

Abstract

The coupling of aryl and aliphatic azides with isocyanides yielding carbodiimides (8–17) were efficiently catalyzed by well-defined structurally characterized trans-(MIC)PdI2(L) [MIC = 1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene, L = NC5H5 (4), MesNC (5)], trans-(MIC)2PdI2 (6), and cis-(MIC)Pd(PPh3)I2 (7) type palladium complexes, which incidentally mark the first instances of the use of mesoionic singlet palladium carbene complexes for the said application. As observed from the product yields, the catalytic activity varied in the order 4 > 5 ∼ 6 > 7 for these complexes. A detailed mechanistic studies indicated that the catalysis proceeded via a palladium(0) (4a–7a) species. Using a representative palladium precatalyst (4), the azide–isocyanide coupling was successfully extended to synthesizing two different bioactive heteroannular benzoxazole (18–22) and benzimidazole (23–27) derivatives, thereby broadening the scope of the catalytic application.
Introduction
Carbodiimides have long evoked interest for over half a century now, primarily for their diverse range of applications varying from peptide synthesis to phosphorylation reactions to dehydration reactions.1−3 Their biocompatibility and nontoxicity make them well-suited for peptide synthesis as cross-linkers providing “zero-length” amide linkages between carboxylic and amine groups.4 In organic synthesis, carbodiimides, belonging to the heterocumulene family of compounds and containing two adjacent C=N bonds, are also of sustained interest as they provide a unique opportunity for synthesizing N-heterocycles, which are known for their pharmaceutical properties.5,6 The N-heterocycles constitute common fragments in marketed drugs and medicinal chemistry targets as a part of the drug discovery initiatives.1 Hence, convenient access routes to these important carbodiimide class of compounds are in demand,7 and catalytically achieving that remains a challenging objective. Carbodiimides, with two adjacent C=N bonds, make synthons for N-heterocycles.1,5
A common approach involves the decarboxylative coupling of isocyanates, leading to the elimination of CO2.3 The method is primarily restricted to preparing symmetrical carbodiimides, as the selectivity between the homocoupled and heterocoupled products becomes an issue in coupling between different isocyanates. Notably, the syntheses of the more synthetically challenging unsymmetrical carbodiimides are obtained in a stoichiometric fashion by aza-Wittig reactions of phosphinimines with isocyanates8 and by the Sn-mediated metathesis of isocyanates and silyl amines.9 Under heterogeneous conditions, the catalytic oxidative coupling of primary amines to isocyanides using gold10 or PdCl2/Ag2O11 produces carbodiimides.
However, an esoteric organometallic approach involving transition metal-mediated nitrene transfer in an azide–isocyanide coupling provides a more efficient atom economic approach to these carbodiimides.12−14 The appeal of this method resides in the easy and convenient synthetic access available for different azides, the simplicity of the process for preparing the symmetrical and unsymmetrical carbodiimides, and the generation of the environmentally benign dinitrogen as the only byproduct of the reaction.
Judicious exploitation of this method allows for the construction of a variety of bioactive heterocycles15 like the benzoxazoles known for their antibiotic, antifungal, antiviral, antitumor, antiulcer, antibacterial, anti-inflammatory, antitubercular, and analgesic properties16,17 that rightfully constitute the core motif of a chiral nonsteroidal anti-inflammatory drug, flunoxaprofen.18 This approach also allows synthesis of other important N-heterocycles like the benzimidazoles having pharmaceutical and industrial applications.19
With our interest in exploring the potentials of the transition metal complexes of the heteroatom stabilized cyclic and acyclic singlet carbene ligands in homogeneous catalysis20−25 and in biomedical applications,26 we study different singlet-carbene ligand classes covering the cyclic normal N-heterocyclic carbenes,27 abnormal mesoionic carbenes25,28 the acyclic diaminocarbenes22,29 and the fused-cyclic ones of the imidazole,30 benzimidazole,31 triazole,23,24,32 oxazolidine,33 bioxazoline,34 and triazolooxazine35 architectures. The mesoionic (MIC) carbenes, featuring less heteroatom stabilization than the more ubiquitous normal N-heterocyclic carbenes,36 come with specific donors and reactivity properties but surprisingly remain less explored.37 Because of the reasons mentioned above, we decided to study the catalytic utility of the transition metal complexes of the 1,2,3-triazole-derived mesoionic carbene in the azide–isocyanide coupling reaction for the synthesis of carbodiimides. In this regard, we decided to design a new type of the 1,2,3-triazole-derived mesoionic carbene ligand bearing phenothiazine moiety. Of late, phenothiazine has received attention for its organic light-emitting diodes (OLEDs), photovoltaic devices, data storage, sensors, and bioimaging applications but with no footprint in chemical catalysis.38 Hence, we decided to pursue the potential of the phenothiazine-based 1,2,3-triazole-derived mesoionic carbene stabilized transition metal complexes in the azide–isocyanide coupling reaction.
Here, in the manuscript, we report a convenient synthetic route to various carbodiimide compounds (8–17) by the azide–isocyanide coupling mediated by palladium complexes (4–7) (Figure 1) of a new phenothiazine-based 1,2,3-triazole derived mesoionic carbene ligand. Further, we demonstrate their utility in synthesizing two bioactive heteroannular benzoxazole (18–22) and benzimidazole (23–27) derivatives.
Figure 1.

Synthesized phenothiazine-derived mesoionic singlet carbene complexes of palladium (4–7).
Results and Discussions
A new phenothiazine-derived mesoionic singlet carbene ligand namely, 1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene, was designed primarily for the reasons of exploring its utility in transition metal mediated homogeneous catalysis applications. This new class of phenothiazine functionalized 1,2,3-triazole derived N-heterocyclic carbene ligand was conveniently obtained from its triazolium iodide salt, 1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazolium iodide (3), by the alkylation reaction of 10-[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]-10H-phenothiazine (2) with MeI in ca. 99% yield (Scheme 1). The formation of 3 was confirmed by the observation of the characteristic (NCHC) resonance at δ ca. 9.52 ppm in the 1H NMR spectrum and at δ ca. 143.1 in 13C{1H} NMR spectrum (Supporting Information, Figures S13–S16). The corresponding NCH3 resonance appeared at δ ca. 4.26 ppm in 1H NMR, and at δ ca. 39.6 ppm in 13C{1H} NMR. The Cu mediated click reaction between 10-(prop-2-yn-1-yl)-10H-phenothiazine (1) and benzyl azide yielded the desired phenothiazine functionalized 1,2,3-triazole derivative (2) in ca. 65% yield.39
Scheme 1. Synthetic Routes to the Compounds (1–3) and the Phenothiazine-Derived Mesoionic Singlet Carbene Complexes of Palladium (4–7).

The 1,2,3-triazolium iodide salt (3) provided a convenient platform for accessing a large number of transition metal complexes. For example, the direct metalation reaction of (3) with PdCl2 in pyridine, in the presence of K2CO3 as base yielded trans-[1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]PdI2(NC5H5) (4) complex in 95% yield (Scheme 1). As expected, the o-NC5H5 resonance of the metal bound pyridine moiety appeared significantly downfield shifted at δ ca. 9.05 ppm as opposed to that of δ ca. 8.62 ppm for free pyridine in the 1H NMR40 (Supporting Information, Figures S20 and S21).
Furthermore, the characteristic Pd–Ccarbene peak at δ ca. 139.0 ppm (Supporting Information, Figures S22–S23) in the 13C{1H} NMR spectra of (4) is comparable to that observed for related trans-[1-Ph-3-Me-4-Ph-1,2,3-triazol-5-ylidene]PdI2(NC5H5) (δ ca.139.8 ppm),41trans-[{(S)-7-CH2Ph-2-Me-6,7-dihydro-4H-[1,2,3]-triazolo[5,1-c][1,4]oxazin-3-ylidene}PdI2(NC5H5) (δ ca.138.5 ppm)35 and the other structurally characterized examples known in the literature (Supporting Information, Table S2).
As expected, the molecular structure of 4 (Figure 2 and Supporting Information, Table S1), as determined by the single crystal X-ray diffraction study, exhibited a nonplanar butterfly structure with the S and N heteroatoms occupying the 1,4 apical positions, while the two aromatic rings are pointed outward from the (2,3) and (5,6) positions of a boat confirmation, consistent with commonly observed conformation of a phenothiazine moiety.42 In agreement with the d8 configuration of a palladium(II) center, a square planar geometry was observed for 4 with the mesoionic 1,2,3-trizole based N-heterocyclic carbene ligand and the “‘throwaway’” pyridine moiety disposed at opposite ends of a near linear angle at the metal center [∠C(1)–Pd(1)–N(5) = 176.9(3)°] while two more iodine atoms occupied the other two trans positions [∠I(2)–Pd(1)–I(1) = 177.06(3)°].
Figure 2.
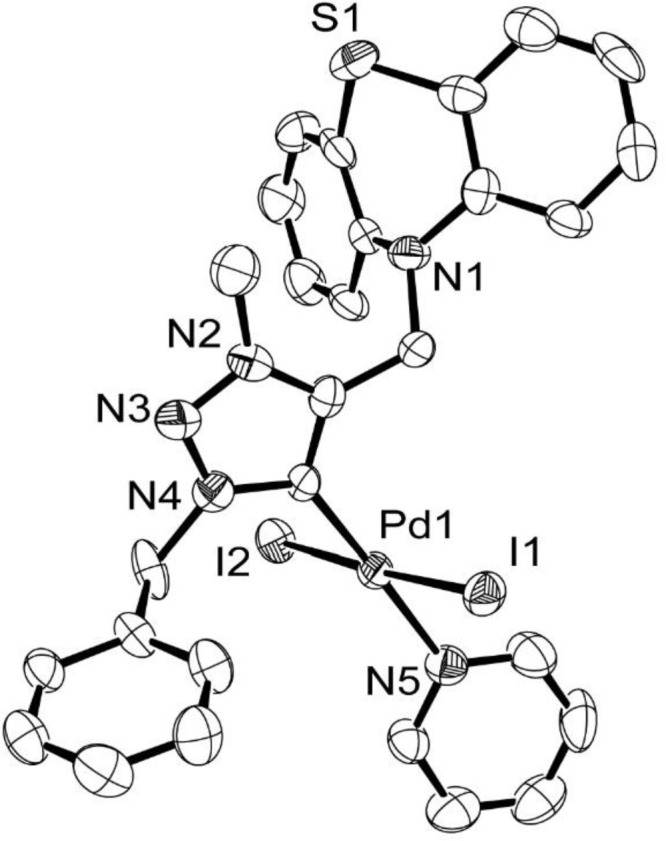
ORTEP diagram of 4 with thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (deg): Pd(1)–C(1) 1.981(5), Pd(1)–I(1) 2.6069(8), Pd(1)–I(2) 2.6104(8), Pd(1)–N(5) 2.105(7), I(2)–Pd(1)–I(1) 177.06(3), N(5)–Pd(1)–I(1) 90.34(19), N(5)–Pd(1)–I(2) 91.55(19), C(1)–Pd(1)–I(1) 88.0(2), C(1)–Pd(1)–I2 90.2(2), and C(1)–Pd(1)–N(5) 176.9(3).
The Pd–Ccarbene bond distance of 1.981(8) Å in (4) compared well with the representative reported analogues, namely trans-[1-Ph-3-Me-4-Ph-1,2,3-triazol-5-ylidene]PdI2(NC5H5) [1.971(6) Å]41 and trans-{(S)-7-CH2Ph-2-Me-6,7-dihydro-4H-[1,2,3]-triazolo[5,1-c][1,4]oxazin-3-ylidene}PdI2(NC5H5) [1.974(9) Å].35 (Supporting Information, Table S2).
A rare isocyanide bound palladium complex, trans-[1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]PdI2(CN–Mes) (5), was prepared by the treatment of (4) with MesNC at ambient temperature in ca. 50% yield (Scheme 1). To the best of our knowledge, the palladium complex (5) represents the only reported example of a (MIC)PdI2(CN–R) type complex of a mesoionic 1,2,3-triazole derived N-heterocyclic carbene. However, a handful of related analogues are known for benzimidazole43,44 and indazole derived N-heterocyclic carbenes.44 (Supporting Information, Table S3). Given the fact that all of these mixed carbene/isocyanide complexes were synthesized from a dimeric halide-bridged complex by treatment with the aliphatic and aromatic isocyanides,43,44 the current method for the preparation of 5 offers a new way for accessing these type of mixed carbene/isocyanide complexes.
Of foremost importance to a mixed carbene/isocyanide themed precatalyst (5) is the metal bound isocyanide moiety. The successful coordination of MesNC to the Pd(II) center was evident from the 13C{1H} NMR spectra for complex 5, where the Pd–CN signal appeared as a broad triplet at δ ca.138.7 ppm and was significantly downfield shifted (Δδ = ca. 14.6 ppm) in comparison to the free MesNC (δ ca.124.1 ppm) (Supporting info Figures S29 and S30). An analogue shift of (Δν) ca. 73 cm–1 was observed for (ν(CN) = 2188 cm–1) in IR spectroscopy with respect to the free MesNC (ν(CN) = 2115 cm–1).45
Single-crystal X-ray diffraction study revealed a square planar geometry for complex 5 (Figure 3 and Supporting Information, Table S1) analogous to that of complex 4. Quite interestingly, an elongation of the Pd–Ccarbene bond length in 5 [2.002(5) Å] with respect to 4 [1.981(8) Å)], suggests a stronger trans-effect of the MesNC moiety in the former than the pyridine moiety in latter. Consistently, the Pd–CNMes bond length in (5) [1.970(5) Å] is significantly shorter than the sum of individual covalent radii of Pd and Csp atoms [2.08 Å].46 This observation is in agreement with a reported mixed indazole-carbene/isocyanide complex, trans-[1,2-Me2-indazol-3-ylidene]PdI2(CN–Xyl), [d/(Pd–Ccarbene) = 2.002(5) Å and d/(Pd–CN) = 1.993(5) Å].44 The benzimidazole derived analogues, trans-[1,3-Me2-benzimidazol-2-ylidene]PdI2(CN–Xyl)44 [d/(Pd–Ccarbene) = 1.996(3) Å and d/(Pd–CN) = 1.986(4) Å], trans-[1,3-Me2-benzimidazol-2-ylidene]PdI2(CN–Cy), [d/(Pd–Ccarbene) = 1.986(4) Å and d/(Pd–CN) = 1.993(5) Å],44 however, do not show such a profound trans-effect.
Figure 3.

ORTEP diagram of 5 with thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (deg): Pd(1)–C(1) 2.002(5), Pd(1)–I(1) 2.5892(5), Pd(1)–I(2) 2.6144(5), Pd(1)–C(24) 1.970(5), I(2)–Pd(1)–I(1) 178.11(2), C(24)–Pd(1)–I(1) 89.85(15), C(24)–Pd(1)–I(2) 90.14(15), C(1)–Pd(1)–I(1) 88.07(14), C(1)–Pd(1)–I2 92.07(14), and C(1)–Pd(1)–C(24) 175.6(2).
The complex (4), when treated with PPh3, gave a mixed carbene/PPh3 type complex cis-[1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]Pd(PPh3)I2 (7) in ca. 80% yield at ambient temperature (Scheme 1).
Quite interestingly, the 1H, 13C{1H} and 31P{1H} NMR, all convincingly pointed at the existence of an equilibrium between cis-(7) and trans-(7′) species in solution at ambient temperature (Scheme 2 and Supporting Information, Figures S41–S45). Specifically, in the 1H NMR spectrum, the NCH3 resonance appeared at δ ca. 3.71 ppm for cis-(7) and at δ ca. 4.05 ppm for trans-(7′) in ca. 3.4:1 ratio. Indeed, in the variable temperature 1H NMR experiment, the ratio of the NCH3 resonances, one that appeared at δ ca. 3.71 ppm for cis-(7) and the other at δ ca. 4.05 ppm for trans-(7′), changed from ca. 3.4:1 at 25 °C to ca. 1:1 at 100 °C. (Supporting Information, Figures S219–S220). In the spin saturation 1H NMR experiment, when the NCH3 moiety of cis-(7) was irradiated at δ ca 3.71 ppm, the intensity of the NCH3 moiety of trans-(7′) at δ ca. 4.05 ppm decreased and vice versa, thereby attesting to the equilibrium present between the two species in solution (Supporting Information, Figure S221).
Scheme 2. 1H NMR Analysis Showing Equilibrium between cis-(7) and trans-(7′) in ca. 3.4:1 Ratio in Solution at Room Temperature.

Furthermore, diastereotopic resonances were observed for both the CH2Ph and NCH2 moieties in cis-(7), with a doublet pair observed at δ ca. 5.94 ppm and δ ca 5.07 ppm and at δ ca. 5.40 ppm and δ ca. 4.19 ppm, exhibiting geminal (2JHH) coupling constants of 14 and 15 Hz respectively. In contrast, both the CH2Ph and NCH2 resonances were singlets at δ ca. 5.86 ppm and at δ ca. 5.43 ppm respectively for the trans-(7′) complex. The 13C{1H} NMR of cis-(7) too showed the complete set of isomeric peaks due to the presence of the minor isomer trans-(7′) in solution. In the 31P{1H} NMR, the Pd–P̲Ph3 resonance at δ ca. 25.0 ppm was attributed to cis-(7), based on the reported examples, cis-[1-Et-3-Me-4-C12H9-1,2,3-triazol-5-ylidene]Pd(PPh3)I2 (δ ca. 25.8 ppm)47 and cis-[1-CH2Ph-3-Me-4-Ph-1,2,3-triazol-5-ylidene]Pd(PPh3)I2 (δ ca. 25.2 ppm).25 (Supporting Information, Table S4), while the other upfield shifted Pd–P̲Ph3 resonance at δ ca. 15.9 ppm was assigned to the trans-(7′), based on the related analogues, trans-[1-CH2Ph-3-Et-4-C6H10OH-1,2,3-triazol-5-ylidene]Pd(PPh3)I225 (δ ca. 16.9 ppm) and trans-[1-CH2Ph-3-Me-4-C6H10OH-1,2,3-triazol-5-ylidene]Pd(PPh3)I2 (δ ca. 17.0 ppm).25
The structural characterization using single crystal X-ray diffraction study revealed a cis complex (7) (Figure 4 and Supporting Information, Table S1), in which the mesoionic 1,2,3-trizole based N-heterocyclic carbene ligand and the PPh3 moieties were in cis disposition around the square planar palladium(II) metal center [∠C(1)–Pd(1)–P(1) = 91.29(19)°] with the other two sites occupied by the iodine atoms [∠I(1)–Pd(1)–I(2) = 91.98(2)°]. Furthermore, the Pd–Ccarbene [2.001(7) Å] and Pd–Pphosphine [2.2950(17) Å] bond distances in cis-(7) were in good agreement with other related complexes, namely cis-[1-Ph-3-Me-4-n-Bu-1,2,3-triazol-5-ylidene]Pd(PPh3)I2 [d/(Pd–Ccarbene) = 2.000(4) Å, d/(Pd–Pphosphine) = 2.2789(10) Å]48 and cis-[1-CH2Ph −3-Me-4-Ph-1,2,3-triazol-5-ylidene]Pd(PPh3)I2 [d/(Pd–Ccarbene) = 2.006(6) Å, d/(Pd–Pphosphine) = 2.2804(17) Å]25 (Supporting Information, Table S4).
Figure 4.

ORTEP diagram of 7 with thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (deg): Pd(1)–C(1) 2.001(7), Pd(1)–I(1) 2.6381(6), Pd(1)–I(2) 2.6633(7), Pd(1)–P(1) 2.2950(17), I(1)–Pd(1)–I(2) 91.98(2), C(1)–Pd(1)–I(1) 87.18(19), C(1)–Pd(1)–I(2) 174.17(19), C(1)–Pd(1)–P(1) 91.29(19), P(1)–Pd(1)–I2 89.81(5), and P(1)–Pd(1)–I(1) 176.93(5).
The metalation of the triazolium iodide salt (3) with Pd(OAc)2 quantitatively yielded the trans-[1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]2PdI2 complex (6) in 93% yield (Scheme 1). In this case too, two sets of resonances were observed in both the 1H and 13C{1H} NMR spectra signifying the presence of two conformers i.e. trans-anti and trans-syn (Supporting Information, Figures S34–S37). In the 1H NMR spectrum, the sterically favored trans-anti isomer appeared in a ca. 1.7:1 ratio over the trans-syn conformer at 25 °C, which changed to ca. 1.5:1 at 50 °C. (Supporting Information, Figure S218).49 In 13C{1H} NMR spectra, two closely spaced Pd–Ccarbene signals for the trans-anti and trans-syn conformers of 6 appeared at 156.7 and 156.4 ppm, respectively.
The complex (6) is among the few structurally characterized examples of the (MIC)2PdI2 type complexes supported over a 1,2,3 triazole-derived mesoionic N-heterocyclic carbene ligand known in the literature (Supporting Information, Table S5). The square planar complex (6) (Figure 5 and Supporting Information, Table S1) was surrounded by two mesoionic carbene units in [∠C(1)–Pd(1)–C(1i) = 180.0°], while two iodide atoms occupied the remaining two diametrically opposite sites [∠I(1)–Pd(1)–I(1i) = 180.0°]. In addition, the observed Pd–Ccarbene bond distance of 2.030(4)Å and the Pd–I bond distance of 2.6107(3) Å were in close range to that reported for similar complexes, namely, trans-[1-Ph-3-Me-4-Ph-1,2,3-triazol-5-ylidene]2PdI2 [d/(Pd–Ccarbene) = 2.026(6) Å, d/(Pd–I) = 2.6174(5) Å]50 and trans-[1-Et-3-Me-4-Ph-1,2,3-triazol-5-ylidene]2PdI2 [d/(Pd–Ccarbene) = 2.049(3) Å, d/(Pd–I) = 2.6181(3) Å].51 (Supporting Information, Table S5).
Figure 5.

ORTEP diagram of 6 with thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (deg): Pd(1)–C(1) 2.030(4), Pd(1)–I(1) 2.6107(3), and C(1)–Pd(1)–I(1) 88.66(10).
The palladium (4–7) complexes efficiently carried out the coupling of aryl and aliphatic azides with different isocyanides yielding carbodiimides in the presence of a base (Table 1, 2, 3 and Supporting Information, Table S6–S12). A series of optimization experiments on the representative pair of substrates, namely phenyl azide and benzyl isocyanide, with a representative precatalyst (4) provided the optimal catalysis conditions. As expected, the blank run performed without the precatalyst (4) and the base, produced no product for the aforementioned substrates (entry 1, Supporting Information, Table S12). The control run, performed only in the presence of the base K2CO3, too yielded no detectable product (entry 2, Supporting Information, Table S12). The control runs performed with the metal precursor Pd2(dba)3 gave ca. 41% product yield (entry 4, Supporting Information, Table S12) and that with 4 yielded no detectable product (entry 9, Supporting Information, Table S12). However, the same run when performed with Pd2(dba)3 and with 4 in the presence of the base K2CO3 produced N-[(benzylimino)methylene]aniline (8) in ca. 37% (entry 5, Supporting Information, Table S12) and ca. 88% (entry 10, Supporting Information, Table S12) yields respectively. Additional control runs involving the ligand precursor (3) in the presence of K2CO3 base showed no detectable product formation, thereby highlighting the significant influence played by the palladium (4–7) precatalysts in the azide–isocyanide coupling reaction (entry 3, Supporting Information, Table S12). Notably, the significant enhancement in the product yield by ca. 51% in case of complex 4, in comparison to Pd2(dba)3, highlights the favorable influence of the mesoionic 1,2,3-triazole derived N-heterocyclic carbene ligand in catalyzing the coupling of phenyl azide and benzyl isocyanide. The Hg drop experiment bore testimony toward the homogeneous nature of the catalysis as near equal yield of ca. 83% was found in the presence of mercury as opposed to 88% in absence of it (entries 10 and 11, Supporting Information, Table S12).
Table 1. Selected Results for Phenothiazine-Derived Mesoionic Singlet Carbene Palladium (4–7) Complexes Catalyzed Azide–Isocyanide Coupling Yielding Carbodiimidesa,b.


Reaction conditions: azide (0.369 mmol), isocyanide (0.443 mmol), complex (4/5/6/7) (0.003 mmol, 1 mol %) and K2CO3 (1.10 mmol) in 5.0 mL of toluene at 50 °C for 12 h.
Isolated yields (%).
Table 2. Comparison of the Reaction Yield and Turnover Number (TON) Calculated with Respect to the Formation of N-[(Xylylimino)methylene]aniline in the Catalyzed Azide–Isocyanide Coupling Reaction of the Representative Phenyl Azide and 2,6-Dimethylphenyl Isocyanide Substrates as Catalyzed by 4–7 and other Transition Metal Complexes Known in the Literature.


Table 3. Selected Results for a Simple Practical Synthesis of a Variety of Benzoxazole Derivatives Using Azide–Isocyanide Coupling Catalyzed by a Phenothiazine-Derived Mesoionic Singlet Carbene Palladium (4) Complexa,b.

Reaction conditions: azide (0.369 mmol), isocyanide (0.443 mmol), complex 4 (0.003 mmol, 1 mol %), and K2CO3 (1.10 mmol) in 5.0 mL of toluene at 50 °C for 12 h.
Isolated yields (%).
Furthermore, the base variation study suggested K2CO3 (Supporting Information, Table S6) and the solvent variation study suggested toluene for optimal catalysis conditions (Supporting Information, Table S7). The variation of the molar ratio of the optimal base K2CO3 with the substrate phenyl azide produced highest yield at ca. 1:3 ratio (Supporting Information, Table S9). A catalysis reaction temperature of 50 °C (entry 2, Supporting Information, Table S8) and a catalyst loading of 1 mol % were found to be suitable based on the temperature dependence and the catalyst loading variation studies (entry 3, Supporting Information, Table S10).
The temperature dependence study with complex 4 showed that the product yield decreased with temperature, which can be attributed to the thermal decomposition of the azide moiety.52 Similar observation was made for the said azide–isocyanide coupling reaction mediated by Cr,13 as starting materials were decomposed, when the reaction mixture was heated beyond 60 °C.
In line with this investigation, the XPS analysis of the 4–7 complexes, before and after heating in toluene at 100 °C for 24 h, were performed. No trace of Pd leaching was observed in that process, and peaks corresponding to Pd(0) was also absent in the data. (Supporting Information, Figures S238–S246). Additionally, the temperature dependent 1H NMR were also analyzed for complexes 4–7, and in this case too, no signature of decomposed peaks were found. (Supporting Information, Figures S216–S220). These experiments clearly showed that these (4–7) catalysts were stable during the catalysis conditions, and the decomposition of the azide substrates at the catalysis temperature was more dominant. It is worth noting that in long reaction time and under prolonged heating, carbodiimides tend to decompose or polymerize.12,53
The time dependence study yielded 12 h to be the optimum reaction time as per the product yield (entry 4, Supporting Information, Table S11). Hence based on the aforementioned results, the catalysis conditions for the azide–isocyanide coupling that were chosen for the further substrate scope studies were at 1 mol % of the catalyst loading in the presence of the 3 equiv of K2CO3 in toluene at 50 °C for 12 h of the reaction time.
It is noteworthy to mention, all of the (4–7) complexes successfully carried out the azide–isocyanide coupling of different aryl azides ranging from phenyl azide to 4-methylphenyl azide, to 2-methoxyphenyl azide to 4-fluorophenyl azide to 3,5-bis(trifluoromethyl)phenyl azide to eventually an aliphatic azide, namely cyclohexyl azide with a host of isocyanides, namely, benzyl isocyanide, 4-methylphenyl isocyanide, 2,6-dimethylphenyl isocyanide, 2,4,6-trimethylphenyl isocyanide, and cyclohexyl isocyanide under the optimized catalysis conditions. Quite expectedly, with the increase in sterics, on going from 4-methylphenyl isocyanide to 2,6-dimethylphenyl isocyanide to 2,4,6-trimethylphenyl isocyanide, a steady decrease in the catalysis yield was observed for all the four (4–7) precatalysts when coupled with phenyl azide (entries 2–4, Table 1). Again, for the same isocyanide, i.e. benzyl isocyanide, the electron deficient 4-fluorophenyl azide and 3,5-bis(trifluoromethyl)phenyl azide performed consistently better than the corresponding electron rich 4-methylphenyl azide for all the (4-7) precatalysts (entries 7, 9, and 10, Table 1). Similar instances of higher reactivity of the electron deficient azides have been reported for an Fe catalyst and has been rationalized as a consequence of metal to azide π-back bonding that stabilizes the metal bound azide species.54 For the coupling with cyclohexyl isocyanide higher yield was consistently observed with phenyl azide compared to its aliphatic counterpart cyclohexyl azide for all the four (4–7) precatalysts.
A careful scrutiny of the substrate scope study brings out an interesting trend of the catalyst reactivity observed along the line, 4 > 5 ∼ 6 > 7, thereby implying that the pyridine bound Pd complex (4) is the most effective, while the PPh3 bound Pd complex (7) is the least effective of all the ten substrates studied for the azide–isocyanide coupling reaction. The other two precatalysts, i.e., the isocyanide bound Pd complex (5) and the bis-mesoionic 1,2,3-triazole based NHC complex (6), lie in between the highly active (4) and the least active (7) precatalysts, and are comparable to each other in terms of the catalysis product yield.
We are not aware of any other structurally characterized molecular palladium catalysts reported for the azide-iscocyanide coupling barring [Pd(PPh3)4].55 Additionally, there exists only one other structurally characterized Ni(I) complex, namely (MesNCHCH3CH2CHCH3NMes)Ni(2-picoline)14 for the azide–isocyanide coupling of the phenyl azide and 2,6-dimethylphenyl isocyanide pair of substrates, similar to that reported for our palladium complexes (4–7) (Table 2). Though the [Pd(PPh3)4]55 complex reported a higher conversion of ca. 82% based on 1H NMR at 5 h of reaction time at room temperature but at higher catalyst loading of 2.5 mol % in comparison to that of 1 mol % of the catalyst loading for our palladium complexes (4–7) showing moderate to good isolated yields of ca. 24–78% at 50 °C after 12 h of reaction time (Table 2). The Ni(I) complex, namely (MesNCHCH3CH2CHCH3NMes)Ni(2-picoline)14 exhibited quantitative conversion by 1H NMR at a longer reaction time of 17 h and also at a higher catalyst loading of 5 mol % at temperatures ranging from −35 °C to room temperature (Table 2). In light of these studies, the catalysis yields exhibited by our palladium (4–7) complexes are indeed promising and would usher further research into the area.
Moreover, in the absence of any prior report of Pd-NHC complex catalyzing the said reaction, a comparison with a benchmark PEPPSI catalyst namely, trans-[1,3-(2,6-(i-Pr)2C6H3)2-imidazol-2-ylidene]PdCl2(NC5H5),56 was made with the palladium (4–7) complexes under analogous catalysis conditions. Specifically, the benchmark PEPPSI complex, trans-[1,3-(2,6-(i-Pr)2C6H3)2-imidazol-2-ylidene]PdCl2(NC5H5) was synthesized according to the literature procedure,56 and when used to carry out the same catalysis, (entry 7, Supporting Information, Table S12) exhibited inferior performance in comparison to our (4–7) catalysts. Another triazolylidene based palladium complex trans-[1-Ph-3-Me-4-CH2Ph-1,2,3-triazol-5-ylidene]PdI2(NC5H5),25 having a phenyl group rather phenothiazine, too exhibited inferior activity than the (4–7) catalysts (entry 8, Supporting Information, Table S12). The study further highlights the significance of these MIC based (4–7) catalysts in the azide–isocyanide coupling reaction.
A unified mechanism involving redox palladium (0/II) oxidation states have been proposed for the (4–7) complexes catalyzing the azide–isocyanide coupling in case of the two representative substrates, phenyl azide and benzyl isocyanide, (Scheme 3 and Supporting Information, Scheme S1 and Figures 6, 7, 8, and S222–S237).55 The proposed catalysis initiates with the formation of the Pd(0) species 4a, 5a, and 7a (Scheme 3) and 6a (Supporting Information, Scheme S1) from the respective Pd(II) (4–7) complexes with the reduction of the metal center occurring in the presence of the base K2CO3.57 With 3 equiv of K2CO3, the catalysis yield was found to be the maximum according to the base and base equivalence variation studies (Supporting Information, Tables S8 and S11), which can be attributed to the fact that K2CO3, being a weak inorganic base, has less solubility in an organic solvent like toluene.58 Quite significantly mass evidence are consistent with the proposed 4a (Figure 6 and Supporting Information, Figure S222), 7a (Supporting Information, Figured S230 and S231) and 6a (Supporting Information, Figures S234 and S235). Upon reaction with phenyl azide, the Pd bound nitrene species, 4b, 5b, and 7b (Scheme 3) and 6b (Supporting Information, Scheme S1) are formed with the elimination of N2, and of which 4b (Figure 7 and Supporting Information, Figure S223) 5b (Supporting Information, Figures S226 and S227) have been observed by mass spectrometry. Subsequently, the insertion of benzyl isocyanide into the Pd–nitrene species 4b, 5b, and 7b (Scheme 3) yields a common intermediate (C) (Figure 8 and Supporting Information, Figures S224, S225, S228, S229, S232, and S233), while 6b generates the intermediate 6c (Supporting Information, Scheme S1 and Figures S236 and S237), and both C and 6c have also been observed by mass spectrometry. Finally, a reductive elimination step regenerates the Pd(0) species 4a, 5a, and 7a (Scheme 3) and 6a (Supporting Information, Scheme S1) along with the elimination of the free carbodiimide product (8).
Scheme 3. Proposed Mechanism Showing the Intermediates 4a,b, 5a,b, 6a,b, and C for the Azide–Isocyanide Coupling as Catalyzed by Phenothiazine-Derived Mesoionic Singlet Carbene Palladium (4, 5, 7) Complexes for the Representative Substrates, Namely Phenyl Azide and Benzyl Isocyanide.

Figure 6.

Expanded ESI-MS data consistent with the proposed Pd (0) species [1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]Pd(NC5H5) (4a), [M-H]− detected in the reaction mixture of K2CO3, 3 mmol and 1 mol % of catalyst (4) in 5.0 mL of toluene heated at 50 °C for 30 min [(a) experimental and (b) simulated pattern of ESI-MS data].
Figure 7.

Expanded ESI-MS data consistent with the proposed Pd bound nitrene species [1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]Pd(=NC6H5)(NC5H5) (4b), [M + H]+ detected in the reaction mixture of phenyl azide, 1 mmol of K2CO3, 3 mmol, 1 mol % of catalyst (4), 5.0 mL of toluene at 50 °C for 1 h [(a) experimental and (b) simulated pattern of ESI-MS data].
Figure 8.

Expanded ESI-MS data consistent with the proposed species [1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]Pd[η2-(Ph)N=C(NCH2Ph)] (C), [M]+ detected in the reaction mixture of phenyl azide, 1 mmol of benzyl isocyanide, 1.2 mmol of K2CO3, 3 mmol, 1 mol % of catalyst (4), 5.0 mL of toluene at 50 °C for 1.5 h [(a) experimental and (b) simulated pattern of ESI-MS data].
In this regard, it is noteworthy that ESI-MS studies59 have been routinely used for screening short-lived, low concentration intermediate species of several homogeneous metal-catalyzed transformations.60 However, since the information about the charged state of a metal, if obtained from the ESI-MS technique, remains inconclusive,61 and so such efforts demands further validation by other experimental techniques. In the present study such validation regarding the oxidation state of the metal center has been obtained using X-ray photoelectron spectroscopy as discussed below.
Additional corroboration of the proposed mechanism came from the X-ray photoelectron spectroscopy studies. In particular, the X-ray photoelectron spectroscopy (XPS) analysis of complexes 4–7 showed the presence of palladium, iodine, nitrogen, sulfur, and carbon in the precatalysts. As expected for the molecular complexes, the binding energies of Pd confirm its +2 oxidation state.62 A comparative XPS analysis of complexes 4–7 after heating in toluene at 100 °C for 24 h showed that the characteristic core level peaks of Pd 3d, I 3d, N 1s, S 2p, and C 1s are at the same binding energies as for the molecular complexes (Figure 9 and see Supporting Information, Table S13 and Figures S238–S246). The 3d core-level palladium of complex 4 was fitted with two main peaks, the peak for Pd 3d5/2 at 341.7 eV and for Pd 3d3/2 at 336.4 eV, which were assignable to the Pd(II) oxidation state.62,63 To characterize the intermediate species generated during the azide–isocyanide coupling reaction X-ray photoelectron spectroscopy (XPS) study was inspected of the reaction mixture using phenyl azide, benzyl isocyanide and complex 4. Two different new peaks at 335.5 and 340.9 eV, attributed to Pd 3d5/2 and Pd 3d3/2 peaks of Pd(0) species, appeared in the analysis.62,63 These peaks confirmed that both Pd(II) and Pd(0) species were involved during the catalytic cycle.
Figure 9.

XPS analysis of the 3d core level peaks of level of Pd 3d: Pd(II) (black), Pd(0) (pink), (a) precatalyst 4, and (b) precatalyst 4 after treatment with PhN3, PhCH2NC, and K2CO3 for 12 h in 5 mL of toluene at 50 °C.
As part of our ongoing efforts to synthesize bioactive heterocycles in a tandem fashion,21,22,24 the azide–isocyanide cross-coupling was extended to accessing benzoxazole motifs from the reaction of o-functionalized aryl azides with different isocyanides. Specifically, the representative catalyst (4) successfully catalyzed the coupling and the cyclization sequence steps producing the 2-aminobenzoxazole derivatives (18–22) from the reaction of 2-azido alcohol and R–NC [R = Cy (18), 2,6-Me2C6H3 (19), t-Bu (20), 2,4,6-Me3C6H2 (21), CH2C6H5 (22)] under optimal catalysis conditions in moderate to excellent yields of 54–89% (Table 3). It is needless to mention that the benzoxazole rings are important for their antimicrobial, anticonvulsant, anti-inflammatory and anticancer properties and are also studied for their DNA topoisomerase inhibitor and cholesterol ester transfer protein (CETP) inhibitor activities.16,64
A similar coupling/cyclization sequence were extended for synthesizing various bioactive 2-substituted benzimidazoles.65 In particular, the representative precatalyst (4) coupled the 2-halidophenyl azide with benzyl and cyclohexyl isocyanides yielding the corresponding o-haloarylcarbodiimides, followed by a cascade cyclization process in the presence of another Cu(I) catalyst in one pot and without the need for isolation of the intermediate to give the N-substituted 2-heterobenzimidazoles derivatives (23–27) in good yields using different N-heterocycles as nucleophiles (Table 4).
Table 4. Selected Results for a Simple Practical Synthesis of a Variety of Benzimidazole Derivatives Using Azide–Isocyanide Coupling Catalyzed by a Phenothiazine-Derived Mesoionic Singlet Carbene Palladium (4) Complexa,b.


Reaction conditions: azide (0.369 mmol), isocyanide (0.443 mmol), complex 4 (0.003 mmol, 1 mol %), and K2CO3 (1.10 mmol) in 5.0 mL of toluene at 50 °C for 12 h. Reaction mixture was cooled to room temperature and then CuI (0.100 mmol, 10 mol %), 1,10-phenanthroline (0.200 mmol, 20 mol %), Cs2CO3 (2.00 mmol), and N-heterocycle (1.10 mmol) were added to it and heated at 80 °C for 20 h.
Isolated yields (%).
Conclusion
In conclusion, several late transition metal complexes of palladium (4–7) were stabilized over a new class of phenothiazine derived mesoionic 1,2,3-triazole based carbene (MIC) ligand (3), thereby highlighting the versatility of this kind of MIC ligand for synthesizing different metal complexes. Significantly enough, the mixed carbene/isocyanide complex (5) represents the only structurally characterized example known until date of a (MIC)PdI2(CN–R) type complex, supported over a mesoionic 1,2,3-triazole derived carbene. The palladium (4–7) complexes efficiently catalyzed the azide–isocyanide coupling reaction yielding various symmetrical and unsymmetrical carbodiimides (8–17). More importantly, the (MIC)PdI2(NC5H5) type complex (4) beats out all other palladium complexes (5–7) in terms of catalysis performance, while the PPh3 bound cis-(MIC)Pd(PPh3)I2 type complex (7) was found to be the least active one. The remaining two complexes, i.e., the mixed carbene/isocyanide (MIC)PdI2(CN–R) type complex (5) and the trans–(MIC)2PdI2 type complex (6) exhibited comparable catalytic activities. The mechanistic study performed on two representative substrates, i.e. the phenyl azide and benzyl isocyanide revealed that the catalysis proceeds through the formation of palladium(0) species of the type (MIC)Pd(L) [L = NC5H5, MesNC, MIC, PPh3] and the palldium bound nitrene species (MIC)]Pd(=NC6H5)(L) [L = NC5H5, MesNC, PPh3, MIC], and of which the following species (4a, 6a, 7a, 4b, and 5b) have been characterized by mass spectrometry. The scope of the azide–isocyanide coupling was successfully extended by demonstrating its application in the formation of different derivatives of two bioactive heteroannular ring systems namely, 2-aminobenzoxazole (18–22) and the N-substituted 2-heterobenzimidazole (23–27). As the 4–7 complexes represent a rare class of well-defined palladium based molecular catalyst for the azide–isocyanide coupling reaction yielding various carbodiimide compounds, the study will provide further impetus to the development of the area.
Experimental Section
General Procedures
All manipulations were carried out using standard Schlenk techniques. Solvents were purified and degassed by standard procedures. Benzyl bromide, sodium azide, and methyl iodide were purchased from Spectrochem Chemicals and phenothiazine and propargyl bromide were purchased from Sigma-Aldrich Chemicals and used without any further purification. 1H NMR and 13C{1H} NMR spectra were recorded on Bruker 400 MHz and Bruker 500 MHz NMR spectrometer. 1H NMR peaks are labeled as singlet (s), doublet (d), triplet (t), quartet (q), doublet of doublet (dd), doublet of triplets (dt), triplets of doublets (td), doublet of triplets of triplets (dtd), and multiplet (m). Variable temperature dependent 1H NMR for complexes 4–6 were recorded on a JEOL ECZR Series 600 MHz NMR spectrometer. Variable Temperature dependent 1H NMR and spin saturation transfer (SST) 1H NMR for complex 7 were recorded on a Bruker 400 MHz spectrometer. The 10-(prop-2-yn-1-yl)-10H-phenothiazine was synthesized by modification of procedures reported in literature.66 The benchmark PEPPSI complex [trans-[1,3-(2,6-(i-Pr)2C6H3)2-imidazol-2-ylidene]PdCl2(NC5H5),] was made according to the literature procedure.56 High-resolution mass spectrometry measurements were done on a Micromass Q–Tof spectrometer and a Bruker maxis impact spectrometer. Infrared spectra were recorded on a PerkinElmer Spectrum One FT-IR spectrometer. Elemental Analysis was carried out on Thermo Quest FLASH 1112 SERIES (CHNS) Elemental Analyzer. X-ray photoelectron spectra were acquired on ULVAC-PHI/PHI 5000 VersaProbe-II spectrometer with a monochromatic Al Kα X-ray source (1486.6 eV) using a pass energy of 58.7 eV. X-ray diffraction data were collected for compounds (4–7) on a Bruker D8 QUEST single crystal diffractometer with Mo Ka (k = 0.71073 Å) radiation. The Bruker D8 QUEST single-crystal X-ray diffractometer were equipped with an Oxford liquid nitrogen cryostream. Crystals were mounted on a nylon loop with paraffin oil. The structures were solved via direct methods using SHELXT and refined via the full matrix least-squares method with SHELXL-2018/3, refining on F2.67 Crystal data collection and refinement parameters were summarized in Supporting Information, Table S1. CCDC-2041824 (for 4), CCDC-2060553 (for 5), CCDC-2123529 (for 6), and CCDC-2069000 (for 7), contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data center via www.ccdc.cam.ac.uk/data_request/cif.
Synthesis of 10-(Prop-2-yn-1-yl)-10H-phenothiazine (1)66
A mixture of phenothiazine (5.00 g, 25.0 mmol) and Na2CO3 (3.98 g, 37.5 mmol) in ca. 50 mL of anhydrous toluene was stirred at room temperature for 30 min, followed by the dropwise addition of propargyl bromide (80 wt % in toluene, 5.57 g, 37.5 mmol). The reaction mixture was refluxed for 24 h, after which it was evaporated invacuo. The resultant residue was dissolved in ca. 100 mL of CH2Cl2, filtered and washed with water (ca. 2 × 50 mL). The collected organic layer was dried over anhydrous Na2SO4 and evaporated invacuo. The crude product was further purified by column chromatography using silica gel as the stationary phase and by elution with petroleum ether to obtain a white solid (1) (2.12 g, 35%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.22 (dd, 2H, 3JHH = 8 Hz, 4JHH = 1 Hz, C12H8NS), 7.18 (td, 2H, 3JHH = 7 Hz, 4JHH = 1 Hz, C12H8NS), 7.13 (dd, 2H, 3JHH = 7 Hz, 4JHH = 1 Hz, C12H8NS), 6.95 (td, 2H, 3JHH = 7 Hz, 4JHH = 1 Hz, C12H8NS), 4.52 (d, 2H, 4JHH = 2 Hz, NCH2C≡CH), 2.46 (t, 1H, 4JHH = 2 Hz, NCH2C≡CH). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 144.1 (C12H8NS), 127.5 (C12H8NS), 127.1 (C12H8NS), 123.4 (C12H8NS), 123.0 (C12H8NS), 114.8 (C12H8NS), 79.3 (NCH2C≡CH), 74.5 (NCH2C≡CH), 38.5 (NCH2CCH). HRMS (ESI): calcd for [C15H11NS + K]+, 276.0244; found, 276.0239.
Synthesis of 10-((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)-10H-phenothiazine (2)
10-(Prop-2-yn-1-yl)-10H-phenothiazine (1) (0.478 g, 2.01 mmol), benzyl azide (0.322 g, 2.41 mmol), CuSO4·5H2O (0.604 g, 2.41 mmol), and sodium ascorbate (0.479 g, 2.41 mmol) were mixed together in 10 mL of DMF and refluxed for 24 h. The reaction mixture was diluted with ca. 50 mL of CH2Cl2 and ca. 150 mL of water and stirred vigorously at room temperature for 30 min. Subsequent separation of the organic layer, followed by repeated washing with water (ca. 3 × 100 mL), drying over anhydrous Na2SO4, and finally evaporating the volatiles invacuo gave the crude product, which was further purified by column chromatography using silica gel as the stationary phase and by elution with CH2Cl2 to get a white solid (2) (0.485 g, 65%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.21–7.17 (m, 4H, C2HN3 and CH2C6H5), 7.01 (dd, 2H, 3JHH = 7 Hz, 4JHH = 1 Hz, C12H8NS), 7.00–6.99 (m, 2H, CH2C6H5), 6.95 (td, 2H, 3JHH = 7 Hz, 4JHH = 1 Hz, C12H8NS), 6.80 (td, 2H, 3JHH = 7 Hz, 4JHH = 1 Hz, C12H8NS), 6.67 (dd, 2H, 3JHH = 7 Hz, 4JHH = 1 Hz, C12H8NS), 5.36 (s, 2H, CH2C6H5), 5.11 (s, 2H, NCH2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 145.3 (C12H8NS), 144.2 (C2HN3), 134.6 (CH2C6H5), 129.0 (CH2C6H5), 128.5 (CH2C6H5), 127.5 (C12H8NS), 127.3 (CH2C6H5), 127.1 (C12H8NS), 124.0 (C12H8NS), 122.8 (C12H8NS), 122.5 (C2HN3), 115.3 (C12H8NS), 54.1 (NCH2), 45.0 (CH2C6H5). IR data (cm–1) KBr pellet: 3445 (m), 3121 (m), 3062 (m), 2946 (m), 1571 (m), 1495 (s), 1440 (s), 1357 (s), 1286 (s), 1251 (s), 1210 (s), 1122 (s), 1050 (s), 857 (s), 749 (s), 718 (s), 643 (s), 616 (s). HRMS (ESI): calcd for [C22H18N4S + H]+, 371.1325; found, 371.1323. Anal. Calcd for C22H18N4S: C, 71.33; H, 4.90; N, 15.12; S, 8.65. Found: C, 71.21; H, 4.58; N, 14.80; S, 8.93%.
Synthesis of 1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazolium Iodide (3)
10-((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)-10H-phenothiazine (2) (0.400 g, 1.07 mmol), and MeI (1.53 g, 10.8 mmol) were mixed in 10 mL of CH3CN and refluxed for 24 h. The reaction mixture was then cooled to room temperature and all the volatiles were evaporated in vaccuo. The crude product thus obtained was further purified through column chromatography using silica gel as the stationary phase and by elution with CHCl3/MeOH (95:5 v/v) mixture to get the product as a yellow solid (3) (0.548 g, 99%). 1H NMR (CDCl3, 500 MHz, 25 °C): δ 9.52 (s, 1H, NCHC of C2HN3), 7.35–7.27 (m, 5H, C12H8NS and CH2C6H5), 7.14 (t, 2H, 3JHH = 7 Hz, CH2C6H5), 7.08 (d, 4H, 3JHH = 8 Hz, C12H8NS), 6.89 (t, 2H, 3JHH = 8 Hz, C12H8NS), 5.73 (s, 2H, CH2C6H5), 5.57 (s, 2H, NCH2), 4.26 (s, 3H, NCH3). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): δ 143.1 (C2HN3), 139.8 (C12H8NS), 131.6 (C2HN3), 131.1 (CH2C6H5), 129.8 (CH2C6H5), 129.4 (CH2C6H5), 129.1 (CH2C6H5), 128.2 (C12H8NS), 127.8 (C12H8NS), 125.7 (C12H8NS), 124.0 (C12H8NS), 116.7 (C12H8NS), 57.5 (NCH2), 41.8 (CH2C6H5), 39.6 (NCH3). IR data (cm–1) KBr pellet: 3433 (m), 3061 (w), 2924 (w), 2851 (w), 1589 (s), 1572 (w), 1485 (w), 1461 (s), 1352 (m), 1320 (m), 1285 (m), 1254 (m), 1224 (m), 1160 (w), 1079 (w), 933 (m), 854 (m), 754 (s), 643 (s), 572 (w), 512 (w), 473 (w). HRMS (ESI): calcd for [C23H21N4S]+, 385.1481; found, 385.1482. Anal. Calcd for C23H21N4IS: C, 53.91; H, 4.13; N, 10.93; S, 6.26. Found: C, 53.94; H, 4.42; N, 10.54; S, 6.62%.
Synthesis of trans-[1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]PdI2(NC5H5) (4)
A mixture of 1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazolium iodide (3) (0.250 g, 0.487 mmol), PdCl2 (0.086 g, 0.487 mmol), K2CO3 (0.337 g, 2.43 mmol), and NaI (0.365 g, 2.43 mmol) was refluxed in pyridine (5 mL, 63.0 mmol) for 16 h. The reaction mixture was cooled to room temperature, diluted with CHCl3 (ca. 50 mL), filtered and subsequently washed with saturated aqueous CuSO4 solution (ca. 3 × 50 mL) and water (ca. 100 mL). The organic layer was separated and dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated under vacuum to give a sticky, brown residue. The residue thus obtained was further triturated using hexane and EtOAc to give the product (4) as a yellow solid (0.381 g, 95%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 9.05 (d, 2H, 3JHH = 5 Hz, NC5H5), 7.75 (t, 1H, 3JHH = 7 Hz, NC5H5), 7.63 (dd, 2H, 3JHH = 7 Hz, 4JHH = 2 Hz, C12H8NS), 7.44–7.39 (m, 3H, CH2C6H5), 7.35 (t, 2H, 3JHH = 7 Hz, CH2C6H5), 7.31–7.29 (m, 4H, C12H8NS and NC5H5), 7.22 (d, 2H, 3JHH = 7 Hz, 4JHH = 2 Hz, C12H8NS), 7.03–6.99 (m, 2H, C12H8NS), 5.96 (s, 2H, CH2C6H5), 5.54 (s, 2H, NCH2), 4.00 (s, 3H, NCH3). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 153.7 (NC5H5), 143.6 (C12H8NS), 139.0 (Pd-C), 137.3 (NC5H5), 134.8 (CH2C6H5), 133.3 (C2N3), 129.8 (CH2C6H5), 128.6 (CH2C6H5), 128.5 (CH2C6H5), 128.2 (C12H8NS), 127.4 (C12H8NS), 125.0 (C12H8NS), 124.2 (C12H8NS), 123.4 (NC5H5), 117.0 (C12H8NS), 59.5 (NCH2), 45.3 (CH2C6H5), 37.8 (NCH3). IR data (cm–1) KBr pellet: 3439 (s), 3031 (w), 2954 (w), 2922 (w), 2851 (w), 2602 (w), 1732 (w), 1602 (w), 1457 (s), 1446 (s), 1364 (m), 1325 (m), 1258 (m), 1225 (m), 1213 (m), 1153 (w), 1129 (w), 1105 (w), 1070 (w), 956 (s), 865 (m), 753 (s), 618 (w), 580 (w), 498 (w). HRMS (ESI): calcd for [C28H25N5I2PdS–C5H5N–I]+, 616.9483; found, 616.9487. Anal. Calcd for C28H25N5I2PdS: C, 40.82; H, 3.06; N, 8.50; S, 3.89. Found: C, 40.59; H, 3.06; N, 7.95; S, 3.30%.
Synthesis of trans-[1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]PdI2(CN–Mes) (5)
A mixture of [1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]PdI2(NC5H5) (4) (0.340 g, 0.413 mmol) and MesNC (0.090 g, 0.619 mmol) was dissolved in a mixed solvent medium of dry toluene (ca. 5 mL) and dry THF (ca. 5 mL) and stirred at room temperature for 24 h. The solvent was evaporated and the crude product thus obtained was further purified by column chromatography using silica gel as stationary phase and by elution with petroleum ether/ethyl acetate (90:10 v/v) mixture to get a yellow solid (5) (0.161 g, 50%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.56 (d, 2H, 3JHH = 7 Hz, C12H8NS), 7.39–7.35 (m, 3H, CH2C6H5), 7.28 (t, 2H, 3JHH = 7 Hz, CH2C6H5), 7.22 (t, 4H, 3JHH = 7 Hz, C12H8NS), 7.00 (t, 2H, 3JHH = 7 Hz, C12H8NS), 6.93 (s, 2H, 2,4,6-(CH3)3C6H2), 5.85 (s, 2H, CH2C6H5), 5.45 (s, 2H, NCH2), 4.01 (s, 3H, NCH3), 2.55 (s, 6H, 2,4,6-(CH3)3C6H2), 2.32 (s, 3H, 2,4,6-(CH3)3C6H2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 147.2 (Pd-C), 143.7 (C12H8NS and 2,4,6-(CH3)3C6H2), 140.2 (C2N3), 139.8 (2,4,6-(CH3)3C6H2), 138.7 (NC), 136.2 (2,4,6-(CH3)3C6H2), 133.6 (CH2C6H5), 129.8 (CH2C6H5), 128.8 (CH2C6H5), 128.7 (CH2C6H5), 128.4 (C12H8NS), 127.6 (C12H8NS), 125.3 (C12H8NS), 123.6 (C12H8NS), 117.3 (C12H8NS), 59.6 (NCH2), 45.0 (CH2C6H5), 37.8 (NCH3), 21.3 (2,4,6-(CH3)3C6H2), 19.0 (2,4,6-(CH3)3C6H2). IR data (cm–1) KBr pellet: 3854(w), 3747 (w), 3444 (m), 3058 (w), 3031 (w), 2950 (w), 2919 (w), 2359 (w), 2188 (s), 1604 (w), 1571 (w), 1455 (s), 1371 (m), 1326 (m), 1284 (w), 1256 (w), 1224 (m), 1140 (w), 1104 (w), 1082 (w), 1038 (w), 933 (w), 889 (w), 837 (w), 757 (s), 708 (w), 645 (w), 597 (w), 524 (w). HRMS (ESI): calcd for [C33H31N5I2PdS–I]+, 762.0374; found, 762.0383. Anal. Calcd for C33H31N5I2PdS: C, 44.54; H, 3.51; N, 7.87; S, 3.60. Found: C, 45.27; H, 3.57; N, 7.49; S, 2.86%.
Synthesis of trans-[1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]2PdI2 (6)
1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazolium iodide (3) (0.256 g, 0.500 mmol) and Pd(OAc)2 (0.056 g, 0.250 mmol) were dissolved in ca. 15 mL of 1,4-dioxane and then refluxed for 24 h. The resulting reaction mixture was filtered through Celite and further purified by column chromatography with silica gel as the stationary phase and by elution with petroleum ether/ethyl acetate (80:20 v/v) mixture to get the product as a yellow solid (6) (0.261 g, 93%). Both the 1H NMR and 13C{1H} NMR spectra showed the presence of two isomers, trans-anti and trans-syn, in ca. 1.7:1 ratio, respectively. 1H NMR (CDCl3, 400 MHz, 25 °C) (major): δ 7.46 (d, 4H, 3JHH = 7 Hz, C12H8NS), 7.35–7.31 (m, 2H, CH2C6H5), 7.21–7.13 (m, 5H, C12H8NS and CH2C6H5), 7.01 (t, 2H, 3JHH = 8 Hz, C12H8NS), 6.04 (s, 2H, CH2C6H5), 5.27 (s, 2H, NCH2), 4.03 (s, 2H, NCH3). (Minor) δ 7.39 (d, 4H, 3JHH = 7 Hz, C12H8NS), 7.31–7.29 (m, 2H, CH2C6H5), 7.26–7.21 (m, 5H, C12H8NS and CH2C6H5), 6.97 (t, 2H, 3JHH = 8 Hz, C12H8NS), 5.85 (s, 2H, CH2C6H5), 5.64 (s, 2H, NCH2), 4.01 (s, 2H, NCH3). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): (major) δ 156.4 (Pd-C), 143.7 (C12H8NS), 140.5 (C2N3), 134.4 (CH2C6H5), 128.9 (CH2C6H5), 128.7 (2 CH2C6H5), 128.3 (C12H8NS), 127.5 (C12H8NS), 125.0 (C12H8NS), 123.4 (C12H8NS), 117.5 (C12H8NS), 58.7 (NCH2), 45.3 (CH2C6H5), 37.4 (NCH3). (Minor) δ 156.7 (Pd-C), 143.9 (C12H8NS), 140.1 (C2N3), 134.5 (CH2C6H5), 129.1 (CH2C6H5), 128.6 (2 CH2C6H5), 128.3 (C12H8NS), 127.7 (C12H8NS), 125.1 (C12H8NS), 123.4 (C12H8NS), 117.2 (C12H8NS), 59.0 (NCH2), 45.5 (CH2C6H5), 37.5 (NCH3). IR data (cm–1) KBr pellet: 3451(m), 3058 (m), 2927 (m), 2851 (w), 2728 (w), 2581 (w), 1943 (w), 1738 (w), 1591 (m), 1571 (m), 1484 (m), 1453 (s), 1369 (m), 1330 (m), 1286 (m), 1257 (w), 1224 (m), 1153 (w), 1105 (w), 1072 (w), 1038 (w), 930 (w), 844 (w), 753 (s), 733 (s), 614 (w), 526 (w), 500 (w). HRMS (ESI): calcd for [C46H40I2N8PdS2–I]+, 1001.0891; found, 1001.0906. Anal. Calcd for C46H40I2N8PdS2: C, 48.93; H, 3.57; N, 9.92; S, 5.68. Found: C, 48.84; H, 3.20; N, 9.62; S, 5.18%.
Synthesis of cis-[1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]Pd(PPh3)I2 (7)
A mixture of [1-CH2Ph-3-Me-4-(CH2N(C6H4)2S)-1,2,3-triazol-5-ylidene]PdI2(NC5H5) (4) (0.300 g, 0.367 mmol) and PPh3 (0.114 g, 0.437 mmol) was dissolved in dry CH2Cl2 (ca. 15 mL) and stirred at room temperature for 16 h. The solvent was evaporated and the crude product thus obtained was further purified by column chromatography using silica gel as stationary phase and by elution with petroleum ether/ethyl acetate (90:10 v/v) mixture to get a yellow solid (7) (0.293 g, 80%). Both the 1H NMR and 13C{1H} NMR spectra showed the presence of two isomers, cis and trans in ca.3.7:1 ratio. 1H NMR (DMSO-d6, 400 MHz, 25 °C) (major): δ 7.63–7.48 (m, 15H, P(C6H5)3), 7.33 (d, 2H, 3JHH = 7 Hz, CH2C6H5), 7.28–7.19 (m, 7H, C12H8NS and CH2C6H5), 7.04 (t, 4H, 3JHH = 7 Hz, C12H8NS), 5.94 (d, 1H, 2JHH = 14 Hz, CH2C6H5), 5.40 (d, 1H, 2JHH = 15 Hz, NCH2), 5.07 (d, 1H, 2JHH = 14 Hz, CH2C6H5), 4.19 (d, 1H, 2JHH = 15 Hz, NCH2), 3.71 (s, 3H, NCH3). (minor): δ 7.63–7.48 (m, 15H, P(C6H5)3), 7.35 (d, 2H, 3JHH = 7 Hz, CH2C6H5), 7.28–7.19 (m, 7H, C12H8NS and CH2C6H5), 7.04 (t, 4H, 3JHH = 7 Hz, C12H8NS), 5.86 (s, 2H, CH2C6H5), 5.43 (s, 2H, NCH2), 4.05 (d, 3H, 4JHH = 6 Hz, NCH3). 13C{1H} NMR (DMSO-d6, 100 MHz, 25 °C) (major): δ 155.1 (Pd-C), 144.2 (C2N3), 143.7 (C12H8NS), 137.5 (P(C6H5)3), 134.8 (P(C6H5)3), 133.4 (P(C6H5)3), 131.5 (P(C6H5)3), 131.1 (CH2C6H5), 130.1 (CH2C6H5), 129.2 (CH2C6H5), 128.9 (CH2C6H5), 128.4 (C12H8NS), 127.9 (C12H8NS), 124.8 (C12H8NS), 124.1 (C12H8NS), 117.9 (C12H8NS), 58.6 (NCH2), 43.1 (CH2C6H5), 38.5 (NCH3). (Minor): δ 155.1 (Pd-C), 144.1 (C2N3), 143.6 (C12H8NS), 137.5 (P(C6H5)3), 135.2 (P(C6H5)3), 132.5 (P(C6H5)3), 132.0 (P(C6H5)3), 130.6 (CH2C6H5), 129.6 (CH2C6H5), 129.1 (CH2C6H5), 128.8 (CH2C6H5), 128.3 (C12H8NS), 127.9 (C12H8NS), 124.9 (C12H8NS), 123.8 (C12H8NS), 117.6 (C12H8NS), 59.1 (NCH2), 44.6 (CH2C6H5), 38.0 (NCH3). 31P{1H} NMR (CDCl3, 162 MHz, 25 °C) (Major): δ 25.0 (Pd–P). (Minor): δ 15.9 (Pd–P) IR data (cm–1) KBr pellet: 3450(s), 3052 (w), 2967 (w), 2925 (w), 1633 (m), 1480 (w), 1456 (s), 1435 (s), 1370 (w), 1314 (m), 1257 (m), 1223 (w), 1159 (w), 1093 (w), 1072 (w), 999 (w), 952 (w), 899 (w), 836 (w), 815 (w), 753 (s), 697 (s), 663 (w), 618 (w), 527 (m), 512 (m). HRMS (ESI): calcd for [C41H35I2N4PPdS–I]+, 879.0394; found, 879.0409. Anal. Calcd for C41H35I2N4PPdS: C, 48.90; H, 3.50; N, 5.56; S, 3.18. Found: C, 49.05; H, 3.66; N, 5.72; S; 3.30%.
General Procedure for the Synthesis of Carbodimides Using Azide–Isocyanide Cross-Coupling as Catalyzed by Phenothiazine-Derived Mesoionic Singlet Carbene Palladium (4–7) Complexes (Table 1, Entries 1–10)
In a typical catalysis run, a 20 mL reaction vial was charged with a mixture of palladium complex 4/5/6/7 (0.003 mmol, 1 mol %) and K2CO3 (1.10 mmol), and then PhN3 (0.369 mmol) and PhCH2NC (0.443 mmol) were added to it, followed by 5 mL of toluene, and the reaction mixture was heated at 50 °C for 12 h. Then it was cooled to room temperature, filtered, and subsequently purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS, and elemental analysis data.
General Procedure for the Blank Experiment
In a typical blank catalysis run, a mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol) in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The reaction mixture was then cooled to room temperature, and the volatiles were evaporated. The crude product thus obtained was analyzed by 1H NMR spectroscopy and GC–MS (entry 1, Supporting Information, Table S12).
General Procedure for the Control Experiment
The following different control runs were performed.
-
(i)
A mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), and K2CO3 (1.10 mmol) in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The crude product thus obtained was analyzed by 1H and 13C{1H} NMR spectroscopy, and GC–MS (entry 2, Supporting Information, Table S12).
-
(ii)
A mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), ligand 3 (0.003 mmol, 1 mol %), and K2CO3 (1.10 mmol) was heated for 12 h at 50 °C. The crude product thus obtained was analyzed by 1H and 13C{1H} NMR spectroscopy, and GC–MS (entry 3, Supporting Information, Table S12).
-
(iii)
A mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), and [Pd2(dba)3] (1 mol %) in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The crude product thus obtained was purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS, and elemental analysis data (entry 4, Supporting Information, Table S12).
-
(iv)
A mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), [Pd2(dba)3] (1 mol %), and K2CO3 (1.10 mmol) in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The crude product thus obtained was purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS, and elemental analysis data (entry 5, Supporting Information, Table S12).
-
(v)
A mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), [(CH3CN)2PdCl2] (1 mol %), and K2CO3 (1.10 mmol) in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The crude product thus obtained was purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS, and elemental analysis data (entry 6, Supporting Information, Table S12).
-
(vi)
A mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), [trans-[1,3-(2,6-(i-Pr)2C6H3)2-imidazol-2-ylidene]PdCl2(NC5H5)] (1 mol %), and K2CO3 (1.10 mmol) in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The crude product thus obtained was purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS, and elemental analysis data (entry 7, Supporting Information, Table S12).
-
(vii)
A mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), trans-[1-Ph-3-Me-4-CH2Ph-1,2,3-triazol-5-ylidene]PdI2(NC5H5) (0.003 mmol, 1 mol %), and K2CO3 (1.10 mmol) in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The crude product thus obtained was purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS, and elemental analysis data (entry 8, Supporting Information, Table S12).
-
(viii)
A mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), and complex 4 (0.003 mmol, 1 mol %) was heated for 12 h at 50 °C. The crude product thus obtained was analyzed by 1H and 13C{1H} NMR spectroscopy and GC–MS (entry 9, Supporting Information, Table S12).
-
(ix)
A mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), complex 4 (0.003 mmol, 1 mol %), and K2CO3 (1.10 mmol) was heated for 12 h at 50 °C. The crude product thus obtained was purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS and elemental analysis data (entry 10, Supporting Information, Table S12).
General Procedure for the Hg Drop Test
-
(i).
In a typical Hg drop catalysis run, a mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), and catalyst 4 (0.003 mmol, 1 mol %), K2CO3 (1.10 mmol) and excess Hg0 in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The reaction mixture was then cooled to room temperature, and the volatiles were evaporated. The crude product thus obtained was purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS and elemental analysis data (entry 11, Supporting Information, Table S12).
-
(ii).
In a typical Hg drop catalysis run, a mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), and catalyst 5 (0.003 mmol, 1 mol %), with K2CO3 (1.10 mmol) and excess Hg0 in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The reaction mixture was then cooled to room temperature, and the volatiles were evaporated. The crude product thus obtained was purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS and elemental analysis data (entry 12, Supporting Information, Table S12).
-
(iii).
In a typical Hg drop catalysis run, a mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), and catalyst 6 (0.003 mmol, 1 mol %), with K2CO3 (1.10 mmol) and excess Hg0 in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The reaction mixture was then cooled to room temperature, and the volatiles were evaporated. The crude product thus obtained was purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS and elemental analysis data (entry 13, Supporting Information, Table S12).
-
(iv).
In a typical Hg drop catalysis run, a mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), and catalyst 7 (0.003 mmol, 1 mol %), with K2CO3 (1.10 mmol) and excess Hg0 in toluene (ca. 5 mL) was heated for 12 h at 50 °C. The reaction mixture was then cooled to room temperature, and the volatiles were evaporated. The crude product thus obtained was purified by column chromatography using silica gel (neutralized by triethylamine) as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS and elemental analysis data (entry 14, Supporting Information, Table S12).
General Procedure for the Mass Experiment
In a typical mass experiment run, a mixture of catalyst 4–7 (0.01 mmol, 1 mol %) and K2CO3 (3 mmol) in 5.0 mL of toluene was heated at 50 °C for 30 min, and then an aliquot from the reaction mixture was analyzed by ESI-MS, where the Pd(0) species 4a (Figure 6 and Supporting Information, Figure S222), 6a (Supporting Information, Figures S234 and S235), and 7a (Supporting Information, Figures S230 and S231) were detected.
In a typical mass experiment run, a mixture of PhN3 (1 mmol), catalyst 4–7 (0.01 mmol, 1 mol %), and K2CO3 (3 mmol) in 5.0 mL of toluene was heated at 50 °C for 30 min, and then an aliquot from the reaction mixture was analyzed by ESI-MS, where the Pd bound nitrene species 4b (Figure 7 and Supporting Information, Figure S223) and 5b (Supporting Information, Figures S226 and S227) were detected.
In a typical mass experiment run, a mixture of PhN3 (1 mmol), PhCH2NC (1.2 mmol), catalyst 4–7 (0.01 mmol, 1 mol %), and K2CO3 (3 mmol) in 5.0 mL of toluene was heated at 50 °C for 1.5 h, and then an aliquot from the reaction mixture was analyzed by ESI-MS, where the species C (Figure 8 and Supporting Information, Figures S224, S225, S228, S229, S232, and S233) and 6c (Supporting Information, Figure S236 and S237) were detected.
General Procedure for the XPS Experiment
The following different XPS runs were performed.
-
(i)
Complex 4 (0.030 mmol) was heated for 24 h in 5 mL of toluene at 100 °C, and then the solvent was evaporated and dried. The crude product thus obtained was analyzed by XPS (Supporting Information, Figure S239).
-
(ii)
Complex 5 (0.030 mmol) was heated for 24 h in 5 mL of toluene at 100 °C, and then the solvent was evaporated and dried. The crude product thus obtained was analyzed by XPS (Supporting Information, Figure S241).
-
(iii)
Complex 6 (0.030 mmol) was heated for 24 h in 5 mL of toluene at 100 °C, and then the solvent was evaporated and dried. The crude product thus obtained was analyzed by XPS (Supporting Information, Figure S243).
-
(iv)
Complex 7 (0.030 mmol) was heated for 24 h in 5 mL of toluene at 100 °C, and then the solvent was evaporated and dried. The crude product thus obtained was analyzed by XPS (Supporting Information, Figure S245).
-
(v)
A mixture of PhN3 (0.369 mmol), PhCH2NC (0.443 mmol), complex 4 (0.030 mmol, 10 mol %), and K2CO3 (1.10 mmol) was heated for 12 h in 5 mL of toluene at 50 °C, and then the solvent was evaporated and dried. The crude product thus obtained was analyzed by XPS (Supporting Information, Figure S246).
Synthesis of N-((Benzylimino)methylene)aniline (8)68

Yields: 0.068 g, 88% (4), 0.060 g, 79% (5), 0.051 g, 66% (6), 0.036 g, 48% (7).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.42–7.40 (m, 4H, CH2C6H5), 7.36–7.33 (m, 1H, CH2C6H5), 7.28 (t, 2H, 3JHH = 7 Hz, C6H5), 7.13 (t, 1H, 3JHH = 7 Hz, C6H5), 7.03 (d, 2H, 3JHH = 7 Hz, C6H5), 4.60 (s, 2H, CH2C6H5). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 140.0 (C6H5), 137.8 (CH2C6H5), 137.3 (NCN), 129.3 (C6H5), 128.8 (C6H5), 127.8 (C6H5), 127.4 (CH2C6H5), 124.9 (CH2C6H5), 123.6 (CH2C6H5), 50.5 (CH2C6H5). GC–MS (ESI): m/z = 208 [M]+. Anal. Calcd for C14H12N2: C, 80.74; H, 5.81; N, 13.45. Found: C, 80.08; H, 5.74; N, 13.05%.
Synthesis of N-((Tolylimino)methylene)aniline (9)69
Yields: 0.072 g, 94% (4), 0.065 g, 85% (5), 0.055 g, 74% (6), 0.040 g, 52% (7).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.33 (t, 2H, 3JHH = 7 Hz, C6H5), 7.19 (d, 3H, 3JHH = 7 Hz, C6H5), 7.14 (d, 2H, 3JHH = 8 Hz, C6H4CH3), 7.09 (d, 2H, 3JHH = 8 Hz, C6H4CH3), 2.34 (s, 3H, C6H4CH3). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 138.7 (C6H5), 135.6 (NCN), 135.5 (C6H4CH3), 130.1 (C6H5), 129.5 (C6H5), 125.5 (C6H5), 124.2 (C6H4CH3), 124.1 (C6H4CH3), 123.9 (C6H4CH3), 21.0 (C6H4CH3). GC–MS (ESI): m/z = 208 [M]+. Anal. Calcd for C14H12N2: C, 80.74; H, 5.81; N, 13.45. Found: C, 80.19; H, 5.96; N, 13.54%.
Synthesis of N-((Xylylimino)methylene)aniline (10)14

Yields: 0.064 g, 78% (4), 0.051 g, 62% (5), 0.041 g, 50% (6), 0.019 g, 24% (7).
1H NMR (CDCl3, 500 MHz, 25 °C): δ 7.33 (t, 2H, 3JHH = 7 Hz, C6H5), 7.18 (d, 2H, 3JHH = 7 Hz, C6H5), 7.14 (t, 1H, 3JHH = 7 Hz, 2,6-(CH3)2C6H3), 7.05 (d, 2H, 3JHH = 7 Hz, 2,6-(CH3)2C6H3), 7.00 (t, 1H, 3JHH = 7 Hz, C6H5), 2.40 (s, 6H, 2,6-(CH3)2C6H3). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): δ 139.8 (C6H5), 134.9 (2,6-(CH3)2C6H3), 132.8 (2,6-(CH3)2C6H3), 131.5 (NCN), 129.5 (C6H5), 128.2 (C6H5), 125.1 (C6H5), 124.7 (2,6-(CH3)2C6H3), 123.7 (2,6-(CH3)2C6H3), 19.0 (2,6-(CH3)2C6H3). GC–MS (ESI): m/z = 222 [M]+. Anal. Calcd for C15H12N2: C, 81.05; H, 6.35; N, 12.60. Found: C, 81.71; H, 6.60; N, 12.37%.
Synthesis of N-((Mesityimino)methylene)aniline (11)70

Yields: 0.053 g, 61% (4), 0.046 g, 53% (5), 0.034 g, 39% (6), 0.014 g, 16% (7).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.32 (t, 2H, 3JHH = 7 Hz, C6H5), 7.18–7.11 (m, 3H, C6H5), 6.87 (s, 2H, 2,4,6-(CH3)3C6H2), 2.36 (s, 6H, 2,4,6-(CH3)3C6H2), 2.27 (s, 3H, 2,4,6-(CH3)3C6H2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 140.2 (C6H5), 134.9 (2,4,6-(CH3)3C6H2), 132.7 (2,4,6-(CH3)3C6H2), 132.1 (2,4,6-(CH3)3C6H2), 131.8 (NCN), 129.4 (C6H5), 128.9 (C6H5), 124.6 (C6H5), 123.6 (2,4,6-(CH3)3C6H2), 20.8 (2,4,6-(CH3)3C6H2), 18.9 (2,4,6-(CH3)3C6H2). GC–MS (ESI): m/z = 236 [M]+. Anal. Calcd for C16H16N2: C, 81.32; H, 6.82; N, 11.85. Found: C, 81.31; H, 6.74; N, 11.68%.
Synthesis of N-((Cyclohexyimino)methylene)aniline (12)71
Yields: 0.069 g, 94% (4), 0.068 g, 92% (5), 0. 056 g, 76% (6), 0.023 g, 32% (7).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.31 (t, 2H, 3JHH = 7 Hz, C6H5), 7.13–7.10 (m, 3H, C6H5), 3.53–3.46 (m, 1H, C6H11), 2.06–2.01 (m, 2H, C6H11), 1.83–1.77 (m, 2H, C6H11), 1.66–1.47 (m, 3H, C6H11), 1.42–1.28 (m, 3H, C6H11). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 140.9 (C6H5), 136.2 (NCN), 129.3 (C6H5), 124.5 (C6H5), 123.3 (C6H5), 56.6 (C6H11), 34.9 (C6H11), 25.3 (C6H11), 24.3 (C6H11). GC–MS (ESI): m/z = 200 [M]+. Anal. Calcd for C13H16N2: C, 77.96; H, 8.05; N, 13.99. Found: C, 78.73; H, 8.28; N, 13.20%.
Synthesis of N-((Cyclohexylimino)methylene)cyclohexylamine (13)71

Yields: 0.064 g, 84% (4), 0.051 g, 67% (5), 0.041 g, 54% (6), 0.028 g, 37% (7).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 3.20–3.14 (m, 1H, C6H11), 1.91–1.89 (m, 2H, C6H11), 1.73–1.70 (m, 2H, C6H11), 1.57–1.53 (m, 1H, C6H11), 1.35–1.16 (m, 5H, C6H11). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): δ 139.8 (NCN), 55.7 (C6H11), 34.9 (C6H11), 25.4 (C6H11), 24.7 (C6H11). GC–MS (ESI): m/z = 206 [M]+. Anal. Calcd for C13H22N2: C, 75.68; H, 10.75; N, 13.58. Found: C, 76.19; H, 10.75; N, 13.56%.
Synthesis of N-((Tolylimino)methylene)cyclohexylamine (14)72

Yields: 0.054 g, 66% (4), 0.043 g, 52% (5), 0. 032 g, 39% (6), 0.023 g, 28% (7).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.37–7.36 (m, 4H, CH2C6H5), 7.32–7.29 (m, 1H, CH2C6H5), 7.04 (d, 2H, 3JHH = 8 Hz, C6H4CH3), 6.88 (d, 2H, 3JHH = 8 Hz, C6H4CH3), 4.55 (s, 2H, CH2C6H5), 2.29 (s, 3H, C6H4CH3). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 137.9 (C6H4CH3), 137.8 ((NCN), 137.0 CH2C6H5), 134.6 (C6H4CH3), 129.9 (C6H4CH3), 128.7 (C6H4CH3), 127.7 (CH2C6H5), 127.4 (CH2C6H5), 123.4 (CH2C6H5), 50.5 (CH2C6H5), 20.9 (C6H4CH3). GC–MS (ESI): m/z = 222 [M]+. Anal. Calcd for C15H14N2: C, 81.05; H, 6.35; N, 12.60. Found: C, 79.80; H, 6.32; N, 12.20%.
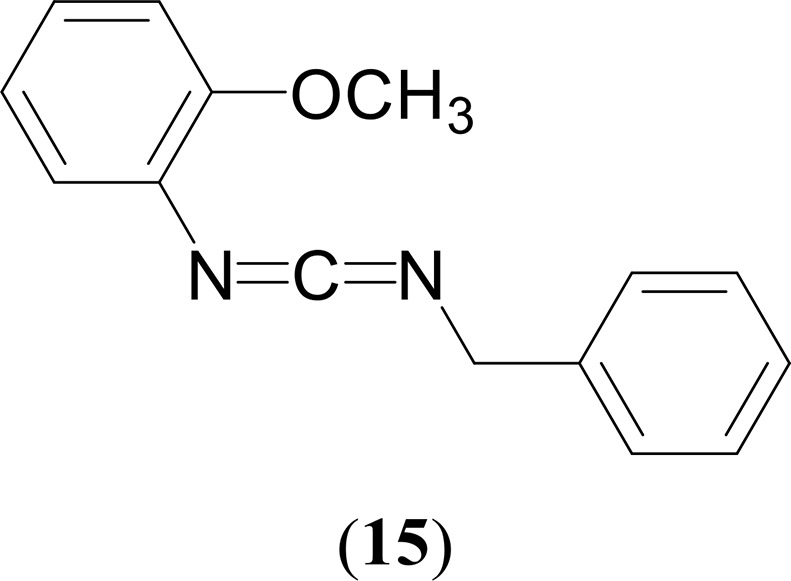
Synthesis of N-((Tolylimino)methylene)cyclohexylamine (15)
Yields: 0.049 g, 56% (4), 0.033 g, 38% (5), 0. 023 g, 26% (6), 0.010 g, 12% (7).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.41–7.34 (m, 4H, CH2C6H5), 7.31–7.28 (m, 1H, CH2C6H5), 7.06 (td, 1H, 3JHH = 8 Hz, 4JHH = 2 Hz, C6H4OCH3), 6.99 (d, 2H, 3JHH = 8 Hz, C6H4OCH3), 6.85 (t, 1H, 3JHH = 8 Hz, C6H4OCH3), 6.84 (d, 2H, 3JHH = 8 Hz, C6H4OCH3), 4.58 (s, 2H, CH2C6H5), 3.76 (s, 3H, C6H4OCH3). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 154.0 (C6H4OCH3), 138.1 (CH2C6H5), 128.6 (C6H4OCH3), 128.4 (NCN), 127.5 (CH2C6H5), 127.4 (2 C6H4OCH3), 125.6 (CH2C6H5), 124.7 (CH2C6H5), 120.9 (C6H4OCH3), 111.0 (C6H4OCH3), 55.8 (C6H4OCH3), 50.6 (CH2C6H5). GC–MS (ESI): m/z = 238 [M]+. Anal. Calcd for C15H14N2O: C, 75.61; H, 5.92; N, 11.76. Found: C, 75.26; H, 5.84; N, 11.45%.
Synthesis of N-((4-Fluorophenylimino)methylene)cyclohexylamine (16)

Yields: 0.082 g, 99% (4), 0.077 g, 93% (5), 0. 067 g, 80% (6), 0.050 g, 61% (7).
1H NMR (C6D6, 400 MHz, 25 °C): δ 7.05–7.04 (m, 3H, CH2C6H5), 7.03–6.94 (m, 2H, CH2C6H5), 6.75–6.71 (m, 2H, C6H4F), 6.60–6.56 (m, 2H, C6H4F), 4.00 (s, 2H, CH2C6H5). 13C{1H} NMR (C6D6, 100 MHz, 25 °C): δ 161.1 (d, 1JCF = 244 Hz, C6H4F), 137.9 (CH2C6H5), 137.1 (NCN), 132.0 (CH2C6H5), 131.4 (d, 4JCF = 4 Hz, C6H4F), 128.5 (CH2C6H5), 127.2 (CH2C6H5), 124.8 (d, 3JCF = 8 Hz, C6H4F), 115.9 (d, 2JCF = 23 Hz, C6H4F), 49.9 (CH2C6H5). 19F{1H} NMR (C6D6, 376 MHz, 25 °C): δ – 117.71 (C6H4F). GC–MS (ESI): m/z = 226 [M]+. Anal. Calcd for C14H11N2F: C, 74.32; H, 4.90; N, 12.38. Found: C, 75.31; H, 5.12; N, 11.74%.
Synthesis of N-((3,5-Bis(trifluoromethyl)phenylimino)methylene)cyclohexylamine (17)

Yields: 0.115 g, 91% (4), 0.095 g, 75% (5), 0. 082 g, 65% (6), 0.050 g, 44% (7).
1H NMR (C6D6, 400 MHz, 25 °C): δ 7.37 (s, 1H, C6H3(CF3)2), 7.08 (s, 2H, C6H3(CF3)2), 7.06–7.01 (m, 3H, CH2C6H5), 6.95 (d, 2H, 3JHH = 7 Hz, CH2C6H5), 3.86 (s, 2H, CH2C6H5). 13C{1H} NMR (C6D6, 100 MHz, 25 °C): δ 143.1 (C6H3(CF3)2), 137.3 (CH2C6H5), 134.6 (NCN), 132.4 (q, 2JCF = 33 Hz, C6H3(CF3)2), 131.7 (CH2C6H5), 128.8 (CH2C6H5), 127.1 (CH2C6H5), 123.4 (d, 4JCF = 3 Hz, C6H3(CF3)2), 123.1 (q, 1JCF = 273 Hz, C6H3(CF3)2), 117.5 (quin, 3JCF = 4 Hz, C6H3(CF3)2), 49.7 (CH2C6H5). 19F{1H} NMR (C6D6, 376 MHz, 25 °C): δ – 62.86 (C6H3(CF3)2). GC–MS (ESI): m/z = 344 [M]+. Anal. Calcd for C16H10N2F6: C, 55.82; H, 2.93; N, 8.14. Found: C, 55.54; H, 3.79; N, 8.98%.
General Procedure for the Synthesis of Benzoxazole Derivatives Using Azide and Isocyanide Cross-Coupling as Catalyzed by a Phenothiazine-Derived Mesoionic Singlet Carbene Palladium (4) Complex (Table 3, Entries 1–5)
In a typical catalysis run, a 20 mL reaction vial was charged with a mixture of palladium complex 4 (0.01 mmol, 1 mol %) and K2CO3 (3.00 mmol), and then the corresponding azide (1.00 mmol) and isocyanide (1.20 mmol) were added to it, followed by 5 mL of toluene, and the reaction mixture was heated at 50 °C for 12 h. The reaction mixture was cooled to room temperature, filtered, and subsequently purified by column chromatography using silica gel as a stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS, and elemental analysis data.
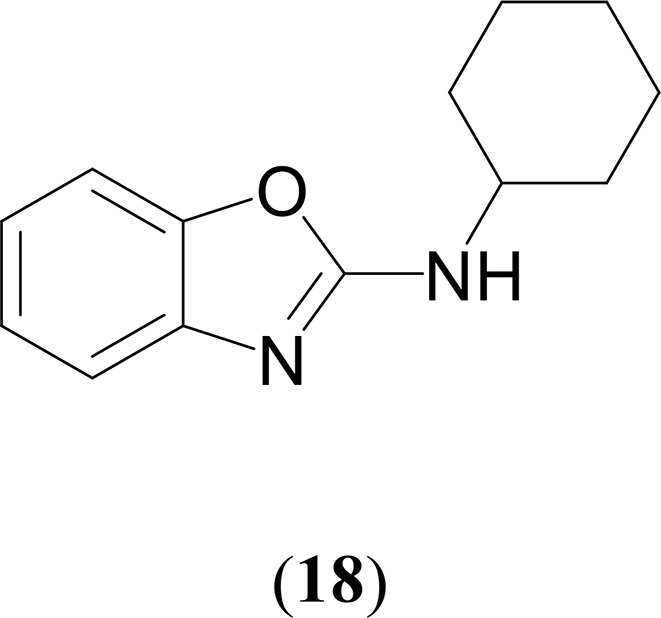
Synthesis of N-Cyclohexylbenzooxazol-2-amine (18)73
Yield: 0.181 g, 84% (4).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.35 (d, 1H, 3JHH = 8 Hz, C7H4NO), 7.22 (d, 1H, 3JHH = 8 Hz, C7H4NO), 7.14 (t, 1H, 3JHH = 8 Hz, C7H4NO), 7.00 (t, 1H, 3JHH = 8 Hz, C7H4NO), 5.08 (br, 1H, NH), 3.80–3.71 (m, 1H, C6H11), 2.14–2.11 (m, 2H, C6H11), 1.79–1.74 (m, 2H, C6H11), 1.67–1.62 (m, 1H, C6H11), 1.48–1.38 (m, 2H, C6H11), 1.34–1.25 (m, 3H, C6H11). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 161.4 (OCN of C7H4NO), 148.3 (C6H4 of C7H4NO), 143.1 (C6H4 of C7H4NO), 123.8 (C6H4 of C7H4NO), 120.6 (C6H4 of C7H4NO), 116.2 (C6H4 of C7H4NO), 108.6 (C6H4 of C7H4NO), 52.0 (C6H11), 33.4 (C6H11), 25.4 (C6H11), 24.7 (C6H11). GC–MS (ESI): m/z = 216 [M]+. Anal. Calcd for C13H16N2O: C, 72.19; H, 7.46; N, 12.95. Found: C, 72.34; H, 7.08; N, 12.87%.
Synthesis of N-(2,6-dimethylphenyl)lbenzooxazol-2-amine (19)74

Yield: 0.128 g, 54% (4).
1H NMR (DMSO-d6, 400 MHz, 25 °C): δ 9.58 (br, 1H, NH), 7.39 (d, 1H, 3JHH = 8 Hz, C7H4NO), 7.22 (d, 1H, 3JHH = 8 Hz, C7H4NO), 7.17–7.10 (m, 4H, C7H4NO and 2,6-(CH3)2C6H3), 7.01 (t, 1H, 3JHH = 8 Hz, C7H4NO), 2.20 (s, 6H, 2,6-(CH3)2C6H3). 13C{1H} NMR (DMSO-d6, 100 MHz, 25 °C): δ 160.3 (OCN of C7H4NO), 148.0 (C6H4 of C7H4NO), 142.8 (C6H4 of C7H4NO), 135.6 (2,6-(CH3)2C6H3), 134.9 (2,6-(CH3)2C6H3), 128.2 (2,6-(CH3)2C6H3), 126.9 (2,6-(CH3)2C6H3), 123.8 (C6H4 of C7H4NO), 120.6 (C6H4 of C7H4NO), 115.7 (C6H4 of C7H4NO), 108.8 (C6H4 of C7H4NO), 17.9 (2,6-(CH3)2C6H3). GC–MS (ESI): m/z = 238 [M]+. Anal. Calcd for C15H14N2O: C, 75.61; H, 5.92; N, 11.76. Found: C, 75.28; H, 6.59; N, 11.43%
Synthesis of N-tert-Butylbenzooxazol-2-amine (20)74

Yield: 0.169 g, 89% (4).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.39 (d, 1H, 3JHH = 8 Hz, C7H4NO), 7.27 (d, 1H, 3JHH = 8 Hz, C7H4NO), 7.18 (t, 1H, 3JHH = 8 Hz, C7H4NO), 7.04 (t, 1H, 3JHH = 8 Hz, C7H4NO), 5.46 (br, 1H, NH), 1.52 (s, 9H, C(CH3)3). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 160.8 (OCN of C7H4NO), 148.0 (C6H4 of C7H4NO), 142.5 (C6H4 of C7H4NO), 123.8 (C6H4 of C7H4NO), 120.7 (C6H4 of C7H4NO), 116.2 (C6H4 of C7H4NO), 108.6 (C6H4 of C7H4NO), 52.1 (C(CH3)3), 29.2 (C(CH3)3). GC–MS (ESI): m/z = 190 [M]+. Anal. Calcd for C11H14N2O: C, 69.45; H, 7.42; N, 14.73. Found: C, 68.76; H, 6.54; N, 14.37%.
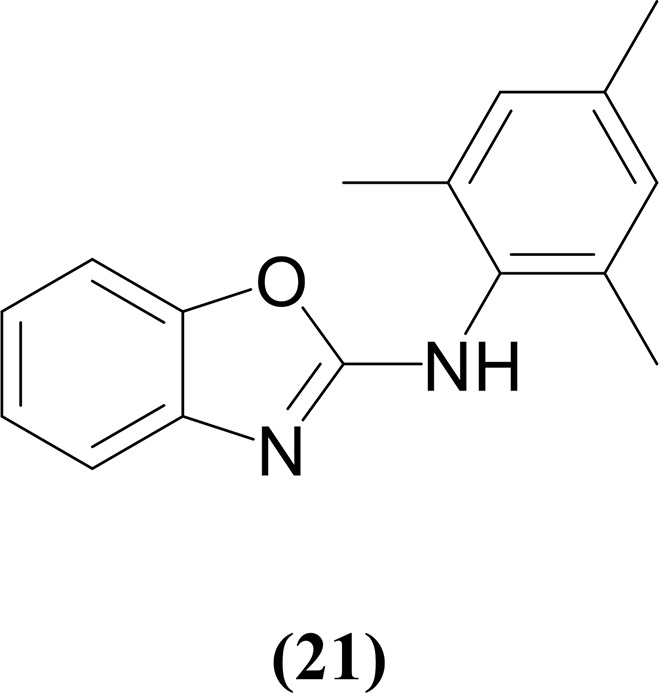
Synthesis of N-Mesiylbenzooxazol-2-amine (21)
Yield: 0.083 g, 33% (4).
1H NMR (DMSO-d6, 400 MHz, 25 °C): δ 9.03 (br, 1H, NH), 6.91 (s, 2H, 2,4,6-(CH3)3C6H2), 6.71 (dd, 1H, 3JHH = 8 Hz, C7H4NO), 6.64 (dd, 1H, 3JHH = 8 Hz, C7H4NO), 6.57 (td, 1H, 3JHH = 8 Hz, C7H4NO), 6.43 (td, 1H, 3JHH = 8 Hz, C7H4NO), 2.26 (s, 6H, 2,4,6-(CH3)3C6H2), 2.22 (s, 3H, 2,6-(CH3)3C6H2). 13C{1H} NMR (DMSO-d6, 100 MHz, 25 °C): δ 167.8 (OCN of C7H4NO), 144.5 (C6H4 of C7H4NO), 139.2 (C6H4 of C7H4NO), 136.9 (2,4,6-(CH3)3C6H2), 134.4 (2,4,6-(CH3)3C6H2), 128.8 2,4,6-(CH3)3C6H2), 123.7 (2,4,6-(CH3)3C6H2), 120.0 (C6H4 of C7H4NO), 117.0 (C6H4 of C7H4NO), 115.0 (C6H4 of C7H4NO), 114.8 (C6H4 of C7H4NO), 21.1 (2,4,6-(CH3)3C6H2), 18.6 (2,4,6-(CH3)3C6H2). GC–MS (ESI): m/z = 252 [M]+. Anal. Calcd for C16H16N2O: C, 76.16; H, 6.39; N, 11.10. Found: C, 76.47; H, 6.59; N, 10.20%.
Synthesis of N-Benzylbenzooxazol-2-amine (22)75

Yield: 0.162 g, 72% (4).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.41–7.24 (m, 6H, C7H4NO and CH2C6H5), 7.16 (t, 1H, 3JHH = 8 Hz, C7H4NO), 7.03 (t, 1H, 3JHH = 8 Hz, C7H4NO), 5.98 (br, 1H, NH), 4.67 (s, 9H, CH2C6H5). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 162.1 (OCN of C7H4NO), 148.5 (C6H4 of C7H4NO), 142.7 (C6H4 of C7H4NO), 137.7 (CH2C6H5), 128.8 (CH2C6H5), 127.8 (CH2C6H5), 127.6 (CH2C6H5), 124.0 (C6H4 of C7H4NO), 120.9 (C6H4 of C7H4NO), 116.3 (C6H4 of C7H4NO), 108.8 (C6H4 of C7H4NO), 47.0 (CH2C6H5). GC–MS (ESI): m/z = 224 [M]+. Anal. Calcd for C14H12N2O: C, 74.98; H, 5.39; N, 12.49. Found: C, 74.30; H, 5.68; N, 13.38%.
General Procedure for the Synthesis of Benzimidazole Derivatives Using Azide and Isocyanide Cross-Coupling as Catalyzed by a Phenothiazine-Derived Mesoionic Singlet Carbene Palladium (4) Complex (Table 4, Entries 1–5)
In a typical catalysis run, a 20 mL reaction vial was charged with a mixture of palladium complex 4 (0.01 mmol, 1 mol %) and K2CO3 (3.00 mmol), and then the corresponding azide (1.00 mmol) and isocyanide (1.20 mmol) were added to it, followed by 5 mL of toluene, and the reaction mixture was heated at 50 °C for 12 h. The reaction mixture was cooled to room temperature, CuI (0.100 mmol, 10 mol %), 1,10-phenanthroline (0.200 mmol, 20 mol %), Cs2CO3 (2.00 mmol), and N-heterocycle (1.1 mmol) were added to it and heated at 80 °C for 20 h. Then it was filtered, the solvent was evaporated, and the result was subsequently purified by column chromatography using silica gel as the stationary phase and by elution with a mixed medium of petroleum ether/EtOAc to give the desired product, as characterized by 1H and 13C{1H} NMR spectroscopy, GC–MS, and elemental analysis data.
Synthesis of 4-(1-Benzyl-1H-benzimidazol-2-yl)morpholine (23)76

Yield: 0.187 g, 64% (4).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.65 (d, 1H, 3JHH = 8 Hz, C7H4N2), 7.34–7.29 (m, 3H, CH2C6H5), 7.19 (td, 1H, 3JHH = 8 Hz, 4JHH = 1 Hz, C7H4N2), 7.14 (d, 2H, 3JHH = 7 Hz, CH2C6H5), 7.09 (td, 1H, 3JHH = 8 Hz, 4JHH = 1 Hz, C7H4N2), 7.02 (d, 1H, 3JHH = 7 Hz, C7H4N2), 5.23 (s, 2H, CH2C6H5), 3.79 (t, 4H, 3JHH = 5 Hz, NC4H8O), 3.24 (t, 4H, 3JHH = 5 Hz, NC4H8O). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 157.6 (NCN of C7H4N2), 141.3 (C6H4 of C7H4N2), 136.0 (C6H4 of C7H4N2), 135.3 (CH2C6H5), 128.9 (CH2C6H5), 127.6 (CH2C6H5), 125.9 (CH2C6H5), 122.0 (C6H4 of C7H4N2), 121.5 (C6H4 of C7H4N2), 118.1 (C6H4 of C7H4N2), 109.3 (C6H4 of C7H4N2), 66.4 (NC4H8O), 50.8 (NC4H8O), 47.4 (CH2C6H5). GC–MS (ESI): m/z = 293 [M]+. Anal. Calcd for C18H19N3O: C, 73.69; H, 6.53; N, 14.32. Found: C, 73.55; H, 6.02; N, 14.02%.
Synthesis of 1-Benzyl-2-(piperidin-1-yl)-1H-benzimidazole (24)77

Yield: 0.197 g, 68% (4).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.63 (d, 1H, 3JHH = 8 Hz, C7H4N2), 7.34–7.27 (m, 3H, CH2C6H5), 7.18 (d, 2H, 3JHH = 7 Hz, CH2C6H5), 7.17 (t, 1H, 3JHH = 8 Hz, C7H4N2), 7.06 (t, 1H, 3JHH = 8 Hz, C7H4N2), 6.98 (d, 1H, 3JHH = 8 Hz, C7H4N2), 5.20 (s, 2H, CH2C6H5), 3.20 (t, 4H, 3JHH = 5 Hz, NC5H10), 1.67–1.60 (m, 6H, NC5H10). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 159.0 (NCN of C7H4N2), 141.6 (C6H4 of C7H4N2), 136.4 (C6H4 of C7H4N2), 135.3 (CH2C6H5), 128.8 (CH2C6H5), 127.5 (CH2C6H5), 126.1 (CH2C6H5), 121.7 (C6H4 of C7H4N2), 121.1 (C6H4 of C7H4N2), 117.8 (C6H4 of C7H4N2), 109.2 (C6H4 of C7H4N2), 51.8 (NC5H10), 47.6 (CH2C6H5), 25.6 (NC5H10), 24.1 (NC5H10). GC–MS (ESI): m/z = 291 [M]+. Anal. Calcd for C19H21N3: C, 78.32; H, 7.26; N, 14.42. Found: C, 78.78; H, 7.37; N, 14.50%.
Synthesis of 1-Benzyl-2-(pyrrolidin-1-yl)-1H-benzimidazole (25)76

Yield: 0.083 g, 33% (4).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.55 (d, 1H, 3JHH = 8 Hz, C7H4N2), 7.34–7.27 (m, 4H, C7H4N2 and CH2C6H5), 7.55 (d, 2H, 3JHH = 7 Hz, CH2C6H5), 7.01 (t, 1H, 3JHH = 8 Hz, C7H4N2), 6.97 (d, 1H, 3JHH = 8 Hz, C7H4N2), 5.28 (s, 2H, CH2C6H5), 3.58 (br, 4H, NC4H8), 1.91 (br, 4H, NC4H8). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 161.0 (NCN of C7H4N2), 141.6 (C6H4 of C7H4N2), 138.6 (C6H4 of C7H4N2), 136.7 (CH2C6H5), 128.9 (CH2C6H5), 127.5 (CH2C6H5), 125.7 (CH2C6H5), 122.0 (C6H4 of C7H4N2), 120.3 (C6H4 of C7H4N2), 116.5 (C6H4 of C7H4N2), 108.4 (C6H4 of C7H4N2), 50.6 (NC4H8), 47.9 (CH2C6H5), 25.6 (NC4H8). GC–MS (ESI): m/z = 277 [M]+. Anal. Calcd for C18H19N3: C, 77.95; H, 6.90; N, 15.15. Found: C, 78.21; H, 6.95; N, 14.68%.
Synthesis of 1-Benzyl-N-cyclohexyl-1H-benzimidazol-2-amine (26)78
Yield: 0.168 g, 55% (4).
1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.81 (d, 1H, 3JHH = 8 Hz, C7H4N2), 7.37–7.30 (m, 4H, CH2C6H5), 7.30–7.27 (m, 1H, CH2C6H5), 7.25 (t, 1H, 3JHH = 8 Hz, 4JHH = 1 Hz, C7H4N2), 6.96 (d, 1H, 3JHH = 8 Hz, 4JHH = 1 Hz, C7H4N2), 6.70 (t, 1H, 3JHH = 7 Hz, C7H4N2), 4.40 (s, 2H, CH2C6H5), 3.14 (br, 1H, C6H11), 1.96–1.93 (m, 3H, C6H11), 1.62–1.59 (m, 3H, C6H11), 1.28–1.27 (m, 2H, C6H11), 1.12–1.06 (m, 2H, C6H11). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 150.9 (NCN of C7H4N2), 139.3 (C6H4 of C7H4N2), 138.6 (CH2C6H5), 129.3 (CH2C6H5), 128.9 (C6H4 of C7H4N2), 128.8 (CH2C6H5), 127.6 (CH2C6H5), 127.4 (C6H4 of C7H4N2), 124.0 (C6H4 of C7H4N2), 123.8 (C6H4 of C7H4N2), 50.4 (C6H11), 46.1 (CH2C6H5), 33.7 (C6H11), 25.4 (C6H11), 24.6 (C6H11). GC–MS (ESI): m/z = 305 [M]+. Anal. Calcd for C20H23N3: C, 78.65; H, 7.59; N, 13.76. Found: C, 77.72; H, 8.47; N, 13.06%.
Synthesis of 4-(1-Cyclohexyl-1H-benzimidazol-2-yl)morpholine (27)
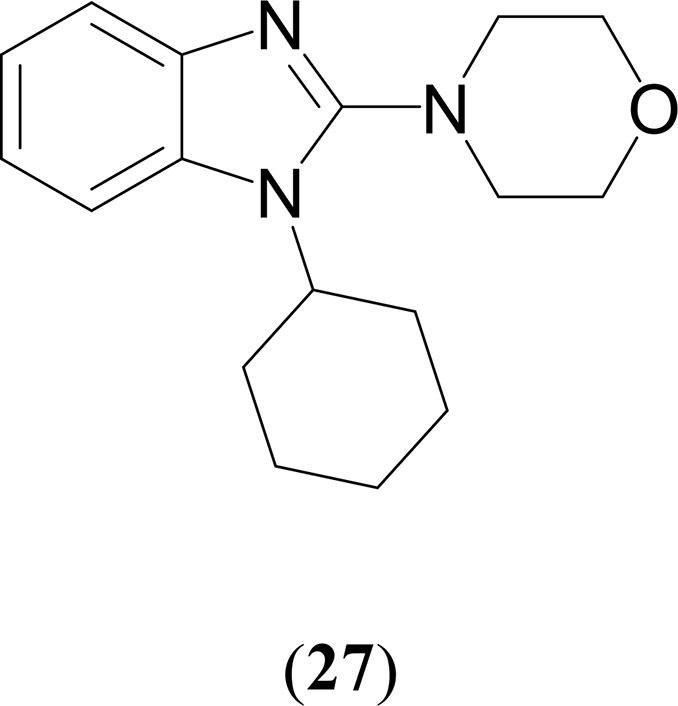
Yield: 0.205 g, 72% (4).
1H NMR (CDCl3, 500 MHz, 25 °C): δ 7.80 (d, 1H, 3JHH = 8 Hz, C7H4N2), 7.26 (t, 1H, 3JHH = 8 Hz, 4JHH = 1 Hz, C7H4N2), 6.91 (d, 1H, 3JHH = 8 Hz, 4JHH = 1 Hz, C7H4N2), 6.74 (t, 1H, 3JHH = 7 Hz, C7H4N2), 3.73 (t, 4H, 3JHH = 5 Hz, NC4H8O), 3.34 (t, 4H, 3JHH = 5 Hz, NC4H8O), 3.14 (br, 1H, C6H11), 1.90–1.88 (m, 2H, C6H11), 1.65–1.62 (m, 2H, C6H11), 1.54–1.51 (m, 1H, C6H11), 1.24–1.18 (m, 3H, C6H11), 1.07–1.05 (m, 2H, C6H11). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): δ 155.2 (NCN of C7H4N2), 139.3 (C6H4 of C7H4N2), 129.2 (C6H4 of C7H4N2), 124.4 (C6H4 of C7H4N2), 123.0 (C6H4 of C7H4N2), 66.5 (NC4H8O), 53.7 (C6H11), 48.5 (NC4H8O), 34.0 (C6H11), 25.2 (C6H11), 25.0 (C6H11). GC–MS (ESI): m/z = 285 [M]+. Anal. Calcd for C17H23N3O: C, 71.55; H, 8.12; N, 14.72;. Found: C, 72.20; H, 9.07; N, 13.74%.
Acknowledgments
We thank the Department of Science and Technology (Grant No: CRG/2019/000029), New Delhi, India, and the Council of Scientific & Industrial Research (CSIR) {01(2880)/17/EMR-II} New Delhi, India, for financial support of this research. We gratefully acknowledge the Single Crystal X-ray Diffraction Facility, Department of Chemistry, IIT Bombay, Mumbai, India, for the crystallographic characterization data. S.R.D. thanks IIT Bombay for a research fellowship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c07875. CCDC-2041824 (for 4), CCDC-2060553 (for 5), CCDC- 2123529 (for 6), and CCDC-2069000 (for 7) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data center via www.ccdc.cam.ac.uk/data_request/cif
1H NMR, 13C{1H} NMR, IR, HRMS, and elemental analysis data of precarbene ligands (1–3), 1H NMR, 13C{1H} NMR, IR, HRMS, elemental analysis, and XPS data of palladium carbene complexes (4–7), and 1H NMR, 13C{1H} NMR, GC–MS chromatograms, and elemental analysis data of the catalysis products (8–17), the benzoxazole derivatives (18–22), and the guanidine derivatives (23–27) (PDF)
X-ray crystallographic data of palladium carbene complexes (4–7) (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wang Y.; Zhang W.-X.; Xi Z. Carbodiimide-based synthesis of N-heterocycles: moving from two classical reactive sites to chemical bond breaking/forming reaction. Chem. Soc. Rev. 2020, 49 (16), 5810–5849. 10.1039/C9CS00478E. [DOI] [PubMed] [Google Scholar]
- Williams A.; Ibrahim I. T. Carbodiimide chemistry: recent advances. Chem. Rev. 1981, 81 (6), 589–636. 10.1021/cr00046a004. [DOI] [Google Scholar]
- Khorana H. G. The Chemistry of Carbodiimides. Chem. Rev. 1953, 53 (2), 145–166. 10.1021/cr60165a001. [DOI] [Google Scholar]
- Alavarse A. C.; Frachini E. C. G.; da Silva R. L. C. G.; Lima V. H.; Shavandi A.; Petri D. F. S. Crosslinkers for polysaccharides and proteins: Synthesis conditions, mechanisms, and crosslinking efficiency, a review. Int. J. Biol. Macromol. 2022, 202, 558–596. 10.1016/j.ijbiomac.2022.01.029. [DOI] [PubMed] [Google Scholar]; Cammarata C. R.; Hughes M. E.; Ofner C. M. Carbodiimide Induced Cross-Linking, Ligand Addition, and Degradation in Gelatin. Mol. Pharmaceutics 2015, 12 (3), 783–793. 10.1021/mp5006118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. H.; Li J.; Qin Y.; Wang H.; Gupta S. Carbodiimide scaffolds: Efficient and versatile reagents in synthesis of heterocycles. Synth. Commun. 2021, 51 (18), 2713–2731. 10.1080/00397911.2021.1953533. [DOI] [Google Scholar]
- Bellucci M. C.; Volonterio A. Carbodiimides-mediated multi component synthesis of biologically relevant structures. Org. Chem. Insights 2012, 4, 1–24. 10.4137/OCI.S9112. [DOI] [Google Scholar]
- Zhang W.-X.; Xu L.; Xi Z. Recent development of synthetic preparation methods for guanidines via transition metal catalysis. Chem. Commun. 2015, 51 (2), 254–265. 10.1039/C4CC05291A. [DOI] [PubMed] [Google Scholar]
- Palacios F.; Alonso C.; Aparicio D.; Rubiales G.; de los Santos J. M. The aza-Wittig reaction: an efficient tool for the construction of carbon–nitrogen double bonds. Tetrahedron 2007, 63 (3), 523–575. 10.1016/j.tet.2006.09.048. [DOI] [Google Scholar]
- Babcock J. R.; Sita L. R. Facile Preparation of Unsymmetric Carbodiimides via in Situ Tin(II)-Mediated Heterocumulene Metathesis. J. Am. Chem. Soc. 1998, 120 (22), 5585–5586. 10.1021/ja980173c. [DOI] [Google Scholar]
- Lazar M.; Angelici R. J. Gold Metal-Catalyzed Reactions of Isocyanides with Primary Amines and Oxygen: Analogies with Reactions of Isocyanides in Transition Metal Complexes. J. Am. Chem. Soc. 2006, 128 (32), 10613–10620. 10.1021/ja0618907. [DOI] [PubMed] [Google Scholar]
- Hine J.; Mookerjee P. K. Structural effects on rates and equilibriums. XIX. Intrinsic hydrophilic character of organic compounds. Correlations in terms of structural contributions. Journal of Organic Chemistry 1975, 40 (3), 292–298. 10.1021/jo00891a006. [DOI] [Google Scholar]
- Beaumier E. P.; McGreal M. E.; Pancoast A. R.; Wilson R. H.; Moore J. T.; Graziano B. J.; Goodpaster J. D.; Tonks I. A. Carbodiimide Synthesis via Ti-Catalyzed Nitrene Transfer from Diazenes to Isocyanides. ACS Catal. 2019, 9 (12), 11753–11762. 10.1021/acscatal.9b04107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousif M.; Tjapkes D. J.; Lord R. L.; Groysman S. Catalytic Formation of Asymmetric Carbodiimides at Mononuclear Chromium(II/IV) Bis(alkoxide) Complexes. Organometallics 2015, 34 (20), 5119–5128. 10.1021/acs.organomet.5b00703. [DOI] [Google Scholar]
- Wiese S.; Aguila M. J. B.; Kogut E.; Warren T. H. β-Diketiminato Nickel Imides in Catalytic Nitrene Transfer to Isocyanides. Organometallics 2013, 32 (8), 2300–2308. 10.1021/om300909n. [DOI] [Google Scholar]
- Maruthamuthu; Rajam S.; Stella P. C. R.; Dileepan A. G. B.; Ranjith R. The chemistry and biological significance of imidazole, benzimidazole, benzoxazole, tetrazole and quinazolinone nucleus. J. Chem. Pharm. Res. 2016, 8 (5), 505–526. [Google Scholar]
- Wong X. K.; Yeong K. Y. A Patent Review on the Current Developments of Benzoxazoles in Drug Discovery. ChemMedChem. 2021, 16 (21), 3237–3262. 10.1002/cmdc.202100370. [DOI] [PubMed] [Google Scholar]
- Rajyalakshmi G.; Rama Narsimha Reddy A.; Sarangapani M. Synthesis and biological activities of some novel 2-amino-(5 or 7-substituted-2-oxoindolin-3-ylidene) benzoxazole-5-carbohydrazide derivatives. Lett. Drug Des. Discovery 2012, 9 (6), 625–632. 10.2174/157018012800673029. [DOI] [Google Scholar]; Jyothi M.; Merugu R. Antibacterial and antifungal activity of some newly substituted benzoxazoles. Int. J. ChemTech Res. 2013, 5 (5), 2425–2428. [Google Scholar]; Šlachtová V.; Brulíková L. Benzoxazole Derivatives as Promising Antitubercular Agents. ChemistrySelect 2018, 3 (17), 4653–4662. 10.1002/slct.201800631. [DOI] [Google Scholar]
- Sattar R.; Mukhtar R.; Atif M.; Hasnain M.; Irfan A. Synthetic transformations and biological screening of benzoxazole derivatives: A review. Journal of Heterocyclic Chemistry 2020, 57 (5), 2079–2107. 10.1002/jhet.3944. [DOI] [Google Scholar]
- Smaili A.; Jebbari S.; Rifai L. A.; Faize L.; Koussa T.; Sir H. A.; Makroum K.; Belfaiza M.; El Kihel A.; Ahbala M.; et al. Synthesis and in planta antibacterial activity of head-to-head bis-benzimidazole and bis-benzoxazole derivatives. Phytoparasitica 2019, 47 (5), 733–741. 10.1007/s12600-019-00764-9. [DOI] [Google Scholar]
- Rao M. N.; Manne R.; Tanski J. M.; Butcher R.; Ghosh P. One pot synthesis of propargylamines by three component amine-aldehyde-acetylene (A3) coupling catalyzed by neutral Ag(I) and Au(I) and cationic Pd(II) and Ni(II) complexes of a pincer N-heterocyclic carbene. Molecular Catalysis 2022, 529, 112515. 10.1016/j.mcat.2022.112515. [DOI] [Google Scholar]; Ramasamy B.; Ghosh P. The Developing Concept of Bifunctional Catalysis with Transition Metal N-Heterocyclic Carbene Complexes. Eur. J. Inorg. Chem. 2016, 2016 (10), 1448–1465. 10.1002/ejic.201501429. [DOI] [Google Scholar]; Prakasham A. P.; Ghosh P. Nickel N-heterocyclic carbene complexes and their utility in homogeneous catalysis. Inorg. Chim. Acta 2015, 431, 61–100. 10.1016/j.ica.2014.11.005. [DOI] [Google Scholar]
- Prakasham A. P.; Ta S.; Dey S.; Ghosh P. One pot tandem dual C=C and C=O bond reductions in the β-alkylation of secondary alcohols with primary alcohols by ruthenium complexes of amido and picolyl functionalized N-heterocyclic carbenes. Dalton Transactions 2021, 50 (43), 15640–15654. 10.1039/D1DT02849A. [DOI] [PubMed] [Google Scholar]
- Singh C.; Prakasham A. P.; Gangwar M. K.; Butcher R. J.; Ghosh P. One-Pot Tandem Hiyama Alkynylation/Cyclizations by Palladium(II) Acyclic Diaminocarbene (ADC) Complexes Yielding Biologically Relevant Benzofuran Scaffolds. ACS Omega 2018, 3 (2), 1740–1756. 10.1021/acsomega.7b01974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R.; Katari M.; Choudhary A.; Rajaraman G.; Ghosh P. Computational Insight Into the Hydroamination of an Activated Olefin, As Catalyzed by a 1,2,4-Triazole-Derived Nickel(II) N-Heterocyclic Carbene Complex. Inorg. Chem. 2017, 56 (24), 14859–14869. 10.1021/acs.inorgchem.7b02097. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Gangwar M. K.; Prakasham A. P.; Mhatre D.; Kalita A. C.; Ghosh P. Accessing a Biologically Relevant Benzofuran Skeleton by a One-Pot Tandem Heck Alkynylation/Cyclization Reaction Using Well-Defined Palladium N-Heterocyclic Carbene Complexes. Inorg. Chem. 2016, 55 (6), 2882–2893. 10.1021/acs.inorgchem.5b02727. [DOI] [PubMed] [Google Scholar]
- Modak S.; Gangwar M. K.; Nageswar Rao M.; Madasu M.; Kalita A. C.; Dorcet V.; Shejale M. A.; Butcher R. J.; Ghosh P. Fluoride-free Hiyama coupling by palladium abnormal N-heterocyclic carbene complexes. Dalton Trans. 2015, 44 (40), 17617–17628. 10.1039/C5DT02317C. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Naaz A.; Prakasham A. P.; Gangwar M. K.; Butcher R. J.; Panda D.; Ghosh P. Potent Anticancer Activity with High Selectivity of a Chiral Palladium N-Heterocyclic Carbene Complex. ACS Omega 2017, 2 (8), 4632–4646. 10.1021/acsomega.7b00688. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ray S.; Mohan R.; Singh J. K.; Samantaray M. K.; Shaikh M. M.; Panda D.; Ghosh P. Anticancer and antimicrobial metallopharmaceutical agents based on palladium, gold, and silver N-heterocyclic carbene complexes. J. Am. Chem. Soc. 2007, 129 (48), 15042–15053. 10.1021/ja075889z. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Prakasham A. P.; Gangwar M. K.; Ghosh P. Solvent-free Cyanosilylation of Aromatic and Heteroaryl Aldehydes Catalyzed by a Cationic Iron N-Heterocyclic Carbene Complex at Ambient Temperature Under UV Irradiation. Inorg. Chim. Acta 2019, 495, 119003. 10.1016/j.ica.2019.119003. [DOI] [Google Scholar]; Singh C.; Gangwar M. K.; Ghosh P. Mass spectrometric support for a bifunctional catalysis mechanism for the base-free Michael addition by a nickel N-heterocyclic carbene complex: Detection of the catalytic intermediates. Inorg. Chim. Acta 2017, 466, 358–369. 10.1016/j.ica.2017.06.030. [DOI] [Google Scholar]; Rao M. N.; Haridas M.; Gangwar M. K.; Rajakannu P.; Kalita A. C.; Ghosh P. Asymmetric base-free michael addition at room temperature with nickel-based bifunctional amido-functionalized N-heterocyclic carbene catalysts. Eur. J. Inorg. Chem. 2015, 2015 (9), 1604–1615. 10.1002/ejic.201403224. [DOI] [Google Scholar]
- Modak S.; Borah S.; Prakasham A. P.; Shaikh M. M.; Butcher R. J.; Gangwar M.; Ghosh P. A comparison between (a/n-NHC)PdX2(pyridine) and (a/n-NHC)2PdX2 (X = I, Cl) type complexes of abnormal fused-bicyclic imidazo[1,2-a]pyridine based N-heterocyclic carbene (a-NHC) and of normal imidazole based N-heterocyclic carbene (n-NHC) ligands in the Suzuki-Miyaura coupling reactions. Inorg. Chim. Acta 2019, 498, 119090. 10.1016/j.ica.2019.119090. [DOI] [Google Scholar]
- Singh C.; Prakasham A. P.; Ghosh P. Palladium Acyclic Diaminocarbene (ADC) Triflate Complexes as Effective Precatalysts for the Hiyama Alkynylation/Cyclization Reaction Yielding Benzofuran Compounds: Probing the Influence of the Triflate Co-Ligand in the One-Pot Tandem Reaction. ChemistrySelect 2019, 4 (1), 329–336. 10.1002/slct.201803292. [DOI] [Google Scholar]
- Prakasham A. P.; Gangwar M. K.; Ghosh P. β-Enaminone Synthesis from 1,3-Dicarbonyl Compounds and Aliphatic and Aromatic Amines Catalyzed by Iron Complexes of Fused Bicyclic Imidazo[1,5-a]pyridine Derived N-Heterocyclic Carbenes. Eur. J. Inorg. Chem. 2019, 2019 (2), 295–313. 10.1002/ejic.201800906. [DOI] [Google Scholar]
- Prakasham A. P.; Gangwar M. K.; Ghosh P. Michael addition of cyclic β-oxo ester and α-methyl cyano ester substrates with activated olefins by iron complexes of benzimidazole derived N-heterocyclic carbene ligands. J. Organomet. Chem. 2018, 859, 106–116. 10.1016/j.jorganchem.2018.01.061. [DOI] [Google Scholar]
- Kumar A.; Prakasham A. P.; Gangwar M. K.; Vishnoi P.; Butcher R. J.; Ghosh P. An Efficient Synthetic Approach to trans-(NHC)2Pd(R)Br Type Complexes and Their Use in Suzuki–Miyaura Cross-Coupling Reactions. Eur. J. Inorg. Chem. 2017, 2017 (14), 2144–2154. 10.1002/ejic.201700017. [DOI] [Google Scholar]; Kumar A.; Bheeter L. P.; Gangwar M. K.; Sortais J.-B.; Darcel C.; Ghosh P. Nickel complexes of 1,2,4-triazole derived amido-functionalized N-heterocyclic carbene ligands: Synthesis, theoretical studies and catalytic application. J. Organomet. Chem. 2015, 786, 63–70. 10.1016/j.jorganchem.2015.03.007. [DOI] [Google Scholar]
- Ramasamy B.; Kumar Gangwar M.; Ghosh P. Chiral Oxazolidine-Fused N-Heterocyclic Carbene Complexes of Rhodium and Iridium and Their Utility in the Asymmetric Transfer Hydrogenation of Ketones. Eur. J. Inorg. Chem. 2017, 2017 (26), 3253–3268. 10.1002/ejic.201700303. [DOI] [Google Scholar]
- Ramasamy B.; Prakasham A. P.; Gangwar M. K.; Ghosh P. 1,4-Conjugate Addition of Aryl boronic Acids on Cyclohexenone as Catalyzed by Rhodium(I) Complexes of C2-Symmetric Bioxazoline Fused N-heterocyclic Carbenes. ChemistrySelect 2019, 4 (29), 8526–8533. 10.1002/slct.201902408. [DOI] [Google Scholar]
- Kumar Gangwar M.; Dey S.; A. P P.; Ghosh P. Palladium(II), silver(I), and gold(I) complexes of a new class of chiral bicyclic [1,2,3]-triazolooxazine derived N-heterocyclic carbenes (NHCs): Synthesis, structure and application studies. Polyhedron 2021, 197, 115011. 10.1016/j.poly.2020.115011. [DOI] [Google Scholar]
- Hopkinson M. N.; Richter C.; Schedler M.; Glorius F. An overview of N-heterocyclic carbenes. Nature (London, U. K.) 2014, 510 (7506), 485–496. 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]
- Vivancos A.; Segarra C.; Albrecht M. Mesoionic and Related Less Heteroatom-Stabilized N-Heterocyclic Carbene Complexes: Synthesis, Catalysis, and Other Applications. Chem. Rev. 2018, 118 (19), 9493–9586. 10.1021/acs.chemrev.8b00148. [DOI] [PubMed] [Google Scholar]; Guisado-Barrios G.; Soleilhavoup M.; Bertrand G. 1H-1,2,3-Triazol-5-ylidenes: Readily Available Mesoionic Carbenes. Acc. Chem. Res. 2018, 51 (12), 3236–3244. 10.1021/acs.accounts.8b00480. [DOI] [PubMed] [Google Scholar]; Crabtree R. H. Abnormal, mesoionic and remote N-heterocyclic carbene complexes. Coord. Chem. Rev. 2013, 257 (3), 755–766. 10.1016/j.ccr.2012.09.006. [DOI] [Google Scholar]; Marichev K. O.; Patil S. A.; Bugarin A. Recent advances in the synthesis, structural diversity, and applications of mesoionic 1,2,3-triazol-5-ylidene metal complexes. Tetrahedron 2018, 74 (21), 2523–2546. 10.1016/j.tet.2018.04.013. [DOI] [Google Scholar]; Aizpurua J. M.; Sagartzazu-Aizpurua M.; Monasterio Z. Mesoionic 1,2,3-triazoles and 1,2,3-triazole carbenes. Top. Heterocycl. Chem. 2014, 40, 211–267. 10.1007/7081_2014_120. [DOI] [Google Scholar]; Maity R.; Sarkar B. Chemistry of Compounds Based on 1,2,3-Triazolylidene-Type Mesoionic Carbenes. JACS Au 2022, 2 (1), 22–57. 10.1021/jacsau.1c00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posso M. C.; Domingues F. C.; Ferreira S.; Silvestre S. Development of Phenothiazine Hybrids with Potential Medicinal Interest: A Review. Molecules 2022, 27 (1), 276. 10.3390/molecules27010276. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wu C.-H.; Bai L.-Y.; Tsai M.-H.; Chu P.-C.; Chiu C.-F.; Chen M. Y.; Chiu S.-J.; Chiang J.-H.; Weng J.-R. Pharmacological exploitation of the phenothiazine antipsychotics to develop novel antitumor agents–A drug repurposing strategy. Sci. Rep. 2016, 6 (1), 27540. 10.1038/srep27540. [DOI] [PMC free article] [PubMed] [Google Scholar]; Al-Busaidi I. J.; Haque A.; Al Rasbi N. K.; Khan M. S. Phenothiazine-based derivatives for optoelectronic applications: A review. Synth. Met. 2019, 257, 116189. 10.1016/j.synthmet.2019.116189. [DOI] [Google Scholar]
- Donnelly K. F.; Petronilho A.; Albrecht M. Application of 1,2,3-triazolylidenes as versatile NHC-type ligands: synthesis, properties, and application in catalysis and beyond. Chem. Commun. (Cambridge, U. K.) 2013, 49 (12), 1145–1159. 10.1039/C2CC37881G. [DOI] [PubMed] [Google Scholar]; Meldal M.; Tornøe C. W. Cu-Catalyzed Azide–Alkyne Cycloaddition. Chem. Rev. 2008, 108 (8), 2952–3015. 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- Fulmer G. R.; Miller A. J. M.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29 (9), 2176–2179. 10.1021/om100106e. [DOI] [Google Scholar]
- Sureshbabu B.; Ramkumar V.; Sankararaman S. A mild and efficient method for the synthesis of structurally diverse 1,2,3-triazolylidene palladium(II) diiodo complexes. Comparison of catalytic activities for Suzuki–Miyaura coupling. J. Organomet. Chem. 2015, 799–800, 232–238. 10.1016/j.jorganchem.2015.10.002. [DOI] [Google Scholar]
- Karuppasamy A.; Udhaya kumar C.; Karthikeyan S.; Velayutham Pillai M. P.; Ramalingan C. Synthesis, spectral, structural prediction and computational studies of octylcarbazole ornamented 3-phenothiazinal. J. Mol. Struct. 2017, 1147, 487–494. 10.1016/j.molstruc.2017.06.136. [DOI] [Google Scholar]; Dutkiewicz G.; Siddaraju B. P.; Yathirajan H. S.; Narayana B.; Kubicki M. The Crystal Structure of Fluphenazinium Dipicrate Dimethylsulfoxide Solvate. J. Chem. Crystallogr. 2010, 40 (11), 970–974. 10.1007/s10870-010-9773-z. [DOI] [Google Scholar]
- Han Y.; Huynh H. V. Mixed carbene-isocyanide Pd(ii) complexes: synthesis, structures and reactivity towards nucleophiles. Dalton Trans. 2009, (12), 2201–2209. 10.1039/b816471a. [DOI] [PubMed] [Google Scholar]; Meier M.; Tan T. T. Y.; Hahn F. E.; Huynh H. V. Donor Strength Determination of Benzoxazolin-2-ylidene, Benzobisoxazolin-2-ylidene, and Their Isocyanide Precursors by 13C NMR Spectroscopy of Their PdII and AuI Complexes. Organometallics 2017, 36 (2), 275–284. 10.1021/acs.organomet.6b00736. [DOI] [Google Scholar]
- Han Y.; Yuan D.; Teng Q.; Huynh H. V. Reactivity Differences of Palladium(II) Dimers Bearing Heterocyclic Carbenes with Two, One, or No α-Nitrogen Atoms toward Isocyanides. Organometallics 2011, 30 (5), 1224–1230. 10.1021/om101169x. [DOI] [Google Scholar]
- Bardsley K.; Hagigeorgiou M.; Lengyel I.; Cesare V. New One-Step Synthesis of Isonitriles. Synth. Commun. 2013, 43 (12), 1727–1733. 10.1080/00397911.2012.666693. [DOI] [Google Scholar]
- Cordero B.; Gomez V.; Platero-Prats A. E.; Reves M.; Echeverria J.; Cremades E.; Barragan F.; Alvarez S. Covalent radii revisited. Dalton Trans. 2008, (21), 2832–2838. 10.1039/b801115j. [DOI] [PubMed] [Google Scholar]
- Naskar R.; Majumder A.; Kundu K.; Biswas S.; Maji M. S.; Maity R. Palladium(II) Complexes Bearing a Mixed Set of aNHC/Py/PR3/I2 Ligands: Applications in α-Arylation of Amide and Suzuki-Miyaura Coupling Reactions. J. Organomet. Chem. 2021, 949, 121925. 10.1016/j.jorganchem.2021.121925. [DOI] [Google Scholar]
- Zhang X.; Yan X.; Zhang B.; Wang R.; Guo S.; Peng S. Mono- and dinuclear palladium(II) complexes incorporating 1,2,3-triazole-derived mesoionic carbenes: syntheses, solid-state structures and catalytic applications. Transition Metal Chemistry 2019, 44 (1), 39–48. 10.1007/s11243-018-0267-8. [DOI] [Google Scholar]
- Teng Q.; Upmann D.; Ng Wijaya S. A. Z.; Huynh H. V. Bis(functionalized NHC) Palladium(II) Complexes via a Postmodification Approach. Organometallics 2014, 33 (13), 3373–3384. 10.1021/om500274c. [DOI] [Google Scholar]; Nguyen V. H.; Ibrahim M. B.; Mansour W. W.; El Ali B. M.; Huynh H. V. Postmodification Approach to Charge-Tagged 1,2,4-Triazole-Derived NHC Palladium(II) Complexes and Their Applications. Organometallics 2017, 36 (12), 2345–2353. 10.1021/acs.organomet.7b00329. [DOI] [Google Scholar]
- Sureshbabu B.; Ramkumar V.; Sankararaman S. Facile base-free in situ generation and palladation of mesoionic and normal N-heterocyclic carbenes at ambient conditions. Dalton Trans. 2014, 43 (28), 10710–10712. 10.1039/C4DT01112K. [DOI] [PubMed] [Google Scholar]
- Poulain A.; Canseco-Gonzalez D.; Hynes-Roche R.; Müller-Bunz H.; Schuster O.; Stoeckli-Evans H.; Neels A.; Albrecht M. Synthesis and Tunability of Abnormal 1,2,3-Triazolylidene Palladium and Rhodium Complexes. Organometallics 2011, 30 (5), 1021–1029. 10.1021/om101076u. [DOI] [Google Scholar]
- Abramovitch R. A.; Kyba E. P.. Decomposition of organic azides. In The Azido Group (1971); PATAI’S Chemistry of Functional Groups; 1971; pp 221–329. [Google Scholar]
- Muthyala R.5.28 - Functions with at Least One Nitrogen and No Chalcogens. In Comprehensive Organic Functional Group Transformations, Katritzky A. R., Meth-Cohn O., Rees C. W., Eds.; Elsevier Science, 1995; pp 1061–1089. [Google Scholar]
- Cowley R. E.; Golder M. R.; Eckert N. A.; Al-Afyouni M. H.; Holland P. L. Mechanism of Catalytic Nitrene Transfer Using Iron(I)–Isocyanide Complexes. Organometallics 2013, 32 (19), 5289–5298. 10.1021/om400379p. [DOI] [Google Scholar]
- Zhang Z.; Li Z.; Fu B.; Zhang Z. Palladium-catalyzed cross-coupling reaction of azides with isocyanides. Chem. Commun. 2015, 51 (91), 16312–16315. 10.1039/C5CC05981J. [DOI] [PubMed] [Google Scholar]
- Shi S.; Szostak M. Pd–PEPPSI: a general Pd–NHC precatalyst for Buchwald–Hartwig cross-coupling of esters and amides (transamidation) under the same reaction conditions. Chem. Commun. 2017, 53 (76), 10584–10587. 10.1039/C7CC06186B. [DOI] [PubMed] [Google Scholar]
- Li H.; Grasa G. A.; Colacot T. J. A Highly Efficient, Practical, and General Route for the Synthesis of (R3P)2Pd(0): Structural Evidence on the Reduction Mechanism of Pd(II) to Pd(0). Org. Lett. 2010, 12 (15), 3332–3335. 10.1021/ol101106z. [DOI] [PubMed] [Google Scholar]; Johansson Seechurn C. C. C.; Sperger T.; Scrase T. G.; Schoenebeck F.; Colacot T. J. Understanding the Unusual Reduction Mechanism of Pd(II) to Pd(I): Uncovering Hidden Species and Implications in Catalytic Cross-Coupling Reactions. J. Am. Chem. Soc. 2017, 139 (14), 5194–5200. 10.1021/jacs.7b01110. [DOI] [PubMed] [Google Scholar]
- Alonso D. A.; Nájera C.; Pacheco M. C. Oxime-Derived Palladium Complexes as Very Efficient Catalysts for the Heck–Mizoroki Reaction. Advanced Synthesis & Catalysis 2002, 344 (2), 172–183. . [DOI] [Google Scholar]
- Kuck D. Reactive Intermediates. MS Investigations in Solution. Edited by Leonardo S. Santos. Angew. Chem., Int. Ed. 2010, 49 (28), 4705–4706. 10.1002/anie.201002610. [DOI] [Google Scholar]
- Wenzel M. N.; Owens P. K.; Bray J. T. W.; Lynam J. M.; Aguiar P. M.; Reed C.; Lee J. D.; Hamilton J. F.; Whitwood A. C.; Fairlamb I. J. S. Redox Couple Involving NOx in Aerobic Pd-Catalyzed Oxidation of sp3-C–H Bonds: Direct Evidence for Pd–NO3–/NO2– Interactions Involved in Oxidation and Reductive Elimination. J. Am. Chem. Soc. 2017, 139 (3), 1177–1190. 10.1021/jacs.6b10853. [DOI] [PubMed] [Google Scholar]; Zheng Q.; Liu Y.; Chen Q.; Hu M.; Helmy R.; Sherer E. C.; Welch C. J.; Chen H. Capture of Reactive Monophosphine-Ligated Palladium(0) Intermediates by Mass Spectrometry. J. Am. Chem. Soc. 2015, 137 (44), 14035–14038. 10.1021/jacs.5b08905. [DOI] [PubMed] [Google Scholar]; Aliprantis A. O.; Canary J. W. Observation of Catalytic Intermediates in the Suzuki Reaction by Electrospray Mass Spectrometry. J. Am. Chem. Soc. 1994, 116 (15), 6985–6986. 10.1021/ja00094a083. [DOI] [Google Scholar]; Sabino A. A.; Machado A. H. L.; Correia C. R. D.; Eberlin M. N. Probing the Mechanism of the Heck Reaction with Arene Diazonium Salts by Electrospray Mass and Tandem Mass Spectrometry. Angew. Chem., Int. Ed. 2004, 43 (19), 2514–2518. 10.1002/anie.200353076. [DOI] [PubMed] [Google Scholar]
- McIndoe J. S.; Vikse K. L. Assigning the ESI mass spectra of organometallic and coordination compounds. Journal of Mass Spectrometry 2019, 54 (5), 466–479. 10.1002/jms.4359. [DOI] [PubMed] [Google Scholar]
- Mollar-Cuni A.; Ventura-Espinosa D.; Martín S.; Mayoral A.; Borja P.; Mata J. A. Stabilization of Nanoparticles Produced by Hydrogenation of Palladium–N-Heterocyclic Carbene Complexes on the Surface of Graphene and Implications in Catalysis. ACS Omega 2018, 3 (11), 15217–15228. 10.1021/acsomega.8b02193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji A.; Singh O.; Singh S.; Mohanty A.; Maji P. K.; Ghosh K. Palladium-Based Catalysts Supported by Unsymmetrical XYC–1 Type Pincer Ligands: C5 Arylation of Imidazoles and Synthesis of Octinoxate Utilizing the Mizoroki–Heck Reaction. Eur. J. Inorg. Chem. 2020, 2020 (17), 1596–1611. 10.1002/ejic.202000211. [DOI] [Google Scholar]
- Kale M.; Chavan V. Exploration of the Biological Potential of Benzoxazoles: An Overview. Mini-Reviews in Organic Chemistry 2019, 16 (2), 111–126. 10.2174/1570193X15666180627125007. [DOI] [Google Scholar]
- Pathare B.; Bansode T. Review- biological active benzimidazole derivatives. Results in Chemistry 2021, 3, 100200. 10.1016/j.rechem.2021.100200. [DOI] [Google Scholar]; Faheem M.; Rathaur A.; Pandey A.; Kumar Singh V.; Tiwari A. K. A Review on the Modern Synthetic Approach of Benzimidazole Candidate. ChemistrySelect 2020, 5 (13), 3981–3994. 10.1002/slct.201904832. [DOI] [Google Scholar]
- Cann J. R.; Cabanetos C.; Welch G. C. Synthesis of Molecular Dyads and Triads Based Upon N-Annulated Perylene Diimide Monomers and Dimers. Eur. J. Org. Chem. 2018, 2018 (48), 6933–6943. 10.1002/ejoc.201801383. [DOI] [Google Scholar]
- Sheldrick G. M. A short history of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64 (1), 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Lu C.; Gong C.; Zhao B.; Hu L.; Yao Y. RE[N(SiMe3)2]3-Catalyzed Guanylation/Cyclization of Amino Acid Esters and Carbodiimides. Journal of Organic Chemistry 2018, 83 (3), 1154–1159. 10.1021/acs.joc.7b02550. [DOI] [PubMed] [Google Scholar]
- Wu H.; Sun Y.-F.; Zhang C.; Miao C.-B.; Yang H.-T. A facile method for the preparation of carbodiimides from thioureas and (Boc)2O. Tetrahedron Lett. 2018, 59 (8), 739–742. 10.1016/j.tetlet.2018.01.025. [DOI] [Google Scholar]
- Peddarao T.; Baishya A.; Barman M. K.; Kumar A.; Nembenna S. Metal-free access of bulky N,N′-diarylcarbodiimides and their reduction: bulky N,N′-diarylformamidines. New J. Chem. 2016, 40 (9), 7627–7636. 10.1039/C6NJ00907G. [DOI] [Google Scholar]
- Duangkamol C.; Pattarawarapan M.; Phakhodee W. Ultrasonic-assisted synthesis of carbodiimides from N,N′-disubstituted thioureas and ureas. Monatshefte für Chemie - Chemical Monthly 2016, 147 (11), 1945–1949. 10.1007/s00706-016-1761-3. [DOI] [Google Scholar]
- Melchor Bañales A. J.; Larsen M. B. Thermal Guanidine Metathesis for Covalent Adaptable Networks. ACS Macro Lett. 2020, 9 (7), 937–943. 10.1021/acsmacrolett.0c00352. [DOI] [PubMed] [Google Scholar]
- Wang X.; Xu D.; Miao C.; Zhang Q.; Sun W. N-Bromosuccinimide as an oxidant for the transition-metal-free synthesis of 2-aminobenzoxazoles from benzoxazoles and secondary amines. Organic & Biomolecular Chemistry 2014, 12 (19), 3108–3113. 10.1039/c4ob00386a. [DOI] [PubMed] [Google Scholar]
- Wang G.-N.; Zhu T.-H.; Wang S.-Y.; Wei T.-Q.; Ji S.-J. NiCl2-catalyzed cascade reaction of isocyanides with functionalized anilines. Tetrahedron 2014, 70 (43), 8079–8083. 10.1016/j.tet.2014.08.032. [DOI] [Google Scholar]
- Zhang J.; Chen L.; Dong Y.; Yang J.; Wu Y. A Cu2O/TBAB-promoted approach to synthesize heteroaromatic 2-amines via one-pot cyclization of aryl isothiocyanates with ortho-substituted amines in water. Organic & Biomolecular Chemistry 2020, 18 (37), 7425–7430. 10.1039/D0OB01431A. [DOI] [PubMed] [Google Scholar]
- Saha P.; Ramana T.; Purkait N.; Ali M. A.; Paul R.; Punniyamurthy T. Ligand-Free Copper-Catalyzed Synthesis of Substituted Benzimidazoles, 2-Aminobenzimidazoles, 2-Aminobenzothiazoles, and Benzoxazoles. Journal of Organic Chemistry 2009, 74 (22), 8719–8725. 10.1021/jo901813g. [DOI] [PubMed] [Google Scholar]
- Lv X.; Bao W. Copper-Catalyzed Cascade Addition/Cyclization: An Efficient and Versatile Synthesis of N-Substituted 2-Heterobenzimidazoles. Journal of Organic Chemistry 2009, 74 (15), 5618–5621. 10.1021/jo900743y. [DOI] [PubMed] [Google Scholar]
- Li F.; Kang Q.; Shan H.; Chen L.; Xie J. Regioselective N-Alkylation of 2-Aminoimidazoles with Alcohols to 2-(N-Alkylamino)imidazoles Catalyzed by the [Cp*IrCl2]2/K2CO3 System. Eur. J. Org. Chem. 2012, 2012 (26), 5085–5092. 10.1002/ejoc.201200698. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.