küçükali F., Jansen I.E., Kleineidam L., Moreno-Grau S., Amin N., Naj A.C., Campos-Martin R., Grenier-Boley B., Andrade V., et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022;54:412–436. doi: 10.1038/s41588-022-01024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uffelmann E., Huang Q.Q., Munung N.S., de Vries J., Okada Y., Martin A.R., Martin H.C., Lappalainen T., Posthuma D. Genome-wide association studies. Nat. Rev. Methods Primers. 2021;1:59. [Google Scholar]
- 8.Zhang F., Lupski J.R. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015;24:R102–R110. doi: 10.1093/hmg/ddv259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khani M., Gibbons E., Bras J., Guerreiro R. Challenge accepted: uncovering the role of rare genetic variants in Alzheimer’s disease. Mol. Neurodegener. 2022;17:3. doi: 10.1186/s13024-021-00505-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corces M.R., Shcherbina A., Kundu S., Gloudemans M.J., Frésard L., Granja J.M., Louie B.H., Eulalio T., Shams S., Bagdatli S.T., et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020;52:1158–1168. doi: 10.1038/s41588-020-00721-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nott A., Holtman I.R., Coufal N.G., Schlachetzki J.C.M., Yu M., Hu R., Han C.Z., Pena M., Xiao J., Wu Y., et al. Brain cell type-specific enhancer-promoter interactome maps and disease risk association. Science. 2019;366:1134–1139. doi: 10.1126/science.aay0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathys H., Davila-Velderrain J., Peng Z., Gao F., Mohammadi S., Young J.Z., Menon M., He L., Abdurrob F., Jiang X., et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–337. doi: 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grubman A., Chew G., Ouyang J.F., Sun G., Choo X.Y., McLean C., Simmons R.K., Buckberry S., Vargas-Landin D.B., Poppe D., et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019;22:2087–2097. doi: 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- 14.Sadick J.S., O’Dea M.R., Hasel P., Dykstra T., Faustin A., Liddelow S.A. Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron. 2022;110:1788–1805.e10. doi: 10.1016/j.neuron.2022.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leng K., Li E., Eser R., Piergies A., Sit R., Tan M., Neff N., Li S.H., Rodriguez R.D., Suemoto C.K., et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021;24:276–287. doi: 10.1038/s41593-020-00764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y., Song W.M., Andhey P.S., Swain A., Levy T., Miller K.R., Poliani P.L., Cominelli M., Grover S., Gilfillan S., et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020;26:131–142. doi: 10.1038/s41591-019-0695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alsema A.M., Jiang Q., Kracht L., Gerrits E., Dubbelaar M.L., Miedema A., Brouwer N., Hol E.M., Middeldorp J., van Dijk R., et al. Profiling microglia from Alzheimer’s disease donors and non-demented elderly in acute human postmortem cortical tissue. Front. Mol. Neurosci. 2020;13:134. doi: 10.3389/fnmol.2020.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lau S.-F., Cao H., Fu A.K.Y., Ip N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 2020;117:25800–25809. doi: 10.1073/pnas.2008762117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morabito S., Miyoshi E., Michael N., Shahin S., Martini A.C., Head E., Silva J., Leavy K., Perez-Rosendahl M., Swarup V. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat. Genet. 2021;53:1143–1155. doi: 10.1038/s41588-021-00894-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kartha V.K., Duarte F.M., Hu Y., Ma S., Chew J.G., Lareau C.A., Earl A., Burkett Z.D., Kohlway A.S., Lebofsky R., et al. Functional inference of gene regulation using single-cell multi-omics. Cell Genom. 2022;2:100166. doi: 10.1016/j.xgen.2022.100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper Y.A., Teyssier N., Dräger N.M., Guo Q., Davis J.E., Sattler S.M., Yang Z., Patel A., Wu S., Kosuri S., et al. Functional regulatory variants implicate distinct transcriptional networks in dementia. Science. 2022;377:eabi8654. doi: 10.1126/science.abi8654. [DOI] [PubMed] [Google Scholar]
- 22.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods. 2021;18:1333–1341. doi: 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belonwu S.A., Li Y., Bunis D., Rao A.A., Solsberg C.W., Tang A., Fragiadakis G.K., Dubal D.B., Oskotsky T., Sirota M. Sex-stratified single-cell RNA-seq analysis identifies sex-specific and cell type-specific transcriptional responses in Alzheimer’s disease across two brain regions. Mol. Neurobiol. 2022;59:276–293. doi: 10.1007/s12035-021-02591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boni C., Laudanna C., Sorio C. A comprehensive Review of receptor-type tyrosine-protein phosphatase gamma (PTPRG) role in health and non-neoplastic disease. Biomolecules. 2022;12:84. doi: 10.3390/biom12010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herold C., Hooli B.V., Mullin K., Liu T., Roehr J.T., Mattheisen M., Parrado A.R., Bertram L., Lange C., Tanzi R.E. Family-based association analyses of imputed genotypes reveal genome-wide significant association of Alzheimer’s disease with OSBPL6, PTPRG, and PDCL3. Mol. Psychiatry. 2016;21:1608–1612. doi: 10.1038/mp.2015.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ceprian M., Fulton D. Glial cell AMPA receptors in nervous system health, injury and disease. Int. J. Mol. Sci. 2019;20:2450. doi: 10.3390/ijms20102450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X.-L., Li L. Cell type-specific potential pathogenic genes and functional pathways in Alzheimer’s Disease. BMC Neurol. 2021;21:381. doi: 10.1186/s12883-021-02407-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connor S.A., Ammendrup-Johnsen I., Chan A.W., Kishimoto Y., Murayama C., Kurihara N., Tada A., Ge Y., Lu H., Yan R., et al. Altered cortical dynamics and cognitive function upon haploinsufficiency of the autism-linked excitatory synaptic suppressor MDGA2. Neuron. 2016;91:1052–1068. doi: 10.1016/j.neuron.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 30.Khrimian L., Obri A., Ramos-Brossier M., Rousseaud A., Moriceau S., Nicot A.-S., Mera P., Kosmidis S., Karnavas T., Saudou F., et al. Gpr158 mediates osteocalcin’s regulation of cognition. J. Exp. Med. 2017;214:2859–2873. doi: 10.1084/jem.20171320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghose U., Sproviero W., Winchester L., Fernandes M., Newby D., Ulm B.S., Shi L., Liu Q., Adams C., Albukhari A., et al. Genome wide association neural networks (GWANN) identify novel genes linked to family history of Alzheimer’s disease in the UK Biobank. medRxiv. 2022 doi: 10.1101/2022.06.10.22276251. Preprint at. [DOI] [Google Scholar]
- 32.DeTomaso D., Yosef N. Hotspot identifies informative gene modules across modalities of single-cell genomics. Cell Syst. 2021;12:446–456.e9. doi: 10.1016/j.cels.2021.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Fulco C.P., Nasser J., Jones T.R., Munson G., Bergman D.T., Subramanian V., Grossman S.R., Anyoha R., Doughty B.R., Patwardhan T.A., et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019;51:1664–1669. doi: 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lettice L.A., Heaney S.J.H., Purdie L.A., Li L., de Beer P., Oostra B.A., Goode D., Elgar G., Hill R.E., de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- 35.ENCODE Project Consortium. Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A., et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710. doi: 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kozlenkov A., Li J., Apontes P., Hurd Y.L., Byne W.M., Koonin E.V., Wegner M., Mukamel E.A., Dracheva S. A unique role for DNA (hydroxy)methylation in epigenetic regulation of human inhibitory neurons. Sci. Adv. 2018;4:eaau6190. doi: 10.1126/sciadv.aau6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Starks R.R., Biswas A., Jain A., Tuteja G. Combined analysis of dissimilar promoter accessibility and gene expression profiles identifies tissue-specific genes and actively repressed networks. Epigenet. Chromatin. 2019;12:16. doi: 10.1186/s13072-019-0260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pastor W.A., Stroud H., Nee K., Liu W., Pezic D., Manakov S., Lee S.A., Moissiard G., Zamudio N., Bourc’his D., et al. MORC1 represses transposable elements in the mouse male germline. Nat. Commun. 2014;5:5795. doi: 10.1038/ncomms6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ampuja M., Rantapero T., Rodriguez-Martinez A., Palmroth M., Alarmo E.L., Nykter M., Kallioniemi A. Integrated RNA-seq and DNase-seq analyses identify phenotype-specific BMP4 signaling in breast cancer. BMC Genom. 2017;18:68. doi: 10.1186/s12864-016-3428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merrill C.B., Montgomery A.B., Pabon M.A., Shabalin A.A., Rodan A.R., Rothenfluh A. Harnessing changes in open chromatin determined by ATAC-seq to generate insulin-responsive reporter constructs. BMC Genom. 2022;23:399. doi: 10.1186/s12864-022-08637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lara-Astiaso D., Weiner A., Lorenzo-Vivas E., Zaretsky I., Jaitin D.A., David E., Keren-Shaul H., Mildner A., Winter D., Jung S., et al. Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014;345:943–949. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sudwarts A., Ramesha S., Gao T., Ponnusamy M., Wang S., Hansen M., Kozlova A., Bitarafan S., Kumar P., Beaulieu-Abdelahad D., et al. BIN1 is a key regulator of proinflammatory and neurodegeneration-related activation in microglia. Mol. Neurodegener. 2022;17:33. doi: 10.1186/s13024-022-00535-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bryois J., Calini D., Macnair W., Foo L., Urich E., Ortmann W., Iglesias V.A., Selvaraj S., Nutma E., Marzin M., et al. Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat. Neurosci. 2022;25:1104–1112. doi: 10.1038/s41593-022-01128-z. [DOI] [PubMed] [Google Scholar]
- 44.Chen J.A., Chen Z., Won H., Huang A.Y., Lowe J.K., Wojta K., Yokoyama J.S., Bensimon G., Leigh P.N., Payan C., et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol. Neurodegener. 2018;13:41. doi: 10.1186/s13024-018-0270-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang Y., Harigaya Y., Zhang Z., Zhang H., Zang C., Zhang N.R. Nonparametric interrogation of transcriptional regulation in single-cell RNA and chromatin accessibility multiomic data. bioRxiv. 2022 doi: 10.1101/2021.09.22.461437. Preprint at. [DOI] [Google Scholar]
- 46.Kigerl K.A., de Rivero Vaccari J.P., Dietrich W.D., Popovich P.G., Keane R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014;258:5–16. doi: 10.1016/j.expneurol.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halbgebauer S., Steinacker P., Hengge S., Oeckl P., Abu Rumeileh S., Anderl-Straub S., Lombardi J., Von Arnim C.A.F., Giese A., Ludolph A.C., et al. CSF levels of SNAP-25 are increased early in Creutzfeldt-Jakob and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 2022;93:1059–1065. doi: 10.1136/jnnp-2021-328646. [DOI] [PubMed] [Google Scholar]
- 48.Furuya T.K., Silva P.N.O., Payão S.L.M., Bertolucci P.H.F., Rasmussen L.T., De Labio R.W., Braga I.L.S., Chen E.S., Turecki G., Mechawar N., et al. Analysis of SNAP25 mRNA expression and promoter DNA methylation in brain areas of Alzheimer’s Disease patients. Neuroscience. 2012;220:41–46. doi: 10.1016/j.neuroscience.2012.06.035. [DOI] [PubMed] [Google Scholar]
- 49.Saijo K., Winner B., Carson C.T., Collier J.G., Boyer L., Rosenfeld M.G., Gage F.H., Glass C.K. A Nurr1/CoREST transrepression pathway attenuates neurotoxic inflammation in activated microglia and astrocytes. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patir A., Shih B., McColl B.W., Freeman T.C. A core transcriptional signature of human microglia: derivation and utility in describing region-dependent alterations associated with Alzheimer’s disease. Glia. 2019;67:1240–1253. doi: 10.1002/glia.23572. [DOI] [PubMed] [Google Scholar]
- 51.Holtman I.R., Skola D., Glass C.K. Transcriptional control of microglia phenotypes in health and disease. J. Clin. Invest. 2017;127:3220–3229. doi: 10.1172/JCI90604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Braunewell K.H. The visinin-like proteins VILIP-1 and VILIP-3 in Alzheimer’s disease—old wine in new bottles. Front. Mol. Neurosci. 2012;5:20. doi: 10.3389/fnmol.2012.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacob T.C. Neurobiology and therapeutic potential of α5-GABA type A receptors. Front. Mol. Neurosci. 2019;12:179. doi: 10.3389/fnmol.2019.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.R., Anttila V., Xu H., Zang C., Farh K., et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tansey K.E., Cameron D., Hill M.J. Genetic risk for Alzheimer’s disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 2018;10:14. doi: 10.1186/s13073-018-0523-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Novikova G., Kapoor M., Tcw J., Abud E.M., Efthymiou A.G., Chen S.X., Cheng H., Fullard J.F., Bendl J., Liu Y., et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun. 2021;12:1610. doi: 10.1038/s41467-021-21823-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.International Multiple Sclerosis Genetics Consortium. Wellcome Trust Case Control Consortium 2. Sawcer S., Hellenthal G., Pirinen M., Spencer C.C.A., Patsopoulos N.A., Moutsianas L., Dilthey A., Su Z., et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Rheenen W., van der Spek R.A.A., Bakker M.K., van Vugt J.J.F.A., Hop P.J., Zwamborn R.A.J., de Klein N., Westra H.-J., Bakker O.B., Deelen P., et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 2021;53:1636–1648. doi: 10.1038/s41588-021-00973-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grove J., Ripke S., Als T.D., Mattheisen M., Walters R.K., Won H., Pallesen J., Agerbo E., Andreassen O.A., Anney R., et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019;51:431–444. doi: 10.1038/s41588-019-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wray N.R., Ripke S., Mattheisen M., Trzaskowski M., Byrne E.M., Abdellaoui A., Adams M.J., Agerbo E., Air T.M., Andlauer T.M.F., et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018;50:668–681. doi: 10.1038/s41588-018-0090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pardiñas A.F., Holmans P., Pocklington A.J., Escott-Price V., Ripke S., Carrera N., Legge S.E., Bishop S., Cameron D., Hamshere M.L., et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018;50:381–389. doi: 10.1038/s41588-018-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., Ripke S., Lee J.C., Jostins L., Shah T., et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okada Y., Wu D., Trynka G., Raj T., Terao C., Ikari K., Kochi Y., Ohmura K., Suzuki A., Yoshida S., et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Julià A., López-Longo F.J., Pérez Venegas J.J., Bonàs-Guarch S., Olivé À., Andreu J.L., Aguirre-Zamorano M.Á., Vela P., Nolla J.M., de la Fuente J.L.M., et al. Genome-wide association study meta-analysis identifies five new loci for systemic lupus erythematosus. Arthritis Res. Ther. 2018;20:100. doi: 10.1186/s13075-018-1604-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Locke A.E., Kahali B., Berndt S.I., Justice A.E., Pers T.H., Day F.R., Powell C., Vedantam S., Buchkovich M.L., Yang J., et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wood A.R., Esko T., Yang J., Vedantam S., Pers T.H., Gustafsson S., Chu A.Y., Estrada K., Luan J., Kutalik Z., et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 2014;46:1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weiss C.V., Harshman L., Inoue F., Fraser H.B., Petrov D.A., Ahituv N., Gokhman D. The cis-regulatory effects of modern human-specific variants. Elife. 2021;10:e63713. doi: 10.7554/eLife.63713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uebbing S., Gockley J., Reilly S.K., Kocher A.A., Geller E., Gandotra N., Scharfe C., Cotney J., Noonan J.P. Massively parallel discovery of human-specific substitutions that alter enhancer activity. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2007049118. e2007049118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Arensbergen J., Pagie L., FitzPatrick V.D., de Haas M., Baltissen M.P., Comoglio F., van der Weide R.H., Teunissen H., Võsa U., Franke L., et al. High-throughput identification of human SNPs affecting regulatory element activity. Nat. Genet. 2019;51:1160–1169. doi: 10.1038/s41588-019-0455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu B., Won H., Mah W., Park R.B., Kassim B., Spiess K., Kozlenkov A., Crowley C.A., Pochareddy S., Li Y., et al. PsychENCODE Consortium Neuronal and glial 3D chromatin architecture informs the cellular etiology of brain disorders. Nat. Commun. 2021;12:3968. doi: 10.1038/s41467-021-24243-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ruzzo E.K., Pérez-Cano L., Jung J.-Y., Wang L.K., Kashef-Haghighi D., Hartl C., Singh C., Xu J., Hoekstra J.N., Leventhal O., et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 2019;178:850–866.e26. doi: 10.1016/j.cell.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garbino A., van Oort R.J., Dixit S.S., Landstrom A.P., Ackerman M.J., Wehrens X.H.T. Molecular evolution of the junctophilin gene family. Physiol. Genomics. 2009;37:175–186. doi: 10.1152/physiolgenomics.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bourinaris T., Athanasiou A., Efthymiou S., Wiethoff S., Salpietro V., Houlden H. Allelic and phenotypic heterogeneity in Junctophillin-3 related neurodevelopmental and movement disorders. Eur. J. Hum. Genet. 2021;29:1027–1031. doi: 10.1038/s41431-021-00866-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schneider S.A., Marshall K.E., Xiao J., LeDoux M.S. JPH3 repeat expansions cause a progressive akinetic-rigid syndrome with severe dementia and putaminal rim in a five-generation african-American family. Neurogenetics. 2012;13:133–140. doi: 10.1007/s10048-012-0318-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adams L., Song M.K., Tanaka Y., Kim Y.-S. Single-nuclei paired multiomic analysis of young, aged, and Parkinson’s disease human midbrain reveals age- and disease-associated glial changes and their contribution to Parkinson’s disease. medRxiv. 2022 doi: 10.1101/2022.01.18.22269350. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alzheimer's Association Calcium Hypothesis Workgroup Calcium Hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017;13:178–182.e17. doi: 10.1016/j.jalz.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 78.Cascella R., Cecchi C. Calcium dyshomeostasis in Alzheimer’s disease pathogenesis. Int. J. Mol. Sci. 2021;22:4914. doi: 10.3390/ijms22094914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schnurra I., Bernstein H.-G., Riederer P., Braunewell K.-H. The neuronal calcium sensor protein VILIP-1 is associated with amyloid plaques and extracellular tangles in Alzheimer’s disease and promotes cell death and tau phosphorylation in vitro: a link between calcium sensors and Alzheimer’s disease? Neurobiol. Dis. 2001;8:900–909. doi: 10.1006/nbdi.2001.0432. [DOI] [PubMed] [Google Scholar]
- 80.Burgoyne R.D., Helassa N., McCue H.V., Haynes L.P. Calcium sensors in neuronal function and dysfunction. Cold Spring Harb. Perspect. Biol. 2019;11:a035154. doi: 10.1101/cshperspect.a035154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen Y., Sun Y., Luo Z., Chen X., Wang Y., Qi B., Lin J., Lin W.-W., Sun C., Zhou Y., et al. Exercise modifies the transcriptional regulatory features of monocytes in Alzheimer’s patients: a multi-omics integration analysis based on single cell technology. Front. Aging Neurosci. 2022;14:881488. doi: 10.3389/fnagi.2022.881488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Borggrewe M., Grit C., Den Dunnen W.F.A., Burm S.M., Bajramovic J.J., Noelle R.J., Eggen B.J.L., Laman J.D. VISTA expression by microglia decreases during inflammation and is differentially regulated in CNS diseases. Glia. 2018;66:2645–2658. doi: 10.1002/glia.23517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Powell D.R., Blasky A.J., Britt S.G., Artinger K.B. Riding the crest of the wave: parallels between the neural crest and cancer in epithelial-to-mesenchymal transition and migration. Wiley Interdiscip. Rev. Syst. Biol. Med. 2013;5:511–522. doi: 10.1002/wsbm.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stemmler M.P., Eccles R.L., Brabletz S., Brabletz T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019;21:102–112. doi: 10.1038/s41556-018-0196-y. [DOI] [PubMed] [Google Scholar]
- 85.Twohig D., Nielsen H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019;14:23. doi: 10.1186/s13024-019-0320-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Numakura Y., Uemura R., Tanaka M., Izawa T., Yamate J., Kuramoto T., Kaneko T., Mashimo T., Yamamoto T., Serikawa T., et al. PHF24 is expressed in the inhibitory interneurons in rats. Exp. Anim. 2021;70:137–143. doi: 10.1538/expanim.20-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Telpoukhovskaia M.A., Hadad N., Gurdon B., Dai M., Ouellette A.R., Neuner S.M., Dunn A.R., Hansen S., Wu Y., Dumitrescu L., et al. Conserved cell-type specific signature of resilience to Alzheimer’s disease nominates role for excitatory cortical neurons. bioRxiv. 2022 doi: 10.1101/2022.04.12.487877. Preprint at. [DOI] [Google Scholar]
- 88.Gurses M.S., Ural M.N., Gulec M.A., Akyol O., Akyol S. Pathophysiological function of ADAMTS enzymes on molecular mechanism of Alzheimer’s disease. Aging Dis. 2016;7:479–490. doi: 10.14336/AD.2016.0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gupta C., Xu J., Jin T., Khullar S., Liu X., Alatkar S., Cheng F., Wang D. Single-cell network biology characterizes cell type gene regulation for drug repurposing and phenotype prediction in Alzheimer’s disease. PLoS Comput. Biol. 2022;18:e1010287. doi: 10.1371/journal.pcbi.1010287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.O’Brien J., Hayder H., Zayed Y., Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018;9:402. doi: 10.3389/fendo.2018.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Statello L., Guo C.-J., Chen L.-L., Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021;22:96–118. doi: 10.1038/s41580-020-00315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fueyo R., Judd J., Feschotte C., Wysocka J. Roles of transposable elements in the regulation of mammalian transcription. Nat. Rev. Mol. Cell Biol. 2022;23:481–497. doi: 10.1038/s41580-022-00457-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Germain P.-L., Lun A., Meixide C.G., Macnair W., Robinson M.D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 2022;10:979. doi: 10.12688/f1000research.73600.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., et al. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 101.Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lawrence M., Gentleman R., Carey V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics. 2009;25:1841–1842. doi: 10.1093/bioinformatics/btp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yu G., Wang L.-G., He Q.-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 104.Huang X., Huang Y. Cellsnp-lite: an efficient tool for genotyping single cells. Bioinformatics. 2021;37:4569–4571. doi: 10.1093/bioinformatics/btab358. [DOI] [PubMed] [Google Scholar]
- 105.Huang Y., McCarthy D.J., Stegle O. Vireo: bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol. 2019;20:273. doi: 10.1186/s13059-019-1865-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhu Y., Wang L., Yin Y., Yang E. Systematic analysis of gene expression patterns associated with postmortem interval in human tissues. Sci. Rep. 2017;7:5435. doi: 10.1038/s41598-017-05882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bakken T.E., Jorstad N.L., Hu Q., Lake B.B., Tian W., Kalmbach B.E., Crow M., Hodge R.D., Krienen F.M., Sorensen S.A., et al. Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature. 2021;598:111–119. doi: 10.1038/s41586-021-03465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xie Z., Bailey A., Kuleshov M.V., Clarke D.J.B., Evangelista J.E., Jenkins S.L., Lachmann A., Wojciechowicz M.L., Kropiwnicki E., Jagodnik K.M., et al. Gene set knowledge discovery with enrichr. Curr. Protoc. 2021;1:e90. doi: 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Castro-Mondragon J.A., Riudavets-Puig R., Rauluseviciute I., Lemma R.B., Turchi L., Blanc-Mathieu R., Lucas J., Boddie P., Khan A., Manosalva Pérez N., et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022;50:D165–D173. doi: 10.1093/nar/gkab1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bulik-Sullivan B.K., Loh P.R., Finucane H.K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics Consortium. Patterson N., Daly M.J., Price A.L., Neale B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium Electronic address douglasruderfer@vanderbiltedu. Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell. 2018;173:1705–1715.e16. doi: 10.1016/j.cell.2018.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P., et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jiang Y., Matevossian A., Huang H.-S., Straubhaar J., Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Savic D., Gertz J., Jain P., Cooper G.M., Myers R.M. Mapping genome-wide transcription factor binding sites in frozen tissues. Epigenet. Chromatin. 2013;6:30. doi: 10.1186/1756-8935-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reddy T.E., Pauli F., Sprouse R.O., Neff N.F., Newberry K.M., Garabedian M.J., Myers R.M. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19:2163–2171. doi: 10.1101/gr.097022.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of samples and data. All QC stats are mean.
DEGs for one subcluster versus all other subclusters of the same cell type. DEGs were found using MAST with age and sex as covariates. Genes must be expressed in at least 10% of the cells in the subcluster. Significant genes are those with an absolute log2(FC) > 0.5 and an adjusted p value < 0.01.
DEGs for AD versus control within each cell type. MAST was used with age and sex as covariates. Genes must be expressed in at least 25% of the cells in that cell type. Significant genes have an adjusted p value < 0.01 and an absolute log2(FC) > 0.25.
GO Biological Process 2021 enrichment for AD versus control DEGs (worksheet 1) split by cell type and effect direction and GO Biological Process 2021 enrichment for genes linked to cell type-specific peaks (worksheet 2). Significant GO terms have adjusted p value < 0.01.
Merged AD and control feature linkages with correlations between peak accessibility and gene expression within each cell type.
Trios that showed significant correlation between peak accessibility-gene expression, peak accessibility-TF expression, and TF expression-gene expression.
GO Biological Process 2021 enrichments are split into genes in MEF2C trios where the linked peak was called in neurons and genes in MEF2C trios where the linked peak was called in microglia.
Partitioned heritability from LDSC analysis. Linked peaks were split by cell type that the peak was called in and by the dataset that the link was called in (AD, control, common).
Genomic location of elements cloned into luciferase reporter vector, genes linked to peaks that are represented by luciferase elements, and individual replicate data for luciferase assays where reference elements are compared with empty vector control.
Genomic location of elements cloned into luciferase reporter vector, genes linked to peaks that are represented by elements tested, SNPs tested, and individual replicate data for luciferase assays performed in HEK293 cells where alternate elements are compared with corresponding reference elements.
Sample ID assignment for 10X lanes demultiplexed with cellSNP/vireo where 2 samples were pooled on the same lane.
Document S2. Transparent peer review records for Anderson et al.
Data Availability Statement
-
•
Single nuclei RNA-seq, single nuclei ATAC-seq, and ChIP-seq data have been deposited at GEO and are publicly available under series accession number GSE214637.
-
•
All original code generated during this study is publicly available at https://github.com/aanderson54/scMultiomics_AD and has been deposited at Zenodo. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.