

Abstract
Cancer is a disease of cells gone off the rails, of biochemical pathways that have escaped the regulatory bounds that define normal homeostatic balance. This balance is maintained through precise spatiotemporal regulation, and the formation of biomolecular condensates via liquid-liquid phase separation (LLPS) has recently emerged as a widespread mechanism underlying the spatiotemporal coordination of biological activities in cells. Biomolecular condensates are widely observed to directly regulate key cellular processes involved in cancer cell pathology, and the dysregulation of LLPS is increasingly implicated as a previously hidden driver of oncogenic activity. Here, we offer our perspective on how LLPS shapes the biochemical landscape of cancer cells.
INTRODUCTION
Cellular processes are tightly coordinated in both space and time to ensure proper biological function. Disrupting this precise spatiotemporal regulation can have devastating pathological consequences, as exemplified by the broad dysregulation of basic cellular processes in cancer, including transcription, genomic integrity, quality control pathways, and intracellular signaling1–6, which underlies the acquisition of disease hallmarks such as uncontrolled cell growth and proliferation, increased cell survival, and metabolic reprogramming7–9. Unraveling the mechanisms of spatiotemporal coordination in biological pathways can therefore shed light on the molecular basis of pathway disruption in cancers.
Spatiotemporal control is often achieved by sequestering biological reactions within discrete subcellular compartments, such as organelles, with distinct biochemical environments. Increasingly, subcellular compartmentation is being found to arise through higher-order molecular assemblies, variously known as membraneless organelles, droplets, puncta, granules, and of late, biomolecular condensates, formed via liquid-liquid phase separation (LLPS)10. Biomolecular condensates lack the lipid membranes of classic intracellular organelles, as well as the fixed stoichiometries of more traditional macromolecular complexes, instead being defined by weak, multivalent interactions between biological polymers (e.g., proteins, RNA, DNA), which coalesce into stable, micron-sized bodies with distinct compositions from the cellular milieu10,11. The past decade has revealed a pervasive role for biomolecular condensates in organizing myriad biochemical pathways throughout the cell, upending established views of cellular compartmentation via membrane bound organelles. Recent evidence has also begun to highlight the particular importance of biomolecular condensates in not only controlling cancer-related processes but also in directly promoting oncogenic dysregulation12–16, thus helping to reconcile conflicting observations and longstanding mysteries in the field while also opening up new avenues for investigating disease pathology and treatment.
Here, we examine how compartmentation via LLPS defines the biochemical landscape of cancer cells. We first summarize the thermodynamic and molecular principles underlying LLPS and the formation of biomolecular condensates. Then, we take a detailed look at several key cellular processes that are critical to cancer development, focusing specifically on the role of LLPS in controlling nuclear function, regulating cellular quality control, and organizing biochemical networks, followed by a discussion of recent studies highlighting the emerging view of dysregulated LLPS as a cryptic driver of cancer pathology. Finally, we look ahead to future strategies for investigating the molecular function and therapeutic potential of LLPS in cancer.
PHASE SEPARATION AT A GLANCE
LLPS describes the spontaneous de-mixing of a homogeneous solution into two or more distinct phases. Phase separation occurs when interactions among groups of like molecules overcome the tendency to remain disordered in solution (i.e., entropy)11,17, causing these molecules to become enriched in the de-mixed (i.e., condensed) phase and depleted from the bulk (i.e., diffuse) solution (Figure 1a). Surface tension causes the condensed phase to adopt a spherical shape and form droplets within the diffuse phase. Molecular interactions also shift the free-energy landscape such that, although individual molecules freely diffuse between phases, there is no chemical potential difference across the phase boundary, and thus no net diffusive flux, despite a dramatic concentration gradient. Hence, the two phases dynamically exchange material over short timescales (e.g., seconds) (Figure 1b) while stably co-existing over long timescales (e.g., minutes or hours).
Figure 1. Basic principles of liquid-liquid phase separation and biomolecular condensates.
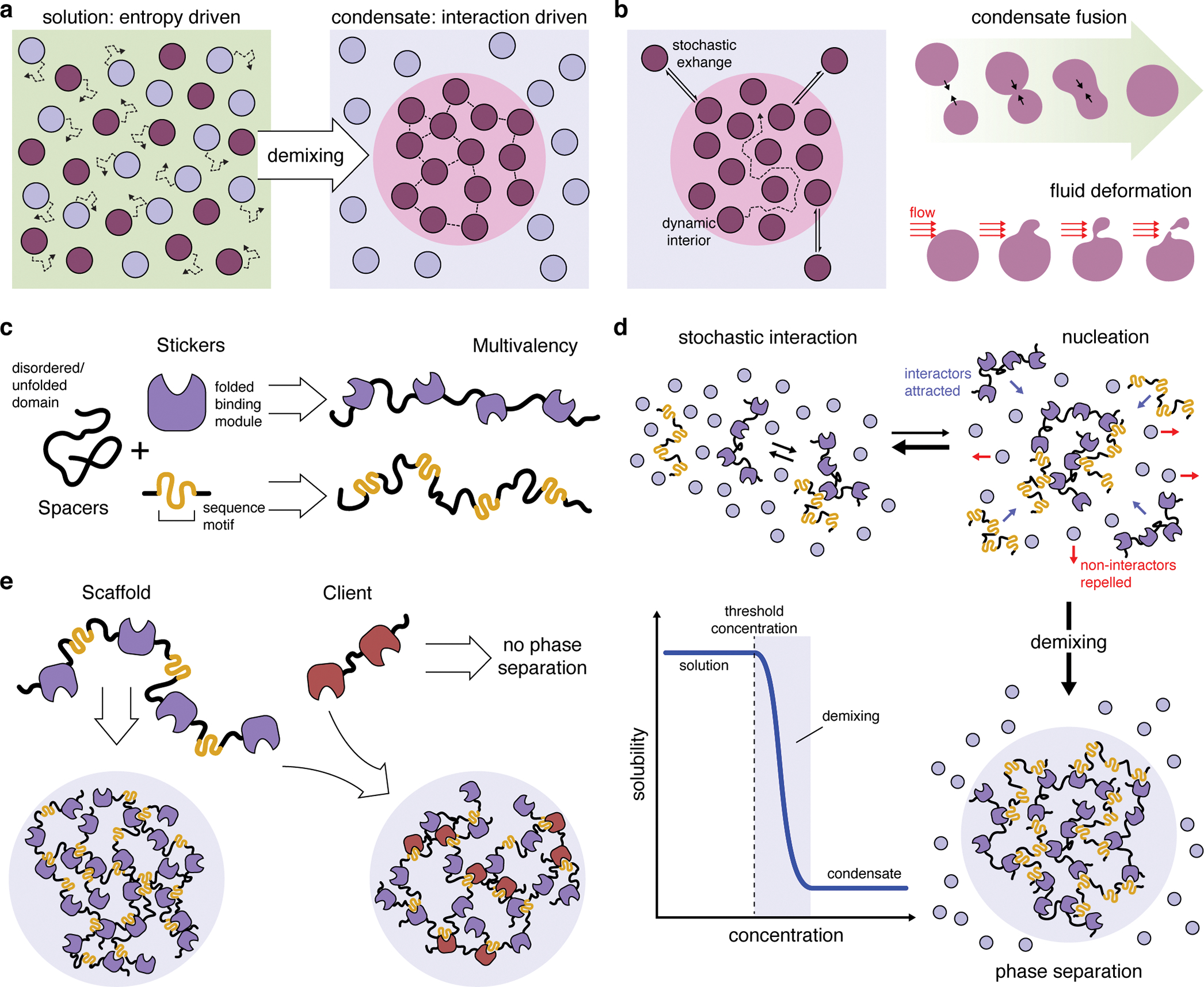
(a) Whereas entropy typically drives molecules to become dispersed in solution, mutual interactions among a subset of molecules can shift the free-energy landscape to favor de-mixing and drive the formation of a separate condensed phase. (b) Biomolecular condensates display liquid-like properties, such as highly dynamic interiors capable of rapid rearrangement and stochastic exchange with the bulk solution, as well as dynamic fusion, deformation, and fission events. (c) Phase separation of biomolecules is driven my multivalent binding interactions. These molecules often exhibit a sticker-and-spacer configuration, in which multiple folded binding modules and/or short interaction sequence (stickers) are separated by unfolded or disordered regions (spacers), thus achieving multivalency while promoting liquid-like behaviors. (d) At low concentrations, molecules capable of phase-separation interact randomly but remain in solution. Increasing the concentration of these molecules promotes nucleation of small complexes, which begin to exclude the surrounding solution. Each new interactor contributes binding sites that attract more interactors. This positive feedback loop results in switch-like de-mixing and phase separation (blue curve) once a critical threshold concentration is reached. (e) Phase-separating biomolecules are often categorized as scaffolds, which are both necessary and sufficient for phase separation, and clients, which can selectively partition into scaffold-containing condensates but cannot independently phase separate.
First suspected over a century ago18, the biological significance of LLPS is evidenced by the many membraneless structures found in cells10: these spherical bodies rapidly form and dissolve, can fuse and be deformed by shear flow, and exhibit dynamic internal organization (Figure 1b), as first revealed through elegant studies of Caenorhabditis elegans germline granules19. Biomolecular LLPS is driven by multivalency, or the ability to engage in multiple weak interactions that rapidly form, break, and reform10,17. Proteins achieve multivalency via tandem binding modules, as well as through repetitive motifs, often containing aromatic or charged residues, within stretches of low sequence complexity (Figure 1c). These interactor domains, called stickers, are typically interspersed between unfolded loops or within intrinsically disordered regions (IDRs), called spacers17,20 (Figure 1c). Nucleic acids often exhibit a similar multivalent sticker-and-spacer configuration and can thus also phase separate in the presence or absence of proteins. Together, these features promote liquid-like behavior by extending the range over which molecules interact, preventing them from becoming too densely packed and incapable of dynamic rearrangement11.
Multivalency gives rise to oligomeric complexes, whose growth decreases solubility while simultaneously promoting additional interactions10. This positive-feedback loop drives the switch-like phase transition that occurs at the critical threshold concentration (Figure 1d). Considering this exquisite concentration dependence, one way that cells can regulate condensate formation is thus by controlling the concentration (e.g., expression) of constituent molecules. These are loosely characterized as scaffolds, which are obligatory for condensate formation, and clients, which bind scaffolds and make up the bulk of the condensate but are largely dispensable for LLPS10,21 (Figure 1e). Cells can further regulate droplet composition by altering the relative stoichiometry and valency of scaffolds and clients. The physical properties of condensates can also vary, as condensates are not exclusively liquid-like. In fact, as the interaction density increases, particularly among IDR-rich constituents, condensates can “mature” from liquid-like to gel- or solid-like materials10. Notably, many of these properties can be modulated via post-translational (or post-transcriptional, in the case of RNA) modifications, which can add or remove interaction sites or alter surface charges, thus altering valency, solubility, and condensate dynamics10.
The ability of biomolecular condensates to concentrate specific molecules within stable, defined structures whose dynamic, liquid-like interiors are conducive of chemical reactions lends itself to the formation of biochemically distinct subcompartments. Indeed, biomolecular condensates are implicated in the spatiotemporal regulation of myriad cellular functions. Below, we highlight several key processes regulated by LLPS that also figure prominently in the pathophysiology of cancer cells.
NUCLEAR FUNCTION
Eukaryotic nuclei comprise a multitude of distinct microenvironments essential for the spatiotemporal regulation of nuclear processes. Visible disruption of this complex architecture due to the sweeping dysregulation of nuclear function is a prominent feature of cellular transformation and is frequently exploited in cancer diagnosis22. LLPS is already known to control the formation of constitutive nuclear substructures including nucleoli, speckles, Cajal bodies, promyelocytic leukemia (PML) bodies23,24, the latter playing a prominent role in cancer25–27, and mounting evidence reveals that LLPS is the primary driver of nuclear subcompartmentation23. Indeed, biomolecular condensates are increasingly found to play an integral role in diverse nuclear processes, such as gene expression28–31, DNA repair32,33, and chromatin organization34–36.
Regulation of gene expression
Activation of oncogenic transcriptional programs causes myriad changes in gene expression, often leading cancer cells to exhibit “transcriptional addiction”4 via the actions of so-called super-enhancers (SEs). These clusters of enhancers were initially identified by their extreme enrichment of transcriptional regulators such as bromodomain-containing protein 4 (BRD4) and the Mediator complex and are essential for maintaining cell-lineage-specific gene expression5. Numerous oncogenes also acquire SEs during tumorigenesis37, likely marking a critical step in oncogene activation. LLPS is predicted to underlie SE function38 (Figure 2a), and indeed, both BRD4 and MED1, a core Mediator subunit, were shown to phase separate via their IDRs upon in vitro reconstitution and to form liquid-like condensates at SE sites in cell nuclei28,29. BRD4 phase separation also requires its tandem bromodomains39, which increase valency through interactions with acetylated histones. Recent evidence also implicates the BRD4 short isoform (BRD4S) in driving BRD4 LLPS and influencing BRD4 function in gene transcription, as BRD4 puncta size mirrors BRD4S expression in cancer cells and changing BRD4S levels alone impacts puncta formation and gene transcription39. SEs are also highly enriched in enhancer RNAs (eRNAs) (Figure 2a), a class of long-noncoding RNA (lncRNA) transcribed from active enhancers and implicated in enhancer function40. RNA-protein interactions promote LLPS by increasing interaction valency41, and although their precise mechanism remains unclear, eRNAs have been implicated in SE phase separation40. Indeed, eRNA-containing ribonucleoprotein condensates were recently shown to drive rapid ligand-induced SE assembly and activation by estrogen receptor α in breast cancer42. Finally, the nucleated assembly of transcriptional regulators via LLPS has been implicated in positive feedback-mediated amplification of transcriptional activity43 (Figure 2a). LLPS thus seem to play an essential role in promoting robust oncogene activation and expression to establish and maintain cancer cell identity.
Figure 2. The regulation of nuclear function by liquid-liquid phase separation.
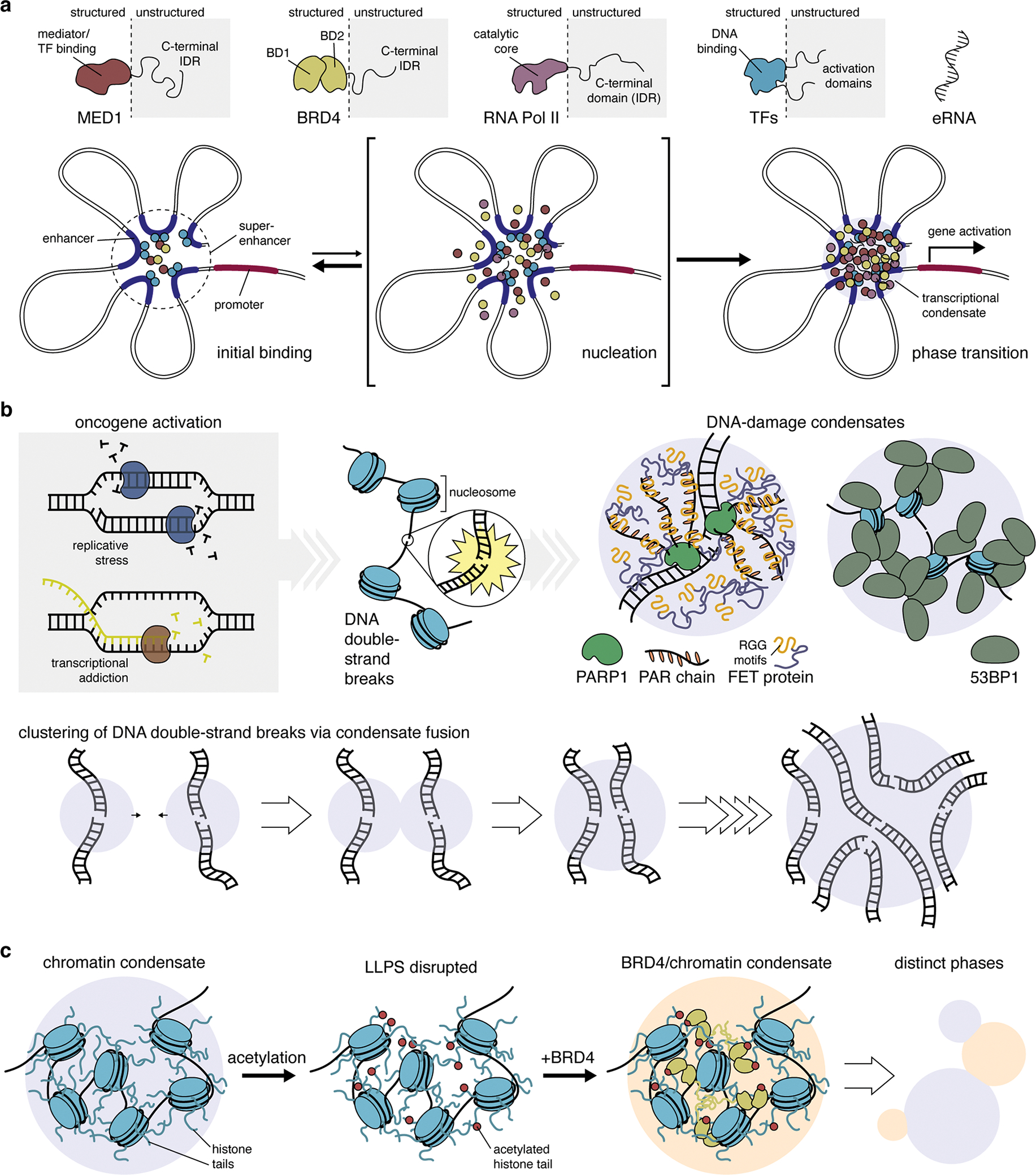
(a) Many transcriptional regulators, such as mediator complex subunit 1 (MED1), bromodomain-containing protein 4 (BRD4), RNA polymerase II (RNA Pol II), and various transcription factors (TFs) feature large intrinsically disordered regions (IDRs) that allow them to undergo phase separation and form biomolecular condensates at super-enhancer sites. Initial binding of TFs and other regulatory proteins at neighboring enhancer sites triggers the nucleation of additional factors via multivalent interactions mediated by their IDRs. This short-lived intermediate rapidly gives rise to a phase transition, yielding a transcriptional condensate that drives gene activation. Thus, LLPS is thought to play a key role in the robust, switch-like regulation of cell-identity genes by super-enhancers. (b) High levels of replicative stress and excessive transcription induced by oncogene activation cause the accumulation of genome damage, including DNA double-strand breaks (DSBs). The binding of poly-(ADP-ribose) polymerase 1 (PARP1) and synthesis of PAR chains at DSB sites leads to the recruitment of disordered FET-family proteins via their RGG repeats, resulting in the formation of phase-separated condensates. Similarly, P53 binding protein 1 (53BP1) binds nucleosome proteins near DSB sites, resulting in the formation of phase-separated condensates via 53BP1 oligomerization. Notably, 53BP1 condensates have been observed to fuse, suggesting a possible mechanism of the clustering of DSBs in cells (lower panel). (c) Chromatin undergoes LLPS as a result of multivalent interactions driven by intrinsically disordered histone tails. LLPS of chromatin can be modulated by post-translational modification of histone tails, such as acetylation, which disrupts condensate formation. The addition of BRD4, which binds acetylated histone tails, restores LLPS of acetylated chromatin. The resulting condensates cannot mix with unmodified chromatin condensates, yielding two distinct phases, similar to distinct chromatin domains in the nucleus.
Aspects of this model have recently been challenged by data suggesting that SE proteins (e.g., MED1, BRD4) are not required for enhancer-promoter interactions44–46, leading some to completely dismiss the role of LLPS in transcriptional regulation47. However, these studies do not rule out a specific role for LLPS in transcriptional activation, and in fact observed that loss of SE proteins disrupted transcription, particularly of lineage-specific genes44–46. Thus, although condensates may not drive all aspects of SE function, LLPS may still play a key role.
The transcriptional co-activator Yes-associated protein (YAP) and its paralogue transcriptional co-activator with PDZ-binding motif (TAZ) are another set of key players linked to cancer cell transcriptional addiction via SEs48. YAP and TAZ are widely activated in cancer cells49, driving proliferation, survival, and invasiveness, as well as stem-like properties. Both YAP and TAZ form condensates in vitro and in cell nuclei through their protein-interaction domains31,50 and extensive intrinsic disorder30,50. YAP/TAZ condensates associate with transcriptionally permissive chromatin regions, recruit transcriptional components such as RNA polymerase II (Pol II), TEA-domain-family transcription factors, BRD4, and MED1, and even co-localize with nascent mRNAs30,31,50, demonstrating that YAP/TAZ condensates represent transcriptionally active SE sites. Disrupting YAP/TAZ condensates abolishes downstream gene expression, further confirming the importance of LLPS in YAP/TAZ transcriptional regulation30,31,50. Curiously, YAP and TAZ phase separate under distinct conditions. Nuclear TAZ condensates appear to form constitutively in proliferating cells and were in fact shown to be elevated in cancer versus healthy tissues31. YAP condensates fail to form under similar conditions but are induced by external stimuli such as hyperosmotic stress30 and interferon (IFN)-γ50. Despite their considerable homology, YAP and TAZ exhibit non-redundant functions, likely mediated by differential interactions with binding partners51. Differences in YAP/TAZ phase separation may further highlight distinct roles in regulating cancer proliferation versus adaptation and survival. Indeed, IFN-γ-induced YAP phase separation confers resistance to α-PD1 immunotherapy in tumor cells, which are sensitized by YAP condensate disruption50.
DNA damage and repair
Aberrant cell cycle regulation triggered by oncogene activation places cancer cells in a constant state of replicative stress, leading to the frequent generation of DNA double-strand breaks (DSBs)52 (Figure 2b). DSBs are a major source of genome instability in cancer1, resulting in persistent activation of the DNA damage response (DDR) pathway52,53. An early step in the DDR is the deposition of poly(ADP-ribose) (PAR) polymers at DSB sites by poly(ADP-ribose) polymerase 1, which has been shown to trigger LLPS of intrinsically disordered FET proteins (fused in sarcoma [FUS]/Ewing sarcoma/TATA box-binding protein-associated factor 15) at DSBs32 (Figure 2b). Another key factor in the DDR is p53-binding protein 1 (53BP1), which localizes at nuclear foci corresponding to DSB sites53. Nuclear 53BP1 foci were recently shown to exhibit properties of biomolecular condensates, including liquid-like internal dynamics and coalescence of individual foci33,54, the latter being consistent with the clustering of DSBs, which may aid chromosomal translocations55 (Figure 2b). Interestingly, FET protein recruitment to PAR chains is mediated by their Arg/Gly-rich (RGG) LC domains32,56, which also participate in RNA binding, and PAR chains appear to compete with mRNAs for FUS binding56. Although FET protein RNA binding has not been directly implicated in LLPS at DSBs, RNA does play a role in forming DSB foci. Specifically, DSBs actively recruit Pol II, MED1, and other factors to transcribe damage-induced lncRNAs, which were recently shown to facilitate 53BP1 condensate formation54. Ironically, transcription itself can induce DNA breaks57 (Figure 2b), and the need for cancer cells to balance repairing DNA damage with feeding transcriptional addiction likely places tremendous strain on their transcriptional apparatus. Further insights into how LLPS regulates DSB repair may thus reveal opportunities to therapeutically exploit this delicate balance.
Chromatin organization
The assembly of eukaryotic nuclear DNA into chromatin is essential for genome function and regulation. Recent work has revealed that nucleosome/DNA arrays, the fundamental unit of chromatin, are intrinsically capable of undergoing LLPS34. Condensate formation depended on intrinsically disordered histone tails and was also sensitive to post-translational modifications, being dispersed by histone acetylation, a typical marker of transcriptionally active, de-condensed chromatin. Strikingly, the addition of multi-bromodomain-containing proteins reformed droplets that were immiscible with un-acetylated chromatin condensates, suggesting that LLPS spontaneously organizes chromatin into distinct physical and functional domains34. Chromatin LLPS was further increased by addition of the linker histone H1, which caused condensates to become more dense and less internally dynamic, consistent with the role of H1 in heterochromatin formation34. Heterochromatin plays major roles in gene silencing and genome stability, and altered heterochromatin is a frequent hallmark of disrupted chromatin organization in cancer22. Another major component, heterochromatin protein 1 α (HP1α), has also been shown to undergo LLPS in vitro and in cells35,36,58. LLPS of human HP1α required DNA binding or phosphorylation of its unstructured N-terminus, either of which promoted multivalency through its disordered hinge domain35. HP1α was also recently shown to promote nucleosome LLPS through conformational rearrangements that expose buried histone tails, thereby increasing overall disorder36. Notably, peptides can be identified to bind the interface between HP1α dimers to either increase or decrease LLPS35, hinting at a potential novel strategy of targeting HP1α LLPS to rescue chromatin alterations in cancers where HP1α expression is upregulated59. However, such enthusiasm is tempered by recent work showing little HP1α LLPS in cells and a largely dispensable role for HP1α maintaining heterochromatin60, highlighting the need for further investigation.
CELLULAR QUALITY CONTROL
Effective quality control mechanisms are essential for maintaining cellular homeostasis61. These pathways rapidly clear damage to proteins and/or organelles incurred through changes in growth, metabolism, or adaptations to environmental stress. Two major quality control processes in cells, the ubiquitin-proteasome system and autophagy, are highly interconnected pathways that also play important roles in cancer6. These and other adaptive pathways are often exploited by cancer cells for survival62–64. Recent studies have begun to uncover evidence strongly linking LLPS to proteasomal degradation and autophagy, deepening our understanding of their pathophysiological mechanisms and potentially illuminating new avenues for therapeutic targeting.
Proteasomal degradation
Proteins bound for the ubiquitin-proteasome system are typically selected and targeted via a series of adaptor proteins and enzymes that ultimately catalyze the addition of poly-ubiquitin chains, marking the target protein for degradation61 (Figure 3a). One such adaptor protein, speckle-type POZ protein (SPOP) is a known tumor suppressor that regulates the degradation of numerous oncoproteins65. While SPOP is known to associate with existing nuclear condensates, SPOP has also been shown to undergo LLPS itself66. SPOP was observed to form phase-separated condensates in conjunction with its targeted protein death-domain-associated protein (DAXX), both in vitro and in cell nuclei. Phase separation was mediated both by SPOP oligomerization through its self-interaction domains and by multivalent interaction with the DAXX C-terminal IDR (Figure 3a). Co-condensation of SPOP and DAXX also triggered DAXX polyubiquitination, which was disrupted by mutants lacking LLPS capability, indicating that SPOP utilizes LLPS for efficient substrate targeting66. More recent work suggests that the proteasome is also capable of undergoing LLPS, as evidenced by the formation of proteasome-containing biomolecular condensates in the nuclei of various cell lines under acute hyperosmotic stress67. Proteasomal condensates were found to be sites of active proteolysis and to form through multivalent interactions between polyubiquitinated substrates and ubiquitin-binding domains in the proteasomal shuttle protein RAD23B67. Although it remains to be seen whether proteasome condensates can be triggered by other stress conditions, cancer cells are known to exhibit severe proteotoxic stress68 and to upregulate proteasomal activity for survival69. Further insights into LLPS within this pathway may therefore yield strategies to augment the growing use of proteasome inhibitors for cancer treatment63.
Figure 3. The role of liquid-liquid phase separation in cellular quality control.
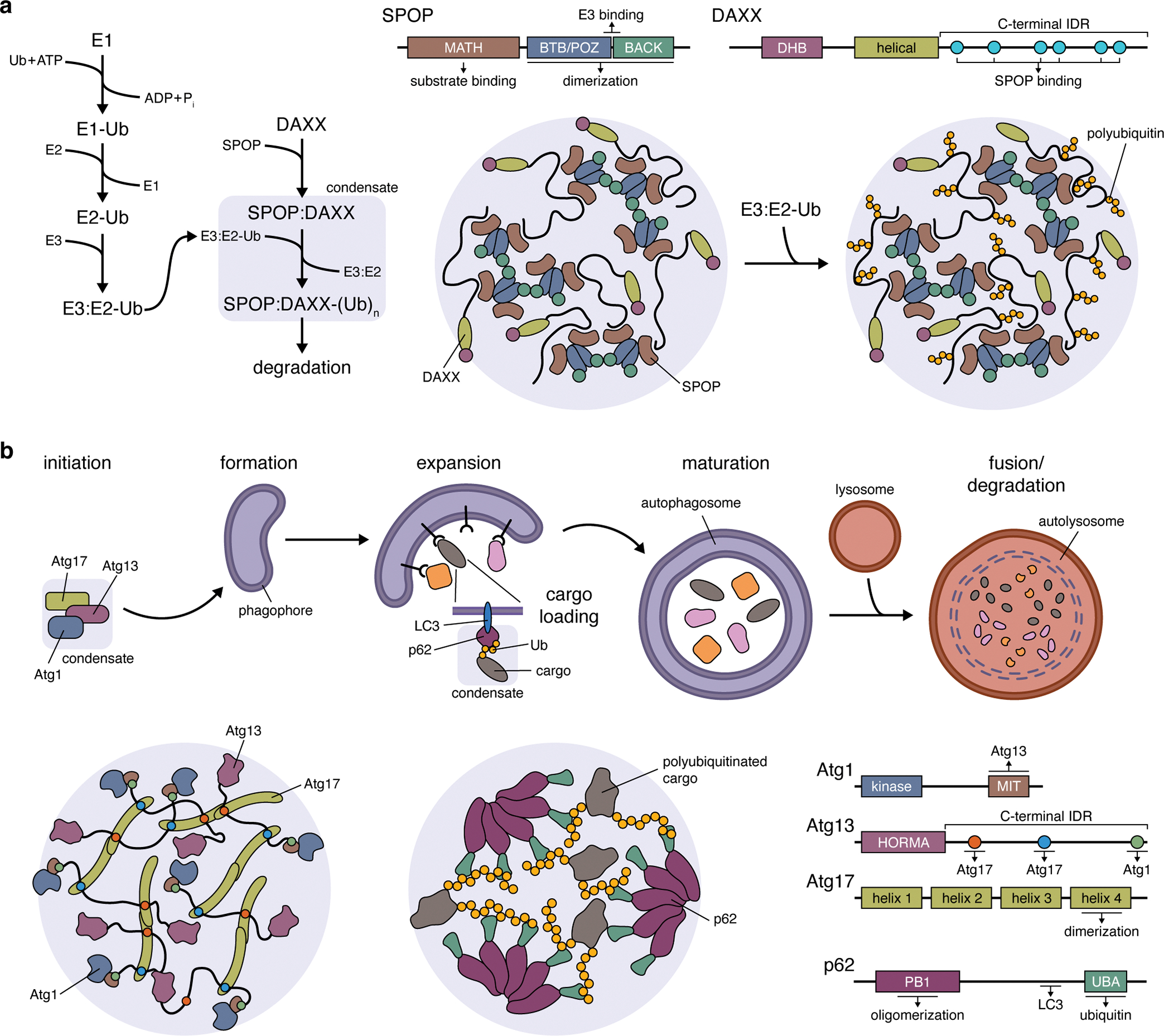
(a) The formation of biomolecular condensates between speckle-type POZ protein (SPOP) and its target death-domain-associated protein (DAXX). Left: Pathway schematic highlighting the role of SPOP:DAXX condensates in facilitating DAXX polyubiquitination. Right: LLPS of SPOP and DAXX is driven by the formation of SPOP dimers through its BR-C/ttk/bab (BTB)/Pox virus and zinc finger (POZ) domain, interactions between SPOP dimers mediated by their BTB and C-terminal Kelch (BACK) domains, as well as interactions between the long, C-terminal IDR of DAXX with multiple copies of the N-terminal meprin and TRAF homology (MATH) domain of SPOP. The BTB/POZ-BACK domain junction in SPOP also binds E3 ubiquitin ligases, recruiting them into condensates (omitted from cartoon) to promote DAXX polyubiquitination. Ub, ubiquitin; (Ub)n, polyubiquitin; E1, Ub-activating enzyme; E2, Ub-conjugating enzyme; E3, Ub-ligase. (b) Top: Simplified schematic of the autophagy pathway, highlighting the involvement of Atg1, Atg13, and Atg17 condensates in autophagy initiation and the role of p62 condensates in cargo loading during phagophore expansion. Bottom: Atg1:Atg13:Atg17 condensates (left panel) are scaffolded by Atg13 and Atg17, with the C-terminal IDR of Atg13 containing a pair of binding sites that crosslink Atg17 dimers. Atg1 is recruited as a client protein via its C-terminal microtubule-interacting and transport (MIT) domain, which binds Atg13. Bottom: LLPS of p62 condensates (middle panel) requires the formation of p62 oligomers, mediated by an N-terminal Phox and Bem1p (PB1) domain, whose C-terminal ubiquitin-associated (UBA) domains then participate in multivalent interactions with the ubiquitin chains on polyubiquitinated substrate proteins.
Autophagy
The autophagy pathway channels aberrant proteins and damaged organelles (e.g., mitochondria, endoplasmic reticulum) to the lysosome for degradation and recycling61. During autophagy, poly-ubiquitinated cellular components are bound and sequestered by adaptor proteins such as p62, followed by engulfment within double-membrane structures known as autophagosomes (Figure 3b). Autophagosome formation is triggered by the activation of a protein complex containing unc51-like kinase 1/2 (ULK1/2), autophagy-related protein 13 (ATG13), and 200 kD focal adhesion kinase family interacting protein (FIP200). Recently, the yeast counterparts of these proteins, Atg1, Atg13, and Atg17, were found to assemble into a dynamic liquid-like condensate both in vitro and in living yeast cells70 (Figure 3b). Condensate formation was primarily scaffolded by interactions between IDRs in Atg13 and specific sites in Atg17 dimers. Atg13 also mediated condensate localization to the yeast vacuole membrane70. ULK1/2 and ATG13 also both contain IDRs and analogously seed autophagosome formation on the ER surface71,72. It thus seems plausible to speculate that ULK/ATG13/FIP200 LLPS occurs in higher eukaryotes. Meanwhile, p62 has previously been shown to form poly-ubiquitin-containing cytosolic inclusions in cells, which were recently revealed to be liquid-like biomolecular condensates73. LLPS was triggered by poly-ubiquitinated proteins, with longer poly-ubiquitin chains more effectively driving condensation, and also required p62 oligomerization (Figure 3b). In cells, p62 bodies were observed to recruit the autophagosome receptor LC3 and could in fact be engulfed by autophagosomes73, suggesting that p62 LLPS plays an active role in cargo trafficking.
Autophagy is important for cell survival not only as a quality control pathway but also as a source for raw materials for metabolic pathways during nutrient-deficient conditions. Thus, autophagy has a complex relationship to tumorigenesis, being implicated in tumor suppression through the elimination of pre-cancerous cells while also being essential for the survival of more advanced tumors by sustaining tumor growth74,75. Mutations in autophagy components, including ULK1/2, have been detected in various cancers74,76, though their role in driving tumorigenesis is debated77. Nevertheless, autophagy is emerging as a promising target for cancer therapy75, and further investigation into the links between LLPS and autophagy in cancer may yet reveal important new insights.
BIOCHEMICAL PATHWAY ORGANIZATION
Subcellular compartmentation through the assembly of higher-order molecular complexes is critical for organizing biochemical pathways within cells78, whether it be to channel metabolic flux or maintain the specificity of intracellular signaling. The accelerating pace of discovery over the last 5 years has unequivocally demonstrated that molecular assembly via LLPS is a major force governing the spatiotemporal organization of biochemical pathways.
Condensates regulate metabolic flux
Cancer cells are typified by dysregulated metabolism. Famously, the Warburg effect was coined almost a century ago to describe the reliance of tumors on glycolysis rather than aerobic respiration to metabolize glucose9. Rather than being a mere symptom of energetic imbalance, the Warburg effect is now understood as a hallmark of the extensive metabolic reprogramming that shifts biosynthetic pathways to meet the demands of uncontrolled proliferation9. For example, increases in glycolysis drive metabolic flux through the pentose phosphate and de novo serine synthesis pathways, both of which are upregulated in cancers and function to supply critical metabolites required to support tumor cell proliferation and survival79,80. These pathways are also both linked to the formation of the glucosome, a multi-enzyme metabolic complex (i.e., metabolon81) that comprises four rate-limiting cytosolic enzymes involved in glycolysis and gluconeogenesis82. Notably, glucosome size has been found to increase as a function of flux through the pentose phosphate and serine synthesis pathways, with cancer cells exhibiting higher numbers of large glucosome puncta compared with normal cells, suggesting a link to cancer metabolism82.
Another well-studied cytosolic metabolon, the purinosome, encompasses all of the enzymes responsible for catalyzing de novo purine biosynthesis83. Itself an offshoot of the pentose phosphate pathway, de novo purine biosynthesis is also upregulated in cancers and has been implicated in therapeutic resistance84 and metastasis85. Purinosomes rapidly form in cells upon purine depletion83, and upregulated purinosome formation was recently observed in highly metastatic breast cancer cells. Emerging evidence also suggests that purinosomes can be induced by hypoxia86, a hallmark of the tumor microenvironment.
The formation of metabolons is thought to enhance the specificity and efficiency of metabolic reactions by insulating metabolic intermediates from the bulk milieu and allowing them to pass directly between enzymes that catalyze sequential reaction steps (i.e., substrate channeling)81, as was elegantly demonstrated for purinosomes using mass spectrometry imaging87. Both glucosomes and purinosomes appear as roughly circular cytoplasmic puncta that exhibit characteristic liquid-like internal dynamics of biomolecular condensates in cells82,88, though additional mechanistic factors (e.g., concentration dependence, sources of multivalency, scaffold-client relationships) will need to be resolved before these structures can be regarded as examples of LLPS. Nevertheless, these liquid-like metabolic assemblies are emerging as potentially central factors in metabolic rewiring and key targets for deeper investigation.
Phase separation in intracellular signaling
Intracellular signaling pathways form a critical regulatory network governing cellular behavior and homeostasis, the dysregulation of which underlies multiple facets of cancer pathology3. The molecular elements that promote LLPS are common features of signaling proteins89, and indeed, a growing body of evidence argues compellingly for biomolecular condensates as pervasive organizers of the signaling machinery. Through these studies, we are also beginning to unravel the direct functional role of LLPS in shaping biochemical dynamics.
Phase separation controls cytosolic DNA sensing and immune signaling
The presence of cytoplasmic DNA is an acute danger signal that triggers innate immunity, cellular senescence, and cell death90. These responses are mediated by pattern recognition receptors such as cyclic 2’,3’ guanosine monophosphate-adenosine monophosphate (cGAMP) synthase (cGAS), which becomes activated upon dsDNA binding to synthesize cGAMP from ATP and GTP. cGAMP binds the ER transmembrane receptor stimulator of interferon (IFN) genes (STING), which then traffics to the Golgi apparatus and activates TBK1 and IKK, triggering type I IFN and proinflammatory cytokine production90.
dsDNA binding by cGAS was recently shown to induce LLPS of cGAS:DNA biomolecular condensates, both in vitro and in living cells91. cGAS binds DNA in a sequence-independent manner through multivalent interactions mediated by its catalytic core and its positively charged, disordered N-terminal domain (Figure 4b). cGAS phase separation not only requires its disordered N-terminus but is also enhanced by longer dsDNA, underscoring the critical role of multivalency in condensate formation. Importantly, cGAS condensates are catalytically active, and their disruption drastically impairs cellular cGAMP production91, directly implicating LLPS in pathway function. cGAMP production may be further enhanced through the exclusion of a negative regulator, the exonuclease TREX1, which degrades cytosolic dsDNA and thus antagonizes cGAS activation. In vitro, TREX1 phase separates to form a shell surrounding cGAS:DNA condensates, thereby restricting its activity and prolonging cGAMP production92. LLPS thus produces a discrete biochemical compartment for efficient DNA sensing and switch-like pathway activation91.
Figure 4. Liquid-liquid phase separation shapes signaling pathway function.
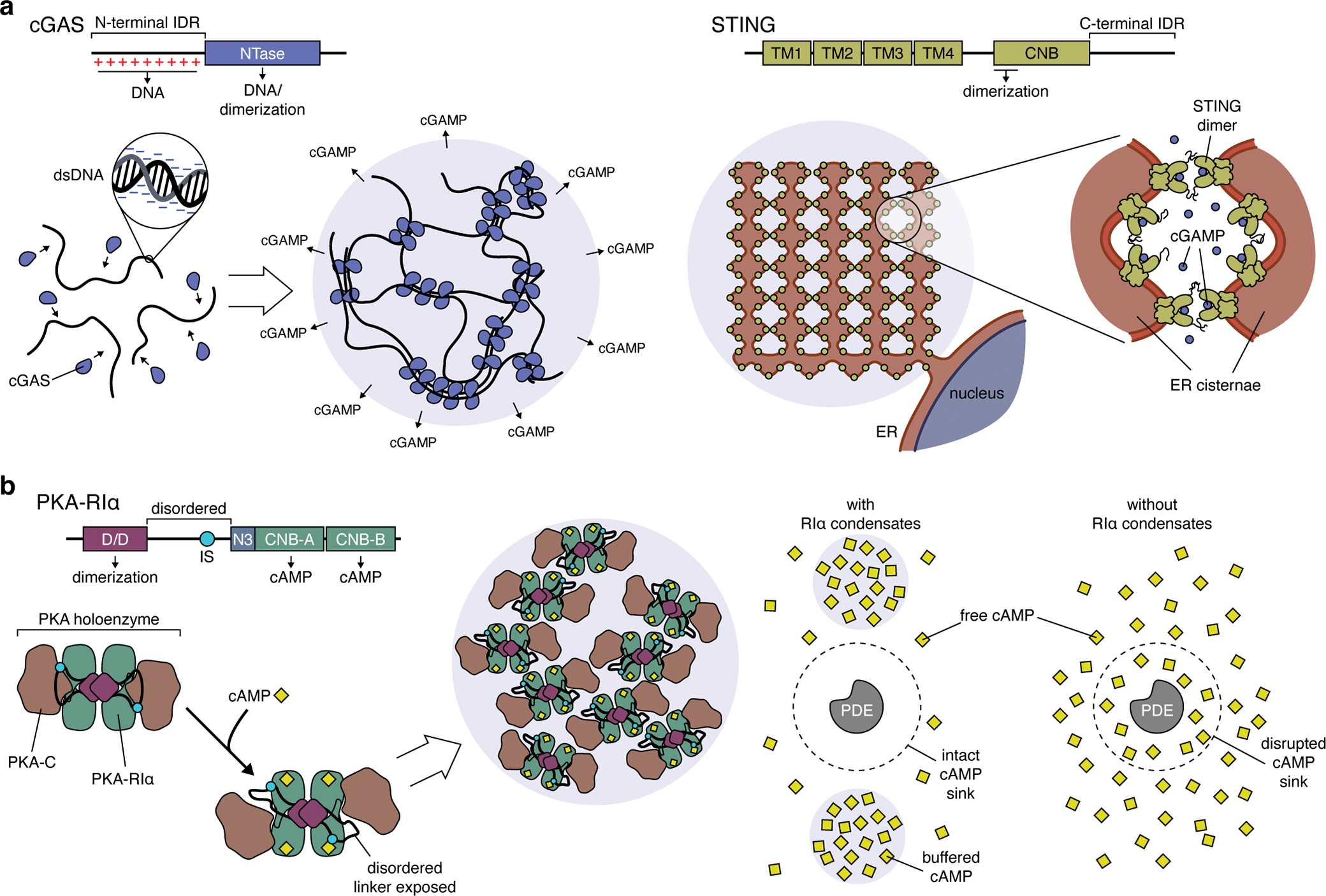
(a) Left: cGAS binds double-stranded DNA (dsDNA) via its positively charged N-terminal IDR and its C-terminal nucleotidyltransferase (NTase) domain, which also mediates cGAS dimerization. These multivalent interactions rapidly nucleate the formation of cGAS:dsDNA condensates, dramatically lowering the concentration threshold for cGAS activation and leading to ultrasensitive dsDNA detection and cGAMP production. Right: However, excess cGAMP production has been shown to trigger LLPS of the ER-resident cGAMP receptor STING, which requires interactions mediated by the C-terminal IDRs in STING dimers and forms condensates containing intricately folded membrane networks. Because LLPS requires cGAMP-bound STING, these condensates may function to buffer excess cGAMP and regulate immune signaling. (b) Left: The formation of PKA-RIα biomolecular condensates requires the docking and dimerization (D/D) domain and a disordered linker region containing an inhibitory site (IS) that binds the PKA-C subunit. The binding of cAMP to RIα triggers a conformational rearrangement in the PKA holoenzyme that exposes the disordered linker to help drive LLPS, though the precise molecular mechanism remains unclear. Right: RIα condensates dynamically buffer cAMP levels, allowing PDEs to efficiently degrade free cAMP and act as cAMP sinks. In the absence of RIα condensates, PDEs catalytic activity is overwhelmed, resulting in the collapse of cAMP sinks and the breakdown of compartmentalized cAMP signaling.
Cytoplasmic DNA is frequently present in cancer cells, in the form of cytoplasmic chromatin fragments and micronuclei generated via DNA damage and other nuclear aberrations, and activation of the cGAS-STING pathway is thought to play a tumor-suppressive role by stimulating senescence and immune-mediated clearance of pre-cancerous cells90. Indeed, many cancers show reduced or loss of cGAS and/or STING expression. cGAS-STING signaling has also been linked to antitumor immunity through host cell uptake of tumor DNA93 and tumor-produced cGAMP94, which triggers host-cell STING activation and immune responses. cGAS-STING activation thus represents an attractive therapeutic target and in fact contributes to the efficacy of established therapies90. However, emerging evidence that cGAS-STING proinflammatory signaling promotes tumorigencity95 represents a potential complication, as does the recent discovery of LLPS by STING to form ER-incorporating spherical condensates that buffer cGAMP and dampen immune responses96 (Figure 4b). Unravelling the dual role LLPS plays in tuning cGAS-STING signaling may thus prove essential for effectively targeting this pathway in cancer.
cAMP/PKA signaling compartmentation by phase separation
The ubiquitous intracellular messenger cyclic 3’5’ adenosine monophosphate (cAMP) is involved in regulating a diverse array of cellular processes, including gene expression, growth, proliferation, and migration97. cAMP is typically produced in response to G protein-coupled receptor-induced activation of adenylyl cyclases via the Gαs heterotrimeric G protein subunit. GNAS, which encodes Gαs, is the most frequently mutated G protein gene in cancer, with key mutational hotspots linked to constitutive Gαs activation98, implicating hyperactive cAMP production in oncogenesis. cAMP signaling is primarily mediated by the cAMP-dependent protein kinase (PKA), a tetrameric holoenzyme composed of a regulatory (R) subunit dimer bound to a pair of catalytic (C) subunits. Of the various R subunit isoforms (RIα, RIβ, RIIα, RIIβ), only RIα is ubiquitously expressed, and RIα loss through various mechanisms is linked to tumorigenesis99,100. RIα has recently been shown to undergo LLPS to form biomolecular condensates in vitro and at endogenous expression levels in cells101 (Figure 4c). LLPS of RIα requires its dimerization domain and a disordered linker region and is promoted by cAMP binding, which is thought to induce a conformational change in the holoenzyme that exposes the disordered linker101.
Functionally, RIα condensates are directly involved in the spatial control of cAMP signaling. The compartmentation of cAMP elevation within discrete subcellular microdomains is essential for encoding specificity in the regulation of downstream targets. Indeed, the idea of compartmentalized signaling was first articulated with respect to the cAMP pathway102. The classic model of cAMP signaling has focused on cAMP-degrading phosphodiesterases (PDEs) as the major drivers of compartmentation through the formation of sinks that consume cAMP and limit its subcellular mobility103. However, restricted diffusion through cAMP buffering is emerging as another critical mechanism underling compartmentation104. RIα condensates are strongly implicated as the source of cAMP buffering, as genetically encoded biosensors targeted directly to endogenous RIα in cells report saturating levels of cAMP inside droplets, which was also confirmed through in vitro droplet reconstitution using purified RIα and a fluorescent cAMP analogue101. In fact, RIα LLPS was shown to be required for effective cAMP compartmentation by PDEs, with loss of phase separation disrupting PDE-mediated cAMP sinks (Figure 4c). Notably, disrupting RIα condensates phenocopies the ability of RIα knock-out to induce cell proliferation and anchorage-independent growth, two markers of cell transformation101. Thus, it may be time to reconsider the role of cAMP and RIα in cancer through the lens of phase separation.
CONDENSATES IN THE DRIVER’S SEAT
Alongside work implicating LLPS in basic cellular processes that are intimately connected to cancer biology, recent studies are increasingly affirming a direct link between LLPS and tumorigenesis. In particular, prominent nuclear condensates such as PML bodies are being shown to play an expansive role in tumorigenesis25,26,105. A striking example comes from the study of paraspeckles105,106, which have been shown to form upon activation of p53, a critical tumor suppressor that directs multiple pathways, including cell cycle regulation, DNA repair, and cell death107,108. p53 induces paraspeckles by directly upregulating the scaffold lncRNA nuclear-enriched abundant transcript 1 (NEAT1) in response to various oncogenic stimuli105,106,109. Knocking out NEAT1 not only disrupts p53-induced paraspeckle formation but also prevents the formation of chemically induced skin tumors in mice, indicating that paraspeckles are required for tumorigenesis105. Cells lacking NEAT1 expression, and thus paraspeckles, were found to show elevated levels of replicative stress, DNA damage, and apoptosis compared with control cells in response to chemical carcinogens. p53 accumulation was also markedly prolonged in the absence of NEAT1105, suggesting that paraspeckle formation triggers a negative-feedback loop to limit DNA damaged and attenuate p53 signaling, allowing precancerous cells to survive and accumulate additional mutations (e.g., loss of p53) to drive cancer progression.
LLPS of the nuclear-localized A-kinase anchoring protein (AKAP) AKAP95, which regulates RNA splicing110, was also recently shown to directly promote tumorigenesis111. AKAP95 is overexpressed in a broad spectrum of cancers111–113, including ovarian, rectal, and breast cancers, and was shown to be required for in vitro proliferation and anchorage-independent growth, as well as in vivo tumor formation, by cancer cells111. Knocking down AKAP95 expression in cancer cells leads to the downregulation of pro-tumorigenic gene expression pathways (e.g., cell proliferation, DNA damage repair) and upregulation of tumor-suppressive pathways (e.g., senescence, apoptosis)111, suggesting that AKAP95 plays a similar role to that of paraspeckles in enabling survival and adaptation to oncogene-induced stress. The pro-tumorigenic effects of AKAP95 largely stem from its role in controlling the alternative splicing of multiple cancer-associated gene transcripts, such as CCNA2 and SMAD6, which was found to depend on AKAP95 phase separation111. AKAP95 LLPS behavior is mediated primarily by a core disordered region (residues 101–210) that is sufficient to induce droplet formation in vitro, whereas deleting this entire region or mutating key residues blocks both in vitro droplet formation and the appearance of AKAP95 nuclear puncta111. Importantly, these LLPS-disrupting mutants lacked RNA splicing activity, despite successfully interacting with key binding partners, and failed to rescue the defects in gene expression, RNA splicing, and tumorigenesis observed in AKAP95-knockout cells111.
Cancers also rely on mutations that disrupt the normal functions of key gene products, either via loss (e.g., inactivation, silencing, deletion) or gain (e.g., amplification, hyperactivation) of function. Yet the mechanistic link between specific cancer-related mutations and their pathological effects is not always clear. For example, the protein tyrosine phosphatase (PTP) SHP2, a major positive regulator of Ras activation, is frequently mutated in cancers and is an emerging therapeutic target114,115. Curiously, mutations that increase and decrease SHP2 phosphatase activity both increase cancer risk116, suggesting that altered catalytic activity is not sufficient to drive disease pathology and raising questions about the actual molecular mechanism. New work is revealing that these and other seemingly cryptic mutations achieve their pathological effects by dysregulating LLPS. Indeed, several disease-associated SHP2 mutants were recently shown to undergo LLPS in vitro and in cells117 (Figure 5a). SHP2 LLPS is driven by charged residues within the PTP domain, which become exposed when SHP2 adopts its open, active conformation, and allosteric modulators that lock SHP2 in its open and closed states can promote and disrupt LLPS, respectively117. The open conformation is also prevalent in disease mutants, regardless of their catalytic activity, suggesting that SHP2 mutants gain LLPS ability through their altered conformations. Importantly, both activating and inactivating SHP2 mutants were found to promote ERK pathway hyperactivation in cells, which required their ability to form condensates117. Yet whereas the intrinsic activity of condensates formed by SHP2-activating mutants was sufficient to induce signaling, condensates formed by SHP2-inactivating mutants were found to acquire catalytic activity by recruiting and activating WT SHP2117 (Figure 5a). Thus, SHP2 disease-causing mutants gain the capacity for constitutive molecular assembly via LLPS, allowing them to induce pathological signaling independent of their intrinsic catalytic activity.
Figure 5. Dysregulation of liquid-liquid phase separation as a tumorigenic driver.
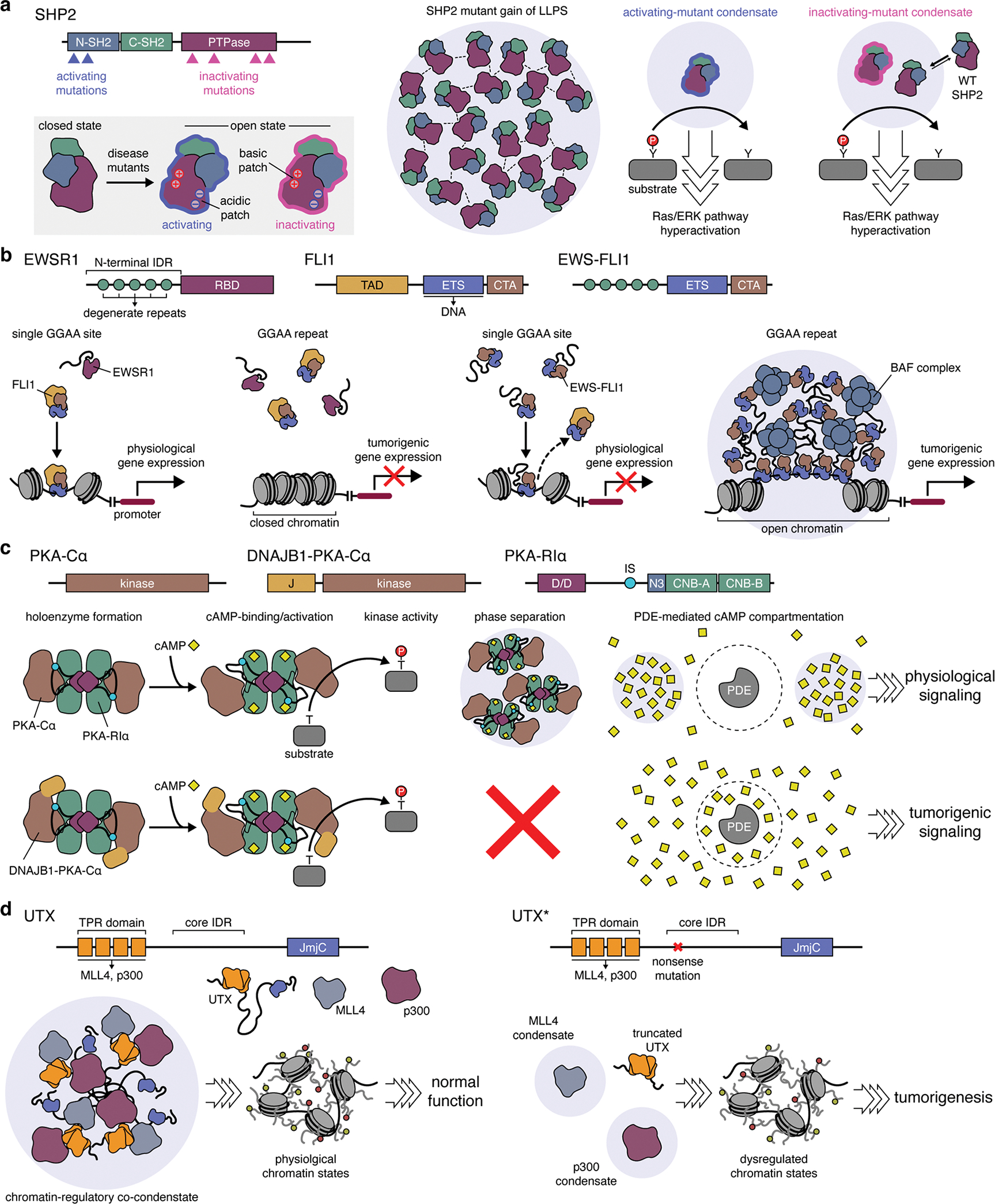
(a) Gain of LLPS ability by disease-causing SHP2 mutants. Both activating and inactivating mutations in SHP2 are able to trigger oncogenic signaling by altering SHP2 conformation to favor the open state. This conformation exposes a patch of positively charged, basic residues on the surface of the SHP2 protein tyrosine phosphatase (PTPase) domain, which can then form electrostatic interactions with negatively charged acidic residues elsewhere on the protein surface, thus driving LLPS by these mutants. Condensates formed by activating SHP2 mutants induce Ras/ERK pathway hyperactivation through their intrinsically high PTPase activity. By contrast, while condensates formed by inactivating SHP2 mutants gain PTPase activity by recruiting and activating wild-type (WT) SHP2 to trigger hyperactive signaling. (b) Gain of LLPS by EWS-FLI1 oncogenic fusion. In Ewing sarcoma, the N-terminal IDR of the EWSR1 becomes fused to the C-terminal DNA-binding domain of FLI1. Left: Native FLI1 binds conserved enhancer sites containing a single GGAA consensus sequence to control physiological gene expression but is unable to bind GGAA microsatellite repeats. Right: The EWS-FLI1 fusion disrupts the binding of native FLI1 to enhancers and also gains the ability to undergo LLPS via its IDR. EWS-FLI1 condensates tightly bind GGAA microsatellites while also strongly recruiting the BAF chromatin remodeling complex, producing open chromatin sites that act as super-enhancers for pro-tumor gene expression. (c) Pathological loss of LLPS by an oncogenic fusion. In the DNAJB1-PKA-Cα chimera, an N-terminal portion of DNAJB1 encompassing the J-domain (J) replaces the native N-terminus of WT PKA-Cα. WT PKA-Cα and DNAJB1-PKA-Cα behave similarly in forming holoenzymes with PKA-R subunits (e.g., RIα), in undergoing cAMP-induced activation, and in phosphorylating substrates. However, DNAJB1-PKA-Cα completely abolishes RIα LLPS, which disrupts the compartmentation of cAMP signaling by PDEs, leading to tumorigenic signaling. (d) Loss of LLPS by UTX mutants disrupts chromatin regulation. Left: In healthy cells, histone demethylase UTX undergoes LLPS mediated by a core IDR between the tetratricopeptide repeat (TPR) and Jumonji C (JmjC) domains. The TPR domain of UTX recruits the MLL4 histone methyl transferase and the p300 histone acetyl transferase to form chromatin-regulating co-condensates that maintain physiological chromatin states. Right: UTX is frequently mutated in cancers, with the most frequent alteration being a nonsense mutation near the start of the core IDR. These mutant UTX variants (UTX*) cannot phase separate with MLL4 and p300, leading to dysregulated chromatin states and tumorigenesis.
Many cancers are specifically driven by chimeric proteins generated by chromosomal rearrangments118–122, and gain of LLPS ability is increasingly being found to underlie the pathology of these oncogenic fusions. For example, multiple fusion oncoproteins contain intact kinase domains from receptor tyrosine kinases (RTKs) but lack transmembrane domains for canonical RTK signaling118, leaving their tumorigenic mechanism unclear. Recently, several variants of the echinoderm microtubule-associated protein-like 4/anaplastic lymphoma kinase (EML4-ALK) fusion found in lung cancers118,123 were shown to form biomolecular condensates, both in vitro and in vivo, spanning a range of liquid- to gel-like properties depending on their precise fusion architecture124,125. Phase separation of EML4-ALK variants appears largely mediated by oligomerization via EML4-derived self-interaction domains, as removing these domains or mutating key residues abolished condensates124,125. EML4-ALK condensates were also shown to recruit a number of Ras-activating factors, such as SOS1 and SHP2, notably triggering their dramatic redistribution from the cytosol into condensates124. Meanwhile, negative regulators of Ras activity, such as GTPase activating proteins, were excluded from EML4-ALK condensates, similar to the behavior of other Ras-activating condensates126. Disrupting the formation of EML4-ALK condensates blocked the hyperactivation of Ras/ERK signaling, which was found to specifically occur through a cytosolic rather than membrane-associated pool of Ras124. Given that similar RTK fusions typically contain oligomerization domains such as coiled-coils118, gain of LLPS may be a general mechanism for hyperactive signaling by these oncogenic chimeras, and indeed, similar behavior was observed with a CCDC6-RET fusion kinase124.
Aside from altering signaling, many oncogenic fusions incorporate elements of transcription factors or other DNA-binding proteins and are directly associated with profound changes in gene regulation119–121,127, yet how these effects are accomplished is unknown. Ewing sarcoma, for instance, is typically driven by fusion of the N-terminus of Ewing sarcoma RNA-binding 1 (EWSR1) and the DNA-binding domain of the E-Twenty Six (ETS) transcription factor Friend leukemia integration 1 (FLI1)120,128,129 (Figure 5b). The resulting EWS-FLI1 chimera binds GGAA microsatellite repeats to induce de novo activation of oncogenic enhancers via chromatin remodeling, while inactivating canonical ETS sites130,131. Both EWS-FLI1 and native FLI1 recognize the GGAA core ETS consensus sequence130,131, but only EWS-FLI1 binds GGAA repeats120, suggesting that DNA-binding per se is not responsible for tumorigenic activity. Similarly, EWS-FLI1 recruits the BRG1/BRM-associated factor (BAF) chromatin remodeling complex to GGAA repeats, yet both EWSR1 and FLI1 interacted with the BAF complex on their own, which was not sufficient to reproduce the actions of the EWS-FLI1 fusion120. Instead, the novel functions of EWS-FLI1 were shown to stem from its ability to undergo LLPS and form liquid-like nuclear condensates. The N-terminus of EWS-FLI1 corresponds to the N-terminal IDR of EWSR1, and IDR-mediated phase separation was found to be required for the tumorigenic activities of EWS-FLI1120. Specifically, mutating key aromatic residues to disrupt LLPS abolished BAF recruitment to GGAA repeats, chromatin remodeling, and enhancer activation by EWS-FLI1, whereas a minimal repeat derived from the EWSR1 IDR was sufficient to recapitulate these effects when fused to the FLI1 DNA-binding domain120.
Recent work on the oncogenic nucleoporin 98/homeobox A9 (NUP98-HOXA9) chimera further demonstrates the role of acquired LLPS as a key molecular alternation driving tumorigenicity121. This chimeric protein found in leukemia combines the NUP98 IDR, containing the characteristic phenylalanine/glycine (FG) repeats132, with a DNA-binding homeodomain and was shown to form liquid-like condensates in vitro and in cell nuclei121. NUP98-HOXA9 was found to exhibit a dense, super-enhancer-like DNA binding pattern and to drive aberrant chromatin looping within genomic regions located near leukemia-associated genes; however, an NUP98-HOXA9 mutant in which FG-repeat phenylalanines were changed to serines, which abolished LLPS, showed dramatically reduced DNA binding and chromatin looping behavior and also lacked tumorigenic activity121. Notably, a similar disruption was observed using NUP98-HOXA9 mutants whose FG repeats were below a threshold number, illustrating a direct link between multivalency and tumorigenicity.
The loss of LLPS can likewise account for the tumorigenic effects of cancer-driving mutations with no clear mechanistic explanation. Fibrolamellar carcinoma (FLC), for instance, is a rare and lethal form of liver cancer that predominantly strikes in the young that is almost universally driven by a single mutation: an ~400 kilobase heterozygous deletion in chromosome 19 joins the 1st exon of DNAJB1, encoding the J domain of an Hsp40-like molecular chaperone, with exon 2 of PRKACA, which encodes the α isoform of PKA-C (PKA-Cα)133,134 (Figure 5c). The resulting fusion is transcribed from the DNAJB1 promoter, resulting in ~10-fold higher accumulation of DNAJB1-PKA-Cα versus WT PKA-Cα133,135. Yet overexpressing PKA-Cα fails to recapitulate FLC tumorigenesis134. DNAJB1-PKA-Cα also differs little from WT PKA-Cα with respect to R-subunit binding, cAMP-induced activation, or intrinsic catalytic activity133,136. Instead, DNAJB1-PKA-Cα was found to completely abolish the LLPS behavior of RIα, both when reconstituted using purified proteins and when expressed in cells101, resulting in the total collapse of PDE-mediated cAMP compartmentation. This effect is identical to that of RIα deletion, and indeed, a small number of FLC cases have been linked to the loss of RIα protein expression, rather than acquisition of DNAJB1-PKA-Cα137. Thus, although the principal genetic alteration in FLC is PKA-Cα fusion and overexpression, the actual molecular defect appears to be loss of RIα LLPS. Similar PKA-C chimeras are recurrent in intraductal oncocytic papillary neoplasms138, and while their effect on RIα LLPS remains unknown, it is tempting to speculate on a general role for loss of LLPS-mediated cAMP compartmentation in tumorigenesis.
Another example of defective LLPS driving tumorigenesis comes from work on ubiquitously transcribed tetratricopeptide repeat (TPR) on chromosome X (UTX), a histone demethylase that plays an important role in developmental gene regulation and is also a major tumor suppressor139–141. Although UTX is mutated in several cancers142, mutations affecting catalytic activity are uncommon, and UTX demethylase activity is often dispensable for developmental regulation and tumor suppression 139–141. Instead, UTX mutations typically produce truncated proteins139,142. A recent study identified a central IDR in UTX spanning residues 549–848 that drives UTX phase separation in vitro and in cell nuclei and is almost completely deleted in the most frequent UTX mutant, which is truncated at residue 555139. The formation of UTX condensates was required for its tumor-suppressive function in cancer cell lines and in vivo, which was abolished by either deleting the IDR or mutating residues enriched in this region and preserved by mutants containing an unrelated IDR, as well as catalytically dead UTX139. LLPS-deficient UTX mutants showed reduced chromatin binding, which also coincided with global changes in histone methylation and dysregulated gene expression. The UTX N-terminal TPR domain was shown to recruit other histone-modifying enzymes, such as multiprotein mixed-lineage leukemia 4 (MLL4) and p300, which also form condensates143,144, suggesting a potential scaffolding role for UTX in the co-condensation of chromatin-modifiers at key genomic sites139 (Figure 5d).
Considering the tight link between biomolecular condensates and multiple fundamental processes that both protect against cancer and drive its progression, it is only to be expected that the dysregulation of LLPS directly contributes to cancer pathology. Nevertheless, it is becoming increasingly clear that LLPS behavior is a critical aspect of protein function that, if aberrantly gained or lost, can fundamentally alter cellular homeostasis and drive tumorigenesis. These examples highlight the need to pay careful attention to LLPS when attempting to decipher the molecular mechanisms underlying seemingly cryptic pathological mutations.
CONCLUDING REMARKS
A clear picture has emerged that places LLPS in direct control of multiple processes that define the hallmarks of cancer. Yet too often, the precise functional link between condensates and cancer cell pathophysiology remains elusive, owing to an incomplete understanding of how condensates actually shape biochemical reactions. Bridging this gap requires expanding the toolkit for studying biomolecular condensates (see Box 1). Several recent studies devised approaches combining in vitro reconstitution of biomolecular condensates with enzyme activity assays117,124,145, while a recent strategy for targeting genetically encoded fluorescent biosensors to endogenously expressed proteins101,146 proved invaluable for dissecting in situ biochemical regulation by native condensates in living cells101. Tools to manipulate LLPS in cells are similarly essential for unraveling the functional impact of condensates. Although recent approaches have enabled light-controlled induction of LLPS by heterologously expressed proteins147,148, strategies to selectively perturb LLPS by specific, endogenously expressed proteins remain lacking. Continued innovations along these lines are eagerly anticipated to achieve greater insights into condensate function.
BOX 1. TOOLS FOR STUDYING BIOMOLECULAR CONDENSATES.
Various strategies have been used to investigate biomolecular condensates157, with new techniques continually emerging and gaining wider use. The chosen technique depends primarily on the question(s) being explored.
Formation and Composition
Observing droplet formation by purified proteins can yield key insights into condensate formation, including detailed phase behavior, as well as composition, although purifying multiple candidate proteins is labor-intensive. Alternatively, condensates can be reconstituted with cell lysates, or directly in live cells, expressing fluorescently tagged proteins. Endogenous tagging via CRISPR/Cas gene-editing29,158,159 can help avoid overexpression117, especially combined with split-fluorescent protein tagging101,124,160. Proximity-labeling techniques161 (e.g., APEX, BioID) are also increasingly facilitating unbiased explorations of condensate composition96,162,163. Similarly, individual RNA/DNA species can be synthesized for in vitro reconstitution studies, while advanced aptamer-based labeling164 and APEX-seq165 promise to enable broader profiling of nucleic acid-containing condensates.
Physical Characteristics
Microscopy is widely used to investigate the physical properties of biomolecular condensates. A classic example involves imaging condensate fusion via standard light microscopy to monitor liquid-like behavior. More sophisticated techniques, such as fluorescence recovery after photobleaching (FRAP)166 and fluorescence correlation spectroscopy, are typically used to obtain greater detail, with FRAP being particularly useful for measuring diffusion within condensates and molecular exchange with the bulk phase. Atomic force microscopy is also being taken up to probe not only condensate fluidity167 but also structure70,168, while other recent studies have adopted various superresolution169,170 imaging approaches29,30,124,171, as well as electron microscopy96,162,172, to extract information on condensate structure.
Function
Disrupting LLPS using mutations or chemical agents (e.g., 1,6-hexanediol173) and measuring cellular responses is often used to infer the function of biomolecular condensates, and optogenetic approaches to spatiotemporally manipulate LLPS147,148 may further elevate these approaches. Meanwhile, strategies based on the enrichment of fluorescently labeled enzyme substrates117 or binding partners145 by condensates reconstituted in solution or on model membranes have recently been used to directly monitor biochemical reactions within condensates, as have biochemical pull-downs from cell lysates124. Genetically encoded biosensors are also being used to probe condensate function101,167, and a new suite of fluorescent sensors targeted to endogenously expressed proteins146 shows promise as a generalizable platform for illuminating condensate biochemical dynamics in situ.
Deeper investigation into the molecular function of condensates enabled by these new technologies will ultimately intersect with and inform budding efforts to therapeutically target dysregulated LLPS. One potential strategy for effective targeting will be to develop drugs tailored towards specific condensates, and indeed, several small molecules that disrupt LLPS by specific proteins have already been identified149–152, which may enable selective targeting of aberrant gain of LLPS without the deleterious effects of broader-acting chemotherapies. Nevertheless, the effectiveness of these compounds, and whether their design principles are readily generalized, remains to be determined. Rescuing the loss of LLPS may also be possible, as illustrated by the case of PML bodies, which are disrupted in acute promyelocytic leukemia by fusion of the PML protein to retinoic acid receptor-α (RARα). LLPS can be recovered by treating with all-trans retinoic acid and inducing degradation of the PML-RARα fusion, leading to complete recovery27. Other condensate-disrupting oncogenic fusions may be similarly druggable, potentially via proteolysis-targeting chimeras153 or RNA therapeutics154,155; however, this has yet to be demonstrated. Successful treatments may also emerge from efforts to identify and target the molecular factors responsible for modulating condensate formation (e.g., through post-translational modification). Further insights may also come from studying how existing cancer drugs interact with condensates156. Of course, all of these efforts remain in their infancy. Regardless, we expect the study of LLPS to foster a deeper understanding of pathological mechanisms in cancer and unlock new therapeutic opportunities.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (R35 CA197622, R01 DK073368, and R01 DE030497 to J.Z.) and the Air Force Office of Scientific Research (FA9500-18-1-0051 to J.Z.).
Footnotes
COMPETING INTERESTS
The authors declare they have no competing interests.
REFERENCES
- 1.Lengauer C, Kinzler KW & Vogelstein B Genetic instabilities in human cancers. Nature 396, 643–649 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Sever R & Brugge JS Signal Transduction in Cancer. Csh Perspect Med 5, a006098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez-Vega F et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradner JE, Hnisz D & Young RA Transcriptional Addiction in Cancer. Cell 168, 629–643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sengupta S & George RE Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends Cancer 3, 269–281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pohl C & Dikic I Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818–822 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D & Weinberg RA Hallmarks of Cancer: The Next Generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Ward PS & Thompson CB Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 21, 297–308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeBerardinis RJ & Chandel NS Fundamentals of cancer metabolism. Sci Adv 2, e1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banani SF, Lee HO, Hyman AA & Rosen MK Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Bio 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hyman AA, Weber CA & Jülicher F Liquid-Liquid Phase Separation in Biology. Annu Rev Cell Dev Bi 30, 39–58 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Boija A, Klein IA & Young RA Biomolecular condensates and cancer. Cancer Cell 39, 174–192 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai D, Liu Z & Lippincott-Schwartz J Biomolecular Condensates and Their Links to Cancer Progression. Trends Biochem Sci (2021) doi: 10.1016/j.tibs.2021.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Lu J et al. Emerging Roles of Liquid–Liquid Phase Separation in Cancer: From Protein Aggregation to Immune-Associated Signaling. Frontiers Cell Dev Biology 9, 631486 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taniue K & Akimitsu N Aberrant phase separation and cancer. Febs J (2021) doi: 10.1111/febs.15765. [DOI] [PubMed] [Google Scholar]
- 16.Jiang S, Fagman JB, Chen C, Alberti S & Liu B Protein phase separation and its role in tumorigenesis. Elife 9, e60264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi J-M, Holehouse AS & Pappu RV Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu Rev Biophys 49, 1–27 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyman AA & Brangwynne CP Beyond Stereospecificity: Liquids and Mesoscale Organization of Cytoplasm. Dev Cell 21, 14–16 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Brangwynne CP et al. Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science 324, 1729–1732 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Harmon TS, Holehouse AS, Rosen MK & Pappu RV Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. Elife 6, e30294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banani SF et al. Compositional Control of Phase-Separated Cellular Bodies. Cell 166, 651–663 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zink D, Fischer AH & Nickerson JA Nuclear structure in cancer cells. Nat Rev Cancer 4, 677–687 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Strom AR & Brangwynne CP The liquid nucleome – phase transitions in the nucleus at a glance. J Cell Sci 132, jcs235093 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lafontaine DLJ, Riback JA, Bascetin R & Brangwynne CP The nucleolus as a multiphase liquid condensate. Nat Rev Mol Cell Bio 22, 165–182 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Min J, Wright WE & Shay JW Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Gene Dev 33, 814–827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H et al. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol Biol Cell 31, 2048–2056 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thé H. de, Pandolfi PP & Chen Z Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 32, 552–560 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Sabari BR et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho W-K et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, eaar4199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai D et al. Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat Cell Biol 21, 1578–1589 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Y et al. Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat Cell Biol 22, 453–464 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altmeyer M et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat Commun 6, 8088 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kilic S et al. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. Embo J 38, e101379 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibson BA et al. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larson AG et al. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547, 236–240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanulli S et al. HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575, 390–394 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia Q, Chen S, Tan Y, Li Y & Tang F Oncogenic super-enhancer formation in tumorigenesis and its molecular mechanisms. Exp Mol Medicine 52, 713–723 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hnisz D, Shrinivas K, Young RA, Chakraborty AK & Sharp PA A Phase Separation Model for Transcriptional Control. Cell 169, 13–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han X et al. Roles of the BRD4 short isoform in phase separation and active gene transcription. Nat Struct Mol Biol 27, 333–341 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Arnold PR, Wells AD & Li XC Diversity and Emerging Roles of Enhancer RNA in Regulation of Gene Expression and Cell Fate. Frontiers Cell Dev Biology 7, 377 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roden C & Gladfelter AS RNA contributions to the form and function of biomolecular condensates. Nat Rev Mol Cell Bio 22, 183–195 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nair SJ et al. Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat Struct Mol Biol 26, 193–203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei M-T et al. Nucleated transcriptional condensates amplify gene expression. Nat Cell Biol 22, 1187–1196 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Jaeger MG et al. Selective Mediator dependence of cell-type-specifying transcription. Nat Genet 52, 719–727 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khattabi LE et al. A Pliable Mediator Acts as a Functional Rather Than an Architectural Bridge between Promoters and Enhancers. Cell 178, 1145–1158.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crump NT et al. BET inhibition disrupts transcription but retains enhancer-promoter contact. Nat Commun 12, 223 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsieh T-HS et al. Enhancer-promoter interactions and transcription are maintained upon acute loss of CTCF, cohesin, WAPL, and YY1. Biorxiv 2021.07.14.452365 (2021) doi: 10.1101/2021.07.14.452365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zanconato F et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med 24, 1599–1610 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zanconato F, Cordenonsi M & Piccolo S YAP/TAZ at the Roots of Cancer. Cancer Cell 29, 783–803 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu M et al. Interferon-γ induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol Cell 81, 1216–1230.e9 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Reggiani F, Gobbi G, Ciarrocchi A & Sancisi V YAP and TAZ Are Not Identical Twins. Trends Biochem Sci 46, 154–168 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Dietlein F, Thelen L & Reinhardt HC Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet 30, 326–339 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Halazonetis TD, Gorgoulis VG & Bartek J An Oncogene-Induced DNA Damage Model for Cancer Development. Science 319, 1352–1355 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Pessina F et al. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat Cell Biol 21, 1286–1299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iarovaia OV et al. Dynamics of double strand breaks and chromosomal translocations. Mol Cancer 13, 249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singatulina AS et al. PARP-1 Activation Directs FUS to DNA Damage Sites to Form PARG-Reversible Compartments Enriched in Damaged DNA. Cell Reports 27, 1809–1821.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Aguilera A The connection between transcription and genomic instability. Embo J 21, 195–201 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strom AR et al. Phase separation drives heterochromatin domain formation. Nature 547, 241–245 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dialynas GK, Vitalini MW & Wallrath LL Linking Heterochromatin Protein 1 (HP1) to cancer progression. Mutat Res Fundam Mol Mech Mutagen 647, 13–20 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Erdel F et al. Mouse Heterochromatin Adopts Digital Compaction States without Showing Hallmarks of HP1-Driven Liquid-Liquid Phase Separation. Mol Cell 78, 236–249.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dikic I Proteasomal and Autophagy Degradation Systems. Annu Rev Biochem 86, 1–32 (2016). [DOI] [PubMed] [Google Scholar]
- 62.White E The role for autophagy in cancer. J Clin Invest 125, 42–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manasanch EE & Orlowski RZ Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol 14, 417–433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim H, Bhattacharya A & Qi L Endoplasmic reticulum quality control in cancer: Friend or foe. Semin Cancer Biol 33, 25–33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clark A & Burleson M SPOP and cancer: a systematic review. Am J Cancer Res 10, 704–726 (2020). [PMC free article] [PubMed] [Google Scholar]
- 66.Bouchard JJ et al. Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol Cell 72, 19–36.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yasuda S et al. Stress- and ubiquitylation-dependent phase separation of the proteasome. Nature 578, 296–300 (2020). [DOI] [PubMed] [Google Scholar]
- 68.Dai C, Whitesell L, Rogers AB & Lindquist S Heat Shock Factor 1 Is a Powerful Multifaceted Modifier of Carcinogenesis. Cell 130, 1005–1018 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen L et al. Enhanced Degradation of Misfolded Proteins Promotes Tumorigenesis. Cell Reports 18, 3143–3154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fujioka Y et al. Phase separation organizes the site of autophagosome formation. Nature 578, 301–305 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Yamamoto H et al. The Intrinsically Disordered Protein Atg13 Mediates Supramolecular Assembly of Autophagy Initiation Complexes. Dev Cell 38, 86–99 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Noda NN & Fujioka Y Atg1 family kinases in autophagy initiation. Cell Mol Life Sci 72, 3083–3096 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sun D, Wu R, Zheng J, Li P & Yu L Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res 28, 405–415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li X, He S & Ma B Autophagy and autophagy-related proteins in cancer. Mol Cancer 19, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M & Aguilar-Cazares D The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Frontiers Oncol 10, 578418 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kumar M & Papaleo E A pan-cancer assessment of alterations of the kinase domain of ULK1, an upstream regulator of autophagy. Sci Rep-uk 10, 14874 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amaravadi R, Kimmelman AC & White E Recent insights into the function of autophagy in cancer. Gene Dev 30, 1913–1930 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mehta S & Zhang J Biochemical Activity Architectures Visualized–Using Genetically Encoded Fluorescent Biosensors to Map the Spatial Boundaries of Signaling Compartments. Accounts Chem Res (2021) doi: 10.1021/acs.accounts.1c00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Patra KC & Hay N The pentose phosphate pathway and cancer. Trends Biochem Sci 39, 347–354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang M & Vousden KH Serine and one-carbon metabolism in cancer. Nat Rev Cancer 16, 650–662 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Sweetlove LJ & Fernie AR The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat Commun 9, 2136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kohnhorst CL et al. Identification of a multienzyme complex for glucose metabolism in living cells. J Biol Chem 292, 9191–9203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.An S, Kumar R, Sheets ED & Benkovic SJ Reversible Compartmentalization of de Novo Purine Biosynthetic Complexes in Living Cells. Science 320, 103–106 (2008). [DOI] [PubMed] [Google Scholar]
- 84.Zhou W et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat Commun 11, 3811 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lv Y et al. Nucleotide de novo synthesis increases breast cancer stemness and metastasis via cGMP-PKG-MAPK signaling pathway. Plos Biol 18, e3000872 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Doigneaux C et al. Hypoxia drives the assembly of the multienzyme purinosome complex. J Biol Chem 295, 9551–9566 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pareek V, Tian H, Winograd N & Benkovic SJ Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 368, 283–290 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kyoung M, Russell SJ, Kohnhorst CL, Esemoto NN & An S Dynamic Architecture of the Purinosome Involved in Human De Novo Purine Biosynthesis. Biochemistry-us 54, 870–880 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wright PE & Dyson HJ Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Bio 16, 18–29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yum S, Li M, Frankel AE & Chen ZJ Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annu Rev Cancer Biology 3, 323–344 (2019). [Google Scholar]
- 91.Du M & Chen ZJ DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, eaat1022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou W, Mohr L, Maciejowski J & Kranzusch PJ cGAS phase separation inhibits TREX1-mediated DNA degradation and enhances cytosolic DNA sensing. Mol Cell 81, 739–755.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Woo S-R et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 41, 830–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schadt L et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Reports 29, 1236–1248.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Ng KW, Marshall EA, Bell JC & Lam WL cGAS–STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends Immunol 39, 44–54 (2018). [DOI] [PubMed] [Google Scholar]
- 96.Yu X et al. The STING phase-separator suppresses innate immune signalling. Nat Cell Biol 23, 330–340 (2021). [DOI] [PubMed] [Google Scholar]
- 97.Zaccolo M Spatial control of cAMP signalling in health and disease. Curr Opin Pharmacol 11, 649–655 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu V et al. Illuminating the Onco-GPCRome: Novel G protein–coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J Biol Chem 294, 11062–11086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deng Z et al. Selective autophagy of AKAP11 activates cAMP/PKA to fuel mitochondrial metabolism and tumor cell growth. Proc National Acad Sci 118, e2020215118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Beristain AG et al. PKA signaling drives mammary tumorigenesis through Src. Oncogene 34, 1160–1173 (2015). [DOI] [PubMed] [Google Scholar]
- 101.Zhang JZ et al. Phase Separation of a PKA Regulatory Subunit Controls cAMP Compartmentation and Oncogenic Signaling. Cell 182, 1531–1544.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Buxton IL & Brunton LL Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J Biological Chem 258, 10233–9 (1983). [PubMed] [Google Scholar]
- 103.Baillie GS Compartmentalized signalling: spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. Febs J 276, 1790–1799 (2009). [DOI] [PubMed] [Google Scholar]
- 104.Bock A et al. Optical Mapping of cAMP Signaling at the Nanometer Scale. Cell 182, 1519–1530.e17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Adriaens C et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat Med 22, 861–868 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Barra J et al. Integrator restrains paraspeckles assembly by promoting isoform switching of the lncRNA NEAT1. Sci Adv 6, eaaz9072 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vogelstein B, Lane D & Levine AJ Surfing the p53 network. Nature 408, 307–310 (2000). [DOI] [PubMed] [Google Scholar]
- 108.Sengupta S & Harris CC p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Bio 6, 44–55 (2005). [DOI] [PubMed] [Google Scholar]
- 109.Blume CJ et al. p53-dependent non-coding RNA networks in chronic lymphocytic leukemia. Leukemia 29, 2015–2023 (2015). [DOI] [PubMed] [Google Scholar]
- 110.Hu J et al. AKAP95 regulates splicing through scaffolding RNAs and RNA processing factors. Nat Commun 7, 13347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li W et al. Biophysical properties of AKAP95 protein condensates regulate splicing and tumorigenesis. Nat Cell Biol 22, 960–972 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Qi F et al. Synergistic effects of AKAP95, Cyclin D1, Cyclin E1, and Cx43 in the development of rectal cancer. Int J Clin Exp Patho 8, 1666–73 (2015). [PMC free article] [PubMed] [Google Scholar]
- 113.Liu W et al. Roles of Cx43 and AKAP95 in ovarian cancer tissues in G1/S phase. Int J Clin Exp Patho 8, 14315–24 (2015). [PMC free article] [PubMed] [Google Scholar]
- 114.Kerr DL, Haderk F & Bivona TG Allosteric SHP2 inhibitors in cancer: Targeting the intersection of RAS, resistance, and the immune microenvironment. Curr Opin Chem Biol 62, 1–12 (2021). [DOI] [PubMed] [Google Scholar]
- 115.Yuan X, Bu H, Zhou J, Yang C-Y & Zhang H Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J Med Chem 63, 11368–11396 (2020). [DOI] [PubMed] [Google Scholar]
- 116.Edouard T et al. How do Shp2 mutations that oppositely influence its biochemical activity result in syndromes with overlapping symptoms? Cell Mol Life Sci 64, 1585–1590 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhu G et al. Phase Separation of Disease-Associated SHP2 Mutants Underlies MAPK Hyperactivation. Cell 183, 490–502.e18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nelson KN, Peiris MN, Meyer AN, Siari A & Donoghue DJ Receptor Tyrosine Kinases: Translocation Partners in Hematopoietic Disorders. Trends Mol Med 23, 59–79 (2017). [DOI] [PubMed] [Google Scholar]
- 119.Kovar H Dr. Jekyll and Mr. Hyde: The Two Faces of the FUS/EWS/TAF15 Protein Family. Sarcoma 2011, 837474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Boulay G et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 171, 163–178.e19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ahn JH et al. Phase separation drives aberrant chromatin looping and cancer development. Nature 595, 591–595 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Du Z & Lovly CM Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer 17, 58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sabir SR, Yeoh S, Jackson G & Bayliss R EML4-ALK Variants: Biological and Molecular Properties, and the Implications for Patients. Cancers 9, 118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tulpule A et al. Kinase-mediated RAS signaling via membraneless cytoplasmic protein granules. Cell (2021) doi: 10.1016/j.cell.2021.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Qin Z et al. Phase separation of EML4–ALK in firing downstream signaling and promoting lung tumorigenesis. Cell Discov 7, 33 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Su X et al. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 352, 595–599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brien GL, Stegmaier K & Armstrong SA Targeting chromatin complexes in fusion protein-driven malignancies. Nat Rev Cancer 19, 255–269 (2019). [DOI] [PubMed] [Google Scholar]
- 128.Arvand A & Denny CT Biology of EWS/ETS fusions in Ewing’s family tumors. Oncogene 20, 5747–5754 (2001). [DOI] [PubMed] [Google Scholar]
- 129.Delattre O et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 359, 162–165 (1992). [DOI] [PubMed] [Google Scholar]
- 130.Johnson KM et al. Role for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growth. Proc National Acad Sci 114, 9870–9875 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Riggi N et al. EWS-FLI1 Utilizes Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell 26, 668–681 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Denning DP, Patel SS, Uversky V, Fink AL & Rexach M Disorder in the nuclear pore complex: The FG repeat regions of nucleoporins are natively unfolded. Proc National Acad Sci 100, 2450–2455 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Honeyman JN et al. Detection of a Recurrent DNAJB1-PRKACA Chimeric Transcript in Fibrolamellar Hepatocellular Carcinoma. Science 343, 1010–1014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kastenhuber ER et al. DNAJB1–PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc National Acad Sci 114, 13076–13084 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Riggle KM et al. Enhanced cAMP-stimulated protein kinase A activity in human fibrolamellar hepatocellular carcinoma. Pediatr Res 80, 110–118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cao B et al. Structures of the PKA RIα Holoenzyme with the FLHCC Driver J-PKAcα or Wild-Type PKAcα. Structure 27, 816–828.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Graham RP et al. Fibrolamellar carcinoma in the Carney complex: PRKAR1A loss instead of the classic DNAJB1-PRKACA fusion. Hepatology 68, 1441–1447 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Singhi AD et al. Recurrent Rearrangements in PRKACA and PRKACB in Intraductal Oncocytic Papillary Neoplasms of the Pancreas and Bile Duct. Gastroenterology 158, 573–582.e2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shi B et al. UTX condensation underlies its tumour-suppressive activity. Nature 1–6 (2021) doi: 10.1038/s41586-021-03903-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Andricovich J et al. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 33, 512–526.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Miller SA, Mohn SE & Weinmann AS Jmjd3 and UTX Play a Demethylase-Independent Role in Chromatin Remodeling to Regulate T-Box Family Member-Dependent Gene Expression. Mol Cell 40, 594–605 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang L & Shilatifard A UTX Mutations in Human Cancer. Cancer Cell 35, 168–176 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fasciani A et al. MLL4-associated condensates counterbalance Polycomb-mediated nuclear mechanical stress in Kabuki syndrome. Nat Genet 52, 1397–1411 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang Y et al. Nuclear condensates of p300 formed though the structured catalytic core can act as a storage pool of p300 with reduced HAT activity. Nat Commun 12, 4618 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Huang WYC et al. A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science 363, 1098–1103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tenner B et al. FluoSTEPs: Fluorescent biosensors for monitoring compartmentalized signaling within endogenous microdomains. Sci Adv 7, eabe4091 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shin Y et al. Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell 168, 159–171.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bracha D et al. Mapping Local and Global Liquid Phase Behavior in Living Cells Using Photo-Oligomerizable Seeds. Cell 175, 1467–1480.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang Z et al. Chemical perturbation of an intrinsically disordered region of TFIID distinguishes two modes of transcription initiation. Elife 4, e07777 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ban D, Iconaru LI, Ramanathan A, Zuo J & Kriwacki RW A Small Molecule Causes a Population Shift in the Conformational Landscape of an Intrinsically Disordered Protein. J Am Chem Soc 139, 13692–13700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Iconaru LI et al. Discovery of Small Molecules that Inhibit the Disordered Protein, p27Kip1. Sci Rep-uk 5, 15686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wheeler RJ et al. Small molecules for modulating protein driven liquid-liquid phase separation in treating neurodegenerative disease. Biorxiv 721001 (2019) doi: 10.1101/721001. [DOI] [Google Scholar]
- 153.Burslem GM & Crews CM Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 181, 102–114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Winkle M, El-Daly SM, Fabbri M & Calin GA Noncoding RNA therapeutics — challenges and potential solutions. Nat Rev Drug Discov 1–23 (2021) doi: 10.1038/s41573-021-00219-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dammes N & Peer D Paving the Road for RNA Therapeutics. Trends Pharmacol Sci 41, 755–775 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Klein IA et al. Partitioning of cancer therapeutics in nuclear condensates. Science 368, 1386–1392 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bracha D, Walls MT & Brangwynne CP Probing and engineering liquid-phase organelles. Nat Biotechnol 37, 1435–1445 (2019). [DOI] [PubMed] [Google Scholar]
- 158.Sander JD & Joung JK CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32, 347–355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Doudna JA & Charpentier E The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 (2014). [DOI] [PubMed] [Google Scholar]
- 160.Kamiyama D et al. Versatile protein tagging in cells with split fluorescent protein. Nat Commun 7, 11046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Qin W, Cho KF, Cavanagh PE & Ting AY Deciphering molecular interactions by proximity labeling. Nat Methods 18, 133–143 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yu H et al. HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 371, eabb4309 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Markmiller S et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 172, 590–604.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen X et al. Visualizing RNA dynamics in live cells with bright and stable fluorescent RNAs. Nat Biotechnol 37, 1287–1293 (2019). [DOI] [PubMed] [Google Scholar]
- 165.Fazal FM et al. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell 178, 473–490.e26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Taylor NO, Wei M-T, Stone HA & Brangwynne CP Quantifying Dynamics in Phase-Separated Condensates Using Fluorescence Recovery after Photobleaching. Biophys J 117, 1285–1300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Quiroz FG et al. Liquid-liquid phase separation drives skin barrier formation. Science 367, eaax9554 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zeng M et al. Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell 166, 1163–1175.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schermelleh L et al. Super-resolution microscopy demystified. Nat Cell Biol 21, 72–84 (2019). [DOI] [PubMed] [Google Scholar]
- 170.Huang B, Bates M & Zhuang X Super-Resolution Fluorescence Microscopy. Biochemistry-us 78, 993–1016 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jain S et al. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 164, 487–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Delarue M et al. mTORC1 Controls Phase Separation and the Biophysical Properties of the Cytoplasm by Tuning Crowding. Cell 174, 338–349.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kroschwald S, Maharana S & Simon A Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters (2017) doi: 10.19185/matters.201702000010. [DOI] [Google Scholar]