

Abstract
Background and Aims:
Hepatocarcinogenesis goes through HCC progenitor cells (HcPCs) to fully established HCC, and the mechanisms driving the development of HcPCs are still largely unknown.
Approach and Results:
Proteomic analysis in nonaggregated hepatocytes and aggregates containing HcPCs from a diethylnitrosamine‐induced HCC mouse model was screened using a quantitative mass spectrometry–based approach to elucidate the dysregulated proteins in HcPCs. The heterotrimeric G stimulating protein α subunit (GαS) protein level was significantly increased in liver cancer progenitor HcPCs, which promotes their response to oncogenic and proinflammatory cytokine IL‐6 and drives premalignant HcPCs to fully established HCC. Mechanistically, GαS was located at the membrane inside of hepatocytes and acetylated at K28 by acetyltransferase lysine acetyltransferase 7 (KAT7) under IL‐6 in HcPCs, causing the acyl protein thioesterase 1–mediated depalmitoylation of GαS and its cytoplasmic translocation, which were determined by GαS K28A mimicking deacetylation or K28Q mimicking acetylation mutant mice and hepatic Kat7 knockout mouse. Then, cytoplasmic acetylated GαS associated with signal transducer and activator of transcription 3 (STAT3) to impede its interaction with suppressor of cytokine signaling 3, thus promoting in a feedforward manner STAT3 phosphorylation and the response to IL‐6 in HcPCs. Clinically, GαS, especially K28‐acetylated GαS, was determined to be increased in human hepatic premalignant dysplastic nodules and positively correlated with the enhanced STAT3 phosphorylation, which were in accordance with the data obtained in mouse models.
Conclusions:
Malignant progression of HcPCs requires increased K28‐acetylated and cytoplasm‐translocated GαS, causing enhanced response to IL‐6 and driving premalignant HcPCs to fully established HCC, which provides mechanistic insight and a potential target for preventing hepatocarcinogenesis.
INTRODUCTION
Among malignant cancers, liver cancer has one of the highest worldwide incidence rates and death tolls every year.1 HCC amounts for 80% of primary liver cancers and occurs mainly in men. Hepatic inflammation, caused by viral infection, alcohol abuse, or lipid accumulation, plays critical roles in hepatocarcinogenesis.2 Sustained hepatocyte injury and death, activation of innate immune cells in the liver, and the subsequent compensatory proliferation of hepatocytes after hepatic injury contribute to the hepatic malignant transformation, which goes through premalignant lesions containing HCC progenitor cells (HcPCs) to the development of fully established HCC.3 HcPC‐containing aggregates isolated from mice treated with the chemical carcinogen diethylnitrosamine (DEN) for 5 months possess tumorigenic properties, as determined by transplantation into major urinary protein–urokinase plasminogen activator–treated or CCl4‐treated mice.4 Among these aggregates, only cluster of differentiation 44–positive (CD44+) cells are HcPCs, which can form established HCC nodules after transplantation.3–5 Moreover, CD44 is up‐regulated in mouse HcPCs, premalignant lesions, and HCC nodules but not expressed in nonaggregated hepatocytes, which demonstrates that CD44 is an HcPC membrane marker.3,6 However, the underlying mechanisms of the vicious hepatic malignant transformation process are still largely elusive, and elucidating the dysregulated molecules in HcPCs and their roles in malignant progression from premalignant HcPCs to established HCC would be of great scientific significance.
Mechanistically, injured and dead hepatocytes release damage‐associated molecular patterns, which activate hepatic resident and recruited macrophages through Toll‐like receptor signaling. Subsequently, the production of proinflammatory cytokines such as IL‐6 initiates hepatic inflammation and the compensatory proliferation of remaining hepatocytes for repairing liver function. However, repeated hepatic injury causes chronic inflammation, leading to inflammation‐induced hepatocarcinogenesis; and proinflammatory cytokine IL‐6 and its downstream oncogenic IL‐6–signal transducer and activator of transcription 3 (STAT3) pathway play the critical role during this process.7 Loss of IL‐6 not only abolishes hepatocarcinogenesis but also diminishes the gender disparity of DEN‐induced carcinogenesis in male and female mice because estrogen‐inhibited hepatocarcinogenesis depends on suppressed IL‐6 production and its effect in female mice.8 Furthermore, during hepatocarcinogenesis, especially from precancerous HcPCs to established HCC, the typical feature of HcPCs is the autocrine IL‐6, which drives their malignant progression to fully established HCC.3 CD44, the membrane marker of HcPCs, induced by autocrine IL‐6, provides proliferative signals by inhibiting p53 genomic surveillance.6 Although autocrine IL‐6 is known to promote HcPC progression, whether the control of IL‐6 effector signaling in HcPCs is dysregulated and the underlying mechanism are still unknown.
IL‐6 effector signaling is strictly controlled by a set of intracellular molecules and mechanisms. The effect of IL‐6 goes through its receptor, consisting of IL‐6 receptor α and membrane glycoprotein 130, to activate the phosphorylation of intracellular Janus kinase 2 (JAK2) and downstream STAT3; and activated STAT3 translocates to the nucleus to initiate the transcription of a series of genes, which are responsible for cell growth, apoptosis inhibition, and cell cycle progression.9 During inflammation and tissue repair, the effect of IL‐6 is strictly controlled by a set of intracellular negative regulators, including protein inhibitor of activated STAT (PIAS) family members in the nucleus, cytoplasmic phosphatase protein tyrosine phosphatase nonreceptor type (PTPN) family members, and the suppressor of cytokine signaling (SOCS) family members.9 The effect of overactivated IL‐6 and downstream STAT3 signaling play critical roles in hepatocarcinogenesis; for example, loss of PTPN11 in hepatocytes promotes STAT3 activation, hepatic inflammation, and spontaneous hepatocarcinogenesis in aged mice.10 Considering the important roles of HcPCs in hepatocarcinogenesis, whether the response to IL‐6 is overactivated in these liver cancer progenitors still needs investigation.
GαS, the α subunit of G stimulating protein, can bind GTP to catalyze adenylyl cyclase (AC) upon G protein–coupled receptor (GPCR) activation and then participates in various cell physiological and pathological processes through cAMP production.11 The overactivation mutations of GαS are highly prevalent in various cancers dependent on cAMP downstream signaling, owing to its GTPase domain inactivation and AC overactivation.11 However, these mutations were identified to account for <1% in patients with HCC.12 Considering the low incidence of GαS overactivation mutations in HCC, we wondered whether GαS is involved in HCC carcinogenesis and progression through a noncanonical way. Currently, the roles of GαS in inflammation‐induced hepatocarcinogenesis are still unknown, especially in the stages from premalignant HcPCs to fully established HCC.
In order to identify the dysregulated molecules in HcPCs, we applied a quantitative mass spectrometry–based approach to detect the proteins with different levels between nonaggregated hepatocytes and aggregates from the liver of mice 5 months post–DEN administration and observed an increased protein level of GαS in aggregates, where HcPCs exist. The increased GαS was then confirmed in HcPCs, suggesting its potential role in HcPC progression and hepatocarcinogenesis. Thus, we constructed GαS hepatocyte‐specific knockin and knockout mice, to focus on the role and underlying mechanism of GαS in inflammation‐induced hepatocarcinogenesis in this study.
MATERIALS AND METHODS
Tissues
Liver tissue samples were obtained from two cohorts of patients with HCC: Cohort 1, 131 patients from Zhongshan Hospital (Shanghai, China); Cohort 2, 129 patients from Eastern Hepatobiliary Surgery Hospital (Shanghai, China). Clinical characteristics of these patients were presented previously,13 and these tissues were made into tissue microarray slides for further staining. Human normal liver tissues were obtained from distal normal liver tissue of patients with liver hemangioma. Liver dysplastic nodule tissues were from the corresponding patients during surgery and were pathologically diagnosed. These tissues were all obtained at the Second Military Medical University (Shanghai, China). All tissue samples in this study were collected with written informed consent from the patients, and the experiments were approved by the institutional research ethics committee of each research center.
Mice
GαSF/F(OVER) mice were constructed by Shanghai Biomodel Organism Science & Technology Development Cooperation (Shanghai, China). GαSF/F(KO) mice were constructed by VIEWSOLID BIOTECH Ltd. (Beijing, China). Micro‐RNA 143 (miR‐143) knockout mice were generated using CRISPR‐CRISPR‐associated 9 technology as described13; they harbor 716‐bp deletion flanking miR‐143 loci. Il6−/− mice (no. 002650), Il6raF/F mice (no. 012944), albumin–cyclization recombination mice (no. 003574), and Socs3F/F mice (no. 010944) were obtained from The Jackson Laboratory. GαS K28A, GαS K28Q, and lysine acetyltransferase 7 (Kat7F/F ) mice were constructed by Cyagen Biosciences Cooperation (Suzhou, China). All animal experiments were undertaken in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals, with the approval of the Scientific Investigation Board of Second Military Medical University.
Statistical analysis
Data are shown as mean ± SD from one representative of three independent experiments. Statistical comparisons between experimental groups were analyzed by unpaired Student t test, paired Student t test, or chi‐squared test in SPSS 17.0 (Chicago, IL); and a two‐tailed p < 0.05 was taken to indicate statistical significance. Correlation analysis was performed by Pearson’s correlation coefficient assay or Spearman’s correlation coefficient assay in SPSS 17.0 with r and p values shown. For analyzing survival of patients with HCC, the log‐rank test in SPSS 17.0 was used with the p values shown. Analysis of univariate or multivariate Cox proportional hazards regression was also conducted using SPSS 17.0 with the HRs and p values shown.
Other methods are described in the Supporting Information.
RESULTS
GαS protein level is increased in premalignant HcPCs and promotes hepatocarcinogenesis
To identify the dysregulated proteins in HcPCs, we used the tandem mass tags (TMT)–based quantitative proteomic technique to detect the proteins with different levels between nonaggregated hepatocytes and aggregates containing HcPCs from the liver of mice 5 months post–DEN treatment3,14 and uncovered eight consistently up‐regulated proteins (>2‐fold change) and eight consistently down‐regulated (<0.5‐fold change) proteins (Figure 1A; Figure S1A). Among the consistently upregulated proteins, some have been suggested to have tumor‐promoting function, such as claudin 3 and solute carrier family 6 member 12, which promote the progression of ovarian cancer,15 and solute carrier family 1 member 2 (SLC1A2), which forms a CD44–SLC1A2 fusion transcript to favor tumor growth of gastric cancer.16 The protein level of GαS was increased most markedly in the aggregates (Figure S1A), suggesting its potential role in HcPC progression and hepatocarcinogenesis. To confirm the characteristics of HcPCs, we examined the expression of the HcPC markers,3 including cell‐surface molecules Cd44, lymphocyte antigen 6 family member D (Ly6d), progenitor cell marker epithelial cell adhesion molecule (Epcam), human HCC marker alpha‐fetoprotein (Afp), and STAT3 phosphorylation, which were all markedly increased in HcPCs (Figure S1B,C). We then confirmed the increased GαS protein level in HcPCs compared to that in nonaggregated hepatocytes, whereas the GαS mRNA level was unaltered (Figure 1B; Figure S1D). Thus, these data suggest that the GαS protein level is increased in HcPCs, which may participate in their malignant progression.
FIGURE 1.
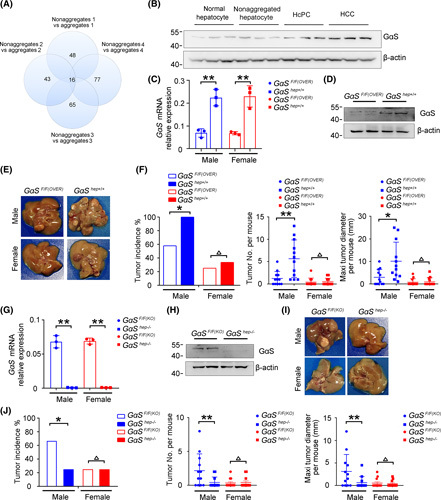
The GαS protein level is increased in premalignant HcPCs and promotes hepatocarcinogenesis. (A) Comparative results of differentially expressed proteins between hepatic aggregates and nonaggregates applying TMT assessment are shown, which were from four groups of male mice 5 months post–initial DEN injection. (B) Protein levels of GαS in normal hepatocytes, nonaggregated hepatocytes, HcPCs, and HCC tissues were detected by western blot. (C) GαS mRNA expression was detected by quantitative RT‐PCR analysis in the livers of GαSF/F(OVER) and GαShep+/+ mice (n = 3, unpaired t test). (D) GαS was detected by western blot in the livers of GαSF/F(OVER) and GαShep+/+ mice. (E) Representative livers of DEN‐induced HCC in GαSF/F(OVER) and GαShep+/+ mice. (F) Tumor incidence (chi‐squared test), number, and maximum diameter (unpaired t test) in (E) were analyzed (n = 12). (G) GαS mRNA expression was detected by quantitative RT‐PCR analysis in the livers of GαSF/F(KO) and GαShep−/− mice (n = 3, unpaired t test). (H) GαS was detected by western blot in the livers of GαSF/F(KO) and GαShep−/− mice. (I) Representative livers of DEN‐induced HCC in GαSF/F(KO) and GαShep−/− mice. (J) Tumor incidence (chi‐squared test), number, and maximum diameter (unpaired t test) in (I) were analyzed (n = 12). Data are shown as mean ± SD or photographs from one representative of three independent experiments. *p < 0.05, **p < 0.01
The corresponding mechanism responsible for the increased protein level of GαS in HcPCs was then examined. The protein level, but not the mRNA level, of GαS is increased in HcPCs, suggesting that the increased GαS protein is promoted posttranscriptionally. Because microRNA (miRNA) is one of the most important posttranscriptional regulators and some dysregulated miRNAs have been determined to promote HCC development,17 we presumed that their dysregulation in HcPCs might be responsible for the increased GαS. The abundant hepatic miRNAs, including miR‐122, miR‐192, miR‐199a‐3p, miR‐101, lethal‐7a, miR‐99a, and miR‐143,17 were examined; and we found that only miR‐143 directly targeted GαS mRNA (Figure S1E,F). Transfection of miR‐143 mimics or inhibitors showed decreased or increased GαS protein levels, respectively, in hepatocyte cell lines (Figure S1G), without affecting GαS mRNA levels (Figure S1H). The expression of miR‐143 was also decreased in HcPCs (Figure S1I). Together, these results determine that decreased miR‐143 expression is responsible for the increased GαS protein level in HcPCs.
To elucidate the potential roles of increased GαS in the malignant progression of HcPCs, we constructed mice that overexpress GαS specifically in the hepatocytes (GαShep+/+ ) (Figure 1C, D; Figure S1J). GαShep+/+ mice seemed normal and showed a similar liver/body weight ratio compared to control mice, and we did not detect spontaneous liver tumors in aged GαShep+/+ mice (data not shown). Using the DEN‐induced hepatocarcinogenesis mouse model that mimics inflammation‐driven HCC and recapitulates the higher incidence in male compared to female mice,8 we found that GαShep+/+ male mice exhibited dramatically aggravated hepatocarcinogenesis, with increased tumor incidence, numbers, and sizes compared to control male mice 8 months post–DEN injection (Figure 1E,F). In contrast, hepatocarcinogenesis was similar between female GαShep+/+ and control mice (Figure 1F). These data suggest that the increased GαS in hepatocytes promotes hepatocarcinogenesis only in male mice. Furthermore, we also constructed GαS hepatocyte‐specific knockout mice (GαShep−/− ) (Figure 1G, H; Figure S1K). Coordinately, GαShep−/− male mice showed significantly reduced tumor incidence, numbers, and sizes compared to control male mice but not female ones (Figure 1I, J). Altogether, these data determine that the GαS protein level is increased in liver cancer progenitor HcPCs and that increased hepatic GαS promotes hepatocarcinogenesis in male mice.
GαS‐promoted hepatocarcinogenesis is dependent on IL‐6
We went further to investigate the underlying mechanisms responsible for the increased GαS‐promoted hepatocarcinogenesis. DEN‐induced liver injury, determined by serum alanine aminotransferase and aspartate aminotransferase, was similar between control and GαShep+/+ male mice (Figure S2A). Next, because the proinflammatory cytokine IL‐6, induced by necrotic or apoptotic hepatocytes, plays critical roles in DEN‐induced hepatocarcinogenesis of male mice8 and given that hepatic GαS overexpression promoted DEN‐induced hepatocarcinogenesis only in male mice, we hypothesized that hepatocarcinogenesis promoted by GαS was dependent on IL‐6. Then, GαShep+/+Il6−/− mice and GαShep+/+Il6rahep−/− mice were generated, and loss of IL‐6 diminished DEN‐induced HCC, while GαShep+/+Il6−/− male mice and Il6−/− male mice displayed a similar reduced induction of HCC by DEN (Figure 2A), suggesting that hepatic GαS overexpression failed to promote hepatocarcinogenesis with IL‐6 deficiency. Similarly, the reduced hepatocarcinogenesis in GαShep+/+Il6rahep−/− male mice was similar to that in hepatic IL‐6 receptor knockout (Il6rahep−/− ) male mice (Figure 2B). Therefore, the hepatocarcinogenesis promoted by hepatic GαS is dependent on the proinflammatory cytokine IL‐6.
FIGURE 2.
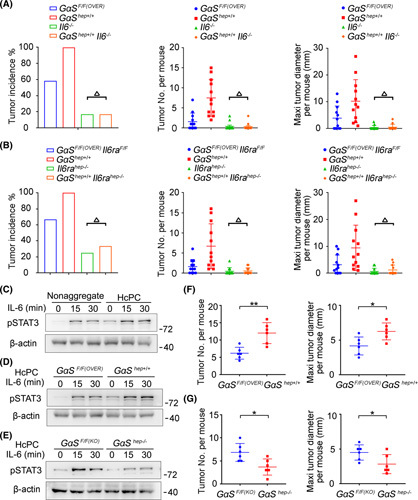
GαS‐promoted hepatocarcinogenesis is dependent on IL‐6. (A) Tumor incidence (chi‐squared test), number, and maximum diameter (unpaired t test) of DEN‐induced HCC in male GαSF/F(OVER) , GαShep+/+ , Il6−/− , and GαShep+/+Il6−/− mice were analyzed (n = 12). (B) Tumor incidence (chi‐squared test), number, and maximum diameter (unpaired t test) of DEN‐induced HCC in male GαSF/F(OVER)Il6raF/F , GαShep+/+ , Il6rahep−/− , and GαShep+/+Il6rahep−/− mice were analyzed (n = 12). (C) Nonaggregated hepatocytes and isolated HcPCs were stimulated with IL‐6 for the indicated time periods, and STAT3 phosphorylation was examined. (D) Isolated HcPCs from male GαSF/F(OVER) and GαShep+/+ mice were stimulated with IL‐6 for the indicated time periods, and STAT3 phosphorylation was examined. (E) Isolated HcPCs from male GαSF/F(KO) and GαShep−/− mice were stimulated with IL‐6 for the indicated time periods, and STAT3 phosphorylation was examined. (F) Tumor number and maximum diameter were analyzed in male mice transplanted by intrasplenic injection with isolated HcPCs from male GαSF/F(OVER) and GαShep+/+ mice (n = 6, unpaired t test). (G) Tumor number and maximum diameter were analyzed in male mice transplanted by intrasplenic injection with isolated HcPCs from male GαSF/F(KO) and GαShep−/− mice (n = 6, unpaired t test). Data are shown as mean ± SD or photographs from one representative of three independent experiments. △ p > 0.05, *p < 0.05, **p < 0.01
Because decreased miR‐143 mediates the increased GαS protein level in HcPCs, we also constructed miR‐143 knockout (miR‐143−/−) mice (Figure S2B,C) and confirmed the increased hepatic GαS protein level, but not mRNA level, by miR‐143 deficiency (Figure S2D,E). The enhanced DEN‐induced hepatocarcinogenesis was also observed in miR‐143−/− male mice, which is dependent on IL‐6 (Figure S2F), further confirming that increased GαS mediated by decreased miR‐143 promotes IL‐6‐mediated and inflammation‐induced hepatocarcinogenesis.
We then analyzed the production of proinflammatory cytokines IL‐6 and TNF‐α and growth factor HGF in the liver following DEN administration. Interestingly, the production of hepatic IL‐6, TNF‐α, and HGF did not show significant differences between control and GαShep+/+ mice following DEN i.p. injection (Figure S2G). Moreover, the autocrine IL‐6 level in HcPCs isolated from GαShep+/+ and control mice also showed little difference (Figure S2H). Thus, the GαS‐promoted hepatocarcinogenesis is less likely to be due to the production of IL‐6 in the liver, which indicates that the response to IL‐6 might be modulated by the increased GαS in HcPCs.
Increased GαS in HcPCs promotes IL‐6 effect and drives hepatocarcinogenesis
Because HcPCs acquire the ability to autocrine IL‐6 and become liver cancer progenitors, we then examined whether the response to IL‐6 was altered in HcPCs. IL‐6 effector STAT3 phosphorylation and activation were found to be significantly promoted in HcPCs (Figure 2C), and HcPCs from GαShep+/+ mice showed enhanced IL‐6–STAT3 signaling compared to those from control mice, while GαShep−/− HcPCs showed a reduced response to IL‐6 (Figure 2D, E). Thus, the enhanced response to IL‐6 in HcPCs is mediated by the increased GαS. Furthermore, to confirm that the increased GαS in HcPCs promotes hepatocarcinogenesis, we isolated CD44+ HcPCs, transplanted them into wild‐type mice, and used CCl4 injection for 5 months to induce liver inflammation and generate HCC.3 HcPCs from GαShep+/+ mice generated more and larger HCC nodules compared to those from control mice (Figure 2F; Figure S2I), while GαShep−/− HcPCs showed reduced ability to induce hepatocarcinogenesis (Figure 2G; Figure S2I). Altogether, we conclude that increased GαS mediates the enhanced IL‐6 effect in HcPCs, which promotes hepatocarcinogenesis from premalignant HcPCs to fully established HCC.
GαS enhances hepatic IL‐6–STAT3 signaling independent of AC activation
The influence of GαS on IL‐6‐induced STAT3 signaling was then analyzed in the liver and in hepatocytes. DEN‐induced STAT3 phosphorylation was significantly promoted in the livers of GαShep+/+ mice but inhibited in GαShep−/− livers (Figure 3A,B). We injected IL‐6 through the hepatic portal vein into the liver and found that STAT3 activation was enhanced in GαShep+/+ mice but inhibited in GαShep−/− mice (Figure 3C). IL‐6‐induced STAT3 activation was also determined to be increased in the primary hepatocytes from GαShep+/+ mice but suppressed in those from GαShep−/− mice (Figure 3D). Simultaneously, DEN‐induced or IL‐6‐induced hepatic STAT3 phosphorylation was significantly elevated in miR‐143−/− mice compared to controls (Figure S3A,B). Additionally, in human HHL5 and mouse BNL CL.2 hepatocyte cell lines, IL‐6‐induced STAT3 activation was promoted by GαS overexpression but suppressed by its knockdown (Figure 3E,F; Figure S3C). Thus, GαS enhanced IL‐6 effector signaling in hepatocytes. Expression of the IL‐6‐induced downstream gene serum amyloid A 1 (Saa1) was also examined,18 and both DEN‐induced and IL‐6‐induced Saa1 expression were promoted in GαShep+/+ mice but inhibited in GαShep−/− mice (Figure 3G,H). Together, we conclude that GαS promotes IL‐6 effector STAT3 signaling in hepatocytes.
FIGURE 3.
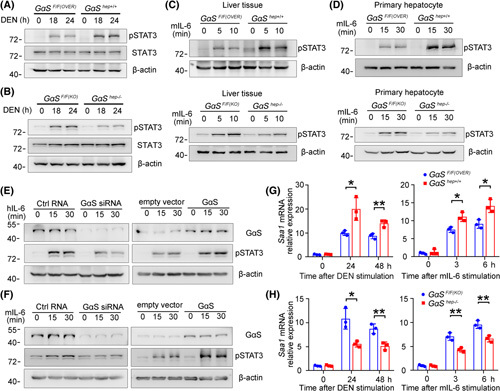
GαS enhances IL‐6–STAT3 effector signaling independent of its GTPase activity. (A) Male GαSF/F(OVER) and GαShep+/+ and (B) GαSF/F(KO) and GαShep−/− mice were i.p.‐injected with DEN for the indicated time periods. STAT3 phosphorylation in the liver tissues was examined by western blot. (C) Male mice in (A) and (B) were injected with recombinant IL‐6 through the hepatic portal vein for the indicated time periods. STAT3 phosphorylation in the liver tissues was examined by western blot. (D) Primary hepatocytes from male mice in (A) and (B) were treated in vitro with recombinant IL‐6 for the indicated time periods. STAT3 phosphorylation was examined by western blot. (E) HHL5 and (F) BNL CL.2 hepatocyte cell lines were transfected with negative control or GαS‐specific small interfering RNA, control vector, or GαS overexpression plasmids and then treated with recombinant IL‐6 for the indicated time periods. STAT3 phosphorylation was examined by western blot. (G) Male GαSF/F(OVER) and GαShep+/+ and (H) GαSF/F(KO) and GαShep−/− mice were injected i.p. with DEN or injected with recombinant IL‐6 through the tail vein for the indicated time periods. Saa1 expression in the liver was examined by quantitative RT‐PCR (n = 3, unpaired t test). Data are shown as mean ± SD or photographs from one representative of three independent experiments. *p < 0.05, **p < 0.01. Abbreviations: h‐, human; m‐, mouse; siRNA, small interfering RNA
Upon binding GTP, GαS activates AC, which promotes cAMP production to regulate cell physiological and pathological processes.11 GαS overactivation mutations are highly prevalent in various cancers but occur in <1% of patients with HCC.12 To determine whether GαS enhances IL‐6–STAT3 signaling by activating AC, we employed NF449, which inhibits GαS‐mediated stimulation of AC activity, and found that NF449 treatment did not influence IL‐6‐induced STAT3 phosphorylation in HHL5 and BNL CL.2 hepatocyte cell lines (Figure S3D,E). Furthermore, GαS overactivation and inactivation mutants were constructed and verified by cAMP ELISA (Figure S3F),19 and these mutants showed similar levels of IL‐6‐induced STAT3 activation as those in wild‐type GαS (Figure S3G). Together, these data suggest that GαS promotes IL‐6–STAT3 signaling independent of its GTPase activity and AC stimulation.
Cytoplasm‐translocated GαS associates with STAT3 upon IL‐6 stimulation to impede SOCS3–STAT3 interaction
To investigate how GαS promotes IL‐6–STAT3 signaling, we evaluated whether GαS directly associates with STAT3. Indeed, using coimmunoprecipitation (co‐IP) assays, GαS was found to interact with STAT3, which was enhanced by IL‐6 stimulation (Figure 4A). Their association was also determined by co‐IP analysis using exogenously expressed tagged constructs (Figure 4B). We also coexpressed V5–STAT3 and Flag–GαS in HHL5 hepatocytes and found that GαS was located at the inner cell membrane and translocated to the cytoplasm to colocalize with STAT3 upon IL‐6 stimulation (Figure 4C). Thus, cytoplasm‐translocated GαS associates with STAT3 upon IL‐6 stimulation, which may promote IL‐6–STAT3 signaling.
FIGURE 4.
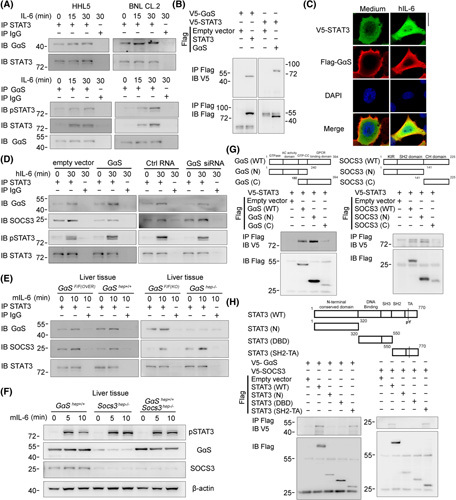
Cytoplasm‐translocated GαS associates with STAT3 upon IL‐6 stimulation to impede SOCS3–STAT3 interaction. (A) The association between GαS and STAT3 was examined by co‐IP in HHL5 and BNL CL.2 cells treated with IL‐6 for the indicated time periods. (B) Tagged STAT3 and GαS constructs were cotransfected into HEK293T cells as indicated, and the association between GαS and STAT3 was examined by co‐IP. (C) V5‐tagged STAT3 and Flag‐tagged GαS constructs were cotransfected into HHL5 cells. Confocal microscopic images of these cells upon IL‐6 stimulation are shown as indicated. Scale bar, 10 μm. (D) HHL5 cells were transfected with control, GαS overexpression plasmids, or GαS small interfering RNA as indicated; the associations between STAT3 and GαS or SOCS3, respectively, were analyzed by co‐IP. (E) Male GαSF/F(OVER) and GαShep+/+ , GαSF/F(KO) and GαShep−/− mice were injected with recombinant IL‐6 through the hepatic portal vein; the associations between STAT3 and GαS or SOCS3, respectively, were analyzed by co‐IP in liver tissue lysates. (F) Male GαShep+/+ , Socs3hep−/− , and GαShep+/+Socs3hep−/− mice were injected with recombinant IL‐6 through the hepatic portal vein for the indicated time periods, and STAT3 phosphorylation was examined by western blot in the liver tissues. (G,H) Tagged STAT3, GαS, SOCS3 and their truncates were constructed and cotransfected into HEK293T cells, and cell lysates were precipitated with Flag antibody and immunoblotted with V5 antibody as indicated. Data are shown as photographs from one representative of three independent experiments. Abbreviations: Ctrl, control; DBD, DNA binding domain; h‐, human; IB, immunoblotting; IP, immunoprecipitation; m‐, mouse; p‐, phosphorylated; SH2‐TA, Src homology 2‐transactivation; WT, wild type
To determine how GαS–STAT3 association promotes STAT3 activation, we screened the kinases and regulators involved in IL‐6–STAT3 signaling. STAT3 could bind to JAK1, JAK2, Src homology domain containing phosphatase 1 (SHP1), SHP2, and SOCS3, but not SOCS1, PIAS1, and PIAS3, following IL‐6 stimulation (Figure S4A); and SOCS3–STAT3 interaction was suppressed by GαS overexpression but enhanced by its knockdown in HHL5 hepatocytes (Figure 4D). Further, IL‐6‐induced SOCS3–STAT3 interaction was reduced in GαShep+/+ livers but increased in GαShep−/− livers (Figure 4E). We also found that GαS inactivation mutant was associated with STAT3 to impede SOCS3–STAT3 interaction (Figure S4B) and that the GαS–STAT3 association was not influenced by GαS AC inhibition (Figure S4C). To determine whether GαS‐promoted STAT3 activation was dependent on SOCS3, we crossed GαShep +/+ mice with Socs3hep−/− mice to generate GαShep +/+ Socs3hep−/− mice and found that GαShep +/+ Socs3hep−/− livers exhibited similar STAT3 activation to those in Socs3hep−/− mice following IL‐6 stimulation (Figure 4F; Figure S4D), demonstrating that GαS failed to promote STAT3 activation under SOCS3 deficiency. Additionally, N‐terminal GαS and SOCS3 were found to, respectively, bind the Src homology 2‐transactivation domain of STAT3, which contains the phosphorylation and activation site Y705 (Figure 4G,H). Together with the known SOCS3‐mediated STAT3 inactivation,9 we conclude that IL‐6‐induced and cytoplasm‐translocated GαS associates with STAT3 to enhance STAT3 activation, which is dependent on the impeded SOCS3–STAT3 interaction.
IL‐6‐induced acetylation of GαS at K28 is responsible for its cytoplasmic translocation and association with STAT3
We then examined the underlying mechanisms responsible for the IL‐6‐induced cytoplasmic translocation of GαS and its association with STAT3. Because the posttranslational modifications (PTMs) of proteins have been identified to play critical roles in their structure, activity, localization, interaction, and function,20 the PTMs of GαS, including phosphorylation, methylation, and acetylation, were then analyzed using mass spectrometry in HHL5 hepatocytes following IL‐6 administration. Methylation at K211 was identified in untreated cells, while acetylation at K28 was identified in IL‐6‐stimulated cells; and methylation at K53 and K181 was identified in both, suggesting four new modified lysine residue sites in the N‐terminal domain of GαS (Figure S5A). The mutants at these sites were constructed; only K28A mutant mimicking deacetylation of evolutionarily conserved K28 significantly decreased GαS–STAT3 association, and GαS K28Q mutant mimicking acetylation enhanced its association with STAT3, while other mutants had little effect (Figure 5A; Figure S5B), suggesting that acetylation of GαS at K28 is important for GαS–STAT3 association. Rabbit polyclonal antibody specific to K28‐acetylated GαS was then generated (Figure S5C), and acetylation of GαS at K28 was determined to be induced by IL‐6 in both mouse liver tissue and whole‐cell lysates precipitated with GαS antibody (Figure 5B,C). Thus, IL‐6‐induced acetylation of GαS at K28 mediates its association with STAT3, which then enhances STAT3 activation.
FIGURE 5.

IL‐6‐induced acetylation of GαS at K28 mediates its cytoplasm translocation and association with STAT3. (A) V5‐tagged STAT3 and Flag‐tagged GαS mutants as indicated were transfected into the HHL5 hepatocyte cell line, and their association was examined by co‐IP analysis. (B) K28‐acetylated GαS was examined using the specific antibody in liver tissue lysates from male mice injected with IL‐6 through the hepatic portal vein, which were immunoprecipitated with total GαS antibody. (C) K28‐acetylated GαS was examined using the specific antibody in the precipitates by total GαS antibody from HHL5 cells following IL‐6 stimulation. (D) IL‐6‐induced cytoplasm translocation of GαS was examined using confocal microscopy in HHL5 cells transfected with Flag‐tagged GαS mutants. Scale bar, 10 μm. (E) Subcellular distribution of GαS was examined in nonaggregated hepatocytes and HcPCs using confocal microscopy. Scale bar, 20 μm. (F) Subcellular distribution of GαS was examined in liver tissues from male GαS K28A or K28Q mutant mice. Scale bar, 20 μm. (G) Tumor incidence (chi‐squared test), number, and maximum diameter (unpaired t test) of DEN‐induced HCC in male wild‐type, GαS K28A, or K28Q mutant mice were analyzed (n = 12). (H) S‐palmitoylation levels of Flag‐tagged GαS mutants expressed in HHL5 cells upon IL‐6 stimulation as indicated were examined by the acyl–biotin exchange assay. (I) V5‐tagged APT1 and Flag‐tagged GαS mutants were cotransfected into HHL5 cells as indicated, and their association was examined by co‐IP analysis. Data are shown as mean ± SD or photographs from one representative of three independent experiments. *p < 0.05, **p < 0.01. Abbreviations: h‐, human; HRP, horseradish peroxidase; IB, immunoblotting; IP, immunoprecipitation; m‐, mouse; WT, wild type
Because GαS was determined to translocate from the inner cell membrane to the cytoplasm and to associate with STAT3 following IL‐6 stimulation, we then examined whether IL‐6‐induced acetylation of GαS at K28 also mediated its cytoplasmic translocation. Confocal and western blot analyses both demonstrated that IL‐6 induced the cytoplasm translocation of Flag‐tagged wild‐type GαS in HHL5 hepatocytes, K28A deacetylation mutant still localized at the membrane after IL‐6 stimulation, and K28Q acetylation mutant constitutively localized at the cytoplasm of untreated hepatocytes (Figure 5D; Figure S5D). Moreover, we analyzed the subcellular localization of GαS in nonaggregated hepatocytes and liver cancer progenitor HcPCs and found that GαS also localized at the cytoplasm of HcPCs (Figure 5E). Thus, IL‐6‐induced acetylation of GαS at K28 mediates its cytoplasm translocation, which then associates with STAT3 and enhances STAT3 activation in HcPCs.
To certify the roles of acetylated GαS in promoting IL‐6–STAT3 activation in vivo, we constructed the K28A mutant mouse mimicking deacetylated GαS and the K28Q mutant mouse mimicking acetylated GαS (Figure S5E). Hepatic STAT3 phosphorylation was inhibited in GαS K28A mice but promoted in GαS K28Q mice upon IL‐6 stimulation in vivo (Figure S5F). Simultaneously, GαS K28A mutant localized at the cell membrane, while K28Q mutant was distributed throughout the cytoplasm in mouse liver tissues (Figure 5F). Similarly, hepatic GαS–STAT3 association was inhibited in K28A mice but increased in K28Q mice, accompanied by increased SOCS3–STAT3 association in K28A mice and decreased association in K28Q mice (Figure S5G). Furthermore, DEN‐induced hepatocarcinogenesis was increased in GαS K28Q mice but decreased in GαS K28A mice (Figure 5G). We conclude that IL‐6 induces acetylation of GαS at K28, which in turn translocates to the cytoplasm and associates with STAT3 to promote IL‐6–STAT3 signaling and hepatocarcinogenesis.
Erased by acyl protein thioesterase 1 associates with K28‐acetylated GαS to erase S‐palmitoylation at C3 and initiate its cytoplasm translocation
We next analyzed the underlying mechanism for the cytoplasm translocation of GαS mediated by K28 acetylation. Previous studies reported that S‐palmitoylation of GαS at N‐terminal cysteine 3 mediated its localization at the inner plasma membrane.21 We found that GαS S‐palmitoylation at C3 was suppressed by GαS K28Q acetylation but enhanced by K28A deacetylation, using C3Y mimicking S‐palmitoylation as a positive control, while C3S mimicked de‐S‐palmitoylation as a negative control (Figure 5H). Thus, the IL‐6‐induced acetylation of GαS at K28 inhibits the S‐palmitoylation at C3 to promote GαS cytoplasm translocation. Furthermore, because the S‐palmitoylation of GαS is written by Asp‐His‐His‐Cys (DHHC) motif–containing palmitoyl acyltransferase but erased by acyl protein thioesterase 1 (APT1),22–24 we found that IL‐6 induced the interaction between GαS and APT1, which was suppressed by K28A mimicking deacetylation but promoted by K28Q mimicking acetylation (Figure 5I). We conclude that IL‐6‐induced acetylation of GαS at K28 promotes the APT1‐mediated erasure of C3 S‐palmitoylation to initiate GαS cytoplasm translocation.
IL‐6‐induced acetylation of GαS at K28 is mediated by KAT7
The mechanism responsible for IL‐6‐induced GαS acetylation was then investigated. We used mass spectrometry to examine the proteins co‐IP with Flag‐tagged GαS in IL‐6‐treated HHL5 hepatocytes, and among the potential GαS‐associated proteins, KAT7 was selected as the candidate because of its acetyltransferase activity (Figure S6A). The IL‐6‐induced GαS–KAT7 association was confirmed by co‐IP both in vivo and in vitro (Figure 6A). The truncates of KAT7 were constructed, and the MYST‐type histone acetyltransferase domain of KAT7 was associated with the N‐terminal domain of GαS, where K28 locates (Figure S6B). Thus, KAT7 associates with GαS following IL‐6 stimulation, which may mediate the IL‐6‐induced acetylation of GαS.
FIGURE 6.
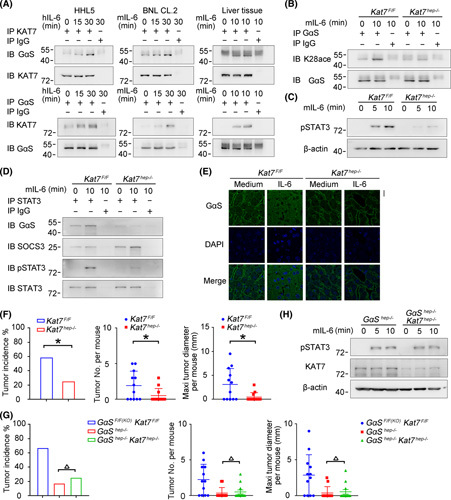
IL‐6‐induced acetylation of GαS at K28 is mediated by KAT7. (A) IL‐6‐induced association between KAT7 and GαS was examined by co‐IP in HHL5 and BNL CL.2 cells in vitro and in mouse liver tissues in vivo. (B–D) Male Kat7F/F and Kat7hep−/− mice were injected with IL‐6 through the hepatic portal vein; acetylated GαS at K28 (B), STAT3 phosphorylation (C), and GαS–STAT3 association (D) were examined as indicated. (E) Confocal microscopy of liver tissues from male Kat7F/F and Kat7hep−/− mice upon IL‐6 injection through the hepatic portal vein. Scale bar, 20 μm. (F) Tumor incidence (chi‐squared test), number, and maximum diameter (unpaired t test) of DEN‐induced HCC in male Kat7F/F and Kat7hep−/− mice were analyzed (n = 12). (G) Tumor incidence (chi‐squared test), number, and maximum diameter (unpaired t test) of DEN‐induced HCC in male Kat7F/FGαSF/F(KO) , GαShep−/− , and Kat7hep−/−GαShep−/− mice were analyzed (n = 12). (H) IL‐6‐induced STAT3 phosphorylation was evaluated in liver tissues from male GαShep−/− and Kat7hep−/−GαShep−/− mice upon IL‐6 stimulation. Data are shown as mean ± SD or photographs from one representative of three independent experiments. △ p > 0.05, *p < 0.05. Abbreviations: h‐, human; IB, immunoblotting; IP, immunoprecipitation; m‐, mouse; p‐, phosphorylated
Thereafter, we constructed hepatocyte‐specific KAT7 knockout mice (Kat7hep−/− ) (Figure S6C,D) and determined that IL‐6‐induced acetylation of GαS at K28 was nearly abolished in Kat7hep−/− livers (Figure 6B). IL‐6‐induced GαS–STAT3 association and STAT3 phosphorylation were also suppressed in Kat7hep−/− livers (Figure 6C,D). Moreover, IL‐6‐induced hepatic GαS cytoplasmic translocation was abolished by KAT7 deficiency (Figure 6E). The DEN‐induced HCC model was applied, and hepatocarcinogenesis was significantly inhibited in Kat7hep−/− livers, which was similar to what occurred in GαShep−/− mice (Figure 6F). Hepatocyte‐specific KAT7 and GαS double knockout mice (Kat7hep−/−GαShep−/− ) were also generated, and both DEN‐induced hepatocarcinogenesis and IL‐6‐induced STAT3 phosphorylation were similar between Kat7hep−/−GαShep−/− mice and GαShep−/− mice (Figure 6G,H), suggesting that hepatic KAT7 deficiency failed to further inhibit hepatocarcinogenesis under GαS deficiency and that the inhibited hepatocarcinogenesis in Kat7hep−/− livers is dependent on GαS. Altogether, these data demonstrate that IL‐6‐induced acetylation of GαS at K28 is mediated by KAT7, which then promotes in a feedforward manner GαS–STAT3 association, IL‐6–STAT3 signaling, and the corresponding hepatocarcinogenesis.
GαS expression and acetylation are correlated to human hepatocarcinogenesis and prognosis
In order to analyze the correlation of GαS with human hepatocarcinogenesis, we detected GαS protein expression in human hepatic dysplastic nodules, which represent premalignant lesions of HCC. Both the GαS protein level and its acetylation at K28 were increased, accompanied by augmented STAT3 phosphorylation in human hepatic dysplastic nodules compared to human normal liver tissues (Figure 7A–C; Figure S7A), which was in accordance with the data obtained in mouse liver cancer progenitors. Additionally, KAT7 expression was moderately increased in dysplastic nodules compared to that in normal livers (Figure 7A,C). Moreover, both GαS expression and its acetylation at K28 were significantly positively correlated with STAT3 phosphorylation in human hepatic dysplastic nodules (Figure 7D,E). Thus, increased GαS in hepatic premalignant dysplastic nodules may mediate the enhanced STAT3 activation and human hepatocarcinogenesis.
FIGURE 7.
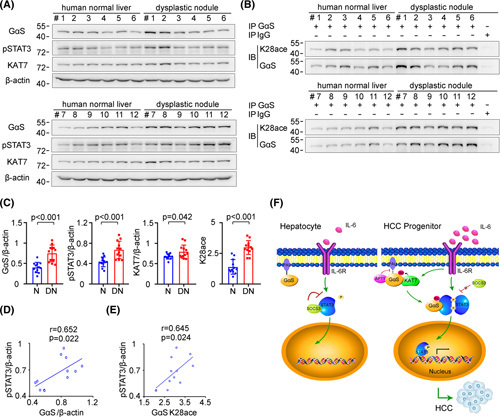
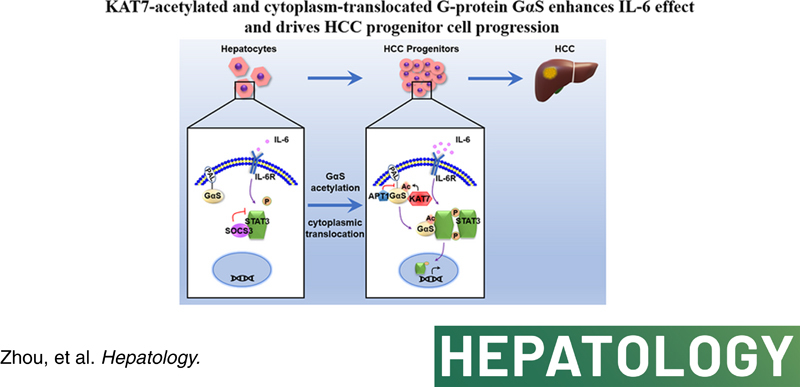
Increased GαS expression and acetylation are correlated with human hepatocarcinogenesis. (A) GαS, KAT7, and STAT3 phosphorylation were examined in human normal liver tissues and dysplastic nodule tissues from the indicated patients. (B) Acetylated GαS at K28 was examined in the precipitates using GαS antibody from human normal liver tissues and dysplastic nodule tissues as in (A). (C) Quantified levels of GαS, KAT7, STAT3 phosphorylation, and GαS acetylation in human normal liver tissues and dysplastic nodule tissues are shown (n = 12, unpaired t test). (D) The correlation between GαS and STAT3 phosphorylation in human dysplastic nodule tissues was analyzed by Pearson’s correlation coefficient assay (n = 12). (E) The correlation between acetylated GαS at K28 and STAT3 phosphorylation in human dysplastic nodule tissues was analyzed by Pearson’s correlation coefficient assay (n = 12). (F) Working model for KAT7‐acetylated and cytoplasm‐translocated GαS feedforward promoting the response to IL‐6 in liver cancer progenitors and driving hepatocarcinogenesis. Data are shown as mean ± SD, dot plots, or photographs directly as indicated. Abbreviations: DN, dysplastic nodule; IB, immunoblotting; IP, immunoprecipitation; N, normal; p‐, phosphorylated
We also examined the expression of GαS in human HCC tissues. The GαS protein level was determined to be increased in human HCC tissues compared to paired nontumor liver tissues from patients of Cohort 1 (n = 131) and Cohort 2 (n = 129) (Figure S7B), and the GαS protein level was positively correlated with STAT3 phosphorylation in HCC tissues of these cohorts (Figure S7C). Moreover, the GαS protein level was positively correlated with both advanced tumor–node–metastasis stage and high histological grade (Figure S7D,E). Additionally, patients with relatively higher GαS protein levels in HCC tissues had shortened overall survival and disease‐free survival than those with lower levels (Figure S7F), as determined in Cohorts 1 and 2 using the median level as the cutoff. Cox proportional hazards regression analysis also determined that higher GαS protein level was an independent predictor for poorer prognosis of patients with HCC (Tables S1 and S2). Thus, increased GαS in HCC tissues may be important for identifying the cancer progression and prognosis of patients with HCC.
DISCUSSION
Many malignant cancers undergo the process from a rare population of cells capable of growth and survival advantages to form a premalignant lesion. These kinds of cancer progenitor cells were first identified in acute myeloid leukemia and subsequently in multiple solid tumors, which promote cancer initiation and maintenance. For example, leucine‐rich repeat containing GPCR 6 marks a rare population of mammary gland progenitor cells which can originate luminal mammary tumors.25 For HCC, EpCAM+ HCC subtype sorted from clinical specimens displays stem cell–like abilities of self‐renewal and differentiation, which initiates highly invasive HCC in immunodeficient mice owing to activation of the Wnt/β‐catenin pathway.26 However, further understanding indicates that stem cell–like traits are nonessential for tumor progenitors, and HcPCs were isolated from mouse HCC models, which give rise to HCC only combined with hepatic chronic damage and inflammation dependent on paracrine and autocrine IL‐6 signaling.3 Interestingly, the transcriptome of aggregated hepatocytes appears closer to that of normal hepatocytes than to the HCC profiles, resulting from the presence of 70% CD44− hepatocytes in aggregates.3 The remaining 30% CD44+ aggregates, identified as HcPCs, exhibit similar markers to HCC like EpCAM, AFP, and Ly6D, which may account for their tumorigenic properties.3 Mechanistically, CD44 inhibits the p53 genomic surveillance response through inducing the phosphorylation and nuclear translocation of Mdm2 proto‐oncogene, facilitating HcPC proliferation and cancer initiation.6 Here, the GαS protein level is determined to be increased in HcPCs but not in the nonaggregates, suggesting that GαS is increased in the carcinogenesis stages starting from liver cancer progenitor HcPCs to fully established HCC. Together with the pivotal roles of enhanced autocrine and paracrine IL‐6 in driving HcPCs to HCC and the present data showing enhanced IL‐6 effector signaling by increased GαS, we conclude that not only the enhanced IL‐6 production but also the increased IL‐6 response are important for hepatocarcinogenesis, especially in the stages from HcPCs to established HCC.
IL‐6 is one of the best‐characterized and most critical protumorigenic cytokines in the liver, and its effector signaling promotes both early carcinogenesis of cancer progenitors and later progression of established HCC.9 It is observed that tumor volumes of the induced HCC in hepatic GαS overexpressing mice are obviously larger than those in controls. Thus, in the IL‐6‐mediated progression of established HCC cells, increased GαS may be also important for their development, which has been suggested recently.27 Moreover, during hepatocarcinogenesis, liver cancer progenitors gain the ability to autocrine IL‐6 and become HcPCs, and then IL‐6 promotes the transformation from premalignant HcPCs to established HCC. Although the response to IL‐6 is tightly controlled by multiple intracellular mechanisms to prevent its overactivation in hepatocytes, the IL‐6 effector signaling in HcPCs was determined here to be significantly enhanced, which drives the malignant progression of liver cancer progenitors. During this process, increased GαS‐induced and IL‐6‐induced acetylation and cytoplasm translocation were suggested to be responsible for the overactivated IL‐6–STAT3 signaling in HcPCs. However, although decreased miR‐143 was determined to mediate the increased protein level of GαS in HcPCs, when and how this mechanism occurs during normal hepatocytes to liver cancer progenitor HcPCs still need to be investigated.
GPCR family members are activated by various agonist ligands, including photons, lipids, and small proteins; and their activation induces the release of GTP‐bound α subunit of G protein (Gα). GαS, as one member of the Gα family, typically stimulates AC to increase intracellular cAMP and activate downstream protein kinase A, which promotes cell growth and survival.28 GPCR activation and downstream cAMP are frequently increased in cancer and promote cancer progression.11 GαS activating mutations and increased cAMP are also oncogenic and trigger cancer cell proliferation.29–31 However, these activation mutations are rarely detected in HCC tissues (<1%),12 indicating that GαS–cAMP signaling is not closely related to HCC carcinogenesis and progression. Here, we determined that GαS promotes HCC carcinogenesis independent of its GTPase activity and increased cAMP but dependent on its cytoplasmic translocation and direct association with STAT3 to promote IL‐6 effector signaling in HcPCs. This noncanonical, cAMP‐independent function of GαS in promoting hepatocarcinogenesis may indicate a more direct style of G‐protein components in the regulation of intracellular signaling, especially inflammatory signaling pathways. Furthermore, the roles of other components of G proteins, including Gβ and Gγ, in inflammation‐induced hepatocarcinogenesis are also unknown. The role of GαS in carcinogenesis may raise interesting future work on the roles of other G‐protein components in cancer development.
PTMs of proteins are important for their structural formation, subcellular localization, and protein–protein interaction, thus modulating their functions. For GαS, PTMs, especially palmitoylation, have been suggested to be important for its subcellular localization and function. S‐palmitoylation of GαS at N‐terminal glycine 2 increases its AC affinity, and S‐palmitoylation at the N‐terminal C3 facilitates its localization at the inner plasma membrane.21 The palmitoylation of GαS is written by DHHC motif–containing palmitoyl acyltransferase but erased by APT1, which is reported to be necessary for the localization of GαS at the inner cell membrane.22–24 Here, we found that KAT7‐acetylated GαS at K28 promoted its association with APT1 and the erasure of S‐palmitoylation at C3, thus causing the cytoplasmic translocation of GαS and its association with STAT3. Hence, the acetylation of GαS at K28 may be important for its interaction with other proteins, and the underlying mechanisms, especially the structural basis, still need further investigation, which may suggest the complete roles of GαS subcellular localization and protein–protein interaction regulated by PTMs.
In the simplest model compatible with our findings, we propose that SOCS3 feedback restrains IL‐6‐induced downstream STAT3 phosphorylation to avoid overactivation of IL‐6 effector signaling in hepatocytes. However, in liver cancer progenitor HcPCs, the increased protein level of GαS promotes IL‐6 effector signaling and drives premalignant HcPCs to fully established HCC. Mechanistically, IL‐6‐induced and KAT7‐acetylated GαS at K28 translocates to the cytoplasm to associate with activated STAT3 and impede SOCS3‐mediated inactivation, which promotes in a feedforward manner IL‐6–STAT3 signaling and drives hepatocarcinogenesis (Figure 7F). Together, our data show that the malignant progression of liver cancer progenitors to fully established HCC requires increased KAT7‐acetylated and cytoplasm‐translocated G protein GαS.
Supplementary Material
Acknowledgments
AUTHOR CONTRIBUTIONS
Ye Zhou, Kaiwei Jia, and Suyuan Wang performed the experiments and contributed equally to the whole study. Zhenyang Li, Yunhui Li, Shan Lu, Yingyun Yang, Liyuan Zhang, Mu Wang, Yue Dong, Luxin Zhang, and Wannian Zhang provided reagents and performed experiments. Nan Li and Yizhi Yu provided reagents and analyzed the data. Xuetao Cao and Jin Hou analyzed the data and wrote the paper. Jin Hou designed and supervised the study. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
Nothing to report.
Footnotes
Abbreviations: AC, adenylyl cyclase; APT1, acyl protein thioesterase 1; CD, cluster of differentiation; co‐IP, coimmunoprecipitation; DEN, diethylnitrosamine; EPCAM, epithelial cell adhesion molecule; GPCR, G protein–coupled receptor; GαS, α subunit of G stimulating protein; HcPC, HCC progenitor cell; JAK, Janus kinase; KAT7, lysine acetyltransferase 7; miRNA/miR, microRNA; PIAS, protein inhibitor of activated STAT; PTM, posttranslational modification; Saa1, serum amyloid A1; SOCS, suppressor of cytokine signaling; STAT3, signal transducer and activator of transcription 3.
Ye Zhou, Kaiwei Jia, and Suyuan Wang share co-first authorship.
Funding information Supported by the National Natural Science Foundation of China (81701566, 91842104, 82171749, 82171755, 81871229, 82073184, 82171811, U20A20136), the Shanghai Sailing Project (17YF1424600), the Program of Shanghai Academic Research Leader (19XD1424900), the Shanghai Chenguang Program (18CG39), and the Shanghai Shuguang Program (20SG39)
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.hepjournal.com.
Contributor Information
Xuetao Cao, Email: caoxt@immunol.org.
Jin Hou, Email: houjin@immunol.org.
REFERENCES
- 1.McGlynn KA, Petrick JL, El‐Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2021;73:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Younossi Z, Tacke F, Arrese M, Sharma BC, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2019;69:2672–82. [DOI] [PubMed] [Google Scholar]
- 3.He G, Dhar D, Nakagawa H, Font‐Burgada J, Ogata H, Jiang Y, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL‐6 signaling. Cell. 2013;155:384–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ichinohe N, Kon J, Sasaki K, Nakamura Y, Ooe H, Tanimizu N, et al. Growth ability and repopulation efficiency of transplanted hepatic stem cells, progenitor cells, and mature hepatocytes in retrorsine‐treated rat livers. Cell Transplant. 2012;21:11–22. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Z, Hao X, Yan M, Yao M, Ge C, Gu J, et al. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. 2010;126:2067–78. [DOI] [PubMed] [Google Scholar]
- 6.Dhar D, Antonucci L, Nakagawa H, Kim JY, Glitzner E, Caruso S, et al. Liver cancer initiation requires p53 inhibition by CD44‐enhanced growth factor signaling. Cancer Cell. 2018;33:1061–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He GB, Karin M. NF‐κB and STAT3—key players in liver inflammation and cancer. Cell Res. 2011;21:159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88‐dependent IL‐6 production. Science. 2007;317:121–4. [DOI] [PubMed] [Google Scholar]
- 9.Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL‐6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bard‐Chapeau E, Li S, Ding J, Zhang S, Zhu H, Princen F, et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell. 2011;19:629–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Hayre M, Vázquez‐Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, et al. The emerging mutational landscape of G‐proteins and G‐protein coupled receptors in cancer. Nat Rev Cancer. 2013;13:412–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nault JC, Fabre M, Couchy G, Pilati C, Jeannot E, Tran Van Nhieu J, et al. GNAS‐activating mutations define a rare subgroup of inflammatory liver tumors characterized by STAT3 activation. J Hepatol. 2012;56:184–91. [DOI] [PubMed] [Google Scholar]
- 13.Han Y, Liu Q, Hou J, Gu Y, Zhang YI, Chen Z, et al. Tumor‐induced generation of splenic erythroblast‐like Ter‐cells promotes tumor progression. Cell. 2018;173:634–48.e12. [DOI] [PubMed] [Google Scholar]
- 14.Mertins P, Tang LC, Krug K, Clark DJ, Gritsenko MA, Chen L, et al. Reproducible workflow for multiplexed deep‐scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography‐mass spectrometry. Nat Protoc. 2018;13:1632–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan M, Chen X, Sun Y, Jiang LI, Xia Z, Ye K, et al. ZDHHC12‐mediated claudin‐3 S‐palmitoylation determines ovarian cancer progression. Acta Pharm Sin B. 2020;10:1426–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sung HY, Yang SD, Park AK, Ju W, Ahn JH. Aberrant hypomethylation of solute carrier family 6 member 12 promoter induces metastasis of ovarian cancer. Yonsei Med J. 2017;58:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shinmura K, Kato H, Igarashi H, Inoue Y, Nakamura S, Du C, et al. CD44–SLC1A2 fusion transcripts in primary colorectal cancer. Pathol Oncol Res. 2015;21:759–64. [DOI] [PubMed] [Google Scholar]
- 18.Hou J, Lin LI, Zhou W, Wang Z, Ding G, Dong Q, et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR‐199a/b‐3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011;19:232–43. [DOI] [PubMed] [Google Scholar]
- 19.Lee JW, Stone ML, Porrett PM, Thomas SK, Komar CA, Li JH, et al. Hepatocytes direct the formation of a pro‐metastatic niche in the liver. Nature. 2019;567:249–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sunahara RK, Tesmer JJ, Gilman AG, Sprang SR. Crystal structure of the adenylyl cyclase activator Gsα. Science. 1997;278:1943–47. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Qian C, Cao X. Post‐translational modification control of innate immunity. Immunity. 2016;45:15–30. [DOI] [PubMed] [Google Scholar]
- 22.Christiane K, Eberhard K. Gαs is palmitoylated at the N‐terminal glycine. EMBO J. 2003;22:826–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen JJ, Marsden AN, Scott CA, Akimzhanov AM, Boehning D. DHHC5 mediates β‐adrenergic signaling in cardiomyocytes by targeting Gα proteins. Biophys J. 2020;118:826–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duncan JA, Gilman AG. A cytoplasmic acyl‐protein thioesterase that removes palmitate from G protein alpha subunits and p21(RAS). J Biol Chem. 1998;273:15830–7. [DOI] [PubMed] [Google Scholar]
- 25.Brent RM, Nevin AL. Activated G protein Gαs samples multiple endomembrane compartments. J Biol Chem. 2016;291:20295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaas L, Pucci F, Messal HA, Andersson AB, Ruiz EJ, Gerling M, et al. Lgr6 labels a rare population of mammary gland progenitor cells that are able to originate luminal mammary tumours. Nat Cell Biol. 2016;18:1346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang H, et al. EpCAM‐positive hepatocellular carcinoma cells are tumor‐initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding H, Zhang X, Su Y, Jia C, Dai C. GNAS promotes inflammation‐related hepatocellular carcinoma progression by promoting STAT3 activation. Cell Mol Biol Lett. 2020;25:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iglesias‐Bartolome R, Torres D, Marone R, Feng X, Martin D, Simaan M, et al. Inactivation of a Gα(s)–PKA tumour suppressor pathway in skin stem cells initiates basal‐cell carcinogenesis. Nat Cell Biol. 2015;17:793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson CH, McIntyre RE, Arends MJ, Adams DJ. The activating mutation R201C in GNAS promotes intestinal tumourigenesis in Apc(Min/+) mice through activation of Wnt and ERK1/2 MAPK pathways. Oncogene. 2010;29:4567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patra KC, Kato Y, Mizukami Y, Widholz S, Boukhali M, Revenco I, et al. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA‐mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol. 2018;20:811–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu C, Xia J, Zhou Y, Lu X, Zhang L, Gou M, et al. Loss of Gsα impairs liver regeneration through a defect in the crosstalk between cAMP and growth factor signaling. J Hepatol. 2016;64:342–51. [DOI] [PubMed] [Google Scholar]