

Abstract
Background
Inclusion body myositis (IBM) is an inflammatory myopathy clinically characterized by proximal and distal muscle weakness, with inflammatory infiltrates, rimmed vacuoles and mitochondrial changes in muscle histopathology. There is scarce knowledge on IBM aetiology, and non‐established biomarkers or effective treatments are available, partly due to the lack of validated disease models.
Methods
We have performed transcriptomics and functional validation of IBM muscle pathological hallmarks in fibroblasts from IBM patients (n = 14) and healthy controls (n = 12), paired by age and sex. The results comprise an mRNA‐seq, together with functional inflammatory, autophagy, mitochondrial and metabolic changes between patients and controls.
Results
Gene expression profile of IBM vs control fibroblasts revealed 778 differentially expressed genes (P‐value adj < 0.05) related to inflammation, mitochondria, cell cycle regulation and metabolism. Functionally, an increased inflammatory profile was observed in IBM fibroblasts with higher supernatant cytokine secretion (three‐fold increase). Autophagy was reduced considering basal protein mediators (18.4% reduced), time‐course autophagosome formation (LC3BII 39% reduced, P‐value < 0.05), and autophagosome microscopic evaluation. Mitochondria displayed reduced genetic content (by 33.9%, P‐value < 0.05) and function (30.2%‐decrease in respiration, 45.6%‐decline in enzymatic activity (P‐value < 0.001), 14.3%‐higher oxidative stress, 135.2%‐increased antioxidant defence (P‐value < 0.05), 11.6%‐reduced mitochondrial membrane potential (P‐value < 0.05) and 42.8%‐reduced mitochondrial elongation (P‐value < 0.05)). In accordance, at the metabolite level, organic acid showed a 1.8‐fold change increase, with conserved amino acid profile. Correlating to disease evolution, oxidative stress and inflammation emerge as potential markers of prognosis.
Conclusions
These findings confirm the presence of molecular disturbances in peripheral tissues from IBM patients and prompt patients' derived fibroblasts as a promising disease model, which may eventually be exported to other neuromuscular disorders. We additionally identify new molecular players in IBM associated with disease progression, setting the path to deepen in disease aetiology, in the identification of novel biomarkers or in the standardization of biomimetic platforms to assay new therapeutic strategies for preclinical studies.
Keywords: Inclusion body myositis, Myopathy, Fibroblasts, Autophagy, Inflammation, Mitochondria
Introduction
Inclusion body myositis (IBM) is an inflammatory myopathy characterized by proximal and distal muscle weakness, inflammation, rimmed vacuoles and mitochondrial alterations in myofibres. 1 This rare disease (OMIM 147421) is the most common acquired muscle disease in patients over 50 years old. 2 Its prevalence ranges from 46 to 84 patients per million, 3 , 4 raising due to the aging of the population. The time from symptoms onset to diagnosis is delayed (around 4–5 years) with a high level of misdiagnosis, due to a lack of specialized diagnostic teams and non‐invasive biomarkers. Despite the progression towards disability being slow, effective treatments are absent, leading to an unmet medical need. 1
Scarce knowledge has been gathered about IBM aetiology. First characterized in 1978, it is currently categorized as an inflammatory myopathy (or myositis), together with polymyositis, dermatomyositis, immune‐mediated necrotizing myopathy and non‐specific myositis. 1 However, IBM was considered the sporadic form (sIBM) of inherited vacuolar myopathies (hIBM, caused by mutations in around 12 genes, mainly VCP), because both shared the presence of rimmed vacuoles. Inflammation is not present in vacuolar myopathies, 5 , 6 whereas it is prominent in IBM. This prompted the classification of IBM among inflammatory myopathies 2 , 5 and, currently, sIBM nomenclature is being moved to IBM.
The first symptoms appear in quadriceps and finger flexor muscles, leading to difficulties for climbing stairs and handling objects. In 60% of the cases, there is dysphagia and swallowing dysfunctions. The diagnosis is based on clinical and muscle histology studies, where inflammatory infiltrate, rimmed vacuoles and mitochondrial abnormalities must be present. Briefly, muscle biopsies show: (i) inflammation (CD8+ T cells, aberrant expression of MHC1 and endomysial mononuclear cell infiltrates in myofibres); (ii) degeneration (rimmed vacuoles, ubiquitin‐positive inclusions and amyloid deposits) and (iii) mitochondrial abnormalities (cytochrome c oxidase negative fibres (COX‐), succinate dehydrogenase positive cells (SDH+) and ragged‐red fibres (RRF), corresponding to an increased number of abnormal mitochondria around myofibres). 1 , 2 , 6
Currently, there is no treatment for IBM. Although being an inflammatory myopathy, corticosteroids have failed, and all trials with immunosuppressants, immunomodulating agents, TGFβ inhibitors and muscle growth factors have been ineffective. Intravenous immunoglobulin treatment has been useful to treat dysphagia, despite this, IBM patients are limited to physical therapy (with difficulties for some patients) and participation in clinical trials. 7
Validated disease models for IBM could help to gain insight into the pathophysiology of this condition, improve diagnosis and test effective treatments. However, previous attempts to develop animal models (MCK‐βAPP transgenic mice, 8 VCP mutant mice models, 9 and a cholesterol rabbit model 10 ) revealed serious limitations, probably because they were mainly based on gene editing, and IBM is an acquired disease with poor (or merely unknown) genetic base. Consequently, most of the advances in IBM have been accomplished directly with samples from affected patients. 5 Previous studies from our group revealed some pathological muscle features in IBM lymphocytes 11 or alternative peripheral tissues, 12 suggesting that IBM hallmarks could be present besides the target tissue (muscle). Considering fibroblasts are proliferative cells, used to diagnose mitochondrial diseases and to model neurodegenerative diseases, 13 , 14 , 15 we tested their usefulness as a potential model to study IBM. Fibroblasts preserve genetic, epigenetic and environmental cues of affected patients, offering a low‐cost maintaining platform to deepen in disease aetiology, discover novel biomarkers and assay new therapeutic strategies for preclinical studies.
In this article, we aim to report a detailed phenotyping of fibroblasts from IBM patients compared with age and sex‐paired healthy controls (CTL), as a promising model for this disease. We offer the transcriptomic and functional assessment of this cell model together with the clinical and pathological data of included patients. Briefly, fibroblasts from IBM patients showed a clear deregulated transcriptomic pattern of 778 differentially expressed genes (DEGs) and a pathological inflammatory, degenerative and mitochondrial profile, resembling the target tissue of the disease. Additionally, the present findings prompt distinct molecular targets as relevant key players in IBM.
Methods
Study design and population
A case–control study was conducted in the Department of Internal Medicine from Hospital Clínic of Barcelona (Barcelona, Spain), including 14 patients and 12 healthy volunteers. IBM patients and healthy volunteers signed the informed consent (ethical code HCB/2015/0562). Additional information is provided in the Supporting Information.
Fibroblast culture
Fibroblasts were obtained from a skin biopsy from cases and controls. Cells were harvested and collected at 80% of confluence, and phenotyped at passages 3 to 10. Briefly, fibroblasts from both groups displayed similar morphology and size. Cell growth rate was slightly reduced in IBM fibroblasts (0.019 ± 0.002 vs. 0.024 ± 0.006), although non‐significantly. Additional information is provided in the Supporting Information.
RNA extraction, mRNA library preparation and sequencing
Total RNA was isolated from cell lysates of three IBM and three CTL fibroblasts. Differential expression analysis was performed with DESeq2 v1.26.0 R package, and genes were considered differentially expressed with an adjusted P‐value < 0.05 and absolute fold change |FC| > 1.5. A detailed description of mRNA libraries, sequencing and analysis is presented in the Supporting Information.
Inflammation: Bio‐Plex assay of secreted inflammatory cytokines
A Bio‐plex assay (Bio‐Plex Pro™ Human Cytokine 27‐plex Assay #M500KCAF0Y) from Bio‐Rad (Hercules, CA, USA) was performed to quantify 27 inflammatory cytokines secreted in the supernatant of IBM and CTL fibroblasts after 96 h in culture (undiluted samples) normalized by cell number.
Autophagy protein array
Cell lysates were obtained from IBM and CTL fibroblasts; 250–500 μg of protein per sample were loaded in the Ray Bio® C Series Human Autophagy Array 1 (Cat#: AAHATG‐1‐8, Ray Biotech, Inc., Atlanta, GA, USA), incubated overnight at 4°C and detected by chemiluminescence. Additional information is provided in Supporting Information.
Autophagy time‐course
Autophagy flux in IBM versus CTL fibroblasts was measured in a time‐course in basal conditions (0 h), and 4 h and 8 h after the addition of 0.1 μM bafilomycin A1 from Streptomyces griseus (Sigma‐Aldrich® #B1793 SIGMA, Missouri, USA), and analysed through western blot analysis. Blots were probed against the anti‐ SQSTM1/p62 (Abcam #ab56416, Cambridge, UK) and anti‐LC3B (Cell Signaling #2775S, Massachusetts, USA) antibodies. More information provided in the Supporting Information.
Immunocytochemistry for autophagosome characterization
Cells were seeded in a 16‐well glass slide (Nunc™ #178599 Lab‐Tek® Chamber Slide™, Austin, USA) at 37°C with 5% CO2 for 24 h. Autophagosomes were stained with anti‐LC3 pAB (MBL International® #PM036, Massachusetts, USA) and images were taken with a Zeiss LSM confocal microscope (63×). A detailed protocol is presented in the Supporting Information.
Mitochondrial respiration
Mitochondrial respiration was measured according to the oxygen consumption of the mitochondrial respiratory chain (MRC). The oxygen consumption rate was detected with the XF Cell Mito Stress Test™ (Seahorse‐XFe24‐Analyser, Agilent Technologies), according to the manufacturer's protocol. Additional information is found in the Supporting Information.
MRC enzymatic activities: COX and citrate synthase (CS) activities
COX and CS enzymatic activities were assessed in 40 μg of total protein from each fibroblast sample according to standardized protocols, 15 at 37°C, and expressed as COX/CS ratio.
Oxidative stress
Oxidative stress was measured through lipid peroxidation (measuring malondialdehyde (MDA) and 4‐hydroxyalkenal (HAE)) and total antioxidant capacity of the cell (TAC). 11 Reagents and protocols are provided in the Supporting Information.
Mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was quantified with tetramethyl rhodamine methyl ester (TMRM) and detected with confocal microscopy, as the number of cells with active mitochondria per total number of cells.
mtDNA genome content
mtDNA content was evaluated with the fragments of the mitochondrially encoded 12S rRNA gene and the nuclear‐encoded RNase‐P gene, amplified by RT‐PCR (Applied Biosystems). The relative mtDNA content was expressed as the ratio mtDNA 12S rRNA:nDNA RNase‐P.
Mitochondrial elongation
Mitochondrial network was immunostained with TOMM20 (St Cruz Bio #sc‐11415 (1:100)), imaged with a Zeiss confocal microscope (63×) and analysed with ImageJ software using a mitochondrial morphology macro to quantify mitochondrial elongation (also called aspect ratio: AR = major axis/minor axis, where a higher AR is linked to healthier mitochondria. 16
Transmission electron microscopy (TEM)
Fibroblasts were fixed and incubated with 1% OsO4 before being sectioned using Leica ultramicrotome (Leica Microsystems). Ultrathin sections (50–70 nm) were stained and observed using a TEM, JEOL JEM‐1010 fitted with a Gatan Orius SC1000 (model 832) digital camera 17 to seek for abnormal organelle structures. Detailed information is presented in the Supporting Information.
Metabolite quantification
Organic acids were extracted in fibroblasts as previously reported. 18 The trimethylsilyl derivatives obtained were separated by gas chromatography (Agilent 7890A, Wilmington, DE, USA) and detected in a mass spectrometer (Agilent 5975C, Wilmington, DE, USA). Amino acids were quantified in fibroblasts by ultra‐performance liquid chromatography coupled to tandem mass spectrometry, as previously reported. 19 Both measurements were normalized by protein content. Additional information is detailed in the Supporting Information.
Statistical analysis
Statistical analysis was performed with the Statistical Package for the Social Sciences software, version 25 (IBM SPSS Statistics; SPSS Inc) and GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). Results were expressed as mean ± SEM or in fold change between conditions, calculated as mean fold change ratio (and deviation) in IBM patients versus CTL fibroblasts, normalized by cell number or by protein content. Obtained data were compared between independent experiments using the non‐parametric Mann–Whitney U test or Kruskal–Walli's test. Statistically significant results (P‐value < 0.05) were highlighted along the manuscript. Additionally, borderline significant results were included according to their biological relevance for better understanding of the biological context.
Results
Clinical and epidemiological data
In this study, IBM patients belonged to Caucasian population between 40 and 83 years old (mean age 66.9 ± 3.6) following a 1:1.7 male to female ratio (n = 14). Control subjects also belonged to the Caucasian population (n = 12), from 35 to 86 years old (mean age 57.4 ± 4.6), following a 1:2.5 male to female ratio. According to the IBM functional rating scale (IBMFRS; ranges from 0 (inability)‐40 (normal behaviour) 20 ), the IBM cohort scored 25.1 ± 2.1, indicating moderate clinical severity. The average time from symptoms onset to diagnosis was 37 months; six patients showed stable IBM prognosis and eight of them had a progressive disease decline. This overview of clinical data is summarized in Table 1, together with patients' muscle biopsy features that aid in IBM diagnosis.
Table 1.
Clinical data of the cohorts and features from IBM patients muscle's biopsies
| Sample code (IBM) | Sex | Age | Time symptoms to diagnosis (months) | IBMFRS | Evolution | Rimmed vacuoles | Inflammation | MHC1 | Auto Ab | CD57 | Mitochondrial Changes | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IBM01 | F | 67 | 26 | 16 | Stability | + | + | NA | All − | Normal | No | |
| IBM02 | M | 51 | 48 | 16 | Progression | ++ | ++ | NA | All − | Increase | No | |
| IBM03 | M | 48 | 48 | 31 | Stability | + | + | NA | All − | NA | Yes | |
| IBM04 | F | 81 | 26 | 17 | Progression | +++ | + | NA | All − | Normal | Yes+ | |
| IBM05 | M | 63 | 36 | 27 | Progression | + | +++ | Universal | All − | NA | Yes + | |
| IBM06 | M | 77 | 48 | 28 | Progression | + | + | Neg | All − | Increase | Yes | |
| IBM07 | F | 61 | 36 | 23 | Progression | +++ | +++ | Universal | All − | NA | Yes | |
| IBM08 | F | 83 | 12 | 8 | Progression | + | +++ | NA | All − | T clonal | Yes + | |
| IBM09 | M | 62 | 84 | 30 | Progression | +++ | ++ | Universal | All − | Increase | Yes | |
| IBM10 | M | 78 | 12 | 28 | Progression | +++ | +++ | + Not universal | All − | NA | No | |
| IBM11 | F | 80 | 24 | 33 | Stability | +++ | + | Universal | Ro + | Normal | Yes + | |
| IBM12 | M | 71 | 36 | 30 | Stability | +++ | +++ | Universal | Ro52+ | Increase | Yes | |
| IBM13 | M | 75 | 72 | 32 | Stability | + | ++ | Universal | All − | Reduced B normal CD57 | Yes | |
| IBM14 | M | 40 | 3 | 33 | Stability | +++ | ++ | Universal | All − | NA | Yes+ | |
| Sample code (CTL) | CTL01 | CTL02 | CTL03 | CTL04 | CTL05 | CTL06 | CTL07 | CTL08 | CTL09 | CTL10 | CTL11 | CTL12 |
| Sex | M | F | F | F | F | F | M | F | F | M | M | M |
| Age | 69 | 66 | 61 | 41 | 48 | 47 | 35 | 45 | 82 | 86 | 57 | 52 |
| Summary | Fibroblasts (n) | Male/female ratio | Mean age (years) | Time to diagnose (months) | IBMFRS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IBM | 14 | 0.6 | 66.9 ± 3.6 | 37 | 25.1 ± 2.1/40 | |||||||
| CTL | 12 | 0.4 | 57.4 ± 4.6 | |||||||||
Note: Data presented as mean ± SEM.
Abbreviations: Sex: F, female; M, male; IBMFRS, Inclusion Body Myositis Functional Rating Scale (from 0 to 40 points). 20 Rimmed vacuoles: from 0 to +++; Inflammation: from 0 to +++; MHC1: Major Histocompatibility Complex: Universal; NA: not available; + not universal: splattered pattern; Auto Ab: Autoantibodies: Myositis specific and associated auto antibodies include: Anti‐Jo‐1, anti‐SRP, anti‐Mi2, anti‐TIF1gamma, anti‐MDA5, anti‐SAE, anti‐NXP2 and anti‐HMGCR (specific) and anti‐PM/Scl, anti‐Ro, anti‐La, and anti‐U1RNP (associated); Mitochondrial changes: Yes or No. Yes: RRF and/or COX neg. Yes + (strong alterations). No: no changes; Age and gender‐paired distribution between cohorts (P = NS).
Transcriptomic phenotyping: Altered gene expression in IBM fibroblasts
RNA sequencing was performed in fibroblasts from IBM patients (n = 3) and genetically unrelated healthy subjects (n = 3). A principal component analysis (PCA) revealed IBM and healthy subjects clustered separately (Figure 1A), suggesting a differential gene expression pattern of disease, comprising 778 DEGs with P‐value adj (FDR) < 0.05; of them, 457 were downregulated and 321 upregulated (Table S1). The top 50 DEGs are represented in the heatmap (Figure 1B). Among these DEGs, some are related to inflammation (MHC components like CD59; T‐cell expansion genes like CLEC2A, CLEC2B, and CLEC3B, and others like CSCS and TNFAIP6); mitochondria (PCK2, MTHFD2, GPAT2, ATP2B4, and CYP2U1); and other processes like cell cycle regulation, metabolism of amino acids and derivatives, and so on. Gene names are found in Table S1.
Figure 1.
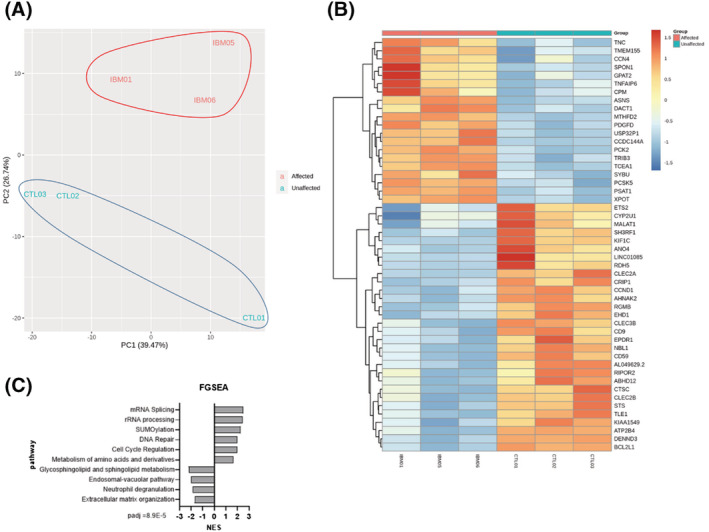
Transcriptome analysis of IBM and CTL fibroblasts (n = 3/group). (A) Principal component analysis (PCA) of affected (IBM) versus unaffected (CTL) subjects. (B) Heatmap of the top 50 differentially expressed genes (DEGs) obtained in a differential expression analysis between both cohorts. All DEGs are represented in Table S1. (C) Top pathways (with lowest P‐value adj) after a functional gene set enrichment analysis (GSEA) and considering their normalized enrichment score (NES). PCA, heatmap and pathway analysis revealed a differential gene expression pattern in IBM patients, with DEGs and pathways related to inflammation, mitochondria and other processes like cell cycle regulation and metabolism of amino acids and derivatives.
These DEGs were clustered with a Gene Ontology enrichment analysis, where the most deregulated clusters were related to cell communication, cell signalling and systems development. Gene set enrichment analysis (GSEA) revealed 358 significantly altered pathways (P‐value < 0.05), the top ones found in Figure 1C.
In addition to GSEA, DEGs were analysed using Ingenuity Pathway Analysis (IPA) software. IPA showed tRNA charging as the top canonical pathway, top networks related to cell cycle, DNA replication, cell‐to‐cell signalling, and interaction, similar to GSEA results, confirming the consistency of reported findings. Overall, these findings point out a clear deregulated transcriptomic profile in IBM fibroblasts and highlight significant pathways and molecular targets in IBM.
Functional phenotyping
Increased inflammatory profile in IBM fibroblasts
Inflammation is one of the hallmarks of IBM in muscle. To explore inflammation in IBM versus CTL fibroblasts, we performed an mRNA‐seq and measured secreted cytokines in cell supernatants.
In the transcriptome, we found a differential inflammatory profile in IBM versus CTL fibroblasts. Briefly, we found 62 inflammatory‐related genes (Table S2) related to MHC1, cytokines and IL receptors, NF‐kB pathway, IFN genes and T‐cell expansion genes, which were grouped in gene clusters (Figure 2A), all involved in IBM pathogenesis.
Figure 2.
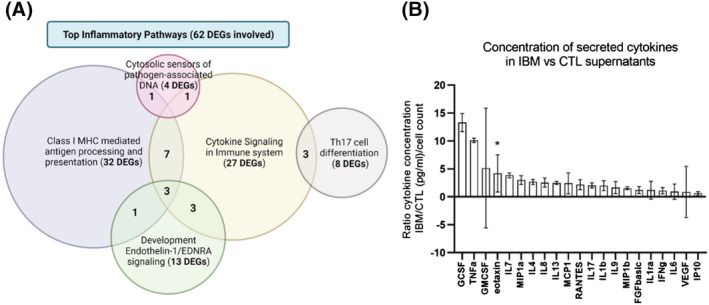
The inflammatory pattern of IBM and CTL fibroblasts. (A) Venn's diagram of the most affected inflammatory clusters and the number of differentially expressed genes (DEGs) involved. Some DEGs are involved in more than one cluster. The list of inflammatory DEGs is represented in Table S2. (B) Mean fold change ratio (and deviation) of secreted cytokines in IBM patients versus CTL fibroblasts, normalized by cell number (n = 13 IBM vs. 12 CTL; *P‐value < 0.05, Mann–Whitney U test) (ratio > 1: Higher concentration in IBM). The concentration of each cytokine and their full names are represented in Table S3. Briefly, an increased inflammatory profile is observed in IBM versus CTL fibroblasts, with differentially expressed genes (DEGs) related to MHC1, cytokines, interleukins, and so on and higher cytokine secretion in fibroblast supernatants.
Considering cytokine secretion in fibroblast supernatants, we quantified 21 pro‐inflammatory cytokines, grouped in chemokines (eotaxin, IP10, MCP1, MIP1a, MIP1b, RANTES, and IL8) and proinflammatory cytokines (GCSF, TNFa, GMCSF, IL7, IL4, IL13, IL17, IL1b, IL9, FGFb, IL1ra, IFNg, IL6, and VEGF). Cytokines drifted towards an upregulated three‐fold expression in IBM patients (Figure 2B and Table S3). In particular, eotaxin (CCL11), involved in a chemotactic activity for eosinophils, 21 showed a statistically significant increase of 4.2 times in IBM versus CTL fibroblasts (P‐value < 0.05, n = 13 IBM vs. 12 CTL), whereas GCSF (that regulate the production, differentiation, and function of granulocytes) showed a high 13.3‐fold change ratio between groups (Figure 2B). These findings revealed a trend towards an increased inflammation in IBM fibroblasts at a transcriptomic and functional level, identifying relevant pathways and targets in this pathology.
Degenerative features and impaired autophagy in IBM fibroblasts
Rimmed vacuoles are another hallmark in the muscle of IBM patients, partly due to misfolded protein depots in the cytoplasm of muscle cells, caused by impaired or insufficient autophagy. Here, we examined autophagy in IBM versus CTL fibroblasts by mRNA‐seq data and analysing target proteins at basal and time‐course manners.
In the gene expression analysis, we found 37 DEGs related to autophagy (Table S2) affecting many steps of the signalling cascade (heat‐shock proteins, stress‐related factors, proteasome genes and regulators of the master AMPK gene), which were grouped in degenerative gene clusters (Figure 3A).
Figure 3.
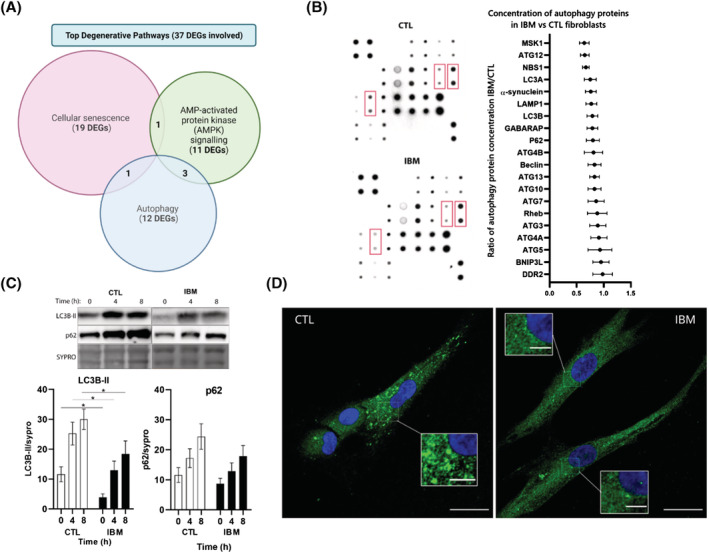
Autophagy in IBM versus CTL fibroblasts. (A) Venn's diagram of the most affected degenerative clusters and the number of differentially expressed genes (DEGs) involved. Some DEGs are involved in more than one cluster. The list of degenerative DEGs is represented in Table S2. (B) Representative images of two autophagy protein arrays of one inclusion body myositis (IBM) and one control (CTL) fibroblast and quantification of n = 8/group autophagy protein arrays. In red, differences in protein concentration (according to spot intensity) among IBM and CTL fibroblasts. In the graph, mean fold change ratio (and deviation) of autophagy proteins in IBM patients versus CTL fibroblasts, normalized by protein content (ratio < 1: Lower concentration in IBM). Each protein concentration is depicted in Table S4. (C) Representative images and graphs of an autophagy time‐course (at basal, 4 h and 8 h under bafilomycin A1 treatment) measured by western blot between IBM (n = 13) and CTL (n = 12) fibroblasts. Results: Mean ± SEM, n = 13 IBM versus 11 CTL; Kruskal–Wallis test, *P‐value < 0.05. SQSTM1/p62: Autophagy substrate, LC3BII: Autophagy receptor, lipidated form, sypro: Total protein content. (D) Representative images of autophagosome accumulation in IBM versus CTL fibroblasts treated with bafilomycin A1 (100 nM) for 6 h obtained by confocal microscope (LC3B in green; nuclei in blue with DAPI, scale 25 μM (inserts 5 μM)). Briefly, autophagy is impaired in terms of differentially expressed genes (DEGs) and decreased expression of basal protein mediators, time‐course autophagosome formation, and microscopic evaluation.
In the functional autophagy analysis, we assessed expression of 20 proteins at steady‐state levels in IBM and CTL fibroblasts. The expression of autophagic proteins was 18.4% reduced in IBM (Table S4 and Figure 3B), leading towards a downregulation of the whole autophagic process. In particular, the following pathways were more affected 22 : Receptor‐mediated mitophagy (ATG5, ATG12, LC3A, and LC3B), PINK1‐PRKN Mediated Mitophagy (ATG12, P62, LC3A, LC3B, and ATG5) and the super pathway Macroautophagy (ATG12, ATG7, Rheb, ATG4A, ATG13, Beclin, P62, GABARAP, LC3B, ATG5, ATG10, LC3A, ATG3, and ATG4B) that displayed a trend of 22.0%, 21.6%, and 17.5% reduction in IBM fibroblasts.
To evaluate if a decreased autophagic activity was due to the inactivation of the process or, to higher consumption of mediators caused by a higher autophagic rate, we performed a time‐course experiment that blocked the process. As observed in Figure 3C, flux rate of both LC3BII and p62 effectors at 4 h and 8 h after bafilomycin A1 treatment was non‐significantly reduced between cohorts when normalized by basal levels. Interestingly, raw data non‐normalized by basal expression showed significantly reduced levels of LC3B‐II marker at 4 h and 8 h after treatment in IBM fibroblasts (44.4% and 33.6% reduced in IBM, P‐value < 0.05), suggesting a lower autophagic performance in the IBM cohort. Overall, a decreased autophagic activity could lead to insufficient cell recycling and accumulation of damaged components in the cytoplasm, increasing the endoplasmic reticulum stress and compromising the homeostatic balance within cells. In the case of p62 expression, we also found a slight reduction in IBM fibroblasts (24.9%, 19.0%, and 20.6% reduced).
To visualize the proportion of autophagic vesicles between cohorts, we performed immunostaining of LC3B after 6 h under bafilomycin A1 treatment (Figure 3D). The results revealed a lower accumulation of autophagosomes in IBM fibroblasts. These findings manifested a deregulated autophagic and degenerative profile in IBM fibroblasts, prompting relevant pathways and targets in this disease.
Mitochondrial abnormalities increased oxidative status in IBM fibroblasts
The third hallmark of IBM in muscle are mitochondrial alterations. In muscle biopsies, there is an increased number of mitochondria across myofibres (creating RRF) and increased complex II expression (SDH+ fibres), probably as an attempt to compensate dysfunctional mitochondria, evidenced as decreased complex IV expression (COX‐fibres). 6 , 11 , 23 , 24 These are features of primary mitochondrial diseases. We aimed to examine the mitochondrial state of IBM versus CTL fibroblasts by analysing mitochondrial deregulated genes, and by functional assessment of bioenergetic and metabolic performance.
Regarding mitochondrial gene expression, we found 42 mitochondrial DEGs compiled in the Mitocarta (version 3.0), 25 in Table S2. Among them, some DEGs were encoded in the mitochondrial genome (NADH:CoQ oxidoreductase subunits, COX and ATP synthase), others related to the MRC function but encoded in the nuclear genome; and tricarboxylic acid cycle (TCA) enzymes. Also, some DEGs were stress‐related or peroxisomal genes. These genes were grouped in gene clusters (Figure 4A).
Figure 4.
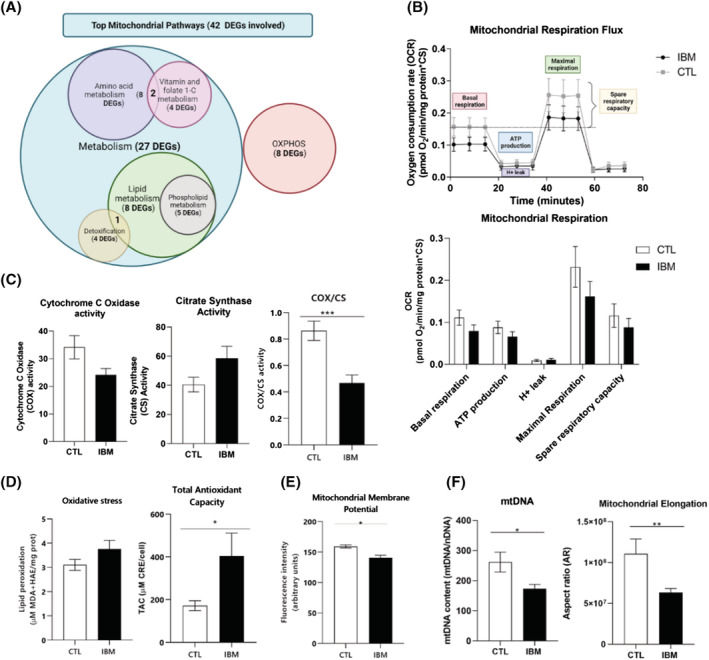
Mitochondrial profile in IBM versus CTL fibroblasts. (A) Venn's diagram of the most affected mitochondrial clusters and the number of differentially expressed genes (DEGs) involved. Many DEGs are involved in more than one cluster. The list of mitochondrial DEGs is represented in Table S2. (B) Mitochondrial respiratory flux and rates of IBM versus CTL fibroblasts. Each sample was seeded in quadruplicate per condition (n = 10/group, Mann–Whitney U test). OCR = oxygen consumption rate. Respiratory control ratios were normalized by total protein content and by citrate synthase (CS) activity as a marker of mitochondrial content. (C) Cytochrome C oxidase (COX), citrate synthase (CS) and COX per CS activity (COX/CS ratio, as a marker of complex IV from the mitochondrial respiratory chain (MRC) per mitochondrial mass (n = 10/group, P‐value < 0.001, Mann–Whitney U test), normalized by protein content. (D) Oxidative stress measured through lipid peroxidation in IBM versus CTL fibroblasts normalized by total protein content (n = 9 IBM vs. 8 CTL, Mann–Whitney U test) (MDA: Malondialdehyde; HAE: Hydroxialkenal); coupled to Total antioxidant capacity (TAC) expressed as μM CRE (copper reducing equivalents) per total cell number (n = 11 IBM vs. 4 CTL, P‐value < 0.05, Mann–Whitney U test). (E) Mitochondrial membrane potential (MMP) measured with TMRM + Arimoclomol in IBM versus CTL fibroblasts (n = 4/group, P‐value < 0.05, Mann–Whitney U test). The results were expressed as mean ± SEM (*P < 0.05). (F) mtDNA genome content in IBM versus CTL fibroblasts expressed as the ratio of mtDNA 12S rRNA:nDNA RNase‐P (mitochondrial vs. nuclear encoded gene) (n = 9 IBM vs. 8 CTL, P‐value < 0.05, Mann–Whitney U test) and mitochondrial elongation (represented by the aspect ratio) in IBM versus CTL fibroblasts (n = 3/group, P‐value < 0.05, Mann–Whitney U test). Briefly, mitochondrial performance is disturbed in IBM versus CTL fibroblasts in terms of genetic content, differentially expressed genes (DEGs) and functionality (reduced respiratory profile, marked COX/CS decline, higher oxidative stress and antioxidant defence activation, reduced MMP and mitochondrial elongation).
At functional level, we measured the respiratory profile of IBM and CTL fibroblasts to quantify the oxygen consumption rate (Figure 4B). IBM fibroblasts leaned towards a decreased respiratory pattern (Figure 4B), especially at basal and maximal respiration levels (28.6% and 31.8% reduced in IBM), suggesting a trend towards less active MRC activity.
These results were supported at the enzymatic level by a 45.6% COX significant decrease, as seen in muscle, normalized by CS activity (P‐value < 0.001) (Figure 4C). Moreover, CS activity shifted towards a 44.6% increase in IBM fibroblasts, leading to an increased mitochondrial mass to compensate for COX reduction, as seen in RRF from IBM muscles.
To assess if MRC downward is correlated to oxidative stress and antioxidant levels, lipid peroxidation markers like MDA and HAE were measured in cell lysates (Figure 4D). Lipid peroxidation tended to increase a 14.3% in IBM versus CTL fibroblasts, suggesting a trend towards higher oxidative stress in IBM cells; coupled with a 135.2% significant increased level of antioxidant defences (TAC) in IBM (P‐value < 0.05) (Figure 4D). Moreover, in the mRNA‐seq we found aldehyde dehydrogenases (ALDH1L2, ALDH1B1, ALDH4A1, and ALDH18A1), enzymes involved in lipid peroxidation, and some antioxidant enzymes (CBR3, CYB5R3, ETHE1, PRXL2A, and SQOR), among the DEGs between IBM and CTL fibroblasts.
Mitochondrial dysfunction is usually linked to the depolarization of MMP, that was significantly decreased by 11.6% in IBM fibroblasts (P‐value < 0.05) (Figure 4E). MMP depolarization may result in impaired proton pumping, leading to the opening of the mitochondrial membrane pore, and potential activation of apoptosis.
mtDNA genome content was significantly decreased by 33.9% in IBM fibroblasts (Figure 4F; P‐value < 0.05), as previously observed in IBM muscle 11 and in accordance to abnormal mitochondrial morphology, either externally (showing a significant 42.8%‐reduced mitochondrial elongation, Figure 4F, Figure 5A), and internally (at subcellular level) (Figure 5B). Briefly, using TEM ultrastructural visualization, 76.2% of IBM fibroblasts revealed abnormal mitochondria (61.9% of them with altered cristae and 42.2% with a round shape, associated with unhealthier mitochondria), compared with 25.1% of CTL fibroblasts with abnormal mitochondria (of which 7.9% had altered cristae and 17.2% a round shape). At mRNA level, we found three DEGs related to mitochondrial dynamics (BCL2L1, FKBP8, and MGARP), all downregulated.
Figure 5.
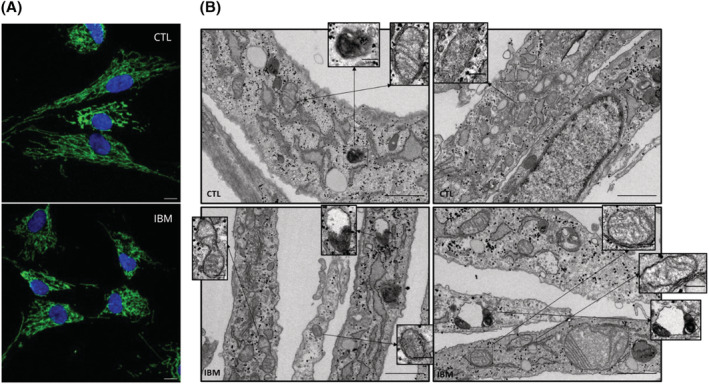
Mitochondrial morphology in IBM versus CTL fibroblasts. (A) Mitochondrial network in a representative image of IBM versus CTL fibroblasts obtained by confocal microscope (TOMM20 in green; nuclei in blue with DAPI, scale 10 μM) displaying reduced mitochondrial elongation in IBM (associated to poor mitochondrial health). (B) Transmission electron microscopy (TEM) images of IBM versus CTL fibroblasts. In IBM fibroblasts, some mitochondria lacked cristae (see inserts) presenting a discontinuous cristae distribution. Also, increased mitochondrial fission (see insert) could explain the smaller and more circular shape of these organelles associated with unhealthier mitochondria. Additionally, the presence of impaired autophagosome‐lysosome structures in IBM patients (see inserts) might be a result of incomplete autophagy. Briefly, these findings highlight a mitochondrial impairment in IBM fibroblasts. Magnification: 30 000×, scale 1 μm for each picture and 0.2 μm for inserts.
Overall, these findings highlight a mitochondrial impairment in IBM fibroblasts and identify contributing pathways and key players in disease.
Targeted metabolites characterization
To examine if mitochondrial dysfunction was linked to metabolic misbalances beyond the MRC, we measured DEGs and metabolite levels related to organic acids and amino acids in IBM and CTL fibroblasts.
At gene expression level, we observed 10 DEGs involved in organic acids and amino acids metabolism (in Table S5). Briefly, increased lactic acid and alanine are related to MT‐ND5, MT‐CO2, and MT‐ATP6, components of the MRC and related to the TCA cycle. ETHE1 is also linked to increased alanine, ethylmalonic and methyl succinic, and related to the SQOR gene. PSAT1 and GPX3, albeit not being mitochondrial genes, are directly related to mitochondrial metabolism, involved in serine and glycine biosynthesis and pyroglutamic acid metabolism, respectively.
At functional level, organic acids showed a 1.8‐fold change increase in IBM, suggesting deregulation of intermediary metabolism. The most relevant organic acids were: lactic acid (biomarker of mitochondrial diseases); succinic acid (part of MRC complex II, increased in SDH+ fibres in muscle); pyroglutamic acid (involved in glutathione synthesis); sebacic acid (involved in the electron transfer from the β‐oxidation pathway to MRC complex II) and above all, 2OH‐glutaric acid (TCA cycle component which accumulates in case of mitochondrial defect), that were significantly deregulated (Figure 6A and Table S6). These components or derivatives of the TCA cycle are related to amino acids or fatty acid metabolism and their increase is frequently linked to MRC dysfunction. Regarding amino acids levels, no relevant differences were observed between IBM and CTL fibroblasts (Figure 6B and Table S7).
Figure 6.
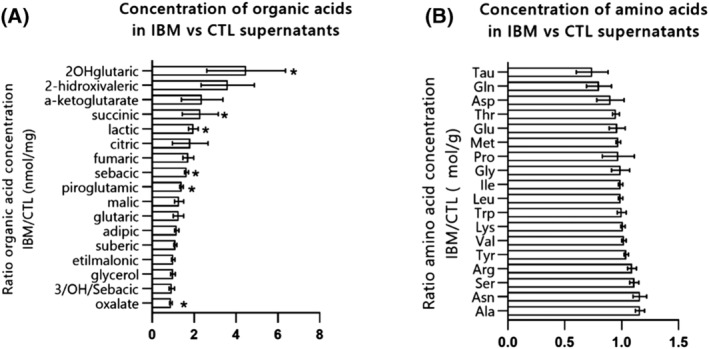
Metabolites concentration in IBM versus CTL fibroblasts. (A) Mean fold change ratio (and deviation) of organic acids in IBM patients versus CTL fibroblasts, normalized by protein content (n = 11 IBM vs. 10 CTL, *P‐value < 0.05, Mann–Whitney U test) (ratio > 1: Higher concentration in IBM). The raw data of each organic acid concentration is represented in Table S6. (B) Mean fold change ratio (and deviation) of amino acids IBM patients versus CTL fibroblasts, normalized by protein content (n = 11 IBM vs. 10 CTL, Mann–Whitney U test) (ratio > 1: Higher concentration in IBM; ratio < 1: Lower concentration in IBM). The raw data of each amino acid concentration is represented in Table S7. Briefly, organic acid levels showed a general increase in IBM, suggesting deregulation of intermediary metabolism related to mitochondrial dysfunction, whereas no relevant differences in amino acid levels were observed between IBM and CTL fibroblasts.
Metabolic reprogramming in IBM fibroblasts supports mitochondrial dysfunction beyond the MRC, and unravels the main pathways and targets involved in IBM.
Association of molecular phenotype and disease progression
Phenotyping IBM vs CTL fibroblasts has revealed an overall differential gene expression and functional profile, especially considering inflammation, degeneration and mitochondrial function. Interestingly, this pattern seems to differ slightly depending on disease evolution.
As observed in Table 1, six patients showed stable IBM prognosis whereas eight followed a progressive disease decline. The correlation of molecular and clinical data yielded to the association of increased pro‐inflammatory and organic acids signature in stable IBM (Table S8 and Figure 7). Contrarily, progressive IBM showed increased oxidative stress and antioxidant capacity. Together, stable and progressive IBM present an evident decrease in LC3BII time‐course and COX/CS with respect to CTLs. 26
Figure 7.
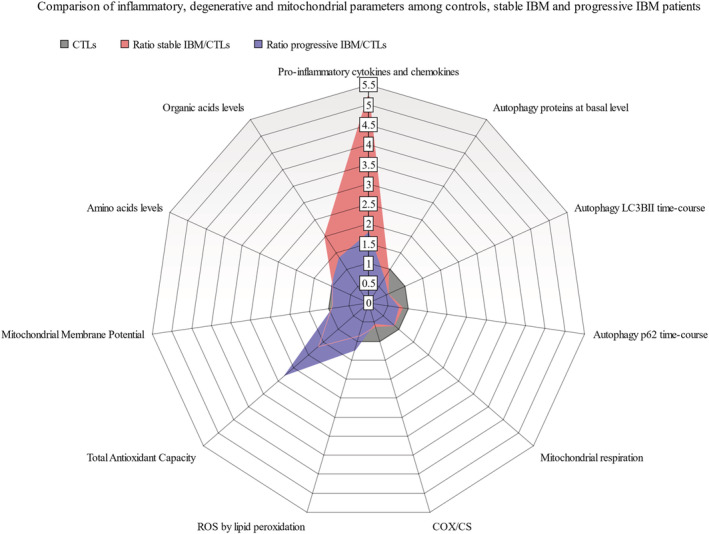
Comparison of inflammatory, degenerative and mitochondrial parameters among controls (CTLs), IBM patients with stable evolution (stable IBM) and IBM patients with disease progression (progressive IBM). Ratios between IBM (stable or progressive) and control fibroblasts were measured to compare inflammatory, degenerative and mitochondrial parameters among the three groups (Table S8). Briefly, stable IBM showed an increased pro‐inflammatory and organic acids signature compared with progressive IBM and controls. Progressive IBM showed increased oxidative stress (ROS) and antioxidant capacity. Together, IBM stable and progressive present an evident decrease in LC3BII time‐course (autophagy marker) and COX/CS (cytochrome c oxidase related to citrate synthase, a marker of mitochondrial mass) with respect to CTLs (ratio > 1: Higher concentration in IBM; ratio < 1: Lower concentration in IBM).
The presence of common and differential molecular profile between stable and progressive IBM patients could help in understanding disease pathogenesis and become potential markers of prognosis.
Discussion
Here we reported the validation of fibroblasts as a disease model for untangling IBM and identifying novel pathogenic molecular players. Briefly, we observed transcriptomic and functional inflammatory, degenerative and mitochondrial changes in IBM fibroblasts, which resemble the alterations found in the target tissue, in muscle.
Previous studies analysed IBM transcriptome 27 , 28 at muscular and blood level, but this is the first IBM RNA‐seq in fibroblasts. Here we found 778 DEGs, supporting a clear pathological pattern of gene expression in IBM.
At the inflammatory level, IBM fibroblast transcriptome revealed altered inflammatory genes resembling muscle features, including MHC1 (HLA genes) and IFN genes. These alterations were found in an IBM deep neural network model. 29 , 30 At the functional scale, cytokines secretion in IBM fibroblasts was increased. Previous studies in IBM muscle detected CD8+ T cell‐secreted cytokines and chemokines related to IFN‐ɣ genes (CXCL9, CXCL10, CCL5, and CCL18) and others, including TNF, IL7, IL15, IL16, and IL32, 1 , 26 some of which were also detected in the sera of IBM patients. 26 Although fibroblasts are not part of the immune system, these results partially mimic the inflammation of IBM muscles.
Regarding degeneration in IBM, impaired autophagy and disturbed proteostasis have been described in muscle. 24 Some degenerative markers like TDP43, p62, and LC3 show an aberrant expression in IBM muscle. 5 , 31 Herein, we found impaired autophagy at gene and functional level, with lower production of autophagy proteins and deficient formation of autophagosomes in IBM fibroblasts, leading to insufficient recycling needs and deposition of cargos within the cell. This phenotype is remarkable in fibroblasts, as their high turnover avoids debris accumulation. In contrast, muscle is a post‐mitotic tissue that accumulates cell debris, forming rimmed vacuoles in IBM, where autophagic deficiencies are more evident.
Related to deficient macroautophagy and mitophagy in IBM, there is a lower turnover of mitochondria, leading to bioenergetic deficits and cell damage due to oxidative stress generation. 24 , 32 The deficient mitochondrial profile observed in IBM fibroblasts revealed altered gene expression, impaired respiratory capacity, increased oxidative stress and mitochondrial mass (CS), reduced MMP, COX activity, mtDNA content and mitochondrial elongation, all previously seen in muscle tissue 11 , 13 , 24 and in mitochondrial myopathies, 13 , 33 suggesting that IBM slightly behaves as a mitochondrial disease.
Metabolic reprogramming could be linked to mitochondrial dysfunction, as proposed in IBM blood metabolomes 33 and primary mitochondrial diseases. Many pathways and genes related to the IBM blood metabolome are altered in IBM fibroblasts. Buzkova et al. demonstrated that IBM blood metabolome clustered more accurately with mitochondrial myopathies than neuromuscular diseases, 33 prompting a higher mitochondrial implication in IBM than previously thought. The current results in fibroblasts support previous findings.
Interestingly, these pathological features of IBM fibroblasts and muscle (inflammation, degeneration and mitochondrial dysfunction), resemble standard physiologic aging, including decreased muscle strength, 23 supporting that IBM involves an accelerated muscle aging.
Considering IBM evolution, it revealed that progressive IBM (22.1 ± 2.7 IBMFRS) is associated to higher imbalances in oxidant and antioxidant capacities whereas stable IBM (29.2 ± 2.7 IBMFRS) is associated to higher inflammatory profile. This increased inflammation could be a protective mechanism in the stable IBM, as these patients perform better at a clinical scale (higher IBMFRS). Considering these findings, oxidative stress and inflammation emerge as potential markers of prognosis.
Overall, this is the first attempt to suggest fibroblasts as a model of IBM pathogenesis, scarcely tested as a model in neuromuscular diseases. 12 , 34 So far, most of the pathological mechanisms in IBM have been discovered in cells and tissue samples from patients. However, although preclinical and omic studies help in understanding disease pathogenesis, the gap in translation to clinical research remains elusive. 35 Currently in IBM, 16 clinical trials are active, 36 in which most treatments were discovered or tested in cell models (e.g. phenylbutyrate in in vitro muscle fibres 37 ), suggesting the relevance of disease models in regulatory settings. In this sense, fibroblasts emerge as a promising model of IBM to discover new targets and perform screening of biomarkers and potential therapies.
Nevertheless, there are some limitations in the present study. First, our limited sample size, mainly due to IBM being a rare disease, even though for a reference unit like ours. In addition, even fibroblasts are associated to cost‐effective maintenance conditions, other cell models, closer to the target tissue, might better reflect IBM pathogenesis. This may be the case of myotubes differentiated from induced pluripotent stem cells, which avoid the invasiveness of a muscle biopsy, but may offer a closer approach to muscle behaviour. However, they require complex protocols, rending fibroblasts to a cost‐effective cell model.
Despite these contingencies, in the present study we validated fibroblasts as a disease model for IBM, showing IBM alterations are present beyond the target tissue of the disease. This model could be exported to other myositis or neuromuscular diseases. Additionally, we unveil numerous pathways and specific molecules that emerge as relevant key players in disease aetiology or evolution, for further evaluation as candidate biomarkers or therapeutic targets.
Funding
This study was supported by Instituto de Salud Carlos III (ISCIII‐FEDER, grants PI18/00498, PI18/00451 and PI21/00935) and co‐funded by the European Union. Additionally, JC‐S is supported by the APIF Programme (University of Barcelona), FJG‐G by the CIBERER (ISCIII‐FEDER), MG‐M by CD21/00019 (ISCIII‐ FSE+), CR by the Serra Húnter Programme (Generalitat de Catalunya), RA by Torrons Vicens, JCF‐C by PID2019‐111669RB‐I00, CG‐R by PID2020‐115055RB‐I00 and AE‐C by PT17/0009/0019 (ISCIII‐MINECO‐FEDER).
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Table S1. Differentially expressed genes between IBM and CTL (778 genes with p‐value adj (FDR) < 0.05, following alphabetical order)
Table S2. Lists of differentially expressed genes (DEGs) related to inflammation (62 DEGs), autophagy (37 DEGs) and mitochondria (42 DEGs)
Table S3. Concentration of secreted inflammatory cytokines in IBM vs CTL supernatants. IBM fibroblasts revealed an increased expression of most of these cytokines.
Table S4. Expression of 20 autophagy proteins in IBM vs. CTL fibroblasts. Most of these proteins displayed a decreased expression in IBM, suggesting a reduced activity of the autophagy process.
Table S5. Differentially expressed genes (DEGs) related to metabolite concentration in fibroblasts. The table depicts the relationship of 10 DEGs involved in metabolite metabolism with their respective alteration at amino acids and organic acids level, to relate expression vs. metabolism patterns in IBM vs. CTL fibroblasts.
Table S6. Organic acids in IBM vs. CTL fibroblasts. Organic acids showed a general increase in IBM, suggesting a deregulation of intermediary metabolism related to mitochondrial function, as many organic acids are involved in tricarboxylic acid cycle (TCA) that further feeds the mitochondrial respiratory chain.
Table S7. Amino acids levels in fibroblasts of IBM patients vs. CTL fibroblasts.
Table S8. Comparison of inflammatory, degenerative, and mitochondrial IBM hallmarks considering the evolution of IBM patients: stable vs progressive prognosis, compared to CTL fibroblasts.
Acknowledgements
We acknowledge the contribution of patients and their families, collaborating to expand the state‐of‐the‐art in rare diseases. We also appreciate the support given by the Advanced Optical Microscopy department of the University of Barcelona. All authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 38
Cantó‐Santos J., Valls‐Roca L., Tobías E., García‐García F. J., Guitart‐Mampel M., Esteve‐Codina A., Martín‐Mur B., Casado M., Artuch R., Solsona‐Vilarrasa E., Fernandez‐Checa J. C., García‐Ruiz C., Rentero C., Enrich C., Moreno‐Lozano P. J., Milisenda J. C., Cardellach F., Grau‐Junyent J. M., and Garrabou G. (2023) Unravelling inclusion body myositis using a patient‐derived fibroblast model, Journal of Cachexia, Sarcopenia and Muscle, 14, 964–977, 10.1002/jcsm.13178
Josep M. Grau‐Junyent and Glòria Garrabou are last signatory.
Contributor Information
Josep M. Grau‐Junyent, Email: jmgrau@clinic.cat.
Glòria Garrabou, Email: garrabou@clinic.cat.
References
- 1. Greenberg SA. Inclusion body myositis: clinical features and pathogenesis. Nat Rev Rheumatol 2019;15:257–272. [DOI] [PubMed] [Google Scholar]
- 2. Weihl CC. Sporadic Inclusion Body Myositis and Other Rimmed Vacuolar Myopathies. CONTINUUM Lifelong Learn Neurol 2019;25:1586–1598. [DOI] [PubMed] [Google Scholar]
- 3. Callan A, Capkun G, Vasanthaprasad V, Freitas R, Needham M. A Systematic Review and Meta‐Analysis of Prevalence Studies of Sporadic Inclusion Body Myositis. J Neuromuscul Dis 2017;4:127–137. [DOI] [PubMed] [Google Scholar]
- 4. Keshishian A, Greenberg SA, Agashivala N, Baser O, Johnson K. Health care costs and comorbidities for patients with inclusion body myositis. Curr Med Res Opin 2018;34:1679–1685. [DOI] [PubMed] [Google Scholar]
- 5. Greenberg SA. Pathogenesis of inclusion body myositis. Curr Opin Rheumatol 2020;32:542–547. [DOI] [PubMed] [Google Scholar]
- 6. Catalán M, Selva‐O'Callaghan A, Grau JM. Diagnosis and classification of sporadic inclusion body myositis (sIBM). Autoimmun Rev 2014;13:363–366. [DOI] [PubMed] [Google Scholar]
- 7. Dalakas MC. Inflammatory myopathies: update on diagnosis, pathogenesis and therapies, and COVID‐19‐related implications. Acta Myologica 2020;39:289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sugarman MC, Yamasaki TR, Oddo S, Echegoyen JC, Murphy MP, Golde TE, et al. Inclusion body myositis‐like phenotype induced by transgenic overexpression of βAPP in skeletal muscle. Proc Natl Acad Sci U S A 2002;99:6334–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahmed M, Machado PM, Miller A, Spicer C, Herbelin L, He J, et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci Transl Med 2016;8:331ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen X, Ghribi O, Geiger JD. Rabbits fed cholesterol‐enriched diets exhibit pathological features of inclusion body myositis. Am J Physiol Regul Integr Comp Physiol 2008;294:829–835. [DOI] [PubMed] [Google Scholar]
- 11. Catalán‐García M, Garrabou G, Morén C, Guitart‐Mampel M, Hernando A, Díaz‐Ramos À, et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin Sci 2016;130:1741–1751. [DOI] [PubMed] [Google Scholar]
- 12. Katsetos CD, Koutzaki S, Melvin JJ. Mitochondrial dysfunction in neuromuscular disorders. Semin Pediatr Neurol 2013;20:202–215. [DOI] [PubMed] [Google Scholar]
- 13. de Paepe B. Sporadic inclusion body myositis: An acquired mitochondrial disease with extras. Biomolecules MDPI AG 2019;9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tokuyama T, Hirai A, Shiiba I, Ito N, Matsuno K, Takeda K, et al. Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients. Biomolecules 2020;10:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. González‐Casacuberta I, Juárez‐Flores DL, Ezquerra M, Fucho R, Catalán‐García M, Guitart‐Mampel M, et al. Mitochondrial and autophagic alterations in skin fibroblasts from Parkinson disease patients with Parkin mutations. Aging 2019;11:3750–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. GitHub ‐ MariaCalvo‐UB/MitochondrialMorphologyAnalysis_Folder.ijm: This macro segments cells and mitochondria network, and measures mitochondria network area and morphology cell by cell.
- 17. Meneses‐Salas E, García‐Melero A, Kanerva K, Blanco‐Muñoz P, Morales‐Paytuvi F, Bonjoch J, et al. Annexin A6 modulates TBC1D15/Rab7/StARD3 axis to control endosomal cholesterol export in NPC1 cells. Cell Mol Life Sci 2020;77:2839–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoffmann GF, Feyh P. Organic Acid Analysis. In: Physician's Guide to the Laboratory Diagnosis of Metabolic Diseases. Berlin, Heidelberg: Springer Berlin Heidelberg; 2003. p 27–44. [Google Scholar]
- 19. Casado M, Sierra C, Batllori M, Artuch R, Ormazabal A. A targeted metabolomic procedure for amino acid analysis in different biological specimens by ultra‐high‐performance liquid chromatography–tandem mass spectrometry. Metabolomics 2018;14:76. [DOI] [PubMed] [Google Scholar]
- 20. Jackson CE, Barohn RJ, Gronseth G, Pandya S, Herbelin L, The Muscle Study Group (MSG) . Inclusion body myositis functional rating scale: A reliable and valid measure of disease severity. Muscle Nerve 2008;37:473–476. [DOI] [PubMed] [Google Scholar]
- 21. Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, et al. GeneCards Version 3: the human gene integrator. Database (Oxford) 2010;2010:baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reactome|Pathway Browser. https://reactome.org/PathwayBrowser/ (accessed December 15, 2021).
- 23. Joshi PR, Vetterke M, Hauburger A, Tacik P, Stoltenburg G, Hanisch F. Functional relevance of mitochondrial abnormalities in sporadic inclusion body myositis. J Clin Neurosci 2014;21:1959–1963. [DOI] [PubMed] [Google Scholar]
- 24. Askanas V, Engel WK, Nogalska A. Sporadic inclusion‐body myositis: A degenerative muscle disease associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. Biochim Biophys Acta Mol Basis Dis 2015;1852:633–643. [DOI] [PubMed] [Google Scholar]
- 25. Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, et al. MitoCarta3.0: An updated mitochondrial proteome now with sub‐organelle localization and pathway annotations. Nucleic Acids Res 2021;49:D1541–D1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Badrising UA, Tsonaka R, Hiller M, Niks EH, Evangelista T, Lochmüller H, et al. Cytokine Profiling of Serum Allows Monitoring of Disease Progression in Inclusion Body Myositis. J Neuromuscul Dis 2017;4:327–335. [DOI] [PubMed] [Google Scholar]
- 27. Pinal‐Fernandez I, Casal‐Dominguez M, Derfoul A, Pak K, Plotz P, Miller FW, et al. Identification of distinctive interferon gene signatures in different types of myositis. Neurology 2019;93:E1193–E1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Amici DR, Pinal‐Fernandez I, Mázala DAG, Lloyd TE, Corse AM, Christopher‐Stine L, et al. Calcium dysregulation, functional calpainopathy, and endoplasmic reticulum stress in sporadic inclusion body myositis. Acta Neuropathol Commun 2017;5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. deAndrés‐Galiana EJ, Fernández‐Ovies FJ, Cernea A, Fernández‐Martínez JL, Kloczkowski A. Deep neural networks for phenotype prediction in rare diseases. In Artificial Intelligence in Precision Health. Elsevier; 2020. p 189–202. [Google Scholar]
- 30. Tadenev ALD, Burgess RW. Model validity for preclinical studies in precision medicine: precisely how precise do we need to be? Mamm Genome 2019;30:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hiniker A, Daniels BH, Lee HS, Margeta M. Comparative utility of LC3, p62 and TDP‐43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol Commun 2013;1:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nogalska A, D’Agostino C, Engel WK, Cacciottolo M, Asada S, Mori K, et al. Activation of the Unfolded Protein Response in Sporadic Inclusion Body Myositis But Not in Hereditary GNE Inclusion Body Myopathy HHS Public Access. J Neuropathol Exp Neurol 2015;74:538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buzkova J, Nikkanen J, Ahola S, Hakonen AH, Sevastianova K, Hovinen T, et al. Metabolomes of mitochondrial diseases and inclusion body myositis patients: treatment targets and biomarkers. EMBO Mol Med 2018;10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kodavati M, Wang H, Hegde ML. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cell 2020;9:1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mak IWY, Evaniew N, Ghert M. Lost in translation: Animal models and clinical trials in cancer treatment. Am J Transl Res 2014;6:114–118. [PMC free article] [PubMed] [Google Scholar]
- 36. Search of: Recruiting, Not yet recruiting, Active, not recruiting Studies |Interventional Studies|Inclusion Body Myositis ‐ List Results ‐ ClinicalTrials.gov. https://clinicaltrials.gov/ (accessed December 10, 2021).
- 37. Nogalska A, D'Agostino C, Engel WK, Askanas V. Sodium phenylbutyrate reverses lysosomal dysfunction and decreases amyloid‐β42 in an in vitro‐model of inclusion‐body myositis. Neurobiol Dis 2014;65:93–101. [DOI] [PubMed] [Google Scholar]
- 38. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed genes between IBM and CTL (778 genes with p‐value adj (FDR) < 0.05, following alphabetical order)
Table S2. Lists of differentially expressed genes (DEGs) related to inflammation (62 DEGs), autophagy (37 DEGs) and mitochondria (42 DEGs)
Table S3. Concentration of secreted inflammatory cytokines in IBM vs CTL supernatants. IBM fibroblasts revealed an increased expression of most of these cytokines.
Table S4. Expression of 20 autophagy proteins in IBM vs. CTL fibroblasts. Most of these proteins displayed a decreased expression in IBM, suggesting a reduced activity of the autophagy process.
Table S5. Differentially expressed genes (DEGs) related to metabolite concentration in fibroblasts. The table depicts the relationship of 10 DEGs involved in metabolite metabolism with their respective alteration at amino acids and organic acids level, to relate expression vs. metabolism patterns in IBM vs. CTL fibroblasts.
Table S6. Organic acids in IBM vs. CTL fibroblasts. Organic acids showed a general increase in IBM, suggesting a deregulation of intermediary metabolism related to mitochondrial function, as many organic acids are involved in tricarboxylic acid cycle (TCA) that further feeds the mitochondrial respiratory chain.
Table S7. Amino acids levels in fibroblasts of IBM patients vs. CTL fibroblasts.
Table S8. Comparison of inflammatory, degenerative, and mitochondrial IBM hallmarks considering the evolution of IBM patients: stable vs progressive prognosis, compared to CTL fibroblasts.