

ABSTRACT
There is considerable interest in the pharmaceutical industry toward development of antibody-based biotherapeutics because they can selectively bind diverse receptors and often possess desirable pharmacology. Here, we studied product characteristics of 89 marketed antibody-based biotherapeutics that were approved from 1986 to mid-2020 by gathering publicly available information. Our analyses revealed major trends in their emergence as the best-selling class of pharmaceuticals. Early on, most therapeutic monoclonal antibodies were developed to treat cancer, with CD20 being the most common target. Thanks to industrialization of antibody manufacturing technologies, their use has now blossomed to include 15 different therapeutic areas and nearly 60 targets, and the field is still growing! Drug manufacturers are solidifying their choices regarding types of antibodies and their molecular formats. IgG1 kappa continues to be the most common molecular format among marketed antibody-based biotherapeutics. Most antibody-based biotherapeutics approved since 2015 are either humanized or fully human, but the data we collected do not show a direct correlation between humanness and reported incidence of anti-drug antibodies. Furthermore, there have also been improvements in terms of drug product stability and high concentration liquid formulations suitable for subcutaneous route of administration, which are being approved more often in recent years. These improvements, however, have not been uniformly adopted across all therapeutic areas, suggesting that multiple options for drug product development are being used to serve diverse therapeutic purposes. Insights gained from this analysis may help us devise better end-to-end antibody-based biotherapeutic drug discovery and development strategies.
KEYWORDS: Antibody, biotherapeutics, developability, drug, formulation, pharmacology
Introduction
Antibody-based biotherapeutics represent one of the fastest-growing segments in the pharmaceutical market (Figure 1(a)) because of their ability to bind with high selectivity along with desirable pharmacological attributes, such as longer half-lives, resulting in safe and effective medicines with convenient dosing schedules. 1,2 By November 21, 2021, a total of 131 antibody-based biotherapeutics were approved under regulatory review in the United States or European Union.3 Moreover, the number of antibodies in clinical development (Phases I-III) grew by over 30% in the past year.4 Representation of antibody-based biotherapeutics in the portfolios of large pharmaceutical companies has also grown significantly in recent years.5 While monoclonal antibodies (mAbs) have been developed to treat infectious diseases previously,6–9 the coronavirus disease 2019 (COVID-19) pandemic has created renewed interest in using antibody-based biotherapeutics as a treatment option for infectious diseases.10–12 In 2021, the global antibody market was valued at 217.3 billion USD and is projected to exhibit an annual growth rate of approximately 15% in the next decade.13–15 Considering the scale at which antibody-based biotherapeutics are now being discovered and developed, it is apparent that the industrialization of mAbs has come of age.
Figure 1.
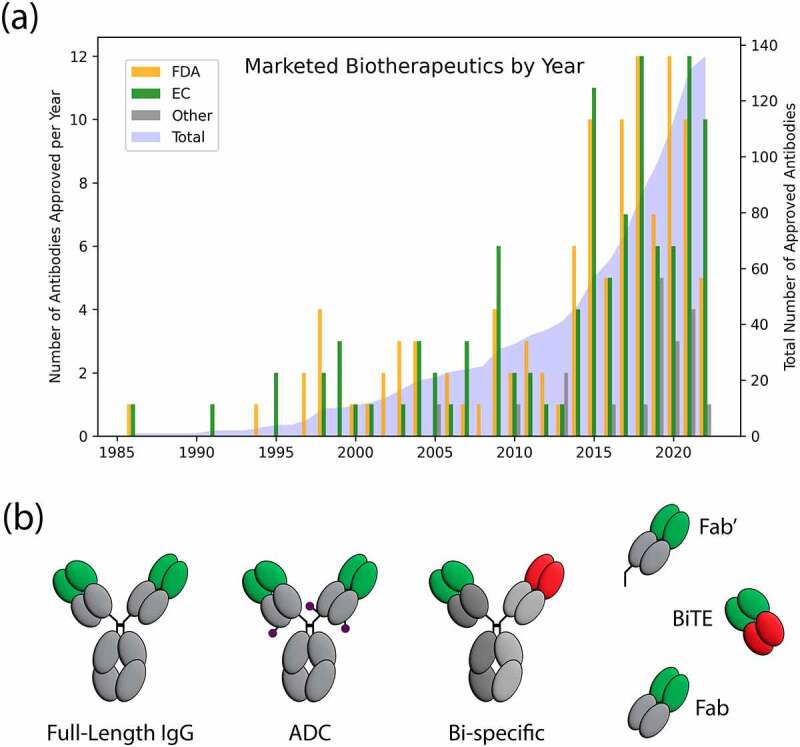
(a) Marketed antibody-based biotherapeutics by year. By June 2022, 111 antibody-based biotherapeutic drug products have been approved by the FDA (shown in orange) and 106 have been approved by the EC (shown in green). 19 total antibody-based biotherapeutic drugs have been approved by India (3), Cuba (2), Russia (4), China (9), and Japan (1), but not by the FDA or EC (shown in gray). The total number of unique marketed antibody-based biotherapeutics as of June 2022 is 136 shown in blue with right y-axis showing growth over time. (b) Molecular formats of marketed antibody-based biotherapeutics. Gray regions represent constant regions. Green regions are variable regions. Red regions are a second variable region in the bispecific therapeutics. The purple circles are examples of where a small molecule drug could attach to an antibody and form antibody-drug conjugates (ADCs).
Early overviews of marketed antibody-based biotherapeutics were limited because very few products had been granted approvals at the time.16–20 As more were approved, growing interest in the scientific community led to broader overviews,21–23 as well as reviews of specific characteristics,24–26 such as cell culture and immunogenicity of marketed and clinical trials stage mAbs.27,28 In 2010, Dr. Janice Reichert started publishing the “Antibodies to Watch” series, which continues to be released yearly.29–41 More recent publications on marketed mAbs have looked at deeply specific characteristics,42,43 such as glycosylation patterns,44,45 or broad concepts like development trajectories.46,47
Given the growth in the field, this is an opportune time to examine how antibody-based biotherapeutics have evolved, since the first approval of muromonab in 1986.48 Specifically, we assessed the evolution of crucial aspects of antibody-based biotherapeutics, such as 1) therapeutic purpose, 2) safety and immunogenicity, 3) developability, and 4) patient convenience, by analyzing the trends in their product characteristics (e.g., types and molecular formats, formulation, routes of administration, pharmacokinetics (PK), and product presentation) over time. Toward this goal, we collected in-depth information on 89 antibody-based biotherapeutic products that were approved by the end of June 2020. We started by collecting publicly available information from the websites of the United States Food and Drug Administration (FDA),49 the European Medicines Agency (EMA),50 and the World Health Organization (WHO),51,52 along with the information available in the package inserts and the reviews of biologics license applications (BLAs) of these drug products. Overall, each antibody in the dataset has more than 80 data points, providing us with multiple ways to compare antibody-based biotherapeutic drug product characteristics and study evolution of the biopharmaceutical industry over time. Insights gained from this study will be useful toward improving the rate of successful translation of biological drug discoveries into the approved drug products available in the market to improve patient lives.
Results
The discovery and development of antibody-based biotherapeutics is highly dynamic. Understanding the current trends in marketed drug products can potentially aid in the discovery and design of more developable drug candidates. Publicly available information, such as package inserts and reviews of BLAs available from regulatory agency websites were used to collect information on 89 antibody-based biotherapeutic drug products approved through June 2020 (see Methods section). Table 1 provides a summary of salient characteristics, such as the year of the first FDA approval, name, antibody format, type, formulation buffer, formulation pH, active pharmaceutical ingredient (API) concentration, product presentation, and route of administration for each of the 89 antibody-based biotherapeutics studied in this work. Further details for each of these can be found in Tables S1 and S2 of the supplementary material. The data we collected were analyzed to reveal useful trends in therapeutic purpose, safety, developability, PK, and patient convenience of antibody-based biotherapeutic drug products. These analyses have improved our understanding of the evolving landscape of antibody-based biotherapeutics.
Table 1.
Salient characteristics of 89 marketed antibody-based biotherapeutics sorted by their first FDA† approval date*.
| Year of first approval by FDA | INN name | Antibody format |
Antibody type | Formulation buffer | Formulation pH | API Conc. mg/mL | Product Presentation | Route of administration for the first approval |
|---|---|---|---|---|---|---|---|---|
| 1986 | Muromonab-CD3 | IgG | Murine | Phosphate | 7.0 | 1 | Vial | Intravenous |
| 1994 | Abciximab | Fab | Chimeric | Phosphate | 7.2 | 2 | Vial | Intravenous |
| 1997 | Daclizumab | IgG | Humanized | Phosphate | 6.9 | 5 | Vial | Intravenous |
| 1997 | Rituximab | IgG | Chimeric | Citrate | 6.5 | 10 | Vial | Intravenous |
| 1998 | Basiliximab | IgG | Chimeric | Phosphate | NA | 4 | Lyo | Intravenous |
| 1998 | Infliximab | IgG | Chimeric | Phosphate | 7.2 | 10 | Lyo | Intravenous |
| 1998 | Palivizumab | IgG | Humanized | Histidine | 6.0 | 100 | Vial | Intramuscular |
| 1998 | Trastuzumab | IgG | Humanized | Histidine | 6.0 | 21 | Lyo | Intravenous |
| 2000 | Gemtuzumab ozogamicin | IgG | Humanized | None | NA | 1 | Lyo | Intravenous |
| 2001 | Alemtuzumab | IgG | Humanized | Phosphate | 7.2 | 10 | Vial | Intravenous |
| 2002 | Adalimumab | IgG | Human | Phosphate, Citrate | 7.2 | 50 | Pen | Subcutaneous |
| 2002 | Ibritumomab tiuxetan | IgG | Murine | None | NA | 1.6 | Vial | Intravenous |
| 2003 | Efalizumab | IgG | Humanized | Histidine | 6.2 | 100 | Lyo | Subcutaneous |
| 2003 | Omalizumab | IgG | Humanized | Histidine | NA | 125 | Lyo/Pen | Subcutaneous |
| 2003 | Tositumomab | IgG | Murine | Phosphate | 7.2 | 14 | Vial | Intravenous |
| 2004 | Bevacizumab | IgG | Humanized | Phosphate | 6.2 | 25 | Vial | Intravenous |
| 2004 | Cetuximab | IgG | Chimeric | Phosphate | 7.2 | 100 | Vial | Intravenous |
| 2004 | Natalizumab | IgG | Humanized | Phosphate | 6.1 | 20 | Vial | Intravenous |
| 2006 | Panitumumab | IgG | Human | Acetate | 5.8 | 20 | Vial | Intravenous |
| 2006 | Ranibizumab | Fab | Humanized | Histidine | 5.5 | 10 | Pen/Vial | Intravitreal |
| 2007 | Eculizumab | IgG | Humanized | Phosphate | 7.0 | 10 | Vial | Intravenous |
| 2008 | Certolizumab pegol | Fab’ | Humanized | Acetate | 5.2 | 200 | Lyo | Subcutaneous |
| 2009 | Canakinumab | IgG | Human | Histidine | 6.5 | 150 | Lyo | Subcutaneous |
| 2009 | Golimumab | IgG | Human | Histidine | 5.5 | 100 | Pen | Subcutaneous |
| 2009 | Ofatumumab | IgG | Human | Acetate | 6.5 | 20 | Vial | Intravenous |
| 2009 | Tocilizumab | IgG | Humanized | Phosphate | 6.5 | 20 | Vial | Intravenous |
| 2009 | Ustekinumab | IgG | Human | Histidine | 6.0 | 90 | Pen/Vial | Subcutaneous |
| 2010 | Denosumab | IgG | Human | Acetate | 5.2 | 60 | Pen | Subcutaneous |
| 2010 | Nimotuzumab† | IgG | Humanized | Phosphate | NA | 5 | Vial | Intravenous |
| 2011 | Belimumab | IgG | Human | Citrate | 6.5 | 80 | Lyo | Intravenous |
| 2011 | Brentuximab vedotin | IgG | Chimeric | Citrate | 6.6 | 5 | Lyo | Intravenous |
| 2011 | Ipilimumab | IgG | Human | Tris | 7.0 | 5 | Vial | Intravenous |
| 2012 | Pertuzumab | IgG | Humanized | Acetate | 6.0 | 30 | Vial | Intravenous |
| 2013 | Itolizumab† | IgG | Humanized | Histidine | 7.0 | 5 | Lyo | Intravenous |
| 2013 | Obinutuzumab | IgG | Humanized | Histidine | 6.0 | 25 | Vial | Intravenous |
| 2013 | Racotumomab† | IgG | Murine | Tris | NA | 1 | Vial | Intradermal |
| 2014 | Blinatumomab | BiTE | Murine | Citrate | 7.0 | 0.0125 | Lyo | Intravenous |
| 2014 | Nivolumab | IgG | Human | Citrate | 6.0 | 10 | Vial | Intravenous |
| 2014 | Pembrolizumab | IgG | Humanized | Histidine | 5.5 | 25 | Vial | Intravenous |
| 2014 | Ramucirumab | IgG | Human | Histidine | 6.0 | 10 | Vial | Intravenous |
| 2014 | Siltuximab | IgG | Chimeric | Histidine | 5.2 | 20 | Lyo | Intravenous |
| 2014 | Vedolizumab | IgG | Humanized | Histidine | 6.3 | 60 | Lyo | Intravenous |
| 2015 | Alirocumab | IgG | Human | Histidine | 6.0 | 150 | Pen | Subcutaneous |
| 2015 | Atezolizumab | IgG | Humanized | Acetate | 5.8 | 60 | Vial | Intravenous |
| 2015 | Daratumumab | IgG | Human | Acetate | 5.5 | 20 | Vial | Intravenous |
| 2015 | Dinutuximab | IgG | Chimeric | Histidine | 6.8 | 3.5 | Vial | Intravenous |
| 2015 | Elotuzumab | IgG | Humanized | Citrate | NA | 25 | Vial | Intravenous |
| 2015 | Evolocumab | IgG | Human | Acetate | 5.0 | 140 | Pen | Subcutaneous |
| 2015 | Idarucizumab | Fab | Humanized | Acetate | 5.5 | 50 | Vial | Intravenous |
| 2015 | Mepolizumab | IgG | Humanized | Phosphate | 7.0 | 100 | Lyo/Pen/Vial | Subcutaneous |
| 2015 | Necitumumab | IgG | Human | Citrate | 6.0 | 16 | Vial | Intravenous |
| 2015 | Secukinumab | IgG | Human | Histidine | 5.8 | 150 | Lyo/Pen | Subcutaneous |
| 2016 | Bezlotoxumab | IgG | Human | Citrate | 6.0 | 25 | Vial | Intravenous |
| 2016 | Ixekizumab | IgG | Humanized | Citrate | 5.7 | 80 | Pen | Subcutaneous |
| 2016 | Obiltoxaximab | IgG | Chimeric | Histidine | 5.5 | 100 | Vial | Intravenous |
| 2016 | Olaratumab | IgG | Human | Histidine | 5.5 | 10 | Vial | Intravenous |
| 2016 | Reslizumab | IgG | Humanized | Acetate | 5.5 | 10 | Vial | Intravenous |
| 2017 | Avelumab | IgG | Human | Acetate | 5.3 | 20 | Vial | Intravenous |
| 2017 | Benralizumab | IgG | Humanized | Histidine | 6.0 | 30 | Pen | Subcutaneous |
| 2017 | Brodalumab | IgG | Human | Glutamate | 4.8 | 140 | Vial | Subcutaneous |
| 2017 | Dupilumab | IgG | Human | Acetate | 5.9 | 150 | Pen | Subcutaneous |
| 2017 | Durvalumab | IgG | Human | Histidine | 5.5 | 50 | Vial | Intravenous |
| 2017 | Emicizumab | BiSpIgG | Humanized | Histidine | 6.0 | 30 | Vial | Subcutaneous |
| 2017 | Guselkumab | IgG | Human | Histidine | 5.8 | 100 | Pen | Subcutaneous |
| 2017 | Inotuzumab ozogamicin | IgG | Humanized | Tris | 8.0 | 0.25 | Lyo | Intravenous |
| 2017 | Ocrelizumab | IgG | Humanized | Acetate | 5.3 | 30 | Vial | Intravenous |
| 2017 | Sarilumab | IgG | Human | Histidine | 6.0 | 175 | Pen | Subcutaneous |
| 2018 | Burosumab | IgG | Human | Histidine | 6.3 | 10 | Vial | Subcutaneous |
| 2018 | Cemiplimab | IgG | Human | Histidine | 6.0 | 50 | Vial | Intravenous |
| 2018 | Emapalumab | IgG | Human | Histidine | 6.0 | 5 | Vial | Intravenous |
| 2018 | Erenumab | IgG | Human | Acetate | 5.2 | 70 | Pen | Subcutaneous |
| 2018 | Fremanezumab | IgG | Humanized | Histidine | 5.5 | 150 | Pen | Subcutaneous |
| 2018 | Galcanezumab | IgG | Humanized | Histidine | 5.8 | 120 | Pen | Subcutaneous |
| 2018 | Ibalizumab | IgG | Humanized | Histidine | 6.0 | 150 | Vial | Intravenous |
| 2018 | Lanadelumab | IgG | Human | Phosphate | 6.0 | 150 | Vial | Subcutaneous |
| 2018 | Mogamulizumab | IgG | Humanized | Citrate | 5.5 | 4 | Vial | Intravenous |
| 2018 | Moxetumomab pasudotox | Fv | Murine | Phosphate | 6.0 | 1 | Lyo | Intravenous |
| 2018 | Ravulizumab | IgG | Humanized | Phosphate | 7.0 | 10 | Vial | Intravenous |
| 2018 | Tildrakizumab | IgG | Humanized | Histidine | 6.0 | 100 | Pen | Subcutaneous |
| 2019 | Crizanlizumab | IgG | Humanized | Citrate | 6.0 | 10 | Vial | Intravenous |
| 2019 | Enfortumab vedotin | IgG | Human | Histidine | 6.0 | 10 | Lyo | Intravenous |
| 2019 | Polatuzumab vedotin | IgG | Humanized | Succinate | 5.3 | 20 | Lyo | Intravenous |
| 2019 | Risankizumab | IgG | Humanized | Succinate | NA | 90 | Pen | Subcutaneous |
| 2019 | Romosozumab | IgG | Humanized | Acetate | 5.2 | 90 | Pen | Subcutaneous |
| 2020 | Eptinezumab | IgG | Humanized | Histidine | 5.8 | 100 | Vial | Intravenous |
| 2020 | Inebilizumab | IgG | Humanized | Histidine | 6.0 | 10 | Vial | Intravenous |
| 2020 | Isatuximab | IgG | Chimeric | Histidine | 6.0 | 20 | Vial | Intravenous |
| 2020 | Sacituzumab govitecan | IgG | Humanized | Sulfate | 6.5 | 10 | Lyo | Intravenous |
| 2020 | Teprotumumab | IgG | Human | Histidine | 5.5 | 48 | Lyo | Intravenous |
*Antibody format shows IgG for full-length antibodies. BiSpIgG is a bispecific full-length antibody. Fv is for variable region, Fab is for antigen binding fragment, Fab’ is for antigen binding fragment which is cleaved below the hinge region, and BiTE is for bispecific t-cell engager. Note that several of these antibody formats are also conjugated with small molecule payloads to form antibody-drug conjugates (ADCs). Such antibody-based biotherapeutics are easily spotted by the presence of two words in their names. API stands for active pharmaceutical ingredient. For product presentation, lyo stands for lyophilized powder. NA stands for not available. The 89 marketed antibody-based biotherapeutics come in diverse ranges of isotypes, formulation, and product presentation which is driven by the therapeutic purpose. The full table can be found in the supplement (Table S1).
†These three products were not approved in the United States by the FDA, but rather in Cuba and India.
Biotherapeutics are being approved at a more rapid rate
The 89 antibody-based biotherapeutics included here were approved from 1986 to June 2020 (Figure 1(a)) by regulatory agencies, including the FDA, and the European Commission (EC). The first antibody-based biotherapeutic to win regulatory approval was muromonab or Orthoclone OKT3 in 1986. Eight years later, the next therapeutic antibody approved in the United States in 1994 was abciximab. Only six antibody-based biotherapeutics were approved between 1994 and 1999, followed by another 19 that were approved from 2000 to 2009. The next decade saw a substantial increase in approvals of antibody-based biotherapeutics, and 57 were approved between 2010 and 2019. The remaining five of the 89 products were approved in the first half of 2020. Three therapeutic antibodies approved in 1997 (daclizumab), 2000 (gemtuzumab ozogamicin), and 2001 (alemtuzumab) were withdrawn, and then reapproved in 2016, 2017, and 2014, respectively. Two products, muromonab, and abciximab, approved in 1986 and 1994, respectively, were voluntarily withdrawn in 2010 and 2019, respectively.53,54 Additionally, efalizumab was withdrawn in 2009 (approved in 2003) and olaratumab (approved in 2016) was withdrawn in 2019.41,53 Three mAbs included in the 89 were not approved by the FDA or EC but were approved by other drug or medical agencies. These three drugs are itolizumab (approved in India),55 nimotuzumab (approved in various countries),56 and racotumomab (approved in Cuba and Argentina).57,58 Furthermore, three antibodies, tositumomab, emapalumab, and teprotumumab, were approved by the FDA, but not by the EC (see Table S1). Tositumomab was granted an orphan drug designation by the EC but was withdrawn in 2015.59 Emapalumab was not approved by the EC because there was no conclusive evidence of its effectiveness.60 Teprotumumab was granted a product-specific waiver for pediatric trials, but a marketing application was not submitted for full review.61,62 It should also be noted that nebacumab and edrecolomab were approved by various European countries in 1991 and 1995, respectively, and catumaxomab was approved by the EC in 2009, but none of these were approved in the US. These antibodies have not been included in this dataset of 89 antibody products because nebacumab and edrecolomab were withdrawn in 1993 and 2002, respectively, due to subsequent trials showing little benefit; and catumaxomab was withdrawn in 2017 at the request of the manufacturer.63–65
In addition to the 89 included in this survey, Figure 1(a) shows the approval years of all marketed antibody-based biotherapeutic drugs through June 2022. The FDA has approved 111 antibody-based biotherapeutic drug products including one cocktail, while the EC has approved 106. Five antibody-based biotherapeutics approved by the EC were not approved by the FDA, and 11 antibody-based biotherapeutics approved by the FDA were not approved by the EC. Between the FDA and EC, 117 unique antibody-based biotherapeutics have been approved. Furthermore, 19 antibody-based biotherapeutic drugs have been approved by India (3, including one cocktail), Cuba (2), Russia (4), China (9), and Japan (1), but not by the FDA or EC. In total, 136 antibody-based biotherapeutic drugs have been approved by at least one government agency up to June 30, 2022. Of these 136, 4 are bispecific antibodies and another two are antibody-based biotherapeutic cocktails that contain more than one antibody. The four bispecific antibodies are catumaxomab, blinatumomab, emicizumab, and amivantamab. The two cocktails with more than one antibody are Inmazeb (atoltivimab, maftivimab, odesivimab) and Twinrab (docaravimab, miromavimab), the former was approved by the FDA and the latter one was approved in India.
International nonproprietary names
The WHO uses their International Nonproprietary Names (INNs) system to give biotherapeutics a generic name that is used publicly to refer to the drug substance.66 As defined by the WHO, INNs identify pharmaceutical substances or APIs. Each INN is unique, is globally recognized, and is public property. The nonproprietary name of a biotherapeutic is also known as its generic name. In this report, we discuss antibody-based biotherapeutics using their INNs.
The INN nomenclature was created in 1953,67 and while the nomenclature for antibody-based biotherapeutics was first proposed in 1990,68 it was not adapted by the WHO until 1997.69 This original WHO nomenclature has undergone three revisions since 1997. The first revision was in 2009 (up to INN proposed list 102),70 the second revision was in 2017 (INN proposed lists 103 to 117),71 and the most recent revision was in 2021 (INN proposed lists 118−125).72 The original nomenclature from 1997 included sub-stems for source (antibody type) of product (rat, chimeric, humanized, or fully human), disease, or target class (e.g., bacterial, cardiovascular, or bone), and organs/tumors (e.g., colon, testis, or ovary).70 Additional guidance was given for adding letters for facilitating pronunciation. The second nomenclature (the first revision proposed in 2009) simplified the source of product/species and disease/target.71 The third nomenclature (the second revision proposed in 2017) provides information about the target. The naming scheme has a prefix, an infix (target class) and a suffix (mab).72 The most recent naming scheme (the third revision proposed in 2021) divides the substances that contain an immunoglobulin variable domain into four groups: unmodified immunoglobulins (−tug), artificial antibodies (−bart), multi-specific immunoglobulins (−mig), and fragments (−ment).73 This represents the first time that the suffix of a monoclonal antibody will not be “mab.” In addition, several infixes were added to indicate the mode of action of the biotherapeutic product. In the proposed list 126, there are already several products using this new naming scheme such as crexavibart and masavibart. Since we evaluated 89 products approved through June 2020, no antibody therapeutics with INN derived from the new naming scheme were included.
Isotypes and molecular format
In addition to different names, marketed antibody-based biotherapeutic products have multiple molecular formats. Figure 1(b) shows the diversity of molecular formats among the 89 marketed antibody-based biotherapeutics. Eighty-two of these 89 (92%) are full-length monospecific mAbs. One of these 89 is a full-length bispecific mab (emicizumab, which binds to FIX and FX in the blood-clotting pathway) made of two unique variable regions that share a common light chain. Of the remaining six, one is a bispecific T-cell engager (BiTE, blinatumomab), which consists of two murine single-chain variable regions (scFvs), binding to CD3 and CD19, connected via a peptide linker. Three antibody-based biotherapeutics are antigen binding fragments (Fab; abciximab, idarucizumab, and ranibizumab). One is Fab’ (certolizumab pegol), which is a Fab that has been cleaved below the hinge region; and the last one contains only a variable region (Fv) of the antibody fused to a 38 kDa exotoxin called PE38 (moxetumomab pasudotox).74 Overall, the 89 antibody-based biotherapeutics studied here contain 91 Fvs in 6 different antibody-based molecular formats. Note that there are other marketed full-length bi-specific antibodies (e.g., amivantamab) that were approved after our cutoff date of June 2020 and consequently were not included in our analysis.
Figure 2.
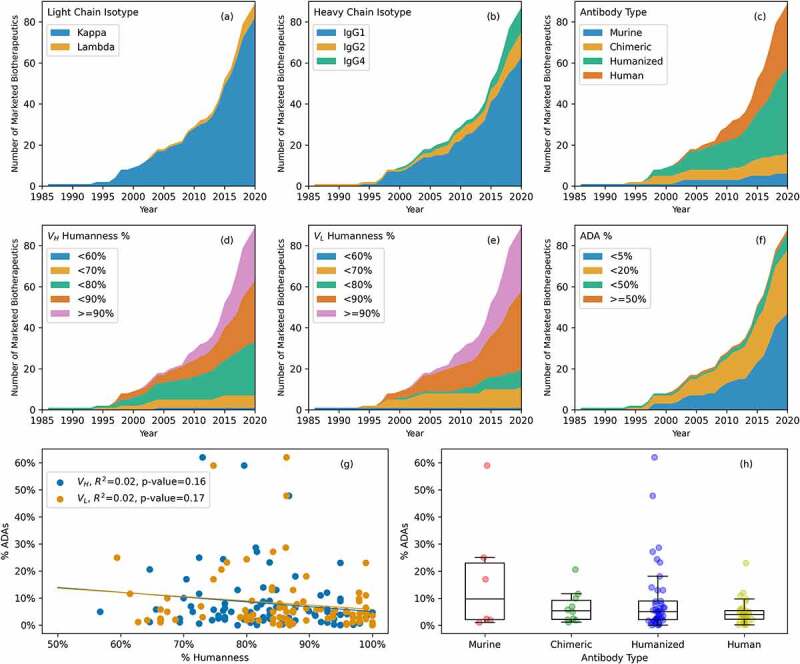
(a) Light chain isotypes of marketed antibody-based biotherapeutics over time. Kappa isotypes are much more common than lambda isotypes. (b) Heavy chain isotypes of marketed antibody-based biotherapeutics over time. IgG1 is the most common heavy isotype, while IgG2 and IgG4 are equally common. (c) Types of marketed antibody-based biotherapeutics over time. Murine and chimeric were common early on, but nearly all approved marketed antibody-based biotherapeutics are now either human or humanized. (d) Heavy chain variable region (VH) percent humanness over time. VH humanness is commonly between 70 and 90%. (e) Light chain variable region (VL) percent humanness over time. VL humanness is commonly between 80 and 90%. (f) Reported percent anti-drug antibodies (% ADAs) over time. In recent years it is much more common to see a reported % ADAs below 5%. (g) Comparison of % Humanness and % ADAs. There is no statistically significant correlation between % Humanness and % ADAs reported for the marketed antibody-based biotherapeutics. (h) % ADAs by antibody type. In general, % ADAs decrease from murine to human, however a direct comparison of % ADAs across the products should be interpreted with caution (see results).
There are seven antibody-drug conjugates (ADCs) among these 89 antibody-based biotherapeutics, in which the antibodies are conjugated to small molecule anti-cancer drug payloads.75 Three of these are conjugated to monomethyl auristatin E,76 a synthetic analog of the marine natural product dolastatin 10.77 Two are conjugated to a cytotoxic calicheamicin antibiotic derivative.78 The remaining two conjugates are conjugated to an isothiocyanatobenzyl derivative of MX-DTPA, a chelating agent,79 and SN-38, the active metabolite of irinotecan.80 All the ADCs are used to deliver small molecule anti-cancer drugs and thus belong to the oncology therapeutic area. Of note, certolizumab pegol is not categorized as an ADC here because it has no anti-cancer drug as payload. Instead, it is pegylated,81 to improve drug PK and bioavailability.82,83 After June 2020, three more ADCs (belantamab mafodotin, tisotumab vedotin, and loncastuximab tesirine) were approved by the FDA. These are not included in our analysis.
The 89 marketed antibody-based biotherapeutics we analyzed contain 91 variable regions (Fvs) because two of these products are bispecific antibodies. The light chains for 84 of the 91 Fvs (92%) are of kappa isotype and the remaining 7 (8%) are of lambda isotype (Figure 2(a)). The majority (5/7; 71%) of the products with lambda light chains were approved after 2015. Figure 2(b) shows that most marketed antibody-based biotherapeutics are IgG1, and this heavy chain isotype is a popular choice. The heavy chain isotypes can be inferred for only 87 antibody-based biotherapeutics (83 full-length IgGs, 3 Fabs, and 1 Fab’) because moxetumomab pasudotox and blinatumomab (a bispecific) contain only the variable regions. Of these 87, the heavy chain isotype for 63 (72.4%) is IgG1. Ten (11.5%) contain IgG2 heavy chains while another 12 (13.8%) contain IgG4 heavy chains and 2 (2.3%) are IgG2/4 hybrids (eculizumab and ravulizumab).84,85 However, the amino acid sequences for both of them show that their heavy chains are of IgG2 isotype. Therefore, eculizumab and ravulizumab were counted as full length IgG2 antibodies in this report. Since 2015, six antibody-based biotherapeutics with IgG2 isotype and eight with IgG4 isotype have been granted approvals, while 22 IgG1 antibody-based biotherapeutics were approved during the same period.
Targets, clinical indications, and therapeutic areas served by marketed antibody-based biotherapeutics
The 89 marketed antibody-based biotherapeutics bind many targets and serve previously unmet medical needs in several therapeutic areas by addressing numerous clinical indications. Tables 2(a) and 2(b) summarize the data on these 89 biotherapeutics regarding their usage in different therapeutic areas, clinical indications, and targets they bind (also see Tables S1 and S2 for more details) as of December 2022. These 89 antibody-based biotherapeutics have received 242 drug approvals for 119 unique clinical indications in 15 therapeutic areas, with oncology, hematology, and immunology accounting for most of them. Moreover, several clinical indications involve multiple therapeutic areas. For example, multiple myeloma (MM) involves immunology, hematology, and oncology. Accounting for clinical indications in multiple therapeutic areas, these 89 antibody-based biotherapeutics have been approved for 140 non-unique clinical indications. Here, we use the term non-unique for clinical indications that are being counted more than once because they fall into multiple therapeutic areas. Thus, oncology has 46 non-unique clinical indications, such as non-small cell lung cancer (NSCLC) and breast cancer. Immunology has 40 non-unique clinical indications and hematology has 15 non-unique clinical indications. The remaining 12 therapeutic areas have 39 non-unique clinical indications. One of the fastest growing therapeutic areas currently is infectious diseases, fueled by the SARS-CoV-2 pandemic, with recent emergency use authorizations (EUAs) for Evusheld (tixagevimab and cilgavimab), etesevimab with bamlanivimab, Xevudy (sotrovimab), and Regen-Cov (casirivimab and imdevimab), and formal approvals for Regkirona (regdanvimab) and Xevudy (sotrovimab) by the EC, as well as a newly approved indication for tocilizumab.86
Table 2a.
Number of clinical indications for the marketed antibody-based biotherapeutics*.
| Marketed Biotherapeutic | Number of Clinical Indications served |
|---|---|
| Pembrolizumab | 18 |
| Nivolumab | 12 |
| Adalimumab | 9 |
| Infliximab | 8 |
| Bevacizumab | 7 |
| Ipilimumab | 7 |
| Tocilizumab | 7 |
| Canakinumab | 6 |
| Certolizumab pegol | 6 |
| Rituximab | 6 |
| Ustekinumab | 6 |
| Atezolizumab | 5 |
| Dupilumab | 5 |
| Golimumab | 5 |
| Ranibizumab | 5 |
| Secukinumab | 5 |
| Brentuximab vedotin | 4 |
| Durvalumab | 4 |
| Eculizumab | 4 |
| Ixekizumab | 4 |
| Mepolizumab | 4 |
| Ramucirumab | 4 |
*This table shows the 89 marketed antibody-based biotherapeutic and how many clinical indications for which it has been approved as of December 2022. For example, pembrolizumab has been approved for most clinical indications, 18 so far. Only antibody-based biotherapeutic products approved for four or more clinical indications were included in this table. Please refer to Table S1 for full details.
The 89 marketed antibody-based biotherapeutics recognize 59 different targets. In Table 2(b), the most common target is B-lymphocyte antigen CD20 (or CD20), with six marketed antibody-based biotherapeutics, followed by tumor necrosis factor alpha (TNF-α), epidermal growth factor receptor (EGFR), and calcitonin gene-related peptide with four approvals each. Moreover, many drugs have been approved for multiple clinical indications,88 sometimes binding to the same target,89 perhaps due to promising clinical data, such as pembrolizumab or adalimumab (see both Tables 2(a) and 2(b).90–95 The same target is also involved in several different clinical indications for multiple approved antibody-based biotherapeutics. For example, programmed cell death 1 ligand 1 (PD-L1) is targeted by pembrolizumab, nivolumab, and cemiplimab, but these three drugs have been approved for 22 unique clinical indications (33 non-unique). Similarly, TNF-α is targeted by adalimumab, certolizumab pegol, golimumab, and infliximab, which serve 12 unique clinical indications (28 non-unique).
Table 2b.
Targets bound by the marketed antibody-based biotherapeutics*.
| Targets Name | UniProt ID87 | Clinical Indications | Drug Products |
|---|---|---|---|
| Programmed Cell Death Protein 1 | Q15116 | 22 | 3 |
| Tumor Necrosis Factor - Alpha | P01375 | 12 | 4 |
| Vascular Endothelial Growth Factor A | P15692 | 12 | 2 |
| B-Lymphocyte Antigen CD20 | P11836 | 9 | 6 |
| Programmed Cell Death 1 Ligand 1 | Q9NZQ7 | 8 | 3 |
| Interleukin-6 Receptor Subunit Alpha | P08887 | 6 | 2 |
| Interleukin-1 Subunit Beta | P01584 | 6 | 1 |
| Interleukin-12 Subunit Beta | P29460 | 6 | 1 |
| Interleukin-17 Subunit Alpha | Q16552 | 5 | 2 |
| Interleukin 5 | P05113 | 5 | 2 |
| Complement C5 | P01031 | 4 | 2 |
| Epidermal Growth Factor Receptor | P00533 | 4 | 4 |
| Tumor Necrosis Factor Receptor Superfamily Member 8 | P28908 | 4 | 1 |
| Proprotein Convertase Subtilisin/Kexin Type 9 | Q8NBP7 | 3 | 2 |
| Receptor Tyrosine-Protein Kinase erbB-2 | P04626 | 3 | 2 |
| Interleukin-2 Receptor Subunit Alpha | P01589 | 3 | 2 |
*This table shows the UniProt87 names and identification numbers of target recognized by the 89 marketed antibody-based biotherapeutics. This table also shows the number of clinical indications that utilize a specific target and how many marketed antibody-based biotherapeutics bind to that target. Only the targets utilized in 3 or more clinical indications were included.
Types and immunogenicity of currently marketed antibody-based biotherapeutics
An antibody-based biotherapeutic’s type depends on how it was discovered and engineered.9 All 89 marketed antibody-based biotherapeutics are one of the following four types: murine, chimeric, humanized, or human. Note that other terms often used in this context are “source” or “origin.” However, these terms fail to account for the fact that human and humanized antibody-based biotherapeutics are not “sourced” or “originated” from humans but are derived via lab-based humanization of the parental murine antibodies in most cases.96–98 Among the 89, there are 6 murine, 10 chimeric, 42 humanized, and 31 human marketed antibody-based biotherapeutics (Figure 2(c)). The first approved antibody in 1986 was of murine origin (muromonab). A murine mAb may elicit undesirable immunogenicity-related side effects due to the activation of the immune system,99 and it has been proposed that many factors, including humanization, can help avoid this immune response.100 The next product to receive approval in 1994 was a chimeric antibody (abciximab). The first humanized antibody was approved in 1997 (daclizumab), and adalimumab became the first human antibody approved in 2002. Since 2003 humanized and human antibodies have been approved at a higher rate than chimeric and murine ones. Furthermore, there is a trend toward an increasing number of approvals for human antibodies, which started around 2005 (see Table 1 and Figure 2(c)). Similarly, Figures 2(d,e) show that the percent humanness of the variable portions of both the heavy and light chains (VHs and VLs, respectively), have increased over time. On average, the VHs are a little less humanized, when compared to their closest human germlines (average humanness = 83 ± 10%) than the VLs (average humanness = 86 ± 11%). Moreover, the VHs of 53 antibody-based biotherapeutics are < 85% human, while only 34 VLs are < 85% human. As expected, the percent humanness of the marketed antibody-based biotherapeutics correlates with their types. For example, percent humanness of human antibodies ranges from 85% to 100% (average 94 ± 3%) for VLs and 87% to 100% (average 97 ± 4%) for VHs. The percent humanness of the humanized antibodies ranges from 73% to 93% (average 84 ± 4%) for VLs and 71% to 88% (average 80 ± 4%) for VHs. Percent humanness of the chimeric and murine antibodies ranges from 59% to 80% (average 68 ± 6%) for VLs and 57% to 80% (average 70 ± 5%) for VHs.
The administration of an antibody-based biotherapeutic can elicit an anti-drug antibody (ADA) response by the immune system, which has important potential consequences for PK, efficacy, and drug safety.101 Consequently, it is a regulatory requirement to measure the presence of ADAs during clinical trials. However, each ADA assay is specific to a given antibody-based biotherapeutic product.102 Therefore, it is not possible to directly compare ADA incidences from one product to another. Keeping this limitation in mind, the reported ADAs of 88 of the 89 marketed antibody-based biotherapeutics were obtained from the package inserts or the pharmacology sections in the reviews of the BLAs (Table S1). No ADA information was available for gemtuzumab ozogamicin in the package insert,103 or pharmacological review section of the drug approval package.104 Figure 2(f) shows the trends of percent ADAs over time. Antibody-based biotherapeutics with lower incidence of ADAs have been approved more frequently in recent years. In 1998, trastuzumab became the first approved antibody-based biotherapeutic with reported ADA incidence below 1%. Ten marketed antibody-based biotherapeutics report ADA incidences above 20% in their package inserts (Table S1). The average reported incidence of ADA across all antibody-based biotherapeutics in this study is 11.3 ± 8.0%; it ranges from 0% for bezlotoxumab to 62% for alemtuzumab. It is important to note that the recommendations for ADA assays have also changed over the years, which hampers our ability to compare overall incidence between older products versus those that follow the 2017 EMA and 2019 FDA guidance.105,106
The data shown in Figures 2(c–f) prompted us to pose the following question: Does % Humanness of the variable regions of marketed antibody-based biotherapeutics directly correlate with their reported % ADAs ? Plots of percent humanness of their VH and VL regions with respect to the incidence of %ADAs reported in the package inserts are shown in Figure 2(g). Although a weak downward trend suggesting a decline in %ADAs with increased % Humanness is observed for both VHs and VLs, the linear correlation coefficients (r2) values for both VHs and VLs are 0.02 and the p-values are 0.16 (VLs) and 0.17 (VHs). Therefore, the correlation between % ADAs and % Humanness is statistically insignificant for the antibody-based biotherapeutics in this dataset. Figure 2(h) explores the same question from the perspective of antibody types. The boxplots shown in this figure suggest that the median values of % ADAs do not decrease significantly among murine, chimeric, humanized, or human antibodies, even though a weak downward trend is observed again. Furthermore, Table S3 shows the average incidence of ADAs among murine, chimeric, humanized, and human antibody-based biotherapeutics. On average, the murine antibodies show an 18% incidence of ADAs along with a wider variation in the ADA incidence than those observed for chimeric, humanized, or human antibodies. The corresponding averages for the chimeric, humanized, and human antibodies are 5%, 9%, and 5%, respectively (see Table S3). Taken together, these observations imply that the immunogenicity of marketed antibody-based biotherapeutics does not depend solely on their humanness.
Generation of immune response among patients receiving immunotherapy is a very complex and multifactorial process.107,108 In addition to the type of the antibody, other factors such as molecular sequence features (e.g., presence or absence of T- and B-cell immune epitopes, stability/aggregation of the molecule under physiological conditions), manufacturing processes, formulation, aggregates already present at the time of drug administration, route of administration, product presentation, as well as genetic and disease history of the patients including presence of preexisting antibodies, can play significant roles.26,109 Furthermore, the real-life experience on immunogenicity of a given antibody-based biotherapeutic product, outside of the clinical trial setting, may be different than the % ADAs reported in the package inserts or BLA reviews.
The absence of correlation between the incidence of ADAs and the humanness of antibody-based biotherapeutics, however, does not imply that humanization of antibody drug candidates discovered using murine and other species is unnecessary. On the contrary, this report shows that technological advances in antibody generation, humanization, and manufacturing along with improved understanding of causes that underpin undesired immunogenicity have resulted in the increased approval of antibody-based biotherapeutics with low incidence of percent ADAs in recent years (see Figure 2(f)). However, as described above, humanization does not fully mitigate the risk of unwanted immunogenicity.
In addition to the humanness of antibody-based biotherapeutics, recent research has also explored the role of formulation in unwanted immunogenicity.110,111 For example, a citrate-free formulation of adalimumab has fewer reports of injection site pain (ISP) and adverse immune reactions.112 This is consistent with reports that use citrate buffer in antibody formulations can lead to stinging and ISPs.113 In general, it is preferable to include fewer inactive ingredients in antibody formulations to lower ISPs.114–118
In addition to the incidence of ADAs, biological drug safety involves several factors, such as target-mediated toxicity, dosing, and formulation.119–121 Target biology, target-mediated toxicity and toxicity induced by dosing levels are out of scope for this analysis because such information is often absent from package inserts and publicly available regulatory submission documents. Nonetheless, package inserts often do contain additional information on the product presentation of antibody-based biotherapeutics, routes of administration, and dosing. These are studied next.
Multiple drug product presentations and routes of administration options for antibody-based biotherapeutics
Antibody-based drug products are packaged as vials, pre-filled syringes or pens containing their liquid formulations, or lyophilized powders for reconstitution of drug product as a liquid injectable at the time of administration (Table 1). Decisions on the product presentation often involve consideration of many factors. Molecular integrity of the API (conformational stability, aggregation propensity or lack thereof, and impact of chemical degradation such as oxidation, deamidation, and isomerization) in a liquid formulation determined by stability studies during early development phases plays a crucial role.122
As of June 2020, there were 96 product presentations for the 89 antibody-based biotherapeutics studied here. Figure 3(a) shows the evolution of the product presentations for the marketed antibody-based biotherapeutics over time. Vial has been the most common drug product presentation for the marketed antibody-based biotherapeutics over the years, accounting for 53 (55.2%) of the 96 product presentations. Of the remainder, 22 (22.9%) products are presented as lyophilized powders and 21 (21.9%) are presented as pre-filled syringes or pens (Table S4). Six antibodies, namely, omalizumab, secukinumab, ranibizumab, ustekinumab, golimumab, and mepolizumab, are marketed in multiple product presentations as described below. Omalizumab and secukinumab are available as lyophilized powders and as pre-filled syringes. Ranbizumab, golimumab, and ustekinumab are available as pre-filled syringes and as vials. Mepolizumab is the only product available as a lyophilized powder, as a pre-filled syringe, and as a vial. Pembrolizumab was originally approved in a lyophilized form in 2014,123 and a vial product presentation was added in 2015.124 As of 2020, pembrolizumab is only available as a vial.125
Figure 3.
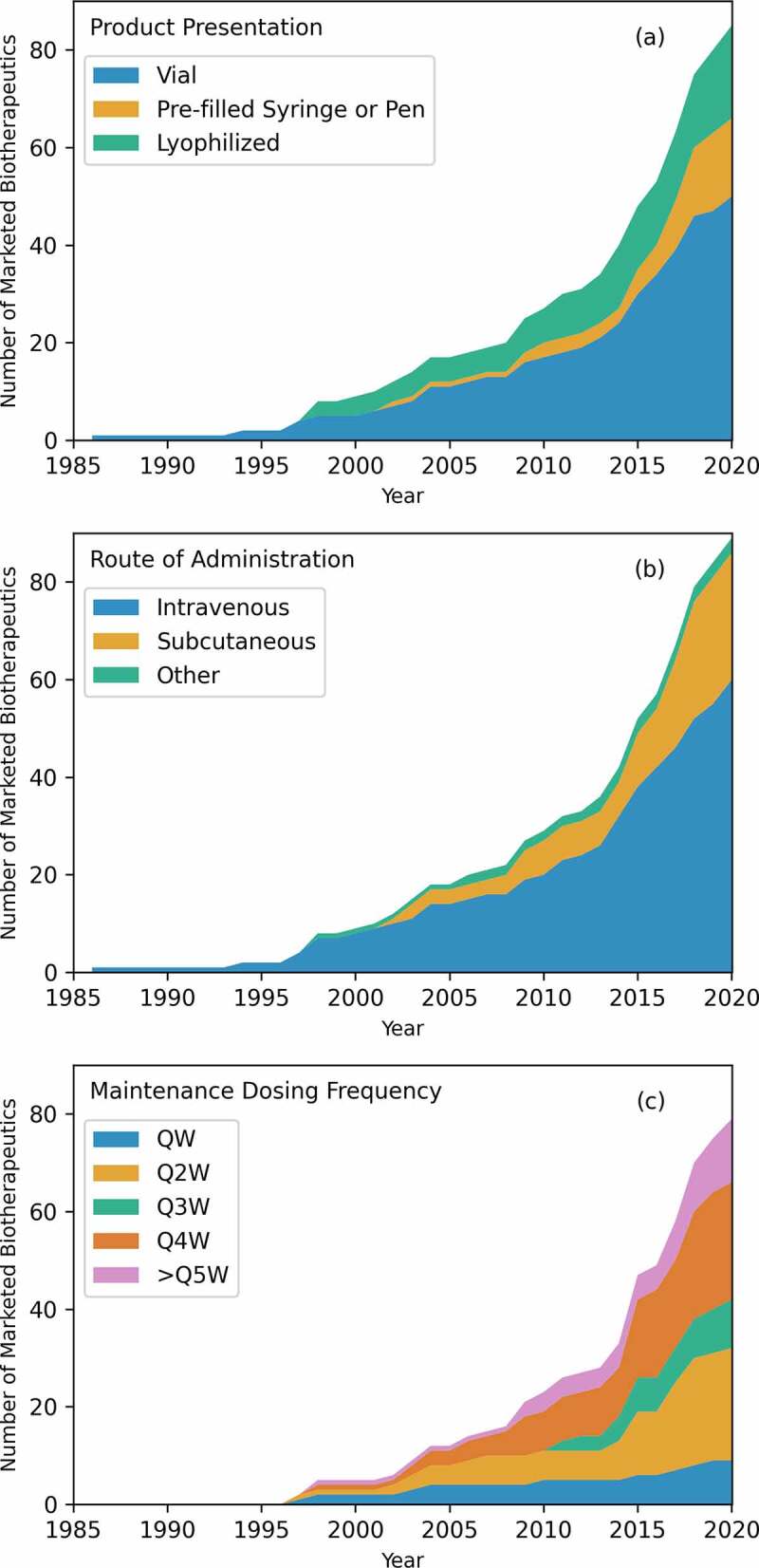
Product information. (a) Product presentation of marketed antibody-based biotherapeutics over time. Current trends show a preference of lyophilized and pre-filled syringe or pen over vial. (b) Route of administration of marketed antibody-based biotherapeutics over time. Intravenous is the most used route of administration, but recently, subcutaneous has seen a similar number of approvals as intravenous. Other routes of administration are intradermal, intramuscular, and intravitreal. (c) Regular maintenance dose frequency for marketed antibody-based biotherapeutics over time. QW stands for once weekly, Q2W stands for once every two weeks, and so on. There are 79 regular dosing regiments. In general, Q2W and Q4W are the most used maintenance dosing regimens.
Routes of administration for antibody-based drug products include intradermal, intramuscular, intravenous (i.v.), intravitreal, and subcutaneous (s.c.).126–129 Of the 89 antibody-based biotherapeutics, approximately two-thirds (59, 66.2%) are i.v. administered and another 27 (30.4%) are s.c. administered. Of the remaining three, one each has been developed for the intradermal (racotumomab), intramuscular (palivizumab), and intravitreal (ranibizumab) routes of administration. Figure 3(b) shows the trends in routes of administration over time for the 89 marketed antibody-based biotherapeutics. The i.v. route of administration has the highest number of approvals over years, but the share of s.c. administered products has also increased from 19% (8 of 42) before 2015 to 40% (19 of 47) from 2015 to 2020. Coincident with this increase in the development of s.c. administered antibody-based biotherapeutics is the use of hyaluronidase PH20 enzyme (rHuPH20, ENHANZE) to help increase the volume available for these injections.130 Four of the 89 marketed antibody-based biotherapeutics (daratumumab, trastuzumab, pertuzumab, and rituximab) have used this technology so far.
Looking at product presentations and routes of administration together, 16 of the 22 lyophilized drug products are for i.v. administration and the remaining six are for s.c. administration. In contrast, 20 of the 21 pre-filled syringes containing high concentration liquid formulations (HCLF) are for s.c. administration and the remaining one is for intravitreal administration. Similarly, 43 of the 52 vial product presentations are i.v. administered, 6 s.c., and 1 each are administered intradermally, intramuscularly, and intravitreally.
Several antibody-based biotherapeutic products available previously only for i.v. administrations are now being marketed in their s.c. form as well. So far, 11 antibodies have been approved for both i.v. and s.c. routes of administration. In the case of s.c. administered antibodies, four are co-administered with PH20 as mentioned previously. Trastuzumab was originally approved by the FDA as a lyophilized powder for i.v. infusions to treat metastatic breast cancer in 1998, then by the EC as an s.c. injection for the same clinical indication in 2013, and by the FDA again in 2019 as an s.c. injection co-administered with PH20. These two routes of administration of trastuzumab have similar efficacy and tolerability, which leads to s.c. administration being preferred by patients.131 The remaining three s.c. administered antibodies approved along with PH20 co-administration are rituximab in 2017 and daratumumab and pertuzumab in 2020. Of the remaining 7 of the 11 antibodies approved for both i.v. and s.c. administrations, belimumab was originally approved in 2011 as a lyophilized powder for i.v. infusions to treat systemic lupus erythematosus then later as an s.c. injection in 2017. Daclizumab was originally approved for i.v. infusions to prevent kidney transplant rejection in 1997. In 2009, Daclizumab was withdrawn due to limited usage as newer products with a less burdensome dosing schedule were available in the market,132 but then it was reapproved for s.c. administration to treat multiple sclerosis in 2016, a change in both routes of administration and clinical indication. Similarly, ofatumumab was approved as an i.v. injection to treat chronic lymphocytic leukemia and as s.c. administration to treat multiple sclerosis in 2009. Interestingly, ofatumumab is marketed as separate drug products with different brand names (Arzerra and Kesimpta) for each of the two routes. Golimumab was approved for s.c. administration in 2009 to treat rheumatoid arthritis (RA), psoriatic arthritis, and ankylosing spondylitis and then for i.v. route of administration in 2013 to also treat RA. Tocilizumab was originally approved for both i.v. and s.c. routes of administration to treat arthritis (RA, polyarticular juvenile idiopathic arthritis, systemic juvenile idiopathic arthritis) in 2009. Ustekinumab was originally approved as an s.c. injection to treat plaque psoriasis in 2009 and as an i.v. infusion to treat Crohn’s disease in 2016. Vedolizumab was approved for i.v. infusion in 2014 by the FDA and then as an s.c. injection in 2020 by the EC.
There are also clear preferences for routes of administration in the different therapeutic areas. For example, there are 46 clinical indications for oncology and all but one of the 38 products for these clinical indications are i.v. administered (97%). The only exception is racotumomab, which is an intradermally administered product to treat NSCLC. Like oncology, hematology has 15 clinical indications, but all except two of the 21 products are i.v. administered (90%). The exceptions are emicizumab and mepolizumab, both s.c. administered. In comparison to oncology and hematology, there are 40 clinical indications for immunology that are served by 38 drug products. Of these 38 drug products, only 20 (53%) are i.v. administered (Tables S1 and S2).
Maintenance dosing frequency
Maintenance dosing frequency refers to how often a maintenance dose is administered to the patient. This is a major consideration from the perspectives of patient convenience and healthcare costs. In the literature, the dosing frequencies are normally represented as QW, Q2W, and so on (from Latin “quaque”; English translation, “each”) where the number represents how many weeks between doses (QW is used instead of Q1W).133 Seventy of the 89 marketed antibody-based biotherapeutics require a regular maintenance dose schedule and 6 of them offer more than one option for dosing regimens. Daratumumab, for example, offers dosing options for QW, Q2W, Q3W, and Q4W. Thus, in total, there are 79 dosing regimens for these 70 antibody-based biotherapeutics. Nine of these 79 dosing regimens are QW (11%), 23 are Q2W (29%), 10 are Q3W (14%), 24 are Q4W (30%), and 13 are ≥Q5W (16%) (Supplementary Figure S1). Figure 3(c) shows the dose frequency over time divided into the following categories: once weekly (QW), once every 2 weeks (Q2W), once every 3 weeks (Q3W), once every 4 weeks (Q4W), and once every 5 or more weeks (≥Q5W). Q2W and Q4W have been the most common dosing regimens for antibody-based biotherapeutics over the years.
There is a relationship between dosing frequency, nature of diseases, and routes of administration. Comparisons between regular dosing regimens for i.v. and s.c. routes of administration and therapeutic areas are shown in Table 3. For example, most s.c. administered products (21 of 31, 68%) developed to treat chronic diseases are prescribed to be dosed Q4W or more. This less frequent dosing regimen is clearly advantageous from the perspective of patient convenience (Table S1).134 However, even with such advantages, the patients and clinicians may not always prefer s.c. over i.v. administration and the option used in the clinical setting may depend on preferences of patient groups and clinicians.135,136
Table 3.
Number of antibody-based biotherapeutic products available for different regular maintenance dose regimens with respect to routes of administration and therapeutic areas*.
| Therapeutic Area | Intravenously Administered |
Subcutaneously Administered |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| QW | Q2W | Q3W | Q4W | ≥Q5W | QW | Q2W | Q3W | Q4W | ≥Q5W | |
| Autoimmunity | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cardiovascular Diseases | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 2 | 0 |
| Cardiology | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 |
| Drug Overdose | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hematology | 3 | 5 | 3 | 4 | 1 | 1 | 0 | 0 | 1 | 0 |
| Hereditary Diseases | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Immunology | 2 | 5 | 2 | 6 | 4 | 1 | 6 | 0 | 5 | 5 |
| Infectious Diseases | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Inflammation | 1 | 2 | 1 | 2 | 1 | 0 | 1 | 0 | 1 | 0 |
| Neurology | 0 | 1 | 0 | 2 | 0 | 0 | 0 | 0 | 3 | 1 |
| Oncology | 6 | 11 | 9 | 4 | 1 | 0 | 0 | 0 | 0 | 0 |
| Ophthalmology | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Osteology | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 |
| Pulmonary | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rheumatology | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
*A comparison of maintenance dose regiments for different therapeutic areas, separated by route of administration. Note that Hematology has 16 i.v. maintenance dosing options while only 2 s.c. options. Oncology has 31 i.v. dosing options and no s.c. options. Also, QW and Q3W are rare for s.c. administered products.
Pharmacokinetics of marketed antibody-based biotherapeutics.
PK data are available consistently for most of the marketed antibody-based biotherapeutics. The data include clearance (the volume of blood cleared of drug per unit time; 80 out of 89, 90%),137 elimination half-life (the time required for plasma/blood concentration to decrease by 50% (after pseudo-equilibrium of distribution is reached; 86 out of 89, 97%)),138,139 and volume of distribution (83 of the 89, 93%) (Table S1).140 Table S1 also contains data on bioavailability (fraction of the dose that reaches systemic circulation as an intact drug),141 which is available for 26 of the 27 (96%) s.c. administered antibody-based biotherapeutics. Bioavailability values for i.v. administered antibody-based biotherapeutics were not included in the analysis because they are assumed to be 100%.141 For volume of distribution, steady state values were used when available and if volume values were given representing different body compartments, these values were added together.
Six other PK parameters also had data, but these were associated with clinical data in the reviews of BLAs, and not included in the package inserts. The parameters are the maximum concentration (Cmax, 89%), time to reach maximum concentration (Tmax, 63% for all; 100% for s.c. administered mAbs), area under the concentration-vs-time curve (AUC, 74%), trough concentration (Ctrough, 66%), time to reach steady state (Tss, 74%), and elimination linearity (89%, of which 68% have linear clearance). Note that the PK values reported in the package inserts and reviews of BLAs are derived from PK data collected from many subjects or patients (sometimes from different patient cohorts) and in general show large biological variability. Furthermore, it is not always clear whether the PK parameters in the package inserts are reported as geometric means, arithmetic means, or median values of the patient PK data. Therefore, caution should be used when analyzing and interpreting PK data across different marketed antibody-based biotherapeutics. Moreover, it may be worth further harmonizing reporting of clinical PK data (and units) to allow better comparisons in the future.
Figures 4(a–d) show time-series analyses for the four common PK parameters mentioned above. Only full-length antibodies were included in the analysis because small molecular size can affect distribution and involve additional renal elimination of a drug product.142 Figure 4(a) shows trends for elimination half-life for 82 full-length antibodies over time. Their half-life ranges from 17 hours (sacituzumab goitecan) to 50 days (ravulizumab) with a geometric mean of 14.5 days (average = 17.8 ± 9.2 days; Median = 18.0 days); and most marketed antibody-based biotherapeutics are eliminated between 15 and 25 days. Furthermore, the elimination half-life of a drug can be calculated using Equation (1) as shown in the data sources and methods section. This equation connects the elimination half-life with the volume of distribution and clearance, which was derived assuming a first-order exponential decay.143 Table 4 shows that the calculated elimination half-life values are usually lower than those reported in the package inserts and BLA reviews even though there is still a significant correlation between the calculated and the reported elimination half-lives for 45 full-length IgG antibodies, excluding the ADCs, that show linear PK, i.e., no target mediated effects (r = 0.84, p-value < 0.001, see table S5). This discrepancy between the calculated and reported elimination half-lives can arise from several sources besides the large scatter in the data obtained from individual patients. Both clearance and volume of distribution are dependent on many factors, such as physical size and molecular characteristics of the antibody, expression of the target and target internalization rate, nature of the target (intracellular, membrane, extracellular), patient characteristics (e.g., body weight, disease history, age, gender, and so on),144 and route of administration.138–147 Clearance varies greatly for all antibody-based biotherapeutics, which is reflected in a wide range of elimination half-lives for reasons mentioned above.148–150
Figure 4.
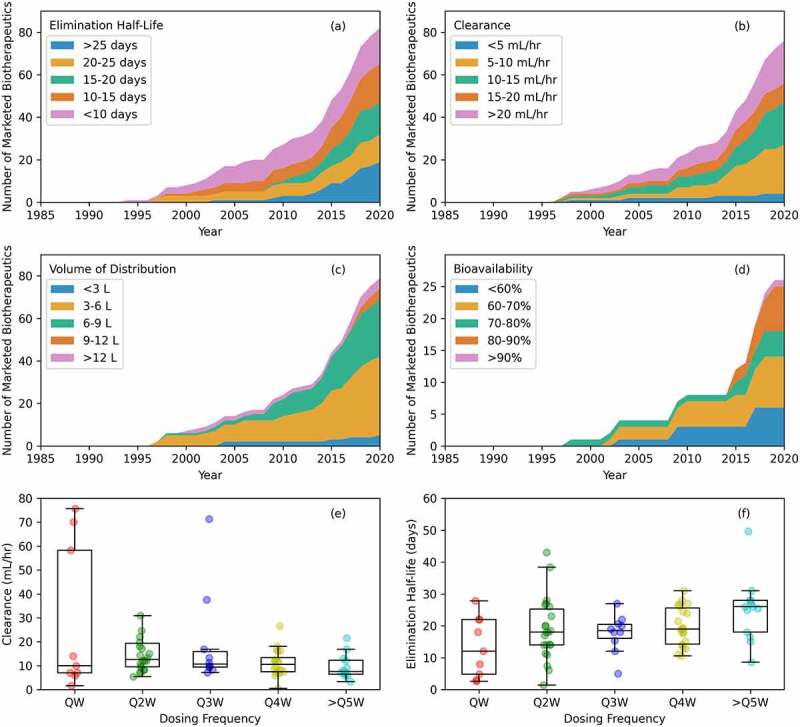
Pharmacokinetic (PK) data over time. (a) Elimination half-life. This varies for all antibody-based biotherapeutics, but most marketed products are eliminated between 15 and 25 days. (b) Clearance. This varies too, but most antibody-based biotherapeutics have a clearance between 5 and 15 mL/hour. (c) Volume of distribution. Values fall mainly between 3 and 9 liters with values between 3 and 6 liters being the most common. (d) Bioavailability. Recent marketed drugs with a subcutaneous route of administration commonly have 90% or more bioavailability. (e) Clearance versus dosing frequency. In general, a higher clearance should result in more frequent dosing, but the median values do not reflect this. (f) Elimination half-life versus dosing frequency. A trend of longer elimination half-lives enabling less frequent dosing is observed.
Table 4.
Pharmacokinetic data for 89 marketed antibody-based biotherapeutics*.
| Subset | Terminal Half-Life (days) | Predicted Half-life (days) | Clearance (mL/hour) | Volume of Distribution (L) | Bioavailability (%) | Variable Regions pIFv3D |
|---|---|---|---|---|---|---|
| All Data (89) | 11.3 (16.8 ± 9.6; 0.0–49.6) | 8.6 (17.5 ± 34,0.0–307.6) | 19.6 (424 ± 2803,0–25000) | 5.8 (6.4 ± 3.1,2.5–21.4) | 69 (70 ± 13,49–100) | 7.7 (7.8 ± 1.3,4.6–9.5) |
| Full-Length (83) | 14.5 (17.8 ± 9.2,0.7–49.6) | 11.4 (18.5 ± 34.9,0.4–307.6) | 14.8 (29.0 ± 58.1,0.5–350.0) | 5.8 (6.4 ± 3.2,2.5–21.4) | 69 (70 ± 13,49–100) | 7.8 (7.89 ± 1.2,4.7 9.5) |
| IgG1 (59) | 14.6 (17.7 ± 8.5,0.7–43) | 11.9 (19.8 ± 40.4,0.6–307.6) | 13.8 (20.7 ± 23.8,0.5–140) | 5.7 (6.2 ± 2.9,2.5–14.9) | 68 (69 ± 14,49–100) | 7.9 (8 ± 1.2,4.7–9.5) |
| IgG2 (12) | 15.8 (19 ± 12.5,7.5–49.6) | 9.5 (14 ± 13.5,2.1–46) | 15.4 (26.4 ± 35.5,3.3–125) | 4.9 (5.2 ± 1.8,3.3–8.9) | 69 (70 ± 10,55–82) | 7.5 (7.7 ± 1.2,5–9.5) |
| IgG4 (14) | 14.7 (17.9 ± 8.6,0.7–43.0) | 11.6 (19.7 ± 41.8,0.6–307.6) | 13.0 (20.4 ± 24.7,0.5–140.0) | 5.5 (6.1 ± 2.7,2.5–14.9) | 69 (70 ± 13,50–100) | 8.0 (8.1 ± 1.2,4.7–9.5) |
| IgG1κ (55) | 14.7 (17.9 ± 8.6,0.7–43.0) | 11.6 (19.7 ± 41.8,0.6–307.6) | 13.8 (21.1 ± 24.6,0.5–140.0) | 5.5 (6.1 ± 2.7,2.5–14.9) | 69 (70 ± 13,50–100) | 8.0 (8.1 ± 1.2,4.7–9.5) |
| IgG1λ (4) | 14.4 (16.0 ± 6.0,6.1–22.0) | 16.0 (20.1 ± 12.7,5.5–40.4) | 13.5 (15.5 ± 7.7,7.0–24.6) | 7.5 (8.3 ± 3.6,4.7–13.5) | 49 (49 ± 0,49–49) | 7.4 (7.4 ± 0.6,6.5–8.3) |
| IgG2κ (9) | 16.3 (19.9 ± 13.6,7.5–49.6) | 9.7 (14.8 ± 14.1,2.1–46.0) | 15.9 (28.2 ± 37.3,3.3–125.0) | 5.3 (5.6 ± 1.8,3.8–8.9) | 65 (66 ± 10,55–81) | 7.5 (7.6 ± 1.4,5.0–9.5) |
| IgG2λ (3) | 14.7 (16.7 ± 8.4,8.0–28.0) | 7.9 (7.9 ± 0,7.9–7.9) | 12.0 (12.0 ± 0.0,12.0–12.0) | 3.6 (3.6 ± 0.3,3.3–3.9) | 77 (77 ± 5,72–82) | 7.7 (7.7 ± 0.5,7.1–8.3) |
| IgG4κ (14) | 13.9 (19.1 ± 12.3,1.4–49.6) | 11.8 (18.0 ± 12.0,0.4–46.0) | 17.6 (60.7 ± 117.6,3.3–350.0) | 7.2 (8.0 ± 4.3,4.8–21.4) | 73 (74 ± 10,64–87) | 7.4 (7.5 ± 1.1,5.5–9.0) |
| IgG1κ Chimeric (9) | 11.6 (14.0 ± 8.2,4.8–28.0) | 8.6 (11.6 ± 7.8,2.1–24.6) | 12.5 (20.9 ± 22.0,1.7–71.3) | 5.1 (5.6 ± 2.3,2.5–8.6) | NA | 8.3 (8.3 ± 0.7,6.8–9.3) |
| IgG1κ Humanized (25) | 15.9 (19.3 ± 8.2,0.7–38.4) | 11.1 (25.5 ± 59.4,0.6–307.6) | 12.7 (23.5 ± 31.3,0.5–140.0) | 4.9 (5.4 ± 2.9,2.9–14.1) | 68 (69 ± 13,50–89) | 8.2 (8.2 ± 1.1,5.7–9.5) |
| IgG1κ Human (19) | 16.7 (18.6 ± 8.0,2.9–43.0) | 14.2 (15.6 ± 6.2,5.0–30.2) | 13.6 (16.1 ± 11.8,7.1–58.3) | 6.8 (7.1 ± 2.4,3.2–14.9) | 69 (71 ± 14,53–100) | 7.5 (7.7 ± 1.4,4.7–9.4) |
| IgG1λ Human (4) | 14.4 (16.0 ± 6.0,6.1–22.0) | 16.0 (20.1 ± 12.7,5.5–40.4) | 13.5 (15.5 ± 7.7,7.0–24.6) | 7.5 (8.3 ± 3.6,4.7–13.5) | 49 (49 ± 0,49–49) | 7.4 (7.4 ± 0.6,6.5–8.3) |
| IgG2κ Humanized (5) | 18.8 (23.1 ± 15.3,10.6–49.6) | 14.4 (20.0 ± 15.5,4.3–46.0) | 10.61 (13.9 ± 9.0,3.3–26.6) | 5.3 (5.4 ± 1.4,3.9–7.7) | 73 (74 ± 7,66–81) | 8.5 (8.5 ± 0.7,7.6–9.5) |
| IgG2κ Human (3) | 12.8 (14.6 ± 7.8,7.5–25.4) | 5.6 (6.5 ± 2.7,2.1–9.3) | 31.1 (52.0 ± 51.6,14.3–125.0) | 5.4 (5.8 ± 2.2,3.8–8.9) | 58 (58 ± 3,55–61) | 6.0 (6.1 ± 1.2,5.0–7.8) |
| IgG4κ Humanized (11) | 12.7 (18.4 ± 13.1,1.4–49.6) | 10.4 (17.3 ± 13.0,0.4–46.0) | 21.3 (74.7 ± 129.2,3.3–350.0) | 7.6 (8.5 ± 4.7,4.8–21.4) | 79 (79 ± 8,71–87) | 7.2 (7.3 ± 1.2,5.5–9.0) |
| IgG4κ Human (3) | 22.5 (22.9 ± 3.9,19.0–26.7) | 19.3 (20.5 ± 6.4,11.5–25.6) | 8.8 (9.4 ± 3.2,5.4–13.3) | 5.9 (6.0 ± 1.4,4.8–8.0) | 64 (64 ± 0,64–64) | 8.1 (8.1 ± 0.3,7.9–8.4) |
| IgG1 i.v. (41) | 12.9 (16.2 ± 8.2,0.7–38.4) | 10.3 (13.6 ± 8.3,0.6–40.4) | 13.8 (21.6 ± 26.5,1.7–140.0) | 5.3 (5.7 ± 2.5,2.5–14.1) | NA | 8.00 (8.1 ± 1.2,4.7–9.5) |
| IgG1 s.c. (16) | 19.7 (21.2 ± 8.1,8.0–43.0) | 13.8 (16.3 ± 7.9,3.2–30.2) | 14.1 (17.5 ± 15.3,7.0–70.0) | 6.8 (7.5 ± 3.4,2.9–14.9) | 67 (69 ± 14,49–100) | 7.8 (7.9 ± 1.2,5.2–9.4) |
| IgG2 i.v. (6) | 12.9 (17.4 ± 16.2,7.5–49.6) | 14.6 (19.0 ± 15.6,9.3–46.0) | 10.5 (12.8 ± 6.7,3.3–22.0) | 5.3 (5.5 ± 1.3,4.3–7.7) | NA | 7.5 (7.6 ± 1.0,5.6–8.6) |
| IgG2 s.c. (6) | 18.7 (20.4 ± 8.0,10.9–31.0) | 6.7 (10.0 ± 9.9,2.0–29.4) | 20.9 (37.3 ± 44.4,5.9–125.0) | 4.7 (5.0 ± 2.0,3.3–8.9) | 69 (70 ± 10,55–82) | 7.5 (7.7 ± 1.5,5.0–9.5) |
| IgG4 i.v. (10) | 12.2 (18.0 ± 13.3,1.4–49.6) | 9.1 (15.4 ± 12.5,0.4–46.0) | 22.9 (81.0 ± 133.9,3.3–350.0) | 7.2 (8.1 ± 4.9,4.8–21.4) | NA | 7.4 (7.5 ± 1.1,5.5–9.0) |
| IgG4 s.c. (4) | 21.4 (22.6 ± 6.8,13.0–27.8) | 23.0 (24.4 ± 7.4,12.6–32.9) | 9.2 (9.9 ± 4.0,5.4–16.3) | 7.3 (7.7 ± 2.4,4.8–11.4) | 73 (74 ± 10,64–87) | 7.2 (7.3 ± 1.0,5.8–8.4) |
| ADCs & Pegylated Fab (8) | 4.0 (6.3 ± 5.1,0.7–14.0) | 3.4 (5.0 ± 3.8,0.6–10.9) | 63.8 (101.1 ± 108.2,17.0–350.0) | 7.5 (9.3 ± 5.9,3.0–21.4) | 80 (80 ± 0,80–80) | 7.6 (7.7 ± 1.3,5.5–9.5) |
| API Conc.<10 mg/mL (14) | 8.0 (10.9 ± 6.9,1.3–22.0) | 7.4 (11.3 ± 10.8,1.8–40.4) | 29.8 (63.3 ± 98.4,7.0–350.0) | 7.6 (8.7 ± 4.9,3.6–21.4) | NA | 7.7 (7.8 ± 1.3,5.2–9.3) |
| API Conc. 10–50 mg/mL (36) | 14.7 (17.8 ± 9.4,0.7–49.6) | 11.7 (14.8 ± 9.2,0.6–46.0) | 13.3 (19.0 ± 24.3,3.3–140.0) | 5.3 (5.8 ± 2.6,2.9–14.1) | 80 (82 ± 18,58–100) | 7.8 (7.9 ± 1.3,4.7–9.5) |
| API Conc. 50–100 mg/mL (18) | 18.5 (20.2 ± 7.1,4.8–28.1) | 14.3 (31.9 ± 69.3,2.1–307.6) | 11.8 (17.1 ± 15.4,0.5–70) | 5.7 (6.3 ± 2.9,2.5–13.5) | 67 (68 ± 13,49–89) | 7.8 (7.9 ± 0.9,5.7–9.4) |
| API Conc.>100 mg/mL (21) | 16.3 (19.8 ± 9.5,1.4–43.0) | 12.1 (29.9 ± 64.4,0.4–307.6) | 14.3 (38.0 ± 76.1,0.5–346.5) | 6.0 (6.7 ± 3.2,2.5–14.9) | 65 (66 ± 11,49–85) | 7.7 (7.8 ± 1.2,5.0–9.5) |
*This table is divided into columns for the four pharmacokinetic values analyzed in this paper. In the first column named as subset, the number in parentheses is how many of the 89 marketed antibody-based biotherapeutic fit into the category. Only subsets with at least 3 antibody-based biotherapeutics were included. For example, IgG4 lambda is not included because none of the 89 marketed antibody-based biotherapeutics are of this isotype combination. In columns 2-7, the first number is the geometric mean, followed by the arithmetic mean, standard deviation, and range in the parentheses. Note that bioavailabity data is available only for subcutaneous products. NA stands for not available. Predicted half-life was calculated from clearance and volume of distribution (see Equation 1) to compare against the observed half-life. The longest half-life is seen in the IgG4κ Human subset and the lowest is in the IgG1κ Chimeric subset. Three subsets have a predicted elimination half-life value greater than its observed elimination half-life value. The slowest clearance is seen in the s.c. administered IgG4 subset and fastest is the IgG2κ Human subset. The lowest volume of distribution is the IgG2λ subset and the highest is IgG1λ. For bioavailability, the highest value is for humanized IgG1κ, and the lowest value is for human IgG1λ.
Figure 4(b) shows clearance trends for 76 full-length antibodies over time. The clearance values range from 0.46 mL/hour (palivizumab) to 350 mL/hour (gemtuzumab ozogamicin) and the geometric mean is 14.8 mL/hour (average = 29.0 ± 58.1 mL/hour; median = 12.1 mL/hour). Despite the enormous range, 73% of the marketed antibody-based biotherapeutics have a clearance of <20 mL/hour and 62% have clearance of <15 mL/hour (Figure 4(b)). All four products with a PH20 option show a decrease in clearance compared to their original i.v. approvals. Clearance for daratumumab decreases from 7.1 mL/hour to 6.8 mL/hour; from 10 mL/hour to 8.3 mL/hour for pertuzumab; 14 mL/hour to 8 mL/hour for rituximab; and from 5.9 mL/hour to 4.6 mL/hour for trastuzumab. This may be partly attributed to slower absorption and a later Tmax for the s.c. administered products (see Table S1). Note that these observations do not separate the impact of ‘use of PH20’ from the ‘change in route of administration’ on the clearance of these products.
Figure 4(c) shows volume of distribution trends for 79 antibody-based biotherapeutics. The volume of distribution ranges from 2.5 L (cetuximab) to 21.4 L (gemtuzumab ozogamicin) with an average of 6.4 ± 3.2 L (5.6 L median, 5.7 L geometric mean). Volume of distribution for 64 of the 79 (81%) antibodies falls between 3 and 9 L and for 38 of them (48%) between 3 and 6 L. This is one of the few trends for antibody-based biotherapeutics that has not changed much, and 3–6 L has been the most common volume of distribution over the years. The main reason is that antibody-based biotherapeutics are mainly confined to the volume of plasma (Vc ~45 mL/kg or ~3 L for a 70 kg patient),151,152 and the interstitial tissue space, as their high molecular weight restricts their uptake by tissues and cells.153
Figure 4(d) shows bioavailability trends for 26 s.c. administered products over time. The average bioavailability for such products approved before 2015 was 60 ± 6% (50–70%) and from 2015 to June 2020 it has increased to 74 ± 13% (49–100%). Therefore, early s.c. products had lower bioavailability and it was not until 2018 that an s.c. administered antibody-based biotherapeutic product had a bioavailability above 90% (burosumab, 100%). Co-administration of PH20 with the formulations of the s.c. administered antibody-based biotherapeutics was expected to increase their bioavailability.154 However, the bioavailability of the four mAb products co-administered with PH20 (daratumumab, pertuzumab, rituximab, and trastuzumab) ranges from 64% to 77% (see Table S1).
The PK of antibody-based biotherapeutics is important to determine appropriate dosing frequencies.155 We studied this relationship by plotting clearance and elimination half-lives with respect to the dosing frequencies. Figures 4(e–f) show boxplots of dosing frequencies versus clearance and versus elimination half-life. There is a slight trend that higher clearance, and therefore shorter elimination half-lives, can lead to shorter dosing intervals, e.g., QW dosing, but in general the data did not reveal a clear correlation, possibly due to the large scatter. However, in general longer elimination half-life values can support less frequent dosing regimen. QW dosing has a geometric mean for elimination half-life of 9.8 days, Q2W of 15.8 days, Q3W of 16.4 days, Q4W of 18.2 days, and ≥Q5W of 22.8 days (Table S6).
The PK of antibody-based biotherapeutics can also vary with their routes of administration. Table 4 compares PK data for s.c. and i.v. routes of administrations (also see Table S7). For the antibody-based biotherapeutics s.c. administered, the average bioavailability was 71 ± 12% and average time to maximum concentration (Tmax) was 5.7 ± 2.4 days. The geometric means of elimination half-life, clearance, and volume of distribution for s.c. administered antibodies were 19.7 days (average = 21.2 ± 8.0 days), 14.2 mL/hour (average = 20.4 ± 25.3 mL/hour), and 6.3 L (average = 7.0 ± 3.2 L), respectively. In comparison to s.c. route, i.v. administered biologics have shorter average elimination half-life (geometric mean = 12.5 days; average = 16.1 ± 9.4 days), faster clearance (geometric mean = 15.2 mL/hour; average = 33.1 ± 68.5 mL/hour) and lower volume of distribution (geometric mean = 5.6 L; average = 6.2 ± 3.2 L). A potential explanation for the longer half-life of s.c. administered antibodies is that their overall clearance is partly limited by a slow rate of absorption.
In Table 4, the PK data is further broken down into several categories depending on the light and heavy chain isotypes, antibody types (chimeric, humanized, or human), and routes of administration. While a large scatter in the data precludes definitive conclusions, a few trends become apparent if one focuses on geometric mean values. For example, antibody-based biotherapeutics with IgG4 heavy chains for all routes of administration have greater volumes of distribution (geometric mean = 7.2 L) and faster clearance rates (geometric mean = 17.6 mL/hour) than IgG1 (geometric means of 5.7 L and 13.8 mL/hour, respectively) and IgG2 (geometric means of 4.9 L and 15.4 mL/hour, respectively) isotypes. Consequently, IgG4 antibody-based biotherapeutics have a shorter elimination half-life (geometric mean = 13.9 days) compared to IgG1 (geometric mean = 14.6 days) and IgG2 (geometric mean = 15.8 days).
When the route administration is considered, we were surprised to find that the s.c. administered IgG4 (4 products) antibody-based biotherapeutics show a geometric mean elimination half-life of 21.4 days, which is longer by nearly 2 days than the geometric mean elimination half-life of 19.7 days for s.c. administered IgG1s (16 products) and 18.7 days for s.c. administered IgG2s (6 products). The s.c. administered IgG4 products also have slower clearance rates overall (geometric mean = 9.2 mL/hour) when compared to s.c. administered IgG1 antibodies (geometric mean = 14.1 mL/hour) and s.c. administered IgG2 antibodies (geometric mean = 20.9 mL/hour), while the volumes of distribution remain similar for IgG4 and IgG1 but are lower for IgG2 (IgG4 = 7.3 L, IgG1 = 6.8 L, IgG2 = 4.6 L). Previously, IgG4 antibodies showed slower clearance compared to the IgG1 antibodies in a cynomolgus monkey study.156 However, the availability of only a small number of data points for the s.c.-administered IgG4 antibodies prevents us from arriving at any definitive conclusions in this regard. Furthermore, in the case of the IgG subclasses that contain fewer than 10 antibodies, some PK parameters might also be influenced by the nature and expression levels of the target and thus non-linearity. Note that data on non-IgG formats such as Fabs, Fvs, and scFvs were not included in Table 4 because of their smaller molecular size. Typically, such molecules have significantly faster clearance rates and shorter elimination half-lives.157 By removing smaller non-IgG, the averages were not dominated by outlier PK data, but values still varied widely for all PK parameters, specifically clearance. Note that IgG-based ADCs were included in the route of administration analysis.
Relationship between the isoelectric point and pharmacokinetics
Intrinsic molecular characteristics of antibody-based biotherapeutics can influence their clearance and pharmacology.158–160 For example, their PK properties have been reported to be influenced by the charge or isoelectric point (pI).161–164 It has been shown that antibodies with a high pI value have a lower bioavailability and a faster clearance rate.165 However, package inserts and reviews of BLAs do not contain information on isoelectric points of antibody-based biotherapeutics. In this work, we have estimated isoelectric points of the variable regions (pIFv3D) of the 89 antibody-based biotherapeutics using their homology-based structural models (see methods and Table S1 for pI data). This is because full-length antibodies demonstrate different solution behaviors and PK properties depending on the electrostatic properties of their variable regions.166,167 Table S8 shows Pearson linear correlation coefficients (r – values) of pIFv3D with the PK parameters reported in the package inserts for different subsets of marketed antibody-based biotherapeutics. As stated above, out of the 89 antibody-based biotherapeutics, only 45 full-length IgG antibodies show linear PK and therefore no target-mediated effects. Moreover, this set excludes ADCs. No statistically significant correlations between pIFv3D and the four PK parameters, namely bioavailability, volume of distribution, clearance, and elimination half-life, were observed for these 45 antibodies. However, when they are grouped into different subsets, like those shown in Table 4, a few statistically significant correlations are found. Interestingly, s.c. administered antibodies showed a significant negative correlation between volume of distribution and pIFv3D (number of observations = 17, r = −0.50, p-value < 0.05, see Table S8), which indicates that the higher the pIFv3D, the lower the volume of distribution.
Formulation characteristics of the marketed antibody-based biotherapeutics
Formulation is a cardinal aspect of biological drug development with important consequences for stability, safety, pharmacology, and product presentation, as well as route of administration of antibody-based biotherapeutic drug products.168 Figures 5(a–d) show time-series plots for four commonly used inactive ingredients, buffer, stabilizer, tonicity modifier and surfactants, for formulation development of the marketed antibody-based biotherapeutics. In several instances, the drug products have multiple formulations (e.g., adalimumab). In this work, we focused on the antibody formulations at the time of the first approval, i.e., the original formulations. Of the 89 antibody-based biotherapeutics, 87 used a buffering agent, 35 a tonicity modifier, 60 a stabilizing agent, and 79 a surfactant. Trends in the usage of each of these inactive ingredients over time are discussed below.
Figure 5.
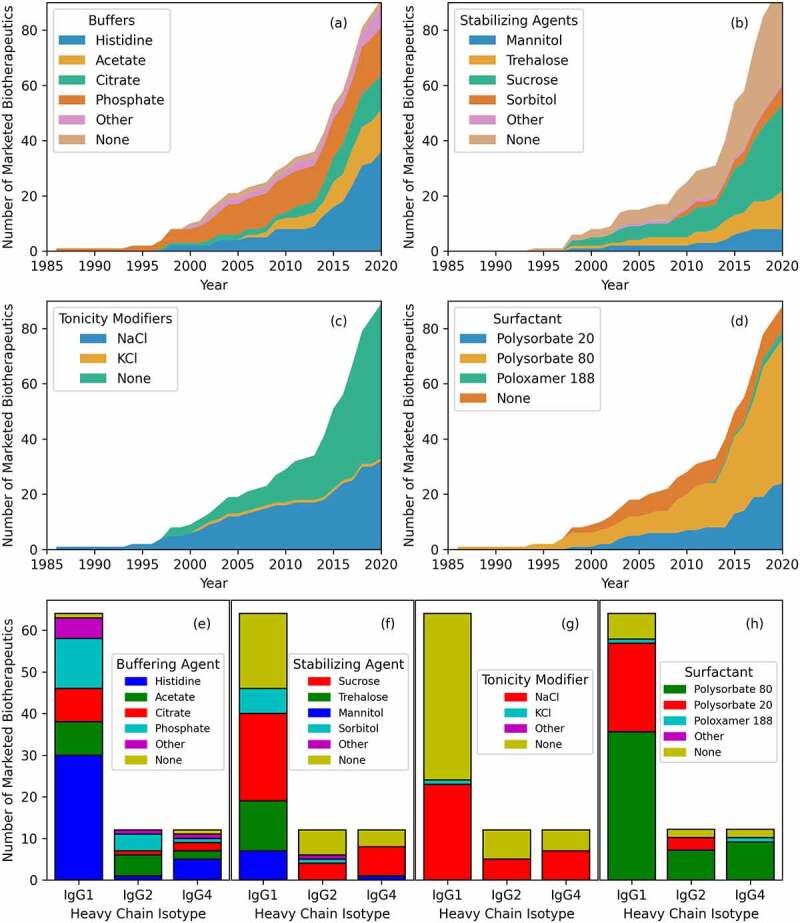
Four of the main inactive ingredient categories for marketed antibody-based biotherapeutics over time (see Table S1 for all). (a) Buffers. Histidine is the most common, included in 36 marketed antibody-based biotherapeutic formulations. (b) Stabilizing agents. No stabilizing agents is the most common, but if one is used, sucrose is the most common. (c) Tonicity modifiers. No tonicity modifier is the most common, but if one is used, NaCl is the most common. (d) Surfactants. Polysorbate 80 is included in 52 formulations. (e) Buffering agent usage versus heavy chain isotype. The four most common buffers are seen in all isotypes, but histidine is much more common in IgG1. (f) Stabilizing agent usage. Sucrose is the most common in all isotypes, but trehalose is not used in IgG2 or IgG4 products. (g) Tonicity modifiers. Sodium chloride and no tonicity modifier have similar representation across all isotypes. (h) Surfactant. Polysorbate 20 is paired mostly with IgG1 formulations while polysorbate 80 is paired with IgG2 and IgG4.
Figure 5(a) shows trends in the buffers used in antibody-based biotherapeutic formulations over time. Of the 87 formulations that contain a buffering agent, histidine, which provides higher colloidal stability compared to other buffers,169 is the most common one. It has been used for 36 (41%) marketed antibodies at an average concentration of 3.0 ± 1.8 mg/mL (range of 0.7–8.9 mg/mL). Before 2015, the top 4 commonly used formulation buffers were histidine (13 of 38, 34%), phosphate (12 of 38, 32%), acetate (5 of 38, 13%), and citrate (5 of 38, 13%). From 2015 through June 2020, the top 4 formulation buffers were histidine (22 of 47, 47%), acetate (10 of 47, 21%), citrate (6 of 47, 12%), and phosphate (4 of 47, 9%). This shows an increase in the use of histidine and acetate as buffering agents, while the use of the phosphate buffer has decreased over time.
Figure 5(b) shows usage of stabilizing agents in drug product formulation over time. Sucrose has been the most used stabilizer over the years. Of the 60 product formulations that contain stabilizers, sucrose is added to 32 (53%) of them at an average concentration of 91.9 ± 134.2 mg/mL (range of 4–822 mg/mL). The top four stabilizers before 2015 were sucrose (11 of 22, 50%), trehalose (6 of 22, 27%), mannitol (3 of 22, 13%), and sorbitol (2 of 22, 9%). From 2015 through June 2020, the top 4 stabilizers were sucrose (19 of 35, 54%), trehalose (7 of 35, 20%), sorbitol (5 of 35, 14%), and mannitol (4 of 35, 11%). Therefore, sucrose and sorbitol have seen an increase in use, and trehalose and mannitol have seen a decrease in recent years. Nonetheless these excipients continue to be used regularly in drug formulations.170,171
Figure 5(c) shows the use of tonicity modifiers over time for antibody formulations. Sodium chloride (NaCl) has been used as tonicity modifier in formulations of 35 marketed antibody-based biotherapeutics at an average concentration of 7.0 ± 6.5 mg/mL. Potassium chloride (KCl) is used with NaCl in the formulation of alemtuzumab, approved in 2001. Since then, NaCl has been the only salt used as a tonicity modifier in marketed antibody-based biotherapeutics formulations. NaCl and KCl are used mainly in product formulations presented as vials (77%) and i.v. administered (83%).172
Surfactants are used in formulations of antibody-based biotherapeutics to prevent aggregation and denaturation of antibodies at the air–water interface.173 Figure 5(d) shows use of surfactants in formulations of antibody-based biotherapeutics over time. Of the 79 formulations that utilize surfactants, 53 use polysorbate 80 (67%) at an average concentration of 0.41 ± 0.48 mg/mL (range of 0.04–2.00 mg/mL). Before 2015, polysorbate 80 was included in 24 of 42 formulations (57%), polysorbate 20 in 8 (19%), poloxamer 188 in 1 (2%), and no surfactant was used in 9 (22%). From 2015 through June 2020, polysorbate 80 accounted for 29 of 47 (62%), polysorbate 20 for 16 (34%), poloxamer 188 for 1 (2%), and no surfactant for 1 (2%). These observations show that the use of polysorbate-based nonionic detergents as surfactants is widespread in antibody formulations. This reliance on polysorbates has been rationalized by their biocompatibility and low toxicity. However, the observation that polysorbates can degrade, leading to particulate formation during long-term storage of protein formulations, has emerged as a major product quality concern and a potential patient safety risk factor for antibody-based biotherapeutics.174,175 It is therefore clear that diversification of surfactants beyond polysorbates would be beneficial toward assuring long-term formulation stability and quality of formulation of antibody-based biotherapeutics.
MAbs with different isotypes show different electrostatic properties that may affect their solution behaviors in different formulation buffers.166 This leads to the following question: Do inactive ingredients of antibody-based biotherapeutic formulations show a relationship with the isotypes of the antibodies? Figures 5(e–h) shows data for the four inactive ingredient types (buffer, stabilizer, tonicity modifier, and surfactant) with respect to heavy chain isotypes (IgG1, IgG2, and IgG4). Note that only the 83 full-length antibodies were used for this analysis. Figure 5(e) shows data for the buffering agents used in the formulations of antibody-based biotherapeutics broken down by their heavy chain isotypes. Histidine is used as the formulation buffer for 29 of 59 (49%) IgG1s and for 5 of 12 (42%) IgG4 ones. However, only one of the 12 (8%) IgG2s has been formulated in histidine buffer. On the other hand, acetate has been used as the formulation buffer for 42% (5 of 12) of IgG2s, but only for 10% (6 of 59) of IgG1s and 17% (2 of 12) of IgG4s. These observations show that heavy chain isotypes can potentially influence the outcomes of pH/buffer screening studies carried out during formulation process development. An analogous analysis exploring the influence of the light chain isotypes was not carried out because of the low representation of lambda light chains among the marketed antibody-based biotherapeutics. Figure 5(f) illustrates the use of stabilizing agents across different antibody heavy chain isotypes. Sucrose is used consistently with all isotypes, but trehalose is not. Trehalose is included only in IgG1 formulations. This is consistent with the reports that IgG1 mAbs are more stable when formulated with trehalose, but other isotypes like IgG2 show instability during freeze-thaw.176,177 Figure 5(g) shows that there is no preference for tonicity modifiers for different antibody heavy chain isotypes. That is, all isotypes use NaCl.151 Lastly, Figure 5(h) shows that Polysorbate 80, the most common surfactant, appears to be equally used across all antibody isotypes (IgG1, (34 of 59, 58%); IgG2 (7 of 12, 58%); and IgG4 (9 of 12, 75%)).152,153 Note that biopharmaceutical companies often use their own preferred discovery pipelines, platform formulations, and manufacturing processes. Such information is often proprietary and typically unavailable in the public domain. Therefore, it is difficult to ascribe the above observations solely to the molecular properties of different IgG isotypes.
In addition to composition of inactive ingredients, the concentration of the API and pH of the formulation are also important considerations.178 Figure 6(a) shows the API concentration of all 89 marketed antibody-based biotherapeutics over time. For our analysis, the API concentration is divided into four categories of 0–10 mg/mL, 10–50 mg/mL, 50–100 mg/mL, and more than 100 mg/mL. High concentration liquid formulations (HCLFs) are finding greater representation among the newer antibody-based biotherapeutic drug products. Seven products approved before 2015 had API concentration > 100 mg/mL, while additional 16 HCLF products were approved from 2015 to June of 2020. A major hurdle to the development of HCLFs of antibody-based biotherapeutics has been that certain concentrated antibody solutions become unacceptably viscous, leading to challenges in drug administration. Several studies have sought to understand this phenomenon,179 developed molecular properties-based screening tools for viscosity of highly concentrated antibody formulations,167,180,181 and have shown that viscosity can be reduced via sequence optimization and use of excipients.182,183
Figure 6.
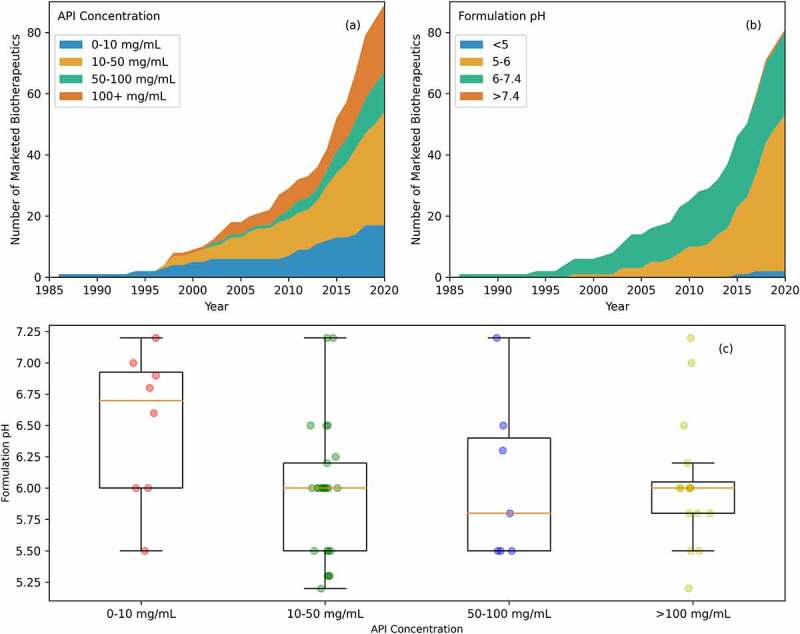
API concentration and formulation pH. (a) API concentration over time. The most common API concentration is between 10 and 50 mg/mL, however, API concentrations≥100 mg/mL are becoming more common in recent years. (b) Formulation pH over time. In recent years, drug products are being more frequently formulated under acidic formulations (≤6.0). (c) API concentration and formulation pH comparison. Antibody-based biotherapeutics with high concentration formulations are often formulated in mildly acidic conditions.
Five antibody-based biotherapeutics have been approved with multiple API concentrations, ranibizumab (6 mg/mL and 10 mg/mL), burosumab (10 mg/mL, 20 mg/mL, and 30 mg/mL), denosumab (60 mg/mL and 70 mg/mL), alirocumab (75 mg/mL and 150 mg/mL) and sarilumab (132 mg/mL and 175 mg/mL). Therefore, there are 95 data points for API concentrations for the 89 marketed antibody-based biotherapeutics. Overall, the API concentrations range from 0.0125 mg/mL (blinatumomab) to 200 mg/mL (certolizumab pegol) with an average of 51 ± 52 mg/mL. There are 18 antibody-based biotherapeutic products with API conc. < 10 mg/mL (19%), 39 with API conc. in range of 10–50 mg/mL (41%), 14 with API conc. between 50 and 100 mg/mL (15%), and the remaining 24 have API conc. > 100 mg/mL (25%). When comparing API concentration and %ADAs, there is no clear significance or correlation between the two, even when i.v. and s.c. values are considered separately (Suplementary Figure S2).
Formulation buffer pH is another important consideration for antibody-based biotherapeutic formulations.166,167,184,185 Figure 6(b) shows the formulation pH over time for the 89 marketed antibody-based biotherapeutics. The pH values of the antibody formulations range from 4.8 (brodulamab) to 8.0 (inotuzumab ozogamicin) with the average pH being 6.1 ± 0.6 for 81 formulations. Information on the pH of formulation buffers is not available for eight antibody-based biotherapeutics in any of their public documentations. Of the 81 marketed antibodies with the known formulation buffer pH values, two (2.5%) have been formulated at a pH ≤ 5, 51 (63%) have been formulated in the range 5<pH ≤ 6, 27 (33%) formulations are in the range of 6<pH ≤ 7.4, and only one (1.2%) has been formulated at pH > 7.4. These observations suggest that most antibody-based biotherapeutics have been formulated in mildly acidic conditions. Before 2015, there were 14 marketed antibodies with formulation buffer pH ≤ 6.0 and 22 with formulation pH > 6.0. In comparison, 39 antibody-based biotherapeutics have been approved with formulation pH ≤ 6.0 since 2015, while only six of them have been approved with formulation pH > 6.0. Moreover, antibody-based biotherapeutics formulated in mildly acidic conditions (5.0<pH ≤ 6) have been granted more approvals every year than those formulated above formulation buffer pH > 6 (Figure 6(b)). These observations are consistent with our recent findings that antibodies with weakly basic isoelectric points are better at balancing the trade-offs required for good colloidal stability of their formulations and the need to minimize off-target binding.186
Is there a correlation between decrease in formulation buffer pH and emergence of high concentration liquid formulations for antibody-based biotherapeutics? Figure 6(c) shows boxplots of API concentration classes with respect formulation buffer pH. There is a downward trend showing that lower formulation buffer pH supports formulations at higher API concentrations. Although this trend is not statistically significant (r2 = 0.11, p-value = 0.002; Figure S2), it does appear to be consistent with the recent rise in approvals for antibody-based biotherapeutics with high concentration formulations.166,185 Furthermore, Table S9 shows that API concentration does increase with a decrease in formulation pH, on average. At high API concentrations, the colloidal stability of the antibody formulations is expected to be reduced because of greater interactions among antibody molecules (solute: solute interactions rather than solute: solvent interactions) due to molecular crowding. However, the effect of molecular crowding is countered by the electrostatic properties of the antibody molecules. MAbs, which constitute most of the marketed antibody-based biotherapeutics, are positively charged when formulated in mildly acidic conditions, promoting repulsive self-interactions.169
Discussion
Antibody-based biotherapeutics have emerged as the most successful class of biopharmaceuticals in recent decades. Starting from the first approval in 1986, we now have more than a hundred marketed antibodies serving patients with previously unmet medical needs. The rapid rise of antibodies as biotherapeutics was facilitated by several technological advances in antibody discovery, manufacturing, product and clinical development, and drug delivery. In this report, we studied the emergence of antibody-based biotherapeutics as the best-selling medicines by analyzing the trends gleaned from publicly available information in their package inserts, reviews of BLAs, and websites of regulatory agencies. We analyzed the data to identify the drivers behind the observed trends. These include: 1) therapeutic purpose; 2) need to improve safety by minimizing adverse effects, e.g., immunogenicity; 3) easier developability via improvements in CMC processes, pharmacokinetics, and dosing frequency; and 4) increased attention to patient convenience. All these drivers are interconnected and will continue to evolve as our collective experience with antibody-based biotherapeutics grows and new therapeutic concepts, molecular formats, and manufacturing technologies emerge. To our knowledge, this report is the first to analyze antibody-based biotherapeutic products from several different perspectives simultaneously and provide a comprehensive understanding of issues involved in their discovery and development. Many useful lessons can be learned from this report. These are broadly summarized as follows:
Notwithstanding the diversification of antibody-based biotherapeutics to different therapeutic areas and recent approvals granted to such molecules for infectious diseases, they continue to primarily serve therapeutic areas of oncology, immunology, and hematology. This observation points to significant opportunities for biotechnology to serve unmet medical needs in other therapeutic areas.
While a diverse range of different antibody-based molecular formats is reaching stages of clinical development (Phases 1–3),187–189 IgG1 kappa remains the most common molecular format among marketed antibody-based biotherapeutics. This is true across different therapeutic areas, indications, formulations, and routes of administration. The literature shows that this format is also the best understood one from several perspectives of drug discovery as well as development. Scientists involved in the early formulation of therapeutic concepts need to be aware of this to assure a successful end-to-end discovery and development strategy with increased efficiency of translation of drug discoveries to antibody-based drug products.
Most of the recently approved antibody-based biotherapeutics are either humanized or human, even though the percent humanness of their variable regions shows no correlation with the incidence of ADAs as reported in the package inserts. This observation, unexpected at first, is consistent with the complexity of the immune pathways and processes that underpin the generation of ADAs.102,190,191 The absence of this correlation does not imply that humanization of antibody-based drug candidates originating from non-human species is not needed before product and clinical development.
Several options are now available when it comes to concentration, formulation, product presentation, and administration of antibody-based biotherapeutic products. The actual usage of these options depends on multiple factors, including molecular stability and formatting, therapeutic purposes, and clinical indications the products need to serve, dosing frequency requirements, as well as patient convenience/preference. Availability of these multiple options has important consequences for immunogenicity and pharmacology of these products, both of which show large variations. For example, for s.c. administration, biotherapeutics needs to be amenable to high concentration formulation, slow clearance, and long elimination half-lives. The converse is also true. Antibody-based biotherapeutic drug candidates that can be formulated at high concentrations, have long elimination half-lives and slow clearance may face fewer hurdles to their development as drug products suitable for s.c. route of administration.
Molecular characteristics, API concentration, and presence/absence of salt in antibody formulations can influence PK properties of antibody-based biotherapeutic products. During the transition from preclinical to clinical stages, clinical pharmacologists rely on optimized molecular characteristics leading to slower clearance, longer half-life, and greater bioavailability in humans. Optimal clinical PK of mAbs allows clinical pharmacologists to choose an optimized therapeutic dose and a convenient dosing interval for patients.
Although comprehensive in its scope, this analysis was limited by several factors. For example, detailed information on discovery, formulation, and biomanufacturing processes followed by manufacturers is often not included in package inserts and is not easily available in the public domain. This prevents a more complete understanding of how such biotherapeutics are developed, tested, and manufactured commercially. There are also limitations to the data reported in the package inserts along with inconsistencies in the format and units used to report similar information. A more consistent reporting of information made available in the package inserts by the manufacturers would help scientists, patients, pharmacists, and medical professionals clearly understand advantages and limitations of these products and make informed choices. Furthermore, different biopharmaceutical companies often use different methods for collecting toxicology, PK, immunogenicity, and other data.192,193 Regulatory agencies have also updated approval policies several times over the years as our understanding of antibody-based biotherapeutics improved.194–196 Due to these changes, it is often difficult to make direct comparisons among marketed antibody-based biotherapeutics.197–199
Despite the above limitations, it is feasible to construct a profile of a therapeutic antibody drug candidate most likely to succeed in product and clinical developments and win regulatory approvals. It is likely to be a full-length human or humanized IgG1 kappa mAb with a liquid formulation in mildly acidic conditions suitable for i.v. or s.c. administration. The formulation would consist of a histidine buffer, contain sucrose as a stabilizer, and polysorbate 80 as a surfactant. Depending on API concentration, the formulation may also contain viscosity-reducing excipients,200,201 and sodium chloride as a tonicity modifier if needed. It must also be emphasized here that discovery, development, and regulation of antibody-based biotherapeutics are inherently multi-factorial, and decisions are often made on a case-to-case basis after critical evaluations of benefits versus risks involved. Therefore, this profile should be taken as advisory rather than as a requirement.
While this report has focused on the trends gleaned from drug product information contained in package inserts and other regulatory documents, it has not escaped our attention that molecular sequences and structures of active ingredients play crucial roles in their discovery, development, and clinical approval. In-silico tools can also be used during discovery, design, and development of antibody-based biotherapeutics.202 For example, computed physiochemical descriptors could potentially be used to assess if a given antibody-based drug candidate is like currently marketed ones. Ahmed et al. previously showed, through sequence and structural analysis, that non-redundant physicochemical descriptors can be used to flag Fv regions of antibodies for medicine-likeness.203 There are numerous ways computer-aided drug design can contribute to the marketed antibody-based biotherapeutic landscape,204 including sequence analysis (for example, see Figures S3 (a, b)),205 structure analysis,206 and machine learning.207
Data sources and methods
Data were compiled from several resources provided by the FDA, EMA, and WHO. Additional resources from The Antibody Society,208 ClinicalTrials.gov,209 The International Immunogenetics Information System (IMGT),210,211 and The Therapeutic Antibody Database212 were used to verify the sequences and provide additional information. Table 1 shows an example of information collected from publicly available resources, with the full table available as a supplement (Table S1), while Table 5 shows examples of where and how these data were collected.
Table 5.
Data collection methods*.
| FDA and EMA information | Package insert location | Example |
|---|---|---|
| Trade Name | The trade name is always located at or near the top of the first page on the package insert for the FDA. It is located on the second page of the product information under “1. NAME OF THE MEDICINAL PRODUCT” for the EMA. | Humira |
| Manufacturer | The current manufacturer name is always located under the “ADVERSE REACTIONS” section on the first page of the package insert for the FDA. The manufacturer name is always located under “Manufacturer” section of the EMA product information. | AbbVie Inc |
| First United States Approval Year | Initial US approval is on the first page of the FDA package information. Later updates to the packaging information will be noted on the first page too. | 2002 |
| First EU Approval Year | The year of approval is found in multiple locations on the EMA website. All licensing information can be found in section 9 “DATE OF FIRST AUTHORIZATION/RENEWAL OF THE AUTHORIZATION” of the EMA product information. | 2003 |
| Indication First Approved | The first approved usage for the FDA package insert will be on the first page under “INDICATIONS AND USAGE.” The first approved usage will be in the product information table of the EMA public assessment next to “Therapeutic indication.” | Rheumatoid arthritis |
| Biosimilars | Biosimilars can be found using the FDA or EMA search function. Biosimilars will only be displayed in the search results if any exist. | Abrilada Amjevita Cyltezo … |
| Targets Sources |
The source and target are in the “INDICATIONS AND USAGE” section of the FDA package document and the “2.1. Introduction” section of the EMA assessment report. | TNF |
| Isotype | The isotype is in the “11 Description” section of the FDA package documents and the “2.2.1. Introduction” section of the EMA report. | IgG1 |
| Lyophilized | Lyophilized powder will only be mentioned in a package or report if that drug comes in that form. This is mentioned in the “2.2.1. Introduction” section of the EMA report and the “11 Description” section of the FDA package documents | Liquid Formulation |
| Dosage Route Dosage Strength pH Formulation |
All four of these items are in the “11 Description” section of the FDA package documents. The order is typically dosage route, dosage strength, formulation, then pH. The EMA information is in the “2.2.1. Introduction” section of the report. | Subcutaneous 80 mg/0.8 mL pH: 5.2 “contains … ” |
| Anti-Drug Antibodies | Anti-drug antibody information comes from the FDA and EMA review documents. For the FDA, the ADA information is in the Clinical Pharmacology and Biopharmaceutics Review(s) document. For the EMA, the ADA information is in the “Antibodies” section of the public assessment report. The ADA value for the longest time passed is taken. If more than one dataset exists, the average ADA value was taken. | “The overall HAHA positivity rate while on treatment was low (5.5%)” |
| Pharmacokinetics Pharmacodynamics |
Pharmacodynamics is in the section “12.2 Pharmacodynamics” and pharmacokinetics is in section “12.3 Pharmacokinetics” of the FDA package document. PKPD data are in “5.1 Pharmacodynamic properties” and “5.2 Pharmacokinetic properties” of the EMA package documents. | “linear”, “bioavailability … 64%”, “Vss ranged from 4.7 to 6.0 L”, “mean terminal half-life was approximately 2 weeks” |
| WHO information | ||
| INN Proposed and recommended lists |
The International Nonpriority Name number can be found in multiple locations, though not with the EMA or FDA. It is issued through the WHO. It can be found through the WHO websites, or other search methods. The IMGT database can be used to look for various data including the INN number. The school of INN, a service provided by the WHO, has resources that can search for INN, proposed list, and recommended list numbers. Utilizing these resources, one can find the proposed and recommended list numbers. These lists can be used to look more in-depth at the name and information of each antibody-based biotherapeutic product. | Adalimumab 85th Proposed List 45th Recommended List |
| Molecular Format | All antibody-based biotherapeutic products that have the -mab suffix are derived from monoclonal antibodies. If they are nonstandard representations, the type can be found in the WHO proposed and recommended lists. For example, the recommended list 41 Abciximab entry states, “immunoglobulin G1, anti-(human integrin αIIbβ3) Fab fragment” and the recommended list 59 Certolizumab pegol entry states, “immunoglobulin, anti-(human tumor necrosis factor α) Fab’ fragment.” Abciximab would be listed as a Fab and Certolizumab would be a Fab’. | immunoglobulin G1 (human monoclonal D2E7 heavy chain anti-human tumor necrosis factor), disulfide with human monoclonal D2E7k-chain, dimer immunomodulator |
| Light Chain Isotype | The light chain isotype is found in the WHO proposed and recommended lists. It can also be found in the IMGT database. The light chain is either a kappa () or lambda (). | kappa |
| Sequences | Sequences are found on WHO resources. INN submissions are not required to give sequences but are found in all recommended lists after 2010. Before that, WHO did not always include the sequence. All older sequences were found by comparing sequences from the Therapeutic Antibody Database and IMGT. None of the sequences differed when compared between Tabs and IMGT. The framework and CDRs were determined using the CCG annotation scheme in MOE. | Light: DIQMTQSPS … … QGTKVEIK Heavy: EVQLVESGG … … QGTLVTVSS |
*The first column shows the type of data collected. The second column shows where the data can be found in the package inserts. The third column shows an example of the data.
In the case of each marketed antibody, percent humanness values are the sequence identity values calculated for heavy and light chains, individually, by aligning them to their respective v-genes in the closest human germlines identified by IMGT,210,211 as implemented in IgBLAST version 1.15.213 Note that the sequence alignments were performed using the amino acid sequences and not the nucleotide ones. Furthermore, HCDR3s were not included in our calculations of percent humanness.
Values of the structure-based pI for the Fv portions of the antibodies (pIFv3D) were calculated by building their homology-based molecular models using Molecular Operating Environment (MOE) version 2022.02214 with the default Amber10: EHT force field. Solvation was included using the Generalized Born implicit model with internal and external dielectric values set, respectively, to 4 and 80 and the non-bonded interaction tapered to zero between 10 and 12 Å. C-termini of the Fv light and heavy chains were amidated to neutralize charges on them. All structures were energy minimized below a 10−6 kcal/mol/Å2 root mean square gradient threshold. The protein property pIFv3D was calculated using energy-minimized homology models.
Equation (1) was used to predict the half-life from the observed clearance and distribution of the full-length antibody-based biotherapeutics. The predicted values are computed for all 89 antibodies (Table S1) and divided into specific groups based on their isotypes, API concentrations, presence/absence of salt in their formulations and routes of administration, wherever sufficient data were available (Table 4).
| (1) |
Supplementary Material
Acknowledgments
KPM thanks Boehringer Ingelheim for postdoctoral fellowship. Authors thank numerous colleagues both within and outside BI for several helpful discussions. Dr. Andrew Nixon is thanked for critical reading of the manuscript. KPM and SK also acknowledge their discussions with Dr. Joschka Bauer, Dr. Anne Karow-Zwick, and Dr. Seema Thakral on the topic of drug formulations. The authors are grateful to anonymous reviewer #1 for very helpful comments on our work.
Funding Statement
This work was funded by Biotherapeutics Discovery, Boehringer Ingelheim Pharmaceuticals, Inc. USA via a postdoctoral fellowship to KPM.
Abbreviations
- ADA
Anti-Drug Antibody
- ADC
Antibody-Drug Conjugate
- API
Active Pharmaceutical Ingredient
- BiTE
Bispecific T-cell Engager
- CDR
Complementarity Determining Region
- CH
Constant region of the heavy chain
- CL
Constant region of the light chain
- EC
European Commission
- EMA
European Medicines Agency
- EUA
Emergency Use Authorization
- Fab
Fragment antigen binding
- Fv
Fragment variable
- FDA
U.S. Food and Drug Administration
- i.v.
Intravenous
- IgG
Immunoglobulin G
- IgM
Immunoglobulin M
- IGMT
The international ImMunoGeneTics information system
- INN
International Nonproprietary Name
- mAb
Monoclonal antibody
- MM
Multiple Myeloma
- NSCLC
Non-small Cell Lung Cancer
- PD
Pharmacodynamics
- PH20
Recombinant human PH20 hyaluronidase (or rHuPH20)
- PJIA
Polyarticular Juvenile Idiopathic Arthritis
- PK
Pharmacokinetics
- PS20
Polysorbate 20
- PS80
Polysorbate 80
- PsA
Psoriatic Arthritis
- QW
Once a Week
- RA
Rheumatoid Arthritis
- s.c.
Subcutaneous
- scFv
Single-chain fragment variable
- sdAb
Single-domain antibody
- SJIA
Systematic Juvenile Idiopathic Arthritis
- TNBC
Triple Negative Breast Cancer
- VH
Variable region of the heavy chain
- VL
Variable region of the light chain
- WHO
World Health Organization
Disclosure statement
All authors were employees of Boehringer-Ingelheim when this research was performed.
Author contributions
SK and CG formulated the research problem. SK provided day-to-day advice to KPM for this postdoctoral research project. KPM collected all the data and performed analyses. RG and SH helped with interpreting the pharmacology data. All authors contributed toward manuscript writing.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2191301.
References
- 1.Leader B, Baca QJ, Golan DE.. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7(1):21–29. doi: 10.1038/nrd2399. PMID: 18097458. [DOI] [PubMed] [Google Scholar]
- 2.Alt N, Zhang TY, Motchnik P, Taticek R, Quarmby V, Schlothauer T, Beck H, Emrich T, Harris RJ.. Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biolog. 2016;44(5):291–305. doi: 10.1016/j.biologicals.2016.06.005. PMID: 27461239. [DOI] [PubMed] [Google Scholar]
- 3.Kaplon H, Chenoweth A, Crescioli S, Reichert JM. Antibodies to watch in 2022. Mabs. 2022;14(1):2014296. doi: 10.1080/19420862.2021.2014296. PMID: 35030985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplon H, Reichert JM. Antibodies to watch in 2021. Mabs. 2021;13(1):1860476. doi: 10.1080/19420862.2020.1860476. PMID: 33459118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinch MS, Kraft Z, Schwartz T. Monoclonal antibodies: trends in therapeutic success and commercial focus. Drug Discov Today. 2023;28(1):103415. doi: 10.1016/j.drudis.2022.103415. PMID: 36280042. [DOI] [PubMed] [Google Scholar]
- 6.Zhu Z, Chakraborti S, He Y, Roberts A, Sheahan T, Xiao X, Hensley LE, Prabakaran P, Rockx B, Sidorov IA, et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc National Acad Sci. 2007;104(29):12123–28. doi: 10.1073/pnas.0701000104. PMID: 17620608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza a hemagglutinins. Sci. 2011;333(6044):850–56. doi: 10.1126/science.1205669. PMID: 21798894. [DOI] [PubMed] [Google Scholar]
- 8.Stettler K, Beltramello M, Espinosa DA, Graham V, Cassotta A, Bianchi S, Vanzetta F, Minola A, Jaconi S, Mele F, et al. Specificity, cross-reactivity, and function of antibodies elicited by zika virus infection. Sci. 2016;353(6301):823–26. doi: 10.1126/science.aaf8505. PMID: 27417494. [DOI] [PubMed] [Google Scholar]
- 9.Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, Wu HC. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27:1. doi: 10.1186/s12929-019-0592-z. PMID: 31894001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu J, Ou J, McHugh KP, Borys MC, Khetan A. Upstream cell culture process characterization and in-process control strategy development at pandemic speed. Mabs. 2022;14(1):2060724. doi: 10.1080/19420862.2022.2060724. PMID: 35380922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pecetta S, Finco O, Seubert A. Quantum leap of monoclonal antibody (mAb) discovery and development in the COVID-19 era. Semin Immunol. 2020;50:101427–101427. doi: 10.1016/j.smim.2020.101427. PMID: 33277154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelley B. Developing therapeutic monoclonal antibodies at pandemic pace. Nat Biotechnol. 2020;38(5):540–45. doi: 10.1038/s41587-020-0512-5. PMID: 32317764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khetan R, Curtis R, Deane CM, Hadsund JT, Kar U, Krawczyk K, Kuroda D, Robinson SA, Sormanni P, Tsumoto K, et al. Current advances in biopharmaceutical informatics: guidelines, impact and challenges in the computational developability assessment of antibody therapeutics. Mabs. 2022;14(1):2020082. doi: 10.1080/19420862.2021.2020082. PMID: 35104168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farid SS, Baron M, Stamatis C, Nie W, Coffman J. Benchmarking biopharmaceutical process development and manufacturing cost contributions to R&D. Mabs. 2020;12(1):1754999. doi: 10.1080/19420862.2020.1754999. PMID: 32449439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.La Merie Publishing . 2021 sales of recombinant therapeutic antibodies, proteins, biosimilars & other biologics; 2022. http://www.lamerie.com.
- 16.Reichert JM, Valge-Archer VE. Development trends for monoclonal antibody cancer therapeutics. Nat Rev Drug Discov. 2007;6(5):349–56. doi: 10.1038/nrd2241. PMID: 17431406. [DOI] [PubMed] [Google Scholar]
- 17.Pavlou AK, Belsey MJ. The therapeutic antibodies market to 2008. Eur J Pharm Biopharm. 2005;59(3):389–96. doi: 10.1016/j.ejpb.2004.11.007. PMID: 15760719. [DOI] [PubMed] [Google Scholar]
- 18.Presta LG. Selection, design, and engineering of therapeutic antibodies. J Allergy Clin Immun. 2005;116(4):731–36. doi: 10.1016/j.jaci.2005.08.003. PMID: 16210043. [DOI] [PubMed] [Google Scholar]
- 19.McNeil C. Monoclonal antibodies to watch. Jnci J National Cancer Inst. 1995;87(22):1659–1659. doi: 10.1093/jnci/87.22.1659. [DOI] [PubMed] [Google Scholar]
- 20.Maggon K. Monoclonal antibody “gold rush. Curr Med Chem. 2007;14(18):1978–87. doi: 10.2174/092986707781368504. PMID: 17691940. [DOI] [PubMed] [Google Scholar]
- 21.Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. Mabs. 2015;7(1):9–14. doi: 10.4161/19420862.2015.989042. PMID: 25529996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui Y, Cui P, Chen B, Li S, Guan H. Monoclonal antibodies: formulations of marketed products and recent advances in novel delivery system. Drug Dev Ind Pharm. 2017;43(4):1–39. doi: 10.1080/03639045.2017.1278768. PMID: 28049357. [DOI] [PubMed] [Google Scholar]
- 23.Wilkinson I, Hale G. Systematic analysis of the varied designs of 819 therapeutic antibodies and Fc fusion proteins assigned international nonproprietary names. Mabs. 2022;14(1):2123299. doi: 10.1080/19420862.2022.2123299. PMID: 36109838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li F, Vijayasankaran N, Shen A, Kiss R, Amanullah A. Cell culture processes for monoclonal antibody production. Mabs. 2014;2(5):466–79. doi: 10.4161/mabs.2.5.12720. PMID: 20622510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raju TS, Jordan RE. Galactosylation variations in marketed therapeutic antibodies. Mabs. 2012;4(3):310–309. doi: 10.4161/mabs.19868. PMID: 22531450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh SK. Impact of product‐related factors on immunogenicity of biotherapeutics. J Pharm Sci. 2011;100(2):354–87. doi: 10.1002/jps.22276. PMID: 20740683. [DOI] [PubMed] [Google Scholar]
- 27.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc National Acad Sci. 2017;114(5):944–49. doi: 10.1073/pnas.1616408114. PMID: 28096333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2018;9(1):86–120. doi: 10.1007/s13238-017-0457-8. PMID: 28822103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reichert JM. Antibodies to watch in 2010. Mabs. 2010;2(1):84–100. doi: 10.4161/mabs.2.1.10677. PMID: 20065640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reichert JM. Antibody-based therapeutics to watch in 2011. Mabs. 2011;3(1):76–99. doi: 10.4161/mabs.3.1.13895. PMID: 21051951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reichert JM. Which are the antibodies to watch in 2012? Mabs. 2012;4(1):1–3. doi: 10.4161/mabs.4.1.18719. PMID: 22327425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reichert JM. Which are the antibodies to watch in 2013? Mabs. 2013;5(1):1–4. doi: 10.4161/mabs.22976. PMID: 23254906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reichert JM. Antibodies to watch in 2013. Mabs. 2013;5(4):513–17. doi: 10.4161/mabs.24990. PMID: 23727858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reichert JM. Antibodies to watch in 2014. Mabs. 2013;6(1):5–14. doi: 10.4161/mabs.27333. PMID: 24284914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reichert JM. Antibodies to watch in 2014. Mabs. 2014;6(4):799–802. doi: 10.4161/mabs.29282. PMID: 24846335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reichert JM. Antibodies to watch in 2015. Mabs. 2015;7(1):1–8. doi: 10.4161/19420862.2015.988944. PMID: 25484055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reichert JM. Antibodies to watch in 2016. Mabs. 2015;8(2):197–204. doi: 10.1080/19420862.2015.1125583. PMID: 26651519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reichert JM. Antibodies to watch in 2017. Mabs. 2017;9(2):00–00. doi: 10.1080/19420862.2016.1269580. PMID: 27960628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaplon H, Reichert JM. Antibodies to watch in 2018. Mabs. 2018;10(2):00–00. doi: 10.1080/19420862.2018.1415671. PMID: 29300693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaplon H, Reichert JM. Antibodies to watch in 2019. Mabs. 2018;11(2):219–38. doi: 10.1080/19420862.2018.1556465. PMID: 30516432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. Mabs. 2019;12(1):1703531. doi: 10.1080/19420862.2019.1703531. PMID: 31847708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaur H. Characterization of glycosylation in monoclonal antibodies and its importance in therapeutic antibody development. Crit Rev Biotechnol. 2021;41(2):300–15. doi: 10.1080/07388551.2020.1869684. PMID: 33430641. [DOI] [PubMed] [Google Scholar]
- 43.Kaur H. Stability testing in monoclonal antibodies. Crit Rev Biotechnol. 2021;41(5):692–714. doi: 10.1080/07388551.2021.1874281. PMID: 33596751. [DOI] [PubMed] [Google Scholar]
- 44.Lai Y, Suo S, Wang R, Kong X, Hu Y, Tang D, Shi H, Chen S, Hu H. Trends involving monoclonal antibody (mAb) research and commercialization: a scientometric analysis of IMS lifecycle R&D focus database (1980–2016). Hum Vacc Immunother. 2018;14(4):00–00. doi: 10.1080/21645515.2017.1420445. PMID: 29293380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abd YE, Tabll A, Smolic R, Smolic M. Mini-review: the market growth of diagnostic and therapeutic monoclonal antibodies – SARS CoV-2 as an example. Hum Antibodies. 2022;30(1):15–24. doi: 10.3233/hab-211513. PMID: 34958012. [DOI] [PubMed] [Google Scholar]
- 46.Focosi D, Tuccori M, Baj A, Maggi F. SARS-CoV-2 variants: a synopsis of in vitro efficacy data of convalescent plasma, currently marketed vaccines, and monoclonal antibodies. Viruses. 2021;13(7):1211. doi: 10.3390/v13071211. PMID: 34201767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Folkman R, Blennow O, Tovatt T, Pettersson K, Nowak P. Treatment of COVID-19 with monoclonal antibodies casirivimab and imdevimab in pregnancy. Infect. 2022;51(1):1–3. doi: 10.1007/s15010-022-01829-4. PMID: 35482208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith S. Ten years of orthoclone OKT3 (muromonab-CD3): a review. J Transpl Coordination. 1996;6(3):109–21. doi: 10.7182/prtr.1.6.3.8145l3u185493182. PMID: 9188368. [DOI] [PubMed] [Google Scholar]
- 49.U.S. Food and Drug Administration (FDA) . Drugs@FDA; 2022. [accessed 2022 Feb 8]. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm.
- 50.European Medicines Agency (EMA) . EMA medicines; 2022. [accessed 2022 Feb 8]. https://www.ema.europa.eu/en/medicines.
- 51.World Health Organization (WHO) . School of international nonproprietary names; 2022. [accessed 2022 Feb 8]. https://extranet.who.int/soinn/.
- 52.World Health Organization (WHO) . INN and biologicals; 2022. [accessed 2022 Feb 8]. https://www.who.int/teams/health-product-and-policy-standards/inn/inn-bio.
- 53.Reichert JM. Marketed therapeutic antibodies compendium. Mabs. 2012;4(3):413–15. doi: 10.4161/mabs.19931. PMID: 22531442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swan D, Loughran N, Makris M, Thachil J. Management of bleeding and procedures in patients on antiplatelet therapy. Blood Rev. 2020;39:100619. doi: 10.1016/j.blre.2019.100619. PMID: 31648803. [DOI] [PubMed] [Google Scholar]
- 55.Jayaraman K. Biocon’s first-in-class anti-CD6 mAb reaches the market. Nat Biotechnol. 2013;31(12):1062–63. doi: 10.1038/nbt1213-1062b. PMID: 24316625. [DOI] [PubMed] [Google Scholar]
- 56.Ramakrishnan MS, Eswaraiah A, Crombet T, Piedra P, Saurez G, Iyer H, Arvind AS. Nimotuzumab, a promising therapeutic monoclonal for treatment of tumors of epithelial origin. Mabs. 2014;1(1):41–48. doi: 10.4161/mabs.1.1.7509. PMID: 20046573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Centro para el Control Estatal de Medicamentos, Equipos y Dispositivos Médicos (CECMED) . Resumen de las características del producto; 2020. [accessed 2022 Feb 9]. https://www.cecmed.cu/registro/rcp/biologicos/vaxirar-racotumomab.
- 58.Administración Nacional de Medicamentos . Alimentos y tecnologia médica (ANMAT). Disposición 9016-16; 2016. [accessed 2022 Feb 9]. www.anmat.gov.ar/boletin_anmat/agosto_2016/Dispo_9016-16.pdf.
- 59.Committee for Orphan Medicinal Products (COMP) . Public summary of opinion on orphan designation Iodine (131I) tositumomab for the treatment of follicular lymphoma; 2015. [accessed 2022 Oct 25]. https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/03/136-public-summary-positive-opinion-orphan-designation-iodine-131i-tositumomab-treatment-follicular_en.pdf.
- 60.European Medicines Agency (EMA) . Refusal of the marketing authorisation for gamifant (emapalumab); 2020. [accessed 2022 Oct 25]. https://www.ema.europa.eu/en/documents/smop-initial/questions-answers-refusal-marketing-authorisation-gamifant-emapalumab_en.pdf.
- 61.European Medicines Agency (EMA) . European medicines agency decision; 2016. [accessed 2022 Oct 25]. https://www.ema.europa.eu/en/documents/pip-decision/p/0345/2016-ema-decision-2-december-2016-granting-product-specific-waiver-teprotumumab-emea-001973-pip01-16_en.pdf.
- 62.Egger GF, Wharton GT, Malli S, Temeck J, Murphy MD, Tomasi P. A comparative review of waivers granted in pediatric drug development by FDA and EMA from 2007-2013. Ther Innov Regul Sci. 2016;50(5):639–47. doi: 10.1177/2168479016646809. PMID: 27274951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mullard A. Drug withdrawal sends critical care specialists back to basics. Lancet. 2011;378(9805):1769. doi: 10.1016/s0140-6736(11)61761-3. PMID: 22106464. [DOI] [PubMed] [Google Scholar]
- 64.Viardot A, Bargou R. Bispecific antibodies in haematological malignancies. Cancer Treat Rev. 2018;65:87–95. doi: 10.1016/j.ctrv.2018.04.002. PMID: 29635163. [DOI] [PubMed] [Google Scholar]
- 65.Goldberg RM. Lessons learned from the edrecolomab story: how a checkered past became a checkered flag for monoclonal antibodies in colorectal cancer therapy. Oncol Res Treat. 2005;28(6–7):311–12. doi: 10.1159/000085570. PMID: 15933417. [DOI] [PubMed] [Google Scholar]
- 66.World Health Organization (WHO) . The use of stems in the selection of international nonproprietary names (INN) for pharmaceutical substances 2018; 2018. [accessed 2022 Feb 9]. https://cdn.who.int/media/docs/default-source/international-nonproprietary-names-inn/stembook-2018.pdf
- 67.Serafini M, Cargnin S, Massarotti A, Tron GC, Pirali T, Genazzani AA. What’s in a name? Drug nomenclature and medicinal chemistry trends using INN publications. J Med Chem. 2021;64(8):4410–29. doi: 10.1021/acs.jmedchem.1c00181. PMID: 33847110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blaine L. The nomenclature of monoclonal antibodies. 1990. pp. 71–84. doi: 10.1007/978-3-642-76011-2_8. [DOI] [Google Scholar]
- 69.World Health Organization (WHO) . Guidelines on the use of international nonproprietary names for pharmaceutical substances; 1997. [accessed 2022 Sep 19]. http://whqlibdoc.who.int/hq/1997/WHO_PHARM_S_NOM_1570.pdf.
- 70.World Health Organization (WHO) . General policies for monoclonal antibodies; 2009. [accessed 2022 Feb 9]. https://www.who.int/medicines/services/inn/generalpoliciesmonoclonalantibodiesjan10.pdf.
- 71.World Health Organization (WHO) . Revised monoclonal antibody (mAb) nomenclature scheme; 2017. [accessed 2022 Feb 9]. https://www.who.int/medicines/services/inn/Revised_mAb_nomenclature_scheme.pdf.
- 72.World Health Organization (WHO) . New INN monoclonal antibody (mAb) nomenclature scheme; 2021. [accessed 2022 Jun 1]. https://cdn.who.int/media/docs/default-source/international-nonproprietary-names-inn/new_mab_-nomenclature-_2021.pdf
- 73.Koch SSG, Thorpe R, Kawasaki N, Lefranc MP, Malan S, Martin ACR, Mignot G, Plückthun A, Rizzi M, Shubat S, et al. International nonproprietary names for monoclonal antibodies: an evolving nomenclature system. Mabs. 2022;14(1):2075078. doi: 10.1080/19420862.2022.2075078. PMID: 35584276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res. 2011;17(20):6398–405. doi: 10.1158/1078-0432.ccr-11-0487. PMID: 22003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McCombs JR, Owen SC. Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. Aaps J. 2015;17(2):339–51. doi: 10.1208/s12248-014-9710-8. PMID: 25604608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, Rejniak SX, Gordon KA, DeBlanc R, Toki BE, et al. CAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102(4):1458–65. doi: 10.1182/blood-2003-01-0039. PMID: 12714494. [DOI] [PubMed] [Google Scholar]
- 77.Younes A, Yasothan U, Kirkpatrick P. Brentuximab vedotin. Nat Rev Drug Discov. 2012;11(1):19–20. doi: 10.1038/nrd3629. PMID: 22212672. [DOI] [PubMed] [Google Scholar]
- 78.Fenton C, Perry CM. Gemtuzumab ozogamicin. Drugs. 2005;65(16):2405–27. doi: 10.2165/00003495-200565160-00014. PMID: 16266206. [DOI] [PubMed] [Google Scholar]
- 79.Wiseman GA, Gordon LI, Multani PS, Witzig TE, Spies S, Bartlett NL, Schilder RJ, Murray JL, Saleh M, Allen RS, et al. Ibritumomab tiuxetan radioimmunotherapy for patients with relapsed or refractory non-hodgHodgkin lymphoma and mild thrombocytopenia: a phase II multicenter trial. Blood. 2002;99(12):4336–42. doi: 10.1182/blood.v99.12.4336. PMID: 12036859. [DOI] [PubMed] [Google Scholar]
- 80.Starodub AN, Ocean AJ, Shah MA, Guarino MJ, Picozzi VJ, Vahdat LT, Thomas SS, Govindan SV, Maliakal PP, Wegener WA, et al. First-in-human trial of a novel anti-trop-2 antibody-SN-38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin Cancer Res. 2015;21(17):3870–78. doi: 10.1158/1078-0432.ccr-14-3321. PMID: 25944802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goel N, Stephens S. Certolizumab pegol. Mabs. 2010;2(2):137–47. doi: 10.4161/mabs.2.2.11271. PMID: 20190560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blick SKA, Curran MP. Certolizumab pegol. Biodrugs. 2007;21(3):195–201. doi: 10.2165/00063030-200721030-00006. PMID: 17516714. [DOI] [PubMed] [Google Scholar]
- 83.Pasut G. Pegylation of biological molecules and potential benefits: pharmacological properties of certolizumab pegol. Biodrugs. 2014;28(S1):15–23. doi: 10.1007/s40259-013-0064-z. PMID: 24687235. [DOI] [PubMed] [Google Scholar]
- 84.Ladwig PM, Barnidge DR, Willrich MAV. Quantification of the IgG2/4 kappa monoclonal therapeutic eculizumab from serum using isotype specific affinity purification and microflow LC-ESI-Q-TOF mass spectrometry. J Am Soc Mass Spectr. 2017;28(5):811–17. doi: 10.1007/s13361-016-1566-y. PMID: 28004336. [DOI] [PubMed] [Google Scholar]
- 85.World Health Organization (WHO) . International nonproprietary names for pharmaceutical substances (INN): proposed INN: list 117; 2017. [accessed 2022 Sep 20]. https://cdn.who.int/media/docs/default-source/international-nonproprietary-names-(inn)/pl117.pdf. [Google Scholar]
- 86.Bateman A, Martin MJ, Orchard S, Magrane M, Agivetova R, Ahmad S, Alpi E, Bowler-Barnett EH, Britto R, Bursteinas B, et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2020;49(D1):D480–9. doi: 10.1093/nar/gkaa1100. PMID: 33237286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Strohl WR, Ku Z, An Z, Carroll SF, Keyt BA, Strohl LM. Passive immunotherapy against SARS-CoV-2: from plasma-based therapy to single potent antibodies in the race to stay ahead of the variants. Biodrugs. 2022;36(3):231–323. doi: 10.1007/s40259-022-00529-7. PMID: 35476216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Aschenbrenner DS. New indications and approvals for anticancer drugs. Ajn Am J Nurs. 2021;121(12):16–17. doi: 10.1097/01.naj.0000803180.17463.0b. PMID: 34792500. [DOI] [PubMed] [Google Scholar]
- 89.Kim MS, Xu A, Haslam A, Prasad V. Quality of biomarker defined subgroups in FDA approvals of PD‐1/PD‐L1 inhibitors 2014 to 2020. Int J Cancer. 2022;150(11):1905–10. doi: 10.1002/ijc.33968. PMID: 35182072. [DOI] [PubMed] [Google Scholar]
- 90.Ahn S, Lee J, Shin DW, Kim J, Hwang JH. High PD-L1 expression is associated with therapeutic response to pembrolizumab in patients with advanced biliary tract cancer. Sci Rep-Uk. 2020;10(1):12348. doi: 10.1038/s41598-020-69366-4. PMID: 32704067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mo DC, Luo PH, Huang SX, Wang HL, Huang JF. Safety and efficacy of pembrolizumab plus lenvatinib versus pembrolizumab and lenvatinib monotherapies in cancers: a systematic review. Int Immunopharmacol. 2021;91:107281. doi: 10.1016/j.intimp.2020.107281. PMID: 33338862. [DOI] [PubMed] [Google Scholar]
- 92.Qin Q, Li B. Pembrolizumab for the treatment of nonsmall cell lung cancer: current status and future directions. J Canc Res Ther. 2019;15(4):743. doi: 10.4103/jcrt.jcrt_903_18. PMID: 31436226. [DOI] [PubMed] [Google Scholar]
- 93.Neri P, Zucchi M, Allegri P, Lettieri M, Mariotti C, Giovannini A. Adalimumab (HumiraTM): a promising monoclonal anti-tumor necrosis factor alpha in ophthalmology. Int Ophthalmol. 2011;31(2):165–73. doi: 10.1007/s10792-011-9430-3. PMID: 21287227. [DOI] [PubMed] [Google Scholar]
- 94.Patel T, Gordon KB. Adalimumab: efficacy and safety in psoriasis and rheumatoid arthritis. Dermatol Ther. 2004;17(5):427–31. doi: 10.1111/j.1396-0296.2004.04045.x. PMID: 15379777. [DOI] [PubMed] [Google Scholar]
- 95.Alwawi EA, Mehlis SL, Gordon KB. Treating psoriasis with adalimumab. Ther Clin Risk Manag. 2008;4:345–51. doi: 10.2147/tcrm.s1265. PMID: 18728850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Almagro JC, Fransson J. Humanization of antibodies. Frontiers Biosci J Virtual Libr. 2007;13:1619–33. doi: 10.2741/2786. PMID: 17981654. [DOI] [PubMed] [Google Scholar]
- 97.Vaswani SK, Hamilton RG. Humanized antibodies as potential therapeutic drugs. Ann Allergy Asthma Immunol. 1998;81(2):105–19. doi: 10.1016/s1081-1206(10)62794-9. PMID: 9723555. [DOI] [PubMed] [Google Scholar]
- 98.Rochere PDL, Guil-Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized mice for the study of immuno-oncology. Trends Immunol. 2018;39(9):748–63. doi: 10.1016/j.it.2018.07.001. PMID: 30077656. [DOI] [PubMed] [Google Scholar]
- 99.Sgro C. Side-effects of a monoclonal antibody, muromonab CD3/orthoclone OKT3: bibliographic review. Toxicol. 1995;105(1):23–29. doi: 10.1016/0300-483x(95)03123-w. PMID: 8638282. [DOI] [PubMed] [Google Scholar]
- 100.Clark M, Clark M. Antibody humanization: a case of the ‘emperor’s new clothes’? Immunol Today. 2000;21(8):397–402. doi: 10.1016/s0167-5699(00)01680-7. PMID: 10916143. [DOI] [PubMed] [Google Scholar]
- 101.Hwang WYK, Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36(1):3–10. doi: 10.1016/j.ymeth.2005.01.001. PMID: 15848070. [DOI] [PubMed] [Google Scholar]
- 102.Kessler M, Goldsmith D, Schellekens H. Immunogenicity of biopharmaceuticals. Nephrol Dial Transpl. 2006;21(suppl_5):v9–12. doi: 10.1093/ndt/gfl476. PMID: 16959792. [DOI] [PubMed] [Google Scholar]
- 103.U.S. Food and Drug Administration (FDA) . Highlights of prescribing information (MYLOTARG); 2020. [accessed 2022 Oct 25]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761060s004lbl.pdf.
- 104.Lee JE, Liu J, John C, Williams G, Rahman NA, Wang Y. Clinical pharmacology and biopharmaceutics (Mylotarg); 2016. [accessed 2022 Oct 25]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761060Orig1s000Orig1Orig2s000ClinPharmR.pdf.
- 105.European Medicines Agency (EMA) . Guideline on immunogenicity assessment of therapeutic proteins; 2017. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf.
- 106.U.S. Food and Drug Administration (FDA) . Immunogenicity testing of therapeutic protein products — developing and validating assays for anti-drug antibody detection; 2019. https://www.fda.gov/media/119788/download.
- 107.Pratt KP. Anti-drug antibodies: emerging approaches to predict, reduce or reverse biotherapeutic immunogenicity. Antibodies. 2018;7(2):19. doi: 10.3390/antib7020019. PMID: 31544871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Myler H, Pedras-Vasconcelos J, Phillips K, Hottenstein CS, Chamberlain P, Devanaryan V, Gleason C, Goodman J, Manning MS, Purushothama S, et al. Anti-drug antibody validation testing and reporting harmonization. Aaps J. 2021;24(1):4. doi: 10.1208/s12248-021-00649-y. PMID: 34853961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schellekens H. Factors influencing the immunogenicity of therapeutic proteins. Nephrol Dial Transpl. 2005;20(suppl_6):vi3–9. doi: 10.1093/ndt/gfh1092. PMID: 15958824. [DOI] [PubMed] [Google Scholar]
- 110.Meis CM, Salzman EE, Maikawa CL, Smith AAA, Mann JL, Grosskopf AK, Appel EA. Self-assembled, dilution-responsive hydrogels for enhanced thermal stability of insulin biopharmaceuticals. Acs Biomater Sci Eng. 2021;7(9):4221–29. doi: 10.1021/acsbiomaterials.0c01306. PMID: 34510910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Buckley LA, Salunke S, Thompson K, Baer G, Fegley D, Turner MA. Challenges and strategies to facilitate formulation development of pediatric drug products: safety qualification of excipients. Int J Pharmaceut. 2018;536(2):563–69. doi: 10.1016/j.ijpharm.2017.07.042. PMID: 28729174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pilunni D, Santuccio C, Sottosanti L, Felicetti P, Navarra P. Relationship between injection site reactions and different adalimumab formulations. Analysis of the adverse events reported in Italy in the period 2016-2019. Eur Rev Med Pharmaco. 2021;25:3300–05. doi: 10.26355/eurrev_202104_25468. PMID: 33928613. [DOI] [PubMed] [Google Scholar]
- 113.Yang YL, Lai TW. Citric acid in drug formulations causes pain by potentiating acid-sensing ion channel 1. J Neurosci. 2021;41(21):4596–606. doi: 10.1523/jneurosci.2087-20.2021. PMID: 33888605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Clair-Jones AS, Prignano F, Goncalves J, Paul M, Sewerin P. Understanding and minimising injection-site pain following subcutaneous administration of biologics: a narrative review. Rheumatology Ther. 2020;7(4):741–57. doi: 10.1007/s40744-020-00245-0. PMID: 33206343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shi GH, Pisupati K, Parker JG, Corvari VJ, Payne CD, Xu W, Collins DS, Felippis MRD. Subcutaneous injection site pain of formulation matrices. Pharmaceut Res. 2021;38(5):779–93. doi: 10.1007/s11095-021-03047-3. PMID: 33942212. [DOI] [PubMed] [Google Scholar]
- 116.Ionova Y, Wilson L, Mallela KMG. Biologic excipients: importance of clinical awareness of inactive ingredients. Plos One. 2020;15(6):e0235076. doi: 10.1371/journal.pone.0235076. PMID: 32584876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nash P, Vanhoof J, Hall S, Arulmani U, Tarzynski-Potempa R, Unnebrink K, Payne AN, Cividino A. Randomized crossover comparison of injection site pain with 40 mg/0.4 or 0.8 mL formulations of adalimumab in patients with rheumatoid arthritis. Rheumatol Ther. 2016;3(2):257–70. doi: 10.1007/s40744-016-0041-3. PMID: 27747583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Usach I, Martinez R, Festini T, Peris J-E. Subcutaneous injection of drugs: literature review of factors influencing pain sensation at the injection site. Adv Ther. 2019;36(11):2986–96. doi: 10.1007/s12325-019-01101-6. PMID: 31587143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJT. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 2010;9(4):325–38. doi: 10.1038/nrd3003. PMID: 20305665. [DOI] [PubMed] [Google Scholar]
- 120.Jiao Z, Wang G, Feng Z, Yan Z, Zhang J, Li G, Wang Q, Feng D. Safety profile of monoclonal antibody compared with traditional anticancer drugs: an analysis of Henan province spontaneous reporting system database. Front Pharmacol. 2022;12:760013. doi: 10.3389/fphar.2021.760013. PMID: 35145400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li Y, Qi L, Bai H, Sun C, Xu S, Wang Y, Han C, Li Y, Liu L, Cheng X, et al. Safety, tolerability, pharmacokinetics, and immunogenicity of a monoclonal antibody (SCTA01) targeting SARS-CoV-2 in healthy adults: a randomized, double-blind, placebo-controlled, phase I study. Antimicrob Agents Ch. 2021;65(11):e01063–21. doi: 10.1128/aac.01063-21. PMID: 34491805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hoe S, Boraey MA, Ivey JW, Finlay WH, Vehring R. Manufacturing and device options for the delivery of biotherapeutics. J Aerosol Med Pulm D. 2014;27(5):315–28. doi: 10.1089/jamp.2013.1090. PMID: 24299502. [DOI] [PubMed] [Google Scholar]
- 123.Keytruda [Package Insert], ORIG-1 . 2014. [accessed 2022 Sep 23]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125514lbl.pdf.
- 124.Keytruda [Package Insert], SUPPL-3 . 2015. [accessed 2022 Sep 23]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125514s003lbl.pdf.
- 125.Keytruda [Package Insert], SUPPL-67 . 2019. [accessed 2022 Sep 23]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125514s067lbl.pdf.
- 126.Fanelli JL. Injection: the third method of drug administration. Review of Optometry. 2012;149:32+. [Google Scholar]
- 127.Dornbos BD, Moliterno CD. Injection procedures encountered in optometry [dissertation]. Big Rapids (MI): Ferris State University; 2012. [Google Scholar]
- 128.Arivazhahan A. Introduction to basics of pharmacology and toxicology, volume 1: general and molecular pharmacology: principles of drug action. 2019. pp. 69–79. doi: 10.1007/978-981-32-9779-1_4. [DOI] [Google Scholar]
- 129.U.S. Food and Drug Administration (FDA) . Route of administration | FDA; 2017. [accessed 2022 Sep 27]. https://www.fda.gov/drugs/data-standards-manual-monographs/route-administration.
- 130.Locke KW, Maneval DC, LaBarre MJ. ENHANZE® drug delivery technology: a novel approach to subcutaneous administration using recombinant human hyaluronidase PH20. Drug Deliv. 2019;26(1):98–106. doi: 10.1080/10717544.2018.1551442. PMID: 30744432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Heo YA, Syed YY. Subcutaneous trastuzumab: a review in HER2-positive breast cancer. Target Oncol. 2019;14(6):749–58. doi: 10.1007/s11523-019-00684-y. PMID: 31686307. [DOI] [PubMed] [Google Scholar]
- 132.Villalba ML. Medical review; 2016. [accessed 2022 Apr 11]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761029Orig1s000MedR.pdf.
- 133.Ribrag V, Dupuis J, Tilly H, Morschhauser F, Laine F, Houot R, Haioun C, Copie C, Varga A, Lambert J, et al. A dose-escalation study of SAR3419, an anti-CD19 antibody maytansinoid conjugate, administered by intravenous infusion once weekly in patients with relapsed/refractory B-cell non-hodgHodgkin lymphoma. Clin Cancer Res. 2014;20(1):213–20. doi: 10.1158/1078-0432.ccr-13-0580. PMID: 24132920. [DOI] [PubMed] [Google Scholar]
- 134.Jonaitis L, Marković S, Farkas K, Gheorghe L, Krznarić Ž, Salupere R, Mokricka V, Spassova Z, Gatev D, Grosu I, et al. Intravenous versus subcutaneous delivery of biotherapeutics in IBD: an expert’s and patient’s perspective. BMC Proc. 2021;15(S17):25. doi: 10.1186/s12919-021-00230-7. PMID: 34879868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Falanga M, Canzona A, Mazzoni D. Preference for subcutaneous injection or intravenous infusion of biological therapy among Italian patients with SLE. J Patient Exp. 2019;6(1):41–45. doi: 10.1177/2374373518770811. PMID: 31236450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Overton PM, Shalet N, Somers F, Allen JA. Patient preferences for subcutaneous versus intravenous administration of treatment for chronic immune system disorders: a systematic review. Patient Prefer Adher. 2021;15:811–34. doi: 10.2147/ppa.s303279. PMID: 33907384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11(1–2):81–88. doi: 10.1016/s1359-6446(05)03638-x. PMID: 16478695. [DOI] [PubMed] [Google Scholar]
- 138.Toutain PL, Bousquet‐mélou A. Plasma terminal half‐life. J Vet Pharmacol Ther. 2004;27(6):427–39. doi: 10.1111/j.1365-2885.2004.00600.x. PMID: 15601438. [DOI] [PubMed] [Google Scholar]
- 139.Birkett DJ. Pocket guide: pharmacokinetics made easy. Maidenhead, England: McGraw-Hill Education; 2009. [Google Scholar]
- 140.Smith DA, Beaumont K, Maurer TS, Di L. Volume of distribution in drug design. J Med Chem. 2015;58(15):5691–98. doi: 10.1021/acs.jmedchem.5b00201. PMID: 25799158. [DOI] [PubMed] [Google Scholar]
- 141.Toutain PL, Bousquet-Mélou A. Bioavailability and its assessment. J Vet Pharmacol Ther. 2004;27(6):455–66. doi: 10.1111/j.1365-2885.2004.00604.x. PMID: 15601440. [DOI] [PubMed] [Google Scholar]
- 142.Kuna M, Mahdi F, Chade AR, Bidwell GL. Molecular size modulates pharmacokinetics, biodistribution, and renal deposition of the drug delivery biopolymer elastin-like polypeptide. Sci Rep-Uk. 2018;8(1):7923. doi: 10.1038/s41598-018-24897-9. PMID: 29784932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Byers JP, Sarver JG. Chapter 10 - pharmacokinetic modeling. In: Hacker M, Messer WBachmann K, editors. Pharmacology: principles and practice. San Diego: Academic Press; 2009. pp. 201–77. doi: 10.1016/b978-0-12-369521-5.00010-5. [DOI] [Google Scholar]
- 144.Andrade C. The practical importance of half-life in psychopharmacology. J Clin Psychiatry. 2022;83:4. doi: 10.4088/jcp.22f14584. PMID: 35900254. [DOI] [PubMed] [Google Scholar]
- 145.Durairaj C, Shah JC, Senapati S, Kompella UB. Prediction of vitreal half-life based on drug physicochemical properties: quantitative structure–pharmacokinetic relationships (QSPKR). Pharmaceut Res. 2008;26(5):1236. doi: 10.1007/s11095-008-9728-7. PMID: 18841448. [DOI] [PubMed] [Google Scholar]
- 146.Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507. doi: 10.2165/11531280-000000000-00000. PMID: 20608753. [DOI] [PubMed] [Google Scholar]
- 147.Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633–59. doi: 10.2165/11535960-000000000-00000. PMID: 20818831. [DOI] [PubMed] [Google Scholar]
- 148.Dall’acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol. 2002;169(9):5171–80. doi: 10.4049/jimmunol.169.9.5171. PMID: 12391234. [DOI] [PubMed] [Google Scholar]
- 149.Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Ward ES. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc National Acad Sci. 2006;103(49):18709–14. doi: 10.1073/pnas.0606304103. PMID: 17116867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang W, Wang E, Balthasar J. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–58. doi: 10.1038/clpt.2008.170. PMID: 18784655. [DOI] [PubMed] [Google Scholar]
- 151.Gibson JG, Evans WA. Clinical studies of the blood volume. II. The relation of plasma and total blood volume to venous pressure, blood velocity rate, physical measurements, age and sex in ninety normal humans. J Clin Invest. 1937;16(3):317–28. doi: 10.1172/jci100860. PMID: 16694481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gibaldi M, McNamara PJ. Apparent volumes of distribution and drug binding to plasma proteins and tissues. Eur J Clin Pharmacol. 1978;13(5):373–78. doi: 10.1007/bf00644611. PMID: 668796. [DOI] [PubMed] [Google Scholar]
- 153.Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. Cpt Pharmacometrics Syst Pharmacol. 2017;6(9):576–88. doi: 10.1002/psp4.12224. PMID: 28653357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bookbinder LH, Hofer A, Haller MF, Zepeda ML, Keller G-A, Lim JE, Edgington TS, Shepard HM, Patton JS, Frost GI. A recombinant human enzyme for enhanced interstitial transport of therapeutics. J Control Release. 2006;114(2):230–41. doi: 10.1016/j.jconrel.2006.05.027. PMID: 16876899. [DOI] [PubMed] [Google Scholar]
- 155.Sime FB, Roberts MS, Roberts JA. Optimization of dosing regimens and dosing in special populations. Clin Microbiol Infec. 2015;21(10):886–93. doi: 10.1016/j.cmi.2015.05.002. PMID: 25980350. [DOI] [PubMed] [Google Scholar]
- 156.Tabrizi M, Neupane D, Elie SE, Shankaran H, Juan V, Zhang S, Hseih S, Fayadat-Dilman L, Zhang D, Song Y, et al. Pharmacokinetic properties of humanized IgG1 and IgG4 antibodies in preclinical species: translational evaluation. Aaps J. 2019;21(3):39. doi: 10.1208/s12248-019-0304-3. PMID: 30868312. [DOI] [PubMed] [Google Scholar]
- 157.Wang W, Prueksaritanont T. Prediction of human clearance of therapeutic proteins: simple allometric scaling method revisited. Biopharm Drug Dispos. 2010;31:253–63. doi: 10.1002/bdd.708. PMID: 20437464. [DOI] [PubMed] [Google Scholar]
- 158.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel. 2010;23(5):385–92. doi: 10.1093/protein/gzq009. PMID: 20159773. [DOI] [PubMed] [Google Scholar]
- 159.Yang D, Kroe-Barrett R, Singh S, Laue T. IgG charge: practical and biological implications. Antibodies. 2019;8(1):24. doi: 10.3390/antib8010024. PMID: 31544830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Piche-Nicholas NM, Avery LB, King AC, Kavosi M, Wang M, O’hara DM, Tchistiakova L, Katragadda M. Changes in complementarity-determining regions significantly alter IgG binding to the neonatal Fc receptor (FcRn) and pharmacokinetics. Mabs. 2018;10(1):81–94. doi: 10.1080/19420862.2017.1389355. PMID: 28991504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjugate Chem. 2010;21(12):2153–63. doi: 10.1021/bc100261d. PMID: 21053952. [DOI] [PubMed] [Google Scholar]
- 162.Yadav DB, Sharma VK, Boswell CA, Hotzel I, Tesar D, Shang Y, Ying Y, Fischer SK, Grogan JL, Chiang EY, et al. Evaluating the use of antibody variable region (Fv) charge as a risk assessment tool for predicting typical cynomolgus monkey pharmacokinetics*. J Biol Chem. 2015;290(50):29732–41. doi: 10.1074/jbc.m115.692434. PMID: 26491012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. Mabs. 2015;7(3):483–93. doi: 10.1080/19420862.2015.1016696. PMID: 25695748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kingsbury JS, Saini A, Auclair SM, Fu L, Lantz MM, Halloran KT, Calero-Rubio C, Schwenger W, Airiau CY, Zhang J, et al. A single molecular descriptor to predict solution behavior of therapeutic antibodies. Sci Adv. 2020;6(32):eabb0372. doi: 10.1126/sciadv.abb0372. PMID: 32923611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Khawli LA, Glasky MS, Alauddin MM, Epstein AL. Improved tumor localization and radioimaging with chemically modified monoclonal antibodies. Cancer Biotherapy Radiopharm. 1996;11(3):203–15. doi: 10.1089/cbr.1996.11.203. PMID: 10851539. [DOI] [PubMed] [Google Scholar]
- 166.Li L, Kumar S, Buck PM, Burns C, Lavoie J, Singh SK, Warne NW, Nichols P, Luksha N, Boardman D. Concentration dependent viscosity of monoclonal antibody solutions: explaining experimental behavior in terms of molecular properties. Pharmaceut Res. 2014;31(11):3161–78. doi: 10.1007/s11095-014-1409-0. PMID: 24906598. [DOI] [PubMed] [Google Scholar]
- 167.Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc National Acad Sci. 2014;111(52):18601–06. doi: 10.1073/pnas.1421779112. PMID: 25512516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Strickley RG, Lambert WJ. A review of formulations of commercially available antibodies. J Pharm Sci. 2021;110(7):e56. doi: 10.1016/j.xphs.2021.03.017. PMID: 33789155. [DOI] [PubMed] [Google Scholar]
- 169.Garidel P, Blume A, Wagner M. Prediction of colloidal stability of high concentration protein formulations. Pharm Dev Technol. 2014;20(3):367–74. doi: 10.3109/10837450.2013.871032. PMID: 24392929. [DOI] [PubMed] [Google Scholar]
- 170.Sun MF, Liao JN, Jing ZY, Gao H, Shen BB, Y-F X, Fang WJ. Effects of polyol excipient stability during storage and use on the quality of biopharmaceutical formulations. J Pharm Analysis. 2022;12(5):774–82. doi: 10.1016/j.jpha.2022.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.James S, McManus JJ. Thermal and solution stability of lysozyme in the presence of sucrose, glucose, and trehalose. J Phys Chem B. 2012;116(34):10182–88. doi: 10.1021/jp303898g. PMID: 22909409. [DOI] [PubMed] [Google Scholar]
- 172.Gervasi V, Agnol RD, Cullen S, McCoy T, Vucen S, Crean A. Parenteral protein formulations: an overview of approved products within the European union. Eur J Pharm Biopharm. 2018;131:8–24. doi: 10.1016/j.ejpb.2018.07.011. PMID: 30006246. [DOI] [PubMed] [Google Scholar]
- 173.Liu GY, Nie S, Zheng X, Li N. Activity-based protein profiling probe for the detection of enzymes catalyzing polysorbate degradation. Anal Chem. 2022;94(24):8625–32. doi: 10.1021/acs.analchem.2c00059. [DOI] [PubMed] [Google Scholar]
- 174.Jones MT, Mahler HC, Yadav S, Bindra D, Corvari V, Fesinmeyer RM, Gupta K, Harmon AM, Hinds KD, Koulov A, et al. Considerations for the use of polysorbates in biopharmaceuticals. Pharmaceut Res. 2018;35(8):148. doi: 10.1007/s11095-018-2430-5. PMID: 29797101. [DOI] [PubMed] [Google Scholar]
- 175.Dwivedi M, Blech M, Presser I, Garidel P. Polysorbate degradation in biotherapeutic formulations: identification and discussion of current root causes. Int J Pharmaceut. 2018;552(1–2):422–36. doi: 10.1016/j.ijpharm.2018.10.008. PMID: 30300706. [DOI] [PubMed] [Google Scholar]
- 176.Singh SK, Kolhe P, Mehta AP, Chico SC, Lary AL, Huang M. Frozen state storage instability of a monoclonal antibody: aggregation as a consequence of trehalose crystallization and protein unfolding. Pharmaceut Res. 2011;28(4):873–85. doi: 10.1007/s11095-010-0343-z. PMID: 21213025. [DOI] [PubMed] [Google Scholar]
- 177.Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci. 2007;96(1):1–26. doi: 10.1002/jps.20727. PMID: 16998873. [DOI] [PubMed] [Google Scholar]
- 178.Neergaard MS, Nielsen AD, Parshad H, Weert MVD. Stability of monoclonal antibodies at high‐Concentration: head‐to‐head comparison of the IgG1 and IgG4 subclass. J Pharm Sci. 2014;103(1):115–27. doi: 10.1002/jps.23788. PMID: 24282022. [DOI] [PubMed] [Google Scholar]
- 179.Garidel P, Kuhn AB, Schäfer LV, Karow-Zwick AR, Blech M. High-concentration protein formulations: how high is high? Eur J Pharm Biopharm. 2017;119:353–60. doi: 10.1016/j.ejpb.2017.06.029. PMID: 28690199. [DOI] [PubMed] [Google Scholar]
- 180.Agrawal NJ, Helk B, Kumar S, Mody N, Sathish HA, Samra HS, Buck PM, Li L, Trout BL. Computational tool for the early screening of monoclonal antibodies for their viscosities. Mabs. 2016;8(1):43–48. doi: 10.1080/19420862.2015.1099773. PMID: 26399600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Neergaard MS, Kalonia DS, Parshad H, Nielsen AD, Møller EH, van de WM. Viscosity of high concentration protein formulations of monoclonal antibodies of the IgG1 and IgG4 subclass – prediction of viscosity through protein–protein interaction measurements. Eur J Pharm Sci. 2013;49(3):400–10. doi: 10.1016/j.ejps.2013.04.019. PMID: 23624326. [DOI] [PubMed] [Google Scholar]
- 182.Wang S, Zhang N, Hu T, Dai W, Feng X, Zhang X, Qian F. Viscosity-lowering effect of amino acids and salts on highly concentrated solutions of two IgG1 monoclonal antibodies. Mol Pharmaceut. 2015;12(12):4478–87. doi: 10.1021/acs.molpharmaceut.5b00643. PMID: 26528726. [DOI] [PubMed] [Google Scholar]
- 183.Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S, Balthazor B, Conley TR, Sek D, Allen MJ. Rational design of viscosity reducing mutants of a monoclonal antibody: hydrophobic versus electrostatic inter-molecular interactions. Mabs. 2015;7(1):212–30. doi: 10.4161/19420862.2014.985504. PMID: 25559441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang W. Advanced protein formulations. Protein Sci. 2015;24:1031–39. doi: 10.1002/pro.2684. PMID: 25858529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Warne NW. Development of high concentration protein biopharmaceuticals: the use of platform approaches in formulation development. Eur J Pharm Biopharm. 2011;78(2):208–12. doi: 10.1016/j.ejpb.2011.03.004. PMID: 21406226. [DOI] [PubMed] [Google Scholar]
- 186.Gupta P, Makowski EK, Kumar S, Zhang Y, Scheer JM, Tessier PM. Antibodies with weakly basic isoelectric points minimize trade-offs between formulation and physiological colloidal properties. Mol Pharmaceut. 2021;19(3):775–87. doi: 10.1021/acs.molpharmaceut.1c00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jäger S, Wagner TR, Rasche N, Kolmar H, Hecht S, Schröter C. Generation and biological evaluation of Fc antigen binding fragment-drug conjugates as a novel antibody-based format for targeted drug delivery. Bioconjugate Chem. 2021;32(8):1699–710. doi: 10.1021/acs.bioconjchem.1c00240. PMID: 34185508. [DOI] [PubMed] [Google Scholar]
- 188.Murer P, Neri D. Antibody-cytokine fusion proteins: a novel class of biopharmaceuticals for the therapy of cancer and of chronic inflammation. New Biotechnol. 2019;52:42–53. doi: 10.1016/j.nbt.2019.04.002. PMID: 30991144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Egan TJ, Diem D, Weldon R, Neumann T, Meyer S, Urech DM. Novel multispecific heterodimeric antibody format allowing modular assembly of variable domain fragments. Mabs. 2017;9(1):68–84. doi: 10.1080/19420862.2016.1248012. PMID: 27786600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Schellekens H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov. 2002;1(6):457–62. doi: 10.1038/nrd818. PMID: 12119747. [DOI] [PubMed] [Google Scholar]
- 191.Deehan M, Garcês S, Kramer D, Baker MP, Rat D, Roettger Y, Kromminga A. Managing unwanted immunogenicity of biologicals. Autoimmun Rev. 2015;14(7):569–74. doi: 10.1016/j.autrev.2015.02.007. PMID: 25742758. [DOI] [PubMed] [Google Scholar]
- 192.Johnson DE. Biotherapeutics: challenges and opportunities for predictive toxicology of monoclonal antibodies. Int J Mol Sci. 2018;19(11):3685. doi: 10.3390/ijms19113685. PMID: 30469350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Gorovits B, Clements-Egan A, Birchler M, Liang M, Myler H, Peng K, Purushothama S, Rajadhyaksha M, Salazar-Fontana L, Sung C, et al. Pre-existing antibody: biotherapeutic modality-based review. Aaps J. 2016;18(2):311–20. doi: 10.1208/s12248-016-9878-1. PMID: 26821802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Knezevic I, Griffiths E. WHO standards for biotherapeutics, including biosimilars: an example of the evaluation of complex biological products. Ann Ny Acad Sci. 2018;1407(1):5–16. doi: 10.1111/nyas.13434. PMID: 28905423. [DOI] [PubMed] [Google Scholar]
- 195.Mehta MU, Uppoor RS, Conner DP, Seo P, Vaidyanathan J, Volpe DA, Stier E, Chilukuri D, Dorantes A, Ghosh T, et al. Impact of the US FDA “biopharmaceutics classification system” (BCS) guidance on global drug development. Mol Pharmaceut. 2017;14(12):4334–38. doi: 10.1021/acs.molpharmaceut.7b00687. PMID: 29076742. [DOI] [PubMed] [Google Scholar]
- 196.Darrow JJ, Avorn J, Kesselheim AS. FDA approval and regulation of pharmaceuticals, 1983-2018. Jama. 2020;323(2):164–76. doi: 10.1001/jama.2019.20288. PMID: 31935033. [DOI] [PubMed] [Google Scholar]
- 197.Ishii-Watabe A, Shibata H, Nishimura K, Hosogi J, Aoyama M, Nishimiya K, Saito Y. Immunogenicity of therapeutic protein products: current considerations for anti-drug antibody assay in Japan. Bioanalysis. 2017;10(2):95–105. doi: 10.4155/bio-2017-0186. PMID: 29243491. [DOI] [PubMed] [Google Scholar]
- 198.Downing NS, Aminawung JA, Shah ND, Krumholz HM, Ross JS. Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005-2012. Jama. 2014;311(4):368–77. doi: 10.1001/jama.2013.282034. PMID: 24449315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dreher-Lesnick SM, Stibitz S, Carlson PE, Britton RA, Cani PD. U.S. regulatory considerations for development of live biotherapeutic products as drugs. Microbiol Spectr. 2017;5:5. doi: 10.1128/microbiolspec.bad-0017-2017. PMID: 28975881. [DOI] [PubMed] [Google Scholar]
- 200.Hung JJ, Dear BJ, Dinin AK, Borwankar AU, Mehta SK, Truskett TT, Johnston KP. Improving viscosity and stability of a highly concentrated monoclonal antibody solution with concentrated proline. Pharmaceut Res. 2018;35(7):133. doi: 10.1007/s11095-018-2398-1. PMID: 29713822. [DOI] [PubMed] [Google Scholar]
- 201.Rodrigues D, Tanenbaum LM, Thirumangalathu R, Somani S, Zhang K, Kumar V, Amin K, Thakkar SV. Product-specific impact of viscosity modulating formulation excipients during ultra-high concentration biotherapeutics drug product development. J Pharm Sci. 2021;110(3):1077–82. doi: 10.1016/j.xphs.2020.12.016. PMID: 33340533. [DOI] [PubMed] [Google Scholar]
- 202.Kumar S, Singh SK. Developability of biotherapeutics: computational approaches. 1st ed. Boca Raton (FL): CRC Press; 2015. [Google Scholar]
- 203.Ahmed L, Gupta P, Martin KP, Scheer JM, Nixon AE, Kumar S. Intrinsic physicochemical profile of marketed antibody-based biotherapeutics. P Natl Acad Sci USA. 2021;118(37):e2020577118. doi: 10.1073/pnas.2020577118. PMID: 34504010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Prabakaran R, Rawat P, Kumar S, Gromiha MM. Evaluation of in silico tools for the prediction of protein and peptide aggregation on diverse datasets. Brief Bioinform. 2021;22(6):PMID: 34181000. doi: 10.1093/bib/bbab240. [DOI] [PubMed] [Google Scholar]
- 205.Liu Y, Tsang K, Mays M, Hansen G, Chiecko J, Crames M, Wei Y, Zhou W, Fredrick C, Hu J, et al. An adapted consensus protein design strategy for identifying globally optimal biotherapeutics. Mabs. 2022;14(1):2073632. doi: 10.1080/19420862.2022.2073632. PMID: 35613320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lin J, Lee SL, Russell AM, Huang RF, Batt MA, Chang SS, Ferrante A, Verdino P, Henry KA. A structure-based engineering approach to abrogate pre-existing antibody binding to biotherapeutics. Plos One. 2021;16(7):e0254944. doi: 10.1371/journal.pone.0254944. PMID: 34297759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Narayanan H, Dingfelder F, Butté A, Lorenzen N, Sokolov M, Arosio P. Machine learning for biologics: opportunities for protein engineering, developability, and formulation. Trends Pharmacol Sci. 2021;42(3):151–65. doi: 10.1016/j.tips.2020.12.004. PMID: 33500170. [DOI] [PubMed] [Google Scholar]
- 208.The Antibody Society . Therapeutic monoclonal antibodies approved or in review in the EU or US; 2022. [accessed 2022 Feb 9]. www.antibodysociety.org.
- 209.Clinical Trials . 2022. [accessed 2022 Feb 9]. https://clinicaltrials.gov/.
- 210.Lefranc MP. IMGT, the international ImMunoGeneTics information system. Cold Spring Harb Protoc. 2011;2011(6):db.top115. doi: 10.1101/pdb.top115. PMID: 21632786. [DOI] [PubMed] [Google Scholar]
- 211.Lefranc MP, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, et al. IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res. 2015;43(D1):D413–22. doi: 10.1093/nar/gku1056. PMID: 25378316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Craic . Therapeutic antibody database; 2022. [accessed 2022 Feb 8]. https://tabs.craic.com.
- 213.Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41:W34–40. doi: 10.1093/nar/gkt382. PMID: 23671333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Molecular Operating Environment (MOE) . Chemical computing group ULC; 2022. [accessed 2023 Feb 15]. https://www.chemcomp.com/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.