

SUMMARY
Color variation is a frequent evolutionary substrate for camouflage in small mammals but the underlying genetics and evolutionary forces that drive color variation in natural populations of large mammals are mostly unexplained. The American black bear, Ursus americanus, exhibits a range of colors including the cinnamon morph which has a similar color to the brown bear, U. arctos, and is found at high frequency in the American southwest. Reflectance and chemical melanin measurements showed little distinction between U. arctos and cinnamon U. americanus individuals. We used a genome-wide association for hair color as a quantitative trait in 151 U. americanus individuals and identified a single major locus (P < 10−13). Additional genomic and functional studies identified a missense alteration (R153C) in Tyrosinase-related protein 1 (TYRP1) that likely affects binding of the zinc cofactor, impairs protein localization, and results in decreased pigment production. Population genetic analyses and demographic modeling indicated that the R153C variant arose 9.36kya in a southwestern population where it likely provided a selective advantage, spreading both northwards and eastwards by gene flow. A different TYRP1 allele, R114C, contributes to the characteristic brown color of U. arctos, but is not fixed across the range.
Keywords: functional phylogeography, GWAS, melan-b, migration, OCA3, TYRP1
eTOC BLURB
Puckett et al. show that a single base pair variant in TYRP1 results in the iconic cinnamon morph of the American black bear. They demonstrate that TYRP1 variants in black and grizzly bears are loss-of-function alleles and impair protein localization to melanosomes. The black bear allele arose 1440 generations ago in the southwestern range.
INTRODUCTION
Variation in animal color is a longstanding system with which to investigate gene action and evolution. In mammals, nearly all pigment is produced by melanocytes via polymerization of dihydroxyindole derivatives to yield insoluble melanins that are transferred to overlying keratinocytes in skin and/or hair. Genetic variation in melanin biosynthesis or transfer gives rise to hair, eye, and skin color differences, and can underlie positive selection1,2. Mutations of melanin biosynthesis underlie a number of conditions associated with impaired fitness or a disease (e.g. human albinism), or unusual color morphs of large mammals that are specifically targeted for trophy hunting3.
Identifying and understanding the genetic causes and consequences of mammalian color variation is of special interest in large mammals for two reasons. First, by contrast to small mammals in which color morphs are often selected as a means of predator avoidance, the evolutionary forces underlying color variation in large mammals are less apparent and occasionally controversial. Second, there are often charismatic and/or cultural values associated with certain species and color morphs such as the black jaguar and white tiger. Despite their common name, American black bears (Ursus americanus) occur in black, cinnamon, brown, blond, grey, and white morphs; similarly, brown bears (U. arctos) occur in blond, brown, dark chocolate, white, and ruddy. The cinnamon morph of U. americanus has a rich history in popular culture4 and systematics, and has been suggested to have adaptive value, either by aiding in thermoregulation in hotter and drier climates of the southwest, or by mimicry of U. arctos in areas where the two species are sympatric. Cinnamon has a distinct latitudinal gradient in western North America, with a high frequency in the southwest that declines moving northwards, and is at low frequency in the east5. This phenotypic variation is consistent with genomic analyses that indicate the deepest phylogeographic structuring occurs between the eastern and western lineages across North America6,7. Further, admixture between the lineages has been identified across the northern range and suggested to be a product of post-glacial range expansion, and east-west gene flow across contiguous habitat in modern Canada7.
The distinct spatial distribution of cinnamon U. americanus presents three alternative phylogeographic hypotheses. First, the causative mutation arose in the western lineage after the split from the eastern lineage. Second, the variant predates lineage divergence, but was lost during eastward range expansion. Third, introgression of the causative variant from U. arctos into U. americanus occurred in a western lineage population.
We used a genome-wide approach and identified the causative variant that produces cinnamon morphs in U. americanus; then applied molecular and population genetic approaches to explore its functional impact and demographic history. We identified a missense alteration in Tyrosinase-related protein 1 (TYRP1) and show that it interferes with melanin synthesis. We found the U. americanus TYRP1 variant was not introgressed from U. arctos; instead, a different TYRP1 variant contributes to their characteristic pigmentation phenotype. Remarkably, the variant responsible for cinnamon U. americanus is identical to one previously described as a cause of oculocutaneous albinism (OCA3) in humans, often associated with nystagmus and reduced visual acuity.
Our analyses indicate that the cinnamon TYRP1 variant arose within the western lineage of U. americanus and likely provided a selective advantage in the southwest. These results illustrate how Mendelian variation in melanogenesis can underlie iconic phenotypes and inform our understanding of color variation and recent evolution in large carnivores.
RESULTS and DISCUSSION
Quantitative and Chemical Characterization of Bear Hair Color
In addition to cinnamon, light-colored morphs of U. americanus have been described as blond, light brown, or chocolate; thus, the results presented reflect all of these color morphs. We measured reflectance of hair samples from 391 U. americanus and 33 U. arctos from across North America. Most U. americanus qualitatively described as black based on photographs exhibited reflectance values < 50 (median = 5), whereas animals scored as cinnamon had broadly ranging values (median = 67; Figure 1A); overall, the distribution of reflectance varied continuously from 0.5–182 (possible range: 0–255; Figures 1A, S1). Reflectance in U. arctos also varied continuously (median = 48). Thus, we consider the reflectance measure a quantitative variable.
Figure 1-. Characterization of bear hair color.
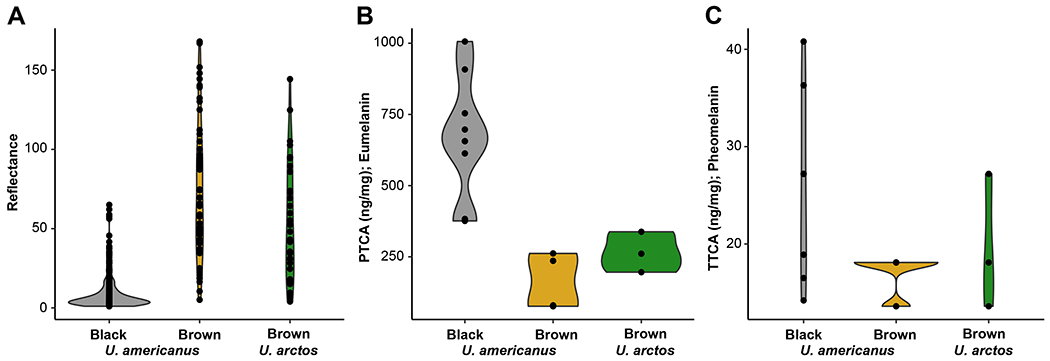
Violin plots of hair reflectance for two bear species (Ursus americanus [n = 327] and U. arctos [n = 33]) categorized by qualitative phenotyping from photos as either black or brown animals (black U. americanus- grey; brown U. americanus- yellow; brown U. arctos- green). Chemical analysis via alkaline hydrogen peroxide oxidation followed by high performance liquid chromatography (HPLC) of 13 U. americanus and three U. arctos individuals’ hair for the concentration of (B) eumelanin (as PTCA; ANOVA F=14.512; P<0.001) or (C) pheomelanin (as TTCA; ANOVA F=2.0297; P=0.17). PTCA concentration in hair ranged from 75–1010 ng mg−1, where TTCA was limited to 13–40 ng mg−1 in U. americanus. Specifically, two-tailed t-tests between U. americanus black and brown animals (P<0.001), and U. americanus black and U. arctos were significantly different (P<0.001); whereas, no difference was observed between species with brown coloration (P=0.17). In U. arctos, PTCA and TTCA ranged respectively from 200–340 ng mg−1 and 13–27 ng mg−1. Both species indicate a dilution of eumelanin, and not an increase in pheomelanin, produces the characteristic hair-lightening.
We determined if hair color of cinnamon U. americanus and U. arctos was caused by dilution of eumelanin or increased production of pheomelanin using HPLC8,9 of the different pigment types in awn hair samples. As depicted in Figures 1B–C, the major determinant of light-colored hair is reduced amounts of eumelanin in both bear species.
Allelic Identification of TYRP1 Variants
We sequenced the genomes of 24 U. americanus to ≥30x depth and an additional 166 animals to 1.3x depth, mapped to the reference genome10, then imputed missing loci in the low coverage samples using the high coverage individuals as a reference panel11. A genome-wide association study (GWAS)12 for quantitative hair reflectance values from 151 bears identified 120 single nucleotide variants (SNVs) above a significance threshold of 10−8 (Figure 2A, Table S2). A single peak had the lowest P-values and spanned the pigmentation gene TYRP1 (Figure 2B), in which we identified a significant (P=1.99 x 10−13) G to A SNV, HiC_scaffold_24:6724152g>a that predicts a missense alteration, p.Arg153Cys, referred to in what follows as TYRP1R153C. This alteration within the luminal domain of TYRP1, a type I transmembrane protein, is within a critical region near the zinc cofactor binding site and thus is likely deleterious (Figure 2C). Nine additional SNVs had lower P-values and were 130kb downstream from the TYRP1 transcription start site (Figure 2B); however, after accounting for TYRP1R153C genotypes as a covariate, the significance of these sites fell below the genome-wide threshold. Further, haplotype analysis13 identified low diversity in the Nevada population, thus we used these samples as input into HAPLOVIEW14 and identified a single 97kb haploblock carrying TYRP1R153C (Figures 2B, S2). Recombination is apparent in populations outside of the southwest, suggesting that TYRP1R153C is derived from a single mutational event.
Figure 2-. Genome-wide association study of American black bear coat color.
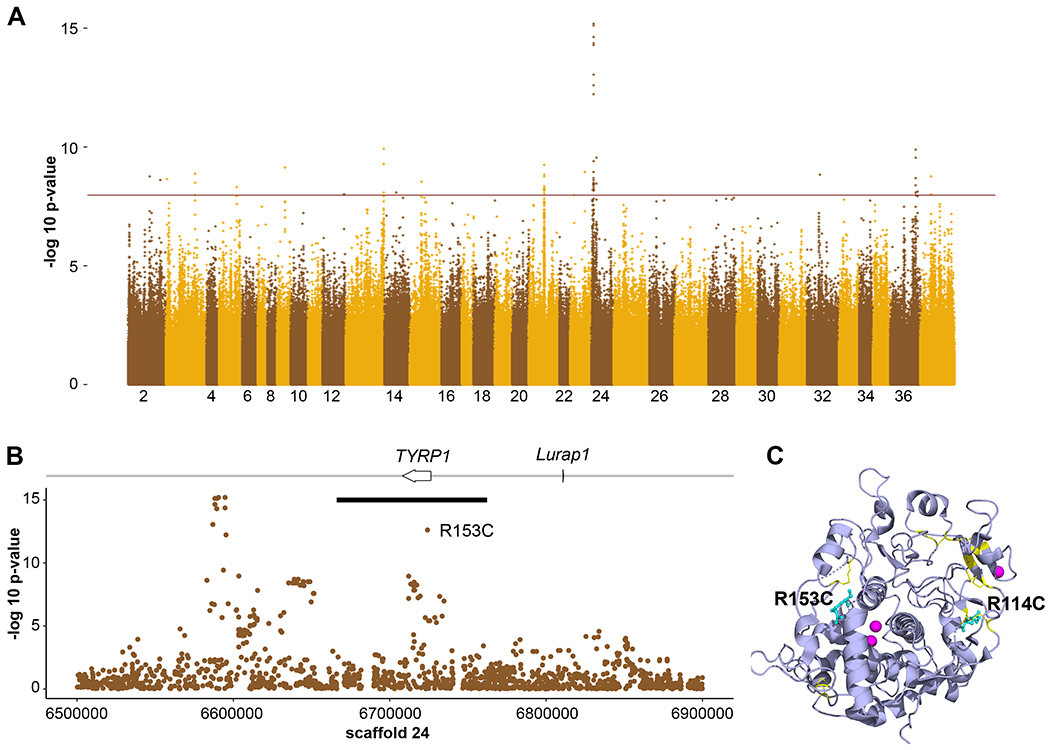
GWAS of both high and low coverage WGS data from Ursus americanus (n = 151) to identify loci associated with coat color. (A) A genome-wide Manhattan plot with a significance cut-off of 10−8 (horizontal brown line) identified a single strong peak on scaffold 24 (additional peaks in Table S6). (B) A detailed view of scaffold 24 surrounding the peak identified two genes, including TYRP1. The black bar denotes the length of the haplotype identified within the Nevada population that contains the R153C derived allele. (C) The locations of R114 and R153 (cyan, shown in atomic format) are shown within a ribbon diagram of the 3D structure of the human TYRP1 luminal domain (purple, from PDB ID 5M8L, 71). Cysteine residues involved in disulfide bonds are yellow and the zinc cofactors are magenta. Note the proximity of R114 to a native disulfide bond and of R153 to a break between two alpha helices that are part of the zinc binding region.
We explored coding sequence variation in TYRP1 and 12 additional pigmentation genes in U. americanus, U. arctos, and U. maritimus (polar bear). For U. americanus, we identified 46 missense or nonsense polymorphisms within 11 genes (Table S3); among these, only the TYRP1R153C variant was correlated with color (Figure S3A) and predicted to be deleterious. We identified 19 missense variants between U. americanus and U. arctos, and 33 missense or nonsense variants between U. arctos and U. maritimus (Table S3). The U. americanus R153C variant was not observed in U. arctos or U. maritimus; however, a different TYRP1 variant was identified in 60% of U. arctos individuals, HiC_scaffold_24:6725331g>a, that predicts a p.Arg114Cys substitution (TYRP1R114C). Both mutations occur in the lumenal domain of TYRP1, which contains 10 evolutionarily conserved cysteine residues that form 5 disulfide bonds; acquisition of an unpaired cysteine residue in TYRP1R153C and TYRP1R114C is likely to interfere with native disulfide bonding and protein folding (Table S3; Figures 2C, S3)34.
We also examined TYRP1 variation at orthologous positions in other species. In humans, loss-of-function for TYRP1 causes a rare form of albinism, rufous oculocutaneous albinism (OCA3), observed mostly in individuals of African or Puerto Rican ancestry, and characterized by reddish skin and hair and frequent visual abnormalities15. Among 15 individuals with clinical albinism and mutations of TYRP1, R114C and R153C were each observed once as compound heterozygotes16. The R153C variant is found at a relatively high frequency in individuals of European ancestry, 7.91x10−4 but is classified as a variant of uncertain significance17,18. In laboratory rats the R114C allele is likely responsible for a spontaneous brown mutation that is fixed in the Brown Norway strain19.
Functional Characterization
To test whether the TYRP1 R153C and R114C variants affect function, we assessed their impact on pigmentation following expression in mouse melan-b cells, which are derived from TYRP1b/b mutant mice and are hypopigmented due to the absence of functional or immunohistochemically detectable TYRP1. Wild-type (WT) mouse or human TYRP1 can rescue pigmentation in melan-b cells. We introduced the TYRP1 R153C or R114C variants into their orthologous positions in mouse and human TYRP1, and expressed WT or variant TYRP1, or mCherry-syntaxin 13 (mCh-STX13) as a negative control, from recombinant retroviruses in melan-b cells. Using a colorimetric melanin content assay (Figure 3A), WT human TYRP1 (hTYRP1) rescued pigmentation relative to control (untransfected or mCh-STX13 transfected) melan-b cells to nearly the same level as WT (melan-Ink4a) melanocytes. By contrast, the R153C and R114C variants rescued pigmentation in melan-b cells respectively only 52.0% and 14.1% as well as WT hTYRP1, and 46.0% and 6.8% as well as WT mouse TYRP1. This correlates with melanin quantification in bear hair detected by HPLC (Figure 1B), and indicates that R153C and R114C are causal variants for reduced pigmentation.
Figure 3-. Functional characterization of TYRP1 alleles.
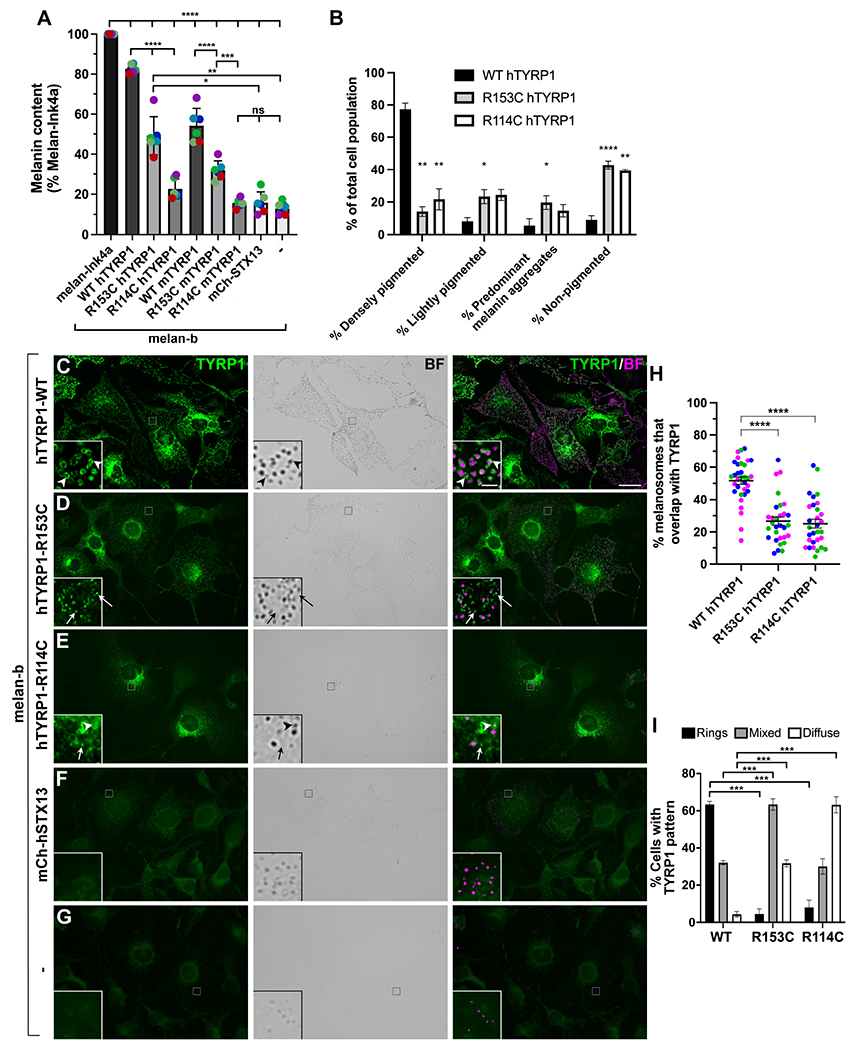
(A) TYRP1-deficient melan-b cells that were untransduced (−) or stably expressed the WT, R153C, or R114C variants of human (h) or mouse (m) TYRP1 or mCh-STX13 as a control were analyzed by quantitative melanin content assay and normalized to protein content. Data represent percent normalized melanin content relative to that of WT melan-Ink4a cells from six experiments (colored dots), each performed in duplicate, and analyzed by two-way ANOVA and Tukey’s multiple comparison test. (B) Stable melan-b transductants expressing WT, R153C, or R114C hTYRP1 variants were analyzed by bright field microscopy, and individual cells were characterized as densely pigmented, lightly pigmented, light with dense aggregates, or non-pigmented. Data from three experiments with 200 of each cell type per experiment were analyzed by a mixed effect analysis with Tukey’s multiple comparison test relative to WT hTYRP1. (C-I). Indicated untransduced (-; G) or stable melan-b transductants (C-F) were analyzed by immunofluorescence microscopy for TYRP1 (left, green) and bright field microscopy (BF) for pigment granules (middle); right, overlay with pigment granules pseudocolored magenta. Insets, 7X magnification of boxed regions (intensities of TYRP1 and pigment granules optimized to better visualized overlap). Arrowheads, TYRP1 in rings around pigment granules; arrows, TYRP1 in separate punctate structures. (H) Quantification of cellular pattern of TYRP1 as predominantly rings, diffuse/punctate, or mixed among transduced cells expressing each of the TYRP1 variants. Data from four experiments with 150 of each cell type per experiment were analyzed by two-way ANOVA with Dunnett’s multiple comparison test relative to WT TYRP1. (I) Quantification of the percent of melanin-containing structures within densely pigmented cells that overlapped with TYRP1. Data from three experiments with 8-14 cells of each type (at least 30 total) per experiment were analyzed by Kruskal-Wallis with Dunn’s multiple comparison tests.
To test if the defect in pigmentation reflected impaired localization of the variants to melanosomes, we examined melan-b cells transfected with the different constructs by bright field microscopy to visualize pigment granules and by immunofluorescence microscopy for TYRP1. The majority of melan-b cells transfected with WT hTYRP1 are densely pigmented. By contrast, most cells expressing hTYRP1R153C or hTYRP1R114C were either non- or lightly pigmented; some of the latter contained large aggregates of pigment, perhaps reflecting melanosome autophagy (Figure 3B). By immunofluorescence microscopy, only background labeling for TYRP1 was detected in melan-b cells that were untransduced (-; Figure 3G) or transduced with mCh-STX13 (Figure 3F). WT hTYRP1 was detected predominantly in “rings” around pigment granules detected by bright field microscopy (Figure 3C, arrowheads; H), representing localization to the limiting membrane of mature melanosomes20. By contrast, hTYRP1R114C primarily localized to punctate and diffuse structures; the puncta did not overlap with pigment granules when present (Figure 3E, arrows; 3H). hTYRP1R153C was mostly detected in a mixture of ring-like and diffuse/punctate patterns (Figure 3D,H). Because melanosome size and shape is determined prior to the onset of pigmentation21, TYRP1 would appear ring-like on both pigmented and non-pigmented melanosomes; thus, the reduced ring structures for hTYRP1R153C and hTYRP1R114 indicate that they do not localize properly to melanosomes. Considering only the cohort of cells that were densely pigmented, detectable pigment granules overlapped significantly less with TYRP1 in cells expressing hTYRP1R153C or hTYRP1R114 than WT hTYRP1 (Figure 3I). Because the TYRP1 lumenal domain is required for proper melanosome localization22, we speculate that eumelanin dilution in bears may be explained by protein misfolding due to substitution of Cys for Arg at residues 114 or 153, leading to enhanced protein degradation and impaired trafficking to and/or retention within melanosomes.
Gene Action and Spatial Distribution of TYRP1R153C
For U. americanus, examining hair reflectance as a function of TYRP1R153C genotype in 317 individuals revealed semidominant gene action, with a median reflectance value for heterozygous G/A (Arg/Cys) individuals, 50.3, that was intermediate between values for homozygous G/G (Arg/Arg) and A/A (Cys/Cys) individuals of 4.7 and 94.0, respectively (inset, Figure 4).
Figure 4-. Spatial distribution of TYRP1R153C across the American black bear range.
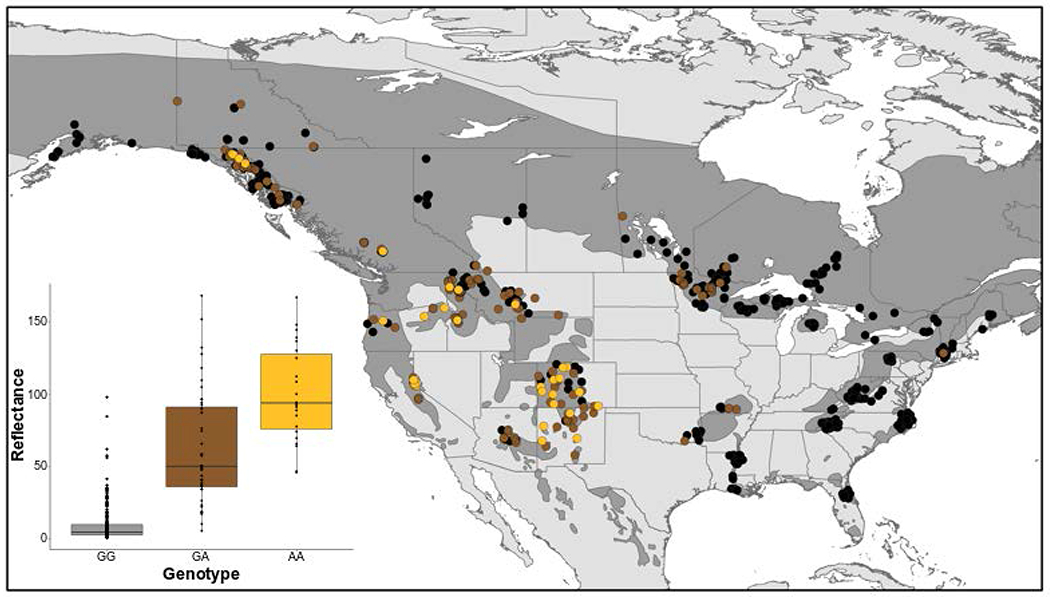
Quantitative and spatial assessment of TYRP1R153C in Ursus americanus. (Inset) Hair color reflectance was compared to the SNP genotype (n = 317). We used t-tests to compare reflectance values between genotypes: homozygous ancestral and heterozygous P=2.02 x 10−11; and heterozygous and homozygous derived P=1.41 x 10−4. These results indicate the allele acts with semidominance. The geospatial pattern of R153C across the range (n = 906) where color denotes genotype (black: homozygous ancestral GG; brown: heterozygous GA; gold: homozygous derived AA), with the species range shown in dark grey.
We determined the spatial distribution of TYRP1R153C in 906 samples across the species’ geographic range. A high frequency of the derived allele was apparent in the southwest, declining northward along the Rocky Mountains into Southeast Alaska and Yukon Territory (Figure 4). By contrast, a low frequency of the derived allele was apparent in the eastern lineage, primarily limited to the Great Lakes region and a single sample in Connecticut, USA. The presence of the derived allele in Missouri, Arkansas, and Oklahoma, USA was concordant with the known translocation history of bears from Minnesota into Arkansas in the 1960s23 and the high proportion of genetic diversity from the Great Lakes region now found in the Central Interior Highlands24. Spatial distribution of TYRP1R153C was consistent with the phenotypic distribution of non-black U. americanus as estimated from a survey of 40,000 animals in the 1980s5. Notably, TYRP1R153C was associated with bears qualitatively described as cinnamon, chocolate, or light brown, and therefore accounts for the majority of color diversity among U. americanus.
Demography
Our TYRP1 haplotype tree (Figure S3A) indicated that introgression of U. arctos alleles into U. americanus did not provide the genetic material for light coloration in the latter species. We investigated the relationship of the TYRP1R153C mutation to U. americanus demographic history. First, we used runtc25 to estimate the first coalescent (a proxy for allele age) of the variant along the scaffold and obtained a time of 9.36kya. Second, we ran MSMC-IM26 from whole genome resequencing (WGS) of six individuals which estimated that divergence between the western and eastern lineages began 100kya with cessation of gene flow 22kya (Figure 5A,B). Taken together with additional coalescent and range expansion analyses (Figures S4–S5), our results suggest that R153C arose in a western lineage population of U. americanus and spread via gene flow across the range (Figure 5C), including contemporary eastward movement too young to be captured using these coalescent estimates.
Figure 5-. American black bear demographic history and TYRP1R153C allele expansion.
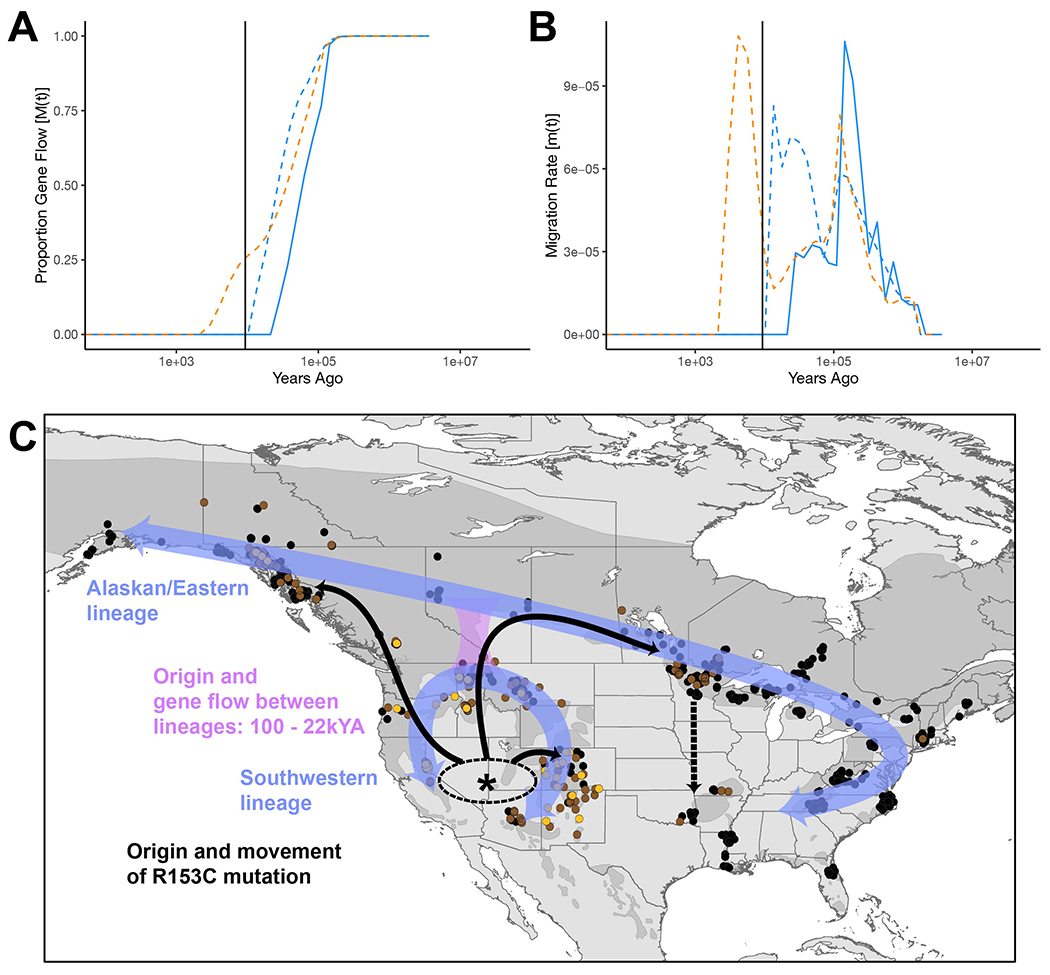
Ursus americanus demographic estimates over time. (A) Approximate completion of divergence and (B) bidirectional migration between lineage pairs including: eastern and western (solid blue line), eastern and Southeast Alaska (dashed blue line), and western and Southeast Alaska (dashed orange line). The vertical black line represents the estimated allele age of TYRP1R153C (also see Figure S4). (C) Our working model of historic lineage divergence (purple arrows), approximate geographic region of TYRP1R153C mutation (asterisks, *), and migration across the species’ geographic range (solid black arrows; dashed black arrow indicating recent translocation).
Selection Analyses
We used population genetic modeling and simulations to investigate whether clinal variation of R153C allele frequency was more likely to be explained by selection or genetic drift. We compared models with a range of dominance (h) and selection (s) coefficients. As depicted in Figure 6, the clinal pattern of allele frequencies was most consistent with weak selection (0.005 < s < 0.01) and was unlikely to be explained by genetic drift (s = 0). Despite the long haplotype carrying the derived TYRP1R153C allele, there was no statistical evidence for a selective sweep (Figure S6–S7), likely due to the combination of weak selection and low effective population size within the southwest population. In humans, SLC24A5 drives the main axis of geographic color variation in Caucasians and has a large s of 0.1627, although other genes, TYRP1 among them, have weaker coefficients of 0.01-0.0527,28.
Figure 6-. Simulation models of population allele frequencies under increasing selection coefficients.
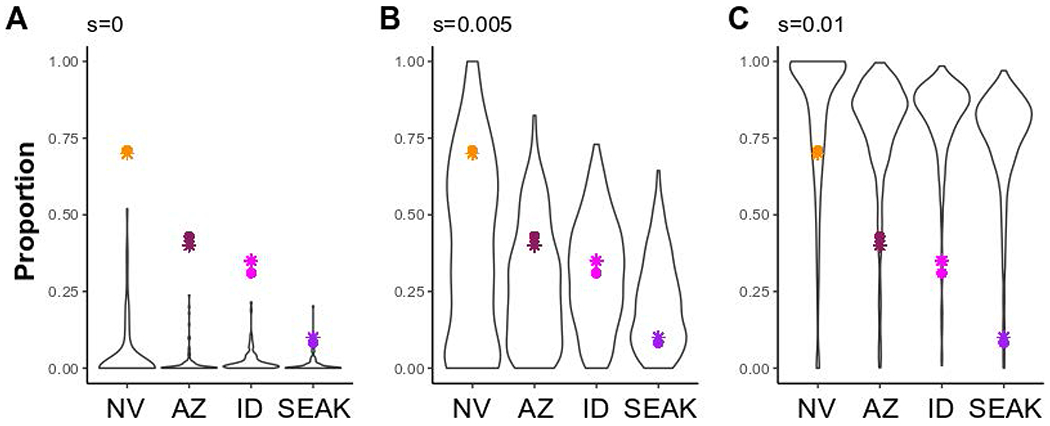
Simulation models for the frequency of a derived allele (h = 0.75) following 1,440 generations in four populations of Ursus americanus (Nevada: NV; Arizona: AZ; Idaho: ID; and Southeast Alaska: SEAK) connected via gene flow (see Figure S8). Models varied by selection coefficients (s; A- 0; B- 0.005; and C- 0.01), and accounted for population expansion and bottlenecks. Each simulation model was run for 1,000 iterations and panels represent runs in which the derived allele did not go extinct within a run; thus, sample size varies (A- 95; B- 394; and C- 614). Colored points represent R153C genotypic frequencies (circles) estimated in this study for each respective population, or phenotypic frequencies (stars) inferred from contour maps5.
Two hypotheses about the selective forces acting upon the cinnamon morph of U. americanus have been proposed. First, lighter coloration aids in thermoregulation of bears in the hotter and/or drier climates of the southwest; second, the cinnamon morph is mimetic with U. arctos where these species are sympatric. We used BayEnv229 to test for correlation between R153C allele frequency and climate, geography, or the presence/absence of U. arctos prior to anthropogenic extirpation, but identified no notable Bayes Factors (Table S4). Although this approach cannot rigorously exclude thermoregulation or mimicry as a force driving selection, it is possible that an additional, as yet, untested mechanism underlies weak selection such as crypsis in open canopy environments. For example, individuals color matched within their environment has been suggested to decrease infanticide and/or predation in the Giant Panda30.
Implications for Infraspecific Taxonomy in American Black Bears
Historically brown coat color has been used as a defining phenotype for delimiting four of the 16 U. americanus subspecies: U. a. amblyceps (southwest/southern Rocky Mountains), U. a. cinnamomum (northern Rocky Mountains), U. a. luteolus (Louisiana), and U. a. machetes (western Mexico)31–33. Further, the original description of U. luteolus from 182132 notes the cinnamon morph ranging from Virginia to Louisiana, where it is not presently found. Our results indicate that the geographic distribution of brown coats is being driven by recent and ongoing gene flow, and suggests that cinnamon morphs will increase in frequency within the eastern lineage in the future. We suggest the young age of the causative variant and rapid spread via gene flow make this a poor character trait for infraspecific delimitation.
Conclusion
Here we show that the cinnamon morph of the American black bear shares phenotypic similarity with brown bears in their coat coloration, and demonstrate that eumelanin dilution causes this similarity. We identified two independently evolved and species-specific alleles within TYRP1 that result in arginine to cysteine residue changes that disrupt protein localization to the melanosome. Our functional assay shows that the U. arctos R114C change has a greater effect on pigment production in melan-b cells than the U. americanus R153C; yet R114C has not fixed within the species (Figure S3B) suggesting it may modify color without being the initial causative locus. In contrast, the U. americanus R153C is responsible for most color variation within the species. Neither variant appears detrimental to fitness in bears unlike in humans with OCA3; instead R153C appears to be under weak positive selection. Crypsis as an adaptive mechanism has generally explained why prey species and ambush predators color match within their environments; here, we suggest crypsis as a broader adaptive mechanism for large bodied species.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests of resources should be directed to the corresponding author Emily E. Puckett (emily.puckett@memphis.edu).
Materials Availability
Data and Code Availability
Sequence data has been deposited onto the NCBI SRA under project number PRJNA867575. This paper does not report original code.
METHOD DETAILS
Sample Collection
We partnered with state, provincial, and federal wildlife agencies, and university partners in North America which collected hair, tissue (i.e., muscle or blood), and photographs from bears that were hunter harvested, vehicle killed, or live animals captured for other research studies or management purposes. Not all samples contained all three sampling elements (hair, tissue, and photographs). Photographs of bears confirmed all samples were adults; cubs may turn from brown to black following a molt, although black to brown transitions have not been observed (personal observation DL Garshelis). Samples were collected from 2015–2020 and georeferenced. The qPCR analysis (see below) incorporated previously collected bear samples64 that were also georeferenced but without phenotypic data.
Hair Analyses
We mounted one awn hair from 391 individuals onto slides, then captured an image from the middle of the sample under 20x magnification using an Olympus BX63 microscope at the Integrated Microscopy Center at the University of Memphis. We viewed all samples with transmitted light brightfield illumination, 488.3 μs exposure time, and 185 LED brightness. We analyzed images in ImageJ65 using the RGBMeasure function on a 46 x 46 pixel section of the hair cortex, and recorded the mean for the red, green, and blue color channels.
We quantified the amount of eumelanin and pheomelanin in 13 U. americanus and three U. arctos individuals. Samples were selected based on both geographic location and visual inspection of hair color under a compound microscope to select for observed variation (U. americanus: black = 9; brown = 4; U. arctos: dark brown = 2, brown = 1). We followed the melanin determination protocols8,9. Eumelanin and pheomelanin concentrations are reported as the concentrations of PTCA (pyrrole-2,3,5-tricarboxylic acid) and TTCA (thiazole-2,4,5-tricarboxylic acid) which are, respectively, the specific degradation products for each melanin.
Whole Genome Sequencing and Variant Calling
High Coverage-
We sequenced 15 U. americanus and six U. arctos individuals to ~30x depth (Key Resources Table). Specifically, 350bp insert libraries were prepared using the NEB Next Ultra II DNA kit, then sequenced (150bp paired-end) on an Illumina NextGen by Novogene (Chula Vista, CA). We sequenced nine additional U. americanus individuals to 50x depth using Illumina HiSeqX at HudsonAlpha (Huntsville, AL). We also downloaded 26 U. arctos and two U. maritimus WGS from the NCBI SRA to use in downstream analyses (Key Resources Table). For all species, we mapped reads to the U. americanus reference genome10 using BWA-MEM v0.7.1744 with default parameters. We marked duplicates, then called variants for each sample using the GenotypeGVCFs function in GATK v4.1.8.045 where we set the heterozygosity parameter to 5.0x10−4. For each species, we combined the samples using CombineGVCFs before joint genotyping using GenotypeGVCFs in GATK. We filtered the dataset using BCFTOOLS v1.947 by requiring minimum and maximum depth (-i ‘MIN (FMT/DP) > 4 & MAX (FMT/DP) < 300’), minimum quality score (-i FMT/GQ >= 30), and biallelic SNPs (-m 2 -M 2 -v snps). The total number of SNPs for U. americanus was 8,885,511. We phased the data from each species separately using BEAGLE v5.148,66.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TA99/Mel-5 | American Type Culture Collection | HB-8704 |
| Alexafluor 488-labeled donkey anti-mouse IgG | Jackson Immunoresearch | 715-545-151 |
| Bacterial and virus strains | ||
| E.coli-Top10 | Invitrogen | C66411 |
| Biological samples | ||
| U. arctos- DNA sample | This study | GYE906 |
| U. arctos- DNA sample | This study | GYE922 |
| U. arctos- DNA sample | This study | GYE953 |
| U. arctos- DNA sample | This study | AK17500 |
| U. arctos- DNA sample | This study | AK17512 |
| U. arctos- DNA sample | This study | AK17578 |
| U. arctos- DNA sample | 34 | RF01 |
| U. arctos- DNA sample | 34 | SJS01 |
| U. arctos- DNA sample | 34 | OFS01 |
| U. arctos- DNA sample | 35 | ALP1 |
| U. arctos- DNA sample | 36 | AKAdmiralty1 |
| U. arctos- DNA sample | 37 | AKAdmiralty2 |
| U. arctos- DNA sample | 36 | AKAdmiralty3 |
| U. arctos- DNA sample | 37 | AKBaranof1 |
| U. arctos- DNA sample | 34 | AKBaranof2 |
| U. arctos- DNA sample | 34 | AKChichagof1 |
| U. arctos- DNA sample | 34 | AKChichagof2 |
| U. arctos- DNA sample | 34 | AKChichagof3 |
| U. arctos- DNA sample | 34 | AKChichagof4 |
| U. arctos- DNA sample | 38 | AKChichagof5 |
| U. arctos- DNA sample | 36 | AKDenali1 |
| U. arctos- DNA sample | 37 | AKKenai |
| U. arctos- DNA sample | 35 | APN2 |
| U. arctos- DNA sample | 35 | GRE2 |
| U. arctos- DNA sample | 39 | JPHc1 |
| U. arctos- DNA sample | 39 | JPHc2 |
| U. arctos- DNA sample | 39 | JPHe1 |
| U. arctos- DNA sample | 39 | JPHe2 |
| U. arctos- DNA sample | 39 | JPHs1 |
| U. arctos- DNA sample | 39 | JPHs2 |
| U. arctos- DNA sample | 34 | MTgnp |
| U. arctos- DNA sample | 38 | Dalarna, SWE |
| U. maritimus- DNA sample | 37 | PB1 |
| U. maritimus- DNA sample | 37 | PB2 |
| U. americanus- DNA sample | This study | AK17023 |
| U. americanus- DNA sample | This study | AK17047 |
| U. americanus- DNA sample | This study | AK17117 |
| U. americanus- DNA sample | This study | AZ12 |
| U. americanus- DNA sample | This study | ID10 |
| U. americanus- DNA sample | This study | MI334 |
| U. americanus- DNA sample | This study | MI335 |
| U. americanus- DNA sample | This study | MN_6083 |
| U. americanus- DNA sample | This study | MS3783 |
| U. americanus- DNA sample | This study | NC00417 |
| U. americanus- DNA sample | This study | NC056 |
| U. americanus- DNA sample | This study | NVb83 |
| U. americanus- DNA sample | This study | NVb99 |
| U. americanus- DNA sample | This study | NVg5 |
| U. americanus- DNA sample | This study | WV17_01 |
| U. americanus- DNA sample | This study | HA1 |
| U. americanus- DNA sample | This study | HA2 |
| U. americanus- DNA sample | This study | HA3 |
| U. americanus- DNA sample | This study | HA4 |
| U. americanus- DNA sample | This study | HA5 |
| U. americanus- DNA sample | This study | HA6 |
| U. americanus- DNA sample | This study | HA7 |
| U. americanus- DNA sample | This study | HA8 |
| U. americanus- DNA sample | This study | HA9 |
| Chemicals, peptides, and recombinant proteins | ||
| BamHI | NEB | R0136L |
| HindIII | NEB | R0104L |
| XholI | NEB | R0146L |
| SnaBI | NEB | R0130S |
| Lipofectamine 2000 | Invitrogen | 11668-019 |
| Hygromycin B | Research Products International | H75020-1.0 |
| RPMI 1640 medium | Gibco | 11875-085 |
| FBS | R&D Systems | S1150 |
| PBS | Corning | 21-031-CM |
| Tris-HCl | Fisher | BP153-1 |
| EDTA | Sigma | E5134-50G |
| diothiothreitol | Sigma | 3483-12-3 |
| DMSO | Sigma | D2660 |
| LB Broth | Fisher | BP1426-2 |
| Ampicillin | Fisher | BP1760-25 |
| Agar | Fisher | BP1423-2 |
| Agarose | GibcoBRL | 15510-027 |
| 2-log DNA Ladder | NEB | N3200L |
| GelGreen Nucleic Acid Stain | Biotium | 41005 |
| Tris-EDTA pH 8.0 | Ambion | AM9858 |
| Terrific Broth | Amresco | J869-5KG |
| Glucose | Sigma | G7528-250G |
| Sodium Hydroxide | Sigma | S8045 |
| SDS | Invitrogen | 15525-017 |
| Potassium Acetate | Sigma | P1190-100G |
| Glacial Acetic Acid | Fisher | BP1185-500 |
| Critical commercial assays | ||
| Prime Time Gene Expression (for qPCR) | Integrated DNA Technologies (IDT) | 10007065 |
| cOmplete Protease Inhibitor Cocktail Tablets | Roche | P8849 |
| Prolong Gold Antifade Mountant | InVitrogen | P36931 |
| Q5 High-Fidelity DNA Polymerase | NEB | M0491L |
| QIAquick Gel Extraction Kit | Qiagen | 28704 |
| BCA Protein Assay Kit | Pierce | 23225 |
| Deposited data | ||
| DNA Sequencing | This study | SRA: PRJNA867575 |
| TYRP1 mouse WT sequence | 40 | NM_031202.3 |
| Crystal structure of the human TYRP1 luminal domain | NCBI Protein Data Base | 5M8L |
| Experimental models: Cell lines | ||
| melan-b | Dorothy Bennett’s laboratory | 41 |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| pBMN vector R153C primer; forward: GGGCCCTGGATATGGCAAAGTGCACAACTCACCCTTTATTTGT | IDT | custom oligo synthesis |
| pBMN vector R153C primer; reverse: ACAAATAAAGGGTGAGTTGTGCACTTTGCCATATCCAGGGCCC | IDT | custom oligo synthesis |
| pBMN vector R114C primer; forward: GACACAACTGTGGGACGTGCTGTCCTGGCTGGAGAGGAGCTGC | IDT | custom oligo synthesis |
| pBMN vector R114C primer; reverse: GCAGCTCCTCTCCAGCCAGGACAGCACGTCCCACAGTTGTGTC | IDT | custom oligo synthesis |
| pBMN flanking primer; forward: CCTCTAGACTGCCGGATCCATTTAAATTCGAATTCCTGCAGG | IDT | custom oligo synthesis |
| pBMN flanking primer; reverse: GGAATCAAAGTTGCTTCTGGATCCCATCAAGTCATCCGTGCAG | IDT | custom oligo synthesis |
| qPCR TYRP1 primer; forward: CCTTGAAGTCAGGAGAAACC | This paper | n/a |
| qPCR TYRP1 primer; reverse: CTGGTCGCAATGACAAAC | This paper | n/a |
| pPCR TYRP1 reference allele probe: FAM-ATGGCGAAGCGCACAATTC-BGQ1 | This paper | n/a |
| pPCR TYRP1 alternative allele probe: TET-ATGGCGAAGTGCACAAT-BHQ1 | This paper | n/a |
| Recombinant DNA | ||
| pCDNA3-TRP1 | Gift from Walter Storkus, Univ. of Pittsburgh | n/a |
| pBMN-I-hygro-mCherry-hSTX13 | Marks laboratory | 42 |
| pBMN-I-hygro | Gift from Andrew Peden, Univ. of Scheffield | 43 |
| Software and algorithms | ||
| BWA v0.7.17 | 44 | https://github.com/lh3/bwa |
| GATK v4.1.8.0 | 45 | https://gatk.broadinstitute.org/hc/en-us |
| SAMTOOLS v1.9 | 46 | https://sourceforge.net/projects/samtools/files/samtools/1.9/ |
| BCFTOOLS v1.9 | 47 | https://sourceforge.net/projects/samtools/files/samtools/1.9/ |
| BEAGLE v5.1 | 48 | https://faculty.washington.edu/browning/beagle/b5_1.html |
| GLIMPSE 1.0.0 | 11 | https://github.com/odelaneau/GLIMPSE/pkgs/container/GLIMPSE%2Fglimpse |
| GEMMA v0.98.1 | 12 | https://github.com/genetics-statistics/GEMMA |
| HAPLOVIEW 4.2 | 14 | https://www.broadinstitute.org/haploview/downloads |
| Haplostrips | 13 | https://bitbucket.org/dmarnetto/haplostrips/src/master/ |
| PROVEAN | 49 | https://www.jcvi.org/research/provean#downloads |
| PolyPhen2 | 50 | http://genetics.bwh.harvard.edu/pph2/ |
| SIFT | 51 | https://sift.bii.a-star.edu.sg/index.html |
| GENEIOUSPRIME | 52 | https://www.geneious.com/prime/ |
| Leica LAS-X | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| Microvolution | Bio-Vision Technologies | NA |
| FIJI - ImageJ | 53 | https://imagej.net/software/fiji/ |
| Adobe Photoshop v. 24.0.0 | Adobe.com | https://www.adobe.com/products/photoshop/ |
| Graphpad Prism v. 9.4.1 | GraphPad.com | https://www.graphpad.com/updates/prism-900-release-notes |
| Pymol | Schrödinger LLC | https://pymol.org |
| program R v4.0.3 | 54 | https://www.r-project.org/ |
| runtc | 25 | https://github.com/jaredgk/runtc |
| STACKS v2.53 | 55 | https://catchenlab.life.illinois.edu/stacks/ |
| PLINK v1.9 | 56 | https://www.cog-genomics.org/plink/ |
| MSMC2 | 57 | https://github.com/stschiff/msmc2 |
| SNPable | 58 | https://lh3lh3.users.sourceforge.net/snpable.shtml |
| rangeexpansion | 59 | https://github.com/BenjaminPeter/rangeexpansion |
| selscan v2 | 60 | https://github.com/szpiech/selscan |
| BayEnv2 | 29 | https://bitbucket.org/tguenther/bayenv2_public/src/master/ |
| PGDSpider | 61 | http://www.cmpg.unibe.ch/software/PGDSpider/ |
| SLiM v3 | 62 | https://messerlab.org/slim/ |
| MSMC-IM | 26 | https://github.com/wangke16/MSMC-IM |
| Unipro UGENE v39.0 | 63 | http://ugene.net/ |
| Other | ||
| Matrigel | Corning | 354248 |
| Electroporator | Bio-Rad | MicroPulser |
| PCR | Bio-Rad Thermal Cycler | T100 |
| Petri Dish | VWR | 25384-088 |
| Sanger Sequencing | Molecular Cloning Laboratories (MCLAB) | service |
| Mouse Tyrp1 WT,R114C and R153C synthesis and cloning | Synbio Technologies | custom gene synthesis |
| Transilluminator | Clare Chemical Research | Dark Reader |
Low Coverage-
We sequenced 166 U. americanus animals to an average of 1.3x depth. Specifically, we followed the manufacturer’s instructions for the Illumina DNA Prep Kit except that we diluted the reactions to 20% volume and started with 20 ng of genomic DNA. Two pooled libraries were constructed and each was sequenced on one lane of an Illumina 4000 at Novogene (Davis, CA). We mapped reads to the U. americanus reference and called variants as described above. The draft U. americanus (2n = 74) genome was assembled into 94,016 scaffolds; we analyzed the longest 36 scaffolds (and removed the X chromosome, HiC_scaffold_1). We used our high coverage WGS samples as a reference panel for joint imputation and phasing of the low coverage data using GLIMPSE11.
GWAS and Haplotype Characterization
To identify loci associated with the brown phenotype, we ran GEMMA v0.98.112. Specifically, we combined our high and imputed low coverage datasets, then used the quantitative hair reflectance values (green channel of RGB measurement) as the phenotype (n = 151). We ran a principal components analysis (PCA) on the relatedness matrix created within GEMMA, and used the first two PC axes as covariates to account for population structure in the analysis. Our GWAS resulted in 23 genomic regions with P-values lower than 10−8, although a single peak stood out and contained a known pigmentation gene, TYRP1 (Table S2). To test if additional loci beyond TYRP1 influenced quantitative color phenotypes in black bears, we reran the analysis using the R153C genotype from the qPCR (see below) as a third covariate. This analysis did not reveal additional loci that contributed to the coloration phenotype.
We estimated the length of the haplotype containing the derived allele within the Nevada population using HAPLOVIEW14 using the default algorithm for estimating linkage disequilibrium (LD)67. We then viewed this region of the genome for all samples using HAPLOSTRIPS13. The Nevada population was selected based on the runtc (see below and Figure S4) analyses which indicated it was geographically near the source population for the R153C TYRP1 population.
Candidate Gene Analysis
To evaluate deleterious protein coding variation in bears, we downloaded coding sequences (CDS) for 13 candidate coat color genes (ASIP, CBD103, EDNRB, KIT, KITLG, MC1R, MLPH, OCA2, PMEL, SLC24A5, SLC45A2, TYR, TYRP1, and TYRP2) from the dog genome (Canis familiaris; CanFam3.1). Exons were queried against genome assemblies of U. americanus, U. arctos, and U. maritimus10,34,68 using the blastn and dc-megablast versions of NCBI BLAST+69,70. We extracted the resulting sequences from the phased genome assemblies using SAMTOOLS v1.9 46 and BCFTOOLS and concatenated exons to form haplotypes. We translated each haplotype and identified missense and nonsense polymorphisms within U. americanus, between U. americanus and four U. arctos samples, and between U. arctos and U. maritimus. We tested each variant for deleterious effects using PROVEAN, PolyPhen2, and SIFT49–51. For any variant where two of the programs predicted a deleterious variant, we identified the individuals and coat color phenotypes of those animals to assess if there was concordance between U. americanus animals with brown coats and the alternate allele. Only the previously identified TYRP1R153C showed such a concordance. We constructed a neighbor joining tree from the haplotypes (with introns) using GENEIOUSPRIME52 to visualize the gene tree.
To understand the location of the arginine to cysteine amino acid changes within the native TYRP1 protein, we input the X-ray crystal structure of the human TYRP1 luminal domain (PDB ID: 5M8L, ref71) into PyMol v2.5, displayed it as a ribbon diagram to emphasize alpha helices, beta sheets, and loops, and then highlighted the following features: the location of R114 and R153 amino acids (expressed in atom format), cysteines and corresponding disulfide bonds throughout the protein, and the zinc cofactors.
Genotyping TYRP1R153C
We genotyped 906 U. americanus samples for the TYRP1R153C variant using a qPCR assay.
The primers included (5’ to 3’):
F: CCTTGAAGTCAGGAGAAACC
R: CTGGTCGCAATGACAAAC
The probes included (5’ to 3’):
reference allele probe: FAM-ATGGCGAAGCGCACAATTC-BGQ1
alternate allele probe: TET-ATGGCGAAGTGCACAAT-BHQ1
Primers and probes were ordered from IDT (Coralville, IA). Reaction conditions had final primer and probe concentrations of 1μM and 0.2 μM, respectively. The thermocycler conditions in a BioRAD CFX96 included: 95°C for 3min, and 40 cycles of 95°C for 15sec then 57.9°C for 30sec. For samples where both hair color phenotypes and R153C genotypes were available (n = 317), we tested if reflectance was significantly different between genotypes using two-sided t-tests in R v3.6.054.
Functional Validation of TYRP1 Alleles
To test if the TYRP1 variants we identified in American black (R153C) and brown (R114C) bears caused a change in pigmentation phenotype, we first used site-specific mutagenesis to generate recombinant retroviruses to co-express wildtype (WT) or variant versions of human and mouse TYRP1 with a drug resistance marker for stable cell line selection. Specifically, we first subcloned a WT TYRP1 cDNA based on the human sequence (pCDNA3-TRP1, a gift to MS Marks from W Storkus, University of Pittsburgh, Pittsburgh, PA, USA) into the pBMN-I-Hygro retroviral vector43. The R153C and R114C variants were generated individually by overlapping PCR using synthetic oligonucleotides bearing the desired mutations (denoted by asterisks [*] in the sequences below).
The oligos for R153C were (5’ to 3’):
F: GGGCCCTGGATATGGCAAAGT*GCACAACTCACCCTTTATTTGT
R: ACAAATAAAGGGTGAGTTGTGCA*CTTTGCCATATCCAGGGCCC
The oligos for R114C were (5’ to 3’):
F: GACACAACTGTGGGACGTGCT*GTCCTGGCTGGAGAGGAGCTGC
R: GCAGCTCCTCTCCAGCCAGGACA*GCACGTCCCACAGTTGTGTC
The flanking oligos for both products were (5’ to 3’):
F: CCTCTAGACTGCCGGATCCATTTAAATTCGAATTCCTGCAGG
R: GGAATCAAAGTTGCTTCTGGATCCCATCAAGTCATCCGTGCAG
The R153C mutation was generated by combining a 707bp and a 386bp product to obtain a 1,050bp product. The R114C mutation was generated by combining a 590bp and a 503bp product to obtain a 1,050bp product. The PCR products bearing the mutations were cloned individually as 1,013bp BamHI fragments into the BamHI site in the pBMN-I-Hygro-hTYRP1 construct after excising the WT BamHI fragment. The clones bearing the correctly oriented fragments were selected by HindIII restriction and confirmed by Sanger sequencing the BamHI junctions. The generated mutations were confirmed by Sanger sequencing.
The WT mouse TYRP1 cDNA sequence (NCBI accession NM_031202.3) was also used to generate the mutant forms. The three versions (WT, R153C, and R114C) were synthesized individually by Synbio Technologies as XhoI/HpaI fragments (of 2270bp) and cloned into the XhoI/SnaBI site of the pBMN-I-Hygro vector. The three sequences were confirmed by Sanger sequencing.
Immortalized melan-b melanocytes derived from mice homozygous for the TYRP1b allele were cultured as described72,73. Melan-b cells were infected with recombinant retroviruses encoding TYRP1 variants (described above). Briefly, Plat-E cells74 were cultured and transfected with the retroviral vectors described above, empty pBMN-I-hygro, or pBMN-I-hygro expressing mCherry-tagged human STX1375 using Lipofectamine 2000. The medium was replaced with fresh melanocyte medium the following day and cell supernatants were harvested 48-72 h post-transfection as described76. The medium, containing recombinant retroviruses, was used directly to infect melan-b cells as described77 except that cells were seeded the previous day in a 10-cm dish. Two days later the medium was supplemented with 500 mg ml−1 hygromycin B to select for infected cells. Antibiotic-resistant cells were used directly as polyclonal cell populations for experiments. Two separate stable lines per construct were selected and included in the analyses.
We performed spectrophotometric assays or melanin and protein content as described77,78. Briefly, melanocytes in a confluent 10-cm dish were harvested by trypsinization. Cells were counted, and duplicate samples of 106 cells for each experimental condition per experiment were washed once with RPMI + 10% FBS and twice with ice-cold PBS, and then resuspended in 50 mM Tris-HCl, pH 7.4, 2 mM EDTA, 150 mM NaCl, 1 mM diothiothreitol, and cOmplete Protease Inhibitor Cocktail Tablets (Roche). Cells were probe-sonicated on ice using the Sonic Dismembrator Model 100 (Fisher Scientific), and sonicates were fractionated into supernatant and pellet fractions by centrifugation at 20,000 x g for 15 min at 4°C. Protein concentrations in supernatants were estimated using the DC colorimetric protein assay (Pierce) with a standard curve derived using differing concentrations of BSA. Melanin-containing pellets were washed in 500 μl 50% ethanol, 50% ether, collected by centrifugation at 20,000 x g, and resuspended in 1 ml of 2 M NaOH, 20% DMSO. Melanin was solubilized by heating at 60°C for 40–60 min and quantified using spectrophotometry to measure Optical Density (OD) at 492 nm. OD values were normalized to total protein content in each lysate. The assay was performed on six different days using samples collected from two different sets of stable cell lines per construct.
We performed imaging and analysis as described79. Briefly, melanocytes were seeded onto Matrigel-(Corning) coated coverslips 48–72 h before fixation. Cells were fixed with 2% formaldehyde in PBS for 20 min, washed briefly with PBS, then labeled with TA99/Mel-5 antibody to TYRP1 (American Type Culture Collection, Rockville, MD) diluted in blocking buffer (PBS, 0.1% BSA, 0.02% saponin) for 1 h at room temperature (RT). After a 15-min wash in PBS, cells were incubated with Alexafluor 488-labeled donkey anti-mouse IgG (Jackson Immunoresearch) diluted in blocking buffer for 30 min at RT. Samples were washed for 15 min in PBS, mounted with Prolong Gold Antifade Mountant (Invitrogen), and analyzed by epifluorescence microscopy on a DMI 6000B microscope (Leica Biosystems) equipped with a 63x plan apochromat (Plan Apo) objective (1.4NA) and a Hamamatsu Photonics ORCA-Flash4.0 sCMOS digital camera. Both fluorescence and bright field images were acquired as a z-series with 0.19-μm steps and were deconvolved using the blind deconvolution algorithm of Microvolution software (Bio-Vision Technologies) and ImageJ. Final images were composed using Adobe Photoshop software77. Images were quantified using ImageJ.
To quantify the percentages of cells with varying levels of pigmentation, bright field images of individual cells within randomly chosen fields were characterized as non-pigmented (no visual pigmentation or fewer than 10 pigment granules per cell), densely pigmented (easily visualized pigmentation throughout the cell with numerous granules), lightly pigmented (signal above background but either very low signal or fewer than 40% of the total number of granules for wild-type cells), or lightly pigmented with prominent pigment aggregates (structures larger than 1 micron in diameter, typically one or two per cell). Data from three experiments with 200 cells per sample type per experiment were analyzed by mixed effect analysis and Tukey’s multiple comparison test.
To quantify the TYRP1 staining pattern, cells in randomly chosen fields were characterized as having predominantly ring-like structures (easily detectable “donuts” of less than 1 micron in diameter), diffuse and/or punctate staining (hazy cytoplasmic fluorescence and/or small puncta with no visible lumen), or a combination of both. Cells were analyzed blinded and without observing pigmentation status. Data from four experiments with 150 cells per sample type per experiment were analyzed by two-way ANOVA (because there were multiple variables) with Dunnett’s multiple comparison test.
The area of overlap between TYRP1 and pigmented melanosomes in the cell periphery of melanocytes was quantified using a threshholding approach in ImageJ on deconvolved fluorescence images as detailed79. Briefly, images were cropped to individual representative cells that had sufficient pigmentation to allow for thresholding. Both the bright field image and the corresponding deconvolved TYRP1 immunofluorescence image were subjected to the following manipulations. Background was subtracted using the Background Subtract function with a rolling ball radius of 2 microns, and then an identical mask corresponding to the perinuclear region (which is typically very densely labeled by TYRP1) was cropped out of both images. The Adjust>Threshold function was used to generate binary images that closely resembled the original images. Structures larger than five pixels were quantified using the Analyze Particles function, the area of overlap between the two images was calculated by using the Math>Multiply function, and values were calculated relative to the total area of thresholded bright field objects. Data from at least 30 cells of each cell type from three independent experiments were analyzed by Kruskal-Wallis test (because the data were not Gaussian) with Dunn’s multiple comparison test.
All statistical analyses were performed using Graphpad Prism 9 for MacOS.
Allele Age
We estimated the age of TYRP1R153C using runtc25, a program that estimates the time of first coalescence to the nearest variant on a chromosome. To increase sample size for this analysis, we used the combined high and imputed low coverage data for HiC_scaffold_24. We reran the GLIMPSE pipeline on the low coverage samples where the high coverage reference panel was not first filtered for MAF, thereby leaving more singletons in the reference dataset for coalescence with the focal allele. We estimated the coalescence time for all variants on the scaffold with all range wide samples together, as well as separately in the western lineage, Southeast Alaska (SEAK), and eastern populations separately, using a mutation rate of 1x10−8 and the recombination rate from the dog genome, 0.97cM Mb−1.
Demography
We inferred the demographic history of U. americanus, then tested if spatial patterns of TYRP1R153C alleles were due to neutral or adaptive processes given the demography. We first assessed U. americanus population structure using PCA. We aligned a previously generated RAD-Seq dataset7 to the U. americanus reference genome and called SNPs using STACKS v2.53 55, including filters for one SNP per RAD-tag, >80% genotyping success per locus, and MAF ≥ 0.05; this resulted in 37,422 SNPs. We pulled down those loci from the high coverage WGS samples to increase the sample size (n = 117). We ran a PCA analyses in PLINK v1.956,80 using the RAD-seq+hcWGS and observed a concordant pattern to the previous analysis7; thus, we felt confident with analyzing lineages and populations as previously determined.
We estimated change in effective population size (Ne) through time using MSMC257. For this analysis we used scaffolds 2–37, and made individual mask files with the bamCaller.py script, then used the phased datasets to make genome-wide masks with SNPable58. We generated input scripts using generate_multihetsep.py. Output was converted to years and number of individuals using a mutation rate of 1x10−8 81 and a generation time of 6.5 years82. We ran MSMC2 on three samples (six haplotypes) from both the eastern and western lineages, and the admixed SEAK population. Historic Ne was approximately 30,000. From 100–10kya Ne declined to 3,000 in the western lineage, yet remained constant in the eastern lineage. Ne also declined in Alaska from 50–10kya to approximately 8,000. MSMC2 plots indicate Ne increased since the Last Glacial Maximum (18–22kya). This demographic history provides an explanation for the low haplotypic diversity in Nevada as the ancestral and causative alleles sit on highly homogeneous haplotypes (Figures S2 and S6). Using the output from MSMC2, we further estimated the cross-coalescent model and input results into MSMC-IM26 to estimate the patterns and timing of population divergence (using MSMC2 estimates), and rate of gene flow over time (using PSMC’ estimates on two haplotypes for the Idaho and Arizona samples). Estimates may be inaccurate in the most recent time lags of MSMC2 analyses warranting caution when estimating contemporary Ne. Our data appear to have a limit for inferences younger than 1–5kya.
While our demographic model infers broad patterns of lineage relatedness, it does not identify the directionality of range expansion; thus, we used the RAD-seq+hcWGS dataset to understand patterns of range expansion via the directionality index (Ψ)59. We removed geographic sites with fewer than three samples for a total of 111 samples across 23 populations. We input genotypes and geographic data into the rangeExpansion package in R, then calculated Ψ for all population pairs using the get.all.psi function. We determined the significance of each pairwise Ψ estimate by calculating the standard error of the upper triangle of the matrix excluding the diagonal, then calculating a Z-score for each pairwise comparison. To visualize the range expansion patterns, we plotted pairwise values where Z ≥ |5| within the western and eastern lineages, and between the two lineages and SEAK.
Selection Tests
We calculated pi (π) using VCFTOOLS83 in 1kb sliding windows with a 100bp step size from the imputed low coverage WGS data using samples from the Nevada (western lineage; n = 38), SEAK (n = 95), and Appalachian Mountains (representative of eastern lineage; n = 38) populations. We further calculated FST between black and brown animals within either the Nevada or SEAK populations using the same sliding window scheme.
We tested for a selective sweep surrounding TYRP1 using both integrated haplotype score (iHS) and extended haplotype homozygosity (EHH) 84 implemented in selscan v260,85. The iHS analysis was run on scaffold 24 of the U. americanus reference genome within the Nevada population, then normalized based on allele frequency using the norm function within selscan v2. No sites within the haplotype block carrying TYRP1R153C had iHS scores greater than |2|. We ran the EHH analysis in both the Nevada and SEAK populations using the --keep-low-freq flag. Further, we identified 15 sites on each of the 36 long scaffolds (or 540 sites each) that matched the derived allele frequency of R153C within each test population and ran EHH on each of those sites. We compared the decay curves of R153C to these allele frequency matched background loci as a test if the decay in haplotype homozygosity was within the range of the background. While decay curves between the ancestral and derived alleles suggested extended haplotype homozygosity for the derived R153C, the background matched samples showed the overall scores were not out of range for the background sites. Both the low Ne within the Nevada population and low selection coefficient complicate the analysis of selection, as simulation studies suggest low power to detect selection under these conditions86. Moreover, we chose not to use cross-population sweep tests due to the decrease in power with increasing divergence, and our estimates that the eastern and western lineage populations diverged ~15.8k generations ago60.
We further tested for possible drivers of selection using BayEnv229,87. Using the combined RAD-Seq, hcWGS, and lcWGS data at the RAD loci, we clustered the data into 24 geographic areas, identified a centroid longitude and latitude coordinate, then extracted values of 11 climate variables and altitude for each coordinate. The climate variables from the CliMond dataset88,89 included: bio1 (annual mean temperature), bio3 (isothermality), bio4 (temperature seasonality), bio5 (max temperature of warmest week), bio6 (min temperature of coldest week), bio10 (mean temperature of warmest quarter), bio11 (mean temperature of coldest quarter), bio12 (annual precipitation), bio19 (precipitation of coldest quarter), and bio20 (annual mean radiation). Beyond the climate variables, altitude, latitude, and longitude were all tested as selective forces. Finally, we used the presence or absence of U. arctos on the landscape as a factor. For each geographic location, we scored the site with the presence of U. arctos before range contraction due to human activity based on maps of the expected species distribution90. We prepared the covariance matrix using PGDSpider61, and the environmental matrix according to the BayEnv2 manual. We ran 100,000 iterations of the model and analyzed the Bayes Factors for the alternative hypothesis that the variable of interest was a selective force on allele frequency of TYRP1R153C.
Gene Flow Simulation
To test if a model of gene flow without selection could produce the spatial gradient of declining frequency of the alternative allele along a south-to-north axis is the western range, we used the forward genetic simulation program SLiM362. We modeled four populations, broadly representative of Nevada, Arizona/New Mexico, Idaho, and SEAK. These population were selected as we have at least one high coverage genome from each population (Key Resources Table). We reran MSMC-IM on one genome from each population for all pairwise combinations to estimate historic gene flow rates to use as input in our model (Figure S8).
Our model seeds a new variant (i.e., R153C) at a frequency of 1% into the Nevada population at generation 0, then tracks allele frequency within each population for 1,440 generations such that we get a simulated estimate to compare to observed data. Our data suggests that R153C has a dominance coefficient (h) greater than 0.5, but not 1; thus, we varied h from 0 to 1 using 0.1 increments. Further, we ran each model with a selection coefficient (s) of either 0, 0.005, or 0.01. MSMC-IM estimates bidirectional gene flow, thus for each population pair we use the peak estimated rate (Figure S8) since the Last Glacial Maximum (LGM). We ran three different models varying the selection coefficients (s = 0, 0.005, or 0.01). From generations 980–1,310, population size increased in each population at a per generation rate of 10−6 which was a rate approximated from our MSMC results. At generation 1,390 (i.e., 50 generations prior to the end of the simulation and meant to represent anthropogenic influences on bear populations), we imposed population specific bottlenecks given estimates from the literature. Specifically, the Nevada population was reduced to 10091, and the Arizona/New Mexico, Idaho, and SEAK populations were reduced to 5,000, 7,000, and 4,000 respectively7. We ran 1,000 iterations of each model. We recorded the number of simulations in which the derived allele went extinct in all populations or was still polymorphic following 1,440 generations (no simulations observed the derived allele fixing in all populations). For simulations where the locus remained polymorphic, we averaged the frequency within each population across simulations, and compared to the observed frequency from our qPCR assay and contour plots of percent of black animals in the population5.
This simulation with 33 different scenarios (h by s combinations) identified that both increasing values of h or s resulted in increased allele frequencies and decreased probability of allele extirpation from the landscape. We reran the simulation with an h of 0.75 for the final model (Figure 6).
Supplementary Material
HIGHLIGHTS.
The cinnamon morph of the American black bear has a missense mutation in TYRP1 that accounts for their coloration
TYRP1 variants in American black bears and brown bears are loss-of-function alleles associated with impaired protein localization to melanosomes
In American black bears, the variant causing the cinnamon morph arose 9,360 years ago in the western lineage where it provides an adaptive advantage, and has spread northwards and eastwards by migration
ACKNOWLEDGEMENTS
We thank Omar Skalli for microscopy training, Jay Puckett for assistance with maps, and two anonymous reviewers for comments that improved the manuscript. MSM and DCH were supported by NIH R01 AR0076241. AP was supported by NIH R35 GM134957-01 and American Diabetes Association Pathway to Stop Diabetes grant 1-19-VSN-02. CBK and GSB were supported by NIH R01 AR067925 and the HudsonAlpha Institute. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Footnotes
DECLARATION of INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, and Crossland JP (2006). A single amino acid mutation contributes to adaptive beach mouse color pattern. Science 313, 101–104. 10.1126/science.1126121. [DOI] [PubMed] [Google Scholar]
- 2.Basu Mallick C, Iliescu FM, Möls M, Hill S, Tamang R, Chaubey G, Goto R, Ho SYW, Gallego Romero I, Crivellaro F, et al. (2013). The light skin allele of SLC24A5 in South Asians and Europeans shares identity by descent. PLoS Genetics 9, e1003912. 10.1371/journal.pgen.1003912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benjamin-Fink N, and Reilly BK (2017). Conservation implications of wildlife translocations; The state’s ability to act as conservation units for wildebeest populations in South Africa. Global Ecology and Conservation 12, 46–58. 10.1016/j.gecco.2017.08.008. [DOI] [Google Scholar]
- 4.Wikipedia (2021). The Cinnamon Bear. https://en.wikipedia.org/wiki/The_Cinnamon_Bear.
- 5.Rounds RC (1987). Distribution and analysis of colourmorphs of the black bear (Ursus americanus). Journal of Biogeography 14, 521–538. [Google Scholar]
- 6.Pedersen MW, De Sanctis B, Saremi NF, Sikora M, Puckett EE, Gu Z, Moon KL, Kapp JD, Vinner L, Vardanyan Z, et al. (2021). Environmental genomics of Late Pleistocene black bears and giant short-faced bears. Current Biology 31, 2728–2736.e2728. 10.1016/j.cub.2021.04.027. [DOI] [PubMed] [Google Scholar]
- 7.Puckett EE, Etter PD, Johnson EA, and Eggert LS (2015). Phylogeographic analyses of American black bears (Ursus americanus) suggest four glacial refugia and complex patterns of post-glacial admixture. Molecular Biology and Evolution 32, 2338–2350. [DOI] [PubMed] [Google Scholar]
- 8.Ito S, Del Bino S, Hirobe T, and Wakamatsu K (2020). Improved HPLC conditions to determine eumelanin and pheomelanin contents in biological samples using an ion pair reagent. International Journal of Molecular Sciences 21, 5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito S, Nakanishi Y, Valenzuela RK, Brilliant MH, Kolbe L, and Wakamatsu K (2011). Usefulness of alkaline hydrogen peroxide oxidation to analyze eumelanin and pheomelanin in various tissue samples: application to chemical analysis of human hair melanins. Pigment Cell & Melanoma Research 24, 605–613. 10.1111/j.1755-148X.2011.00864.x. [DOI] [PubMed] [Google Scholar]
- 10.Srivastava A, Kumar Sarsani V, Fiddes I, Sheehan SM, Seger RL, Barter ME, Neptune-Bear S, Lindqvist C, and Korstanje R (2019). Genome assembly and gene expression in the American black bear provides new insights into the renal response to hibernation. DNA Research 26, 37–44. 10.1093/dnares/dsy036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubinacci S, Ribeiro DM, Hofmeister RJ, and Delaneau O (2021). Efficient phasing and imputation of low-coverage sequencing data using large reference panels. Nature Genetics 53, 120–126. 10.1038/s41588-020-00756-0. [DOI] [PubMed] [Google Scholar]
- 12.Zhou X, and Stephens M (2012). Genome-wide efficient mixed-model analysis for association studies. Nature Genetics 44, 821–824. 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marnetto D, and Huerta-Sánchez E (2017). Haplostrips: revealing population structure through haplotype visualization. Methods in Ecology and Evolution 8, 1389–1392. 10.1111/2041-210X.12747. [DOI] [Google Scholar]
- 14.Barrett JC, Fry B, Maller J, and Daly MJ (2004). Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265. 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 15.Manga P, Kromberg JGR, Box NF, Sturm RA, Jenkins T, and Ramsay M (1997). Rufous Oculocutaneous Albinism in Southern African Blacks Is Caused by Mutations in the TYRP1 Gene. The American Journal of Human Genetics 61, 1095–1101. 10.1086/301603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lasseaux E, Plaisant C, Michaud V, Pennamen P, Trimouille A, Gaston L, Monfermé S, Lacombe D, Rooryck C, Morice-Picard F, and Arveiler B (2018). Molecular characterization of a series of 990 index patients with albinism. Pigment Cell & Melanoma Research 31, 466–474. 10.1111/pcmr.12688. [DOI] [PubMed] [Google Scholar]
- 17.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. (2017). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Research 46, D1062–D1067. 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith JR, Hayman GT, Wang S-J, Laulederkind SJF, Hoffman MJ, Kaldunski ML, Tutaj M, Thota J, Nalabolu HS, Ellanki SLR, et al. (2019). The Year of the Rat: The Rat Genome Database at 20: a multi-species knowledgebase and analysis platform. Nucleic Acids Research 48, D731–D742. 10.1093/nar/gkz1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raposo G, Tenza D, Murphy DM, Berson JF, and Marks MS (2001). Distinct Protein Sorting and Localization to Premelanosomes, Melanosomes, and Lysosomes in Pigmented Melanocytic Cells✪. Journal of Cell Biology 152, 809–824. 10.1083/jcb.152.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seiji M, Fitzpatric T, and Birbeck M (1961). The melanosome: a distinctive subcellular particle of mammalian melanocytes and the site of melanogenesis. Journal of Investigative Dermatology 36, 243–252. [DOI] [PubMed] [Google Scholar]
- 22.Groux-Degroote S, Van Dijk SM, Wolthoorn J, Neumann S, Theos AC, De Mazière AM, Klumperman J, Van Meer G, and Sprong H (2008). Glycolipid-dependent sorting of melanosomal from lysosomal membrane proteins by lumenal determinants. Traffic 9, 951–963. 10.1111/j.1600-0854.2008.00740.x. [DOI] [PubMed] [Google Scholar]
- 23.Smith KG, and Clark JD (1994). Black bears in Arkansas: Characteristics of a successful translocation. Journal of Mammalogy 75, 309–320. 10.2307/1382549. [DOI] [Google Scholar]
- 24.Puckett EE, Kristensen TV, Wilton CM, Lyda SB, Noyce KV, Holahan PM, Leslie DM, Beringer J, Belant JL, White D Jr., and Eggert LS (2014). Influence of drift and admixture on population structure of American black bears (Ursus americanus) in the Central Interior Highlands, USA 50 years after translocation. Molecular Ecology 23, 2414–2427. [DOI] [PubMed] [Google Scholar]
- 25.Platt A, Pivirotto A, Knoblauch J, and Hey J (2019). An estimator of first coalescent time reveals selection on young variants and large heterogeneity in rare allele ages among human populations. PLOS Genetics 15, e1008340. 10.1371/journal.pgen.1008340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang K, Mathieson I, O’Connell J, and Schiffels S (2020). Tracking human population structure through time from whole genome sequences. PLOS Genetics 16, e1008552. 10.1371/journal.pgen.1008552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beleza S, Johnson NA, Candille SI, Absher DM, Coram MA, Lopes J, Campos J, Araújo II, Anderson TM, Vilhjálmsson BJ, et al. (2013). Genetic Architecture of Skin and Eye Color in an African-European Admixed Population. PLOS Genetics 9, e1003372. 10.1371/journal.pgen.1003372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilde S, Timpson A, Kirsanow K, Kaiser E, Kayser M, Unterländer M, Hollfelder N, Potekhina ID, Schier W, Thomas MG, and Burger J (2014). Direct evidence for positive selection of skin, hair, and eye pigmentation in Europeans during the last 5,000 y. Proceedings of the National Academy of Sciences 111, 4832. 10.1073/pnas.1316513111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Günther T, and Coop G (2013). Robust identification of local adaptation from allele frequencies. Genetics 195, 205–220. 10.1534/genetics.113.152462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nokelainen O, Scott-Samuel NE, Nie Y, Wei F, and Caro T (2021). The giant panda is cryptic. Scientific Reports 11, 21287. 10.1038/s41598-021-00742-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baird SF (1859). Mammals of the boundary. In Report on the United States and Mexican boundary survey: made under the direction of the secretary of the Interior, pp. 1–62.
- 32.Griffith E. (1821). General and Particular Descriptions of the Vertebrated Animals: Order Carnivora (Baldwin, Cradock, and Joy; ). [Google Scholar]
- 33.Elliot DG (1903). Descriptions of apparently new species of mammals of the genera Heteromys and Ursus from Mexico and Washington. Field Columbian Museum Publications, Zoology Series 3 80, 233–237. [Google Scholar]
- 34.Liu S, Lorenzen Eline D., Fumagalli M, Li B, Harris K, Xiong Z, Zhou L, Korneliussen Thorfinn S., Somel M, Babbitt C, et al. (2014). Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157, 785–794. 10.1016/j.cell.2014.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benazzo A, Trucchi E, Cahill JA, Maisano Delser P, Mona S, Fumagalli M, Bunnefeld L, Cornetti L, Ghirotto S, Girardi M, et al. (2017). Survival and divergence in a small group: The extraordinary genomic history of the endangered Apennine brown bear stragglers. Proceedings of the National Academy of Sciences 114, E9589–E9597. 10.1073/pnas.1707279114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cahill JA, Green RE, Fulton TL, Stiller M, Jay F, Ovsyanikov N, Salamzade R, St. John J, Stirling I, Slatkin M, and Shapiro B (2013). Genomic evidence for island population conversion resolves conflicting theories of polar bear evolution. PLoS Genetics 9, e1003345. 10.1371/journal.pgen.1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller W, Schuster SC, Welch AJ, Ratan A, Bedoya-Reina OC, Zhao F, Kim HL, Burhans RC, Drautz DI, Wittekindt NE, et al. (2012). Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proceedings of the National Academy of Sciences 109, E2382–E2390. 10.1073/pnas.1210506109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cahill JA, Stirling I, Kistler L, Salamzade R, Ersmark E, Fulton TL, Stiller M, Green RE, and Shapiro B (2015). Genomic evidence of geographically widespread effect of gene flow from polar bears into brown bears. Molecular Ecology 24, 1205–1217. 10.1111/mec.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Endo Y, Osada N, Mano T, and Masuda R (2021). Demographic History of the Brown Bear (Ursus arctos) on Hokkaido Island, Japan, Based on Whole-Genomic Sequence Analysis. Genome Biology and Evolution 13. 10.1093/gbe/evab195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Chi W, Tao L, Wang G, Deepak RNVK, Sheng L, Chen T, Feng Y, Cao X, Cheng L, et al. (2022). Ablation of Proton/Glucose Exporter SLC45A2 Enhances Melanosomal Glycolysis to Inhibit Melanin Biosynthesis and Promote Melanoma Metastasis. Journal of Investigative Dermatology 142, 2744–2755.e2749. 10.1016/j.jid.2022.04.008. [DOI] [PubMed] [Google Scholar]
- 41.Bennett DC, Cooper PJ, and Hart IR (1987). A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. International Journal of Cancer 39, 414–418. 10.1002/ijc.2910390324. [DOI] [PubMed] [Google Scholar]
- 42.Dennis MK, Mantegazza AR, Snir OL, Tenza D, Acosta-Ruiz A, Delevoye C, Zorger R, Sitaram A, de Jesus-Rojas W, Ravichandran K, et al. (2015). BLOC-2 targets recycling endosomal tubules to melanosomes for cargo delivery. Journal of Cell Biology 209, 563–577. 10.1083/jcb.201410026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Setty SRG, Tenza D, Truschel ST, Chou E, Sviderskaya EV, Theos AC, Lamoreux ML, Pietro SMD, Starcevic M, Bennett DC, et al. (2007). BLOC-1 Is Required for Cargo-specific Sorting from Vacuolar Early Endosomes toward Lysosome-related Organelles. Molecular Biology of the Cell 18, 768–780. 10.1091/mbc.e06-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20, 1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Browning SR, and Browning BL (2007). Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. The American Journal of Human Genetics 81, 1084–1097. 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi Y, Sims GE, Murphy S, Miller JR, and Chan AP (2012). Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLOS ONE 7, e46688. 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nature Methods 7, 248–249. 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ng PC, and Henikoff S (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31, 3812–3814. 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geneious Prime 2019.0.4 (2019). Geneious Prime.
- 53.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.R Core Team (2020). R: A language and environment for statistical computing (R Foundation for Statistical Computing; ). [Google Scholar]
- 55.Catchen J, Hohenlohe PA, Bassham S, Amores A, and Cresko WA (2013). STACKS: an analysis tool set for population genomics. Molecular Ecology 22, 3124–3140. 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, and Sham PC (2007). PLINK: A tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics 81, 559–575. 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schiffels S, and Durbin R (2014). Inferring human population size and separation history from multiple genome sequences. Nature Genetics 46, 919–925. 10.1038/ng.3015 http://www.nature.com/ng/journal/v46/n8/abs/ng.3015.html#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H. (2009). SNPable Regions.
- 59.Peter BM, and Slatkin M (2013). Detecting range expansions from genetic data. Evolution 67, 3274–3289. doi: 10.1111/evo.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szpiech ZA (2022). selscan 2.0: scanning for sweeps in unphased data. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lischer HEL, and Excoffier L (2012). PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299. 10.1093/bioinformatics/btr642. [DOI] [PubMed] [Google Scholar]
- 62.Haller BC, and Messer PW (2019). SLiM 3: Forward genetic simulations beyond the Wright–Fisher model. Molecular Biology and Evolution 36, 632–637. 10.1093/molbev/msy228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okonechnikov K, Golosova O, Fursov M, and team, t.U. (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. 10.1093/bioinformatics/bts091. [DOI] [PubMed] [Google Scholar]
- 64.Puckett EE (2015). Phylogeography and Population Genomics of the American Black Bear (Ursus americanus). PhD (University of Missouri; ). [Google Scholar]
- 65.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Browning BL, Zhou Y, and Browning SR (2018). A One-Penny Imputed Genome from Next-Generation Reference Panels. The American Journal of Human Genetics 103, 338–348. 10.1016/j.ajhg.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, et al. (2002). The Structure of Haplotype Blocks in the Human Genome. Science 296, 2225. 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 68.Taylor G, Kirk H, Coombe L, Jackman S, Chu J, Tse K, Cheng D, Chuah E, Pandoh P, Carlsen R, et al. (2018). The Genome of the North American Brown Bear or Grizzly: Ursus arctos ssp. horribilis. Genes 9, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. Journal of Molecular Biology 215, 403–410. 10.1016/s0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Z, Schwartz S, Wagner L, and Miller W (2000). A greedy algorithm for aligning DNA sequences. Journal of Computational Biology 7, 203–214. 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 71.Lai X, Wichers HJ, Soler-Lopez M, and Dijkstra BW (2017). Structure of Human Tyrosinase Related Protein 1 Reveals a Binuclear Zinc Active Site Important for Melanogenesis. Angewandte Chemie International Edition 56, 9812–9815. 10.1002/anie.201704616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bennett DC, Cooper PJ, Dexter TJ, Devlin LM, Heasman J, and Nester B (1989). Cloned mouse melanocyte lines carrying the germline mutations albino and brown: complementation in culture. Development 105, 379–385. [DOI] [PubMed] [Google Scholar]
- 73.Bennett DC, Huszar D, Laipis PJ, Jaenisch R, and Jackson IJ (1990). Phenotypic rescue of mutant brown melanocytes by a retrovirus carrying a wild-type tyrosinase-related protein gene. Development 110, 471. [DOI] [PubMed] [Google Scholar]
- 74.Morita S, Kojima T, and Kitamura T (2000). Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7, 1063–1066. [DOI] [PubMed] [Google Scholar]
- 75.Dennis MK, Delevoye C, Acosta-Ruiz A, Hurbain I, Romao M, Hesketh GG, Goff PS, Sviderskaya EV, Bennett DC, Luzio JP, et al. (2016). BLOC-1 and BLOC-3 regulate VAMP7 cycling to and from melanosomes via distinct tubular transport carriers. Journal of Cell Biology 214, 293–308. 10.1083/jcb.201605090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meng R, Wang Y, Yao Y, Zhang Z, Harper DC, Heijnen HFG, Sitaram A, Li W, Raposo G, Weiss MJ, et al. (2012). SLC35D3 delivery from megakaryocyte early endosomes is required for platelet dense granule biogenesis and differentially defective in Hermansky-Pudlak syndrome models. Blood 120, 404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bowman SL, Le L, Zhu Y, Harper DC, Sitaram A, Theos AC, Sviderskaya EV, Bennett DC, Raposo G, Owen DJ, et al. (2021). A BLOC-1-AP-3 super-complex sorts a cis-SNARE complex into endosome-derived tubular transport carriers. J. Cell Biol 220, e202005173, 33886957. 10.1083/jcb.202005173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Delevoye C, Hurbain I, Tenza D, Sibarita J-B, Uzan-Gafsou S, Ohno H, Geerts WJC, Verkleij AJ, Salamero J, Marks MS, and Raposo G (2009). AP-1 and KIF13A coordinate endosomal sorting and positioning during melanosome biogenesis. J. Cell Biol 187, 247–264. 10.1083/jcb.200907122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dennis MK, Delevoye C, Acosta-Ruiz A, Hurbain I, Romao M, Hesketh GG, Goff PS, Sviderskaya EV, Bennett DC, Luzio JP, et al. (2016). BLOC-1 and BLOC-3 regulate VAMP7 cycling to and from melanosomes via distinct tubular transport carriers. J. Cell Biol 214, 293–308. 10.1083/jcb.201605090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, and Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7. 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kumar S, and Subramanian S (2002). Mutation rates in mammalian genomes. Proceedings of the National Academy of Sciences 99, 803. 10.1073/pnas.022629899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Onorato DP, Hellgren EC, Van Den Bussche RA, and Doan-Crider DL (2004). Phylogeographic patterns within a metapopulation of black bears (Ursus americanus) in the American Southwest. Journal of Mammalogy 85, 140–147. [Google Scholar]
- 83.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Voight BF, Kudaravalli S, Wen X, and Pritchard JK (2006). A map of recent positive selection in the human genome. PLoS Biology 4, e72. 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Szpiech ZA, and Hernandez RD (2014). selscan: An Efficient Multithreaded Program to Perform EHH-Based Scans for Positive Selection. Molecular Biology and Evolution 31, 2824–2827. 10.1093/molbev/msu211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vatsiou AI, Bazin E, and Gaggiotti OE (2016). Detection of selective sweeps in structured populations: a comparison of recent methods. Molecular Ecology 25, 89–103. 10.1111/mec.13360. [DOI] [PubMed] [Google Scholar]
- 87.Coop G, Witonsky D, Di Rienzo A, and Pritchard JK (2010). Using Environmental Correlations to Identify Loci Underlying Local Adaptation. Genetics 185, 1411–1423. 10.1534/genetics.110.114819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kriticos DJ, Jarošik V, and Ota N (2014). Extending the suite of bioclim variables: a proposed registry system and case study using principal components analysis. Methods in Ecology and Evolution 5, 956–960. 10.1111/2041-210X.12244. [DOI] [Google Scholar]
- 89.Hutchinson M, Xu T, Houlder D, Nix H, and McMahon J (2009). ANUCLIM 6.0 User’s Guide. Australian National University, Fenner School of Environment and Society. [Google Scholar]
- 90.Haroldson MA, Clapham M, Costello CM, Gunther KA, Kendall KC, Miller SD, Pigeon KE, Proctor MF, Rode KD, Servheen C, et al. (2021). Brown Bear (Ursus arctos; North America). In Bears of the World, Melletti V.P.a.M., ed. (Cambridge University Press; ). [Google Scholar]
- 91.Malaney JL, Lackey CW, Beckmann JP, and Matocq MD (2017). Natural rewilding of the Great Basin: Genetic consequences of recolonization by black bears (Ursus americanus). Diversity and Distributions 24, 168–178. 10.1111/ddi.12666. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data has been deposited onto the NCBI SRA under project number PRJNA867575. This paper does not report original code.