

Summary
Histone variant H3.3 is encoded by two genes, H3f3a and H3f3b, which can be expressed differentially depending on tissue type. Previous work in our lab has shown that knockout of H3f3b causes some neonatal lethality and infertility in mice, and chromosomal defects in mouse embryonic fibroblasts (MEFs). Studies of H3f3a and H3f3b null mice by others have produced generally similar phenotypes to what we found in our H3f3b nulls, but the relative impacts of the loss of either H3f3a or H3f3b have varied depending on the approach and genetic background. Here we used a knockout-first approach to target the H3f3a gene for inactivation in C57BL6 mice. Homozygous H3f3a targeting produced a lethal phenotype at or before birth. E13.5 null embryos had some potential morphological differences from WT littermates including smaller size and reduced head size. An E18.5 null embryo was smaller than its control littermates with several potential defects including small head and brain size as well as small lungs, which would be consistent with a late gestation lethal phenotype. Despite a reduction in H3.3 and total H3 protein levels, the only histone H3 post-translational modification in the small panel assessed that was significantly altered was the unique H3.3 mark phospho-Serine31, which was consistently increased in null neurospheres. H3f3a null neurospheres also exhibited consistent gene expression changes including in protocadherins. Overall, our findings are consistent with the model that there are differential, cell-type-specific contributions of H3f3a and H3f3b to H3.3 functions in epigenetic and developmental processes.
Keywords: development, H3f3a, H3f3b, histone H3.3, mouse knockouts, neurospheres
1 |. INTRODUCTION
The specific histone molecules making up nucleosomal octamers along with their unique combinations of post-translational modifications (PTMs) are major regulators of chromatin structure and function. Histone H3.3 is a variant member of the histone H3 family that is unusual in a number of ways. Unlike canonical H3 variants H3.1 and H3.2, which are encoded by many genes within gene clusters, H3.3 is encoded by just two separate genes, H3F3A/H3f3a and H3F3B/H3f3b (in humans and mice, respectively), which differ in chromosomal location and untranslated regions, but encode the identical H3.3 protein (Elsaesser, Goldberg, & Allis, 2010). While the expression of H3.1 and H3.2 largely coincides specifically with S phase, H3.3 is expressed throughout the cell cycle. Fitting with that broader expression pattern, H3.3 is also a replication-independent variant uniquely deposited on chromatin at any phase of the cell cycle (Ahmad & Henikoff, 2002), while the deposition of H3.1 and H3.2 is replication-dependent. H3.3 is thus linked to a broader range of epigenetic functions than H3.1/H3.2, including more dynamic regulation of transcriptional control and facilitation of DNA repair (Luijsterburg et al., 2016).
The H3.3 protein differs from canonical H3.1 by only 5 amino acids, and from H3.2 by 4 amino acids (Elsaesser et al., 2010; Filipescu, Szenker, & Almouzni, 2013), but these differences confer its distinct functions. For example, the unique residues within H3.3 mediate its binding to and deposition by its novel chaperones, HIRA in genic regions, and ATRX/DAXX at regions of heterochromatin including within telomeric regions (Banaszynski et al., 2013; Elsaesser & Allis, 2010; Lewis, Elsaesser, Noh, Stadler, & Allis, 2010), while H3.1/H3.2 have their own distinct chaperones. H3.3 also has somewhat unique patterns of PTMs compared to H3.1/H3.2, and H3.3 tends to be enriched at active chromatin and actively transcribed genes (Ahmad & Henikoff, 2002; McKittrick, Gafken, Ahmad, & Henikoff, 2004). The distinctive localization and functions of H3.3 in centromeric and telomeric regions are likely important as well (Wong et al., 2010).
Phenotypes from H3f3a and H3f3b loss-of-function studies in mice consistently indicate roles for H3.3 in normal mammalian development (Bush et al., 2013; Couldrey, Carlton, Nolan, Colledge, & Evans, 1999; Jang, Shibata, Starmer, Yee, & Magnuson, 2015; Tang et al., 2015; Tang, Jacobs, Wong, & Mann, 2013; Yuen, Bush, Barrilleaux, Cotterman, & Knoepfler, 2014). The phenotypes associated with the separate single knockouts of H3f3a and H3f3b have varied in the different published gene targeting studies, likely mainly attributable to differences in both genetic backgrounds and perhaps to some extent to the different methods of gene disruption. However, reduced levels of H3.3 protein have been consistently associated with infertility, genomic instability, and with some degree of embryonic lethality. While H3f3a and H3f3b encode the same H3.3 protein, there is interest in how the function of these genes may differ. The phenotypes from the aforementioned knockout studies suggest that the relative contributions of H3f3a and H3f3b to total cellular H3.3 protein levels are in some cases cell-type specific (Bush et al., 2013; Couldrey et al., 1999; Jang et al., 2015; Tang et al., 2013; Tang et al., 2015; Yuen et al., 2014). More specifically, the 2015 data from Tang, et al. suggested a relatively higher contribution of H3f3a to H3.3 protein levels in the brain, while we have previously found that H3f3b likely contributes more to total H3.3 levels in mouse embryonic fibroblasts (MEFs) and possibly male germ cells (Bush et al., 2013; Yuen et al., 2014). Even so, notably a more recent gene epitope-tagging study in mice reported that the patterns of relative H3f3a and H3f3b expression in diverse tissues were remarkably similar overall (Bachu et al., 2019).
Here, we developed a murine model of disrupted H3f3a expression in C57BL/6 mice. Applying a knockout-first, conditional-ready targeting approach, disruption of H3f3a resulted in developmental lethality, manifesting sometime after mid-gestation to birth. Analyses of both H3f3a targeted neurospheres and MEFs indicate a greater contribution of H3f3a to H3.3 protein levels in the former and of H3f3b in the latter types of cells. We generally did not see compensatory increases in H3f3b expression with disruption of H3f3a in either cell type. The main histone PTM altered in H3f3a deficient cells at a global level in these cells was the H3.3 specific H3.3S31 phosphorylation mark. Overall, these H3f3a targeted mice and cells are likely to be useful tools for deciphering this gene’s function and advancing knowledge of the functions of the H3.3 protein in normal development and disease.
2 |. RESULTS
2.1 |. Production of H3f3a targeted mice
To determine the functions of the H3f3a gene and H3.3 protein in murine development and cell biology, we generated a knock-out first conditional-ready allele (H3f3atm1a; https://www.ncbi.nlm.nih.gov/nucleotide/JN957085.1?report=genbank&log$=nuclalign&blast_rank=1&RID=8ZRU6GXM015) in C57BL/6 N mice using targeted mouse embryonic stem cells (mESCs) available from the Knockout Mouse Project or KOMP Repository (Figure 1a) at the UC Davis Mouse Biology Program (UCD MBP). The rationale for using the knockout-first approach was to efficiently generate data on phenotypes due to H3f3a loss-of-function. The knockout-first allele features an in-frame expression cassette (consisting of an Engrailed2 splice acceptor [En2 SA], lacZ, B-actin promoter, and neomycin) inserted into intron 2 of H3f3a, causing a functional gene trap that disrupts H3f3a expression. Nearest neighbor protein-coding genes of H3f3a (itself located at mm10 chr1:180,800,832–180,813,632) are Acbd3 (mm10 chr1:180,726,043–180,754,204) >50 kb away, and Sde2 (mm10 chr1:180,851,127–180,868,113) almost 38 kb away, making any aberrant transcriptional effects on neighboring genes unlikely.
FIGURE 1.
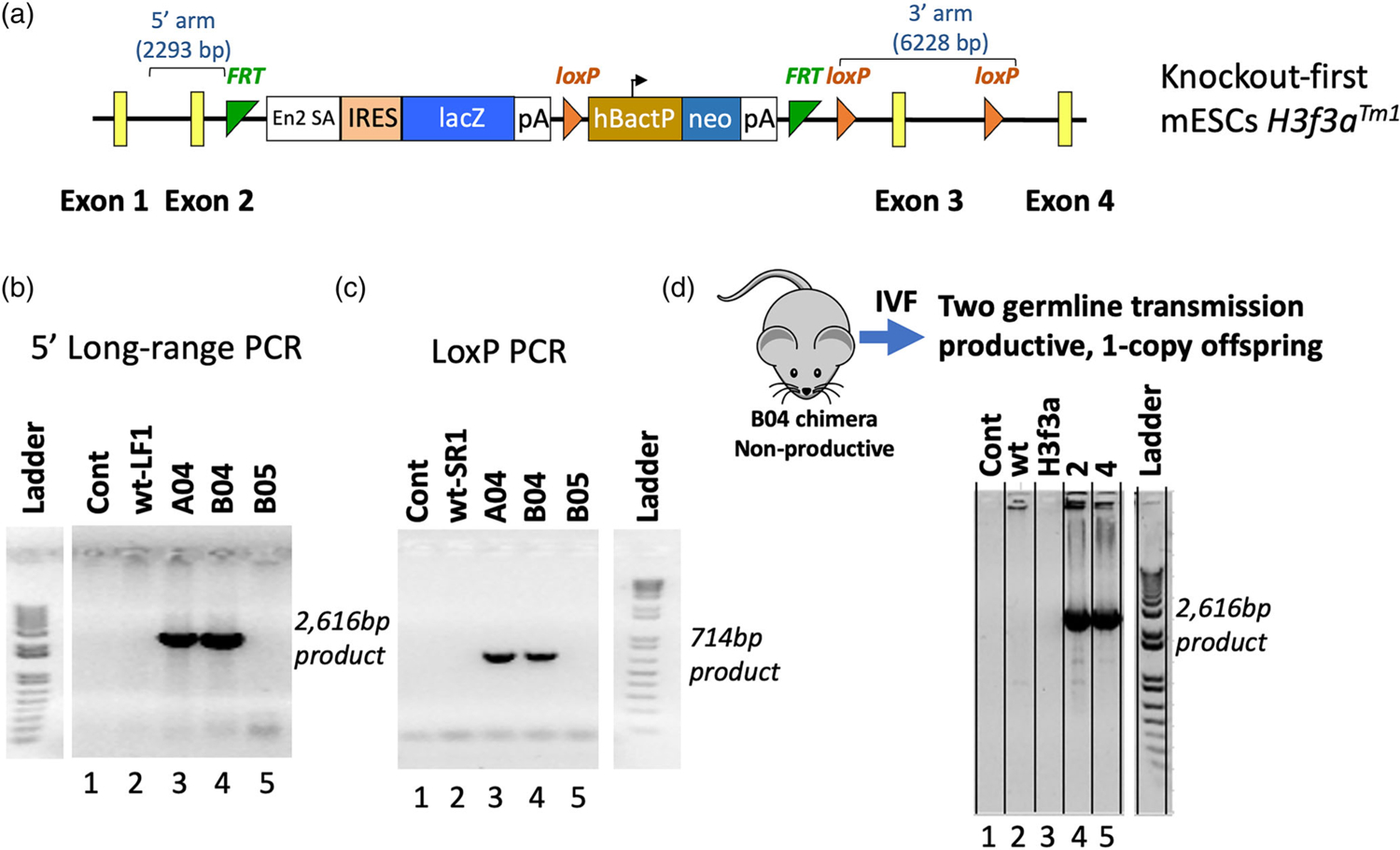
Production and validation of the H3f3a targeted mice. A knock-out first conditional-ready allele (H3f3atm1a) was generated in C57BL/6 N mice using targeted mouse embryonic stem cells (mESCs) from the KOMP Repository. (a) Map of the targeted allele. (b) and (c) Long-range and Lox-specific genomic PCR validation assays, respectively. Gene targeting of the H3f3atm1a allele in mESC by homologous recombination rather than by random insertion was validated via long-range genomic PCR using one primer in the H3f3a region but outside the homology arms of the targeting vector paired with an En2 SA primer inside the knockout-first cassette, producing the specific approximately 2.6 kb product. The retention and presence of the most distal LoxP site was confirmed by the LoxP PCR assay. (d) After IVF, the correct H3f3atm1a allele was detected in offspring. Data in panels b-d are from the UC Davis Mouse Biology Program as they validated the H3f3a targeted ES cells and mice. Because they run a variety of samples from different projects on the same gels, the data not relevant to this project were cropped out. We have included the ladder from the same gel in each case but only with the data relevant to the H3f3a project. Areas of cropping are indicated by white gaps between lanes. The Controls “Cont” in b-d contain no template negative controls, and in each case lane 2 is another negative control
Gene targeting of the H3f3atm1a knockout-first allele in mESC by homologous recombination rather than by random insertion was validated by the UCD MBP via long-range genomic PCR using one primer in the H3f3a region but outside the homology arms of the targeting vector paired with an En2 SA primer inside the knockout-first cassette, generating only the expected approximately 2.6 kb product (Figure 1b). The retention and presence of the most distal LoxP site was confirmed by a LoxP PCR assay (Figure 1c).
Germline transmission of H3f3atm1a was attempted via breeding the high-percentage somatic chimera B04, but it failed to be productive (Figure 1d). IVF was conducted using sperm from B04, leading to germline transmission and stable inheritance of the mutant allele. Throughout the study, WT H3f3a and knockout-first alleles were detected via gene-specific genomic PCR and for H3f3atm1a by the presence of the knockout-first cassette (Figure 2a,b).
FIGURE 2.
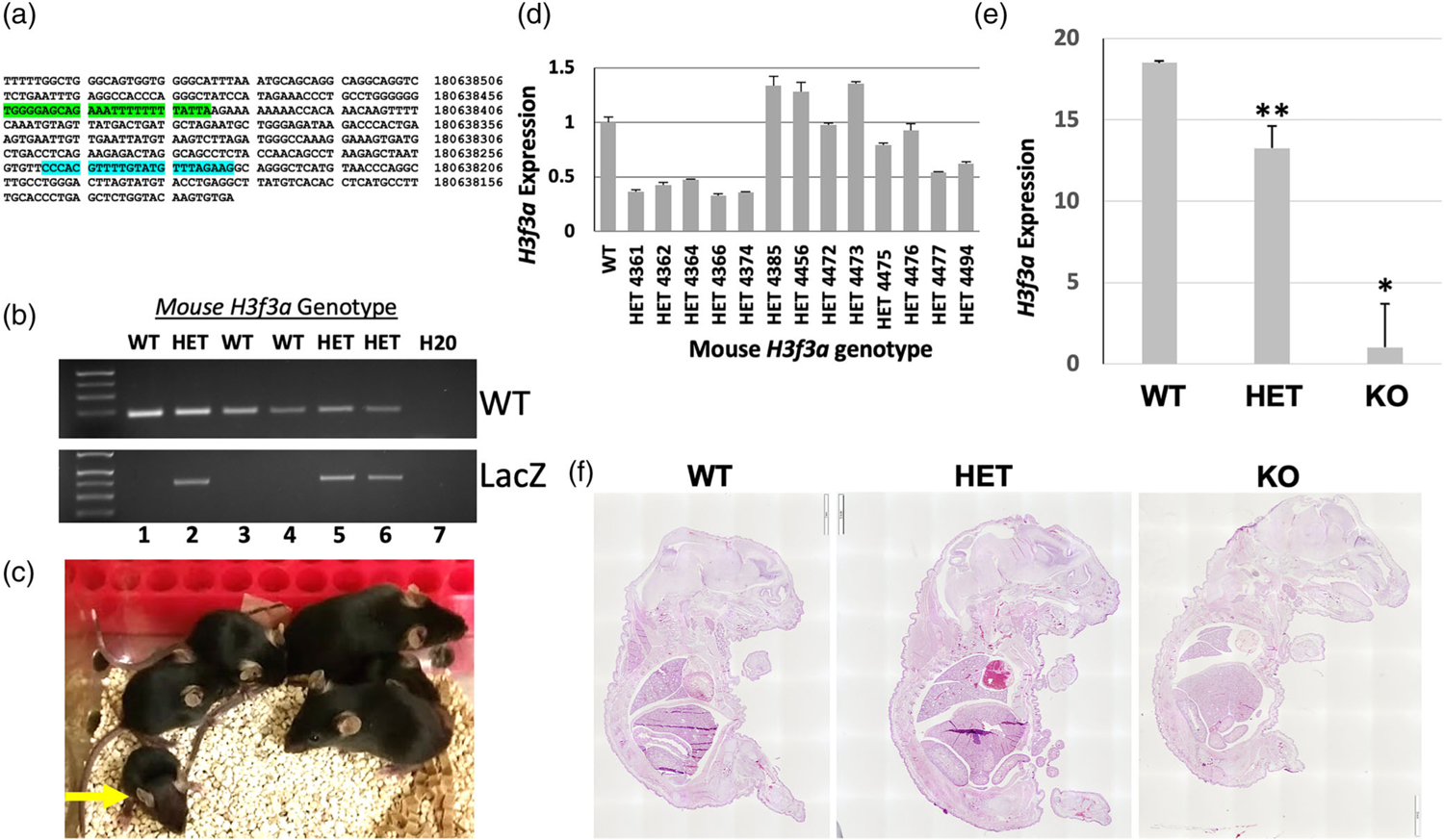
Phenotypes of H3f3a targeted mice and evidence of embryonic lethality. (a) Sequence of the WT H3f3a allele with WT genotyping primer binding sites highlighted from UCSC Browser (mm39). (b) Example of WT and knockout-first allele (LacZ) amplicons detected via gene-specific genomic PCR. (c) A representative litter of pups generated from heterozygous crosses. All surviving pups were WT or heterozygous. A small heterozygous pup at the left is indicated by the yellow arrow. (d) H3f3a RNA levels by RT-qPCR in WT and heterozygous pups; heterozygous H3f3a RNA levels are heterogeneous. (e) H3f3a RNA levels in WT, heterozygous, and null animal ear tissue samples. (f) Representative H&E stained sections of E18.5 littermate embryos from heterozygous intercrosses are shown. Error bars are the standard deviation of triplicate samples in (d–e) and p values for WT versus heterozygous and null are = .0002 and .002, respectively for ** and *
2.2 |. Homozygous lethal and variable heterozygous targeted phenotypes
Numerous H3f3aWT/tm1a × H3f3aWT/tm1a crosses produced only one surviving homozygous H3f3atm1a/tm1a knockout mouse at birth, indicating a generally lethal phenotype. This surviving knockout appears generally healthy but is infertile. Notably, about half of the H3f3aWT/tm1a pups had low body weight (see small heterozygous pup at left in Figure 2c indicated by yellow arrow). This highly variable phenotype in some heterozygous pups included in some cases neonatal lethality, retarded growth, and reduced size, while other heterozygous pups seemed outwardly normal. Despite these partially penetrant initial heterozygous phenotypes, nearly all surviving H3f3aWT/tm1a mice eventually grew to normal size and all those that were bred were fertile.
Since there were apparent phenotypic differences between H3f3aWT/tm1a pups, as some closely resembled H3f3aWT/WT whereas others exhibited early growth retardation or prenatal or neonatal death, we used RT-qPCR to analyze H3f3a mRNA expression in surviving H3f3aWT/tm1a mice relative to H3f3aWT/WT littermates. Approximately half of analyzed H3f3aWT/tm1a mice displayed the expected roughly 50% reduction in H3f3a expression, while notably, the other half exhibited approximately equal or slightly higher than WT levels of H3f3a (Figure 2d). These data suggest that in heterozygotes the H3f3atm1a allele has quite variable rates of transcription, likely correlating with the observed variable heterozygous phenotypes. The one surviving H3f3atm1a/tm1a null mouse exhibited only background levels of H3f3a expression relative to WT and heterozygous levels (Figure 2e).
Of the 30 liters analyzed from H3f3aWT/tm1a × H3f3aWT/tm1a pairs, we observed lower than expected Mendelian ratios of H3f3aWT/tm1a mice, which is consistent with some lethality (Table 1). No H3f3atm1a/tm1a pups were found dead at birth or shortly after, consistent with homozygous disruption of H3f3a being late embryonic lethal. To further investigate this possibility, we established H3f3aWT/tm1a× H3f3aWT/tm1a timed matings and isolated embryos at 13.5 and 18.5 days post-conception (E13.5 and E18.5).
TABLE 1.
H3f3a knockout-first mice are underrepresented from heterozygous intercrosses after E13.5
| H3f3aWT/WT | H3f3aWT/t | H3f3at/t | |
|---|---|---|---|
| >p21 (30) | 46 | 63 | 1 |
| Ratio | 0.42 | 0.57 | 0.01 |
| E18.5 (2) | 2 | 5 | 1 |
| Ratio | 0.25 | 0.63 | 0.13 |
| E13.5 (3) | 3 | 9 | 5 |
| Ratio | 0.18 | 0.53 | 0.29 |
At E18.5, one null embryo was isolated along with two WT and two heterozygous control littermates. These embryos were embedded, sagittally sectioned, and stained with H&E (Figure 2f). The null was generally smaller than its littermates, and had a small head and brain as well as small lungs. Notably, while the pleural cavities appeared of similar volumes in each E18.5 embryo regardless of genotype, the lungs in the null uniquely fill only a small portion of this cavity.
At E13.5, we observed close-to-expected ratios of H3f3aWT/WT, H3f3aWT/tm1a, and H3f3atm1a/tm1a embryos with no underrepresentation of homozygous targeted embryos, suggesting that H3f3a null embryos are mostly viable until at least E13.5 (Table 1). The E13.5 H3f3atm1a/tm1a embryos were slightly smaller on average than littermates with small heads that tended to be flatter, consistent with a smaller brain (Figure 3a).
FIGURE 3.
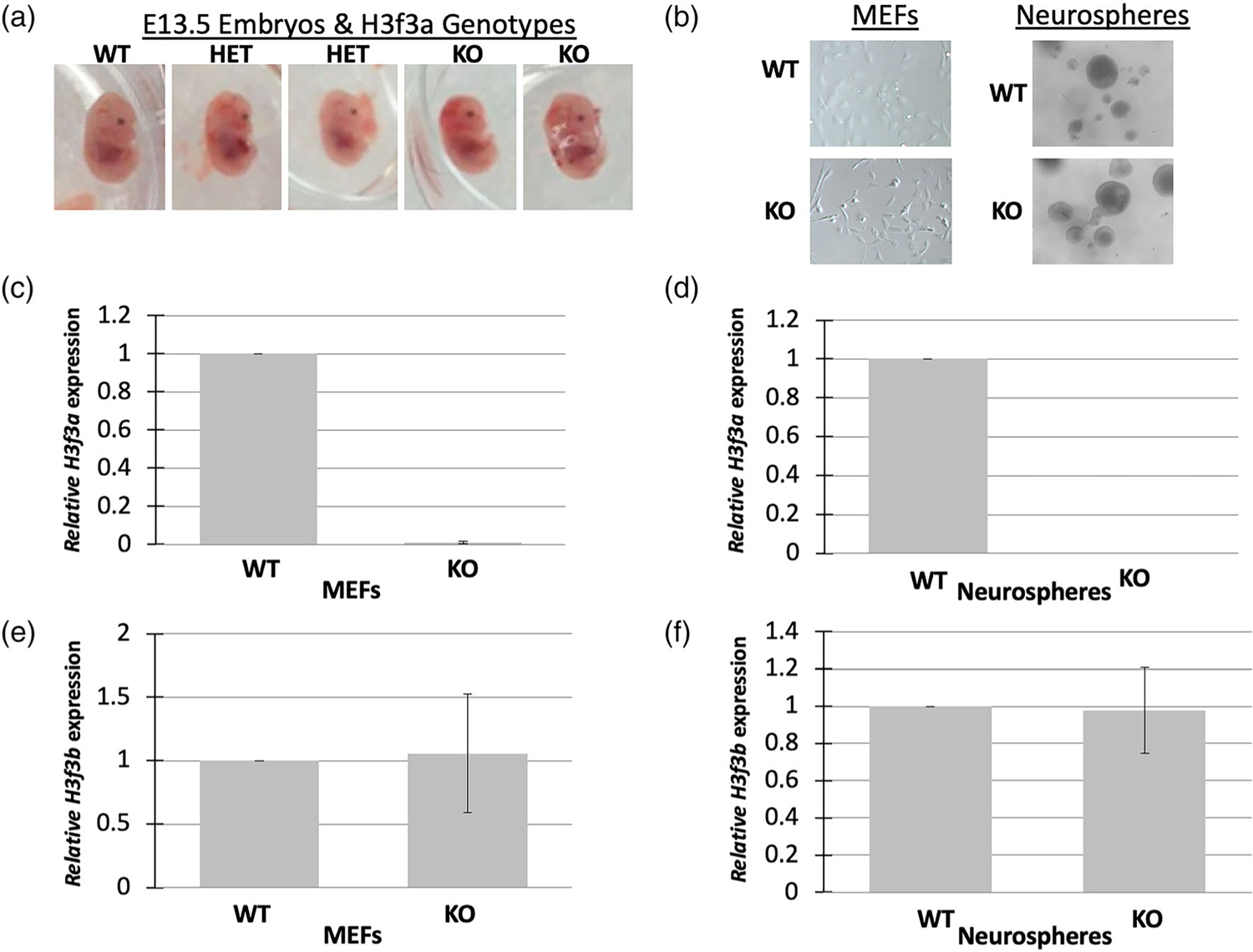
H3f3a targeted MEFs and neurospheres. (a) Heterozygous and null mid-gestational embryos exhibit aberrant phenotypes including small heads. Timed matings were conducted and E13.5 embryos were isolated from heterozygous crosses. Representative embryos are shown. (b) MEFs and neurospheres were isolated from the embryos and cultures established. There were no obvious phenotypes of the cells of different genotypes. (c–d) H3f3a null MEFs and neurospheres exhibited nearly undetectable levels of H3f3a RNA, and (e–f) no significant changes in H3f3b RNA. MEFs and neurospheres were produced from one WT, two heterozygous, and two null embryos for RNA analysis
2.3 |. Generation of E13.5 targeted cell lines
The same E13.5 embryos as described above were used to generate primary cell lines (Figure 3a). The head was removed to establish neural stem cell and precursor cell lines (neurospheres), internal organs were removed, and the remaining body tissue was used to form lines of MEFs. Despite homozygous disruption of H3f3a, H3f3atm1a/tm1a MEFs and neurospheres generally displayed cellular morphology and growth similar to cells from wildtype littermates (Figure 3b).
To validate the disruption of H3f3a expression in H3f3atm1a/tm1a cell lines, we conducted RT-qPCR using Taqman primers specific to H3f3a. Only minimal levels of H3f3a were detected in H3f3atm1a/tm1a MEFs and neurospheres, likely reflecting background signal (Figure 3c,d). We also analyzed H3f3b levels to assess the possibility of compensation in our embryonic cell lines lacking H3f3a. While H3f3b levels varied between lines for both H3f3a null MEFs and neurospheres, on average they displayed levels similar to H3f3b expression in control H3f3aWT/WT lines (Figure 3e,f), suggesting there are no consistent compensatory increases in H3f3b in H3f3a null cultured cells.
2.4 |. Impact of H3f3a knockout on histone H3 marks in primary cells
To examine the effects of H3f3a disruption on H3.3 protein levels and potential effects on overall H3 epigenetic marks in a neural precursor context, we performed Western blots on acid histone extracts isolated from neurospheres (Figure 4a,b). Both total H3.3 and H3 protein levels were significantly reduced in H3f3atm1a/tm1a neurospheres relative to control WT cells isolated from the same litters, suggesting a high percentage of total histone H3 is comprised of H3.3 in these cells. Note that the total H3 antibody recognizes all forms of H3 family members including H3.3.
FIGURE 4.
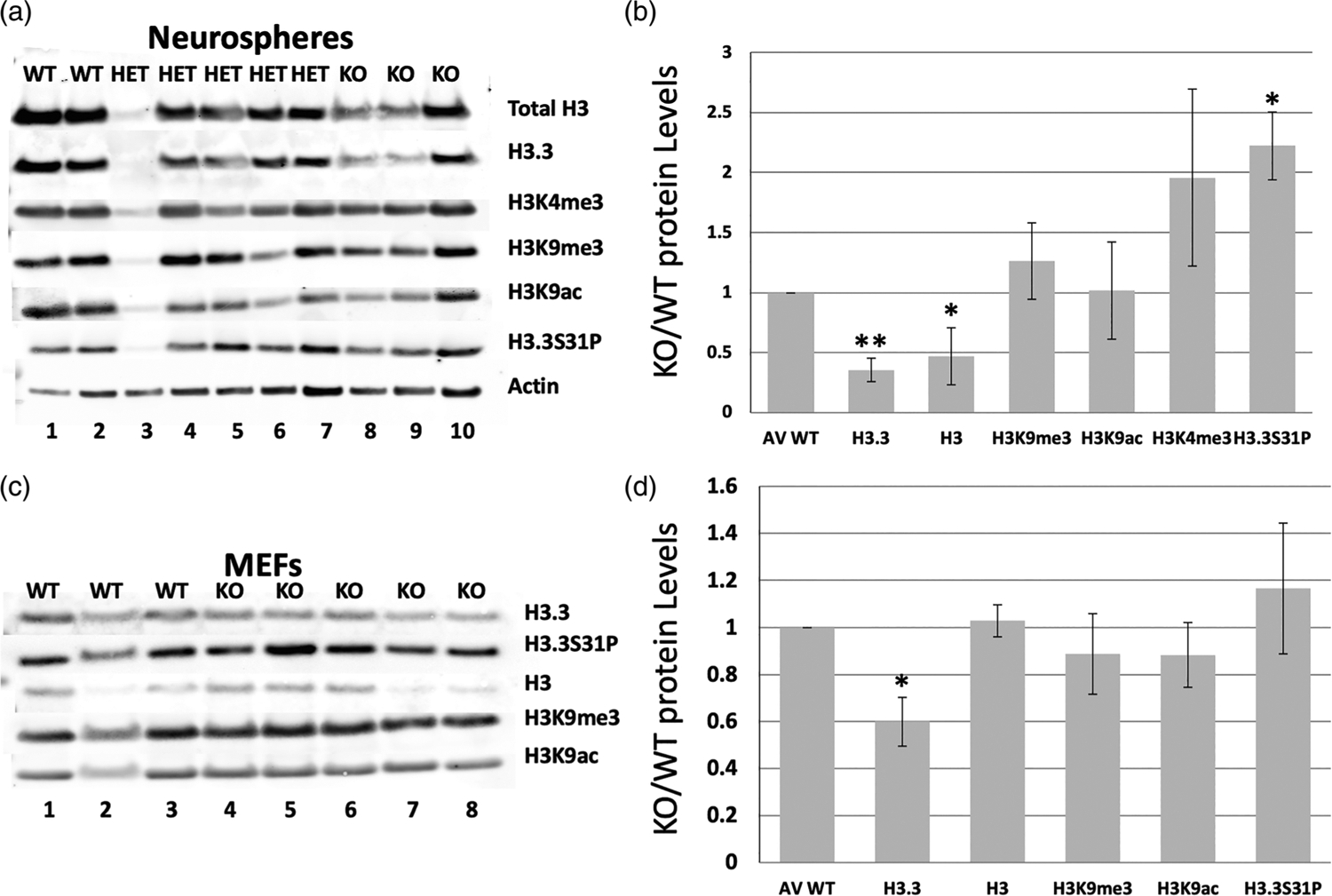
Loss of H3f3a strongly reduces both total H3 and H3.3 protein levels, but only impacts some histone H3 family marks. Histone extracts were isolated from H3f3a null MEFs and neurospheres, and analyzed by Western blot for total H3, H3.3, and a panel of histone marks. (a–b) Western blots for the indicated histone marks and controls of total H3, total H3.3, and b-Actin levels in neurospheres. (c–d) Western blots for the indicated histone marks and controls of total H3, total H3.3, and b-Actin levels in MEFs. Error bars are standard deviations. P values are <.05 and <.005 for * and ** marked samples. MEFs and neurospheres were derived from two WT, five heterozygous, and three null embryos here for histone mark protein level analysis
While levels of H3K9 tri-methylation and acetylation remained largely unchanged relative to total H3 levels in null neurospheres as compared to controls, there was an overall two-fold increase in the active mark H3K4me3 in nulls, although not reaching statistical significance. Notably, the H3.3-specific mitotic mark H3.3S31P was elevated in H3f3atm1a/tm1a lines when normalized to H3.3 protein levels (Figure 4a,b).
In parallel, we performed Western blots in control and H3f3atm1a/tm1a MEFs isolated from the same embryos as the neurosphere derivation (Figure 4c,d). Overall, MEFs appeared less affected by the loss of H3f3a in terms of histone marks as compared to neurospheres. For instance, while we had seen a marked increase in H3.3S31P levels in H3f3atm1a/tm1a neurospheres, the levels were only slightly elevated in MEFs. Further, although we observed a significant decrease in H3.3 levels in H3f3atm1a/tm1a MEFs, total H3 levels remained relatively unchanged in those cells. Previous studies from our lab suggest that H3f3b contributes more to H3.3 protein production in MEFs, which could potentially explain the only moderate epigenetic changes observed here with loss of H3f3a (Bush et al., 2013).
2.5 |. Analysis of impacts of loss of H3f3a on the transcriptome
In order to assess the impact of loss of H3f3a on the transcriptome, RNA was isolated from control and knockout neurospheres and MEFs, and analyzed by 3′-Tag-Seq. A small number of genes were up- or downregulated in null neurospheres (Table 2). As expected, H3f3a itself was one of the most strongly downregulated genes in null neurospheres. The expression of several protocadherin genes was significantly changed in the nulls as well. Null MEFs exhibited almost no consistent and significant changes in gene expression, perhaps due to a more dominant role for H3f3b in MEFs.
TABLE 2.
Genes with the most significantly altered expression from Tag-Seq analysis of H3f3a knockout neurospheres versus wild-type neurospheres. H3f3a itself, bolded in the table, was the gene with the second biggest drop in expression
| Official gene symbol or designation | logFC | p value | FDR |
|---|---|---|---|
| ENSMUST00000080170 | −6.6280 | 2.38 E-06 | 0.0175 |
| h3f3a | 6.5807 | 3.43 E-06 | 0.0219 |
| hmcn1 | −5.0349 | 1.16 E-11 | 3.70 E-07 |
| fras1 | −4.0930 | 5.56 E-17 | 2.66 E-12 |
| tusc1 | −3.8684 | 2.25 E-07 | 0.0027 |
| irs4 | −3.8113 | 2.00 E-06 | 0.0159 |
| pcdhga3 | −3.2228 | 3.83 E-09 | 7.34 E-05 |
| slc6a11 | 1.2595 | 5.26 E-06 | 0.0296 |
| pcdhb7 | 2.4457 | 1.21 E-07 | 0.0019 |
| gsta4 | 2.5102 | 2.75 E-06 | 0.0188 |
| cd38 | 3.2081 | 2.01 E-10 | 4.81 E-06 |
| kcnk1 | 3.2697 | 1.47 E-06 | 0.0145 |
| g530011o06rik | 3.5724 | 1.67 E-07 | 0.0022 |
| nrn1 | 5.2609 | 2.34 E-18 | 2.24 E-13 |
| pcdh15 | 6.5435 | 4.40 E-06 | 0.0263 |
| gmll549 | 6.6440 | 1.67 E-06 | 0.01456 |
| trim30d | 7.2172 | 1.63 E-06 | 0.01456 |
3 |. DISCUSSION
In recent years more has been learned about the roles of the H3.3-coding genes H3f3a and H3f3b in mammalian development. An early gene trap study reported targeting of H3f3a led to murine infertility (Couldrey et al., 1999). Single knockouts of the two H3.3 coding genes (H3.3A and H3.3B) in Drosophila did not strongly affect development and the double H3.3A/H3.3B knockout in the fly was compatible with life but led to infertility (Hodl & Basler, 2009; Sakai, Schwartz, Goldstein, & Ahmad, 2009). Single mouse knockout studies have also found that H3f3a and H3f3b have roles in mammalian fertility and more specifically in gamete and early embryonic states including specifying appropriate chromatin states (Bush et al., 2013; Jang et al., 2015; Tang et al., 2015; Tang, Binos, Ong, Wong, & Mann, 2014; Yuen et al., 2014). Thus, in a broad sense, loss of H3.3 likely leads to infertility due to perturbed chromatin in gametes and early embryos. In mice, double knockout led to a severe, early lethal phenotype and substantial changes in primary H3.3 null cells (Jang et al., 2015).
Our previous work with the H3f3b conditional KO system found that some H3f3b null mice survive even if they were not fertile (Bush et al., 2013), while in our new studies we found only one surviving H3f3a homozygous mutant mouse in years of breeding and found no dead null pups early after birth, most consistent with a late embryonic lethal phenotype. This pattern of H3f3a and H3f3b phenotypes is generally the opposite of that observed by the Mann group, where they found more severe effects of loss of H3f3b versus H3f3a (Tang et al., 2014; Tang et al., 2015). One potentially straightforward explanation for these differences is the distinct genetic backgrounds of the mice in these studies (C57BL/6 in our work versus 129 or 129 mixed with C57BL/6 in mice from the other labs). Furthermore, given that different methods were used for gene targeting, such technological differences may contribute to different phenotypes reported by the various labs as well. More specifically, while we used a knockout-first gene trapping approach for disruption of H3f3a, other labs deleted exonic portions of the gene.
We also observed that some heterozygously targeted H3f3a mice exhibited only temporary impaired growth phenotypes early on. The variably reduced H3f3a expression in our H3f3aWT/tm1a mice likely explains part of their variable phenotype. It is also possible that in H3f3aWT/tm1a mice that the targeted allele is sometimes expressed at low but sufficient levels to avoid an obvious phenotype.
Transient and variable phenotypes with homozygous loss of one of the two H3.3-coding genes could be due to variable compensation by the remaining H3.3-coding gene or other compensatory mechanisms in cells and organisms such as changes in H3.3 chaperone function. In fully H3.3 deficient, double knockout flies, canonical H3 was also reported to have elevated expression and begin functioning in a partially replication-independent manner compensating for loss of H3.3 (Sakai et al., 2009), perhaps explaining the mechanism by which some double knockouts could survive. It is unknown whether loss of H3.3 induces H3.1 or H3.2 to function in a more H3.3-like manner in mammals, but that also could contribute to variability in knockout phenotypes.
In humans, mutations in H3F3A and H3F3B are associated with different kinds of tumors, which may in part reflect their differences in expression patterns. H3F3A mutations are commonly found in high-grade childhood gliomas (reviewed in [Yuen & Knoepfler, 2013]), fitting with a proposed role for H3F3A expression more strongly contributing to H3.3 protein levels in the brain, while H3F3B mutations are by contrast most often present in childhood bone and cartilage tumors (Behjati et al., 2013). Although the specific molecular roles of distinct mutant H3.3 proteins in these tumors are not fully understood, the findings so far support a model in which key normal epigenetic functions of H3.3 in neurodevelopmental programs are perturbed by each of the tumor-associated H3.3 mutant proteins, with some overlap in H3.3 K27M and G34R mutation mechanisms (Chen et al., 2020). This model is consistent with the murine loss-of-function studies pointing to H3.3 epigenetic regulation of normal development via impact on gene expression. In glioma, mutant H3.3 is also associated with perturbed Polycomb repressive complex 2 (PRC2) activity and aberrant patterns of histone modifications including in particular H3K27me3 (Jain et al., 2020). It is unclear if H3.3 normally functions broadly in the developing embryo in part via PRC2 specifically in stem cells but work in mESC indicating a central role for HIRA-H3.3 in PRC2 function (Banaszynski et al., 2013) makes this likely.
More recent work has identified distinct mutations in H3F3A and H3F3B in people with neurodevelopmental disorders as well, pointing to specific nervous system functions (Bryant et al., 2020). Our analysis of the H3f3a null E18.5 embryo suggested a small brain phenotype, which could be consistent with the mutant phenotypes in humans and warrants further exploration. The small lungs in the H3f3a null E18.5 embryo are potentially consistent with findings from H3.3-coding gene knockouts from others who reported finding targeted animals with respiratory problems (Jang et al., 2015).
We found that loss of H3f3a and the accompanying decrease in H3.3 protein levels had moderate effects on epigenetic marks in MEFs or neurospheres. The most pronounced change was an increase in the H3.3-specific mark H3.3S31P in null neurospheres and our previous work found changes in this mark in H3f3b null MEFs (Bush et al., 2013) as well. Effects on histone marks of partial loss of H3.3 by targeting either H3f3a or H3f3b appear to be highly cell-type dependent.
Loss of H3f3a also only had modest effects on the transcriptome. Few effects were found in null MEFs, while null neurospheres exhibited relatively more changes, but still only moderate overall. Only moderate changes in the transcriptome with loss of H3.3-coding genes has been reported before (Jang et al., 2015), suggesting the presence of compensatory or redundant mechanisms. It is also possible that the primary normal function of H3.3 is not to regulate the transcriptome.
Overall, additional genetic tools for studying H3f3a, H3f3b, and H3.3 will shed more light on their functions in normal development and in diseases such as childhood cancer and neurodevelopmental disorders.
4 |. METHODS
4.1 |. Data sharing
The Tag-Seq data from our neurosphere analysis has GEO accession number GSE216047.
4.1.1 |. Transgenic H3f3a knockout-first mouse production
Details of the production of targeted mice are described in the Results section. While the mice were originally generated as C57BL6/N they were also at some point bred with C57BL6/J leading to a mixed C57BL6 background. Long-range PCR for genotyping mESCs and mice to determine homologous versus non-homologous recombination was conducted using the following primers:
CSD-en2frt-R 5′-GGTGGTGTGGGAAAGGGTTCGAAG-3′.
CSD-H3f3a-LF1 5′-TCCTCTTTCTTCGGTGAAATCCCTCC-3′.
The LoxP PCR assay used the following primers:
CSD-H3f3a-SR1 5′-AGAAACAGCTCAACCAATCACACACC-3′.
CSD-loxPcomF 5′-GAGATGGCGCAACGCAATTAAT-3′.
4.1.2 |. Genotyping
All genotyping was performed using Bioneer AccuPower pre-mixed tubes (http://www.bioneer.com) and primers targeting LacZ for the transgenic allele, and regions 5′ and 3′ to the transgenic insertion site for WT. The amplicon sizes are 228 bp and 389 bp for WT and transgenic, respectively. See Figures 1a–c and 2a–b for details.
WT F: 5′-TGG GGA GCA GAA ATT TTT TTT ATT A-3′.
WT R: 5′-CTT CTA AAC ATA CAA AAC GTG GG-3′.
LacZ F: 5′-GTT GCA GTG CAC GGC AGA TAC ACT TGC TGA-3′.
LacZ R: 5′-GCC ACT GGT GTG GGC CAT AAT TCA ATT CGC-3′.
4.1.3 |. Creation of primary cell lines
H3f3aWT/tm1a × H3f3aWT/tm1a crosses were time mated, and upon observance of a plug, embryos were isolated at E13.5. The visceral tissue was removed and used for genotyping, while the head region was used to establish the neurosphere lines as described (Knoepfler et al., 2006), and the body was used to produce MEFs. Neurospheres were initially cultured on low-bind plates (Corning; 3261) in N+ media (Neurobasal media, 1x B27, 1% Pen/Strep/Glutamine), bFGF (20 ng/ml), EGF (20 ng/ml) and cultured routinely in low-bind dishes or suspension flasks (Olympus Plastics; 25–213). MEFs were cultured in DMEM enriched with 10% FBS, 1% non-essential amino acids, and 1% Glutamine. All cells were incubated at 37°C with 5% CO2.
4.1.4 |. RNA and cDNA production and qPCR
RNA was extracted from primary cell cultures or tail tips using the NucleoSpin RNA kit produced by Macherey-Nagel (Macherey-Nagel; 740955.50). 1 μg of RNA was transcribed into cDNA using an iScript cDNA synthesis kit (Bio-Rad; 1708891). cDNA was further diluted and used in qPCR experiments.
qPCR for H3f3a, H3f3b and house-keeping control Eif4g2 was performed using Taqman probes (Applied biosystems; mm01612808_g1, mm00787223_s1 and mm00469036_m1, respectively) and associated protocol. All other genes were analyzed using Sybr Blue master-mix (Abgene; AB4162) or PowerUp SYBR mix (Thermo; A25741) with primers designed in lab. 96-well plates were run on Roche Light Cycler 480, and results were analyzed statistically using the 2−δδCt method.
4.1.5 |. Protein analysis
Histone extracts were prepared from H3f3a MEFs and neurospheres using the Abcam protocol with a few modifications. Briefly, cells were washed in PBS with 5 mM Sodium Butyrate, and lysed in TBE buffer (0.5% Triton X-100, 0.02% sodium azide plus Protease inhibitor: Roche; 11836170001). Cells were then pelleted, re-suspended in 0.2 N HCl, and rocked overnight at 4°C. Supernatant was neutralized using 1 N NaOH and frozen at 80°C. Samples were processed with LDS sample buffer (Life Technologies; NP0008) and DTT and run on 6–12% Bis-Tris SDS Nupage gels (Life Technologies; NP0335BOX). Proteins were then transferred onto PVDF membranes and probed with the antibodies (Table 3). Results were normalized to actin for H3/H3.3 and histone-marks were normalized to actin and H3 or H3.3. For statistical analysis, p-values were calculated using an unpaired t test.
TABLE 3.
Antibodies used
| Target | Vendor | Cat # | Dilution |
|---|---|---|---|
| Total H3 | Millipore | 05–499 | 1:2,000 |
| H3.3 | Abnova | H00003021-M01 | 1:2,500 |
| H3.3 | Millipore | 09–838 | 1:1,000 |
| H3K4me3 | Millipore | 07–523 | 1:2,000 |
| H3K9me3 | Millipore | 04–745 | 1:3,000 |
| H3K9ac | Abcam | ab12179 | 1:2,000 |
| H3.3S31P | Abcam | ab92628 | 1:1,000 |
| B-Actin | Sigma | A1978 | 1:5,000 |
4.2 |. Tag-Seq (RNA-Seq) analysis
RNA was extracted from WT and KO cells using the NucleoSpin RNA Kit (Macherey-Nagel). Samples were submitted to UC Davis Genome Center DNA Technologies Core where RNA quality was assessed by Agilent Bioanalyzer and libraries were prepared using QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina. Sequencing was performed on three biological replicates each for WT MEFs, WT neurospheres, and KO neurospheres and five biological replicates for KO MEFs on Illumina HiSeq 4,000. Transcript abundance was quantified by Salmon (version 1.1.0) based on the ensemble-release87 (Mus_musculus.GRCm38.cdna.all.fa) and imported by Tximport (Bioconductor version 3.10). Downstream differential expression was then determined by EdgeR (Bioconductor version 3.10). Data was subject to a threshold of FDR <0.05 and p-value ≤.01. Raw and analyzed 3′-Tag-seq data are accessible via GEO.
ACKNOWLEDGMENTS
This work was supported by the following NIH grants to Paul S. Knoepfler: 1R01GM116919 and 1R01NS106878. We want to thank the University of California, Davis Mouse Biology Program for their help with the generation and analysis of the mice. We thank Kent Lloyd and Louise Lanoue for providing feedback on the manuscript and data.
Funding information
National Institute of General Medical Sciences, Grant/Award Number: 1R01GM116919; National Institute of Neurological Disorders and Stroke, Grant/Award Number: 1R01NS106878
DATA AVAILABILITY STATEMENT
GEO accession number is GSE216047.
REFERENCES
- Ahmad K, & Henikoff S (2002). The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Molecular Cell, 9(6), 1191–1200 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12086617 [DOI] [PubMed] [Google Scholar]
- Bachu M, Tamura T, Chen C, Narain A, Nehru V, Sarai N, … Ozato K (2019). A versatile mouse model of epitope-tagged histone H3.3 to study epigenome dynamics. Journal of Biological Chemistry, 294(6), 1904–1914. 10.1074/jbc.RA118.005550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski LA, Wen D, Dewell S, Whitcomb SJ, Lin M, Diaz N, … Allis CD (2013). Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell, 155(1), 107–120. 10.1016/j.cell.2013.08.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, … Flanagan AM (2013). Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nature Genetics, 45(12), 1479–1482. 10.1038/ng.2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant L, Li D, Cox SG, Marchione D, Joiner EF, Wilson K, … Bhoj EJ (2020). Histone H3.3 beyond cancer: Germline mutations in histone 3 family 3A and 3B cause a previously unidentified neurodegenerative disorder in 46 patients. Science Advances, 6(49), eabc9207. 10.1126/sciadv.abc9207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush KM, Yuen BT, Barrilleaux BL, Riggs JW, O’Geen H, Cotterman RF, & Knoepfler PS (2013). Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics & Chromatin, 6(1), 7. 10.1186/1756-8935-6-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KY, Bush K, Klein RH, Cervantes V, Lewis N, Naqvi A, … Knoepfler PS (2020). Reciprocal H3.3 gene editing identifies K27M and G34R mechanisms in pediatric glioma including NOTCH signaling. Communications Biology, 3(1), 363. 10.1038/s42003-020-1076-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couldrey C, Carlton MB, Nolan PM, Colledge WH, & Evans MJ (1999). A retroviral gene trap insertion into the histone 3.3A gene causes partial neonatal lethality, stunted growth, neuromuscular deficits and male sub-fertility in transgenic mice. Human Molecular Genetics, 8(13), 2489–2495 https://www.ncbi.nlm.nih.gov/pubmed/10556297 [DOI] [PubMed] [Google Scholar]
- Elsaesser SJ, & Allis CD (2010). HIRA and Daxx constitute two independent histone H3.3-containing predeposition complexes. Cold Spring Harbor Symposia on Quantitative Biology, 75, 27–34. 10.1101/sqb.2010.75.008 [DOI] [PubMed] [Google Scholar]
- Elsaesser SJ, Goldberg AD, & Allis CD (2010). New functions for an old variant: no substitute for histone H3.3. Current Opinion in Genetics and Development, 20(2), 110–117. 10.1016/j.gde.2010.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipescu D, Szenker E, & Almouzni G (2013). Developmental roles of histone H3 variants and their chaperones. Trends in Genetics, 29(11), 630–640. 10.1016/j.tig.2013.06.002 [DOI] [PubMed] [Google Scholar]
- Hodl M, & Basler K (2009). Transcription in the absence of histone H3.3. Current Biology, 19(14), 1221–1226. 10.1016/j.cub.2009.05.048 [DOI] [PubMed] [Google Scholar]
- Jain SU, Khazaei S, Marchione DM, Lundgren SM, Wang X, Weinberg DN, … Lewis PW (2020). Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proceedings of the National Academy of Sciences of the United States of America, 117(44), 27354–27364. 10.1073/pnas.2006076117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang CW, Shibata Y, Starmer J, Yee D, & Magnuson T (2015). Histone H3.3 maintains genome integrity during mammalian development. Genes and Development, 29(13), 1377–1392. 10.1101/gad.264150.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, & Eisenman RN (2006). Myc influences global chromatin structure. EMBO Journal, 25(12), 2723–2734 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16724113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, & Allis CD (2010). Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proceedings of the National Academy of Sciences of the United States of America, 107(32), 14075–14080. 10.1073/pnas.1008850107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, de Krijger I, Wiegant WW, Shah RG, Smeenk G, de Groot AJL, … van Attikum H (2016). PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Molecular Cell, 61(4), 547–562. 10.1016/j.molcel.2016.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKittrick E, Gafken PR, Ahmad K, & Henikoff S (2004). Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proceedings of the National Academy of Sciences of the United States of America, 101(6), 1525–1530 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14732680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai A, Schwartz BE, Goldstein S, & Ahmad K (2009). Transcriptional and developmental functions of the H3.3 histone variant in drosophila. Current Biology, 19(21), 1816–1820. 10.1016/j.cub.2009.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang MC, Binos S, Ong EK, Wong LH, & Mann JR (2014). High histone variant H3.3 content in mouse prospermatogonia suggests a role in epigenetic reformatting. Chromosoma, 123(6), 587–595. 10.1007/s00412-014-0475-8 [DOI] [PubMed] [Google Scholar]
- Tang MC, Jacobs SA, Mattiske DM, Soh YM, Graham AN, Tran A, … Mann JR (2015). Contribution of the two genes encoding histone variant h3.3 to viability and fertility in mice. PLoS Genetics, 11(2), e1004964. 10.1371/journal.pgen.1004964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang MC, Jacobs SA, Wong LH, & Mann JR (2013). Conditional allelic replacement applied to genes encoding the histone variant H3.3 in the mouse. Genesis, 51(2), 142–146. 10.1002/dvg.22366 [DOI] [PubMed] [Google Scholar]
- Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, Hannan RD, … Choo KH (2010). ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Research, 20(3), 351–360. 10.1101/gr.101477.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen BT, Bush KM, Barrilleaux BL, Cotterman R, & Knoepfler PS (2014). Histone H3.3 regulates dynamic chromatin states during spermatogenesis. Development, 141(18), 3483–3494. 10.1242/dev.106450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen BT, & Knoepfler PS (2013). Histone H3.3 mutations: A variant path to cancer. Cancer Cell, 24(5), 567–574. 10.1016/j.ccr.2013.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
GEO accession number is GSE216047.