

Abstract
Next-generation sequencing (NGS) has failed to detect mesenchymal epithelial transition factor gene (MET) polysomy in previous studies. We included three non–small cell lung cancer (NSCLC) cohorts in this retrospective study to establish new criteria for detecting MET polysomy and to explore the clinical relevance of MET polysomy. Cohort 1 included 53 patients whose tissues were available for both FISH and NGS assays. Paired plasma and tissue samples were obtained from 261 patients with NSCLC as cohort 2. Cohort 3 included 46 patients with metastatic NSCLC, who presented with MET copy-number gain assessed by NGS. ROC analysis demonstrated that a cut-off point of 2.3 copies achieved the maximum Youden index in discriminating polysomy from normal copy number. Compared with the FISH test for MET polysomy, the sensitivity, specificity, and agreement of NGS were 90%, 90%, and 96.2%, respectively. Following optimization using maximum somatic allele frequency, the sensitivity and specificity of NGS for defining polysomy using plasma samples according to different circulating tumor DNA mutation frequencies were 42% and 63%. The concordance rate between tissue and plasma samples for detecting polysomy was 85%. Regarding the response to MET inhibitor, the median progression-free survival (PFS) of the MET amplification group was significantly higher than that of the polysomy group. The median PFS was similar between the polysomy and normal groups. Our results indicated that NGS may serve as an alternative method for detecting MET polysomy in NSCLC tissues. Moreover, patients with MET polysomy may not benefit from MET inhibitors.
Significance:
In this study, we established a methodology to differentiate polysomy from normal copy numbers and amplification using NGS. Moreover, this study suggests that it is critical to discriminate MET polysomy from amplification, for the former may dilute the clinical benefit of MET inhibitors.
Introduction
Alterations in the mesenchymal epithelial transition factor gene (MET), such as exon 14 skipping mutation and amplification, have been regarded as driver oncogenic mutations and are potentially targetable (1–4). Moreover, MET copy-number gain (CNG) has been described as one of the mechanisms of acquired resistance to EGFR tyrosine kinase inhibitors (TKI; refs. 2, 5, 6). In the majority of clinical trials, a range of FISH criteria was used to identify patients with MET CNG, which could provide information on the absolute MET copy number and the MET/centromeric enumeration of chromosome 7 (CEP7) ratios (7). However, several limitations exist: this method allows us to investigate only the regions for which FISH probes are available, and multiple FISH probes are needed to be comprehensive, with each probe requiring a resource-consuming validation. Other assays, such as IHC or droplet digital PCR, were neither in poor agreement with FISH nor feasible in the clinic (8–10). In contrast, next-generation sequencing (NGS) technology has improved gene mutation detection and shows many advantages. However, an appropriate threshold for the aforementioned methodology remains to be determined, on the basis of which a MET-target therapy would likely be effective (7).
A proportion of cases with MET CNG is actually harboring polysomy rather than amplification (11). According to current theories, polysomy is not regarded as a driver oncogenic mutation (12). MET/CEP7 ratios were used to discriminate amplification from polysomy and normal copy number using a FISH test. Meanwhile, NGS has failed to separate MET amplification from polysomy in previous studies (12, 13), and the detection of polysomy by NGS represents a clinical gray zone. The threshold of MET copy numbers to distinguish polysomy from normal copy numbers has not yet been established. Therefore, it remains unclear whether NGS can serve as an alternative method to identify different MET abnormalities.
In this study, we aimed to establish an optimal copy-number cut-off value to differentiate polysomy from normal copy numbers using NGS. In addition, we proposed a specific algorithm to detect the MET status following NGS, which is capable of discriminating polysomy from MET amplification. Moreover, the detection performance of NGS for MET polysomy was investigated using both tissue and plasma samples. Furthermore, we explored the response to MET inhibitors in patients with different MET statuses to determine which type of MET status may benefit from MET inhibitors.
Materials and Methods
Study Participants and Design
Cohort 1 was used to establish the cut-off value of MET CNG to distinguish polysomy from normal copy numbers. Between October 2018 and July 2021, 53 patients with NSCLC harboring MET CNG were included in this study. Tissue samples of all patients were available for both FISH and NGS assays. An in-house cohort of 155 patients with untreated EGFR-mutated NSCLC was used to verify the rationality of the proposed method. In this cohort, 155 patients were diagnosed with metastatic EGFR-mutated NSCLC and were untreated previously. All patients harbored a sensitizing EGFR mutation, including 67 with EGFR 19del, 71 with EGFR L858R, 7 with EGFR G719A, 5 with EGFR S768I, and 5 with EGFR L861Q.
Cohort 2 was used to evaluate the concordance of MET status between tissue and plasma samples using NGS. Paired plasma and tissue samples were obtained from 261 patients with NSCLC at the same time. The MET status was divided into three groups: amplification, polysomy, and normal copy number, according to the cut-off value described above. All 261 patients were allocated to one of three groups based on their MET status.
Cohort 3 was used to determine the correlation between the response to MET inhibitors and different MET statuses. Patients in cohort 3 were diagnosed with advanced NSCLC with a high MET copy number (assessed using NGS with tissue samples) and progressed after at least one line of therapy. In total, 46 patients were included in this retrospective study from June 2018 to November 2021.
Supplementary Figure (Supplementary Fig. S1) shows a flowchart of the study design. Written informed consent was obtained from all the patients, and the study was approved by the Ethics Committee of the National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences, and Peking Union Medical College.
FISH
FISH was performed using the Vysis MET Spectrum Red FISH Probe Kit and Vysis CEP7 Spectrum Green FISH probe kit. According to the present criteria, MET amplification was defined as gene copy number ≥5 and/or MET/CEP7 ratio > 2.0.
Next-generation Sequencing
DNA Extraction and Sequencing
DNA was extracted from fresh-frozen tissues or formalin-fixed, paraffin-embedded (FFPE) tumor tissue specimens using the QIAamp DNA Mini Kit (Qiagen) and the ReliaPrep FFPE gDNA Miniprep System (Promega). DNA was extracted from plasma specimens using the QIAamp Circulating Nucleic Acid Kit (Qiagen). All samples were obtained from patients after signing informed consent. Prior to pooling, the samples’ concentrations were quantified by Qbit. A minimum of 15 ng of cell-free DNA was required for NGS library construction. Next, the DNA was sheared into fragments at a 200–250 bp peak using a Covaris S2 ultrasonicator (Covaris, Inc.), and indexed NGS libraries were prepared using the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB). Finally, the 1,021 panel (Integrated DNA Technologies, Inc.), covering approximately 1.5 Mbp of the genome and targeting 1,021 cancer-related genes, was used for hybridization enrichment. The indexed libraries were sequenced using a 100-bp paired-end configuration on a DNBSEQ-T7RS sequencer (MGI Tech) or Gene+Seq-2000 sequencing system (GenePlus-Suzhou), producing 3 Gb of data for fresh specimens/FFPE.
Quality Control
Standard quality control was performed on all sequencing data for all samples. A coverage depth of not less than 100X in more than 98% of exons, sequencing depth of at least 200X, and mapping rate of not less than 95% must be achieved.
MET Amplification and Polysomy Analysis
Copy-number Variation Analysis
Variant calling and copy-number variation analysis of each capture and non-capture regions were performed using the cnvkit (https://github.com/etal/cnvkit). Genomic DNA extracted from peripheral blood leukocytes in the same batch was used as a background control. On average, we set a bin size of 120 bp for the capture regions and 150 kb for the non-capture regions. Default settings were used for all remaining parameters.
Identification of MET Amplification and Polysomy
First, the MET gene copy number was calculated to determine whether the copy number was increased. MET amplification was assessed according to the ratio of CNG to baseline obtained from pooled data from normal samples. Amplified regions on chromosome 7q, including 7q32.1, were searched using the copy number of non-capture regions. A t test was used to assess the differences in copy numbers between adjacent chromosomal bands in the non-capture regions on chromosome 7q. P values ≥ 0.1 were regarded as not statistically significant. Continuous chromosomal regions were obtained in which there were no significant differences between any two chromosomal bands. Similar adjacent regions were merged when any of the following conditions were met: (i) The proportion of chromosomal bands without significant difference in copy number between smaller and larger regions was higher than 50%. (ii) The larger region contained more than seven chromosomal bands, and the proportion of chromosomal bands without significant difference was higher than 30% in smaller and larger regions. The mean copy number of each region was calculated. CNG was defined as a mean copy number of the region higher than 2.3. The length of the amplified region was estimated as a percentage of the total length of chromosome 7q.
Distinguishing MET Amplification from Polysomy
If the MET copy number was increased and the copy number of the region including 7q32.1 did not increase or the region with increased copy number accounted for <10% of the length of 7q, the MET gene was defined as amplified. If the copy number of the region including 7q32.1 was increased and the region with increased copy number accounted for >10% of the length of 7q, it may be defined as MET gene amplification or polysomy, that is, pan-MET gene amplification. If the copy number of the region including 7q32.1 was increased and the region with increased copy number accounted for >80% of the length of 7q, it was defined as polysomy. The detection process is provided in the Supplementary Data (Supplementary Fig. S2).
Data Analysis and Statistical Analysis
Progression-free survival (PFS) was analyzed using the Kaplan–Meier method, and patients were censored if their status was not available at the last follow-up.
To differentiate polysomy from normal cases by copy number using NGS, the Youden index was calculated, and the point at the maximum Youden index was considered as the cut-off value. In addition, ROC analysis demonstrated that a cut-off value of 2.3 copies achieved the maximum Youden index in differentiating polysomy from normal copy number.
On the basis of the established cut-off value, the kappa coefficient was calculated for concordance between tissue and plasma assessments of MET status using NGS.
Statistical significance was set at P < 0.05. All analyses were performed using the IBM SPSS Statistics 24.0 and R 4.2.1.
Data Availability Statement
Variant Call Format files supporting the findings of this study have been deposited in the Genome Variation Map (GVM) database of National Genomics Data Center under accession number GVM000490 (https://ngdc.cncb.ac.cn/gvm). Other data generated in this study are available within the article and its Supplementary Data. All data generated and analyzed are available from the corresponding author upon reasonable request.
Results
Threshold of MET Copy Number to Determine Polysomy using NGS
Of these 53 patients in cohort 1, 60% (32/53) were male, and the rest (40%, 21/53) were female. The median age of the patients was 57 years, ranging from 31 to 75 years. The predominant histology type was adenocarcinoma (90.6%, 48/53), while 5 patients had adenosquamous carcinoma. Most of the patients were diagnosed with stage IIIA or IIIB (47.2%, 25/53), and the remaining patients had stage II (28.3%, 15/53) and stage IV (24.5%, 13/53). Among the 53 patients with paired tissue FISH and NGS results, 29 cases were classified as normal copy number and 10 cases were classified as polysomy according to FISH status. The distribution of MET copy numbers for the two groups is shown in Fig. 1A. ROC analysis also demonstrated that a cut-off point of 2.3 copies achieved the maximum Youden index for discriminating polysomy from normal copy number [Youden index = 0.866 with a sensitivity of 90% and specificity of 96.6%, AUC = 0.976, and 95% confidence interval (CI) = 0.934–1.000, P < 0.0001] (Fig. 1B).
FIGURE 1.

A, The distribution of MET copy number for FISH polysomy group and FISH negative group. B, ROC analysis demonstrated that a cut-off point of 2.3 copies achieved the maximum Youden index for discriminating polysomy from normal copy number (Youden index = 0.866 with a sensitivity of 90% and specificity of 96.6%, AUC = 0.976, and 95% CI = 0.934–1.000, P < 0.0001).
Concordance Between FISH and NGS for Assessment of MET Status in Tissue
In total, 53 patients whose tissues were available for both FISH and NGS were enrolled. The FISH and NGS results are presented in Table 1. FISH analysis identified 14 cases of MET amplification, 10 cases of polysomy, and 29 cases with normal copy numbers. Using FISH as the gold standard, NGS correctly identified 13 cases with MET amplification, nine cases with polysomy, and 28 cases with normal copy numbers. Compared with the FISH test for MET amplification, the sensitivity, specificity, and agreement of NGS were 92.9%, 100%, and 98.1%, respectively. Compared with the FISH test for MET polysomy, the sensitivity, specificity, and agreement of NGS were 90%, 90%, and 96.2%, respectively. A kappa value of 0.886 also revealed high congruence.
TABLE 1.
The paired results of FISH and NGS in 53 patients with NSCLC
| MET FISH | ||||
|---|---|---|---|---|
| Amplification | Polysomy | Negative | ||
| NGS | Amplification | 13 | 0 | 0 |
| Polysomy | 0 | 9 | 1 | |
| Negative | 1 | 1 | 28 | |
Abbreviations: FISH: fluorescence in situ hybridization; NGS: next-generation sequencing; MET: mesenchymal epithelial transition factor gene.
One MET-FISH–positive tumor was identified as negative by NGS. The mean MET/CEP7 ratio (10.5/3.9) was 2.7. The copy number was 2.26 copies and no MET amplification signal was observed in the CNV map (Supplementary Fig. S3). One MET-FISH polysomy tumor was classified as negative using the NGS assay. The MET/CEP7 ratio (5.1/2.7) was 1.9, while the copy number derived from NGS was 2.24 (Supplementary Fig. S4). One MET-FISH–negative patient was classified as having MET polysomy by NGS. The MET/CEP7 ratio (3.6/2.2) was 1.6, and copy number was 2.5596 (Supplementary Fig. S5). The tumor cell content was 15%, 11%, and 37%, respectively. Additional details are provided in the Supplementary Materials and Methods.
Gene Mutation Profiles in Patients with NSCLC with Different MET Status
Figure 2 shows the MET status of 53 patients determined by FISH and NGS, overlapping oncogenes, and mutation burden. Fewer comutations of oncogenes were detected in the MET amplification group than in the other two groups. The frequencies of overlapping oncogenes were similar in the polysomy and normal copy number groups.
FIGURE 2.
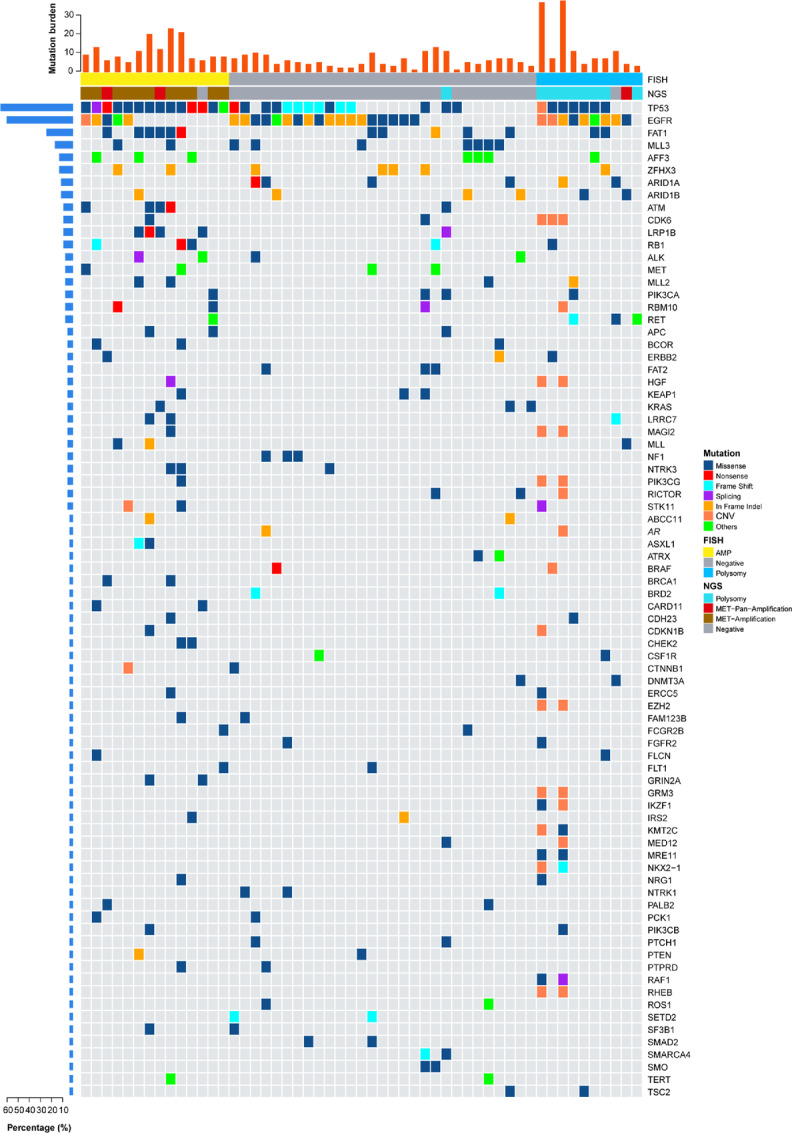
MET status of 53 patients determined by FISH and NGS, overlapping oncogenes, and mutation burden.
Concordance Between Tissue and Plasma-based Testing for Assessment of MET Status Using NGS
The plasma and tissue NGS results were compared for 261 patients who underwent concurrent testing, and the details are listed in Table 2. On the basis of the established standard, NGS identified 19 cases of MET amplification, eight cases of pan-MET amplification, 48 cases of polysomy, and 186 cases of normal copy number in tissue. The results of NGS in the plasma were consistent with those of gene detection in tissue from 10 patients with MET amplification, 2 patients with pan-MET amplification, 6 patients with polysomy, and 181 patients with normal copy number (Table 2). The sensitivity and specificity of NGS for detecting MET amplification using NGS were 53% and 100%, respectively. The concordance between the amplification detection rates in circulating tumor DNA (ctDNA) and tissue samples was 96.6%. For polysomy detection using NGS, sensitivity and specificity were 12.5% and 50%, respectively. The concordance rate between the two sample types was 90.7%.
TABLE 2.
The plasma and tissue NGS results of 261 patients who underwent concurrent testing
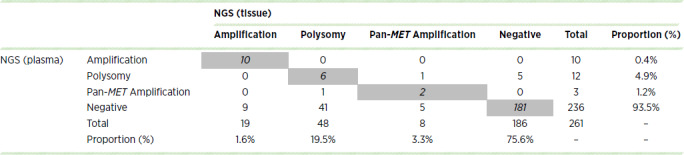
Figure 3 shows the 7q copy numbers in tissue and ratio of maximum somatic allele frequency (MSAF) in plasma to those in the tissues of each polysomy case determined by NGS. In 48 patients, polysomy was consistently detected in the plasma of 6 patients. All 6 patients presented a relatively high copy number of 7q (>2.5) in tissues, and their ratio of MSAF in plasma to that in tissue was relatively high (>0.35). Among the other 42 patients who were negative, 38 presented with a low plasma mutation frequencies/tissue mutation frequency ratio (<0.25).
FIGURE 3.
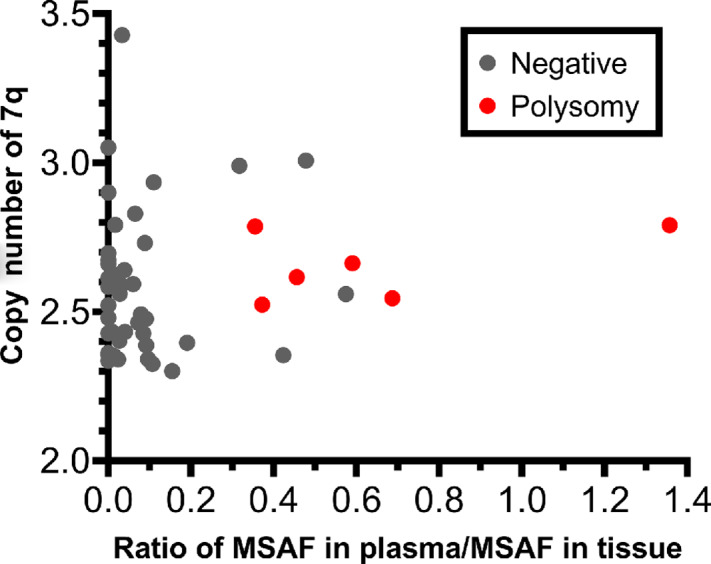
Distribution of the 7q copy numbers in tissue and ratio of MSAF in plasma to those in the tissues of each polysomy case determined by NGS.
On the basis of these results, we calculated the detection rates of liquid biopsies in selected patients. First, the EGFR mutation rate was higher than 5% in 45 patients. Among the 45 patients, MET amplification was identified by NGS in the tissues of 6 patients. Among the 6 patients, 3 presented with MET amplification in plasma ctDNA. When limited to 10 patients with a MSAF higher than 5%, MET amplification was detected in plasma ctDNA from 7 patients. Comparison of ctDNA with tissue in detecting MET amplification using NGS identified a 70% positive percent agreement (PPA) and 95% negative percent agreement (NPA) after optimization.
Of the 48 patients in whom polysomy was identified by NGS in tissue, EGFR in ctDNA was found to be mutated at a frequency higher than 0.5% in 8 patients. Among the 8 patients, only 1 presented with polysomy in plasma ctDNA. Among the 12 patients with MSAF > 5%, polysomy was detected in plasma ctDNA from 5 patients. Following our optimization, the sensitivity and specificity of NGS for defining polysomy with plasma according to different ctDNA mutation frequencies were 42% and 63%, respectively. The concordance rate between the two sample types for detecting polysomy was 85% after optimization.
MET Status Identified Using NGS in MET-high Patients and its Clinical Relevance for MET Inhibitors
Between June 2018 and November 2021, 46 patients with NSCLC with a high MET copy number assessed by NGS were enrolled in the study. Detailed characteristics of the recruited patients are listed in Supplementary Materials and Methods (Supplementary Table S1). All patients received MET inhibitors, such as crizotinib or savolitinib, as second- or third-line therapies. The partial response (PR) rate was 35% and the median PFS was 2.6 months. The PR rate was significantly higher in the MET amplification group than in the polysomy group (50% vs. 21.4%, P < 0.001), whereas there was no significant difference in the PR rate between the polysomy and normal groups (21.4% vs. 25%, P = 0.77). The median PFS was 5.9 months in the MET amplification group, which was significantly higher than that in the polysomy group (2 months, P < 0.001). The median PFS was similar between the polysomy and normal groups (2 vs. 1.9 months, P = 0.79; Fig. 5). Summary of MET status tested by NGS, MET inhibitors with clinical outcome and dynamic changes in gene alterations was listed in Supplementary Table S2.
FIGURE 5.
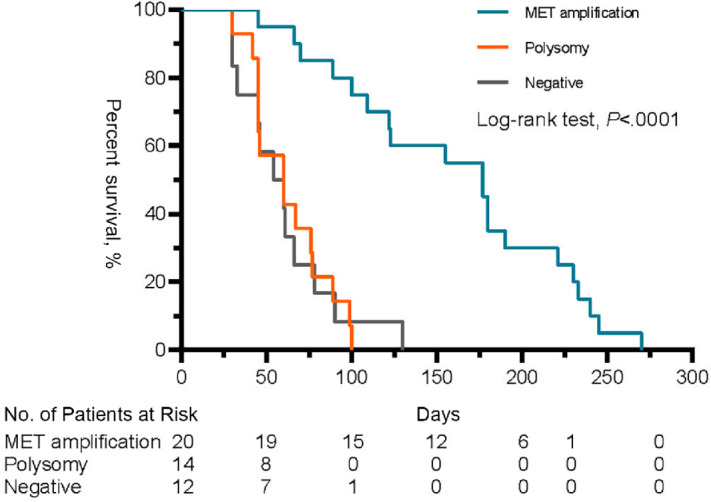
PFS between MET amplification, polysomy, and negative by NGS (n = 46).
Discussion
MET amplification is regarded as an oncogenic driver and represents a therapeutic target (2, 3, 14). However, MET amplification comprises a minority of patients with positive FISH results or high MET. Polysomy was detected on chromosome 7 in other patients with MET CNG. However, to date, no unified criteria have been established to define polysomy using copy numbers derived from NGS. In several studies, the FISH and NGS detection results of MET status were in poor concordance with each other (13, 15). There is no established cut-off value to define polysomy, and whether it is correlated with the response to MET inhibitors remains to be investigated. In this study, we established a new methodology for detecting MET polysomy using NGS. Moreover, our study showed the poor efficacy of MET inhibitors in the treatment of patients with MET polysomy.
NGS technology has improved gene mutation detection and enables more appropriate use of targeted drug therapies (11, 16). However, for MET detection, some issues remain unresolved with NGS. For instance, due to methodologic problems in previous NGS assays, it is challenging to discriminate between MET amplification and polysomy in MET-high patients. Polysomy is occasionally misclassified as MET amplification, and this would include more patients; therefore, some patients might show little clinical benefit to MET-targeted therapy, leading to a diluted clinical benefit. In our study, we proposed a specific algorithm after sequencing to detect the MET status, which is capable of discriminating polysomy from MET amplification. As the first exploratory study on the topic, the optimum cut-off value on which future research can be based should be determined. The threshold copy number for polysomy was determined using the ROC curve. Using the Youden index, we found that the appropriate cutoff was 2.3 with a sensitivity of 90% and specificity of 96.6%.
On the basis of this cut-off value, MET-high and MET-low accounted for 24.3% and 75.7% in our in-house cohort of 691 patients with untreated NSCLC (Fig. 4A.) MET amplification, pan-MET amplification, and polysomy accounted for 1%, 3%, and 19%, respectively, in our in-house cohort of 155 patients with untreated EGFR-mutated NSCLC (Fig. 4B). This proportion was similar to that of patients with untreated EGFR-mutated NSCLC reported in previous studies (12), which verified the rationality of the threshold to some extent.
FIGURE 4.

Prevalence of MET high in treatment-naïve metastatic NSCLC. Prevalence of MET statuses in treatment-naïve metastatic EGFR-mutant NSCLC.
FISH testing for tissue is commonly used in the detection of MET abnormalities (17). Both MET amplification and polysomy can be detected by FISH, and it has been considered the gold standard for MET amplification detection. In the current study, the algorithm and copy-number cut-off value for polysomy were applied to distinguish the different MET statuses in tissue using NGS. Fortunately, we found satisfactory concordance between FISH and NGS assays for testing both MET amplification and polysomy, especially in the former. Our results showed higher PPA (86% vs. 4%), positive predictive value (PPV; 86% vs. 50%), and negative predictive value (NPV; 96% vs. 68%) in detecting polysomy than those reported in the TATTON study, demonstrating that the proposed approach has better detection performance (18). In general, there was substantial concordance between the two methods for all three groups, with a kappa value of 0.886. Up to now, FISH is considered to be the gold standard for detecting MET amplification. There is no uniform interpretation standard for MET gene amplification detected by FISH. NGS is constantly developing and becoming more accessible. The limitation is that the data quality is highly dependent on the platform used, and the lower copy number (especially when the tumor cell content is low) may not be detected. In our study, the FISH and NGS detection results of MET status were in good concordance with each other, which lay the foundation for NGS as routine method to detect MET aberrant molecular alterations. What's more, NGS based on liquid biopsy may be an alternative when tissue biopsy is not available.
Liquid biopsy can overcome many limitations of conventional solid biopsy and has become a valid alternative to tissue biopsy (19). Liquid biopsy is noninvasive and safe and can be repeated easily. Moreover, it is expected to reveal the entire molecular profile of a patient's malignancy. It has been widely believed that liquid biopsy has the potential to change the paradigm in the management of patients with cancer (20). There is growing evidence to support the clinical use of plasma ctDNA to detect multiple gene aberrations by NGS. Recently, the International Association for the Study of Lung Cancer updated consensus for liquid biopsy use in NSCLC. It recommends that plasma ctDNA be considered a valid method for genotyping of newly diagnosed patients with advanced NSCLC. At the time of acquired resistance after TKI therapy, initial use of ctDNA is preferred to evaluate the mechanisms of resistance (21). Our study also explored the diagnostic performance of liquid biopsy in detecting MET abnormalities in patients with NSCLC. The results showed that liquid biopsy had a high NPV and high specificity for detecting MET amplification. NPV and specificity were lower for polysomy detection. In agreement with previous studies, liquid biopsy has a moderate PPV for detecting polysomy (15). In patients with MSAF higher than 5%, the detection rates of both MET amplification and polysomy were higher than those in unselected patients.
Following a case-by-case analysis, we found that a higher copy number of MET in tissues and higher ctDNA content can increase the detection rate of polysomy in plasma. In addition, we suppose that the degree of intratumoral heterogeneity can also influence the detection rate of polysomy. Therefore, the generally good concordance between tissue and plasma for testing MET amplification based on NGS in this study highlights the feasibility of liquid biopsy as an alternative to tissue testing for MET detection.
It has been previously suggested that the response to crizotinib, an inhibitor of c-MET activity, may determine which type of testing is the most relevant in predicting the response to MET inhibition (22). For reliable means of detection, the response to crizotinib could distinguish which type of MET status acts as a tumor driver gene. According to our study, we found significant survival benefits among patients with MET amplification detected using NGS with a MET inhibitor; however, there was no significant benefit in both polysomy and normal copy groups with similar PR rates and PFS. This finding demonstrates that polysomy may not be a therapeutic target for MET inhibitors. A few oncogene overlaps were observed in the MET amplification group, whereas gene mutation profiles were comparable between the polysomy and negative groups. This finding is consistent with polysomy, which does not act as an oncogenic driver. Multivariate analysis revealed that PFS was associated only with MET status, other than MET copy number, which is consistent with a previous study (12).
In summary, the reasonable cut-off value to distinguish polysomy and normal copy number may be 2.3 copies. Identification of amplification and polysomy depends on the different proportion of the amplified region on the chromosome 7q. Table 3 shows the definition/cut-off value of the different MET aberrations. Our results indicate that NGS may serve as an alternative method for detecting MET amplification or polysomy in NSCLC tissues. Liquid biopsy could serve as a substitute for tissue biopsy for detecting MET amplification using NGS. It appears that polysomy does not act as a true oncogenic driver, even though it has a high MET copy number. These findings guaranteed further testing and validation in cohort with enlarged sample size.
TABLE 3.
Cut-off value of the different MET aberrations
| MET GCN | |||
|---|---|---|---|
| <2.3 | ≥2.3 | ||
| Negative | Proportion of the amplified region on the chromosome 7q | ||
| ≤10% | 10%∼80% | ≥80% | |
| MET-amplification | pan-MET amplification | Polysomy | |
Abbreviations: GCN: gene copy number; MET: mesenchymal epithelial transition factor gene.
As the first exploratory study to establish a methodology for detecting polysomy using NGS, our results lay the foundation for future studies on the same topic. Furthermore, our study also demonstrated the feasibility of detecting MET status using NGS. However, this study has some limitations. First, it had a relatively small sample size. Second, its retrospective nature and the fact that the results were from one institution without an external validation group are obvious limitations. Third, most MET inhibitors used in our cohort were crizotinib, and whether the poor response in patients with MET polysomy might be related to drug accessibility needs to be explored.
Supplementary Material
Clinicopathologic characteristics of patients in cohort 3.
Flowchart of the study design
Summary of MET status tested by NGS, MET inhibitors with clinical outcome and dynamic changes in gene alterations in cohort 3
The detection process using NGS of the study.
One MET-FISH-positive tumor was identified as negative by NGS
One MET-FISH polysomy tumor was classified as negative using the NGS assay.
One MET-FISH-negative patient was classified as having MET polysomy by NGS
Acknowledgments
This work was supported by National key research and development project (2022YFC2505004, 2022YFC2505000 to, Z. Wang and J. Wang); the National Natural Science Foundation (grants 81871889 and 82072586, to Z. Wang); CAMS Innovation Fund for Medical Sciences (grants 2021-I2M-1-012, to Z. Wang); Beijing Natural Science Foundation (grant 7212084, to Z. Wang); the National Key Research and Development Project (grant 2019YFC1315700, to J. Wang); the Special Research Fund for Central Universities, Peking Union Medical College (3332021029, to J. Xu); the National Natural Sciences Foundation Key Program (grant 81630071, to J. Wang); Aiyou Foundation (grant KY201701, to J. Wang); Beijing Natural Science Foundation (7222144, to J. Zhong); National Natural Sciences Foundation (82102886, to J. Xu); Guangdong Association of Clinical Trials (GACT)/Chinese Thoracic Oncology Group (CTONG) and Guangdong Provincial Key Lab of Translational Medicine in Lung Cancer (grant no. 2017B030314120). Z. Wang had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Communications Online (https://aacrjournals.org/cancerrescommun/).
Authors’ Disclosures
X. Zeng reports a patent to A method and device for predicting MET gene amplification or polyploid pending. No disclosures were reported by the other authors.
Authors’ Contributions
B. Sun: Conceptualization, data curation, formal analysis, investigation, writing-original draft. T. Qiu: Conceptualization, resources, data curation, methodology. X. Zeng: methodology. J. Duan: Resources, writing-review and editing. H. Bai: Resources, writing-review and editing. J. Xu: Data curation, funding acquisition. J. Li: Resources, methodology. J. Li: Resources, data curation. X. Hao: Resources, data curation. Y. Liu: Resources, data curation. L. Lin: Resources, data curation. H. Wang: Resources, data curation. X. Zhang: Resources, data curation. J. Zhong: Resources, data curation. J. Wang: Resources, data curation, supervision, funding acquisition, writing-review and editing. J. Ying: Conceptualization, resources, methodology, writing-review and editing. Z. Wang: Conceptualization, supervision, funding acquisition, writing-review and editing.
References
- 1. Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression. J Clin Oncol 2016;34:721–30. [DOI] [PubMed] [Google Scholar]
- 2. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039–43. [DOI] [PubMed] [Google Scholar]
- 3. Majeed U, Manochakian R, Zhao Y, Lou Y. Targeted therapy in advanced non-small cell lung cancer: current advances and future trends. J Hematol Oncol 2021;14:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aguado C, Teixido C, Román R, Reyes R, Giménez-Capitán A, Marin E, et al. Multiplex RNA-based detection of clinically relevant MET alterations in advanced non-small cell lung cancer. Mol Oncol 2021;15:350–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park S, Choi YL, Sung CO, An J, Seo J, Ahn MJ, et al. High MET copy number and MET overexpression: poor outcome in non-small cell lung cancer patients. Histol Histopathol 2012;27:197–207. [DOI] [PubMed] [Google Scholar]
- 6. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19:2240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Camidge DR, Davies KD. MET copy number as a secondary driver of epidermal growth factor receptor tyrosine kinase inhibitor resistance in EGFR-mutant non-small-cell lung cancer. J Clin Oncol 2019;37:855–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y, Tang ET, Du Z. Detection of MET gene copy number in cancer samples using the droplet digital PCR method. PLoS One 2016;11:e0146784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guo R, Berry LD, Aisner DL, Sheren J, Boyle T, Bunn PA Jr, et al. MET IHC is a poor screen for MET amplification or MET exon 14 mutations in lung adenocarcinomas: data from a tri-institutional cohort of the lung cancer mutation consortium. J Thorac Oncol 2019;14:1666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoshimura K, Inoue Y, Tsuchiya K, Karayama M, Yamada H, Iwashita Y, et al. Elucidation of the relationships of MET protein expression and gene copy number status with PD-L1 expression and the immune microenvironment in non-small cell lung cancer. Lung Cancer 2020;141:21–31. [DOI] [PubMed] [Google Scholar]
- 11. Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in lung cancer: will expectations finally be MET? J Thorac Oncol 2017;12:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lai GGY, Lim TH, Lim J, Liew PJR, Kwang XL, Nahar R, et al. Clonal MET amplification as a determinant of tyrosine kinase inhibitor resistance in epidermal growth factor receptor-mutant non-small-cell lung cancer. J Clin Oncol 2019;37:876–84. [DOI] [PubMed] [Google Scholar]
- 13. Peng LX, Jie GL, Li AN, Liu SY, Sun H, Zheng MM, et al. MET amplification identified by next-generation sequencing and its clinical relevance for MET inhibitors. Exp Hematol Oncol 2021;10:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cappuzzo F, Jänne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol 2009;20:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hartmaier R, Han JY, Cho BC, Markovets A, Janne PA. Abstract CT127: Tumor response and MET-detection methods exploratory biomarker analysis of Part B of the Ph 1b TATTON study. Proceedings: AACR Annual Meeting 2021; April 10-15, 2021 and May 17-21, 2021; Philadelphia, PA. [Google Scholar]
- 16. Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J Thorac Oncol 2008;3:331–9. [DOI] [PubMed] [Google Scholar]
- 17. Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol 2009;27:1667–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oxnard GR, Yang JC, Yu H, Kim SW, Saka H, Horn L, et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol 2020;31:507–16. [DOI] [PubMed] [Google Scholar]
- 19. Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med 2017;9:eaan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Comino-Mendez I, Turner N. Predicting relapse with circulating tumor DNA analysis in lung cancer. Cancer Discov 2017;7:1368–70. [DOI] [PubMed] [Google Scholar]
- 21. Rolfo C, Mack P, Scagliotti GV, Aggarwal C, Arcila ME, Barlesi F, et al. Liquid biopsy for advanced NSCLC: a consensus statement from the international association for the study of lung cancer. J Thorac Oncol 2021;16:1647–62. [DOI] [PubMed] [Google Scholar]
- 22. Camidge DR, Otterson GA, Clark JW, Ou SHI, Weiss J, Ades S, et al. Crizotinib in patients with MET-amplified NSCLC. J Thorac Oncol 2021;16:1017–29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinicopathologic characteristics of patients in cohort 3.
Flowchart of the study design
Summary of MET status tested by NGS, MET inhibitors with clinical outcome and dynamic changes in gene alterations in cohort 3
The detection process using NGS of the study.
One MET-FISH-positive tumor was identified as negative by NGS
One MET-FISH polysomy tumor was classified as negative using the NGS assay.
One MET-FISH-negative patient was classified as having MET polysomy by NGS
Data Availability Statement
Variant Call Format files supporting the findings of this study have been deposited in the Genome Variation Map (GVM) database of National Genomics Data Center under accession number GVM000490 (https://ngdc.cncb.ac.cn/gvm). Other data generated in this study are available within the article and its Supplementary Data. All data generated and analyzed are available from the corresponding author upon reasonable request.