

Abstract
Epilepsy is a neurological disorder caused by the pathological hyper-synchronization of neuronal discharges. The fundamental research of epilepsy mechanisms and the targets of drug design options for its treatment have focused on neurons. However, approximately 30% of patients suffering from epilepsy show resistance to standard anti-epileptic chemotherapeutic agents while the symptoms of the remaining 70% of patients can be alleviated but not completely removed by the current medications. Thus, new strategies for the treatment of epilepsy are in urgent demand. Over the past decades, with the increase in knowledge on the role of glia in the genesis and development of epilepsy, glial cells are receiving renewed attention. In a normal brain, glial cells maintain neuronal health and in partnership with neurons regulate virtually every aspect of brain function. In epilepsy, however, the supportive roles of glial cells are compromised, and their interaction with neurons is altered, which disrupts brain function. In this review, we will focus on the role of glia-related processes in epileptogenesis and their contribution to abnormal neuronal activity, with the major focus on the dysfunction of astroglial potassium channels, water channels, gap junctions, glutamate transporters, purinergic signaling, synaptogenesis, on the roles of microglial inflammatory cytokines, microglia-astrocyte interactions in epilepsy, and on the oligodendroglial potassium channels and myelin abnormalities in the epileptic brain. These recent findings suggest that glia should be considered as the promising next-generation targets for designing anti-epileptic drugs that may improve epilepsy and drug-resistant epilepsy.
Key Words: astrocyte, cytokines, glutamate, ion channel, microglia, myelin, neuron, oligodendrocyte, purinergic signaling, seizures
Introduction
The past decades brought upon us an incredible increase in knowledge about the properties and functions of glia in health and disease. Thanks to the progress ramped up by the power of modern tools, we now know that glial cells, including astrocytes, microglia, and oligodendrocytes, are critical participants in virtually all aspects of normal brain development and function (Stadelmann et al., 2019; Augusto-Oliveira et al., 2020; Clayton and Tesar, 2021; Ferro et al., 2021). Far more than once thought, glial cells that constitute at least half of the healthy brain not only closely interact with neurons to support their activity, but also mutually interact and shape neuronal circuits. These advances present a tremendous challenge to the neuro-centric doctrine and point toward a reshaping of our perception of brain activity in health and disease, which should not only consider neurons but instead their partnership with glial cells.
Here, we focus on the contribution of astrocytes, microglia and oligodendrocytes to epilepsy, one of the most common serious brain conditions. This condition affects over 70 million people worldwide (Thijs et al., 2019), with the estimate that one in 100 people may develop this disease in their lifetime. In most of the drug treatments for epilepsy, neurons are exclusively targeted, for instance using drugs that block neuronal receptors and voltage-gated ion channels (Bialer et al., 2018). Yet, approximately 30% of patients do not respond to the antiseizure drugs that are currently in use (Janmohamed et al., 2020). On the other hand, the extent to which glial cells undergo profound alterations in morphological and functional aspects and become damaged or die in neurodegenerative processes of epilepsy did not receive significant attention. If the glial cells that support neurons change their function or die in epilepsy, how can the neurons be saved just by targeting neurons? Targeting and preserving glial function, therefore, holds a promise to be an effective therapeutic strategy to prevent brain dysfunction in epilepsy. Throughout this review, we will present research showing that glial cells participate in the pathophysiological processes of epilepsy from its initial stages and that altered glial interaction with neurons might contribute to the disease progression. We conceptualize that the new effective treatments for epilepsy will come once we have a deep mechanistic understanding of disease processes considering both neurons and glia.
Search Strategy
We searched PubMed and Google Scholar for publications in English using terms “microglia and synapses,” “astrocytes,” “astrocytes and epilepsy,” “microglia and epilepsy,” “oligodendrocytes and epilepsy,” “epilepsy treatment.” All years were chosen in the search and particular attention was given to the relatively recent articles published over the years 2018–2022. We included some earlier articles and reviews, if particularly pertinent to the discussion.
Epilepsy: Pathophysiology of Neurons and Glia, and Available Treatments
Epileptogenesis is the chronically persistent process of changing a healthy brain into one capable of generating spontaneous, recurrent seizures (Fisher et al., 2005). This process is described as to result from an imbalance between excitatory and inhibitory activity within a neuronal network that leads to excessive, hypersynchronous neuronal activity that disrupts normal brain function. The most common and severe form of adult epilepsy is termed mesial temporal lobe epilepsy (MTLE), which is frequently characterized by tissue sclerosis – an epileptogenic brain lesion in the hippocampal region. In the sclerotic tissue, the loss of neurons is pronounced, while glial cells undergo profound functional and structural alterations (Thijs et al., 2019). In experimental epilepsy, not all neurons are equally vulnerable. In vivo electrophysiology and transcriptional profiling recently revealed the existence of heterogeneity in the neuronal patterns of activity in the sclerotic hippocampus (Cid et al., 2021). Indeed, superficial pyramidal CA1 neurons were overactive and displayed significantly larger transcriptional changes associated with inflammatory processes and programmed cell death compared to the deep pyramidal CA1 sublayers. Strikingly, the sublayer differences in the gene expression are also found to be pertinent to the glial cells, oligodendrocytes, astrocytes and microglia.
Typically, functional changes and loss of glial cells is considered accompanying phenomenon to neuronal death in epilepsy, however, recent studies using animal models called for rethinking the causal role of glial cells in epileptogenesis. For example, it was found that reactive microglia and astrocytes collaboratively promoted epileptogenesis in a drug-induced status epilepticus model (Sano et al., 2021). The study showed that reactive microglia stained by ionized calcium-binding adaptor molecule 1 marker increased area one day after the status epilepticus, while the signal for reactive astrocytes stained for glial fibrillary acidic protein (GFAP) increased 3 days after the status epilepticus. This sequence of glial activation in the hippocampus was strongly associated with the epileptogenic process as immediate, pharmacological inhibition of microglial activation by minocycline prevented subsequent astrocyte activation and their aberrant Ca2+ signaling. This attenuation of glial activity altogether prevented the increased seizure susceptibility. Another study, using an animal model of kainic acid (KA)-induced status epilepticus that recapitulates chronic human MTLE, identified even earlier time point changes of glial cells during epileptogenesis (Wu et al., 2021). Indeed, early death of astrocytes was observed within four hours after the status epilepticus in the stratum radiatum, and subsequent proliferation led to the repopulation of astrocytes during the first 3 days after the status epilepticus. This early pathological remodeling of astrocytes could impact many homeostatic processes in the brain such as synaptogenesis, concentrations of extracellular ions and transmitters, and the balance between excitation and inhibition. Disruption of all these processes that are maintained by astrocytes, even transient, could impact brain function and contribute to the initiation of epilepsy, which remains to be investigated in future research.
Antiepileptic drugs are the first-line treatment for epilepsy. Although their number has increased over the years, the principles governing drug treatments are in many ways similar to that established decades ago. Despite a large number of available drugs, the proportion of people that do not respond to the treatment has so far remained notably at ~30% (Bialer et al., 2018; Janmohamed et al., 2020). Indeed, epilepsy research has mainly focused on how to inhibit abnormal electrical brain discharges, which consequently resulted in treatment options targeting neurons. Various antiepileptic drugs work via modulating neuronal voltage-dependent sodium, calcium and potassium ion channels by enhancing inhibitory GABA or P1 adenosine transmission systems to suppress excessive neuronal activity (Stafstrom, 2010; Bialer et al., 2018). Overall, the drugs considered the first line in best scenario can control and not cure epilepsy. The reason for this is that we still do not understand many basic aspects of epilepsy pathophysiology. Furthermore, the important role of glial cells in epileptogenesis and their pathology in epilepsy is quite often neglected in drug research and design strategies. We will briefly summarize the functions of glial cells that are essential for maintaining neuronal health and present research showing that these safeguarding glial roles are compromised in epilepsy. Such a concept provides a framework for considering glial cells as promising candidates for developing innovative therapeutic strategies.
What Do Glial Cells Do for Neurons? Powerful Players with Multiple Roles
Astrocytes are the most abundant type of glial cells that contiguously tile the entire central nervous system (CNS). Strategic location of astrocytes between blood vessels and synapses allows them to regulate synaptogenesis, fulfill metabolic and homeostatic maintenance functions, and keep an optimal milieu for the proper function of neurons (Parpura et al., 2012; Patel et al., 2019; Augusto-Oliveira et al., 2020). Astrocytes express water channel aquaporin AQP4, which is important for maintaining water balance in the CNS (MacAulay, 2021). In astroglia, AQP4 is co-localized with the inwardly rectifying K+ channel Kir4.1, indicating that astrocytic regulation of water and K+ homeostasis are closely associated processes (Binder et al., 2012; Verhoog et al., 2020). Indeed, astrocytes are the principal controllers of extracellular K+ in the brain which, if left unchecked, can impair regular neuronal activity. They efficiently clear the excess of K+ that is built up by neuronal firing from the extracellular space. According to the spatial buffering model, astrocytes uptake extracellular K+ mainly by Kir4.1 channels at sites of high neuronal activity, redistribute it through the astrocytic syncytium and release it at regions of lower K+ (Orkand et al., 1966). This spatial buffering model relies on the fact that astrocytes are electrically interconnected via gap junction (GJ) channels and form a functional syncytium. Considering the abundance of astroglia in the CNS, this syncytium allows control of extracellular space on a large volume. Furthermore, astrocytes are an integral part of synapses and directly contribute to neuronal information processing through reciprocal interactions (Araque et al., 2014). These interactions rely on the astroglial expression of various receptors for transmitters that neurons release. Astrocytes can uptake and also respond to these neurotransmitters. For example, astrocytic transporters GLAST (EAAT1) and GLT1 (EAAT2) rapidly uptake glutamate from extracellular space which is essential in preventing excitotoxicity in the brain (Verkhratsky and Nedergaard, 2018). Glutamate in astrocytes is then rapidly turned into glutamine by glutamine synthetase (GS). Activation of the astrocytic receptors by neurotransmitters promotes an increase in intracellular Ca2+ which further downstream induces the release of gliotransmitters from astrocytes such as glutamate and adenosine triphosphate (ATP) that contribute to the neuronal firing pattern (Shen et al., 2017).
Microglial cells are brain resident immune cells and represent the major mechanism of self-defense against brain injury (Ferro et al., 2021). Usage of two-photon imaging in the neocortex in vivo showed that microglial cells are highly active and continually patrol a healthy brain. Indeed, while the soma remains static, microglial processes are remarkably motile, continuously undergoing cycles of de novo formation and withdrawal in a healthy brain (Nimmerjahn et al., 2005). Usage of genetically modified Ca2+ indicators in awake mice further demonstrated that microglial process motility and in particular their extensions could be correlated with neuronal activity (Umpierre et al., 2020). It has been shown that an increase or decrease in neuronal activity results in the elevation of microglial Ca2+ signaling some minutes later, and this delay was attributed to the process extension. Microglia play the roles that are essential for proper neuronal activity, including interactions with neuronal networks, phagocytosis (Peri and Nüsslein-Volhard, 2008), synapse formation and pruning (Paolicelli et al., 2011; Miyamoto et al., 2016).
Oligodendrocytes are generated from oligodendrocyte precursor cells (NG2 glia) and are active participants in the CNS nervous system function including sculpting of the structural and electrical properties of neuronal axons by controlling their diameter (Stadelmann et al., 2019). These glial cells wrap the membranes around neuronal axons to form myelin insulation that prevents ion current leakage and enables fast and efficient signal conduction in the CNS. Additionally, myelinating oligodendrocytes are embedded in a network of interconnected glial and neuronal cells and, within this assembly, provide metabolic support to neurons. Previous research revealed that connexins GJs on oligodendrocytes and astrocytes interconnect these two glial cell types in the panglial syncytium (Fasciani et al., 2018). This coupling occurs at the paranodes where GJs connect the outer layer of the myelin to the astrocytic processes. It has been proposed that panglial syncytium constitutes the principal pathway for the long-distance siphoning of K+ ions from the paranodal periaxonal space to blood vessels or the cerebrospinal fluid (Rash, 2010). Oligodendrocytes, like astrocytes, express Kir4.1 channel and Kir4.1 in oligodendrocytes plays an important role in extracellular K+ homeostasis in the white matter (Larson et al., 2018). In addition to their myelinating role, it has been shown that oligodendrocytes participate in excitatory glutamatergic transmission in the CNS and express GS enzyme (Xin et al., 2019).
Astrocytes and Neuronal Activity in Epilepsy
Due to status epilepticus-induced brain damage, the number of astrocytes increases and the morphology of astrocytes changes, a process called reactive astrogliosis, which is normally characterized by substantial cell body and process enlargement, up-regulation of GFAP expression, and by the dynamic changes in the gene expression and the cellular function (Verhoog et al., 2020; Escartin et al., 2021). In animal and human epilepsy, reactive astrocytes have been shown to down-regulate the expression of K+ channels, GS, glutamate transporter and AQPs, to decrease their coupling, to display changes in the cellular metabolism, to change their purinergic signaling and to perturb synaptogenesis (Patel et al., 2019; Verhoog et al., 2020; Binder and Steinhauser, 2021; Twible et al., 2021; Hayatdavoudi et al., 2022).
Astroglial K+ channels and aquaporin-4 water channels
Astrocytes express different types of K+ channels. Among them, Kir4.1 channels play a vital role in buffering redundant extracellular K+ during repeated neuronal firing. A previous study been has reported a significant loss of Kir4.1 expression and function in multiple animal models of epilepsy and patients with TLE (Kinboshi et al., 2020). General genetic deletion of Kir4.1 displays a higher rate of mortality, while glia-specific inactivation of Kir4.1 results in a lowered threshold of epilepsy (Kinboshi et al., 2020). These observations indicate that reduced Kir4.1 expression causes the epileptic condition. However, it is still unclear whether dysfunction of Kir4.1 is the consequence of epileptic activity. It is worth mentioning here that Kir4.1 dysfunction has been reported in other pathological conditions, for instance, Huntington’s disease (Tong et al., 2014), in which symptoms could be rescued by viral delivery of Kir4.1 protein into astrocytes to enhance the expression of Kir4.1 channel. It would be interesting to test whether these strategies could alleviate epileptic activity in further experiments.
The water channel AQP4 is highly expressed in astrocytes, mainly in the end-feet and neuropil (Binder et al., 2012). The main function of AQP4 is to keep water homeostasis and regulate the extracellular space. Absorption of water by AQP4 decreases the volume of extracellular space, which results in increased extracellular K+ concentration that facilitates the uptake of extracellular K+. Further, immunohistochemistry and ultrastructural analyses showed colocalization of AQP4 and Kir4.1, indicating that the two proteins may functionally interact with each other to maintain the normal function of astrocytes (Binder et al., 2012; Verhoog et al., 2020). Dysregulation of AQP4 expression has been observed in animal models of epilepsy and in patients with mesial temporal sclerosis (MTS) (Eid et al., 2005; Alvestad et al., 2013). Also, it has been reported that mislocalization or loss of perivascular AQP4 channels of astrocytes preceding the onset of seizure results in impaired K+ buffering and contributes to seizure generation in an animal with TLE (Alvestad et al., 2013). These results indicate that AQP4 plays a crucial role in K+ clearance and may become a novel antiepileptogenic target on astrocytes.
Gap junctions
Astrocytes are tightly connected via GJs and form the astrocytic syncytium. This formation plays a key role in relocating second messengers, such as inositol triphosphate, Ca2+, and metabolites such as nucleotides (ATP and ADP), glucose, and is also important to remove redundant [K+] and glutamate from the uptake site to the site with lower concentration (Li et al., 2019). The major GJ proteins in astrocytes are Cx43 and Cx30. Several studies have reported the upregulation of connexin mRNA and protein levels in tissues from animal models and patients with epilepsy (Yang et al., 2021). Indeed, administration of GJ blockers including carbenoxolone, mefloquine, quinine, and quinidine alleviated severe seizure activity (Verhoog et al., 2020). These observations may be explained by the astrocytic intercellular network that facilitates the diffusion of metabolites and second messengers, which could contribute to neuronal firing activity in epileptogenesis. On the other hand, an elegant study found GJs loss in the sclerotic tissue of patients with MTLE (Bedner et al., 2015). The same study showed that uncoupling of astrocytic network occurs at an early time point, preceding status epilepticus and neurodegeneration in an intracortical KA mouse model of TLE. These findings indicate that uncoupling of astrocytes is a cause of TLE. Moreover, the same research group found that febrile seizures also impair interastrocytic GJ coupling in juvenile mice (Khan et al., 2016). Interestingly, impairment of GJ coupling among astrocytes through transgenic techniques was found to promote epileptogenesis accompanied by impaired K+ and glutamate clearance (Wallraff et al., 2006). Overall, these studies indicate that GJs have a complex role in epileptogenesis and that they may serve as a novel astrocyte-related therapeutic target for the treatment of epilepsy.
Astroglial glutamate, glutamate transporters, and glutamate synthetase
Glutamate is the main excitatory transmitter of the CNS released from neurons and astrocytes. Indeed, we and others have reported that glutamate released from the hyperactive astrocytes increases synaptic transmission in the hippocampal slices from animal models of epilepsy (Alvarez-Ferradas et al., 2015; Nikolic et al., 2018). Glutamate release was inhibited when astrocytic P2Y1 receptors are blocked, or Ca2+ mobilization was blocked by the Ca2+ chelator in the astrocytic syncytium, indicating that astrocytic purinergic signaling responds to the hyperactivity of astrocytic Ca2+ transients during epileptogenesis. Astrocytic excitatory amino acid transporters EAAT1 and EAAT2 partially uptake redundant extracellular glutamate and thereby regulate its homeostasis. Downregulation of EAAT1 and EAAT2 has been observed in various animal models of epilepsy and patients with epilepsy (Green et al., 2021). Another elegant study showed that early treatment with ceftriaxone increases EAAT2 expression and decreases extracellular glutamate levels, neuronal cell death, and seizures in Tsc1flox/flox-GFAP-Cre knock-out mice (Zeng et al., 2010). Recently, Zaitsev et al. (2019) found that treatment with ceftriaxone has an anticonvulsant effect. These results indicate that targeting astrocytic glutamate transporter could be a potential therapeutic target for the treatment of epilepsy. In addition, glutamate in astrocytes is rapidly turned into glutamine by GS. Dysfunction of GS results in the accumulation of glutamate in astrocytes and impedes transportation of extracellular glutamate. Moreover, a high concentration of glutamate in astrocytes could induce glutamate release. Thus, dysfunction of GS could contribute to astrocytic hyperexcitability. Indeed, GS is often observed to be downregulated in astrocytes in animal models of epilepsy and in patients with temporal epilepsy, which results in glutamate accumulation in astrocytes and extracellular space and consequently induces neuronal hyperactivity (Eid et al., 2019). Accordingly, it has been shown that pharmacologic inhibition or genetic deletion of GS increases extracellular glutamate and causes recurrent seizures in rats (Binder and Steinhauser, 2021). Based on these data, increasing the activity of astrocytic GS may become a novel anti-epileptic strategy.
Adenosine triphosphate, adenosine, and adenosine kinase
Astrocytic ATP is a predominant extracellular molecule that has complex and controversial roles in epilepsy, which might be due to the activation of different types of receptors and cells and different extracellular ATP concentrations (for review see Nikolic et al. (2020) and Nobili et al. (2022)). In a previous study, we have shown that ATP, serving as an autocrine signaling molecule, activates P2Y1 receptors and increases Ca2+ signals and further triggers glutamate release from astrocytes in an animal model of TLE (Nikolic et al., 2018). Recently, an excellent work from Rouach’s group showed that ATP released from pannexin-1 channels contributes to epileptic activity in the tissue obtained from the epileptogenic zones in patients (Dossi et al., 2018), however, whether there is a cross-talk between increased ATP and astrocytic P2Y1 receptors needs to be further investigated. Once ATP is released from neurons and glia, it is rapidly hydrolyzed into ADP and adenosine by a set of extracellular ectonucleotidases. Among them, adenosine plays an important role in inhibiting excitatory synaptic transmission by activating presynaptic A1 adenosine receptors (Shen et al., 2017, 2022). Indeed, it has been frequently reported that adenosine and adenosine receptor agonist have anticonvulsant action (Tescarollo et al., 2020). Thus, unlike ATP, it is widely accepted that adenosine has anticonvulsant effects. On the other hand, extracellular adenosine is re-captured by nucleoside transporters and rapidly degraded in astrocytes by an astroglia-specific enzyme adenosine kinase. Several groups have reported that increased expression of adenosine kinase decreases the concentration of extracellular adenosine and correlates to seizure activity in various animal models of epilepsy and in patients (Tescarollo et al., 2020). In addition, by using an animal model of epilepsy, it is well established that adenosine kinase inhibitor has an anticonvulsant action. Thus, adenosine kinase may also have the potential to serve as an astrocytic therapeutic target in epilepsy.
Cell death of astrocytes during epileptogenesis
Brain injuries change the number, morphology, and function of astrocytes. Astrogliosis and cell proliferation are often observed in patients with TLE. Steinhauser’s group recently demonstrated a significant reduction in the density of astrocytes in the CA1 stratum radiatum four hours after the status epilepticus induction in a mouse model of epilepsy that mimics human MTLE, however, the reduction was transient (Wu et al., 2021). Three days after status epilepticus, the density of astrocytes recovered to the initial level due to the proliferation of astrocytes. Moreover, based on the immunohistochemistry and expression data, the authors suggested that necroptosis and autophagy are related to astrocytic death. Astroglial apoptosis and autophagy have also been observed in the CA1 and the dentate gyrus following the status epilepticus in various animal models of epilepsy (Ryu et al., 2011; Ko et al., 2016; Hyun et al., 2017). Kim’s group (Hyun et al., 2017) showed that inhibitors of CDK5, an upstream regulator of mitochondrial fission protein (dynamin-related protein 1), substantially reduced astroglial apoptosis in the hippocampus 3 days after the status epilepticus, however, this pharmacological intervention did not alter the onset time of seizures after pilocarpine injection. The importance of astrocytic death in the initial time points after the status epilepticus should be carefully anticipated, considering that astrocytes play prominent roles in the regulation of synaptogenesis and maintenance of extracellular K+ and glutamate homeostasis. Consequently, the loss of such supportive functions of astrocytes in epilepsy, even transient, could make neurons more vulnerable and this could have an impact on the balance between excitation and inhibition. It would be beneficial for future studies to better understand if this early and transient astrocyte death in the epileptic brain has the potential to serve as a disease-modifying entry point.
Astroglial regulation of synaptogenesis and synaptic plasticity in epilepsy
The process of synaptogenesis persists in a healthy brain to remodel and modify its cellular networks throughout life. It is well documented that this process is not only neuronal responsibility and that astrocytes are essential players in mediating synapse formation. Many small substances are involved in synaptogenesis. Among them, thrombospondins have been demonstrated to participate in the maturation of the brain, and their expression is downregulated in mature brains (Christopherson et al., 2005; Patel et al., 2019). More recent studies have identified that thrombospondins secreted from reactive astrocytes bind to α2δ1, an auxiliary calcium channel subunit, and promote aberrant synaptogenesis which alters the balance between excitation and inhibition of neuronal networks and consequently promotes epileptiform activity (Eroglu et al., 2009). Interestingly, the usage of the antiepileptic drug gabapentin that inhibits the binding of thrombospondins to α2δ1 reduced the aberrant formation of excitatory synapses and attenuated hyperexcitability in the freeze-lesion model of developmental cortical malformation (Andresen et al., 2014). More recently, Risher et al. (2018) identified Rac1 as a downstream molecule of α2δ1 that regulates synaptogenesis and Rac1 is a small Rho GTPase that is primarily involved in the control of the actin cytoskeleton. These observations indicate that abnormal synaptogenesis through thrombospondin-α2δ1-Rac1 pathways may promote seizures in some types of epilepsy, especially in CNS injury-induced epilepsy. Moreover, described data suggest that astrocytes are an important cell type for controlling aberrant synaptogenesis in epilepsy by targeting their thrombospondin-dependent pathway.
Astrocytes have also been shown to participate in synapse elimination. For example, a complement cascade protein C1q is expressed at synapses in response to astrocyte presence, and activation of the C1q pathway attracts and signals microglia to phagocyte synapses (Stevens et al., 2007). Moreover, Chun et al. (2010) have shown that C1q knockout mice exhibit defects in the synapse elimination that result in enhanced excitatory synaptic connectivity and epileptiform activity. These results indicate that dysfunctional astrocytes could destabilize neuronal networks and induce seizure activity by aberrant signaling for regulating synaptogenesis and pruning.
A few studies have described that abnormal synaptogenesis during the development of the brain generates excessive neural activation (Neniskyte and Gross, 2017). For instance, it is commonly believed that in fragile X syndrome, the impaired regulating function of astrocytes, resulting in abnormal dendritic morphology and synapse density, is tightly associated with epileptic seizures (Patel et al., 2019). Notably, dendritic spines loss has been commonly observed in epilepsy patients and in animal models of epilepsy (Wong and Guo, 2013); however, the precise contribution of astrocytes to these pathological processes remains to be established. Therefore, future studies are necessary to better understand the function of astrocytes in synaptogenesis and spine loss in epilepsy. These studies could provide insight into whether rescuing normal synaptogenesis can be used to design a therapy for the treatment of epilepsy.
Microglia in Epilepsy: Two Sides of the Same Coin
In the last decades, different studies addressed important clues on the crucial role played by microglia in neurological disorders, including epilepsy. Microglia have primarily the protective role to survey the surrounding environment and to rapidly activate in response to excitotoxic insults or inflammatory conditions. This activation process, also called “microgliosis,” is characterized by: (i) morphological changes, prevalently represented by the retraction of processes and the conversion of the cell body to an amoeboid shape, (ii) changes in gene expression profile, and (iii) alterations of their functional properties and behavior with increased proliferation and migration. Once activated, the main function of microglial cells is to restore tissue homeostasis and so, once again, to exert their protective role. In the effort to protect the environment in pathological conditions, microglia can act as a double-edged sword by exerting both good and bad effects on surrounding cells. Indeed, by trying to protect surviving cells, the effects of microglia activation can be detrimental for injured neurons that are phagocyted to facilitate tissue repair. Moreover, hyperactivation of microglial cells can lead to a high level of oxidative stress, due to the release of nitric oxide and reactive oxygen species, determining neurodegeneration in nearby cells and neuroinflammation with the recruitment of peripheral immune cells through a damaged blood-brain-barrier. This dual role of microglial cells is observed also in MTLE, one of the most common forms of epilepsy in which seizures are associated with hippocampal sclerosis (with neuronal degeneration in CA regions and gliosis) and microglia exert both pro and anti-epileptic effects.
The different role of microglia is also related to their location in the sclerotic or non-sclerotic brain areas. In a recent study (Morin-Brureau et al., 2018), the difference in microglia properties was investigated in MTLE patients taking into consideration both sclerotic and non-sclerotic regions. Indeed, profound differences were highlighted in microglial cells in terms of morphology, functional properties, and genetic profile. Their data revealed two distinct microglial phenotypes, with a prominent amoeboid morphology and few processes in sclerotic CA1 and CA3 areas, while morphologically non-activated microglia was found in non-sclerotic tissues. The transcriptomic analysis accordingly revealed that pro-inflammatory cytokines were expressed at a higher level in microglia localized in sclerotic areas.
Microglia and inflammatory cytokines
In the epileptic tissue, activated microglial cells are one of the main sources of release of pro-inflammatory cytokines such as interleukins-1β, interleukin-6, and tumor necrosis factor-α (TNFα). An increase in interleukin-1β levels can be responsible for neuronal dysfunctions and synaptic plasticity impairments (Han et al., 2016) and can exert a pro-epileptic effect through the upregulation of NMDA receptors at the post-synaptic level (Viviani et al., 2003), and can have an impact on hyperexcitability by decreasing GABA mediated neurotransmission (Roseti et al., 2015). Indeed, blocking interleukin-1β activity by means of anakinra, an interleukin-1 receptor antagonist, was sufficient to reduce seizure occurrence in MTLE models (Ravizza and Vezzani, 2018) and in patients enrolled in a clinical study to treat febrile infection-related epilepsy (Dilena et al., 2019).
TNFα is an inflammatory molecule released prevalently by microglia during inflammation. Like interleukin-1β, TNFα also acts on both excitatory and inhibitory transmission. Indeed, it has been shown that TNFα can inhibit glutamate uptake and increase microglial glutamate release by acting on glutaminase and GJ regulation (Takeuchi et al., 2006), as well as reduce GABAergic transmission by increasing GABA receptor endocytosis (Stellwagen et al., 2005). The upregulation of interleukin-1β and TNFα leads to the release of interleukin-6, another pro-inflammatory cytokine that is supposed to contribute to epileptogenesis by increasing microgliosis and decreasing hippocampal neurogenesis (Levin and Godukhin, 2017). The expression of all these pro-inflammatory cytokines, primarily released by activated microglia, was investigated by flow cytometry and real-time PCR and these analyses revealed clearly increased expression levels 3 days after pilocarpine-induced status epilepticus as well as a reduction by day 21 (Benson et al., 2015).
The role played by microglia in the epileptic brain is controverted and particularly complex considering that the same study described above also reported an increase in microglial expression of anti-inflammatory cytokines such as arginase-1, interleukin-4, and interleukin-10. The fact that microglia can exert both pro- and anti-epileptic roles could also depend on the different stages of the epileptogenic process. In transgenic mice, where microglia were conditionally ablated, the preconditioning with LPS 24 hours before pilocarpine-induced seizures was sufficient to increase seizure severity suggesting that microglia could be protective during seizure induction (Mirrione et al., 2010). On the other hand, a pro-epileptic role of microglia was highlighted by studies in which microglia depletion by means of minocycline was protective against neuronal degeneration after seizures, and minocycline treatment post-status epilepticus was indeed able to reduce spontaneous recurrent seizure number and severity, supporting a role of microglia in the propagation of seizures (Wang et al., 2015).
It has been recently shown that a selective elevation of mTOR signaling in microglia of Tsc1flox/flox-Cx3cr1-Cre knock-out mice contributes to epileptogenesis without inflammatory activation (Zhao et al., 2018). The conditional knock-out microglia were characterized by reactive morphology and increased proliferation but their gene expression profile showed a decreased expression of pro-inflammatory cytokines, although pro-inflammatory markers were highly expressed in the hippocampus, indicating that in this model microglia is not contributing to the inflammatory response, which is probably sustained by neurons and astrocytes. Moreover, Tsc1flox/flox- Cx3cr1-Cre knock-out mice were more susceptible to the development of spontaneous seizures, suggesting that mTOR signaling upregulation in microglia could be involved in the induction of spontaneous seizures without the contribution of microglia-derived pro-inflammatory signaling.
Microglia-neuron and microglia-astrocyte interactions in epilepsy
Interactions between microglia and neurons in physiological conditions have been deeply investigated, however, less is known about how microglia interact with hyperexcitable neurons during epilepsy. Increased interaction of microglial cells with the apical dendrites of neurons in the CA1 region of the hippocampus was found 3 days after KA-induced status epilepticus (Hasegawa et al., 2007). ATP and purinergic receptors play a key role in microglia-neuron interactions. Indeed, after the status epilepticus, ATP released by neurons can increase microglia motility and their interaction with neurons, and this process is mediated by the P2Y12 receptor activation on microglia (Avignone et al., 2008). Moreover, after KA-induced status epilepticus, P2Y12 KO mice showed a decreased seizure threshold, suggesting that P2Y12 receptor on microglia can exert antiepileptic effects (Eyo et al., 2014).
A new concept of “somatic microglial junctions” was introduced recently and it refers to the identification of direct contacts of microglial processes with neuronal membrane at the somatic level (Cserép et al., 2020). According to the authors, the contacts created by microglial processes with soma are more stable compared to the ones with dendrites and their interactions occur prevalently where clusters of voltage-gated Kv 2.1 and Kv 2.2 K+ channels are located. These clusters are usually involved in exocytosis through the formation of junctions between the endoplasmic reticulum and the membrane and, considering the location of microglial junctions, it is believed that their role is to allow microglia to check the “healthy” state of neurons through specific molecules released by exocytosis. Once again, ATP and purinergic signaling are crucial for microglia-neuron interactions since the release of ATP occurring as a consequence of neuronal activity activates P2Y12 receptors on microglia facilitating the formation of somatic junctions by stimulating microglial process growth.
Another important factor influencing microglial activity that should be taken into account is astrocyte-microglia interaction. Astrocytes take part in the formation of the tripartite synapses and their interactions with neurons and microglia in epilepsy have been widely investigated. Astrocyte-microglia interaction can take place in both directions: neuronal activity during KA-induced status epilepticus leads to ATP release from astrocytes and this ATP is able, on one side, to modulate neuronal activity (Scemes et al., 2019), and on the other side is responsible for the attraction of microglial processes close to synapses. On the other way around, LPS-activated microglia are able to release ATP and activate astrocytes through P2Y1 receptor activation. The release of glutamate from activated astrocytes is then responsible for enhancing neuronal excitability (Pascual et al., 2012). Moreover, pro-inflammatory factors released by activated microglia can activate astrocytes that, on their side, promote microglia activation, generating a kind of self-sustaining cycle that impacts neuronal activity (Alibhai et al., 2018).
Our knowledge of the role of microglia in pathological conditions is still limited, in spite of growing interest in elucidating the role of microglia in acute and chronic seizures. This is in particular caused by the evidence that the same cells exert both pro- and anti-epileptic effects depending on the stage of the disease, the brain areas involved, and the interactions with other cell types. Understanding how functional changes of microglia can be protective or detrimental for the surrounding tissue is of fundamental importance to shed more light on the mechanisms underlying the generation of seizures, going beyond the imbalance between excitation and inhibition. Understanding these mechanisms and how microglia are able to impact the functionality of neuronal networks will allow for the generation of new therapeutic strategies that could represent a potential and innovative tool for the treatment of epilepsy.
Limited Understanding of the Safeguarding Role of Oligodendrocytes in Epilepsy
The role of oligodendrocytes has become recognized as a part of the functionally integrated neuron-glia network in seizure pathogenesis and epilepsy. Although epilepsy is considered primarily to be a gray matter disease, growing evidence highlights the importance of the white matter, in the pathogenesis of both focal and generalized epilepsy. In the white matter, oligodendrocytes are extensively associated with neuronal axons and they wrap neuronal membranes to form myelin. A recent neuroimaging study including a very large sample size, consistent definitions of epilepsy syndromes, and standardized post-image acquisition processing algorithms for analysis of results, definitively confirmed abnormalities of white matter in patients with epilepsy (Hatton et al., 2020).
There are many studies to date, both experimental and clinical, that reported connection between the myelin content and abnormal myelination with epilepsy, epileptic seizures, and epileptogenesis. Reduced myelin content was observed in animal models of focal and multifocal experimental epilepsy utilizing pharmacological treatments with convulsants and electrical stimulation, as recently reviewed in (de Curtis et al., 2021). New research using Wag/Rij rats and Scn8a+/mut mice found that activity-dependent myelination resulting from absence seizures may contribute to epilepsy progression. Authors found increased oligodendrogenesis and myelination specifically within the seizure network in the two models of generalized epilepsy with absence seizures, evident only after epilepsy onset (Knowles et al., 2022). The far-reaching implications of these findings to generalized epilepsy in humans, however, remain to be fully elucidated.
Oligodendrocytes, as a part of the panglial syncytium, also play a critical role in maintaining K+ homeostasis inside white matter tracts (Larson et al., 2018). By using transgenic mouse lines, the authors found that specific interruption of oligodendrocyte K+ buffering leads to seizures. Indeed, selective deletion of Kir4.1 from oligodendrocyte precursor cells or mature oligodendrocytes impairs slower clearance of extracellular K+, delays recovery of axons from repetitive stimulation in the white matter, and promotes seizures. This research expands a basic mechanism of seizure activity and provides opportunities for the development of new approaches for epilepsy treatment that may include oligodendrocytes.
Dysregulation of glutamate, the major excitatory neurotransmitter in the brain, has been identified in a number of pathological states, including epilepsy. In addition to the important role of astrocytes in regulating glutamatergic transmission, oligodendrocytes have been shown to participate in the regulation of glutamate levels in the brain. Xin et al. (2019) showed that mature oligodendrocytes express GS. Furthermore, they show that selective removal of oligodendrocyte GS in mice leads to reduced brain glutamate and glutamine levels and disrupts neuronal glutamatergic transmission. This new role for oligodendrocytes in supporting excitatory glutamate signaling in the brain may be of therapeutic value in epilepsy, however, the molecular mechanisms regulating the activity of GS within oligodendrocytes remain to be explored.
Could Glial Cells Be Important Drug Targets?
As described above, glial cells are highly involved in the genesis, development, and progression of epilepsy pathophysiology. Therefore, the answer to every important question about epilepsy treatment will also involve glia. Based on research advances so far, discovery and synthesis of anti-epileptic drugs targeting glia may provide novel therapeutic interventions for the treatment of epilepsy at its early stages and with fewer side effects. Additionally, the newly designed glial cell-targeting drugs may also be used in the treatment of drug-resistant epilepsy. As described, each type of CNS glia has several properties that could be targeted to preserve neuronal health (Figure 1). Furthermore, the same proteins can be co-expressed by different glial cell types and thus can be powerful drug candidates (for example Kir4.1, GS, purinergic receptors, see Figure 1). Such therapeutic strategies would target more than one glial cell type and can be more efficient in preserving normal neuronal activity in the epileptic brain. Further studies are necessary to dissect carefully the potential molecular mechanisms of how glial cells, each type or their combined activity, participate in epileptogenesis. Results from such studies will provide new insights into how glia can save neurons in epilepsy.
Figure 1.
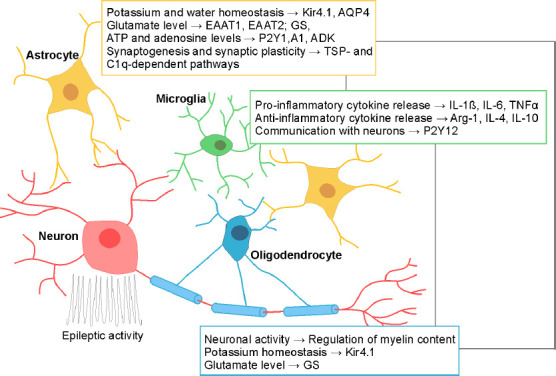
Possible glial therapeutic targets in epilepsy.
The potential candidates targeting astrocytes, microglia, and oligodendrocytes are depicted in color-coded rectangles. For each depicted glial candidate, literature data have shown its contribution to epilepsy and the abnormal pattern of neuronal activity. Grey connecting lines depict the combined targets of different glial cell types for more effective therapeutic strategy. A1: Adenosine receptors; ADK: adenosine kinase; AQP4: aquaporin; Arg-1: arginase-1; ATP: adenosine 5′-triphosphate; C1q: complement cascade protein; EAAT1, EAAT2: excitatory amino acid transporters 1 and 2; GS: glutamine synthetase; IL-1β, 4, 6, 10: Interleukins-1β, 4, 6, 10; Kir4.1: inwardly rectifying K+ channel; P2Y1, P2Y12: purinergic receptors 1 and 12; TNFα: tumor necrosis factor α; TSP: thrombospondins.
Acknowledgments:
The authors thank Danijela Kostić for language editing of the manuscript.
Footnotes
Funding: This work was supported by the Ministry of Education, Science and Technological Development of Republic of Serbia, Nos. 451-03-68/2022-14/200007 and 451-03-68/2022-14/200053 (to LN and JBP), Grants from European Commission (H2020 MSCA-ITN EU-GliaPhD No. 72205) (to PN), Agence Nationale de la Recherche, Nos. ANR-19-CE16-0018-03 and ANR-20CE16-0003-02 (to PN), Grants from Science and Technology Department of Zhejiang Province, China, No. 2021RC051 (to WS), and Scientific Research Foundation for Returned Scholars of Hangzhou City, China, No. 2019 (to WS).
Conflicts of interest: The authors declare that there is no conflict of interest regarding the publication of this manuscript.
C-Editors: Zhao M, Liu WJ, Wang L; T-Editor: Jia Y
References
- 1.Alibhai JD, Diack AB, Manson JC. Unravelling the glial response in the pathogenesis of Alzheimer’s disease. FASEB J. 2018;32:5766–5777. doi: 10.1096/fj.201801360R. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez-Ferradas C, Morales JC, Wellmann M, Nualart F, Roncagliolo M, Fuenzalida M, Bonansco C. Enhanced astroglial Ca2+signaling increases excitatory synaptic strength in the epileptic brain. Glia. 2015;63:1507–1521. doi: 10.1002/glia.22817. [DOI] [PubMed] [Google Scholar]
- 3.Alvestad S, Hammer J, Hoddevik EH, Skare O, Sonnewald U, Amiry-Moghaddam M, Ottersen OP. Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res. 2013;105:30–41. doi: 10.1016/j.eplepsyres.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Andresen L, Hampton D, Taylor-Weiner A, Morel L, Yang Y, Maguire J, Dulla CG. Gabapentin attenuates hyperexcitability in the freeze-lesion model of developmental cortical malformation. Neurobiol Dis. 2014;71:305–316. doi: 10.1016/j.nbd.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Araque A, Carmignoto G, Haydon PG, Oliet SHR, Robitaille R, Volterra A. Gliotransmitters travel in time and space. Neuron. 2014;81:728–739. doi: 10.1016/j.neuron.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Augusto-Oliveira M, Arrifano GP, Takeda PY, Lopes-Araújo A, Santos-Sacramento L, Anthony DC, Verkhratsky A, Crespo-Lopez ME. Astroglia-specific contributions to the regulation of synapses, cognition and behaviour. Neurosci Biobehav Rev. 2020;118:331–357. doi: 10.1016/j.neubiorev.2020.07.039. [DOI] [PubMed] [Google Scholar]
- 7.Avignone E, Ulmann L, Levavasseur F, Rassendren F, Audinat E. Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling. J Neurosci. 2008;28:9133–9144. doi: 10.1523/JNEUROSCI.1820-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bedner P, Dupper A, Huttmann K, Muller J, Herde MK, Dublin P, Deshpande T, Schramm J, Haussler U, Haas CA, Henneberger C, Theis M, Steinhauser C. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain. 2015;138:1208–1222. doi: 10.1093/brain/awv067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benson MJ, Manzanero S, Borges K. Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia. 2015;56:895–905. doi: 10.1111/epi.12960. [DOI] [PubMed] [Google Scholar]
- 10.Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Tomson T, White HS. Progress report on new antiepileptic drugs:A summary of the Fourteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIV). I. Drugs in preclinical and early clinical development. Epilepsia. 2018;59:1811–1841. doi: 10.1111/epi.14557. [DOI] [PubMed] [Google Scholar]
- 11.Binder DK, Nagelhus EA, Ottersen OP. Aquaporin-4 and epilepsy. Glia. 2012;60:1203–1214. doi: 10.1002/glia.22317. [DOI] [PubMed] [Google Scholar]
- 12.Binder DK, Steinhauser C. Astrocytes and epilepsy. Neurochem Res. 2021;46:2687–2695. doi: 10.1007/s11064-021-03236-x. [DOI] [PubMed] [Google Scholar]
- 13.Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 14.Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A. 2010;107:7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cid E, Marquez-Galera A, Valero M, Gal B, Medeiros DC, Navarron CM, Ballesteros-Esteban L, Reig-Viader R, Morales AV, Fernandez-Lamo I, Gomez-Dominguez D, Sato M, Hayashi Y, Bayés À, Barco A, Lopez-Atalaya JP, de la Prida LM. Sublayer- and cell-type-specific neurodegenerative transcriptional trajectories in hippocampal sclerosis. Cell Rep. 2021;35:109229. doi: 10.1016/j.celrep.2021.109229. [DOI] [PubMed] [Google Scholar]
- 16.Clayton BLL, Tesar PJ. Oligodendrocyte progenitor cell fate and function in development and disease. Curr Opin Cell Biol. 2021;73:35–40. doi: 10.1016/j.ceb.2021.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cserép C, et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 2020;367:528–537. doi: 10.1126/science.aax6752. [DOI] [PubMed] [Google Scholar]
- 18.de Curtis M, Garbelli R, Uva L. A hypothesis for the role of axon demyelination in seizure generation. Epilepsia. 2021;62:583–595. doi: 10.1111/epi.16824. [DOI] [PubMed] [Google Scholar]
- 19.Dilena R, Mauri E, Aronica E, Bernasconi P, Bana C, Cappelletti C, Carrabba G, Ferrero S, Giorda R, Guez S, Scalia Catenacci S, Triulzi F, Barbieri S, Calderini E, Vezzani A. Therapeutic effect of Anakinra in the relapsing chronic phase of febrile infection-related epilepsy syndrome. Epilepsia Open. 2019;4:344–350. doi: 10.1002/epi4.12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dossi E, Blauwblomme T, Moulard J, Chever O, Vasile F, Guinard E, Le Bert M, Couillin I, Pallud J, Capelle L, Huberfeld G, Rouach N. Pannexin-1 channels contribute to seizure generation in human epileptic brain tissue and in a mouse model of epilepsy. Sci Transl Med. 2018;10:eaar3796. doi: 10.1126/scitranslmed.aar3796. [DOI] [PubMed] [Google Scholar]
- 21.Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjornsen LP, Spencer DD, Agre P, Ottersen OP, de Lanerolle NC. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci U S A. 2005;102:1193–1198. doi: 10.1073/pnas.0409308102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eid T, Lee TW, Patrylo P, Zaveri HP. Astrocytes and glutamine synthetase in epileptogenesis. J Neurosci Res. 2019;97:1345–1362. doi: 10.1002/jnr.24267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen WT, Cohen-Salmon M, Cunningham C, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24:312–325. doi: 10.1038/s41593-020-00783-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci. 2014;34:10528–10540. doi: 10.1523/JNEUROSCI.0416-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fasciani I, Pluta P, González-Nieto D, Martínez-Montero P, Molano J, Paíno CL, Millet O, Barrio LC. Directional coupling of oligodendrocyte connexin-47 and astrocyte connexin-43 gap junctions. Glia. 2018;66:2340–2352. doi: 10.1002/glia.23471. [DOI] [PubMed] [Google Scholar]
- 27.Ferro A, Auguste YSS, Cheadle L. Microglia, cytokines , and neural activity:unexpected interactions in brain development and function. Front Immunol. 2021;12:1–14. doi: 10.3389/fimmu.2021.703527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fisher RS, Van Emde Boas W, Blume W, Elger C, Genton P, Lee P, Engel J. Response:Definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) Epilepsia. 2005;46:1701–1702. doi: 10.1111/j.0013-9580.2005.66104.x. [DOI] [PubMed] [Google Scholar]
- 29.Green JL, Dos Santos WF, Fontana ACK. Role of glutamate excitotoxicity and glutamate transporter EAAT2 in epilepsy:opportunities for novel therapeutics development. Biochem Pharmacol. 2021;193:114786. doi: 10.1016/j.bcp.2021.114786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han T, Qin Y, Mou C, Wang M, Jiang M, Liu B. Seizure induced synaptic plasticity alteration in hippocampus is mediated by IL-1βreceptor through PI3K/Akt pathway. Am J Transl Res. 2016;8:4499–4509. [PMC free article] [PubMed] [Google Scholar]
- 31.Hasegawa S, Yamaguchi M, Nagao H, Mishina M, Mori K. Enhanced cell-to-cell contacts between activated microglia and pyramidal cell dendrites following kainic acid-induced neurotoxicity in the hippocampus. J Neuroimmunol. 2007;186:75–85. doi: 10.1016/j.jneuroim.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 32.Hatton SN, Huynh KH, Bonilha L, Abela E, Alhusaini S, Altmann A, Alvim MKM, Balachandra AR, Bartolini E, Bender B, Bernasconi N, Bernasconi A, Bernhardt B, Bargallo N, Caldairou B, Caligiuri ME, Carr SJA, Cavalleri GL, Cendes F, Concha L, et al. White matter abnormalities across different epilepsy syndromes in adults:an ENIGMA-Epilepsy study. Brain. 2020;143:2454–2473. doi: 10.1093/brain/awaa200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayatdavoudi P, Hosseini M, Hajali V, Hosseini A, Rajabian A. The role of astrocytes in epileptic disorders. Physiol Rep. 2022;10:e15239. doi: 10.14814/phy2.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyun HW, Min SJ, Kim JE. CDK5 inhibitors prevent astroglial apoptosis and reactive astrogliosis by regulating PKA and DRP1 phosphorylations in the rat hippocampus. Neurosci Res. 2017;119:24–37. doi: 10.1016/j.neures.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 35.Janmohamed M, Brodie MJ, Kwan P. Pharmacoresistance - Epidemiology, mechanisms , and impact on epilepsy treatment. Neuropharmacology. 2020;168:107790. doi: 10.1016/j.neuropharm.2019.107790. [DOI] [PubMed] [Google Scholar]
- 36.Khan D, Dupper A, Deshpande T, Graan PN, Steinhauser C, Bedner P. Experimental febrile seizures impair interastrocytic gap junction coupling in juvenile mice. J Neurosci Res. 2016;94:804–813. doi: 10.1002/jnr.23726. [DOI] [PubMed] [Google Scholar]
- 37.Kinboshi M, Ikeda A, Ohno Y. Role of astrocytic inwardly rectifying potassium (Kir) 4.1 channels in epileptogenesis. Front Neurol. 2020;11:626658. doi: 10.3389/fneur.2020.626658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knowles JK, Xu H, Soane C, Batra A, Saucedo T, Frost E, Tam LT, Fraga D, Ni L, Villar K, Talmi S, Huguenard JR, Monje M. Maladaptive myelination promotes generalized epilepsy progression. Nat Neurosci. 2022;25:596–606. doi: 10.1038/s41593-022-01052-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ko AR, Hyun HW, Min SJ, Kim JE. The differential DRP1 phosphorylation and mitochondrial dynamics in the regional specific astroglial death induced by status epilepticus. Front Cell Neurosci. 2016;10:124. doi: 10.3389/fncel.2016.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larson VA, Mironova Y, Vanderpool KG, Waisman A, Rash JE, Agarwal A, Bergles DE. Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. Elife. 2018;7:e34829. doi: 10.7554/eLife.34829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levin SG, Godukhin OV. Modulating effect of cytokines on mechanisms of synaptic plasticity in the brain. Biochemistry (Mosc) 2017;82:264–274. doi: 10.1134/S000629791703004X. [DOI] [PubMed] [Google Scholar]
- 42.Li Q, Li QQ, Jia JN, Liu ZQ, Zhou HH, Mao XY. Targeting gap junction in epilepsy:perspectives and challenges. Biomed Pharmacother. 2019;109:57–65. doi: 10.1016/j.biopha.2018.10.068. [DOI] [PubMed] [Google Scholar]
- 43.MacAulay N. Molecular mechanisms of brain water transport. Nat Rev Neurosci. 2021;22:326–344. doi: 10.1038/s41583-021-00454-8. [DOI] [PubMed] [Google Scholar]
- 44.Mirrione MM, Konomos DK, Gravanis I, Dewey SL, Aguzzi A, Heppner FL, Tsirka SE. Microglial ablation and lipopolysaccharide preconditioning affects pilocarpine-induced seizures in mice. Neurobiol Dis. 2010;39:85–97. doi: 10.1016/j.nbd.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, Koizumi S, Moorhouse AJ, Yoshimura Y, Nabekura J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun. 2016;7:12540. doi: 10.1038/ncomms12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morin-Brureau M, Milior G, Royer J, Chali F, LeDuigou C, Savary E, Blugeon C, Jourdren L, Akbar D, Dupont S, Navarro V, Baulac M, Bielle F, Mathon B, Clemenceau S, Miles R. Microglial phenotypes in the human epileptic temporal lobe. Brain. 2018;141:3343–3360. doi: 10.1093/brain/awy276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neniskyte U, Gross CT. Errant gardeners:glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat Rev Neurosci. 2017;18:658–670. doi: 10.1038/nrn.2017.110. [DOI] [PubMed] [Google Scholar]
- 48.Nikolic L, Shen W, Nobili P, Virenque A, Ulmann L, Audinat E. Blocking TNFalpha-driven astrocyte purinergic signaling restores normal synaptic activity during epileptogenesis. Glia. 2018;66:2673–2683. doi: 10.1002/glia.23519. [DOI] [PubMed] [Google Scholar]
- 49.Nikolic L, Nobili P, Shen W, Audinat E. Role of astrocyte purinergic signaling in epilepsy. Glia. 2020;68:1677–1691. doi: 10.1002/glia.23747. [DOI] [PubMed] [Google Scholar]
- 50.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Neuroforum. 2005;11:95–96. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 51.Nobili P, Shen W, Milicevic K, Bogdanovic Pristov J, Audinat E, Nikolic L. Therapeutic potential of astrocyte purinergic signalling in epilepsy and multiple sclerosis. Front Pharmacol. 2022;13:900337. doi: 10.3389/fphar.2022.900337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- 53.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 54.Parpura V, Heneka MT, Montana V, Oliet SHR, Schousboe A, Haydon PG, Stout RF, Spray DC, Reichenbach A, Pannicke T, Pekny M, Pekna M, Zorec R, Verkhratsky A. Glial cells in (patho)physiology. J Neurochem. 2012;121:4–27. doi: 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pascual O, Achour SB, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A. 2012;109:E197–205. doi: 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patel DC, Tewari BP, Chaunsali L, Sontheimer H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat Rev Neurosci. 2019;20:282–297. doi: 10.1038/s41583-019-0126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peri F, Nüsslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008;133:916–927. doi: 10.1016/j.cell.2008.04.037. [DOI] [PubMed] [Google Scholar]
- 58.Rash JE. Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction:pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience. 2010;168:982–1008. doi: 10.1016/j.neuroscience.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ravizza T, Vezzani A. Pharmacological targeting of brain inflammation in epilepsy:Therapeutic perspectives from experimental and clinical studies. Epilepsia Open. 2018;3:133–142. doi: 10.1002/epi4.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Risher WC, Kim N, Koh S, Choi JE, Mitev P, Spence EF, Pilaz LJ, Wang D, Feng G, Silver DL, Soderling SH, Yin HH, Eroglu C. Thrombospondin receptor alpha2delta-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J Cell Biol. 2018;217:3747–3765. doi: 10.1083/jcb.201802057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roseti C, van Vliet EA, Cifelli P, Ruffolo G, Baayen JC, Di Castro MA, Bertollini C, Limatola C, Aronica E, Vezzani A, Palma E. GABAA currents are decreased by IL-1βin epileptogenic tissue of patients with temporal lobe epilepsy:Implications for ictogenesis. Neurobiol Dis. 2015;82:311–320. doi: 10.1016/j.nbd.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 62.Ryu HJ, Kim JE, Yeo SI, Kang TC. p65/RelA-Ser529 NF-kappaB subunit phosphorylation induces autophagic astroglial death (Clasmatodendrosis) following status epilepticus. Cell Mol Neurobiol. 2011;31:1071–1078. doi: 10.1007/s10571-011-9706-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sano F, Shigetomi E, Shinozaki Y, Tsuzukiyama H, Saito K, Mikoshiba K, Horiuchi H, Cheung DL, Nabekura J, Sugita K, Aihara M, Koizumi S. Reactive astrocyte-driven epileptogenesis is induced by microglia initially activated following status epilepticus. JCI Insight. 2021;6:e135391. doi: 10.1172/jci.insight.135391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scemes E, Velíšek L, Velíšková J. Astrocyte and neuronal pannexin1 contribute distinctly to seizures. ASN Neuro. 2019;11:1759091419833502. doi: 10.1177/1759091419833502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shen W, Nikolic L, Meunier C, Pfrieger F, Audinat E. An autocrine purinergic signaling controls astrocyte-induced neuronal excitation. Sci Rep. 2017;7:11280. doi: 10.1038/s41598-017-11793-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen W, Li Z, Tang Y, Han P, Zhu F, Dong J, Ma T, Zhao K, Zhang X, Xie Y, Zeng LH. Somatostatin interneurons inhibit excitatory transmission mediated by astrocytic GABAB and presynaptic GABAB and adenosine A1 receptors in the hippocampus. J Neurochem. 2022 doi: 10.1111/jnc.15662. doi: 10-1111/jnc.15662. [DOI] [PubMed] [Google Scholar]
- 67.Stadelmann C, Timmler S, Barrantes-Freer A, Simons M. Myelin in the central nervous system:Structure, function , and pathology. Physiol Rev. 2019;99:1381–1431. doi: 10.1152/physrev.00031.2018. [DOI] [PubMed] [Google Scholar]
- 68.Stafstrom CE. Mechanisms of action of antiepileptic drugs:The search for synergy. Curr Opin Neurol. 2010;23:157–163. doi: 10.1097/WCO.0b013e32833735b5. [DOI] [PubMed] [Google Scholar]
- 69.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 71.Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-αinduces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- 72.Tescarollo FC, Rombo DM, DeLiberto LK, Fedele DE, Alharfoush E, Tome AR, Cunha RA, Sebastiao AM, Boison D. Role of adenosine in epilepsy and seizures. J Caffeine Adenosine Res. 2020;10:45–60. doi: 10.1089/caff.2019.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thijs RD, Surges R, O’Brien TJ, Sander JW. Epilepsy in adults. Lancet. 2019;393:689–701. doi: 10.1016/S0140-6736(18)32596-0. [DOI] [PubMed] [Google Scholar]
- 74.Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci. 2014;17:694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Twible C, Abdo R, Zhang Q. Astrocyte role in temporal lobe epilepsy and development of mossy fiber sprouting. Front Cell Neurosci. 2021;15:725693. doi: 10.3389/fncel.2021.725693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Umpierre AD, Bystrom LL, Ying Y, Liu YU, Worrell G, Wu LJ. Microglial calcium signaling is attuned to neuronal activity in awake mice. ELife. 2020;9:1–24. doi: 10.7554/eLife.56502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Verhoog QP, Holtman L, Aronica E, van Vliet EA. Astrocytes as guardians of neuronal excitability:mechanisms underlying epileptogenesis. Front Neurol. 2020;11:591690. doi: 10.3389/fneur.2020.591690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev. 2018;98:239–389. doi: 10.1152/physrev.00042.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1βenhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang N, Mi X, Gao B, Gu J, Wang W, Zhang Y, Wang X. Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience. 2015;287:144–156. doi: 10.1016/j.neuroscience.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 82.Wong M, Guo D. Dendritic spine pathology in epilepsy:cause or consequence? Neuroscience. 2013;251:141–150. doi: 10.1016/j.neuroscience.2012.03.048. [DOI] [PubMed] [Google Scholar]
- 83.Wu Z, Deshpande T, Henning L, Bedner P, Seifert G, Steinhauser C. Cell death of hippocampal CA1 astrocytes during early epileptogenesis. Epilepsia. 2021;62:1569–1583. doi: 10.1111/epi.16910. [DOI] [PubMed] [Google Scholar]
- 84.Xin W, Mironova YA, Shen H, Marino RAM, Waisman A, Lamers WH, Bergles DE, Bonci A. Oligodendrocytes support neuronal glutamatergic transmission via expression of glutamine synthetase. Cell Rep. 2019;27:2262–2271.e2265. doi: 10.1016/j.celrep.2019.04.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang TT, Qian F, Liu L, Peng XC, Huang JR, Ren BX, Tang FR. Astroglial connexins in epileptogenesis. Seizure. 2021;84:122–128. doi: 10.1016/j.seizure.2020.11.022. [DOI] [PubMed] [Google Scholar]
- 86.Zaitsev AV, Malkin SL, Postnikova TY, Smolensky IV, Zubareva OE, Romanova IV, Zakharova MV, Karyakin VB, Zavyalov V. Ceftriaxone treatment affects EAAT2 expression and glutamatergic neurotransmission and exerts a weak anticonvulsant effect in young rats. Int J Mol Sci. 2019;20:5852. doi: 10.3390/ijms20235852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zeng LH, Bero AW, Zhang B, Holtzman DM, Wong M. Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of Tuberous Sclerosis Complex. Neurobiol Dis. 2010;37:764–771. doi: 10.1016/j.nbd.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao X, Liao Y, Morgan S, Mathur R, Feustel P, Mazurkiewicz J, Qian J, Chang J, Mathern GW, Adamo MA, Ritaccio AL, Gruenthal M, Zhu X, Huang Y. Noninflammatory changes of microglia are sufficient to cause epilepsy. Cell Rep. 2018;22:2080–2093. doi: 10.1016/j.celrep.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]