

Abstract
Hepatocellular carcinoma (HCC) is a leading cause of cancer deaths, and the most common primary liver malignancy to present in the clinic. With the exception of liver transplant, treatment options for advanced HCC are limited, but improved tumor stratification could open the door to new treatment options. Previously, we demonstrated that the circadian regulator Aryl Hydrocarbon-Like Receptor Like 1 (ARNTL, or Bmal1) and the liver-enriched nuclear factor 4 alpha (HNF4α) are robustly co-expressed in healthy liver but incompatible in the context of HCC. Faulty circadian expression of HNF4α– either by isoform switching, or loss of expression- results in an increased risk for HCC, while BMAL1 gain-of-function in HNF4α-positive HCC results in apoptosis and tumor regression. We hypothesize that the transcriptional programs of HNF4α and BMAL1 are antagonistic in liver disease and HCC. Here, we study this antagonism by generating a mouse model with inducible loss of hepatic HNF4α and BMAL1 expression. The results reveal that simultaneous loss of HNF4α and BMAL1 is protective against fatty liver and HCC in carcinogen-induced liver injury and in the “STAM” model of liver disease. Furthermore, our results suggest that targeting Bmal1 expression in the absence of HNF4α inhibits HCC growth and progression. Specifically, pharmacological suppression of Bmal1 in HNF4α-deficient, BMAL1-positive HCC with REV-ERB agonist SR9009 impairs tumor cell proliferation and migration in a REV-ERB-dependent manner, while having no effect on healthy hepatocytes. Collectively, our results suggest that stratification of HCC based on HNF4α and BMAL1 expression may provide a new perspective on HCC properties and potential targeted therapeutics.
Keywords: BMAL1, circadian, HCC, hepatocellular carcinoma, HNF4α, SR9009
1 |. INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common liver cancer to present in the clinic.1 Though viral infection of the liver and excess alcohol consumption have been primary risk factors for HCC, a recent rise in non-alcoholic fatty liver disease (NAFLD)-associated HCC has been observed.2 While some patients with NAFLD progress to HCC through the normal sequence of liver disease stages, including fibrosis and cirrhosis, some patients progress from fatty liver disease to HCC, without evidence of intermediate stages. Thus, HCC risk prediction remains challenging. The need to identify biomarkers that assist with risk stratification is pressing, as is the need to identify new treatment options when tumor removal is not possible.
The circadian clock is an internal, 24-h biological time-keeping system that is functional in most cells of the body, and orchestrates rhythmicity at the cellular, tissue, and organism-wide level. At the cellular level, a well-defined core transcriptional feedback loop drives these 24-h rhythms. This loop consists of two transcriptional activators, Circadian Locomotor Output Cycles Protein Kaput (Clock) and Aryl Hydrocarbon-Like Receptor Like 1 (ARNTL1, Bmal1), that heterodimerize to drive 24-h waves of gene expression.3 Their transcriptional activity is inhibited in a 24-h manner by the expression of the target genes, period (Per) and cryptochrome (Cry). Epidemiological studies have revealed that circadian disruption (such as occurs in the form of night or rotating shift work, for example) increases the risk of several forms of cancer.4,5 Preclinical studies have validated these findings, and in the context of HCC, simply exposing an animal to jet lag for an extended period of time is sufficient to cause fatty liver and early onset HCC.6 Similar to environmental disruption of the circadian clock, genetic disruption of the clock also results in fatty liver and early onset HCC in rodent models.6 For example, period 2 (Per2) mutant mice have an increased incidence of irradiation-induced cancers.7 Similarly, Clock mutant mice (ClkΔ19/Δ19) develop nonalcoholic fatty liver disease.8 Additionally, Bmal1−/+ mice and mice with cryptochrome 1 and 2 (Cry1−/−/Cry2−/−) deficiency have early onset liver and ovarian cancers as well as lymphoma, the incidence of which is amplified by irradiation.9
Just as disruption of genes central to circadian clock function leads to liver disease progression, disruption in the 24-h activity of specific nuclear receptors can also lead to fatty liver and early onset HCC.6,8,10–12 One of these receptors is hepatocyte nuclear factor four alpha (HNF4α), an abundantly expressed transcription factor enriched in the liver, and necessary for hepatocyte cell fate and liver function.13,14 Binding to thousands of genomic loci,15–20 HNF4α transcriptionally coordinates genes involved in gluconeogenesis, bile acid synthesis, and xenobiotic and fatty acid metabolism in the liver. Interestingly, HNF4α has potent circadian activity in the liver, repressing many genes involved in cell cycle and tumor progression in a circadian manner.12,21 While global loss of HNF4α results in embryonic lethality, and mice with hepatic HNF4α deficiency die within several weeks of age,22–25 an inducible knockout strategy to delete HNF4α in adult animals has been a tractable solution to better understand its role in hepatic function and health.26
The Hnf4a gene has two promoters, “P1” and “P2,” that drive expression of two groups of isoforms that differ in their 5′ exon and hence N-terminal domain. The “P1-HNF4α” isoform(s) are the predominant ones expressed in the normal adult liver while the “P2-HNF4α” isoform(s) were originally thought to be expressed only in fetal liver or liver cancer.13,14,27 Previous work has demonstrated that unlike P1-HNF4α, which suppresses the circadian activation of proliferation and inflammation genes in the liver,15,28–30 P2-HNF4α is upregulated in HCC, as well as colon and gastric carcinomas.31–35 This isoform emerges at much earlier stages of liver disease, showing increased expression under conditions of diet-induced obesity and hyperinsulinemia, as well as alcohol-induced liver damage.10,36,37 Recently, P2-HNF4α was also shown to contribute to hepatic glucose production in the context of fasting.38 Unlike the tumor suppressive isoform, P1-HNF4α, P2-HNF4α appears to be pro-proliferative, and to directly repress Bmal1.12 Loss of BMAL1 alters the expression of numerous metabolic genes including those involved in fatty acid metabolism, lipoprotein production, cholesterol export, and atherosclerosis.39–43 Liver-specific BMAL1 knockout (BKO) mice have a fatty liver, dyslipidemia, and hypoglycemia during fasting.43,44 Thus, induction of P2-HNF4α and loss of BMAL1 are two inter-related but distinct mechanism that promote fatty liver disease and HCC.
In the context of HCC, forced P1-HNF4α expression inhibits cell proliferation and suppresses epithelial–mesenchymal transition (EMT).29,45–47 On the other hand, complete HNF4α deficiency promotes cancer cell proliferation and diethylnitrosamine (DEN)-induced HCC formation.48,49 Interestingly, in combination with a high fat diet (HFD), we have shown that the loss of HNF4α induces early onset HCC similarly in male and female mice.11 We have also shown that approximately 50% of human HCC is HNF4α-positive, expressing the P2 isoform either in combination with the P1 isoform, or by itself. The P2-HNF4α isoform not only suppresses the expression of Bmal1, but induces cytoplasmic localization of the tumor suppressive P1-HNF4α isoform,10 an event that could be due to phosphorylation of P1-HNF4α by SRC tyrosine kinase and/or protein kinase C, both of which are implicated in liver cancer.35,50–52
Though loss of P1-HNF4α has negligible effects on Bmal1 expression in normal hepatocytes and HCC cells alike,12 its cytoplasmic localization in HCC means a loss of circadian restraint of many genes involved in cell proliferation and tumor progression. In contrast to HNF4α-positive tumors, HNF4α-deficient HCC express high levels of BMAL1. This inverse relationship is only present in HCC and not in the context of healthy hepatocytes, where both proteins are robustly co-expressed. Forced expression of BMAL1 in HNF4α-positive HCC cells causes apoptosis and inhibits tumor growth in vivo.10 Similarly, forced expression of P1-HNF4α in HNF4α-deficient but BMAL1-positive cells also inhibits HCC growth and proliferation. This leads to the hypothesis that while BMAL1 and HNF4α coordinate gene transcription programs that are compatible in a healthy hepatocyte, they are incompatible in the context of HCC, where the tumor repressive form of HNF4α (P1-HNF4α) is replaced at least partially by alternate P2 isoforms in HNF4α-positive tumors.
In this study, we examined whether concomitant reduction of both BMAL1 and HNF4α could alter liver disease progression and HCC risk. Our results suggest that in the absence of these transcription factors, the liver is protected from the rapid progression of liver disease in the context of chemical carcinogens and HFD feeding, and in the STAM model, a mouse model of progressive liver disease and HCC, though single deletion of these genes results in accelerated liver disease. We identify numerous cancer-related genes that are dysregulated in the inducible liver-specific HNF4α/BMAL1 double knockout (BHLivDKO) mice, which could be responsible for the protection against HFD-induced HCC. Finally, our results indicate that a pharmacological inhibitor of Bmal1 expression could be effective against HNF4α-negative/BMAL1-positive HCC, underscoring the importance of stratification of HCC in terms of HNF4α and BMAL1 expression for effective treatment in the clinic.
2 |. MATERIALS AND METHODS
2.1 |. Animals
Animal use was approved by the UT Health Institutional Animal Care and Use Committee. Mice were group housed in pathogen-free conditions and fed ad libitum with chow (PicoLab Rodent Diet 5053). Animals were entrained in 12-h light/12-h dark cycles. To generate the conditional double knock out mice, Hnf4F/F,AlbERT2cre (H4LivKO) mice were crossed with the Arntltm1witF/F,AlbERT2cre (Bmal1LivKO) mice. Albumin Cre [SA+/Cre-ERT] mice were kindly gifted by Dr. Daniel Metzger. Cre+ mice enabling inducible loss of Hnf4a and Arntl (Bmal1) are referred to as “BHLivDKO.”
2.2 |. Tamoxifen injections
H4LivKO, BmalLivKO, BHLivDKO, and wild-type (WT) littermates were injected intraperitoneally with tamoxifen (10 mg/ml) in corn oil for 5 consecutive days (days 1–5). Tamoxifen injections were performed at approximately ZT4 each day.
2.3 |. STAM mouse model
The NASH-HCC model (the so-called “STAM” mouse) was generated by a single subcutaneous injection of 200 μg streptozotocin (STZ) (Sigma) 2 days after birth for both BHLivDKO and Bmal1LivKO genetic background mice. At 4 weeks of age, mice were placed on HFD (Research Diet Cat# D12492). Mice were injected with tamoxifen to delete Bmal1 and Hnf4a in the liver at 8 weeks of age.
2.4 |. Metabolic phenotyping
Mice were placed in metabolic cages (Comprehensive Lab Animal Monitoring System-CLAMS, Columbus Instruments) at 15 weeks of age and provided ground diet ad libitum. Data were collected for 4 days, averaged, and analyzed using Oxymax V 4.87. Energy expenditure data are normalized to body weight.
2.5 |. Basal glucose measurement
Glucose was measured using a glucose meter (ACCU-CHEK Nano). Glucose was measured from tail blood 5 h after fasting.
2.6 |. Serum and hepatic triglyceride measurements
Liver and serum triglycerides (TGs) were examined using a kit from Cayman (Cat# 10010303) according to the manufacturer’s protocol. Briefly, 400 mg of liver tissue was homogenized in NP40 substrate assay reagent containing protease inhibitors. Ten μl of liver samples or standards were added to reactions. Levels of total hepatic cholesterol, LDL, and HDL were measured using a BioVison kit (Cat#k613–100).
2.7 |. Immunoblotting
Liver tissue was homogenized using a TissueLyserLT (QIAGEN). Cultured cells were sonicated (Qsonica, Newton, USA) for 10 s at 30% amplitude in RIPA buffer. Samples were spun at 10,000 × g for 10 min and supernatant protein concentration was determined using the bicinchoninic acid (BCA) method. Lysates were analyzed using 8% SDS polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. For antibodies, see Table S2.
2.8 |. RNA extraction and qRT-PCR
Total RNA was isolated using TRIzol reagent according to the manufacturer’s protocol (Thermo Fisher Scientific). One μg of total RNA was used for cDNA synthesis using an iScript cDNA Synthesis Kit (Bio-Rad). Advanced Universal SYBR Green Super mix (Bio-Rad) was used for qPCR amplification using a Bio-Rad C1000. PCR protocol settings were as follows: 95°C for 30 s, 95°C for 10 s, 62°C for 30 s, and then 39 cycles at 65°C for 31 s and 65°C for 5 s. Ribosomal subunit 18S expression was used as normalization for gene expression. Fold change in mRNA expression for each gene was calculated using 2−ΔΔC. For primers, see Table S3. For multiple time point experiments, expression is normalized to the control condition at ZT0.
2.9 |. Transcriptomic profiling (RNA-seq) and analysis
Livers were rinsed in PBS and immediately frozen in liquid nitrogen prior to RNA preparation. Total liver RNA was submitted to the Cancer Genomics Center at The University of Texas Health Science Center at Houston. Total RNA was quality-checked using Agilent RNA 6000 Pico kit (#5067–1513) by Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, USA). RNA with an integrity number of greater than 7 was used for library preparation. Libraries were prepared with KAPA mRNA HyperPrep (KK8581, Roche) and KAPA UDI Adapter Kit 15 μM (KK8727, Roche) following the manufacturer’s instructions. The quality of the final libraries was examined using Agilent High Sensitive DNA Kit (#5067–4626) by Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, USA), and the library concentrations were determined by qPCR using Collibri Library Quantification kit (#A38524500, Thermo Fisher Scientific). The libraries were pooled evenly and went for the paired-end 75-cycle sequencing on an Illumina NextSeq 550 System (Illumina, Inc., USA) using High Output Kit v2.5 (#20024907, Illumina, Inc., USA).
The raw data were pre-processed using Cutadapt (v1.15). (Cutadapt removes adapter sequences from high-throughput sequencing reads to remove the adapter sequences and bases with quality scores <20.) We used ultrafast universal RNA-seq aligner STAR (v2.5.3a) to map the RNA-seq reads to mouse the reference genome GRCm38. Gene read counts were calculated through setting the argument –quantMode to “GeneCounts,” which would allow STAR to quantify uniquely-mapped reads per gene from the GencodeM15 reference. We filtered out those genes with <5 reads in all samples and conducted the differential expression analysis by DESeq2 software [cite Differential expression analysis for sequence count data]. The p-values of genes were adjusted using the Benjamini and Hochberg’s procedure to control the false discovery rate (FDR). And the differentially expressed genes (DEGs) were defined as the genes with fold change >2 and FDR <0.05. Venn diagram summarizing the overlap between total DEGs, with p-values >.01 and fold change >1. None redundant gene ontology and KEGG pathway enrichment analysis were performed using WebGestalt (v0.4.3) software [cite WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs]. All raw data and processed read count have been submitted to Gene Expression Omnibus (GSE173968).
2.10 |. Cell culture
Cell lines were obtained from the American Tissue Culture Collection (ATCC, Manassas, VA). Hepa1c1c7 and HepG2 Cells were grown in Eagle’s Minimal Essential Medium, and AML12 cells were grown in a 1:1 mixture of Dulbecco’s Modified Eagle’s Medium and Ham’s F12 medium supplemented with 10% fetal bovine serum, 100 units ml−1 penicillin G sodium, and 100 μg ml−1 streptomycin.
2.11 |. Transient transfection and siRNA
Cells were transfected using Lipofectamine RNAiMAX Transfection reagent according to the manufacturer’s manual. Knockdown was performed using mouse Bmal1 (THERMOFISHER Cat# 4390771) and HNF4α (sense: CGCUUGAGGAAGACCUACUdTdT, antisense: UCAUCCAGAAGGAGUUCGCdTdT) siRNA. Briefly, 1 × 106 cells were transfected with 25 pmol of siRNA and 5 μl of Lipofectamine. Scrambled siRNA with similar GC content served as control.
2.12 |. MTT assay
Cell viability was assessed by MTT proliferation assay using a CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT) Kit (Promega) according to manufacturer’s instructions. Absorbance was read at 570 nm using a Cytation 5 reader.
2.13 |. Transwell migration assays
Cells were seeded into the upper chamber of a Transwell insert in serum-free medium at a density of 50 000 cells/well. For migration assays, medium containing 20% FBS was placed in the lower chamber as a chemoattractant, and cells were incubated for 48 h in a CO2 incubator. Non-migrating cells were removed from the upper chamber by scraping. Migrated cells were fixed with methanol for 10 min and stained with 0.1% crystal violet. Cells were imaged using a Cytation 5 and 4 randomly chosen fields were used for quantification.
2.14 |. Histology
Mouse livers harvested at specific time points were fixed in 10% neutral buffered formalin for 48 h and embedded in paraffin. Sections were washed twice with xylene (3 min) and rehydrated in ethanol gradients (100%, 2 × 5 min; 90%, 1 × 5 min; 80%, 1 × 5 min; and 70%, 1 × 5 min). Five μm sections were mounted on glass slides and stained with hematoxylin and eosin (H&E) or Oil Red O. For Oil Red O staining, sections were counterstained in hematoxylin, mounted and cover-slipped, and imaged using a Cytation 5. (For scoring, see Table S1.)
2.15 |. Quantification and statistical analysis
Kaplan–Meier is a non-parametric statistic used to estimate the survival function from lifetime data. Survival curves of patients with HCC were analyzed according to gene expression using the tumor liver hepatocellular carcinoma (TCGA) LIHC dataset using the R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl). Survival time measured from the time of initial diagnosis to the date of death or the date of the last follow-up. The survival distribution was estimated by the Kaplan–Meier method. p-values <.05 were considered to be statistically significant.
Results are expressed as mean ± SEM. Experiments involving two variables were analyzed by two-way ANOVA using Sidak’s multiple comparisons test (Prism 8.0). A sample size calculator (https://clincalc.com/stats/samplesize.aspx) assisted with number approximations.
3 |. RESULTS
3.1 |. Inducible loss of hepatic BMAL1 and HNF4α prevents hepatic steatosis
Since BMAL1 or HNF4α, but not both, are expressed in HCC, we examined whether concomitant reduction of BMAL1 and HNF4α delays liver disease. To this end, we generated an inducible DKO mouse model whereby tamoxifen treatment results in loss of the floxed Bmal1 and Hnf4a alleles (“BHLivDKO”). BHLivDKO mice were generated by mating Hnf4F/F,AlbERT2cre (H4LivKO) mice12 with the Arntltm1witF/F,AlbERT2cre (Bmal1LivKO)10,28 mice (Figure 1A). The extent of BMAL1 and HNF4α depletion in both male and female mice was analyzed by Western blot and qPCR at two zeitgeber times (ZT), ZT8 (8 h following “lights on” in the animal room) and ZT20 (8 h following “lights off” in the animal room) (Figure 1B,C and Figure S1).
FIGURE 1.
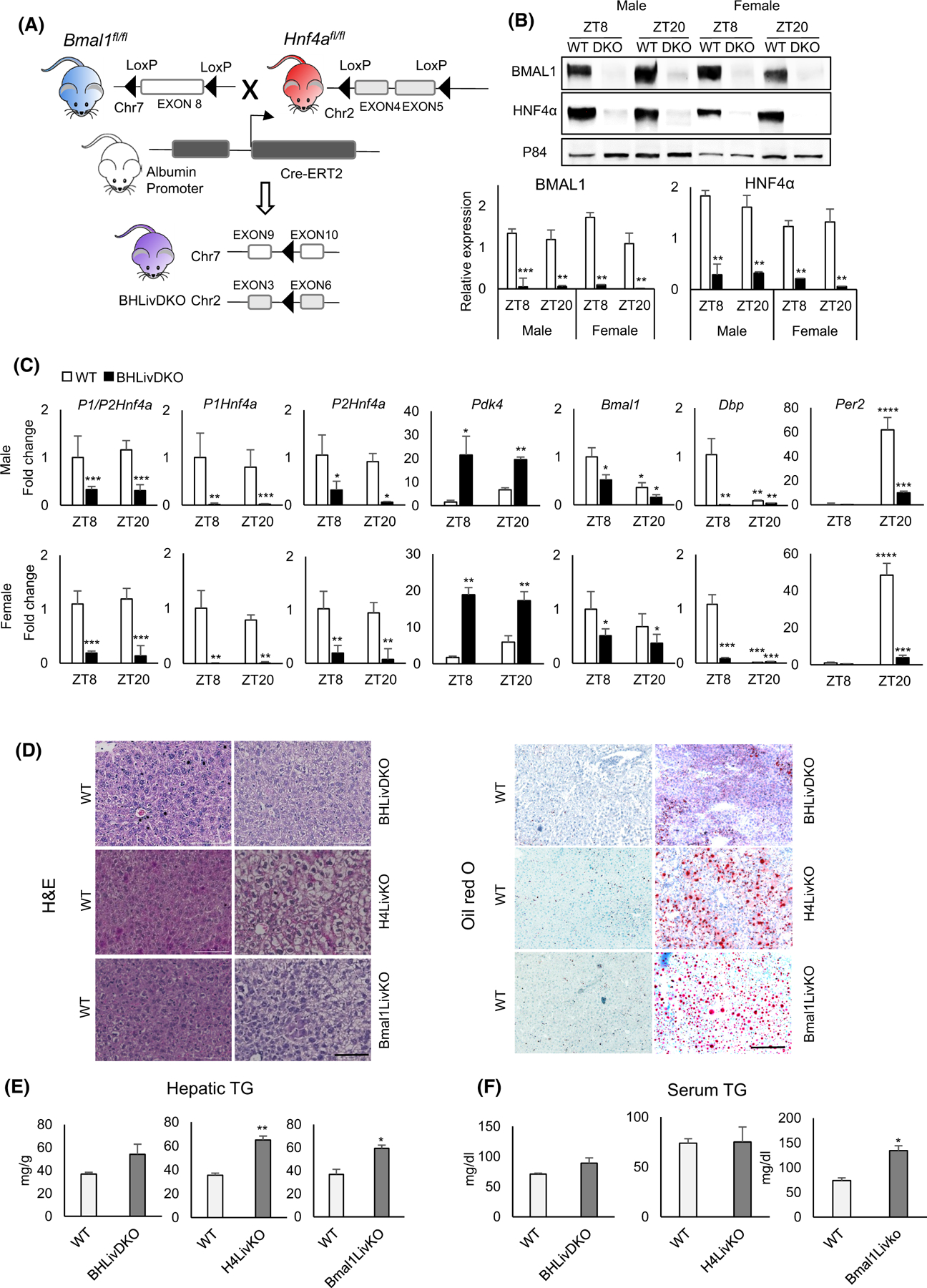
Loss of hepatic BMAL1 and HNF4α is protective against ectopic lipid deposition in the liver. (A) Mouse model of the hepatic Bmal1 and Hnf4a inducible double knockout (BHLivDKO). (B) Western blot of BMAL1 and HNF4α in the liver of BHLivDKO mice 10 days post-tamoxifen injection. Quantification of the immunoblots normalized to P84 (bottom panels). Student’s t-test. **P ≤ 0.01; ***P ≤ 0.001. (C) Hepatic gene expression in WT and BHLivDKO measured by qPCR. Two-way ANOVA, Sidak’s multiple comparisons test (N = 6–8): *p < .03; **p < .005; ***p < .0005. (D) H&E (left panel) and Oil Red O (right panel) staining of WT, BHLivDKO, H4LivKO and Bmal1LivKO (Scale bar = 100 μm). (E and F) Hepatic (E) and serum (F) triglyceride levels in WT, BHLivDKO and H4LivKO mice. Significance (p < 0.05) was determined by Mann–Whitney U-test.
While this strategy prevents the expression of the tumor suppressive isoform, it also prevents expression of the P2 isoform, which has positive effects on HCC proliferation.12 The use of primers that recognize both HNF4α isoforms as well as those targeting the specific isoforms, revealed a pronounced depletion in mRNA expression for the P1- and P2-HNF4α isoforms (Figure 1C). Expression of Pdk4 (normally repressed by HNF4α) was elevated, while Bmal1 and its E box-containing target genes Dbp and Per2 were both significantly downregulated at the level of mRNA expression in BHLivDKO (Figure 1C). Interestingly, BHLivDKO mice have less lipid deposition in the liver compared with the single Hnf4a and Bmal1 inducible knockout mice, as measured by H&E (left) and Oil Red O staining (right) (Figure 1D). To test for changes in circulating and hepatic TGs in BHLivDKO mice, we measured serum and liver TGs in WT and BHLivDKO littermate mice (Figure 1E,F). Liver and serum TGs were somewhat elevated in BHLivDKO compared to their littermate controls but did not reach significance. In contrast, both single knockout mice had significantly higher TG levels in the liver (Figure 1E), while only Bmal1LivKO mice also showed increased serum TG compared to their WT littermates (Figure 1F). BHLivDKO mice were also slightly hypoglycemic compared to controls (Figure S1), similar to single Hnf4a and Bmal1 KO mice.28,43,44 No significant changes were observed in diurnal oxygen consumption, carbon dioxide emission, body weight, or energy intake between the two genotypes (Figure S1). While BHLivDKO mice showed normal diurnal patterns of movement and no changes in body weight, their home cage locomotion was slightly reduced compared to WT littermate controls (Figure S1).
3.2 |. Concomitant loss of BMAL1 and HNF4α suppresses DEN-induced HCC formation and IL6 activation
Previous studies reveal that non-inducible, liver-specific Bmal1 and inducible liver-specific Hnf4a individual knockout mice are more susceptible to developing HCC.6,11 Based on the reduced lipid deposition in BHLivDKO mice, we examined whether the elimination of BMAL1 and HNF4α together was protective in the context of HCC. Using Hnf4a individual knockout mice (H4LivKO) as positive controls, we used DEN injection followed by HFD feeding to induce HCC, a combination of insults that has been shown to accelerate the growth of HCC53,54 (Figure 2A). WT male and female mice were treated with DEN at day 15 and injected with tamoxifen for 5 days starting at 8 weeks of age. When switched to HFD 2 weeks later, male WT mice showed reduced weight gain over time, as is commonly seen in mice with tumor burdens (Figure 2B). At 45 weeks of age, BHLivDKO mice were partially protected from HFD- and DEN-induced HCC compared to single H4LivKO mice (Figure 2C, quantification right). Female mice have been reported to be partially protected from DEN-induced HCC,55 and our results were consistent with this finding, with only 50% of WT females showing tumor incidence (vs. 100% of males), though females of the BHLivDKO genotype were further protected from HCC incidence, showing only 15% tumor incidence. The number of tumors per liver were also reduced in the BHLivDKO in both sexes compared to WT and H4LivKO controls (Figure 2C). In contrast to BHLivDKO, we found 100% tumor incidence in the single H4LivKO liver following DEN and HFD treatment, consistent with earlier reports.11
FIGURE 2.
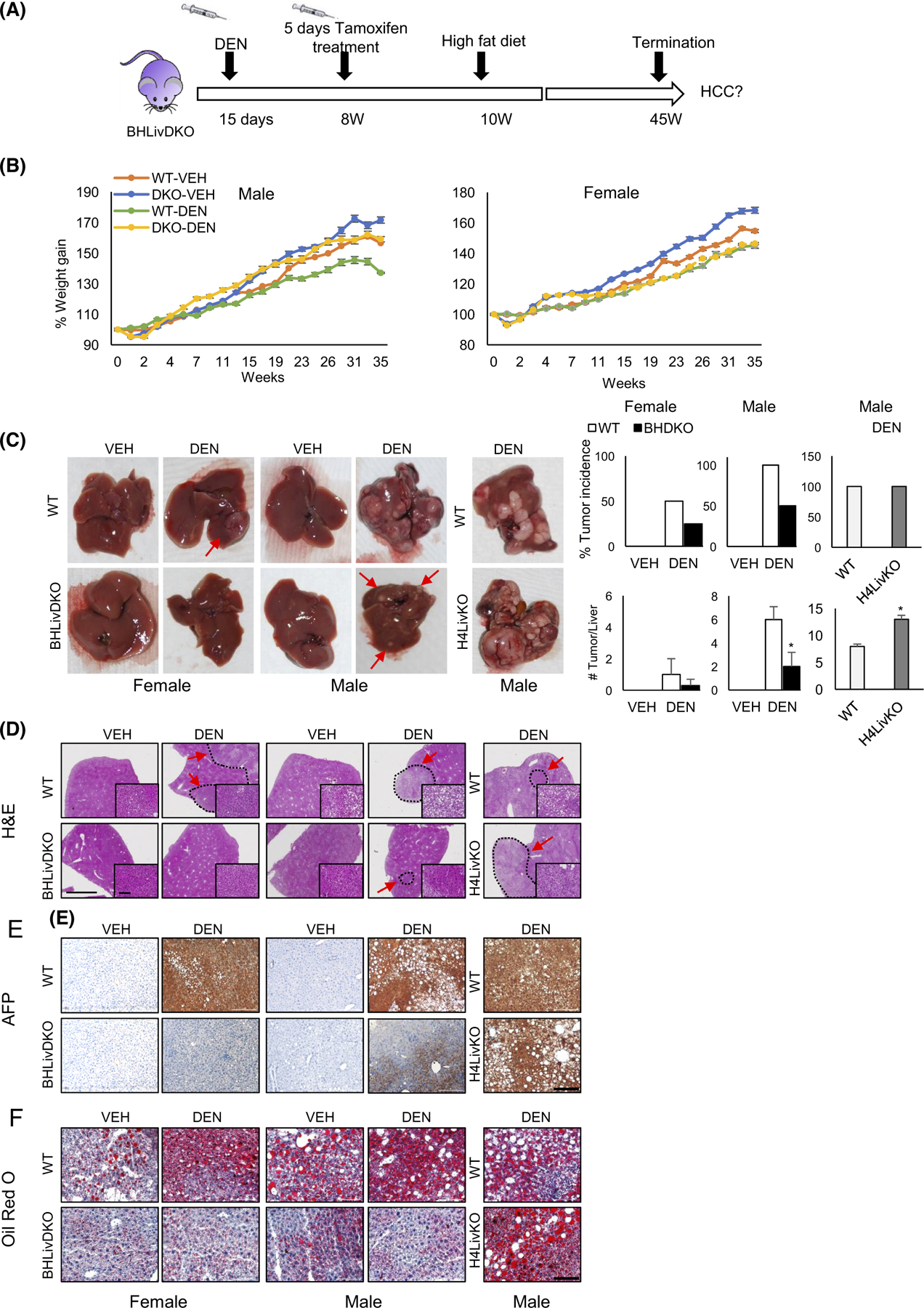
Mice with hepatic BMAL1and HNF4α deficiency are less prone to develop hepatocellular carcinoma (HCC). (A) Experimental timeline for carcinogen (DEN)-induced HCC in WT and BHLivKO mice. (B) Body weights of WT and BHLivDKO mice fed high fat diet (HFD) for 35 weeks with VEH versus DEN treatment. (C) Whole livers taken from mice fed HFD following VEH or DEN injection, left panel. Percent tumor incidence and the number of tumors per liver per animal group (right panel). Two-way ANOVA, Sidak’s multiple comparisons test (N = 8–12): *p < .03. (D–F) H&E (D), AFP (E), and Oil-Red O (F) staining of WT and BHLivDKO, and H4LivKO mouse livers after VEH or DEN injection and after 35 weeks of HFD. (Scale bar = 2000 μm, magnified scale bar = 100 μm, 200 μm).
To further phenotype the livers of WT and BHLivDKO with or without DEN treatment, we performed pathological analysis using H&E staining and histological scoring (Figure 2D and Table S1). Collectively, male and female BHLivDKO livers showed reduced lobular inflammation, fewer tumors, and reduced tumor size for tumor-bearing livers (Table S1). Staining of the liver for the protein alpha fetoprotein (AFP), a marker of HCC, revealed robust AFP expression in WT male and female mice treated with DEN, but not BHLivDKO mice (Figure 2E). Single knockout H4LivKO mice also showed robust AFP expression, similar to WT mice treated with DEN, consistent with previous reports. To test for lipid deposition in the different mouse models, liver sections were stained with Oil Red O. Oil Red O staining revealed significant lipid deposition in male and female WT mice treated with DEN, as well as single H4LivKO mice, but not BHLivDKO mice (Figure 2F). These data support the idea that while having both BMAL1 and HNF4α is incompatible in the context of HCC,12 deficiency of both of these proteins delays HCC formation and growth, and loss of both proteins provides a protective environment from liver-induced injury and subsequent HCC incidence.
Though we have previously reported that single knock-out of HNF4α combined with a HFD causes accelerated liver disease and HCC in both male and female mice,56 we also used the STAM model on the genetic background of an inducible hepatic Bmal1-specific knockout mouse, “Bmal1LivKO” (Figure S2A). In the context of liver-specific loss of BMAL1, Bmal1LivKO mice showed rapidly accelerated liver disease and decline in health, with a greatly reduced probability of survival compared to CRE-deficient WT littermates also treated with tamoxifen (Figure S2B). When animals that survived to the 16-week time point were euthanized, STZ-treated Bmal1LivKO livers showed advanced HCC in both male and female mice, compared to vehicle-treated littermates (Figure S2C). Though tumor incidence was high in both WT and Bmal1LivKO mice, the number of tumors in BmalLivKO mice surpassed that of the CRE-negative control mice (Figure S2C). Consistent with the number of visible tumors, H&E staining revealed prominent tumors in the STZ-treated Bmal1LivKO livers, which also showed pronounced AFP expression and lipid deposition as measured by immunohistochemistry and Oil Red O staining, respectively (Figure S2D–F). To determine how inducible loss of BMAL1 affected HCC-associated gene expression, we analyzed the expression of several cyclins, including Ccna2, Ccnd1, Ccnb1, and the inflammatory cytokine, interleukein-6 (Il6) 10 days following tamoxifen treatment in male mice. Though only Ccnd1 was elevated among the cyclins, hepatic Il6 trended higher but was not significant (Figure S3A). Though Bmal1 mRNA expression was reduced by tamoxifen treatment, western blot analysis revealed an almost complete loss of BMAL1 protein in the liver, 10 days following the last tamoxifen injection (Figure S3B). To determine how cyclin and Il6 gene expression was altered in Bmal1LivKO mice after the development of HCC, we performed RT-PCR on the liver of mice euthanized at 16 weeks of age. At this stage, Ccna2, Ccnd1, Ccnb1, and hepatic Il6 showed a pronounced induction in Bmal1LivKO mice, with, levels highest in STZ-treated Bmal1LivKO knockout mice compared to the vehicle-treated controls (Figure S3C) in both male and female mice (Figure S3C). We compared Rev-erba (Nr1d1) and Rev-erbb (Nr1d2) expression in BHLivDKO mice compared to single H4LivKO and Bmal1LivKO mice. Surprisingly, Nr1d1 and Nr1d2 expression were only moderately altered in BHLivDKO and Bmal1LivKO at ZT8 or ZT20, in contrast to H4LivKO mice, in which loss of HNF4α at either ZT tested resulted in greatly reduced Nr1d1 and Nr1d2 (Figure S3D).
To further characterize BHLivDKO livers, we examined the expression of several HCC-related genes in the liver after DEN or vehicle (Veh) administration followed by HFD. R-spondin (Rspond), a regulator of WNT signaling, Paternally Expressed 10 (Peg10) and Glypican 3 (Gpc3) are all markers of HCC.57–59 Our results revealed a significant reduction in all three genes in DEN-treated BHLivDKO liver compared to controls (Figure 3A), though no changes were noted in BHLivDKO livers compared to controls under HFD alone (Veh). In contrast to the males, significant changes were not detected in BHLivDKO DEN-treated female mice compared to their controls (Figure 3A, bottom panels). While H4LivKO mice showed elevated TG levels in the liver compared to WT DEN-treated mice, liver and serum TG content was comparable between WT and BHLivDKO mice following DEN injection and HFD feeding (Figure 3B).
FIGURE 3.
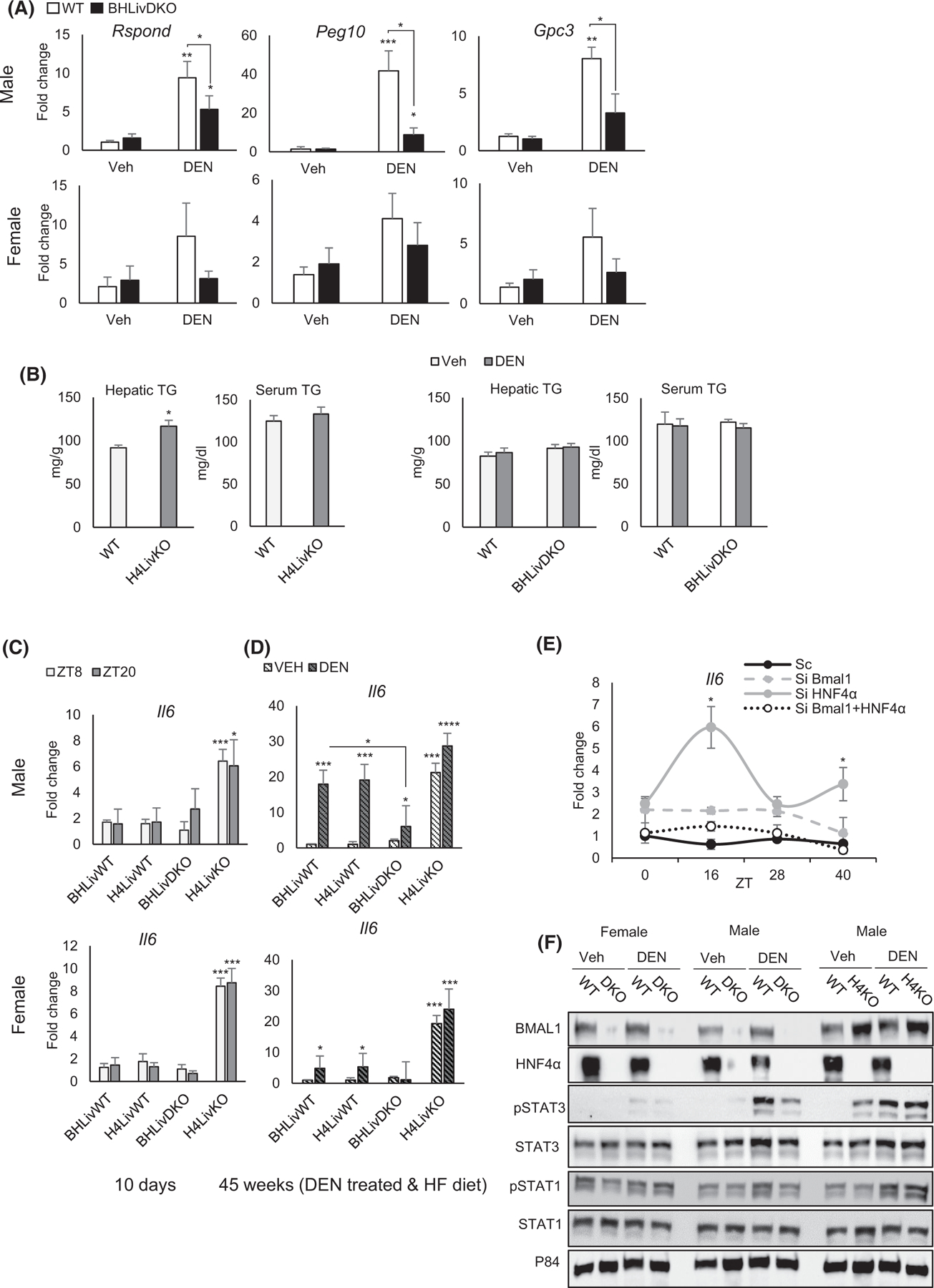
Loss of hepatic BMAL1 and HNF4α protects against high fat diet-induced hepatocellular carcinoma (HCC). (A) Expression of HCC-specific genes in the livers of WT and BHLivDKO mice as measured by qPCR. (B) Hepatic and serum triglyceride levels in the WT, H4LivKO and BHLivKO mice following VEH or DEN administration and 35 weeks of HFD feeding. (C and D) RT-PCR reveals mRNA abundance of Il6 in WT and BHLivDKO mice 10 days after tamoxifen treatment (C) and 45 weeks after DEN plus HFD (D). (E) qPCR reveals expression of Il6 following Hnf4α and Bmal1 siRNA or scrambled control treatment. Two-way ANOVA, Sidak’s multiple comparisons test (N = 6–10):*p < .03; **p < .005; ***p < .0005, ****p < .0001. (F) Western blot of whole cell liver lysates from WT, BHLivDKO and H4LivKO mice fed with HFD following VEH/DEN injections, using antibodies to STAT1, STAT3, total STAT protein, and P84. Two-way ANOVA, Sidak’s multiple comparisons test (N = 8–12): *p < .03; **p < .005; ***p < .0005, ****p < .0001.
In HCC, elevated IL-6 involves the downstream phosphorylation of Signal Transducer and Activator of Transcription 3 (STAT3), while phosphorylation of STAT1 is associated with fatty liver.60–62 We found that baseline levels of Il6 mRNA expression in BHLivDKO was not changed in 8-week old mice, 10 days following tamoxifen injection, while single H4LivKO mice showed de-repressed Il6 expression, as previously described20 (Figure 3C). Hepatic Il6 expression was greatly augmented following DEN injection and HFD feeding in WT male mice (Figure 3D), as reported previously.36 However, BHLivDKO mice were largely protected from this increase. Since both vehicle- and DEN-treated H4LivKO mice showed significantly elevated Il6 in male and female mice, Il6 expression in control and BHLivDKO mice treated with DEN was reduced from that observed in male mice, consistent with their protected state from DEN-induced HCC (Figure 3D). To parse out whether induction of Il6 might be a direct consequence of transcriptional manipulation of BMAL1 or HNF4α, we performed siRNA experiments in serum shocked (circadian synchronized) AML12 cells. While reduced expression of HNF4α increased Il6 dramatically and in a circadian fashion (Figure 3E and Figure S3E), reduced BMAL1 did not significantly increase Il6 expression. In addition, reduced expression of both BMAL1 and HNF4α resulted in negligible changes in Il6 expression (Figure 3E), indicating that HNF4α controls Il6 expression even in the absence of liver injury.
To further address short-term changes in vivo as a result of BMAL1 and HNF4α loss, we measured total cholesterol (TC), and high and low-density lipoproteins (HDL and LDL) in the serum of BHlivDKO, H4LivKO, and Bmal1LivKO. While total TC, HDL, and LDL did not change between BHLivKO and controls, single knockout mice showed significant changes in TC, HDL, or LDL, thought to different degrees (Figure S4A). This is consistent with prior reports of lipids being trapped in the liver for H4LivKO mice, likely due to dysregulation of apolipoprotein gene expression.25
To determine whether Il6 expression was consistent with levels of STAT3 phosphorylation, we analyzed total and phosphorylated STAT3 (pSTAT3) in the livers of DEN- or vehicle-treated male and female mice of each genotype. Consistent with the Il6 expression results, H4LivKO livers had elevated levels of pSTAT3 in both DEN and Veh conditions. In contrast, BHLivDKO mice showed lower levels of pSTAT3 compared to H4LivKO (Figure 3F and Figure S4B,C). Phosphorylated STAT1 (pSTAT1), which is associated with fatty liver but not HCC,63 was not changed by DEN treatment, perhaps due to the fact that all groups were on HFD (Figure 3F and Figure S4B,C). Taken together these results indicate that while HNF4a appears to repress the IL6/STAT3 pathway, BMAL1 may be essential for it, at least in the absence of HNF4a and in the presence of DEN and HFD.
3.3 |. Concomitant loss of hepatic BMAL1and HNF4α alters expression of genes involved in lipogenesis
To identify biological pathways that might confer protection against HCC in the context of BHLivDKO livers, we performed RNA-seq analysis on the livers of BHLivDKO and H4LivKO male mice and their corresponding WT littermate controls. As anticipated, RNA-seq data revealed vastly variable gene expression profiles in H4LivKO mice compared to WT controls. This is evident in the heatmap of the top five percent of genes ranked by the highest median absolute deviation (Figure 4A). Venn diagrams reveal the extent to which genes are altered in each genotype versus similarities (Figure 4B). Specifically, ∼200 genes were differentially expressed between the two genotypes, with 1012 upregulated and 1411 downregulated genes in H4LivKO livers (Figure 4A,B and S5A). BHLivDKO livers also showed a significantly different gene expression profile, with ∼2500 genes differentially expressed between WT and BHLivDKO mice, 807 being upregulated and 1716 being downregulated (Figure 4A,B and Figure S5A) Because analysis of WT control data showed 99.68% similarity across WT cohorts, we also compared gene expression patterns in BHLivDKO versus H4LivKO livers. This comparison resulted in ∼1000 genes differentially expressed in BHLivDKO compared to H4LivKO (Figure S5A). Fewer changes in gene expression were noted between WT and BHLivDKO fed HFD, with 611 being elevated and 382 being downregulated (Figure S5B). These results suggest that the concomitant loss of the circadian protein BMAL1 and differentiation factor HNF4α conferred significantly different transcriptional regulation in hepatocytes compared to the loss of HNF4α alone.
FIGURE 4.
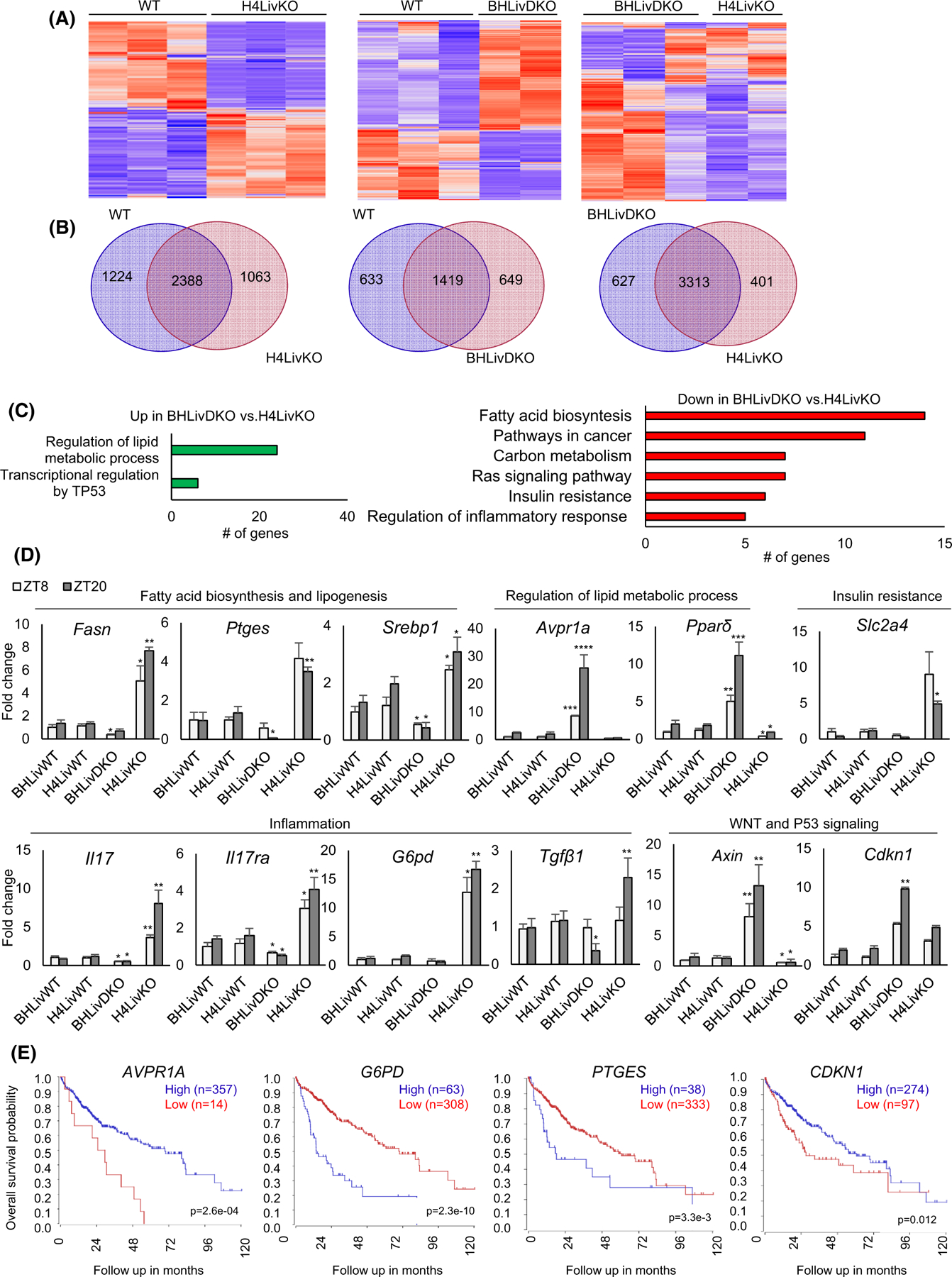
Dual loss of BMAL1and HNF4α alters the expression of genes involved in lipogenesis and cancer progression. (A) Heat maps of RNA-seq data reveal changes in gene expression of the top 5% of genes expressed between WT versus H4LivKO, WT versus BHLivDKO and H4LivKO versus BHLivDKO mice. (B) Venn diagrams for the total number of differentially expressed genes between WT versus H4LivKO, WT versus BHLivDKO and H4LivKO versus BHLivDKO genotypes. (C). Gene annotation of altered genes in BHLivDKO versus H4LivKO mice shows pathways both upregulated (red) or downregulated (green) in the BHLivDKO. (D) RT-PCR reveals altered expression of genes involved in lipid metabolism, inflammation, and inhibition of WNT/β-catenin signaling. Two-way ANOVA, Sidak’s multiple comparisons test (N = 6–8): *p < .03; **p < .005; ***p < .0005, ****p < .0001. (E) Kaplan–Meier survival curves of patients with HCC separated according to AVPR1A, G6P, PTGES and CDKN1 gene expression using the tumor liver hepatocellular carcinoma (TCGA) LIHC dataset in the R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl). Survival time measured from the time of initial diagnosis to the date of death or the date of last follow up. The survival distribution estimated by Kapan–Meier method. p-values <.05 were considered to be statistically significant.
Pathway analysis of genes altered in BHLivDKO mice versus H4LivKO livers revealed a large number of genes involved in “fatty acid biosynthesis,” “pathways in cancer,” “insulin resistance,” “lipid catabolic process,” and “regulation of inflammatory response” (Figure 4C). Within the “lipid catabolic process” category, peroxisome proliferator activated receptor delta (Ppard), fatty acid amide hydrolase (Faah), and carboxylesterase 1 g (Ces1g) (all implicated in protection from fatty liver or HCC64–66) were significantly downregulated in H4LivKO compared to the BHLivDKOs, while Enoyl-CoA Hydratase and 3-Hydroxyacyl CoA Dehydrogenase (Ehhadh), a gene upregulated in a subset of patients with HCC,67 was significantly elevated in H4LivKO (Figure S6). Genes involved in “regulation of lipid metabolic processes” and “transcriptional regulation by TP53” were generally up-regulated in BHLivDKO livers. We validated several of these genes by qPCR in all genotypes, and because some of these genes are known to be expressed diurnally, we tested expression at two different Zeitgeber times (ZT8 and ZT20) (Figure 4D). Genes involved in “fatty acid biosynthesis” included fatty acid synthase (Fasn), prostaglandin E2 (Ptges), and sterol regulatory element-binding transcription factor 1 (Srebp1). Levels of FASN mRNA are significantly higher in human NAFLD as is the expression of SREBP1, which is the key transcriptional regulator of FASN. Furthermore, FASN is the key metabolic enzyme of de novo lipogenesis, providing proliferative and metastatic advantage to cancer cells.68 Upregulation of FASN occurs in most human cancers, including HCC.69–73 Therefore, FASN provides a potential oncologic target and diagnostic marker for NAFLD formation.68–70,72–75 PGE2/PTGES2 is induced in human and rodent NASH livers,76–78 and is up-regulated in several types of cancer, including colorectal cancer,79,80 breast cancer,81,82 and HCC.83,84 Deregulated genes involved in “insulin resistance” or “regulation of inflammatory response” included solute carrier family 2 member 4 (Slc2a4), interleukin 17 (Il17), and interleukin 17 receptor A (Il17ra). Glucose-6-phosphate dehydrogenase (G6PD), involved in carbon metabolism, was greatly reduced in BHLivDKO compared to H4LivKO mice, as was integrin subunit alpha 2 (Itga2), in the “pathways in cancer” classification. IL17 signaling stimulates fibrosis by activating inflammatory and resident hepatocytes and amplifies production of IL6, IL1, TNFα, TGF1, as well as IL17, and promotes lipogenesis.85 In addition, elevation of IL17 and IL17RA is associated with poor prognosis of HCC.86,87 Of note, pathways that were significantly altered in H4LiKO compared to WT but not differentially altered between BHLivKO and WT were “maturity onset diabetes of the young,” “glycolysis/gluconeogenesis,” and “primary bile acid biosynthesis” (Figure S6). The bile acid receptor farnesoid x receptor (FXR) has been shown to play an important role in jet-lag-induced HCC.6
Genes upregulated in BHLivKO mice versus the single H4LivKO are involved in “WNT signaling” (Axin), “p53 signaling” (cyclin-dependent kinase inhibitor 1 [Cdkn1]), and “regulation of lipid metabolic process” (type arginine vasopressin receptor [Avpr1a] and Ppard). Deficiencies in different components of the WNT signaling pathway including Axin promote progression of HCC, and Axin prevents the proliferation and invasion of several types of cancer cells.88–90 Avpr1a is involved in hepatic glucose release, fatty acid esterification, and ureagenesis, and reduction of Avpr1a mRNA is an indicator of NAFLD progression.91–94 An anti-inflammatory role of PPARβ/δ has been demonstrated using PPARβ/δ knockout mice, and the PPARβ/δ agonist GW501516 was shown to protect mice fed a methionine/choline-deficient diet from hepatic inflammation,95,96 PPARβ/δ also plays a role in parenchymal and non-parenchymal hepatic cells and progression of liver fibrosis and inflammation.95,97,98 Thus, elevated Ppard in the BHLivDKO may be indicative of protection at the level of hepatic inflammation. Collectively, these genes both up and down regulated in BHLivDKO versus H4LivKO are consistent with the protected status of the BHLivDKO mice in terms of developing HCC, and may even play a casual role. Genes upregulated at ZT20 may be reflective of the potent circadian repression that HNF4α confers at this particular ZT in the liver.12,21
To gain insight as to which transcriptional changes might confer the greatest protection in BHLivDKO mice, we analyzed the survival probability for individuals with HCC using the TCGA data set, which contains survival data for 371 individuals (250 male and 121 female) (Figure 4E and Figure S7). Noteworthy were survival curves for AVPR1A and CDNK1A, where low levels of AVPR1A or CDNK1A correlated with low survival (Figure 4E). In contrast, low levels of G6PD and PTGES correlated with improved overall survival probability (Figure 4E).
3.4 |. Hepatic BMAL1and HNF4α deficiency protects against the derepression of cyclin genes caused by loss of HNF4α alone
An essential feature of cancer is dysregulated cell cycle control. Cyclin activation of the cyclin-dependent kinases is a well-recognized mechanism for uncontrolled proliferation of cancer cells.99,100 HNF4α represses cyclin genes and HNF4α-deficient mice express cyclins robustly and in a circadian manner.10,28,101 Consistent with this, we observed a robust induction of Cyclin A2 (Ccna2), Cyclin D1 (Ccnd1), and Cyclin B1 (Ccnb1) in H4LivKO liver in both males and females ten days following tamoxifen injections (Figure 5A). Ccnb1 acts as an essential regulatory subunit for Cdk1. Overexpression of Ccnb1 has been described in several cancers, including HCC.71,102–106 We evaluated expression of Ccnb1 in the BHLivDKO liver 10 days post-tamoxifen, and found the levels of Ccnb1 mRNA and protein to be significantly lower than in H4LivKO livers (Figure 5A and Figure S8A,B). Ccnd1, a central controller of cell cycle and cancer progression, was also significantly lower in the BHLivDKO mice compared with H4LivKO (Figure 5A and Figure S8). To determine how the different cyclins affect overall survival probability in humans, we again analyzed the TCGA LIHC group of 371 individuals. While low levels of CCNA2 and CCNB1 both correlated with significantly improved survival probability compared to individuals with high levels of expression, there was no significant correlation with CCND1 expression (Figure 5D), though CCDN1 levels have been reported to be elevated in the context of HCC.107 Thus, we conclude that decreases in Ccna2 and Ccnb1 may be providing the protective effects seen in BHLivDKO compared to H4LivKO mouse livers; Ccnd1 may play a less important role.
FIGURE 5.
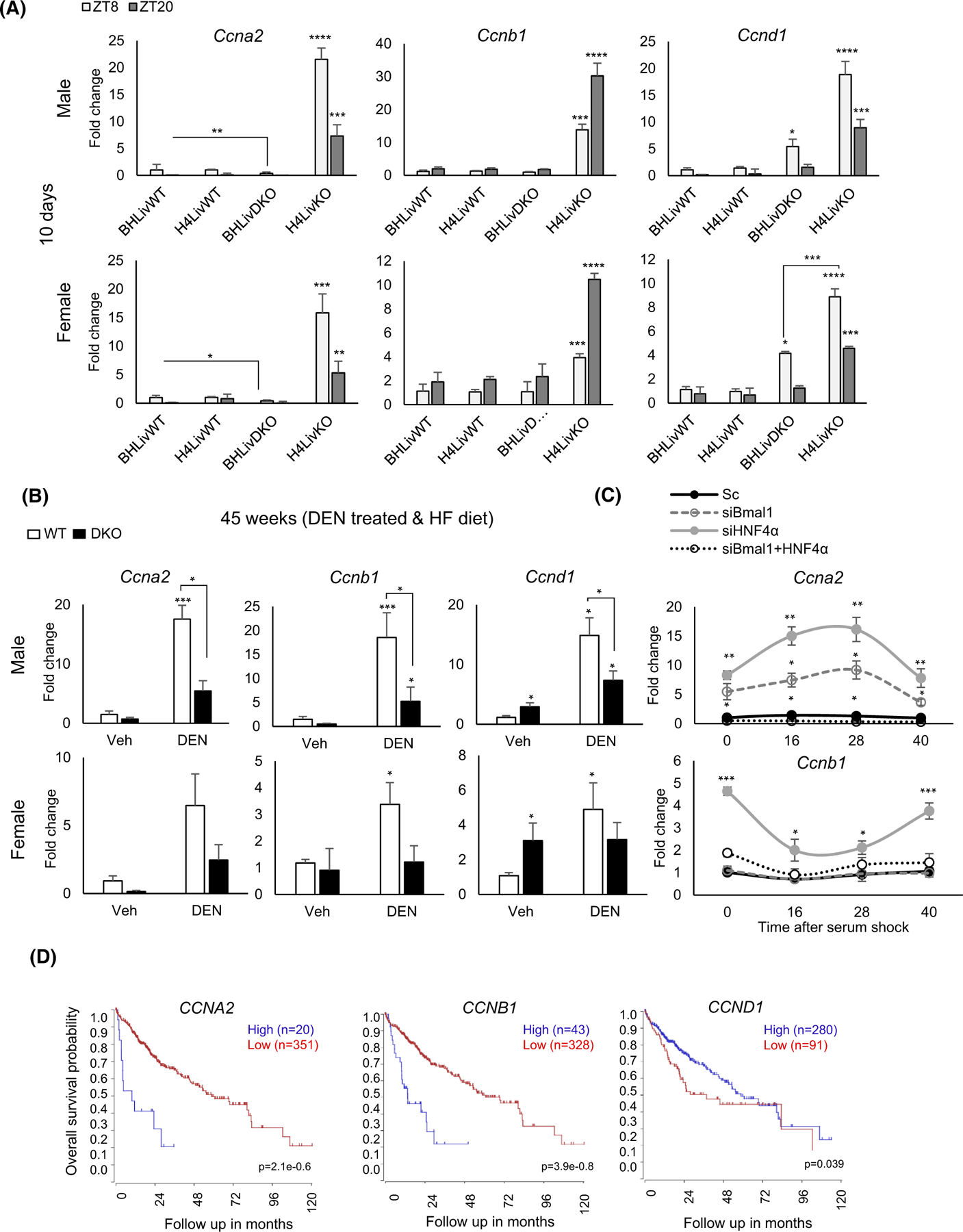
Expression of cyclin genes is significantly reduced in BHLivDKO mice compared to H4LivKO mice. (A) qPCR analysis shows expression of cyclin genes Ccnd1, Ccnb1 and Ccna2 10 days post-tamoxifen injection. Two-way ANOVA, Sidak’s multiple comparisons test (N = 6–8): *p < .03; **p < .005; ***p < .0005, ****p < .0001. (B) qPCR analysis of cyclin genes after DEN and HFD treatment, Two-way ANOVA, Sidak’s multiple comparisons test (N = 8–12): *p < .03; ***p < .0005. (C) Cyclin A2 (Ccna2) expression in Aml12 cells following knockdown of Bmal1, Hnf4α, or both Bmal1 and Hnf4α using siRNA at 0,16, 20, and 40 h post-serum shock. Scrambled oligonucleotide (Sc) was used as a control. Two-way ANOVA, Sidak’s multiple comparisons test (N = 10): *p < .03; **p < .005; ***p < .0005, ****p < .0001. (D) Survival curves show CCNA2, CCNB1, CCND1 genes expression using the tumor liver hepatocellular carcinoma (TCGA) LIHC dataset using the R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl). Survival time measured from the time of initial diagnosis to the date of death or the date of last follow up. The survival distribution estimated by Kaplan–Meier method. p-values <.05 were considered to be statistically significant.
We next analyzed Ccna1, Ccnb1, and Ccnd1 expression in the DEN-treated mice, and found that their expression was also significantly lower in the BHLivDKO versus WT animal at this late stage of liver disease (Figure 5B and Figure S8B). In addition to the rodent models, we performed knockdown experiments using siRNA to HNF4α and BMAL1 in a normal liver cell line to test the effects of HNF4α versus BMAL1 on cyclin gene expression following circadian synchronization of the cells by serum shock. Ccna2 and Ccnb1 expression was significantly lower after knockdown of both HNF4α and BMAL1 compared to single knockdown siRNA approaches and scrambled siRNA, consistent with our in vivo experiments (Figure 5C and Figure S3E). In addition to the cyclins, we also analyzed Pcna and Ki67 in all genotypes. These proliferation genes were also significantly elevated in all genotypes with exception of the BHLivDKO (Figure S8C).
3.5 |. BMAL1 and HNF4α deficiency alters EMT of HCC
Epithelial plasticity is necessary for the development of HCC. EMT inducers such as TGF-β or Wnt/β-catenin signaling are capable of driving fibrogenesis and, ultimately, HCC in the liver.108,109 Forced expression of HNF4α can reduce tumor invasion and growth by increasing extracellular matrix contacts.110–112 Similarly, Bmal1 overexpression has been reported to inhibit EMT in cancer cells.113 To determine the effect of BMAL1 and HNF4α deficiency in EMT in liver cells, we used siRNA against one or both genes followed by MTT cell proliferation and migration assays in AML12 liver cells. Interestingly, knockdown of BMAL1 and HNF4α separately increased cell proliferation and migration. In contrast, knockdown of both BMAL1 and HNF4α expression rescued the proliferation and migration effects, consistent with our in vivo results (Figure 6A,B). Furthermore, our data revealed that EMT-related gene expression, including the expression of Ctnnb1 and Snail1, was significantly reduced to control levels by concomitant inhibition of Bmal1 and Hnf4a expression, compared to single knockdown conditions (Figure 6C). Based on these in vitro siRNA data, we examined EMT-related gene expression in the BHLivDKO mice 10 days following tamoxifen treatment. Interestingly, we found a significant reduction of Ctnnb1, 10 days following knockdown (Figure 6D). We also observed a significant reduction in Ctnnb1 and Snai1 expression post DEN and HFD treatment in both male and female livers. There was also an increase in Cdh1 expression but only in the DKO males (Figure 6E). Taken together, these results reveal another pathway by which BHLivDKO mice are protected from HCC.
FIGURE 6.
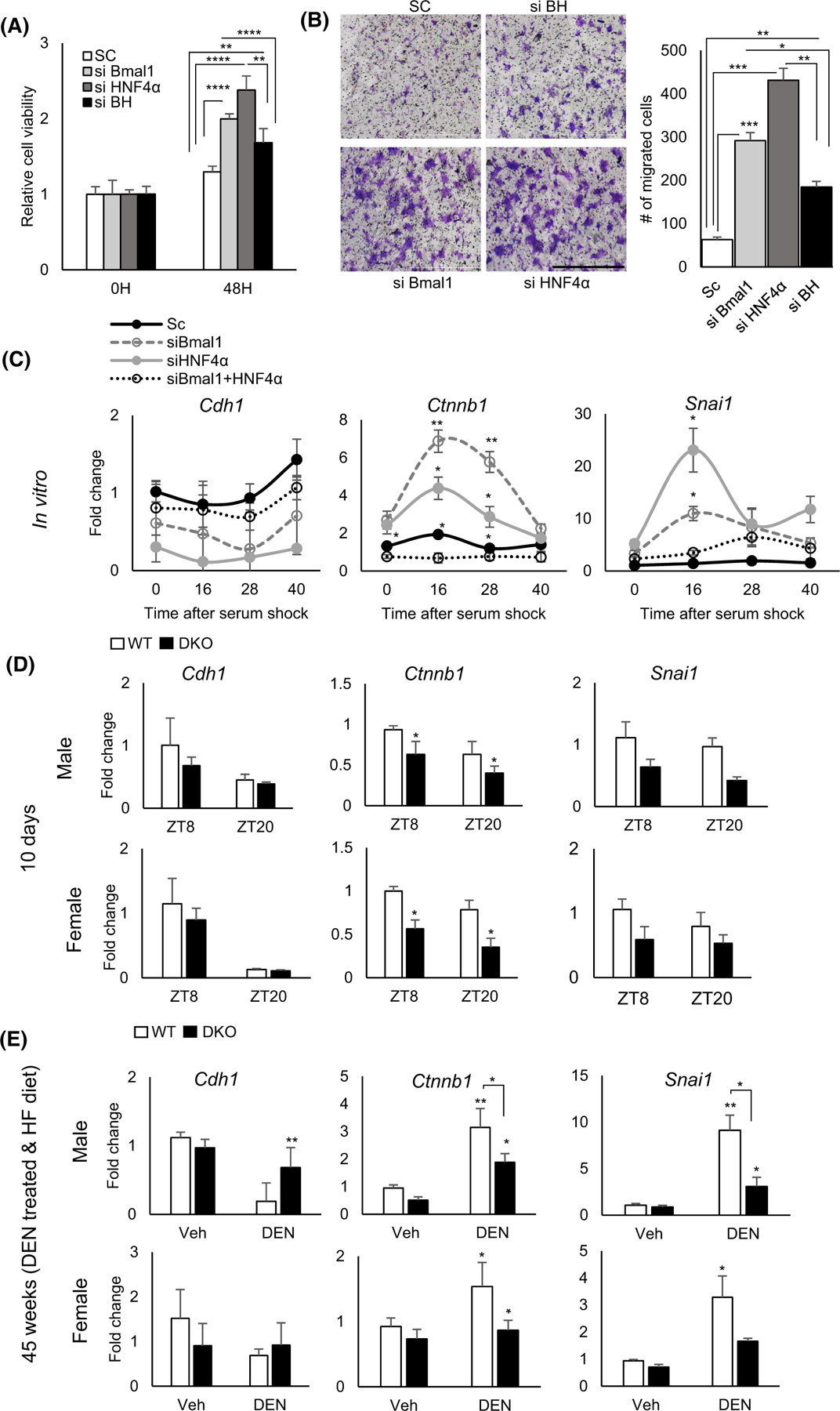
The Epithelial–Mesenchyme Transition (EMT) is impaired by dual loss of BMAL1 and HNF4α in the liver. (A) Fold change in proliferating Aml12 cells following HNF4α, Bmal1 or both Bmal1 and HNF4α knockdown using siRNA or scrambled control at 48 hr using MTT assay. (B) Migrated Aml12 cells following Hnf4α, Bmal1 or both Bmal1 and Hnf4α knockdown using siRNA or scrambled control. Quantification, left panel. (Scale bar = 1000 μm, N = 8). (C) qPCR reveals expression of EMT-related genes. in synchronized cells following Hnf4α, Bmal1 or both Bmal1 and Hnf4α using siRNA or scrambled control Two-way ANOVA, Sidak’s multiple comparisons test (N = 6–10): *p < .03; **p < .005; ***p < .0005, ****p < .0001 (D and E) Expression of EMT-related genes in BHLivDKO livers 10 days post tamoxifen injection (D) (Two-way ANOVA, Sidak’s multiple comparisons test [N = 6–8]: *p < .03) and in liver of BHLivDKO mice post VEH/DEN injection and 35 weeks of high fat diet feeding (E). Two-way ANOVA, Sidak’s multiple comparisons test (N = 6–8): *p < .03; **p < .005.
3.6 |. BMAL1 and HNF4α deficiency is protective in the STAM model of HCC
To examine whether reduced HNF4α and BMAL1 protected from HCC in another mouse model, which recapitulates the liver disease progression often seen in human patients, we superimposed the so-called “STAM” model onto the genetic background of our BHLivDKO mice (Figure 7A). Depletion of pancreatic beta cells with STZ combined with HFD in the context of BHLivDKO conditions resulted in fewer tumor-bearing livers as well as a reduction in the number of tumors per liver in BHLivDKO compared to WT controls under the same conditions (Figure 7B). When livers were analyzed for the presence of HCC, H&E staining validated excess liver damage and the presence of additional tumors in both male and female WT mice treated with STZ and HFD (Figure 7C). In addition to H&E staining, we performed immunohistochemistry to look at AFP expression. STZ-treated male and female showed pronounced AFP expression, which was abrogated in the BHLivDKO (Figure 7D). Similarly, Oil Red O staining revealed reduced lipid deposition in the BHLivDKO compared to STZ-treated control mice (Figure 7E). Thus, loss of HNF4α and BMAL1 concomitantly is protective at all stages of liver disease progression, and in the context of HCC in two different mouse models.
FIGURE 7.
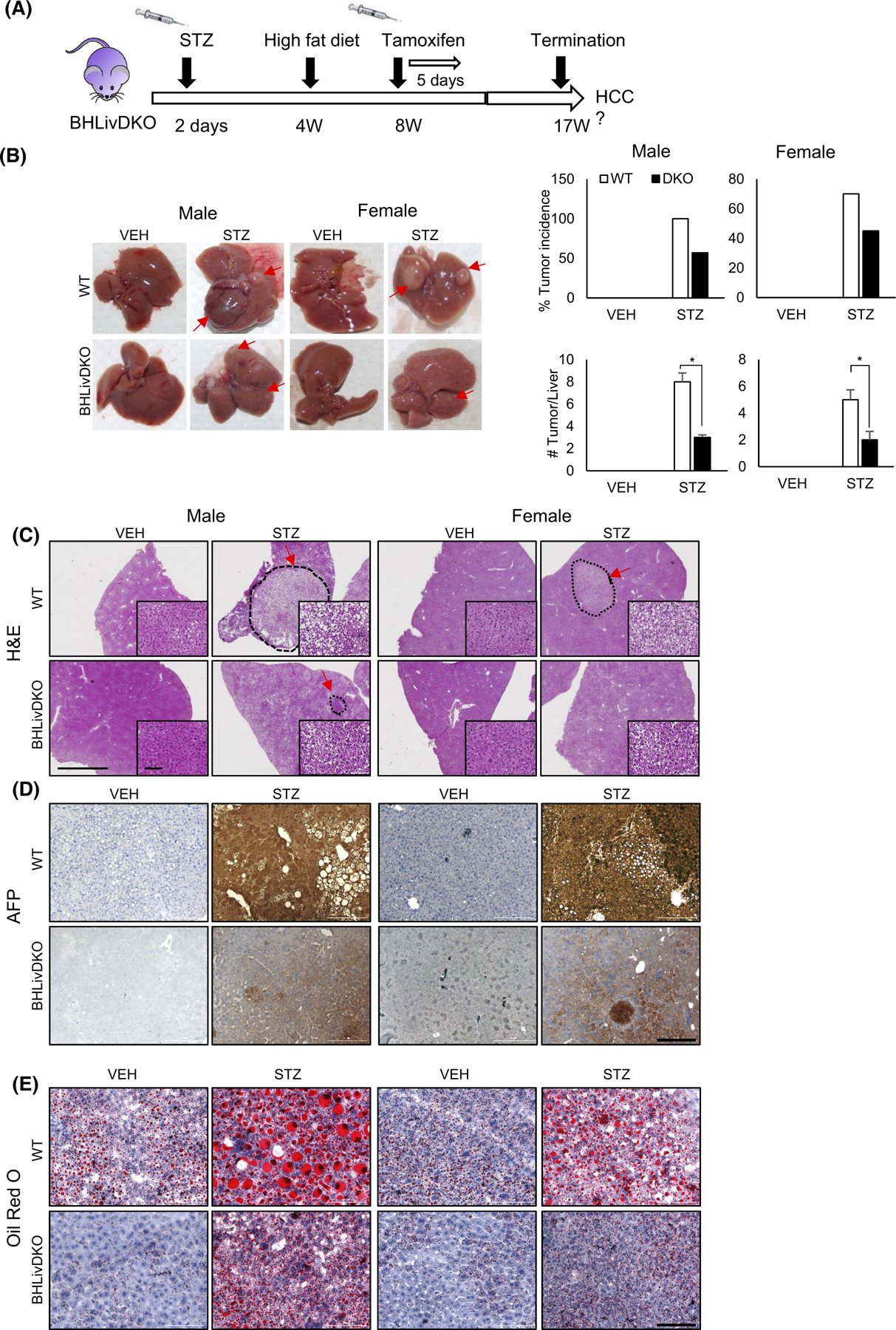
BMAL1 and HNF4α loss is protective in the STAM model and SR9009 blocks invasion of BMAL1-expressing tumor cells. (A) Experimental timeline for STAM model on the BHLivDKO background. (B) Whole livers of VEH and STZ treated STAM mice (WT/ BHLivDKO) at 17 weeks of age (left panel), and percent tumor incidence and number of tumors per liver (right panel). (C–E) H&E, AFP, and oil-Red O staining of WT and, BHLivDKO, and H4LivKO mouse livers after VEH or STZ injection followed by HFD AT 16 weeks (scale bar = 2000 μm, magnified scale bar = 100 μm, 200 μm).
3.7. REV-ERB agonist prevents migration exclusively in BMAL1-expressing, HNF4α-deficient HCC cells
The nuclear receptors REV-ERBα and REV-ERBβ (NR1D1/2, respectively) are BMAL1-regulating clock components, which also control the expression of a large number of metabolic genes in the liver and other tissues.114,115 REV-ERB agonists exhibit a variety of actions on lipid and glucose metabolism and circadian rhythms116 and have been shown to be specifically fatal to certain types of cancer cells, while having little impact on the viability of normal cells.117 It also has been reported that REV-ERB agonism represses Ccna2 expression causing cell cycle arrest in cancer cells.117,118 To interrogate the association of BMAL1 expression and HCC outcomes, we examined cell viability and migration in normal versus HCC lines with or without BMAL1 expression and in the presence and absence of REV-ERB agonist SR9009. The results show that SR9009 treatment impairs cell viability and migration of HCC cells containing high levels of BMAL1 expression such as Hepa1c1c cells (Figure 8A,B). In contrast, migration of normal AML12 liver cells and HCC cells deficient in BMAL1 expression (HepG2) was not altered with SR9009 (Figure 8A,B). To test whether SR9009 worked predominantly in a REV-ERB-dependent manner, we used siRNA to Reverbs and applied SR9009 in vitro (Figure S8D). We first measured Bmal1 expression in all lines with and without SR9009 and siRNA to the Reverb genes. Across the different cell lines, Bmal1 expression tended to increase consistent with REV-ERBs ability to transcriptionally repress Bmal1 (Figure 8C). Though SR9009 has been reported to have some REV-ERB-independent effects, in our experiments we found that SR9009 predominantly worked in a REV-ERB-dependent manner in Hepa1c1c cells (Figure 8D), though SR9009 had minimal effects on the low BMAL1-expressing HCC lines and normal AML12 cells in both MTT and invasion assays (Figure 8D,E).
FIGURE 8.
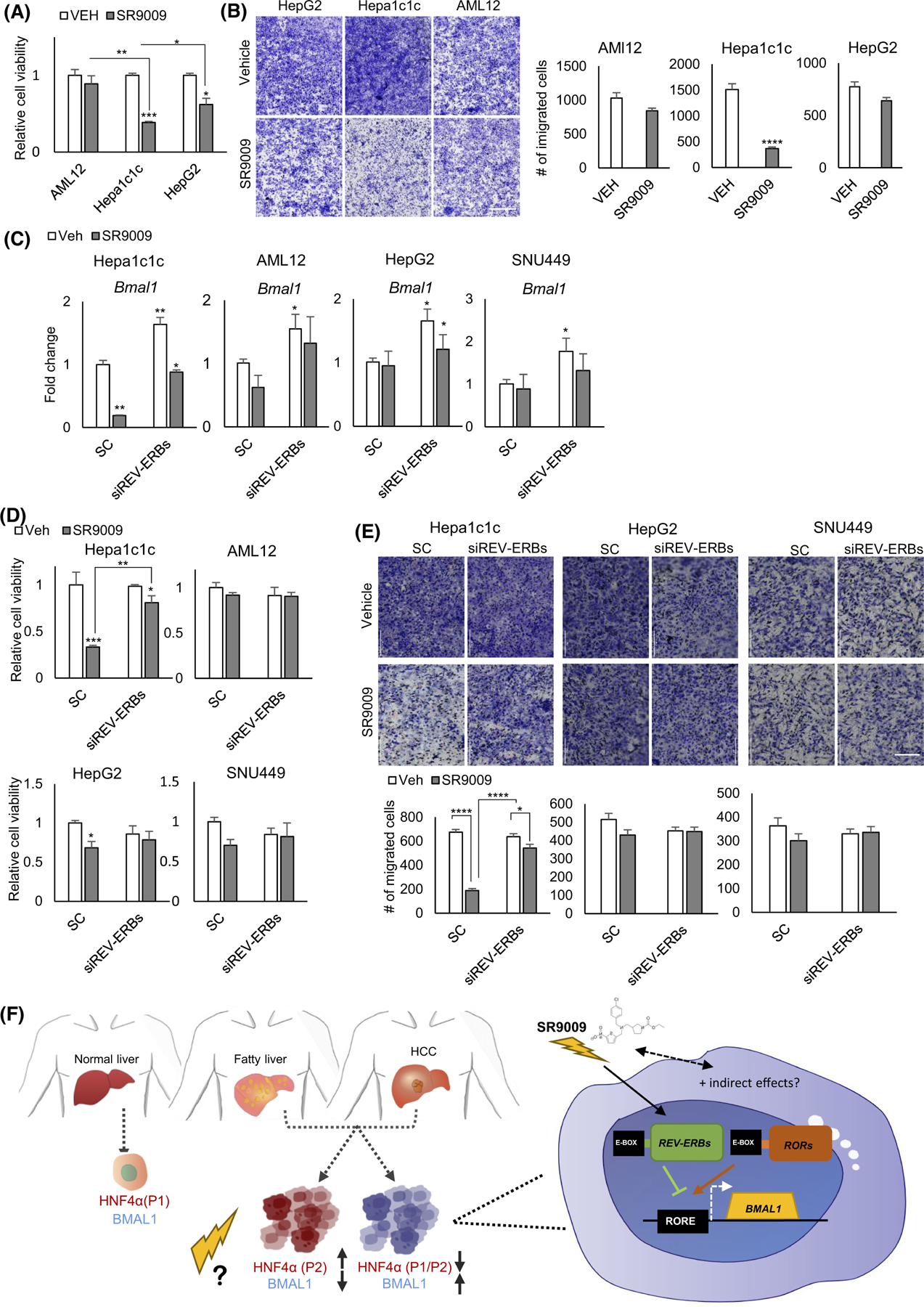
SR9009 blocks proliferation and migration of Bmal1 expressing tumor cells. (A) MTT assay shows normalized cell viability following SR9009 or vehicle treatment. Two-way ANOVA, Sidak’s multiple comparisons test (N = 8): *p < .03; **p < .005; ***p < .0005. (B) Migration assay reveals effects of SR9009 versus vehicle on AMl12, Hepa-1c1c, and HepG2 cells 48-h post-treatment (left panel). Quantification, right panel. ****p < .0001 was determined by Mann–Whitney U-test (N = 6) (Scale bar = 1000 μm). (C) Bmal1 gene expression in Aml12, Hepa-1c1c, HepG2, and SNU449 cells post-VEH/SR9009 treatment following REV-ERBs siRNA or the scrambled control. (D) Cell viability measurement using MTT assay at 48 h following VEH/SR9009and SC/REV-ERBs siRNA treatment. (E) Cell migration assays show effects of SR9009 versus vehicle and scrambled (sc) versus siREV-ERB oligonucleotides on Aml12, Hepa-1c1c, HepG2, and SNU449 cells 48-h post-treatment (top panel). Quantification, bottom panel. ****p < .0001 was determined by Mann–Whitney U-test (N = 6) (Scale bar = 200 μm). (F) Model of HCC stratification. Previous results indicate that while healthy hepatocytes co-express BMAL1 and HNF4α, human HCC with high levels of HNF4α contain the “fetal” form of the protein, which is not tumor suppressive and transcriptionally inhibits BMAL1 expression. Conversely, HCC which shows high levels of BMAL1 is deficient in HNF4α. Our data suggest that treating these tumor types with modulators of BMAL1 may provide opportunities for improved therapy.
4 |. DISCUSSION
Hepatocellular carcinoma is one of a handful of cancers whose incidence is increasing in the US and for which there are few effective treatment options (ACS Cancer Facts & Figures 2020). The increase appears to be due to the rise in fatty liver disease fueled by the obesity epidemic and the increasing disruption of circadian rhythms in the modern lifestyle. Indeed, we recently showed that master regulators of lipid metabolism and the circadian clock in the liver– HNF4α and BMAL1, respectively – are implicated in HCC in a complex, inerdependent fashion in which either one or the other but not both are expressed in HCC. Here, we use inducible DKO mice and two models of HFD-induced HCC to show that concomitant loss of both HNF4α and BMAL1 decreases HCC incidence in male and female mice. Transcriptomics analysis reveals a number of genes involved in the cell cycle, fatty acid metabolism, inflammation, and the WNT/β-catenin pathway that could be driving the decrease. Finally, preliminary studies suggest how the HNF4α-BMAL1 nexus could be targeted to treat this deadly disease.
The ultimate goal of this study was to determine whether stratification of diseased liver by expression of HNF4α and BMAL1 proteins could provide insights into future opportunities for more effective therapies (Figure 8F). Exploitation of pharmacological modulators of BMAL1 (such as Nobiletin, a ROR agonist, or SR9009) in HCC based on the presence of absence of BMAL1 versus HNF4α might provide benefits to standard forms of therapy. Though a greater understanding of how HNF4α isoform switching occurs in the liver is needed, we propose that an understanding of the expression patterns of these two proteins either in severely diseased liver or HCC might provide tractable opportunities for new combination therapies to improve liver function. SR9009 might be one of these compounds, based on its ability to repress BMAL1 expression in HNF4α-deficient but BMAL1-positive HCC.
One finding in our study that is consistent with previous studies looking at the sex disparity of HCC, is that in both DEN-treated WT and BHLivDKO mice, males acquire HCC more frequently than females. When it does occur, the number of tumors in female mice compared to male mice is also lower (see Figure 1C). Contrast this to single H4LivKO mice, where we have previously shown HCC incidence to be equal in aged male and female mice following HFD feeding, due to robust derepression of Il6 in both sexes.11 The data here can be at least partly explained by the robust changes in Il6 expression in male mice treated with DEN compared to female mice treated with DEN in WT and BHLivDKO mice (see Figure 2D).
While our previous gain and loss of function experiments have demonstrated that forced co-expression of BMAL1 and P1-HNF4α in HCC cells results in apoptosis and inhibition of tumor growth in vivo, this study demonstrates that concomitant inhibition of their function is also protective in the process of liver disease progression. Because the induction of P2-HNF4α results in direct transcriptional repression at the Bmal1 locus, either loss of HNF4α altogether, or isoform switching to the fetal P2-driven isoform results in a hepatocyte devoid of concomitant expression. Compare this to healthy hepatocytes, where both transcription factors are robustly expressed, and drive gene expression programs necessary for hepatocyte cell fate as well as metabolic pathways necessary for liver function.
Based on this information, it can be argued that one paradox of the study is that while both P1-HNF4α and BMAL1 appear to function as tumor suppressors (in that loss of one causes early onset HCC), loss of both together was protective in this context of HCC formation and progression. One reason for this could be that loss of HNF4α prevents the induction of the predominantly fetal form of the protein, P2-HNF4α, which transcriptionally represses BMAL1, while not having tumor suppressive activity itself.
A second reason for this could be that at genetic loci critically involved in HCC proliferation and progression (i.e., CCNA2) where the tumor suppressive form of HNF4α potently represses, BMAL1 cannot now activate based on its absence in the DKO mice.119 Thus, compared to single HNF4α-deficient mice, Ccna2 expression remains low. The same concept could apply to single Bmal1 knockout mice compared to the DKO situation.
In summary, HNF4α and BMAL1 have competing roles in HCC, and loss of both prior to disease onset partially protects against liver disease progression and delays HCC onset. The hope is that further stratification of liver tumors by HNF4α and BMAL1 expression might provide additional options for new treatment, similar to what has occurred in breast cancer in the context of estrogen receptor and Erb-B2 Receptor Tyrosine Kinase 2 (HER2) expression, where expression levels of these genes guides treatment decisions.120 In addition to Nobiletin and SR9009, HNF4α has a ligand binding pocket and a known endogenous ligand (linoleic acid) that binds reversibly.121 Thus, HNF4α is a druggable target, yet an appropriate drug target has not yet been identified. Due to the isoform similarities, in this region, further targeting may be necessary in the context of P1-versus P2 expression. Nevertheless, in the context of HCC our data suggest that pharmacological exploitation of BMAL1 might be a tractable strategy to treat tumors based on stratification by HNF4α expression.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr. Pierre Chambon, Dr. Daniel Metzger, and Dr. Frank Gonzalez for providing Albtm1(cre/ERT2)Mtz mice for inducible knockout studies. We would also like to thank Zhengmei Mao for help with tissue processing. We thank Dr. Katherine Brannick, Cneshia Traylor, Christine Obregon, and Carolina Cuatzo for help with animal care and husbandry. The sequencing data were generated by the UTHealthCancer Genomics Core, which is supported by the Cancer Prevention and Research Institute of Texas (CPRIT RP180734). This work was also supported by ACS grant RSG-17-215-01-C (to K.E.-M.) and DK114037 (to K.E.-M.).
FUNDING INFORMATION
This study was supported by grants DK114037 (K.E.M.) and ACS grant RSG-17-215-01-C (K.E.M.). Additional funds were provided by McGovern Medical School, part of UT Health Science Center at Houston. The sequencing data was generated by the UTHealth Cancer Genomics Core, which is supported by the Cancer Prevention and Research Institute of Texas (CPRIT RP180734). This work was also supported by the USDA National Institute of Food and Agriculture, Hatch Project CA-R-NEU-5680-H (F.M.S.). Y.D. and Z.Z. were partially supported by CPRIT grant RP210045. Y.D. and Z.Z. were partially supported by CPRIT grant RP210045.
American Cancer Society (ACS), Grant/Award Number: RSG-17-215-01-C; Cancer Prevention and Research Institute of Texas (CPRIT), Grant/Award Number: RP180734; National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), Grant/Award Number: DK114037
Abbreviations:
- ARNTL
Aryl hydrocarbon receptor nuclear translocator
- BMAL1
Brain and muscle ARNT-Like
- DEN
Diethylnitrosamine
- DKO
Double knock out
- EMT
Epithelial-mesenchymal transition
- FBS
Fetal bovine serum
- HCC
Hepatocellular carcinoma
- HDL
High-density lipoprotein
- H&E
Hematoxylin and eosin
- HFD
High fat diet
- HNF4α
Hepatocyte nuclear factor-4 alpha
- IL
Interleukin
- KO
Knockout
- LDL
Low-density lipoprotein
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NALFD
Nonalcoholic fatty liver disease
- NASH
Nonalcoholic steatohepatitis
- STA
TSignal transducer and activator of transcription
- STAM
Stelic Animal Model
- STZ
Streptozotocin
- TC
Total cholesterol
- TGs
Triglycerides
- WT
Wild-type
- ZT
Zeitgeber time
Footnotes
DISCLOSURES
The authors declare no potential conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
Included in article: The data that support the findings of this study are available in the methods and Supporting Information of this article. Included publically: All genomics data generated and analyzed for this manuscript will be publically available on the Gene Expression Omnibus (GEO) of NCBI.
REFERENCES
- 1.Kulik L, El-Serag HB. Epidemiology and management of hepatocellular carcinoma. Gastroenterology 2019;156(2):477–491 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanwal F, Kramer JR, Mapakshi S, et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018;155(6):1828–1837 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol 2014;24(2):90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens RG. Light-at-night, circadian disruption and breast cancer: assessment of existing evidence. Int J Epidemiol 2009;38(4):963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schernhammer ES, Kroenke CH, Laden F, Hankinson SE. Night work and risk of breast cancer. Epidemiology 2006;17(1):108–111. [DOI] [PubMed] [Google Scholar]
- 6.Kettner NM, Voicu H, Finegold MJ, et al. Circadian homeostasis of liver metabolism suppresses hepatocarcinogenesis. Cancer Cell 2016;30(6):909–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fu L, Pelicano H, Liu J, Huang P, Lee CC. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002;111(1):41–50. [DOI] [PubMed] [Google Scholar]
- 8.Pan X, Queiroz J, Hussain MM. Nonalcoholic fatty liver disease in CLOCK mutant mice. J Clin Invest 2020;130(8):4282–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS One 2010;5(6):e10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fekry B, Ribas-Latre A, Baumgartner C, et al. Incompatibility of the circadian protein BMAL1 and HNF4α in hepatocellular carcinoma. Nat Commun 2018;9(1):4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fekry B, Ribas-Latre A, Baumgartner C, et al. HNF4α-deficient fatty liver provides a permissive environment for sex-independent hepatocellular carcinoma. Cancer Res 2019;79(22):5860–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fekry B, Ribas-Latre A, Baumgartner C, et al. Incompatibility of the circadian protein BMAL1 and HNF4alpha in hepatocellular carcinoma. Nat Commun 2018;9(1):4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sladek FM, Zhong WM, Lai E, Darnell JE. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev 1990;4(12B):2353–2365. [DOI] [PubMed] [Google Scholar]
- 14.Dubois V, Staels B, Lefebvre P, Verzi M, Eeckhoute J. Control of cell identity by the nuclear receptor HNF4 in organ pathophysiology. Cell 2020;9(10):2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walesky C, Apte U. Role of hepatocyte nuclear factor 4α (HNF4α) in cell proliferation and cancer. Gene Expr 2015;16(3):101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolotin E, Chellappa K, Hwang-Verslues W, Schnabl JM, Yang C, Sladek FM. Nuclear receptor HNF4α binding sequences are widespread in Alu repeats. BMC Genomics 2011;12(1):560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaestner KH. Making the liver what it is: the many targets of the transcriptional regulator HNF4alpha. Hepatology 2010;51(2):376–377. [DOI] [PubMed] [Google Scholar]
- 18.Battle MA, Konopka G, Parviz F, et al. Hepatocyte nuclear factor 4α orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc Natl Acad Sci 2006;103(22):8419–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang B, Mane-Padros D, Bolotin E, Jiang T, Sladek FM. Identification of a binding motif specific to HNF4 by comparative analysis of multiple nuclear receptors. Nucleic Acids Res 2012;40(12):5343–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Odom DT, Zizlsperger N, Gordon DB, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science 2004;303(5662):1378–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qu M, Duffy T, Hirota T, Kay SA. Nuclear receptor HNF4A transrepresses CLOCK:BMAL1 and modulates tissue-specific circadian networks. Proc Natl Acad Sci U S A 2018;115(52):E12 305-E12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Jimenez CP, Kyrmizi I, Cardot P, Gonzalez FJ, Talianidis I. Hepatocyte nuclear factor 4alpha coordinates a transcription factor network regulating hepatic fatty acid metabolism. Mol Cell Biol 2010;30(3):565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inoue Y, Yu AM, Inoue J, Gonzalez FJ. Hepatocyte nuclear factor 4alpha is a central regulator of bile acid conjugation. J Biol Chem 2004;279(4):2480–2489. [DOI] [PubMed] [Google Scholar]
- 24.Rhee J, Inoue Y, Yoon JC, et al. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc Natl Acad Sci U S A 2003;100(7):4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol 2001;21(4):1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonzo JA, Ferry CH, Matsubara T, Kim JH, Gonzalez FJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J Biol Chem 2012;287(10):7345–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dean S, Tang JI, Seckl JR, Nyirenda MJ. Developmental and tissue-specific regulation of hepatocyte nuclear factor 4-alpha (HNF4-alpha) isoforms in rodents. Gene Expr 2010;14(6):337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonzo JA, Ferry CH, Matsubara T, Kim JH, Gonzalez FJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4α in adult mice. J Biol Chem 2012;287(10):7345–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ning BF, Ding J, Yin C, et al. Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res 2010;70(19):7640–7651. [DOI] [PubMed] [Google Scholar]
- 30.Hatziapostolou M, Polytarchou C, Aggelidou E, et al. An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011;147(6):1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka T, Jiang S, Hotta H, et al. Dysregulated expression of P1 and P2 promoter-driven hepatocyte nuclear factor-4alpha in the pathogenesis of human cancer. J Pathol 2006;208(5):662–672. [DOI] [PubMed] [Google Scholar]
- 32.Oshima T, Kawasaki T, Ohashi R, et al. Downregulated P1 promoter-driven hepatocyte nuclear factor-4alpha expression in human colorectal carcinoma is a new prognostic factor against liver metastasis. Pathol Int 2007;57(2):82–90. [DOI] [PubMed] [Google Scholar]
- 33.Takano K, Hasegawa G, Jiang S, et al. Immunohistochemical staining for P1 and P2 promoter-driven hepatocyte nuclear factor-4alpha may complement mucin phenotype of differentiated-type early gastric carcinoma. Pathol Int 2009;59(7):462–470. [DOI] [PubMed] [Google Scholar]
- 34.Chellappa K, Deol P, Evans JR, et al. Opposing roles of nuclear receptor HNF4α isoforms in colitis and colitis-associated colon cancer. Elife 2016;5:e10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chellappa K, Jankova L, Schnabl JM, et al. Src tyrosine kinase phosphorylation of nuclear receptor HNF4α correlates with isoform-specific loss of HNF4α in human colon cancer. Proc Natl Acad Sci U S A 2012;109(7):2302–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie X, Liao H, Dang H, et al. Down-regulation of hepatic HNF4alpha gene expression during hyperinsulinemia via SREBPs. Mol Endocrinol 2009;23(4):434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Argemi J, Latasa MU, Atkinson SR, et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun 2019;10(1):3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li D, Cao T, Sun X, et al. Hepatic TET3 contributes to type-2 diabetes by inducing the HNF4alpha fetal isoform. Nat Commun 2020;11(1):342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bunger MK, Wilsbacher LD, Moran SM, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 2000;103(7):1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet 2008;9(10):764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turek FW, Joshu C, Kohsaka A, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005;308(5724):1043–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lipton JO, Yuan ED, Boyle LM, et al. The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell 2015;161(5):1138–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan X, Bradfield CA, Hussain MM. Global and hepatocyte-specific ablation of Bmal1 induces hyperlipidaemia and enhances atherosclerosis. Nat Commun 2016;7(1):13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi D, Chen J, Wang J, et al. Circadian Clock Genes in the Metabolism of Non-alcoholic Fatty Liver Disease. Front Physiol 2019;10:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiba H, Itoh T, Satohisa S, et al. Activation of p21CIP1/WAF1 gene expression and inhibition of cell proliferation by over-expression of hepatocyte nuclear factor-4alpha. Exp Cell Res 2005;302(1):11–21. [DOI] [PubMed] [Google Scholar]
- 46.Wang Z, Li Y, Wu D, Yu S, Wang Y, Leung Chan F. Nuclear receptor HNF4α performs a tumor suppressor function in prostate cancer via its induction of p21-driven cellular senescence. Oncogene 2020;39(7):1572–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erdmann S, Senkel S, Arndt T, et al. Tissue-specific transcription factor HNF4alpha inhibits cell proliferation and induces apoptosis in the pancreatic INS-1 beta-cell line. Biol Chem 2007;388(1):91–106. [DOI] [PubMed] [Google Scholar]
- 48.Walesky C, Gunewardena S, Terwilliger EF, Edwards G, Borude P, Apte U. Hepatocyte-specific deletion of hepatocyte nuclear factor-4α in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol 2013;304(1):G26–G37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walesky C, Edwards G, Borude P, et al. Hepatocyte nuclear factor 4 alpha deletion promotes diethylnitrosamine-induced hepatocellular carcinoma in rodents. Hepatology 2013;57(6):2480–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun K, Montana V, Chellappa K, et al. Phosphorylation of a conserved serine in the deoxyribonucleic acid binding domain of nuclear receptors alters intracellular localization. Mol Endocrinol 2007;21(6):1297–1311. [DOI] [PubMed] [Google Scholar]
- 51.Walker S, Wankell M, Ho V, et al. Targeting mTOR and Src restricts hepatocellular carcinoma growth in a novel murine liver cancer model. PLoS One 2019;14(2):e0212860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshiji H, Kuriyama S, Ways DK, et al. Protein kinase C lies on the signaling pathway for vascular endothelial growth factor-mediated tumor development and angiogenesis. Cancer Res 1999;59(17):4413–4418. [PubMed] [Google Scholar]
- 53.Wang Y, Ausman LM, Greenberg AS, Russell RM, Wang XD. Nonalcoholic steatohepatitis induced by a high-fat diet promotes diethylnitrosamine-initiated early hepatocarcinogenesis in rats. Int J Cancer 2009;124(3):540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arboatti AS, Lambertucci F, Sedlmeier MG, et al. Diethylnitrosamine enhances hepatic tumorigenic pathways in mice fed with high fat diet (Hfd). Chem Biol Interact 2019;303:70–78. [DOI] [PubMed] [Google Scholar]
- 55.Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol 2009;90(4):367–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fekry B, Ribas-Latre A, Baumgartner C, et al. HNF4alpha-deficient fatty liver provides a permissive environment for sex-independent hepatocellular carcinoma. Cancer Res 2019;79(22):5860–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monga SP. β-Catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology 2015;148(7):1294–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo J-H, Ren B, Keryanov S, et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology 2006;44(4):1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jia H-L, Ye QH, Qin LX, et al. Gene expression profiling reveals potential biomarkers of human hepatocellular carcinoma. Clin Cancer Res 2007;13(4):1133–1139. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol 2016;64(6):1403–1415. [DOI] [PubMed] [Google Scholar]
- 61.Johnson C, Han Y, Hughart N, McCarra J, Alpini G, Meng F. Interleukin-6 and its receptor, key players in hepatobiliary inflammation and cancer. Transl Gastrointest Cancer 2012;1(1):58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/ JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 2018;15(4):234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grohmann M, Wiede F, Dodd GT, et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018;175(5):1289–1306 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim MJ, Choi YK, Park SY, et al. PPARdelta reprograms glutamine metabolism in sorafenib-resistant HCC. Mol Cancer Res 2017;15(9):1230–1242. [DOI] [PubMed] [Google Scholar]
- 65.Bahitham W, Watts R, Nelson R, Lian J, Lehner R. Liver-specific expression of carboxylesterase 1g/esterase-x reduces hepatic steatosis, counteracts dyslipidemia and improves insulin signaling. Biochim Biophys Acta 2016;1861(5):482–490. [DOI] [PubMed] [Google Scholar]
- 66.Wang XF, Galaj E, Bi GH, et al. Different receptor mechanisms underlying phytocannabinoidversus synthetic cannabinoid-induced tetrad effects: Opposite roles of CB1 /CB2 versus GPR55 receptors. Br J Pharmacol 2020;177(8):1865–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Björnson E, Mukhopadhyay B, Asplund A, et al. Stratification of hepatocellular carcinoma patients based on acetate utilization. Cell Rep 2015;13(9):2014–2026. [DOI] [PubMed] [Google Scholar]
- 68.Dorn C, Riener MO, Kirovski G, et al. Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int J Clin Exp Pathol 2010;3(5):505–514. [PMC free article] [PubMed] [Google Scholar]
- 69.Jones SF, Infante JR. Molecular pathways: fatty acid synthase. Clin Cancer Res 2015;21(24):5434–5438. [DOI] [PubMed] [Google Scholar]
- 70.Lupien LE, Dunkley EM, Maloy MJ, et al. An inhibitor of fatty acid synthase thioesterase domain with improved cytotoxicity against breast cancer cells and stability in plasma. J Pharmacol Exp Ther 2019;371(1):171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Soria JC, Jang SJ, Khuri FR, et al. Overexpression of cyclin B1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res 2000;60(15):4000–4004. [PubMed] [Google Scholar]
- 72.Bauerschlag DO, Maass N, Leonhardt P, et al. Fatty acid synthase overexpression: target for therapy and reversal of chemo-resistance in ovarian cancer. J Transl Med 2015;13:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007;7(10):763–777. [DOI] [PubMed] [Google Scholar]
- 74.Jiang L, Fang X, Wang H, Li D, Wang X. Ovarian cancer-intrinsic fatty acid synthase prevents anti-tumor immunity by disrupting tumor-infiltrating dendritic cells. Front Immunol 2018;9:2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu S, Archer MC. Fatty acid synthase is a potential molecular target for the chemoprevention of breast cancer. Carcinogenesis 2005;26(1):153–157. [DOI] [PubMed] [Google Scholar]
- 76.Pérez S, Aspichueta P, Ochoa B, Chico Y. The 2-series prostaglandins suppress VLDL secretion in an inflammatory condition-dependent manner in primary rat hepatocytes. Biochim Biophys Acta 2006;1761(2):160–171. [DOI] [PubMed] [Google Scholar]
- 77.Henkel J, Frede K, Schanze N, et al. Stimulation of fat accumulation in hepatocytes by PGE2-dependent repression of hepatic lipolysis, β-oxidation and VLDL-synthesis. Lab Invest 2012;92(11):1597–1606. [DOI] [PubMed] [Google Scholar]
- 78.Henkel J, Coleman CD, Schraplau A, et al. Augmented liver inflammation in a microsomal prostaglandin E synthase 1 (mPGES-1)-deficient diet-induced mouse NASH model. Sci Rep 2018;8(1):16127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sano H, Kawahito Y, Wilder RL, et al. Expression of cyclooxygenase-1 and −2 in human colorectal cancer. Cancer Res 1995;55(17):3785–3789. [PubMed] [Google Scholar]
- 80.Roelofs HM, Te Morsche RH, van Heumen BW, Nagengast FM, Peters WH. Over-expression of COX-2 mRNA in colorectal cancer. BMC Gastroenterol 2014;14:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Half E, Tang XM, Gwyn K, Sahin A, Wathen K, Sinicrope FA. Cyclooxygenase-2 expression in human breast cancers and adjacent ductal carcinoma in situ. Cancer Res 2002;62(6):1676–1681. [PubMed] [Google Scholar]
- 82.Mehrotra S, Morimiya A, Agarwal B, Konger R, Badve S. Microsomal prostaglandin E2 synthase-1 in breast cancer: a potential target for therapy. J Pathol 2006;208(3):356–363. [DOI] [PubMed] [Google Scholar]
- 83.Zang S, Ni M, Lian Y, Zhang Y, Liu J, Huang A. Expression of microsomal prostaglandin E2 synthase-1 and its role in human hepatocellular carcinoma. Hum Pathol 2013;44(8):1681–1687. [DOI] [PubMed] [Google Scholar]
- 84.Koga H, Sakisaka S, Ohishi M, et al. Expression of cyclooxygenase-2 in human hepatocellular carcinoma: relevance to tumor dedifferentiation. Hepatology 1999;29(3):688–696. [DOI] [PubMed] [Google Scholar]
- 85.Zhangdi H-J, Su SB, Wang F, et al. Crosstalk network among multiple inflammatory mediators in liver fibrosis. World J Gastroenterol 2019;25(33):4835–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liao R, Sun J, Wu H, et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J Exp Clin Cancer Res 2013;32(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ma S, Cheng Q, Cai Y, et al. IL-17A produced by γδ T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res 2014;74(7):1969–1982. [DOI] [PubMed] [Google Scholar]
- 88.Salahshor S, Woodgett JR. The links between axin and carcinogenesis. J Clin Pathol 2005;58(3):225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang LH, Xu HT, Han Y, et al. Axin downregulates TCF-4 transcription via beta-catenin, but not p53, and inhibits the proliferation and invasion of lung cancer cells. Mol Cancer 2010;9:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abitbol S, Dahmani R, Coulouarn C, et al. AXIN deficiency in human and mouse hepatocytes induces hepatocellular carcinoma in the absence of β-catenin activation. J Hepatol 2018;68(6):1203–1213. [DOI] [PubMed] [Google Scholar]
- 91.Lede V, Meusel A, Garten A, et al. Altered hepatic lipid metabolism in mice lacking both the melanocortin type 4 receptor and low density lipoprotein receptor. PLoS One 2017;12(2):e0172000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dunser MW, Westphal M. Arginine vasopressin in vasodilatory shock: effects on metabolism and beyond. Curr Opin Anaesthesiol 2008;21(2):122–127. [DOI] [PubMed] [Google Scholar]
- 93.Zhang W, Shibamoto T, Kuda Y, Shinomiya S, Kurata Y. The responses of the hepatic and splanchnic vascular beds to vasopressin in rats. Biomed Res 2012;33(2):83–88. [DOI] [PubMed] [Google Scholar]
- 94.Hui ST, Kurt Z, Tuominen I, et al. The Genetic Architecture of Diet-Induced Hepatic Fibrosis in Mice. Hepatology 2018;68(6):2182–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagasawa T, Inada Y, Nakano S, et al. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol 2006;536(1–2):182–191. [DOI] [PubMed] [Google Scholar]
- 96.Sanderson LM, Boekschoten MV, Desvergne B, Müller M, Kersten S. Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver. Physiol Genomics 2010;41(1):42–52. [DOI] [PubMed] [Google Scholar]
- 97.Peyrou M, Ramadori P, Bourgoin L, Foti M. PPARs in liver diseases and cancer: epigenetic regulation by microRNAs. PPAR Res 2012;2012:757803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kang K, Reilly SM, Karabacak V, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab 2008;7(6):485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene 2005;24(17):2909–2915. [DOI] [PubMed] [Google Scholar]
- 100.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011;11(8):558–572. [DOI] [PubMed] [Google Scholar]
- 101.Gonzalez FJ. Regulation of hepatocyte nuclear factor 4α-mediated transcription. Drug Metab Pharmacokinet 2008;23(1):2–7. [DOI] [PubMed] [Google Scholar]
- 102.Wang A, Yoshimi N, Ino N, Tanaka T, Mori H. Overexpression of cyclin B1 in human colorectal cancers. J Cancer Res Clin Oncol 1997;123(2):124–127. [DOI] [PubMed] [Google Scholar]
- 103.Zhao M, Kim YT, Yoon BS, et al. Expression profiling of cyclin B1 and D1 in cervical carcinoma. Exp Oncol 2006;28(1):44–48. [PubMed] [Google Scholar]
- 104.Kawamoto H, Koizumi H, Uchikoshi T. Expression of the G2-M checkpoint regulators cyclin B1 and cdc2 in nonmalignant and malignant human breast lesions: immunocytochemical and quantitative image analyses. Am J Pathol 1997;150(1):15–23. [PMC free article] [PubMed] [Google Scholar]
- 105.Banerjee SK, Weston AP, Zoubine MN, Campbell DR, Cherian R. Expression of cdc2 and cyclin B1 in Helicobacter pylori-associated gastric MALT and MALT lymphoma: relationship to cell death, proliferation, and transformation. Am J Pathol 2000;156(1):217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Park TJ, Kim JY, Oh SP, et al. TIS21 negatively regulates hepatocarcinogenesis by disruption of cyclin B1-Forkhead box M1 regulation loop. Hepatology 2008;47(5):1533–1543. [DOI] [PubMed] [Google Scholar]
- 107.Joo M, Kang YK, Kim MR, Lee HK, Jang JJ. Cyclin D1 over-expression in hepatocellular carcinoma. Liver 2001;21(2): 89–95. [DOI] [PubMed] [Google Scholar]
- 108.van Zijl F, Zulehner G, Petz M, et al. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol 2009;5(8):1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol 2016;65(4):798–808. [DOI] [PubMed] [Google Scholar]
- 110.Lazarevich NL, Cheremnova OA, Varga EV, et al. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology 2004;39(4):1038–1047. [DOI] [PubMed] [Google Scholar]
- 111.Cicchini C, Amicone L, Alonzi T, Marchetti A, Mancone C, Tripodi M. Molecular mechanisms controlling the phenotype and the EMT/MET dynamics of hepatocyte. Liver Int 2015;35(2):302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chang H, Liu Y, Xue M, et al. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res 2016;44(6):2514–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tang Q, Cheng B, Xie M, et al. Circadian clock gene Bmal1 inhibits tumorigenesis and increases paclitaxel sensitivity in tongue squamous cell carcinoma. Cancer Res 2017;77(2):532–544. [DOI] [PubMed] [Google Scholar]
- 114.Everett LJ, Lazar MA. Nuclear receptor Rev-erbα: up, down, and all around. Trends Endocrinol Metab 2014;25(11):586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cho H, Zhao X, Hatori M, et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 2012;485(7396):123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Solt LA, Kojetin DJ, Burris TP. The REV-ERBs and RORs: molecular links between circadian rhythms and lipid homeostasis. Future Med Chem 2011;3(5):623–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sulli G, Rommel A, Wang X, et al. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018;553(7688):351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang Y, Kojetin D, Burris TP. Anti-proliferative actions of a synthetic REV-ERBα/β agonist in breast cancer cells. Biochem Pharmacol 2015;96(4):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Farshadi E, Yan J, Leclere P, Goldbeter A, Chaves I, van der Horst GTJ. The positive circadian regulators CLOCK and BMAL1 control G2/M cell cycle transition through Cyclin B1. Cell Cycle 2019;18(1):16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344(11):783–792. [DOI] [PubMed] [Google Scholar]
- 121.Yuan X, Ta TC, Lin M, et al. Identification of an endogenous ligand bound to a native orphan nuclear receptor. PLoS One 2009;4(5):e5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Included in article: The data that support the findings of this study are available in the methods and Supporting Information of this article. Included publically: All genomics data generated and analyzed for this manuscript will be publically available on the Gene Expression Omnibus (GEO) of NCBI.