

Macrophages hold promise as indicators of disease progression and therapeutic targets. Using a single-cell multi-omic approach, the authors uncover the heterogeneity of macrophages in human colorectal liver metastasis and identify distinct populations with opposite clinical relevance.
Abstract
Patients with colorectal liver metastasis (CLM) present with heterogenous clinical outcomes and improved classification is needed to ameliorate the therapeutic output. Macrophages (Mϕ) hold promise as prognostic classifiers and therapeutic targets. Here, stemming from a single-cell analysis of mononuclear phagocytes infiltrating human CLM, we identified two Mϕ markers associated with distinct populations with opposite clinical relevance. The invasive margin of CLM was enriched in pro-inflammatory monocyte-derived Mϕ (MoMϕ) expressing the monocytic marker SERPINB2, and a more differentiated population, tumor-associated Mϕ (TAM), expressing glycoprotein nonmetastatic melanoma protein B (GPNMB). SERPINB2+ MoMϕ had an early inflammatory profile, whereas GPNMB+ TAMs were enriched in pathways of matrix degradation, angiogenesis, and lipid metabolism and were found closer to the tumor margin, as confirmed by spatial transcriptomics on CLM specimens. In a cohort of patients, a high infiltration of SERPINB2+ cells independently associated with longer disease-free survival (DFS; P = 0.033), whereas a high density of GPNMB+ cells correlated with shorter DFS (P = 0.012) and overall survival (P = 0.002). Cell–cell interaction analysis defined opposing roles for MoMϕ and TAMs, suggesting that SERPINB2+ and GPNMB+ cells are discrete populations of Mϕ and may be exploited for further translation to an immune-based stratification tool. This study provides evidence of how multi-omics approaches can identify nonredundant, clinically relevant markers for further translation to immune-based patient stratification tools and therapeutic targets. GPNMB has been shown to set Mϕ in an immunosuppressive mode. Our high dimensional analyses provide further evidence that GPNMB is a negative prognostic indicator and a potential player in the protumor function of Mϕ populations.
Introduction
Macrophages (Mϕ) are key elements of the tumor microenvironment (TME) and they are the most abundant mononuclear phagocytes (MP) in human cancers (1–5). The high density of Mϕ in tumor tissues, coupled to a predominant protumor role, have fostered studies aimed at evaluating Mϕ-related variables as indicators of disease, to complement the current staging system in cancer and achieve a more refined patient stratification (5, 6), as well as translating preclinical knowledge into actionable Mϕ-based therapeutic targets (2, 3, 7). Several Mϕ-intrinsic properties have so far prevented an unequivocal definition of the role of Mϕ across cancers, and a clear association with tumor clinical progression has not been consistently shown. Plasticity, whereby Mϕ adapt to microenvironmental cues (8–10), accounts for a remarkable diversity of Mϕ in tumor tissues and hampers the identification of definite Mϕ populations and reliable Mϕ markers. In most tissues, including liver, brain, lung, and omentum, a mixture of resident and recruited Mϕ exists and their relative contribution to the pathogenesis and progression of cancer is difficult to grasp (11–14). Mϕ spatial localization and their interplay with stromal and architectural components in tissues can control the functional profile of these phagocytes. Over the years, in fact, the peculiar characteristics of vascular (15), perineural (16), stromal (17), and perinecrotic (18) Mϕ and their association with prognosis have been reported.
Recent multidimensional studies have shed light on the variety of MP cells in human cancer tissues (12, 13, 19–21), including colorectal liver metastases (CLM; ref. 22), and have contributed to the identification of several myeloid subsets populating a complex landscape. Despite the amount of information generated by multi-omics approaches, translating this knowledge into clinically relevant immune-based tools still represents an important challenge (23). Moreover, Mϕ represent only a portion of the various immune cell types infiltrating the TME, and their protumoral or antitumoral action greatly depends on the interaction with other leukocytes in organizing a response.
Many of the above considerations apply to the liver and CLM, where the dichotomy between tissue-resident and inflammatory Mϕ has previously been shown (24, 25). Despite substantial efforts, identifying robust prognostic classifiers for CLM remains a current clinical challenge. In a recent publication (6), we introduced the morphology of Mϕ as a proxy of their function and metabolic orientation with prognostic significance. Mϕ infiltrating human CLM tissues were characterized by different morphologic features and transcriptional signatures, and in particular large Mϕ correlated with worse clinical outcome (6). Here, we further investigated and reduced the complexity of Mϕ populations in human CLM to identify unique and robust markers. By exploiting multiparametric digital pathology and single-cell analysis of MP infiltrating CLM tissues, we performed a deep analysis of spatial localization, transcriptional profiles and trajectories of Mϕ and identified two nonredundant population-specific markers, glycoprotein nonmetastatic melanoma protein B (GPNMB) and SERPINB2, which we validated as prognostic indicators in a cohort of patients with CLM.
Materials and Methods
Patients and study design
Single-cell RNA sequencing (scRNA-seq) was performed on 3 patients aged older than 18 with histologically proven CLM that underwent hepatectomy at IRCCS Humanitas Research Hospital. Matched specimens from the invasive margin (IM) and normal adjacent (NA) areas of 3 patients with CLM were collected during surgery and kept at 4°C until further processing for scRNA-seq. Patients were selected irrespective of KRAS status and therapeutic regimen. Written informed consent was obtained from each patient included in the study. The study protocol was in accordance with the ethical guidelines established in the 1975 Declaration of Helsinki and compliant to the procedures of the ethical committee of IRCCS Humanitas Research Hospital (protocol numbers 168/18 and 1683).
The retrospective cohort study included 48 patients aged older than 18 with histologically proven CLM that underwent hepatectomy at IRCCS Humanitas Research Hospital. Formalin-fixed, paraffin-embedded (FFPE) blocks of CLM surgical specimens (n = 48) were retrieved from the Pathology archive. Written informed consent was obtained from each patient included in the study. The study protocol was in accordance with the ethical guidelines established in the 1975 Declaration of Helsinki and compliant to the procedures of the ethical committee of IRCCS Humanitas Research Hospital (protocol number 168/18 and 1683). Patients’ demographics, clinical, surgical and histopathologic data (detailed in Supplementary Table S1) from the institutional intranet were assembled in a clinical retrospective database for analyses. Following the clinical practice, only patients with partial response to preoperative therapy (for details see Supplementary Table S1) or stable disease were included in the study. Patients with the following criteria were excluded from the study: progressive disease; combination of hepatectomy with radiofrequency or microwave ablation; nonradical hepatectomy. The preoperative workup consisted of total-body contrast-enhanced CT and liver-specific MRI, performed maximum 30 days prior to surgery. Patient postoperative follow-up was performed every 3 months and included an office visit, serum oncological markers, abdominal ultrasonography, and CT or MRI.
Isolation of liver MP and CD45+ cells
Surgically resected matched specimens from the IM and NA areas of 3 patients with CLM (Supplementary Fig. S1A) were chopped into small fragments, subjected to GentleMACS (Miltenyi, catalog no. 130-096-427) dissociation in HBSS +/+ (Euroclone, catalog no. ECB4006L) containing 2 mg/mL of Collagenase D solution (Sigma-Aldrich, catalog no. 11088882001), 2% FBS (Sigma-Aldrich, catalog no. F7524), 50 μg/mL DNAse I (Sigma-Aldrich, catalog no. 10104159001), and 10 mmol/L HEPES (Lonza, catalog no. BE17–737E), and subsequently incubated for 35 minutes at 37°C in the same solution. The resulting single-cell suspension was filtered through a 100-μm cell strainer and erythrocytes were lysed with ACK (Lonza, catalog no. BP10–548E).
Flow cytometry staining and cell sorting
Single-cell suspensions were then preincubated with a blocking solution containing 1% human serum (Sigma-Aldrich, catalog no. H3667) in saline solution and stained with fluorophore-conjugated primary antibodies at room temperature in the dark for 15 minutes: anti-CD45 (BD Biosciences, clone HI30, RRID:AB_1645452), anti-CD66b (BD Biosciences, clone G10F5, RRID:AB_396067), and anti-CD163 (BD Biosciences, clone GHI/61, RRID:AB_2737697). Cell viability was assessed using Zombie Aqua Fixable Viability Kit (BioLegend, catalog no. 423101). MP and CD45+ cells were sorted on a FACSAria III (BD Biosciences) as shown in Supplementary Fig. S1B.
scRNA-seq
MP and CD45+ cells sorted from nonadjacent and IM region of 3 CLM specimens (Supplementary Fig. S1A) were subjected to scRNA-seq analysis. Single-cell suspensions were prepared by tissue mincing and enzyme digestion. FACS-sorted cells were resuspended in 0.5 mL PBS 1X plus 0.04% BSA and washed once by centrifugation at 450 rcf for 7 minutes. Cells were then resuspended in 50 μL and counted with an automatic cell counter (Countess II, Thermo Fisher). Approximately 10,000 cells for each sample were loaded into the Chromium Chip B using the Single Cell Reagent Kit v3 (10X Genomics, catalog no. 1000128) for Gel bead Emulsion generation into the Chromium system. Following capture and lysis, cDNA was synthesized and amplified for 14 cycles following the manufacturer's protocol. 50 ng of the amplified cDNA were then used for each sample to construct barcoded sequencing libraries using the Chromium Single Cell 3′ Reagent Kit v3 (10X Genomics, catalog no. 1000128) following the manufacturer's instructions. Sequencing was performed on the NextSeq550 Illumina sequencing platform following manufacturer's instruction for read generation, reaching at least 35,000 reads as mean reads per cell.
Spatial transcriptomics
Four-μm FFPE tissue sections were stained with hematoxylin and eosin (H&E) and the appropriate tissue regions (inclusive of tumor and IM) were selected for further processing. Briefly, after deparaffinization and rehydration, sections were stained with hematoxylin (Histo-Line, catalog no. 01HEMH1000) for 15 minutes, followed by eosin (Histo-Line, catalog no. 01EOY101000) for 7 minutes. RNA quality of the FFPE tissue blocks was determined before performing spatial transcriptomics assay using Visium Spatial for FFPE Gene Expression Kit (10X Genomics, catalog no. 1000336) according to the manufacturer's instructions (10X Genomics, Tissue Preparation Guide, CG000408). RNA was extracted using RNeasy FFPE Kit (Qiagen, catalog no. 73504) and DV200 evaluation was performed on TapeStation (Agilent) using High Sensitivity RNA ScreenTape Kit (Agilent, catalog no. 5067–5579). Only FFPE tumor blocks with a value of DV200 > 50 were considered for the spatial transcriptomics assay.
FFPE tumor samples were prepared according to manufacturer instructions (10X Genomics, Tissue preparation guide, CG000408). H&E image preparation was performed according to protocol (10X Genomics, Deparaffinization, H&E staining, Imaging and decrosslinking, CG000408). Brightfield histologic images were acquired using the Axio Scan.Z1 (ZEISS). Libraries were prepared using Visium Spatial for FFPE Gene Expression Kit (10X Genomics, catalog no. 1000336) following the manufacturer's protocol (10X Genomics, Visium User Guide, CG000407) and sequenced on a NextSeq2000 (Illumina) at a minimum sequencing depth of 50,000 read pairs per spatial spot. Sequencing was performed with the recommended protocol (read 1: 28 cycles; i7 index read: 10 cycles; i5 index read: 10 cycles; and read 2: 50 cycles), yielding between 200 million and 230 million sequenced reads.
scRNA-seq analysis
Raw sequencing data in the format of BCL files were converted in fastq files and aligned to the human reference genome GRCh38, taking advantage of the Cell Ranger Pipeline version 3.0.1 provided by 10X Genomics. After quality check, we obtained a total of 12,181 and 12,568 cells from MP distal and peritumor tissue, and a total of 8,459 and 5,622 from for CD45+ cells, respectively. Filtered gene expression matrices from Cell Ranger were used as input for clustering analysis by Seurat R package (version 3.1.1; R version 3.6.1) (26). We first processed each individual data set separately, considering the thresholds of 200, 20,000 and 0.2 for number of genes, number of unique molecular identifiers (UMI) and mitochondrial content respectively. For each data set, we selected the 2,000 most variable genes. Subsequently, we used the ‘FindIntegrationAnchors’ function to combine the datasets together, choosing 2,000 anchor genes for integration. After integration, we ran principal component analysis (PCA) and used the first 67 principal components (PC) for MP and the first 51 PCs for CD45+ cells to perform Louvain clustering and Uniform Manifold Approximation and Projection (UMAP) embedding. Finally, we obtained a total of 18 clusters in MP (Supplementary Table S2) and S20 clusters for CD45+ cells (resolution level = 0.6). Markers gene analysis was performed using the ‘FindAllMarkers’ function (setting default parameters). Full list of markers per cluster are provided in Supplementary Table S2. Functional analysis was performed using the gene set variation analysis (GSVA) R package (27). Enriched processes were selected from the Human Reactome annotation (Human_Reactome_August_01_2020_symbol.gmt). Enrichment of selected gene signatures was generated using the ‘AddModuleScore’ function of Seurat package. Complete GSVA results are provided in Supplementary Table S3. Pathway enrichment analysis of tumor-associated Mϕ (TAM; c_9) marker genes was performed using PANTHER 13.1 (pantherdb.org). Full list of significantly (P < 0.05) enriched pathways is provided in Supplementary Table S3.
Integration analysis of public single-cell gene expression datasets
To compare the transcriptional profile of our TAM population with other described Mϕ populations, we performed an integration analysis.
Comparison of TAM versus scar-associated Mϕ (SAMϕ) (28). Cells belonging to MP and cDCs clusters (as indicated by their lineage profile) were selected in a total of 10 samples (5 cirrhotic samples and 5 healthy controls). Raw counts were used as input for the integration analysis. We first processed each individual data set separately, considering the thresholds of 200, 20,000 and 0.2 for number of genes, number of UMI and mitochondrial content respectively. For each data set, we selected the 2,000 most variable genes. After filtering, we retrieved a total of 10,080 cells. Feature genes were selected as those genes shared by the two datasets. Subsequently, we used the FindIntegrationAnchors function to combine the two datasets together, choosing 2,000 anchor genes for integration and obtaining a total of 34,829 cells. We ran PCA and used the first 65 PCs to perform Louvain clustering and UMAP embedding. Clustering was generated choosing a resolution level of 0.5. Comparison analysis of TAM versus SAMϕ was generated selecting cells by their tag ID, as annotated in the two datasets. Cluster-specific genes were selected as differentially expressed genes according to adjusted P value < 0.05 and log2fold change > |0.25|.
Comparison of TAM versus lipid-associated Mϕ (LAM; ref. 29). Cell gene count matrix of myeloid cells in a total of 11 liver samples was downloaded and used as input for the integration analysis. We first processed each individual data set separately, considering the thresholds of 200, 20,000 and 0.2 for number of genes, number of UMI and mitochondrial content respectively. For each data set, we selected the 2,000 most variable genes. After filtering, we retrieved a total of 22,526 cells. Features genes were selected as those genes shared by the two datasets. Subsequently, we used the FindIntegrationAnchors function to combine the two datasets together, choosing 2,000 anchor genes for integration and obtaining a total of 42,275 cells. We ran PCA and used the first 72 PCs to perform Louvain clustering and UMAP embedding. Clustering was generated choosing a resolution level of 0.5. Comparison analysis of TAM versus LAM populations was generated selecting cells by their tag ID, as annotated in the two datasets. Cluster-specific genes were selected as differentially expressed genes according to adjusted P value < 0.05 and log2fold change > |0.25|.
Reclustering of MP
Myeloid populations (clusters 0,3,8,9) from CD45+ clustering were selected for myeloid reclustering. Deriving cells were reclustered using a resolution level of 0.8, producing a total of 12 clusters. For interaction analysis clusters 1,2,4,5,11 were selected. Identities of reclustered cells were reassigned to the total CD45+ dataset.
Single-cell trajectory reconstruction
Single-cell pseudotime trajectory was generated using Monocle version 2.8 (Seurat version 2.5.1; ref. 30). Expression data coming from Seurat (RNA slot) were uploaded in Monocle with their cluster annotation. Marker genes for each cluster were chosen to define cell progression and the ‘DDRTree’ method (max components = 2) to reduce data dimensionality. After trajectory generation, the root state parameter was set to State 8 to define the starting point. The ‘differentialGeneTest’ function was used to estimate differentially expressed genes during psuedotime. Resulting genes (q value < 0.0001 and ‘use for ordering’ = TRUE) were represented in the heat map. To better characterize genes that define cell fate decisions, the branched expression analysis modeling (BEAM) was applied on branch number 3. Full list of differentially expressed genes is provided in Supplementary Table S3.
NicheNet interaction analysis
We applied NicheNet method (NicheNet version 1.0.0, Seurat version 3.6; ref. 31) to predict ligand–receptors interactions existing between Mϕ populations (senders) and CD45+ populations (receivers).
Seurat normalized data was imputed of missed counts with ALRA function (32), with a predicted rank-k approximation of 35. We restricted the analysis to the following gene sets: only ligands and receptors expressed in at least 30% of sender and receivers’ cells clusters were considered; cluster gene markers of each receiver cell population (adjusted P value _0.05 and avglogFC _0.5) were set as target genes set and background genes were chosen as expressed in all cell populations with a cluster frequency above 50%. Ligand activity scores was the calculated, only ligands with a positive Pearson correlation coefficient were considered.
Heat maps of ligand activity show positive ligands ordered according to their z-scores. For those ligands, expression is represented along senders.
For circular visualization of the ligand–receptors interaction pairs, the circlize R (version 0.4.12; ref. 33) package was used. We selected common ligands and assigned them to each sender according to their expression (mean + SD). For each receptor, the width of the sector is proportional to the ligand–receptor interaction weights. Ligand-to-target signaling path for IL15 was generated using the ‘get_ligand_signaling_path’ function.
CellPhoneDB interaction analysis
To analyze cellular interactions between MP in the CD45+ dataset and other CD45+ cell populations, CellPhoneDB (version 2.0.0; ref. 34), was used with statistical_analysis method. Normalized gene expression matrices together with their cluster annotations were used as input (‘data’ slot from Seurat after ALRA imputation step).
The enriched ligand–receptor interactions between two cell subsets were calculated on the basis of permutation test. We selected as significant ligand–receptors pairs those interactions annotated as manually curated and with P value < 0.05. Full list of significant cell–cell interactions is provided in Supplementary Table S4.
Gene signature enrichment analysis
Enrichment analysis of gene signatures related to murine embryonic healthy Kupffer cells [(KC (KC_H)], nonalcoholic steatohepatitis (NASH)-associated KC (KC_N), and monocyte-derived Mϕ (MoMϕ) repopulating the KC niche in NASH (KN_RM) was performed using the Seurat function ‘AddModuleScore’. Gene signatures, as defined by the authors, were used as input to compute a module score for each cell. Module scores along clusters were represented by violin plots.
Gene regulatory network analysis
The analysis of regulon activity was performed with SCENIC R package (version 1.1.2; ref. 35) starting from normalized and imputed (ALRA assay) gene expression matrices. The genes with at least 3 UMIs in at least 10% of the cells and detected in at least in 10% of samples were selected as the input genes. The expression matrix was loaded into GENIE3 (36) and the coexpressed genes to each transcription factor (TF) was constructed. The TF coexpression modules were then analyzed by RcisTarget R package (version 1.12.0). The normalized enrichment score (NES) of the TF binding motifs was calculated and NES > 3 was considered as significantly enriched. The filtered potential targets by RcisTarget human database (hg19–500bp-upstream- 7species.mc9nr.feather; hg19-tss-centered-10kb-7species.mc9nr.feather) from the coexpression module were used to build the regulons. The regulon activity was then analyzed by AUCell R package (version 1.14.0; ref. 35) and the active regulons were determined by AUCell default threshold. Full list of differentially modulated gene regulatory networks is provided in Supplementary Table S4.
Spatial transcriptomics analysis
Visium spatial gene expression data of FFPE tissue samples from patients with CLM in the format of BCL files were converted into fastqs using Illumina BCL Convert software (version 3.8.2). Raw data from Visium slides were processed and aligned to the human reference genome GRCh38 with Space Ranger (10X Genomics) version 1.3.1 using the human probe set v1 (Visium_Human_Transcriptome_Probe_Set_v1.0_GRCh38–2020-A.csv). The generated filtered feature matrices were analyzed individually using Scanpy package version 1.9.1 (python version 3.9.12; ref. 37). Spots with less than 200 expressed genes and 800 UMIs and genes detected in less than 3 spots were filtered out. Mitochondrial and ribosomal genes were not included in the Visium probe set. After quality filters, we obtained a total of 6,907 spots and a mean number of 2,845 genes detected per spot. Count data were normalized (size factor of 10,000) and log-transformed for downstream analyses using normalize_total and log1p functions.
Tumor–stroma interface was manually annotated by a pathologist. Using Loupe Browser, we then divided the normal and tumor regions into continuous zones parallel to the shape of the interface line at intervals of five spots.
Features’ expression projection onto spatial coordinates was obtained with scanpy spatial plotting function and used to evaluate the consistency of the expression patterns of known adjacent liver (APOC3), stromal (COL1A2), and tumor (EPCAM) marker genes in spatial transcriptomics data with the H&E images.
Signatures from TAM (PLA2G7, SPP1, ACP5, TREM2, GPNMB, LPL, RGCC, FABP5, FABP4, MMP19, MGLL) and MoMϕ (S100A8, S100A9, S100A12, SERPINB2, SELL, MCEMP1, IL1R2, RETN, MNDA, FCN1) populations were scored in each spot as the average of log-normalized expression of the genes within each list.
Because SERPINB2 gene was detected in a low number of spots (∼13 on average) in the spatial expression data, Pearson correlation coefficients with SERPINB2 expression were computed for all genes from the scRNA-seq MP dataset to identify the top coexpressing gene (S100A12, ρ = 0.55).
Heat map and Line plot showing the average signature expression levels along the defined contiguous zones from adjacent liver to tumor regions were generated using seaborn package version 0.11.2 and matplotlib version 3.5.1 (38, 39). One-sided Mann–Whitney U rank test was computed using mannwhitneyu function from scipy.stats package version 1.8.0 (40) to compare MoMϕ and TAM score distributions between adjacent normal and tumor regions of each patient.
Analysis of zone similarity
Similarities of continuous spatial zones across patients were assessed by performing PCA and hierarchical clustering. Starting from the intersection of the features detected from the 2 patients, gene expression profile of each tissue zone was measured as the mean log-normalized expression of the relative spots. Python scikit-learn package version 1.0.2 (41) was used to run PCA on scaled and centered region's expression data. The result of this unsupervised dimensionality reduction was visualized as scatter plot of the first two PCs to evaluate the distribution pattern of the manually defined adjacent liver and tumor regions. Hierarchical clustering of the spatial tissue zones on the first five PCs was performed using the Ward's method inside the clustermap function of python seaborn package.
Multiplex fluorescent IHC
Two-μm thick consecutive tissue sections were prepared from FFPE tissues, provided by the Pathology Department of IRCCS Humanitas Research Hospital, and processed for multiplex fluorescent IHC (mf-IHC). Prior to mf-IHC, all primary antibodies were first tested as monoplex staining according to the recommendations given by the manufacturers. mf-IHC was performed using Opal 7-Color Manual IHC Kit (Akoya Biosciences, catalog no. NEL811001KT) following the manufacturer's protocol. Briefly, after deparaffinization and rehydration, antigen retrieval was performed by heat treatment using AR9 buffer (Akoya Biosciences) in water bath at 98°C for 20 minutes. After cooling, nonspecific binding was blocked using Antibody Diluent/Block (Akoya Biosciences) for 10 minutes, followed by incubation with primary antibodies for 1 hour. Slides were then incubated with Opal polymer HRP (Akoya Biosciences) for 10 minutes, followed by incubation with Opal fluorophores (Akoya Biosciences) for 10 minutes. Antibody stripping was then performed by heat treatment in water bath at 98°C for 15 minutes. Protocol was repeated starting from nonspecific blocking step for each subsequent antibody. Slides were then counterstained with Spectral DAPI (Akoya Biosciences, catalog no. FP1490) for 5 minutes and mounted using mounting medium (Abcam, catalog no. ab104135). Except when specified, all steps were performed in the dark at room temperature. The following antibodies were used: anti-CD68 (Agilent, clone KP1, catalog no. M081401–2), anti-GPNMB (Abcam, clone EPR22011–11, catalog no. ab222109), anti-SERPINB2 (Thermo Fisher Scientific, clone OTI1G3, RRID:AB_2725325), anti-LAMP3 (Sigma-Aldrich, polyclonal, catalog no. HPA051467), anti-IL15 (Abcam, clone 3A3, catalog no. ab55276), anti-LYVE1 (Abcam, clone EPR21857, catalog no. ab219556), anti-CD8α (Thermo Fisher Scientific, clone C8/144B, RRID:AB_11000353), anti-IL10 (Thermo Fisher Scientific, clone JES3–9D7, catalog no. 604–950).
Scanning and image analysis
Whole slides were scanned using the Axio Scan.Z1 (ZEISS) at 20x magnification. Image analysis was performed using QuPath (version 0.2.3; ref. 42). Briefly, the tumor core (TC) was manually annotated and expanded by 1 mm to define the IM area. Within the IM, individual cells were identified on the basis of DAPI-stained nucleus size and signal intensity. Thresholds for the analyzed fluorescence-labelled markers were set on the basis of the staining intensity of the entire cell. Cell positivity for each marker (CD68, GPNMB, SERPINB2, LAMP3) was assessed independently. On the basis of cell markers, we identified three different phenotypes: CD68+GPNMB+ Mϕ (referred to as GPNMB+ TAMs in the main text), SERPINB2+ MoMϕ, and LAMP3+ dendritic cells (DC). All phenotypes in the IM were automatically counted for each patient and normalized on the area of the IM to obtain the density (number of positive cells/mm2) for each phenotype, used for subsequent analyses. To assess the number of CD68+GPNMB+ cells and SERPINB2+ cells in the adjacent region (AD) and in the TC, three to five noncontiguous regions of interest (ROI) of 1 mm2 each were analyzed using the same approach described above. The mean value from the different ROIs was calculated. SERPINB2+ and CD68+GPNMB+ median density values were used to stratify patients in survival analyses. The variables were combined to calculate a ratio (SERPINB2+ cells/GPNMB+ cells) or a score (SERPINB2>median/GPNMB<median and SERPINB2<median/GPNMB>median). Spatial assessment of SERPINB2+ and CD68+GPNMB+ cells was performed using Qupath (version 0.2.3). For each phenotype, the distance between each individual cell in the IM and the TC was calculated and averaged to obtain the mean distance for each patient.
Spatial assessment of LAMP3+ and CD68+ cells was performed using MATLAB (version R2020a; MathWorks, RRID:SCR_001622). For each patient, high cell density (HD) regions were identified as the portions of the IM where the coarse-grained nuclear number density 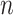 was above the threshold value of 1 nucleus per 100
was above the threshold value of 1 nucleus per 100 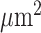 . Coarse-grained number density maps were obtained from the spatial distribution of the centroids of the nuclei by applying a two-dimensional Gaussian filter of width
. Coarse-grained number density maps were obtained from the spatial distribution of the centroids of the nuclei by applying a two-dimensional Gaussian filter of width 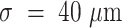 . For each considered marker (CD68, LAMP3), we calculated the mean value
. For each considered marker (CD68, LAMP3), we calculated the mean value 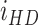 of the intensity in the corresponding fluorescence channel within the HD region and its mean value
of the intensity in the corresponding fluorescence channel within the HD region and its mean value 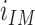 within the whole IM. The
within the whole IM. The 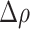 index, corresponding to the number density corrected-percent difference in the average intensity between HD regions and the whole IM, was calculated as
index, corresponding to the number density corrected-percent difference in the average intensity between HD regions and the whole IM, was calculated as
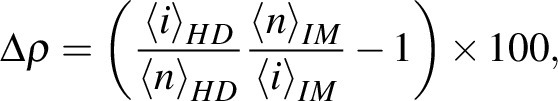 |
where 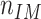 and
and 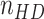 are the average nuclear number density in the HD region and in the whole IM, respectively. According to the definition above, in the absence of any bias in the spatial distribution of cells that are positive for the considered marker, the expectation value of
are the average nuclear number density in the HD region and in the whole IM, respectively. According to the definition above, in the absence of any bias in the spatial distribution of cells that are positive for the considered marker, the expectation value of 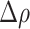 is zero. Positive values of
is zero. Positive values of 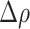 indicate an increased propensity of positive cells to be localized in the HD regions. For example,
indicate an increased propensity of positive cells to be localized in the HD regions. For example, 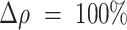 indicates that the fraction of positive cells in HD regions is twice as large as its reference value evaluated over the whole IM. In contrast, negative values of
indicates that the fraction of positive cells in HD regions is twice as large as its reference value evaluated over the whole IM. In contrast, negative values of 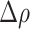 indicate that positive cells tend to systematically avoid HD regions.
indicate that positive cells tend to systematically avoid HD regions.
Statistical analysis
Statistical computations were performed using the software Stata (version 16; College Station, TX, StataCorp LLC, RRID: SCR_012763) and GraphPad Prism 8 (GraphPad Software Inc., San Diego, RRID: SCR_002798). Differences between two groups were calculated by Mann–Whitney test. When comparing immune variables from the same patient paired t test was used. Statistical significance of signature enrichment score between multiple cell types was determined using one-way ANOVA by Kruskal–Wallis test. For each test, only two-sided P values lower than 0.05 were considered statistically significant. The categorical variables were reported as a number and percentage, while continuous variables were reported as the median and range or as mean and SD. Only patients with complete data were considered. Univariate analysis, by log-rank test or Mann–Whitney test as appropriate, was performed to examine association among different covariates and patients' outcome. All surgical, clinical, histologic, and molecular available factors deemed to be prognostically relevant were considered as covariates. Covariates that showed a tendency of association with patients’ outcome were inserted into a multivariate model based on the Cox regression analysis to investigate independent predictors of outcome. The median value was used as cutoff for SERPINB2+ MoMϕ and GPNMB+ TAM density. Both the overall survival (OS) and the disease-free survival (DFS) were considered as outcome measures. All time-to-event endpoints were summarized using the Kaplan–Meier method. Differences in these endpoints between groups were examined using a log-rank test.
The Cancer Genome Atlas survival analysis
Survival analysis of The Cancer Genome Atlas (TCGA) colon cancer gene expression dataset (TCGA-COAD; n = 270) based on the expression of c0_MoMϕ(1) and c9_TAM gene signatures or individual genes (GPNMB) was performed using GEPIA2 (ref. 43; http://gepia2.cancer-pku.cn/). Median value of gene expression values was used as group cutoff to categorize patients in high- and low-expression groups. Differences in survival outcomes between groups were examined using a log-rank test.
Data and materials availability
The raw scRNA-seq data from MP and CD45+ cells have been deposited in Gene Expression Omnibus under the accession numbers GSE200068 and GSE200253. All other data presented in this manuscript are available in the main text or the supplementary materials or from the corresponding authors upon reasonable request.
Results
GPNMB+ TAMs accumulate in the IM of human CLM
We performed scRNA-seq on MP isolated as CD163+CD66b– cells from the NA and IM portions of 3 human CLM tissues (ref. 6; Supplementary Fig. S1A–S1C). Eighteen clusters (Fig. 1A), highly reproducible among patients (Supplementary Fig. S1D; Supplementary Table S2), were obtained by unbiased clustering and annotated on the basis of differentially expressed genes (Supplementary Fig. S1E and S1F; Supplementary Table S2) and published lineage genes (21, 28, 44). We identified monocytes (classical and nonclassical), MoMϕ, DC (including cDC2, cycling DCs, mature LAMP3+ DCs, and monocyte-derived DCs), M2-like TAMs and KCs (Fig. 1A). To exploit this granular description of MP populations in human CLM from the perspective of identifying tumor-associated population markers, we focused on clusters prevalently enriched in IM tissues compared with NA liver (Fig. 1B). These included several monocyte clusters [Mono (HSP+), Mono CD16+ and Mono CD14+], three abundant MoMϕ clusters [MoMϕ (1–3)], characterized by high expression of inflammatory genes (S100A8, THBS1, VCAN) and genes related to leukocyte recruitment (SELL and FPR1; Supplementary Fig. S1E and S1F) and a cluster of more mature Mϕ annotated as TAMs based on enrichment of a TAM gene signature (Fig.1B and C). Compared with MoMϕ clusters, TAMs had lower expression of monocyte genes (ITGAM, SERPINB2), while expressed higher CD68, MSR1, and HLA genes (Supplementary Fig. S1F), suggesting a more mature and differentiated phenotype. Compared with DCs, higher expression of CD68 and lower expression of canonical DC genes, such as CD1C, CLEC10A, CLEC9A, and FCE1R, was considered indicative of Mϕ lineage (Supplementary Fig. S1F). TAMs resembled dysfunctional Mϕ with a profibrotic profile and altered lipid metabolism reported in different liver diseases, including fibrosis, NASH (28, 45), hepatocellular carcinoma and liver metastasis (refs. 6, 24; Fig. 1D). Pathway enrichment analysis showed ontology terms related to matrix degradation and angiogenesis, but also liver-specific pathways, such as lipid and cholesterol metabolism and negative regulation of immune effector processes (Fig. 1D; Supplementary Table S3). In fact, TAMs seemed transcriptionally similar to KC (Fig. 1A), however with lower or no expression of IL10 and other lineage genes (VSIG4, CD5L, and MARCO; Supplementary Fig. S1F). Among the genes that most differentiated TAMs from KC clusters were GPNMB, TREM2, LGALS3, and FABP4 (Supplementary Fig. S1G), which were quite uniquely expressed by TAMs (Fig. 1E; Supplementary Table S3). To further investigate how TAMs were related to Mϕ populations identified in other liver diseases, we compared them with SAMϕ from patients with NASH (28) and LAM from patients with steatotic livers (ref. 29; Fig. 1F). The low number of differentially expressed genes, particularly with LAM, suggested that TAMs shared a similar profile, however with a higher expression of a few signature genes including GPNMB, SPP1, FABP5, and IL1RN (Fig. 1F).
Figure 1.
![Figure 1. Identification of GPNMB+ TAMs in the IM of human CLM. scRNA-seq was performed on MP isolated as CD163+CD66b– cells from the NA and IM portions of 3 human CLM tissues. A, UMAP plot from integrated NA and IM analysis of 24,749 cells, color-coded by cluster assignment. B, Prevalence of each cluster in tissue (NA and IM) calculated by RO/E score. C, Dot plot showing scaled enrichment score of TAM gene signature across MP clusters in CLM. Circle size represents the proportion of cells expressing the TAM gene signature; color represents average signature expression in each cluster. D, Pathway enrichment analysis of TAM marker genes showing selected Gene Ontology annotations. Fisher exact test. E, UMAP representing gene expression of selected TAM marker genes across MP clusters. Clusters are colored according to normalized gene expression values. F, Volcano plot showing differentially expressed genes between TAMs vs. SAMϕs (ref. 28; top) and TAMs versus LAM (ref. 29; bottom). Exemplar genes are labelled. Purple genes represent up regulated genes and blue and yellow genes represent down regulated genes [log2(fold change) > |0.25| and adjusted P value < 0.05]. G, mf-IHC image of CLM section stained for CD68 (green) and GPNMB (magenta). Dashed line shows tumor margin. Picture is representative of n = 48 slides stained. Scale bar: 300 μm. H, mf-IHC image of CLM section stained for CD68 (green) and GPNMB (magenta). Picture is representative of n = 48 slides stained. Scale bar: 30 μm.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10070171_405fig1.jpg)
Identification of GPNMB+ TAMs in the IM of human CLM. scRNA-seq was performed on MP isolated as CD163+CD66b– cells from the NA and IM portions of 3 human CLM tissues. A, UMAP plot from integrated NA and IM analysis of 24,749 cells, color-coded by cluster assignment. B, Prevalence of each cluster in tissue (NA and IM) calculated by RO/E score. C, Dot plot showing scaled enrichment score of TAM gene signature across MP clusters in CLM. Circle size represents the proportion of cells expressing the TAM gene signature; color represents average signature expression in each cluster. D, Pathway enrichment analysis of TAM marker genes showing selected Gene Ontology annotations. Fisher exact test. E, UMAP representing gene expression of selected TAM marker genes across MP clusters. Clusters are colored according to normalized gene expression values. F, Volcano plot showing differentially expressed genes between TAMs vs. SAMϕs (ref. 28; top) and TAMs versus LAM (ref. 29; bottom). Exemplar genes are labelled. Purple genes represent up regulated genes and blue and yellow genes represent down regulated genes [log2(fold change) > |0.25| and adjusted P value < 0.05]. G, mf-IHC image of CLM section stained for CD68 (green) and GPNMB (magenta). Dashed line shows tumor margin. Picture is representative of n = 48 slides stained. Scale bar: 300 μm. H, mf-IHC image of CLM section stained for CD68 (green) and GPNMB (magenta). Picture is representative of n = 48 slides stained. Scale bar: 30 μm.
Figure 2.

In-depth characterization of inflammatory MoMϕ subsets and TAMs in CLM. Functional characterization of selected scRNA-seq MP clusters. A, Heat map of differentially expressed genes in MoMϕ(1–5) clusters, color-coded by cluster. Exemplar genes are labelled. Rows represent marker genes. Columns represent individual cells. B, Violin plots of marker gene expression and abundance across MoMϕ clusters. C, Enrichment score in MoMϕ(1–3) and KC(1–2) clusters of selected pathways from GSVA analysis. D, Monocle pseudotemporal trajectory (light blue to dark blue) of Mono CD14+ and MoMϕ(1–3) clusters in a two-dimensional state-space. Dashed lines mark the different transcriptional states. Labels represent prevalent clusters within transcriptional state. E, Density plot representing cluster frequency along pseudotime. F, UMAP plot of the 18 clusters color-coded by SERPINB2 expression. Clusters are colored according to normalized gene expression values. G, Volcano plot showing differentially expressed genes between SERPINB2pos and SERPINB2neg cells from clusters MoMϕ(1) and MoMϕ(2). Exemplar genes are labelled. Red and orange dots represent genes up regulated in SERPINB2pos and SERPINB2neg, respectively, with log2(fold change) ≥ 0.25 and adjusted P value < 0.05. H, mf-IHC image of CLM section stained for CD68 (green), GPNMB (magenta), and SERPINB2 (red). Arrows indicate SERPINB2+CD68+ cells. Representative picture of n = 5 slides stained. Scale bar: 50 μm. I, Violin plots showing enrichment score of KC-H, KC-N, KN-RM gene signatures (47) across selected MP clusters. One-way ANOVA P < 0.0001 by Kruskal–Wallis test. J, mf-IHC image of CLM section stained for CD68 (green), GPNMB (magenta), and LYVE1 (yellow). Representative picture of n = 3 slides stained. Scale bar: 20 μm. K, Heat map of gene regulatory network activity of selected MP clusters computed by SCENIC. VLDL, very-low-density lipoprotein.
Translation of multidimensional information obtained via transcriptomic analyses into clinically relevant tools relies on the possibility to assess by histology specific markers for different immune subpopulations in cancer tissues. We therefore tested selected signature marker genes prevalently expressed by TAMs in human sections of CLM. GPNMB was highly and selectively expressed in TAMs (Supplementary Fig. S1G–S1I) and clearly identified a population of CD68+ Mϕ strongly infiltrating the IM (Fig. 1G). These cells also displayed a peculiar round-shaped morphology (Fig. 1H), suggesting that they could represent protumor Mϕ already described in CLM (6, 22).
Distinct features of tumor-infiltrating MoMϕ and TAMs
The IM of human CLM was also enriched in MoMϕ subpopulations comprising 3 major clusters, MoMϕ(1–3), exhibiting variable expression of monocytic (VCAN, S100A8) and phagocytic genes (MRC1). A less frequent cluster, MoMϕ(4), displayed increased expression of genes related to early inflammatory activation (JUN, FOS, NFKBIZ; Fig. 2A and B; Supplementary Table S3). Consistent with their monocytic features, MoMϕ were quite distinct from resident populations; KC(1) and KC(2) expressed the liver resident Mϕ genes MARCO, CD5L, LILRB5, and genes related to maintaining homeostasis in the liver (CETP, SLC40A1, HMOX1), as well as IL10 (Supplementary Fig. S1E and S1F). In fact, while KC were enriched for pathways related to VLDL assembly and Heme degradation, MoMϕ clusters showed increased activation of pathways such as induction of chemokines, cell migration and biosynthesis of D-resolvins (Fig. 2C; Supplementary Table S3). To uncover why, despite their similarity, the three predominant clusters MoMϕ(1–3) represented distinct populations, we computed a Monocle-guided transcriptional trajectory, considering Mono CD14+ as the starting point. The clusters were differentially distributed along the pseudotemporal trajectory in 8 functional states (Fig. 2D; Supplementary Fig. S2A), and their density evidenced a higher transcriptional similarity of MoMϕ(1) to Mono CD14+, while MoMϕ(2) and MoMϕ(3) were found to be more divergent (Fig. 2D and E). Trajectory branchpoint analysis showed modules of coexpressed genes selectively upregulated in MoMϕ(1), including several classic neutrophil markers (CSF3R, S100A12, GCA) and neutrophil chemotactic genes (FPR1, FPR2; Supplementary Fig. S2B; Supplementary Table S3), resembling the profile of neutrophil-like monocytes recently described in other pathologic contexts (46). These findings suggest that the three MoMϕ may represent different subsets of the same population captured at diverse activation or maturation states. To translate our findings to a histologic setting, we searched for a candidate marker for MoMϕ assessment in CLM tissues. We considered SERPINB2 for its selective expression in MoMϕ clusters (Fig. 2F). Because we observed a heterogeneous expression among MoMϕ(1) and MoMϕ(2) (Supplementary Fig. S2C and S2D), we further investigated the profile of cells highly expressing SERPINB2 by stratifying the cells in these clusters according to SERPINB2 gene expression [“SERPINB2pos” (n = 3,103 cells) and “SERPINB2neg” (n = 4,229 cells); (Fig. 2G)]. SERPINB2pos cells showed higher expression of genes related to leukocyte recruitment, in particular neutrophil recruitment, and cytokine production (CXCL8, CXCL3, S100A8, S100A9, S100A12; Fig. 2G). We next confirmed the existence of a SERPINB2+CD68+ Mϕ subset by mf-IHC and demonstrated that SERPINB2 and GPNMB identified two distinct Mϕ subsets. SERPINB2+ MoMϕ also appeared smaller in size compared with GPNMB+ TAMs (Fig. 2H), suggestive of their less mature profile.
Understanding the ontogeny of the different Mϕ subsets is critical for both prognostic analyses and the design of strategies depleting tumor-promoting Mϕ without causing off-target effects. GPNMB+ TAMs showed a distinct transcriptional profile from MoMϕ (Supplementary Fig. S1E and S1F), possibly suggesting a different ontogeny. To probe if this cluster represented a functionally deranged KC, or a monocyte-derived tumor-conditioned Mϕ, we exploited published gene expression signatures of murine embryonic healthy KC (KC_H), NASH-associated KC (KC_N), and MoMϕ repopulating the KC niche in NASH (KN_RM; ref. 47). Signature enrichment comparing our MP clusters with murine NASH clusters showed a strong overlap between TAMs and KN_RM (Fig. 2I), suggesting that TAMs may derive from monocytes repopulating the KC niche following embryonic KC loss in the TME (Fig. 2I). In line with these results, CD68+ Mϕ expressing GPNMB+ were sometimes found lining LYVE1+ sinusoids (Fig. 2J), resembling the topographical distribution of KC (45), suggesting that they may represent repopulated KC.
Gene regulatory network analysis confirmed the unique features of the Mϕ subpopulations identified so far. While MoMϕ presented increased activity of inflammatory pathways, such as p65 subunit (RELA), and regulators of MoMϕ differentiation pathways (MAFB, NFIL3), TAMs and KC displayed similar TF activity. They shared with KC(1) and (2) high activity of PPARG, NR1H3, MITF, and exclusively with KC(1), ER-stress related TFs ATF3 and ATF6 (Fig. 2K). Moreover, TAMs uniquely expressed GATA2, which was previously linked to efferocytosis defects in atherosclerotic plaque Mϕ (48). KC, TAMs, and MoMϕ exhibited opposite activation of sterol regulatory element-binding protein genes SREBF1 and SREBF2, involved in cholesterol homeostasis, a pathway linked to inflammatory response in liver phagocytes (49, 50; Fig. 2K; Supplementary Table S4). SREBF1 was higher in KC and TAMs, indicating the activation of lipogenic pathways.
SERPINB2+ MoMϕ and GPNMB+ TAMs correlate with opposite clinical outcomes in CLM
We next put to test our hypothesis that the diversity of MP populations that we observed through scRNA-seq could aid in the identification of nonredundant cell-specific markers with clinical relevance in human CLM tissues. We set up a mf-IHC pipeline (Supplementary Fig. S2E) in a cohort of 48 patients with CLM (Supplementary Table S1), and we probed the clinical relevance of SERPINB2 and GPNMB, identifying distinct Mϕ populations, by correlating their density with DFS and OS. High infiltration of SERPINB2+ cells was associated with longer DFS (P = 0.033), but not OS (Fig. 3A). In contrast, a high density of GPNMB+ cells correlated with shorter DFS (P = 0.012) and OS (P = 0.002; Fig. 3B). The prognostic significance was not further improved when the ratio of GPNMB+/SERPINB2+ cells or a combined score of the variables were considered (Fig. 3C). Multivariate analysis set for DFS confirmed a significant and opposite correlation between SERPINB2+ MoMϕ (HR = 0.51; 95% confidence interval (CI), 0.26–0.99; P = 0.046) and GPNMB+ TAMs (HR = 2.27; 95% CI, 1.11–4.66; P = 0.025; Fig. 3D and E; Supplementary Table S5). Indeed, a high density of SERPINB2+ MoMϕ was found to be protective, whereas a high density of GPNMB+ TAMs was found to be detrimental for DFS. Of note, such findings were independent both of the use of neoadjuvant systemic chemotherapy and of RAS status.
Figure 3.

Clinical relevance of SERPINB2+ MoMϕ and GPNMB+ TAMs in CLM and primary CRC. Association of SERPINB2+ MoMϕ and GPNMB+ TAMs with clinical outcome in a restrospective CLM cohort by mf-IHC and survival analysis of a TCGA colon cancer dataset. A, Kaplan–Meier curve showing DFS (left; P = 0.033 by log-rank Mantel–Cox test) and OS (right; P = not significant by log-rank Mantel–Cox test), according to SERPINB2+ cell density. n = 48. B, Kaplan–Meier curve showing DFS (left; P = 0.012 by log-rank Mantel–Cox test) and OS (right; P = 0.002 by log-rank Mantel–Cox test), according to GPNMB+CD68+ cell density. n = 48. C, Kaplan–Meier curve showing OS according to GPNMB+SERPINB2+ ratio (left; P = 0.058 by log-rank Mantel–Cox test; n = 48) and OS according to GPNMB+SERPINB2+ score (right; P = 0.033 by log-rank Mantel–Cox test; n = 23). D, Forest plot showing the results of multivariate regression analysis for DFS. Reference line of HR for recurrence (dashed line), HRs (circles), and 95% CI (whiskers) are shown. *, P < 0.05. E, Table showing Cox multivariate regression analysis of prognostic factors for DFS. F, DFS and OS curves on TCGA colon cancer (TCGA-COAD; n = 270) dataset based on median expression of gene signatures from c0_MoMϕ(1) and c9_TAM. Dotted lines show 95% CI. P value by log-rank Mantel–Cox test. G, DFS and OS curves on TCGA colon cancer (TCGA-COAD; n = 270) dataset based on median expression of GPNMB. Dotted lines show 95% CI. P value by log-rank Mantel–Cox test.
One of the key issues in metastasis is whether primary tumors already present relevant targets that could prevent metastatic spread. We therefore tested in silico the association of gene signatures from distinct clusters of MP identified in human CLM with progression of primary CRC (Fig. 3F). TCGA survival analysis on CRC indicated a significant correlation between high expression of a MoMϕ(1) signature and favorable prognosis. The opposite was observed for the TAM signature, which strongly correlated with a negative clinical outcome, consistent with their tumor-imprinted profile (Fig. 3F), and for the individual gene GPNMB, highly expressed by TAMs (Fig. 3G). Collectively, these results demonstrate the heterogeneous association of SERPINB2+ MoMϕ and GPNMB+ TAMs, characterized by dissimilar transcriptional profiles, with patient clinical outcome.
Spatial distribution defines opposing roles for MoMϕ and TAMs in the TME
Prompted by these results, we set out to explore the mechanisms behind the distinct prognostic significance of SERPINB2+ MoMϕ and GPNMB+ TAMs. Spatial localization of immune cells is critically linked to their function; therefore, we first analyzed the distribution of Mϕ populations within the IM (Fig. 4A). Considering the IM, antitumor inflammatory SERPINB2+ cells were evenly spread out, whereas GPNMB+ TAMs more often accumulated at the very edge of the tumor, as shown by distance to tumor analysis (Fig. 4B and C). This result suggests that GPNMB+ TAMs may be more exposed to tumor immunosuppressive signals and in a critical position to promote tumor growth. To further corroborate this hypothesis, we quantified the number of GPNMB+ TAMs and SERPINB2+ cells in three distinct regions, NA tissue, the IM and the TC (Fig. 4D and E). SERPINB2+ MoMϕ were present in the adjacent region and at a significantly increased density in the IM with no further increase in the TC (Fig. 4D and E). In contrast, GPNMB+ TAMs were almost absent in the NA region and progressively increased in number in the tumor region (Fig. 4D and E). Because TAMs accumulated at the IM, we also tested their spatial distribution, i.e., whether they displayed a peculiar tendency to localize in aggregates, similarly to other myeloid populations (such as LAMP3+ DCs) that are frequently found in small aggregates, resembling tertiary lymphoid structures (TLS; ref. 51). However, this feature was not displayed by any of the Mϕ populations, which were randomly distributed, compared with DCs organized in lymphoid structures (Supplementary Fig. S2F). In fact, when we quantified by the 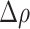 index the propensity of CD68+ and LAMP3+ cells to localize in regions of HD (Supplementary Fig. S2G), CD68+ cells, comprising all Mϕ subsets, displayed an almost random distribution, whereas LAMP3+ cells demonstrated a marked tendency to localize in very HD areas, in line with their prevalent distribution within TLS.
index the propensity of CD68+ and LAMP3+ cells to localize in regions of HD (Supplementary Fig. S2G), CD68+ cells, comprising all Mϕ subsets, displayed an almost random distribution, whereas LAMP3+ cells demonstrated a marked tendency to localize in very HD areas, in line with their prevalent distribution within TLS.
Figure 4.

Spatial assessment of MoMϕ and TAMs in the TME. A–E, Distribution of SERPINB2+ MoMϕ and GPNMB+ TAMs in NA, IM, and TC areas and distance to tumor analysis in a restrospective CLM cohort by mf-IHC. A, mf-IHC images showing the distribution of rendered GPNMB+CD68+ and SERPINB2+ cells in the IM of CLM specimens. Representative picture of n = 48 slides stained. Scale bar: 400 μm. B, Representative images showing the different distribution of rendered GPNMB+CD68+ (top) and SERPINB2+ cell annotations (bottom) in the IM. Cells are colored according to distance from the TC. Scale bar: 300 μm. C, Quantification of the average distance of SERPINB2+ and GPNMB+CD68+ cells from TC. Each dot represents a patient (n = 48). ****, P < 0.0001 by paired t test. D, Representative images of GPNMB+ and SERPINB2+ Mϕ in the NA, IM and TC regions of human CLM. n = 20. E, Violin plots showing the number of SERPINB2+ and GPNMB+CD68+ in the NA, IM and TC. Dashed lines represent median; dotted lines represent quartiles. n = 20; *, P < 0.05; ****, P < 0.0001 by paired t test. (F–I) Spatial assessment of SERPINB2+ MoMϕ and GPNMB+ TAMs by spatial transcriptomics analysis on FFPE CLM samples (n = 2). F, H&E staining of CLM samples analyzed by Visium spatial transcriptomics (left) and tissue segmentation of tumor and normal regions in parallel contiguous zones from the tumor-adjacent liver interface (dashed line; right). Scale bars: 1 mm. G, Spatial feature plots showing signature score for MoMϕ (right) and TAMs (left). White line shows tumor-adjacent liver interface; blue line shows MoMϕ score enrichment in proximity of blood vessels; yellow lines show TAM score enrichment in proximity of bile ducts. H, Heat map showing the z-score of the average expression for each zone of MoMϕ and TAM signature along the adjacent liver (N1–N8) and tumor regions (T1–T7). I, Line plot of MoMϕ (green line) and TAM (purple line) average signature expression along the adjacent liver and tumor regions (N1–N8, T1–T7). ****, P < 0.0001 by one-sided Wilcoxon rank sum test.
To further validate these results, we performed spatial transcriptomics on two CLM samples, by Visium, which allows the integration of histologic and transcriptomic information. Expression of canonical lineage genes such as APOC3, COL1A2, and EPCAM was consistent with histologic information and correctly identified the adjacent liver, stromal and tumor regions of the lesions (Supplementary Fig. S3A). To investigate the localization of TAMs and MoMϕ in the tissue, we segmented the normal and tumor regions into continuous parallel zones starting from the tumor-adjacent liver boundary in either direction (i.e., towards the TC and away from the tumor edge; Fig. 4F). PCA on the manually segmented zones highlighted the similarity of the normal region between CLM#1 and CLM#2, whereas the tumor regions clustered separately (Supplementary Fig. S3B and SC). We next tested the enrichment of TAMs and MoMϕ using a signature-based approach (ref. 52; Fig. 4G). This showed a clear pattern of enrichment of MoMϕ at the IM of both lesions (N4–N1; Fig. 4H and I). Of note, in CLM#1, enrichment of MoMϕ was very evident in proximity to blood vessels (Fig. 4G), possibly suggesting recent recruitment to the tissue. The TAM signature on the other hand, showed a preferential localization in stromal regions of the IM and of the tumor center (T1–T7; Fig. 4H and I), and in the adjacent liver surrounding bile ducts (Fig. 4H and I), confirming the likeness between TAMs and the previously mentioned bile-duct LAMs (29). This pattern was also confirmed by GPNMB expression in the same areas (Supplementary Fig. S3D), further supporting its role as a candidate marker. As to SERPINB2, because its expression was not consistently detected, we searched for the most closely correlated gene (S100A12; Supplementary Fig. S3E) and confirmed its expression to be consistent with signature enrichment (Supplementary Fig. S3F).
Cell–cell interactions of MoMϕ and TAMs in the TME
Mϕ in the TME interact with other neighboring immune cells to orchestrate a proficient or unproductive response. We hypothesized that different interactions could contribute to define opposing roles for MoMϕ and TAMs in CLM. To address this point, CD45+ cells were isolated from the same CLM specimens and subjected to scRNA-seq, generating 20 distinct clusters, including myeloid cells, T and B cells, innate lymphoid cells (Fig. 5A; Supplementary Fig. S4A; Supplementary Table S2). Within CD45+ cell clusters, MP were identified on the basis of marker genes and reclustered to generate 13 distinct subpopulations, comprehensive of the original MP clusters (Supplementary Fig. S4B and S4C). A preliminary analysis using CellPhoneDB (34) allowed us to investigate the bidirectional ligand–receptor interactions between selected MP clusters [MoMϕ(1–3), KC(1), and TAMs] and other CD45+ clusters. MoMϕ(1) and MoMϕ(2) showed a low number of autocrine and paracrine interactions, consistent with their earlier maturation state, whereas KC(1), TAMs, and MoMϕ(3) were the cells most engaged in intercellular interplay (Supplementary Fig. S4D and S4E; Supplementary Table S4). We next performed a deeper analysis into cell–cell communication using NicheNet (31). The top predicted ligands for MoMϕ, including IL15, IL1B, and HMGB2, were expressed by all the sender populations and strongly affected gene expression in most lymphoid receiver cells (Fig. 5B). TAMs instead, expressed high levels of unique predicted ligands, such as IL20 and IL12B, and others shared with KC(1), including C3, CALR, and CD274 (Fig. 5B). We next focused specifically on the ligand–receptor interactions between MP clusters and the most abundant lymphoid cluster, CD8+ T cells (c1; Fig. 5C). Although MoMϕ were mainly predicted to interact with CD8+ T cells via IL15 and its cognate receptors (Fig. 5D), a key signaling and activation axis for T-cell biology, TAMs and KC strongly engaged CD8+ T cells through IL20 and IL10, major immunosuppressive cytokines (Fig. 5D). In keeping with NicheNet results, tissue staining confirmed IL15 expression on MoMϕ expressing SERPINB2 (Fig. 5E) and IL10 expression on TAMs (Fig. 5F), often found in proximity of CD8+ T cells. Altogether, these results indicate that MoMϕ and TAMs affect other immune cells in the TME in an opposite manner, suggesting their ability to skew the immune response towards an anti- or protumoral role, respectively.
Figure 5.

Cell–cell interaction networks of MoMϕ and TAMs in the TME. Interaction analysis on scRNA-seq data between MP and other CD45+ cell clusters and validation by mf-IHC on CLM samples. A, UMAP plot from integrated NA and IM datasets of 14,081 CD45+ cells, color-coded by cluster assignment. B, Heat map of NicheNet analysis showing the potential active ligands, expressed by MP clusters. Rows show active ligands, columns show CD45+ clusters chosen as receiver populations (left). Violin plots show the expression of the potential active ligands across the sender MP clusters (right). C, Schematic representation of NicheNet interaction analysis between MP clusters and CD8+ T cells (c1). D, Circle plot showing the potential active ligands expressed by MP clusters (color-coded by cluster) and the cognate receptors expressed on CD8+ T cells (c1). The opacity of the connecting bands is proportional to the strength of the interaction. E, mf-IHC image of CLM section stained for SERPINB2 (red), IL15 (yellow), and CD8 (cyan). Arrows indicate IL15+SERPINB2+ cells. Picture is representative of n = 3 slides stained. Scale bar: 50 μm. F, mf-IHC image of CLM section stained for GPNMB (magenta), IL10 (yellow), and CD8 (cyan). Arrows indicate IL10+GPNMB+ cells. Picture is representative of n = 3 slides stained. Scale bar: 50 μm.
Discussion
To date, single-cell analyses dissecting the diversity of Mϕ in different cancers have contributed to the identification of several myeloid subsets populating a complex landscape. These studies have broadened our understanding of cancer tissue dynamics, by providing an atlas of immune and nonimmune cells and confirming their relevance in silico. However, the question remains as to whether the multidimensional data generated can be translated into clinically relevant markers for patients with cancer. Here, starting from a single-cell analysis of human CLM, we identified two putative Mϕ markers as prognostic in a real-life cohort of metastatic patients surgically resected. In contrast to extensively studied protumor Mϕ populations, signatures of inflammatory MoMϕ had been previously associated to favorable prognosis in hepatocellular carcinoma (24) and colorectal metastases (6), but a specific marker uniquely identifying these cells had not been provided. In this work, we exploited a single-cell analysis to identify two robust markers that captured discrete populations of Mϕ and could be exploited for further translation to an immune-based stratification tool. The first marker, SERPINB2, is a molecule more commonly known as inhibitor of the urokinase plasminogen activator, although its role in the fibrinolytic cascade has not been confirmed (53). Increased expression of SERPINB2 is associated with pro-inflammatory activation of monocytes and Mϕ, in line with the association of this marker with MoMϕ populations in our study. The transcriptional profile of SERPINB2+ MoMϕ, in fact, suggests that they are early activated and recently derived from recruited monocytes. This evidence suggests that these cells could have been limitedly exposed to the TME and this could account for their antitumor orientation. A distinct population of SERPINB2+ Mϕ has not been reported to our knowledge in previous single-cell works dissecting the variety of myeloid cells in cancer tissues, except for one study, in which it was associated to tumor-infiltrating monocytes (14). This is not surprising, because SERPINB2 was expressed by monocytes also in our dataset, but to a lesser extent compared with MoMϕ. Moreover, we confirmed by mf-IHC that SERPINB2+ cells coexpressed CD68, therefore it could be an ideal marker to identify Mϕ with antitumor potential. In fact, to our knowledge, this is the first time a Mϕ marker has been shown to have a favorable correlation with survival of patients with CLM. In contrast, TAMs, transcriptionally similar to KC, but uniquely expressing GPNMB, were strongly associated with a negative clinical outcome, independently of neoadjuvant therapy administration and KRAS status. Preclinical evidence has shown that pathologic conditions of the liver, such as fibrosis, NASH and cancer, are associated to a progressive derangement of KC (45, 47). Although it is hard to trace a clear ontogeny of these cells in human settings, TAMs in CLM shared with KC both TFs and localization close to sinusoidal endothelial cells, which is instrumental for the execution of specific metabolic and immune functions of liver phagocytes (45). Of note, the transcriptome of GPNMB+ TAMs was very similar to that of LAMs found in steatotic liver (29) and SAMϕ described in liver fibrosis (28).
Spatial distribution in the tumor tissue was one of the key elements differentiating the distinct MP populations. In fact, while the antitumor inflammatory MoMϕ were distributed throughout the IM, TAMs were more often accumulated at the tumor margin and in the tumor center, therefore more exposed to immunosuppressive signals and in a key position to favor tumor growth. This peculiar localization raises a question as to whether increased infiltration of GPNMB+ TAMs may correlate with liver metastasis growth patterns, important histopathologic classifiers currently under evaluation as novel biomarkers (54).
Our analysis uncovered GPNMB as a key marker in human CLM. GPNMB, also known as Osteoactivin and DC-HIL, is a highly glycosylated transmembrane protein that can be cleaved into a soluble isoform. Originally discovered in melanoma cells, osteoblasts and DCs (55), its expression was found increased in Mϕ exposed to tumor-conditioned medium in vitro (56). The soluble form of GPNMB triggers cellular responses, including activation of stemness programs in fibrosarcoma cells (57), increased production of anti-inflammatory cytokines in Mϕ (58) and inhibition of T-cell responses (59). More recently, a single-cell study on hepatocellular carcinoma described a population of GPNMB+ Mϕ associated with worse patient prognosis in silico (24). In addition, GPNMB has been measured in the plasma of patients with cancer and correlated with decreased response to checkpoint inhibitors (60). Overall, GPNMB could thus orchestrate a pathogenetic protumor circuit involving TAMs, tumor cells and possibly other immune cells and could represent a measurable marker in prognostic studies.
In conclusion, we probed and confirmed the clinical relevance of distinct Mϕ signatures in primary colorectal cancer, suggesting that the protumor orientation of Mϕ populations could impact on metastasis. This could provide the basis for the identification of relevant targets that could prevent metastatic spread. Moreover, identifying distinct markers associated with myeloid diversity could provide more specific therapeutic targets and indicators of response to anticancer drugs. Therapeutic strategies aimed at depleting, reeducating or engineering myelomonocytic cells into antitumor effectors are in the spotlight (2–4, 7, 61). However, current approaches have been primarily designed to target the Mϕ population as a whole, possibly affecting those Mϕ that retain an antitumor potential. In conclusion, this study provides an example of how multi-omic approaches can be exploited to characterize the diversity of clinically relevant MP populations for further translation to an immune-based patient stratification tool and therapeutic targets.
Supplementary Material
Supplementary Figures 1-4, Tables S1 and S5
Supplementary figure legends
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Acknowledgments
We thank Javier Cibella from the Genomic Unit of the IRCCS Humanitas Research Hospital for technical assistance and development of methodology. We thank Domenico Supino for helpful discussion and advice on CD45+ scRNA-seq dataset cluster annotation.
This work was supported by Associazione Italiana per la ricerca sul cancro (AIRC): AIRC 5×1000 21147 to A. Mantovani, AIRC MFAG 22083 to F. Giavazzi, and by the Italian Ministry of Health: RF-2019-12369142 to F. Marchesi. The funding agency had no role in design of the study, collection, and analysis of data.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
Authors' Disclosures
A. Mantovani reports personal fees from Ventana, Pierre Fabre, Verily, AbbVie, AstraZeneca, Verseau Therapeutics, Myeloid Therapeutics, Third Rock Venture, Imcheck Therapeutics, Ellipses, Novartis, Roche, Macrophage Pharma, Biovelocita, Merck, Principia, BioLegend; grants from Novartis; other support from Cedarlane Laboratories Ltd, HyCult Biotechnology, eBioscience, BioLegend, ABCAM Plc, Novus Biologicals, Enzo Life, Affymetrix; and personal fees from Olatec Therapeutics outside the submitted work; in addition, A. Mantovani has a patent for WO2019057780 "Anti-human migration stimulating factor (MSF) and uses thereof" pending and issued, a patent for WO2019081591 "NK or T cells and uses thereof" pending and issued, a patent for WO2020127471 "Use of SAP for the treatment of Euromycetes fungi infections" pending and issued, and a patent for EP20182182.6 "PTX3 as a prognostic marker in Covid-19" pending and licensed to Diasorin. F. Marchesi reports grants from Italian Ministry of Health during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
N. Cortese: Formal analysis, investigation, methodology, writing–original draft, writing–review and editing. R. Carriero: Formal analysis, investigation, writing–review and editing. M. Barbagallo: Investigation, methodology. A.R. Putignano: Formal analysis, investigation, methodology. G. Costa: Resources, data curation. F. Giavazzi: Formal analysis, funding acquisition, methodology. F. Grizzi: Methodology. F. Pasqualini: Methodology. C. Peano: Methodology. G. Basso: Methodology. S. Marchini: Resources. F.S. Colombo: Methodology. C. Soldani: Methodology. B. Franceschini: Methodology. L. Di Tommaso: Resources. L. Terracciano: Resources. M. Donadon: Resources, data curation. G. Torzilli: Resources. P. Kunderfranco: Formal analysis, supervision, writing–review and editing. A. Mantovani: Conceptualization, supervision, funding acquisition, writing–review and editing. F. Marchesi: Conceptualization, supervision, funding acquisition, writing–original draft, writing–review and editing.
References
- 1. Ruffell B. Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015;27:462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumor-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017;14:399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov 2018;17:887–904. [DOI] [PubMed] [Google Scholar]
- 4. DeNardo DG, Ruffell B. Macrophages as regulators of tumor immunity and immunotherapy. Nat Rev Immunol 2019;19:369–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cortese N, Carriero R, Laghi L, Mantovani A, Marchesi F. Prognostic significance of tumor-associated macrophages: past, present, and future. Semin Immunol 2020;48:101408. [DOI] [PubMed] [Google Scholar]
- 6. Donadon M, Torzilli G, Cortese N, Soldani C, Di Tommaso L, Franceschini B, et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J Exp Med 2020;217:e20191847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov 2022;21:799–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010;11:889–96. [DOI] [PubMed] [Google Scholar]
- 9. Murray PJ. Macrophage polarization. Annu Rev Physiol 2017;79:541–66. [DOI] [PubMed] [Google Scholar]
- 10. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol 2020;15:123–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blériot C, Chakarov S, Ginhoux F. Determinants of resident tissue macrophage identity and function. Immunity 2020;52:957–70. [DOI] [PubMed] [Google Scholar]
- 12. Friebel E, Kapolou K, Unger S, Núñez NG, Utz S, Rushing EJ, et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 2020;181:1626–42. [DOI] [PubMed] [Google Scholar]
- 13. Antunes ARP, Scheyltjens I, Lodi F, Messiaen J, Antoranz A, Duerinck J, et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat Neurosci 2021;24:595–610. [DOI] [PubMed] [Google Scholar]
- 14. Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 2021;595:578–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lapenna A, De PM, Lewis CE. Perivascular macrophages in health and disease. Nat Rev Immunol 2018;18:689–702. [DOI] [PubMed] [Google Scholar]
- 16. Ydens E, Amann L, Asselbergh B, Scott CL, Martens L, Sichien D, et al. Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury. Nat Neurosci 2020;23:676–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martinek J, Lin J, Kim KI, Wang VG, Wu TC, Chiorazzi M, et al. Transcriptional profiling of macrophages in situ in metastatic melanoma reveals localization-dependent phenotypes and function. Cell Rep Med 2022;3:100621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res 2006;66:605–12. [DOI] [PubMed] [Google Scholar]
- 19. Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity 2021;54:1883–900. [DOI] [PubMed] [Google Scholar]
- 20. Geeraerts X, Fernández-Garcia J, Hartmann FJ, de Goede KE, Martens L, Elkrim Y, et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Rep 2021;37:110171. [DOI] [PubMed] [Google Scholar]
- 21. Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 2021;184:792–809. [DOI] [PubMed] [Google Scholar]
- 22. Wu Y, Yang S, Ma J, Chen Z, Song G, Rao D, et al. Spatiotemporal immune landscape of colorectal cancer liver metastasis at single-cell level. Cancer Discov 2022;12:134–53. [DOI] [PubMed] [Google Scholar]
- 23. Fridman WH. The tumor microenvironment: prognostic and theranostic impact. Recent advances and trends. Semin Immunol 2020;48:101416. [DOI] [PubMed] [Google Scholar]
- 24. Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 2019;179:829–45. [DOI] [PubMed] [Google Scholar]
- 25. Cortese N, Soldani C, Franceschini B, Barbagallo M, Marchesi F, Torzilli G, et al. Macrophages in colorectal cancer liver metastases. Cancers 2019;11:633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell. 2019;177:1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019;575:512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022;185:379–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods 2020;17:159–62. [DOI] [PubMed] [Google Scholar]
- 32. Linderman GC, Zhao J, Roulis M, Bielecki P, Flavell RA, Nadler B, et al. Zero-preserving imputation of single-cell RNA-seq data. Nat Commun 2022;13:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gu Z, Gu L, Eils R, Schlesner M, Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics 2014;30:2811–2. [DOI] [PubMed] [Google Scholar]
- 34. Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand-- receptor complexes. Nat Protoc 2020;15:1484–506. [DOI] [PubMed] [Google Scholar]
- 35. Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 2017;14:1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One 2010;5:e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 2018;19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Waskom ML. seaborn: statistical data visualization. J Open Source Softw 2021;6:3021. [Google Scholar]
- 39. Hunter JD. Matplotlib: A 2D graphics environment. Comput Sci Eng 2007;9:90–5. [Google Scholar]
- 40. Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 2020;17:261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pedregosa F, Varoquaux G, Gramfort A, Michel V, B T. Scikit-learn: Machine learning is python. J Mach Learn Res 2011;12:2825–30. [Google Scholar]
- 42. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res 2019;47:W556–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single-cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 2018;9:4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 2017;17:306–21. [DOI] [PubMed] [Google Scholar]
- 46. Yáñez A, Coetzee SG, Olsson A, Muench DE, Berman BP, Hazelett DJ, et al. Granulocyte-monocyte progenitors and monocyte-dendritic cell progenitors independently produce functionally distinct monocytes. Immunity 2017;47:890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity 2020;52:1057–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yin C, Vrieze AM, Rosoga M, Akingbasote J, Pawlak EN, Jacob RA, et al. Efferocytic defects in early atherosclerosis are driven by GATA2 overexpression in macrophages. Front Immunol 2020;11:594136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol 2015;15:104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol 2017;13:710–30. [DOI] [PubMed] [Google Scholar]
- 51. Fridman WH, Meylan M, Petitprez F, Sun CM, Italiano A, Sautès-Fridman C. B cells and tertiary lymphoid structures as determinants of tumor immune contexture and clinical outcome. Nat Rev Clin Oncol 2022;19:441–57. [DOI] [PubMed] [Google Scholar]
- 52. Wu R, Guo W, Qiu X, Wang S, Sui C, Lian Q, et al. Comprehensive analysis of spatial architecture in primary liver cancer. Sci Adv 2021;7:eabg3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schroder WA, Hirata TD, Le TT, Gardner J, Boyle GM, Ellis J, et al. SerpinB2 inhibits migration and promotes a resolution phase signature in large peritoneal macrophages. Sci Rep 2019;9:12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van Dam PJ, Daelemans S, Ross E, Waumans Y, Van Laere S, Latacz E, et al. Histopathological growth patterns as a candidate biomarker for immunomodulatory therapy. Semin Cancer Biol 2018;52:86–93. [DOI] [PubMed] [Google Scholar]
- 55. Kobayashi M, Chung JS, Beg M, Arriaga Y, Verma U, Courtney K, et al. Blocking monocytic Myeloid-derived suppressor cell function via anti–DC-HIL/GPNMB antibody restores the in vitro integrity of T cells from cancer patients. Clin Cancer Res 2019;25:828–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Solinas G, Schiarea S, Liguori M, Fabbri M, Pesce S, Zammataro L, et al. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J Immunol 2010;185:642–52. [DOI] [PubMed] [Google Scholar]
- 57. Liguori M, Digifico E, Vacchini A, Avigni R, Colombo FS, Borroni EM, et al. The soluble glycoprotein NMB (GPNMB) produced by macrophages induces cancer stemness and metastasis via CD44 and IL33. Cell Mol Immunol 2020;18:711–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Saade M, Araujo de Souza G, Scavone C, Kinoshita PF. The Role of GPNMB in Inflammation. Front Immunol 2021;12:674739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chung JS, Sato K, Dougherty II, Cruz PD, Ariizumi K. DC-HIL is a negative regulator of T lymphocyte activation. Blood 2007;109:4320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chung JS, Ramani V, Kobayashi M, Fattah F, Popat V, Zhang S, et al. DC-HIL/Gpnmb is a negative regulator of tumor response to immune checkpoint inhibitors. Clin Cancer Res 2020;26:1449–59. [DOI] [PubMed] [Google Scholar]
- 61. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer 2016;16:447–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-4, Tables S1 and S5
Supplementary figure legends
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4