

ABSTRACT
Chromatin regions that interact with the nuclear lamina are often heterochromatic, repressed in gene expression, and in the spatial B compartment. However, exceptions to this trend allow us to examine the relative impact of lamin association and spatial compartment on gene regulation. Here, we compared lamin association, gene expression, Hi-C, and histone mark datasets from cell lines representing different states of differentiation across different cell-type lineages. With these data, we compare, for example, gene expression differences when a B compartment region is associated with the nuclear lamina in one cell type but not in another. In general, we observed an additive rather than redundant effect of lamin association and compartment status. But, whether compartment status or lamin association had a dominant influence on gene expression varied by cell type. Finally, we identified how compartment and lamin association influence the likelihood of gene induction or repression in response to physicochemical treatment.
KEYWORDS: Gene regulation, Nuclear lamina, Nucleus architecture, Hi-C, cell differentiation
Introduction
Inside the nucleus, chromosomes interact among themselves, with other different nuclear substructures and the nuclear lamina, leading to the formation of 3D spatial genome organization [1]. Several aspects of this organization have been implicated in gene regulation [2–6], including a repressive role of both chromatin tethering to the nuclear lamina and spatial segregation into the B compartment [5,6]. The nuclear lamina is a protein meshwork underneath the nuclear membrane, which contributes to the structural stability of the nucleus [7,8]. The chromatin regions tethered to the lamina (lamina-associated domains; LADs) are typically between 0.1 and 10 Mb in size and are generally gene-poor, transcriptionally repressed, heterochromatin-rich regions [6]. Several studies have shown that tethering an active region to the lamina tends to lead to decreased expression of the genes located in that region [9–13]. Targeted activation or inactivation of genes can promote chromosome region detachment from or attachment to the nuclear lamina, respectively [14]. However, it is not clear what purpose would be served by relocating an already inactive region to the lamina or an already active region toward the interior. Genome-wide chromosome conformation capture experiments (Hi-C) have demonstrated that chromosomal regions spatially segregate into different compartments in the nucleus [15,16]. The regions classified as belonging to the B compartment are enriched in heterochromatic marks, gene-poor regions, and inactive genes and are often associated with the nuclear lamina. Regions that switch into the B compartment often experience gene repression and vice versa [17]. However, despite the frequently reported correlations between compartment status, lamin association, and gene expression, the combinatorial effects of different repressive or activating environments on gene expression are not fully understood. How are genes affected when a typically active A compartment region is located at the lamina? Is a B compartment region more stably repressed when located at the nuclear periphery as compared to the interior? Detailed analyses of the relationships between lamina association, gene expression, and compartmentalization are often performed in a single cell type, but does the relative contribution of these factors vary by cell type or cell lineage?
One approach to shed light on these questions is to consider cells across the spectrum of differentiation status in different cell-type lineages. As a stem-like cell progresses through differentiation, finally committing to a particular fate, gene expression, genomic compartmentalization, and LAD organization also change [18–24]. In this work, we integrated publicly available genomic data to compare healthy and cancer cell lines derived from different lineages and stages of differentiation to investigate the relationship between compartment identity, LAD reorganization, and gene expression differences between cell lines.
Genome structure and lamina association can influence not only the stable expression or repression of genes in a particular cell type but also how likely a gene is to be induced by a stimulus [25,26]. Given the same perturbation, different cell types will activate or repress different genes [27], and we hypothesize that the preexisting compartment and lamin organization state of the chromatin contributes to this differential gene expression. Thus, we also examined the impact of initial compartment status and lamina association on gene expression changes after stimulus or perturbation.
In this study, we present both conserved and lineage-specific trends of the relationship between LAD and compartment status and gene expression. We found that there is most frequently an additive effect of LAD and compartment status on gene expression. However, for specific regions, depending on their underlying compartment identity and lamina association in different cell-type lineages and states, we observed differences in whether compartment or LAD status dominates the gene expression effect. Further, across different cell types and perturbations, genes in the non-LAD A compartment state were most likely to show altered gene expression in both the induced and repressed directions.
Results
Chromosome compartmentalization and lamina association segregate cells by lineage
To investigate the relationship between LAD and gene expression for genes located in different spatial compartments, we selected cell types for which publicly available lamina association (mainly DamID data, unless otherwise mentioned), genome organization (Hi-C) and gene expression (RNA-seq) data were available. In total, 11 different cell types consisting of both primary cells and cell lines were collected for this study and classified based on developmental lineage, cell type, and malignancy status based on known characteristics (Supplementary Table 1). Considering embryonic stem cells (H1ESC) as the node of origin, cells were separated into three lineages: a) mesoderm, derived from mesenchymal stem cells (HMSC), b) endoderm, primarily composed of epithelial cells and c) hematopoietic cells (Figure 1a) [28,29]. The mesoderm-derived cell lines were sub-divided into two families: fibroblasts (WI38, IMR90, Tig3, and Hffc6) and osteoblast-derived osteosarcoma (U2OS) [30–33]. Among the fibroblast cells, WI38, IMR90, and Tig3 are all fetal lung fibroblast cell lines and, for the purpose of this analysis, considered as a set [31–33]. The epithelial cell lines were classified as either potentially secretory (colon carcinoma – HCT116) or non-secretory (RPE) [34–36]. A special case of epithelial fibrosarcoma (HT1080) was considered a potential midpoint between a bona fide sarcoma (U2OS) and an epithelial line [37]. Finally, the hematopoietic cell lines were classified as either derived from a myeloid progenitor (K562 and HAP1) or a lymphoid progenitor (Jurkat) [38–40]. Notably, these blood-related cell lines all represent cancerous derivatives of their cell type of origin. Finally, it is important to note that the relative relationships classified in Figure 1a do not imply that these cell types are directly derived from one another.
Figure 1.
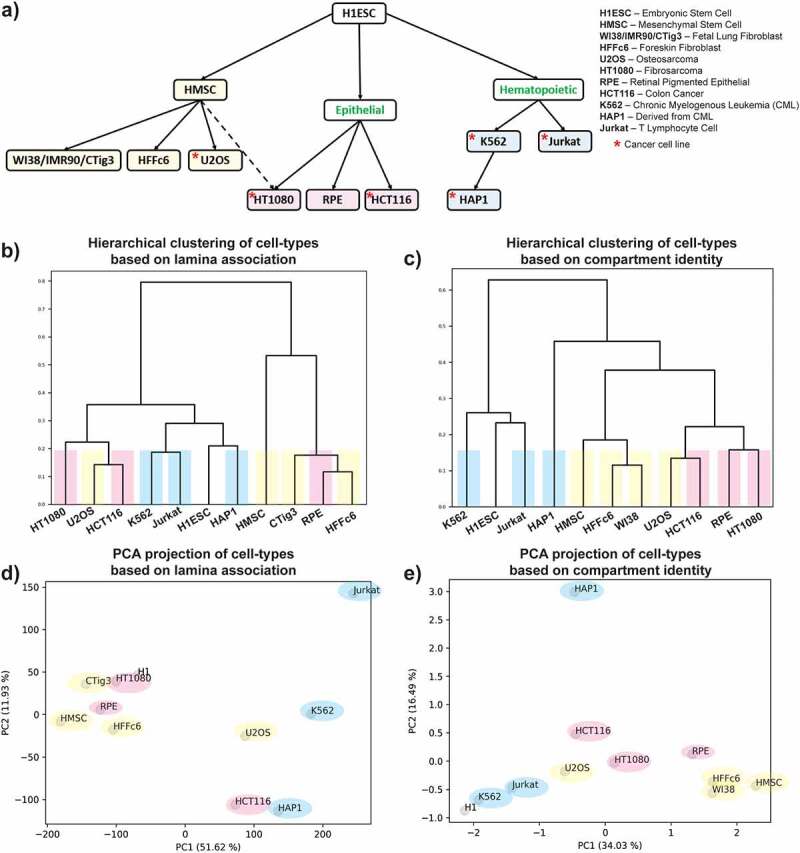
Cell lineage relationships are detected in both compartmentalization and lamin association patterns a) Human primary cells and cell lines are classified based on their developmental lineage relationships and malignancy status based on known characteristics. Yellow: mesenchymal, pink: epithelial, blue: hematopoietic, green labels indicate intermediate states in lineages which do not have specific samples representing them. b, c) Hierarchical clustering of genome-wide lamina association (b) and compartment identity (c) data for different cell types at 40 kb and 250 kb resolutions respectively. Cell lines are colored as in (a). d, e) Principal component analysis of genome-wide lamina association (d) and compartment identity (e) data for different cell types at 40 kb and 250 kb resolutions respectively. Cells lines are colored as in (a).
Once we had ordered the cell types based on prior domain knowledge, we compared this ordering to the unsupervised classification of the cell types based on either the lamina association or compartment identity data as described in the Methods. Briefly, for lamina association data, each 40 kb genomic region was assigned a lamina association value, where a value greater than zero signifies lamina association and vice versa. For compartmental organization, positive and negative values assigned to each 250 kb region indicate A and B compartment status, respectively. In the case of the lamina association, hierarchical clustering produced a cell type grouping similar to what we anticipated based on published cell characteristics, with a few interesting exceptions (Figure 1b). We found that U2OS, which is considered epithelioid-like osteosarcoma, presents a similar type of lamina association to bona fide epithelial cells. Surprisingly, the RPE (retinal pigment epithelial) cells are grouped with the mesenchymal lineage cells rather than with other epithelial cells. This may be explained by the relatively small nuclear size and the incidence of polynucleated cells in RPE [41,42]. Compartment organization clustering produced similar cell type ordering trends, though in these data RPE cells are grouped as expected with the epithelial lineage, suggesting that genome compartmentalization of these cells matches cell lineage trends more than the lamina association (Figure 1c). In both datatypes, we observed H1ESC clustering with the hematopoietic lineage cells (Figure 1b,c). Interestingly, principal component analysis (PCA) on both types of genomic data showed that the principal component 1 (PC1) axis generally ordered the cell types based on their lineages, where the mesenchymal and hematopoietic lineages can be found at the ends of the spectrum, with cells of epithelial lineage in the middle (Figure 1d,e). This recapitulates the relationships observed by unsupervised clustering and suggests that mesenchymal and hematopoietic lineages differ significantly in both lamina and compartmental organization. Further, we observed that cell types segregated according to the overall proportion of their genome associated with the lamina or B compartment (Fig. S1). Notably, H1ESC cells show a similar proportion of genomic regions with peripheral localization as the hematopoietic lineage cells. Similarly, in terms of the fraction of the genome that is lamin associated, U2OS cells are more similar to other epithelial cell types, while RPE cells are more similar to mesenchymal cell types (Fig. S1a). There is a general overall tendency for lamina association and genomic compartmentalization to cluster by lineage, but there are exceptions which vary by cell type. Such discrepancies may reflect cell-type-specific discordance level between compartmentalization and lamina association or could relate to much available data being from cancer cell lines, thus exhibiting aberrant genome organization.
Lamina association adds another layer of gene regulation to compartment identity
We next compared compartment, lamin association, and gene expression status between pairs of cell types. We started by comparing regions that have unaltered compartment identity but change their lamina association between the compared cell types. We then checked the gene expression change between cell types for genes within those regions (Figure 2 and Fig. S2). We found that, for several comparisons, genomic regions that have a similar compartment identity between cell types tend to exhibit decreased or increased expression concordant with their lamina association differences. Further, as seen for the mesenchymal vs. embryonic stem cell comparison, the association between lamin state and gene expression sometimes follows more strongly in the direction that would typically be associated with the underlying compartment state: stronger gene upregulation accompanies a loss of lamina association for regions already in the A compartment and, conversely, gene repression is more likely to accompany relocation to the nuclear lamina for regions already in the B compartment. In contrast, only some of the regions that stay in the B compartment but shift away from the lamina show gene activation.
Figure 2.
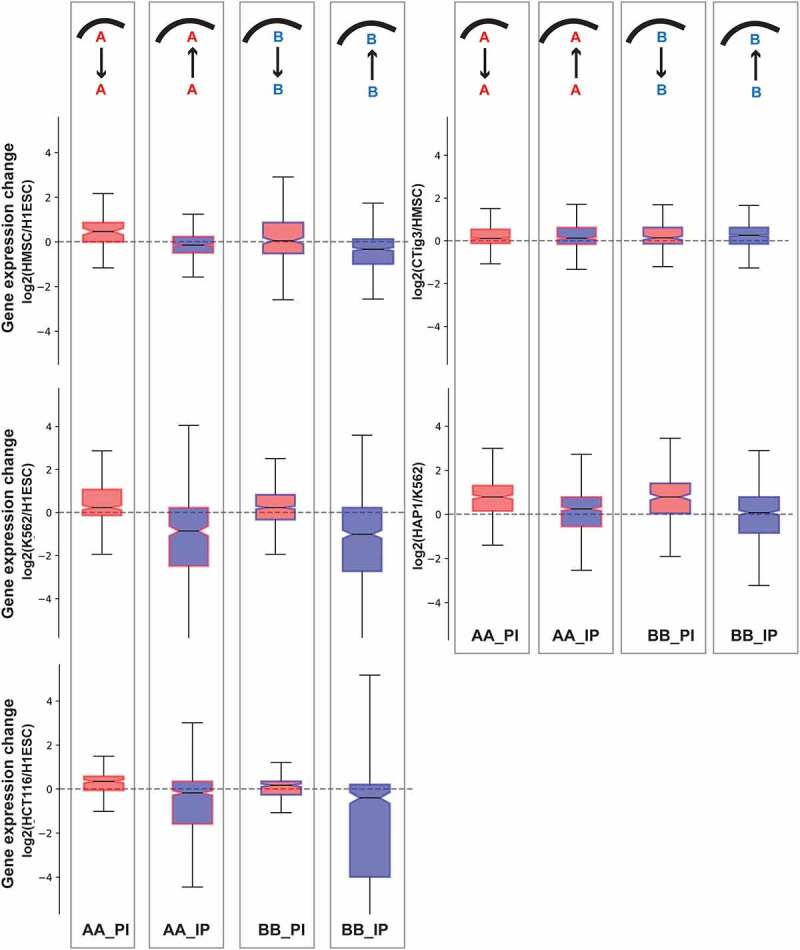
Gene expression change between cell types compared to lamina association alterations for genomic regions with unchanged compartment identity. Cell types being compared are indicated in y-axis labels. Comparisons are represented using acronyms. For example, ‘AA_IP’ indicates regions which belong to a compartment in both cell types but switch from no lamina association (‘Internal’) to lamin associated (‘Peripheral’) from the first to the second cell type. The compartment identity of each set of regions is also shown by box plot border color (red = A, blue = B) while the internal shading indicates the direction of lamin association switch (red = shift to non-LAD, blue = shift to LAD). The transition from one cell type to another for the set of regions is also shown in a cartoon over the boxplot where the curved line indicates the nuclear lamina. Boxplots show Q1–1.5 * IQR, 1st quartile, median, 3rd quartile, and Q3 + 1.5 * IQR. Outliers that fall beyond the±1.5*IQR are not shown.
This trend in which the lamin association status further modulates the gene expression profile of a region that does not change its compartment status is also observed for K562, HCT116, HT1080, RPE, and Jurkat cells compared to ESCs, and in HAP1 compared to K562 (Figure 2 and Fig. S2). This relationship between lamin association alteration and gene expression alteration can also be quantitatively observed independent of what threshold is used to classify a region as ‘lamin associated’. The change in raw DamID signal between these cell-type comparisons is negatively correlated with the change in gene expression (Fig. S3a).
In every comparison, there are genes that do not follow the expected trend, for example, genes that are strongly upregulated despite a gain in lamin association signal (Fig. S3). We next asked whether genes with certain functions were more likely to follow certain lamin association relationships to gene expression. Based on known differences between cell-type pairs, we predicted functional categories of genes that would be up or downregulated in the comparison. We found that differentially regulated genes with these expected cell type-specific functions could be found in both the A and B compartments, but were more likely to be enriched in gene sets that follow the expected relationship between LAD and gene expression change (Fig. S4, Supplementary Data 1). For example, extracellular matrix and cytokeratin functions are enriched among the genes that are both upregulated and lose lamin association in HT1080 cells compared to ES cells. In contrast, genes related to pluripotency are both downregulated and gain LAD status in HT1080 when compared to ES cells (Fig. S4b). When comparing Jurkat vs. ES cells, genes related to blood and immune-response become upregulated and lose LAD status. In this comparison, downregulated mesenchymal lineage genes gain LADs (Fig. S4c). We find that genes with discordant LAD and expression changes – upregulated but increasing in lamin association or downregulated with a loss of LAD – are less likely to be enriched for cell type-specific gene categories.
Other cell-type comparisons do not strongly follow these relationships between lamin association and gene expression. In fibroblasts or U2OS osteosarcoma cells vs. mesenchymal stem cells, for example, gene expression changes are often decoupled from lamin association changes (Figure 2, SFig. S2, SFig. S3b, and SFig. S4). In the U2OS cell comparison in particular, enriched functional categories of genes do not follow expected trends. For example, while extracellular matrix genes would be expected to be upregulated in this bone-related cell type vs. a precursor cell, we observe downregulation of this functional category instead, and further note that the downregulation occurs in the A compartment with a loss of LAD status, opposite from expected (SFig. S4d). These exceptions to the common trend may reflect true differences in the mesenchymal lineage, epigenetic dysregulation in cancer (in the case of U2OS), or particular experimental factors regarding the data available from these cell types. Cancer cells do typically show a loss of distinctive cell-type identity [43] and can show different trends in the influence of LAD and compartment status on gene expression [5,44]. Other differences in the mesenchymal lineage could arise from the fact that the HMSCs and fibroblasts used to collect different types of data derived from somewhat different sources, and HMSC lamin association is measured by proximity to emerin rather than Lamin B1 [45] (see Table S1).
Coupling and decoupling of lamina association and compartmentalization effects on gene expression
We next extended our gene expression analysis to all possible combinations of lamina association and compartment identity alterations for each of the cell-type comparisons (Figure 3, SFig. S5, and SFig. S6). For convenience, we refer to comparisons by acronyms such as BA_PI, indicating the compartment identity in each cell type (BA = B in the first cell type and A in the second cell type) as well as the LAD status in each cell type (PI = peripheral, LAD associated in the first cell type and internal, non-LAD associated in the second cell type). These comparisons further confirm that when compartment identity remains unaltered, changes in gene expression tend to follow the lamina association change. Since B compartment regions are often lamin associated and vice versa, we refer to B/P and A/I associations as ‘congruent’. In most cases, if the second cell type in the comparison showed congruent compartment and LAD status (e.g., AA_PI, BA_PI, AB_IP, and BB_IP), gene expression changes follow LAD status changes. On the contrary, in regions for which lamina association is incongruent with compartment identity (e.g., AB_II, BA_PP, AA_IP, BB_PI, AA_PP, AB_PI, BA_IP, and BB_II) some cell types appear to be more governed by LAD type while others favor compartment identity control. For example, in the HMSC vs. ESC comparison, the gene expression of these incongruent regions matches the compartment identity more than the lamina association (Figure 3). In contrast, the incongruent regions in the HCT116 vs. ESC comparison show gene expression changes matching the lamin association status (Figure 3).
Figure 3.
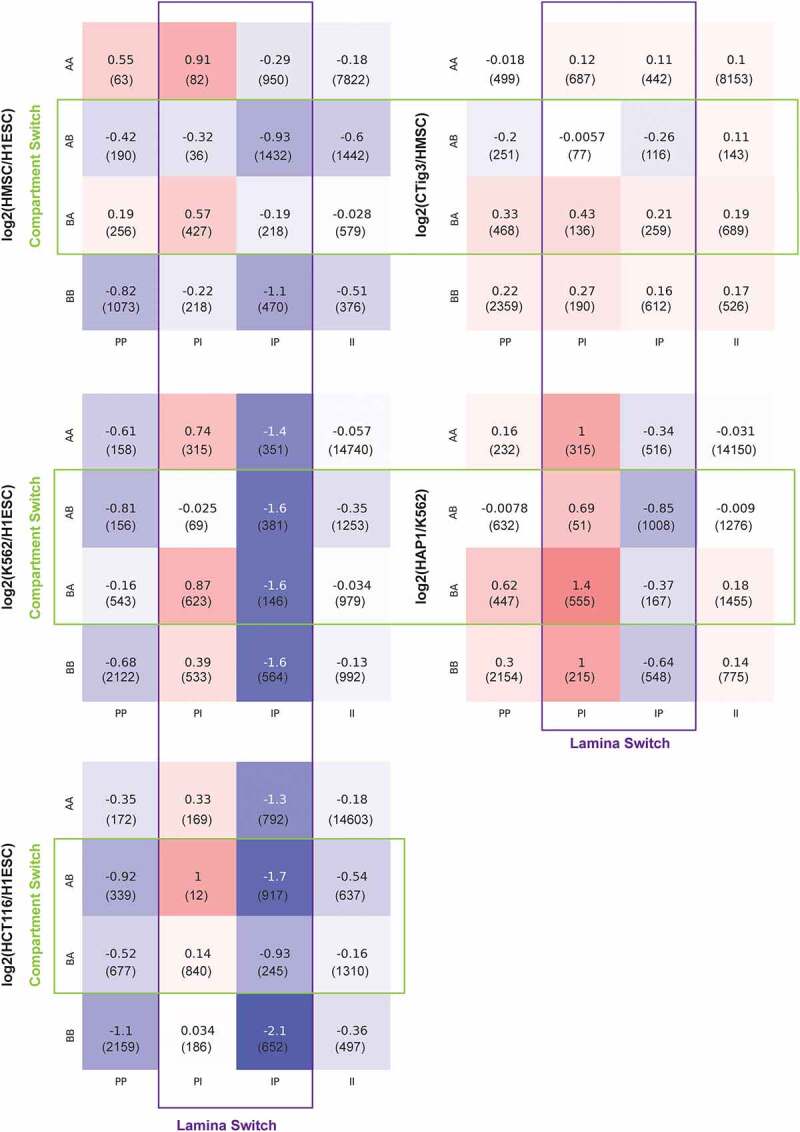
Mean gene expression change for different categories of genomic regions based on compartment identity and lamina association changes. Color shading corresponds to mean gene expression log2 fold change as indicated by the numbers in each box. Numbers in parentheses indicate how many genes fall into each category. Acronyms AB and PI are as defined in Figure 2. Cell type comparisons are indicated to the left of each panel. Green box highlights comparisons where regions switch compartments between cell types while purple box highlights regions that change lamina status.
The strength of compartment identity correlates with lamina association and vice versa
Through the combined analysis of lamina association and compartmental organization data for different cell types, we identified several megabases of genomic regions which change their lamina association, while their compartment identity remains unaltered. We next used these regions to ask how the compartment identity strength (CIS), represented by the magnitude of the compartment analysis eigenvector, is affected by nuclear positioning (Figure 4 and SFig. S7). We found that A compartment regions become more strongly defined as A (more positive CIS) when moved away from the lamina, while B compartment regions become more strongly defined as B (more negative CIS) when located at the lamina.
Figure 4.
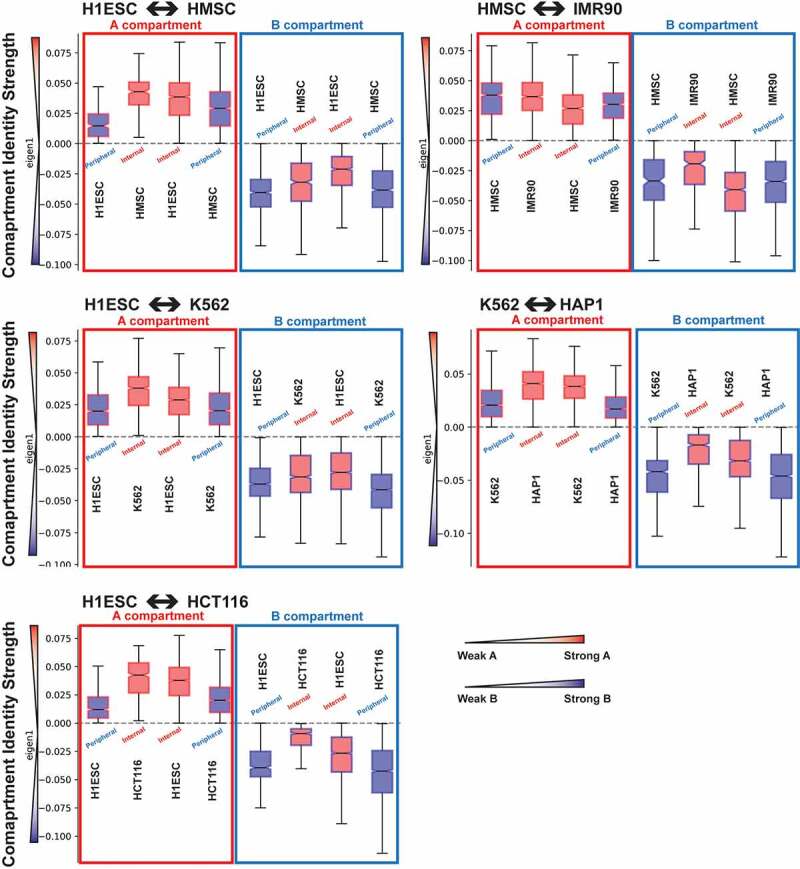
Compartment identity strength shifts with changes in LAD status between cell types for genomic regions having unchanged compartment identity. The state of each set of regions is indicated by box plot border color (compartment identity: A = red, B = blue) and shading (LAD = blue, non-LAD = red) as well as by labels – all a compartment regions are shown on the left, with LAD status and cell type indicated next to each boxplot. The y-axis represents the compartment eigenvector, where higher positive values are stronger a compartments and more negative values are stronger B compartments, as indicated by the color scale on the left of the axis.
In the converse analysis, we considered the strength of the lamin association signal (DamID-seq signal) for regions that change compartment status, while their LAD status remains constant between two cell types (SFig. S8). In the majority of cases, the LAD signal becomes stronger when the compartment shifts from A to B. Especially if the region was already classified as lamina associated in the first cell type, a shift to the B compartment often resulted in shifts to stronger lamina association (see H1->HT1080 comparison in SFig S8a). In contrast, in some cell-type comparisons, non-LAD regions shifted to even weaker lamina signal despite a change to the B compartment (see H1->Jurkat or K562 comparison in SFig. S8c), suggesting that these B compartments are not mediated by lamin association. For B to A compartment shifts, a similar phenomenon was observed. For regions that were already internal, the compartment shift to A tended to reinforce the lamin status with a shift to even lower lamin association values (see, for example, comparisons in SFig. S8c). However, LAD-associated regions were less likely to show a decrease in lamin association signal with a shift to the A compartment (see comparisons in SFig. S8a).
Lamina-associated regions with similar compartment identity exhibit differential histone modifications between cell types
Epigenetic marks are known to be associated with both compartment identity and lamina association [25,46–48]. Therefore, we looked into the enrichment of different histone marks in those regions which exhibit similar compartment identity but differential peripheral organization between different cell types (Figure 5 and SFig. S9). To this end, we focused on three different types of broad histone marks: H3K4me1 (active; enhancer), H3K27me3 (facultative heterochromatin), and H3K9me3 (constitutive heterochromatin). Details regarding the calculation of the enrichment of these different histone marks can be found in the Methods. We found that genomic regions with similar compartment identity but different lamina association status between cell-types exhibit differential histone modifications in a lineage-specific manner (Figure 5). For example, in the case of mesenchymal lineage comparisons, we noticed an enrichment of H3K4me1 active mark as the regions move toward the periphery, which holds for both A and B compartment regions. In contrast, for the other cell lineage comparisons, we observed a decrease in the H3K4me1 level as the regions move toward the periphery. Likewise, we noticed a lineage-specific trend for H3K9me3, where a decreased level of this constitutive heterochromatin mark was associated with the regions that move toward the periphery in most of the cell-type comparisons except mesenchymal lineage cells, which show the opposite trend. And finally, the H3K27me3 facultative heterochromatin mark most often increases in regions that show a relocation to the periphery while maintaining the same compartment status.
Figure 5.
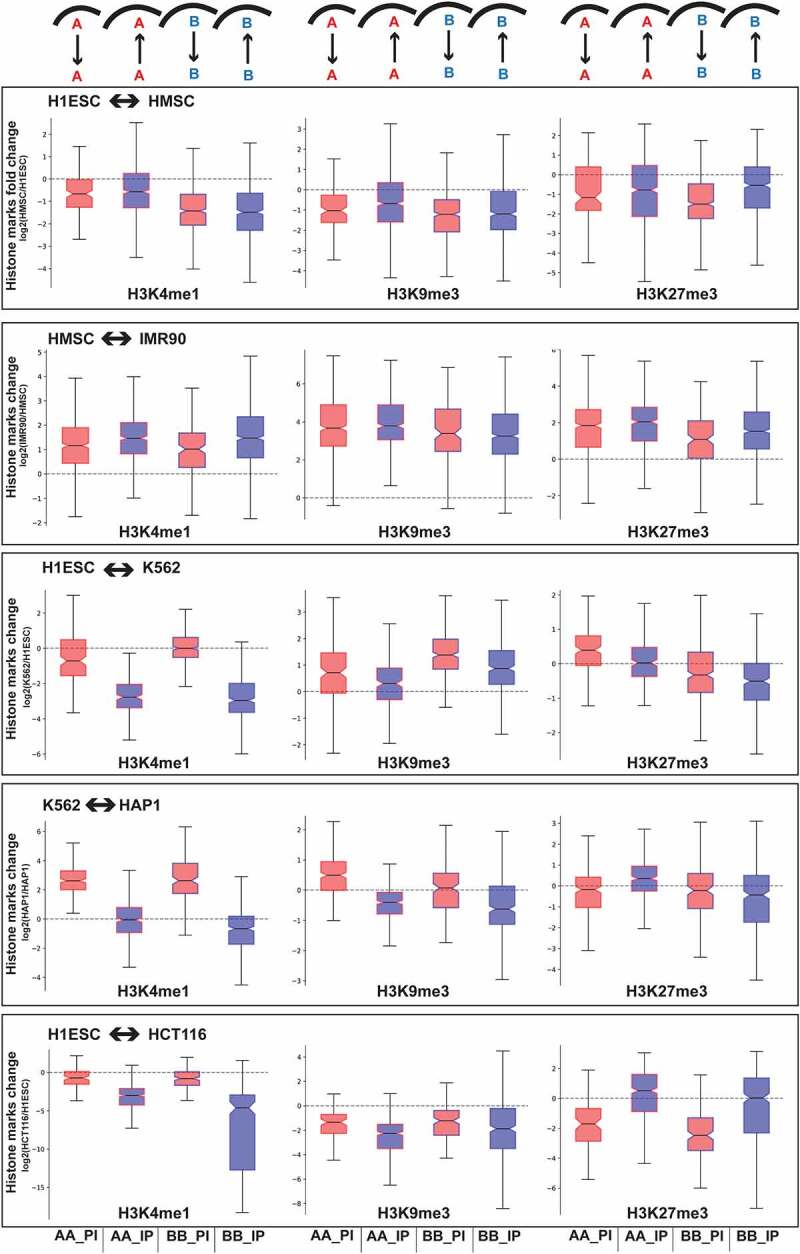
Change of different histone marks for the similar compartment identity regions which exhibit differential lamina association between cell types. Y-axis shows the log ratio of histone mark ChIP-seq signal between the indicated cell types for regions classified by compartment and LAD status changes shown. Shading and labels are as defined in Figure 2.
Lamina association reduces gene expression inducibility after physicochemical treatment
To examine the combined effect of compartmental and spatial localization on gene expression regulation in response to a stimulus, we analyzed transcriptomics data before and after different physicochemical treatments for different cell types with corresponding initial lamina association (either DamID or laminB1 ChIP-seq), and compartment identity (Hi-C) data available from other experiments on these cell types. Here, we are no longer evaluating which regions change their compartment or LAD status, but instead considering the impact of the initial compartment and LAD location of a genomic region on the gene expression response to a stimulus. Therefore, using data derived from an acute heat shock exposure of epithelial cells (HCT116), we asked how gene expression regulation was affected by preexisting compartment and lamin association in this well-characterized model for rapid and widespread transcriptional response [49]. We found that a preexisting internal positioning and A compartment status combinatorically increase the likelihood that genes will be upregulated after heat shock (Figure 6a). In contrast, downregulation occurred most frequently in lamina-associated A compartment regions.
Figure 6.
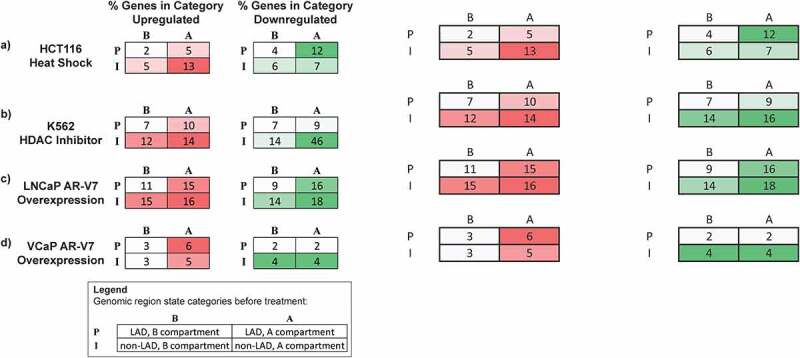
Effect of initial compartment identity and lamina association state on differential gene regulation after treatments. Tables divide genomic regions for each cell type into categories according to baseline compartment and LAD status in that cell type. Colors and numbers inside the box indicate the percentage of genes in that category that are differentially upregulated (red) or downregulated (green) after a) heat shock, b) histone deacetylase inhibitor, or c) and d) androgen receptor variant 7 (AR-V7) overexpression in the indicated cell types. The differentially regulated genes are selected based on an adjusted p-value cutoff of 0.01.
To compare this environmental condition change to an epigenetic alteration, we analyzed gene expression regulation in K562 cells treated with suberoylanilide hydroxamic acid (SAHA), an inhibitor of histone deacetylation (Figure 6b) [50]. In this condition, internal positioning, and A compartment identity combinatorically contributed to differential regulation, for both up- and down-regulation.
Finally, we analyzed the effect of exogenous expression of androgen receptor splice variant 7 (AR-V7) in prostate adenocarcinoma LNCaP and bone metastatic VCaP cells [51]. As with heat shock and histone deacetylase inhibitor treatment, both internal positioning and A compartment status increased the amount of gene activation or repression observed in LNCaP cells after AR-V7 induction (Figure 6c). Gene expression changes were more modest in VCaP cells, with the percentage of upregulated genes correlated with the compartment status. In contrast, the fraction of genes downregulated was more associated with positioning relative to the lamina (Figure 6d).
Discussion
The nuclear lamina has a well-recognized function of providing mechanical support to the nucleus and acting as a backbone to genome organization [52–59]. Functionally, chromatin contacts with the nuclear lamina have primarily been regarded as a mechanism for gene expression suppression. By creating a heterochromatic microenvironment underneath the nuclear membrane, the nuclear lamina contributes to the silencing of the chromatin regions [6,60,61]. Accordingly, genomic regions with heterochromatic properties are often localized to the nuclear periphery, and euchromatin regions are found to be primarily located in the nuclear interior. Moreover, following that trend, when a region switches from a euchromatic to a heterochromatic state, it is more likely to assume a more peripheral localization. However, if we set aside the global trends and look systematically along the genome, at significant numbers of genomic regions, switches in either category do not follow the changes in the other. By looking into those different types of incongruent regions, here, we uncovered additional observations about the relationship between genome compartmentalization, lamina association, and gene expression. For example, the genomic regions that do not alter their compartment identity between cell types exhibit gene expression changes concordant with their lamina association. Further, this effect is strongest when the lamina association changes in a direction concordant with the preexisting compartment identity, suggesting that lamina association can reinforce preexisting chromatin state. In further agreement with this idea, we found a relationship between lamin association strength and compartment identity strength such that, for example, increased lamina association associates with increased compartment identity strength for B compartment regions.
In every cell-type comparison, there are genes whose expression changes do not match the overall trends in lamin association change. By examining functional categories of genes, we found that genes associated with known cell type-specific functions are more likely to change expression concordantly with lamin association changes.
When compartment identity and final lamin association are discordant, some cell types show a strong association between lamin association and gene expression while others do not. This echoes previous observations that radial positioning can be uncoupled from gene expression in some cases [62–65]. Specifically, in the case of the mesenchymal lineage cells, compartment identity is found to be more strongly associated with gene expression regardless of lamin state. For other cell types, the changes in lamina association of those incongruent regions show a stronger association with gene expression. It is possible that in the case of the mesenchymal lineage cells, the genome compartmentalization is relatively decoupled from the lamina association compared to other cell types, as a result of the nuclear geometry in this cell lineage. It can be speculated that due to the flat ellipsoidal geometry of the mesenchymal nuclei, a higher proportion of the genome must be positioned near the periphery regardless of chromatin or compartment state. A nuclear geometry effect may also influence the relationships we observe for the RPE cell type, which behaves more like a mesenchymal cell type in our analyses despite its classification as an epithelial cell. This cell type is often observed to have a small and segmented nucleus, which may again lead to more and different chromosome regions being brought close to the nuclear periphery by geometric constraints [41,42].
The interpretation of the trends we observe here is limited by caveats in the original data sources. The available datasets we compile include isolated experiments that we arranged into potential relationships rather than an experimentally derived differentiation trajectory. Further, often only cancerous derivatives of the desired cell or tissue type were available. Thus, our analyses represent comparisons between different states, rather than actual cell state transitions over time. In the future, there is a need for coherent sets of multi-modal data collected from coherent differentiation trajectories.
When comparing the lamin association status between cell types, it is also important to note that different cell types express different ratios of lamin proteins [52]. In particular, embryonic stem cells express lamin B but not lamin A/C proteins, and then lamin A/C expression increases during differentiation [66,67]. Thus, proximity to the lamina may have different biological effects on chromatin in different cell types depending on lamina composition differences.
The relationships we observe here do not indicate the directionality of the relationship between these changes. For example, while it could be that a shift of a B compartment region away from the nuclear lamina enables the gene upregulation that we observe in a given cell state transition [68,69], it could instead be that the genes in that region were activated first and this led to relocation away from the nuclear lamina, as has been documented in some gene activation experiments [14,69,70]. Or, both factors could work in tandem to produce the changes between cell types: the activation of some genes relocates chromatin away from the lamina, which in turn makes the activation of other genes more likely.
Our analyses of gene expression response to physicochemical treatments provide evidence that preexisting lamina association and compartmentalization state influence the ability to alter gene expression in a region. However, while nuclear lamina contacts are often correlated with gene repression, we found that both induction and repression of genes were generally more likely in genomic regions that were located in the A compartment and away from the nuclear lamina. However, specifically in the case of heat shock treatment, peripheral A compartment regions exhibit a higher level of downregulation compared to other regions. This may represent regions that are actively transcribed and accessible under normal conditions (thus in the A compartment), but which are poised to be shut down by lamina associated factors in the case of a stimulus.
In our results, while relationships between compartmentalization, lamin association, and gene expression tended to follow principles that often applied across different cell-type comparisons, shifts in histone modifications were less consistent between cell types. Some cell lineages showed active and inactive histone modifications changing in concert with lamin association and dissociation, while others did not. This may reflect the different relationships that can exist between chromatin modifications and lamin contacts in different regions and contexts. The nuclear lamina can promote alterations in chromatin modifications, but also alterations in chromatin state can influence the likelihood of lamina tethering [6]. Since several of our cell lines are cancers derived from the lineages we analyze, it is also important to note that the differences in histone modification relationships we observe may relate to the dysregulation of epigenetic factors common to many cancers [71–73]. Likewise, in comparisons with ESCs, it is important to note that histone modifications which typically associate with active or inactive genes in differentiated cells may coexist in a bivalent or poised state in ESCs [74]. It appears that regardless of the divergence of particular combinations of histone marks that are involved in compartment and lamin association status in a given cell type, the resulting spatial organization state produces consistent gene regulatory principles.
Methods
Cell culture
Prostate adenocarcinoma (LNCaP), and bone metastatic (VCaP) cell lines were acquired from the Physical Sciences Oncology Network Bioresource Core Facility, through ATCC (Manassas, VA). These cell lines were cultured according to standard protocols, with subculturing at 80% confluence. LNCaP media was made of RPMI (Gibco;11835030), supplemented to match the ATCC formula (4.5 g/L glucose, 2.383 g/L HEPES, 0.11 g/L sodium pyruvate, 10% FBS). VCaP were cultured in DMEM F12: Ham 1:1 (Gibco; 11-320-033) containing 10% FBS. Media for all cell lines was supplemented with 100 mg/ml penicillin streptomycin (Gibco; 15-140-122).
Prostate cancer cell line Lamin B1 ChIP-seq
LNCaP and VCaP cells growing under continuous culture conditions in T75 flasks were allowed to reach 90% confluency. LNCaP cells were grown in RPMI (Gibco; 11835030) supplemented with 4.5 g/liter glucose, 2.383 g/liter HEPES, 0.11 g/liter sodium pyruvate and 10% FBS (Corning; 35–010-CV). VCaP cells were cultured in DMEM F12: Ham 1:1 (Gibco; 11–320–033) with 10% FBS. After media removal, the cell monolayer was washed once with HBSS, which was promptly removed by aspiration. Cells were fixed in-situ by incubation in 1% formaldehyde (37% formaldehyde diluted in HBSS) for 10 minutes, under gentle shaking. Crosslinking was quenched by adding glycine to a final concentration of 0.14 M (MP Biomedicals ICN19482591), followed by a 5 min incubation at room temperature, with shaking. Flasks were then cooled down on ice for 15 min, after which the formaldehyde solution was aspirated from the plate and substituted with 10 ml of ice-cold HBSS, supplemented with 1× Halt Protease Inhibitor cocktail (Thermo PI78438). Cells were then collected by centrifugation and snap frozen in liquid nitrogen. A replicate flask, cultured under identical conditions, was trypsinized and cells counted, using trypan blue exclusion, to estimate the number of cells per pellet.
Cell pellets were lysed and chromatin was fragmented using a Covaris M220 Sonicator, and the Covaris truChIP Chromatin shearing kit (Covaris; 520154), according to the manufacturers’ instructions, using 1 ml AFA tubes (Covaris). Briefly, cell pellets were thawed on ice and resuspended in 1 ml of 1× lysis buffer, supplemented with protease inhibitors. Samples were incubated on ice for 10 minutes, with gentle vortexing every 2 min. Nuclei were collected by centrifugation at 1700 g for 5 min (at 4 o C). The nuclei pellet was then resuspended in 1 ml of 1× wash buffer (supplemented with protease inhibitors) and collected by centrifugation as before. Samples were then washed twice, using centrifugation as before, with 1 ml of complete shearing buffer. Pellets were resuspended in 1 ml of fresh shearing buffer and transferred into a cold 1 ml AFA tube (Covaris). Sonication conditions were as follows: PIP 75, duty factor 10%, CBP 200, 4 o C, 12 minutes.
To verify sonication, aliquots of time-zero and sonicated samples were treated with RNase A (30 min at 37 o C) and reverse crosslinked overnight with proteinase K via incubation at 65 o C. After reverse crosslinking, DNA was purified using QIAquick PCR Purification Kit (Qiagen; 28104), according to the manufacturer’s instructions. The shearing efficiency was verified via agarose gel electrophoresis.
Chromatin immunoprecipitation was done using the MAGnify Chromatin immunoprecipitation system (Life Technologies, 49–2024), according to the manufacturer’s protocol, diluting the sheared chromatin at a 1:3 ratio in the kit’s dilution buffer and using three reactions per sample. Three micrograms of rabbit polyclonal antibody against Lamin B1 were used, per reaction (Abcam; ab16048). After pulldown and purification, DNA fragments were sonicated for a second time, using the following parameters in a Covaris M220 sonicator: 130-µl AFA tube, 10 o C, PIP 50%, duty factor 20%, CPB 200, for 45 seconds.
Library preparation and sequencing services were provided by Azenta/Genewiz (South Plainfield, NJ).
DamID-seq data processing
DamID-seq data were processed as reported in Leemans et al. 2019, except the trimming step [25]. Briefly, at first, the bwa mem tool (https://github.com/lh3/bwa) was used to map gDNA reads starting with GATC to a combination of hg19 reference genome with a ribosomal model. For further processing, only the mapped reads having a mapping quality of at least 10 is considered as GATC fragments. Next, the reads were combined into the bins of 40 kb resolution depending on the middle of the GATC fragments and then scaled to 1 M reads. For normalization, the log2-ratio of the scaled target over the scaled Dam-only bins was calculated with a pseudo count of 1. Furthermore, a Hidden Markov Model was used to address LADs from the normalized values using HMMt R package (https://github.com/gui11aume/HMMt).
Hi-C data processing
In this study, the HiC-Pro pipeline (https://github.com/nservant/HiC-Pro) was used with default parameters to process all the Hi-C sequencing data. The reads were aligned to hg19 human reference genome [75]. To improve the quality of the contact data, reads from all the replicates of the same cell type or condition were combined together and used for further downstream processing. For all the Hi-C datasets, for each individual chromosome, the contacts were binned at 250 kb resolution and represented in a symmetric square matrix format. Each of the matrices was then further analyzed to obtain the compartmental identity of the chromosome structures at 250 kb resolution using the ‘matrix2compartment.pl’ script from the cworld-dekker (https://github.com/dekkerlab/cworld-dekker) tool suite. Briefly, principal component analysis was performed on the distance-decay normalized contact matrix, and A/B compartment identities were assigned based on the sign of the eigenvectors (positive – A and negative – B).
RNA-seq data processing
For all the RNA-seq data, the fastq reads were first processed for adapter trimming and then for quality trimming using the BBDuk tool (https://github.com/kbaseapps/BBTools) [76]. The parameters used for those adapter and quality trimming steps were ‘ktrim=r k = 23 mink = 11 hdist = 1’ and ‘qtrim=r trimq = 28’, respectively. By performing that quality trimming step, reads with quality score lower than 28 were discarded. After having the adapter trimmed and low-quality reads removed, the remaining reads were aligned to human reference genome hg19 with the help of STAR aligner (https://github.com/alexdobin/STAR) and for that step both the ‘–outFilterScoreMinOverLread’ and ‘–outFilterMatchNminOverLread’ parameter values set to 0.2 [77]. Finally, the mapped aligned reads were sorted based on their genomic coordinates and then gene level feature count step was performed on those reads using HTSeq-Counts (https://github.com/simon-anders/htseq) [78].
Batch effect removal
The RNA-seq files used in this study were generated at different laboratories using various types of transcriptome profiling techniques. As a result of that the raw gene counts calculated by the HTSeq-Counts tool suffer from batch effects, where the samples segregate based on their origin lab and experiment types without showing the underlying true biological signal, when analyzed further for downstream processing. To uncover the biological signal, the ComBat-seq tool (https://github.com/zhangyuqing/ComBat-seq) was applied on the raw counts for the batch effect adjustments [79]. In this step, the files originated from the same laboratory were assigned the same batch number.
Differential expression analysis
Differential gene expression analysis was performed between different condition samples of the same cell type (for example, before and after AR7 induction in LNCaP and VCaP cells) using DESeq2 (https://bioconductor.org/packages/release/bioc/html/DESeq2.html) [80]. To identify significantly differentially regulated genes, we use an adjusted p-value cutoff of 0.01.
Histone ChIP-seq data processing
BBDuk tool (https://github.com/kbaseapps/BBTools) was used to process all the histone ChIP-seq data for adapter removal and quality trimming [76]. After performing those steps, the reads were mapped to hg19 human reference genome using STAR aligner (https://github.com/alexdobin/STAR) [77]. Finally, the peaks were called from the aligned data using MACS2 (https://pypi.org/project/MACS2/) peak calling software [81]. Since, in this study, we are mainly focusing on broad histone marks, here we used MACS2 broad peak calling parameters ‘-p 1e-2 –broad – nomodel –shift 0 –keep-dup all -B – SPMR’. In addition to all those parameters, we also supplied the estimated fragment length obtained from the ChIP-seq data as ‘–extsize’ parameter. To calculate the estimated fragment length, ‘run_spp.R’ script from the phandompeakqualtools (https://github.com/kundajelab/phantompeakqualtools) was used [82,83]. Once we had the broad peak locations for a particular histone mark, the enrichment of that mark for different genomic regions was measured by calculating the base pair overlaps using the ‘intersect’ function of bedtools (https://bedtools.readthedocs.io/en/latest/index.html) [84].
LaminB1 ChIP-seq data processing
LaminB1 ChIP-seq files were first processed using the BBDuk tool (https://github.com/kbaseapps/BBTools) for adapter removal and quality trimming [76]. After that, STAR aligner (https://github.com/alexdobin/STAR) [77] was used to map the reads to the hg19 genome. Once both the target and input mapped reads were mapped, they were binned at 40 kb resolution, and the log2-ratio of the target over the input bins was calculated using ‘bamCompare’ function of deepTools (https://deeptools.readthedocs.io/) with parameters ‘–operation log2 bs 40,000 – ignoreDuplicates – minMappingQuality 30 – scaleFactorsMethod SES – effectiveGenomeSize 2,864,785,220’ [85]. The signal is then further smoothed using a rolling window averaging (120 kb) followed by LAD prediction by a hidden Markov model as described in the DamID-seq data processing.
Hierarchical clustering of genome-wide lamina association and compartmental organization data
For both types of data, we performed hierarchical clustering to cluster the cell types based on similarity of the respective data type. To do that, we first calculated the pairwise similarity between all the 11 cell types using Pearson’s correlation. We further converted the similarity data into distance data by subtracting the values from 1. Following that, we finally used the distance data for hierarchical clustering using ‘ward’ linkage.
Combine lamina association and compartmental organization data
We combined the LAD and compartment identity data together for each cell type. For that, we used the ‘intersect’ function of bedtools (https://bedtools.readthedocs.io/en/latest/index.html) to combine genome-wide LAD and compartment identity data together with a minimum 50% overlap as a fraction of LAD data [84]. This allowed us to categorize the genomic regions based on whether they belong to LAD or not and also simultaneously whether those regions are part of A or B compartment.
Supplementary Material
Acknowledgments
The authors would like to thank all the members of McCord lab for their fruitful suggestions in developing the research project. We thank Campbell Maben for his assistance with gene ontology enrichment analysis.
Funding Statement
This work was supported by the National Institutes of Health NIGMS grant R35GM133557 to R.P.M. and American Cancer Society postdoctoral fellowship 134060-PF-19-183-01-CSM to R.S.M.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability Statement
Datasets used for this project were obtained from NCBI GEO, EMBL ENA, 4DN Data Portal and ENCODE repositories and their accession IDs are given in Supplementary Table 1. Newly generated LaminB1 ChIP data for VCaP and LNCaP are available at GEO with accession number GSE213032 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE213032).
Author contributions
P.D. conceived the study and performed data analysis and prepared figures. R.S.M. performed the Lamin B1 ChIP experiments, assigned cell types to lineages, and performed gene ontology enrichment analysis. R.P.M. supervised the project, provided funding, and prepared figures. P.D. and R.P.M. wrote the manuscript with input from R.S.M.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19491034.2023.2197693
References
- [1].McCord RP, Kaplan N, Giorgetti L.. Chromosome conformation capture and beyond: toward an integrative view of chromosome structure and function. Mol Cell. 2020;77(4):688–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bersaglieri C, Kresoja-Rakic J, Gupta S, et al. Genome-wide maps of nucleolus interactions reveal distinct layers of repressive chromatin domains. Nat Commun. 2022;13(1): bioRxiv 2020:2020.11.17.386797. DOI: 10.1038/s41467-022-29146-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gupta S, Santoro R. Regulation and roles of the nucleolus in embryonic stem cells: from ribosome biogenesis to genome organization. Stem Cell Rep. 2020;15(6):1206–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Morimoto M, Boerkoel CF. The role of nuclear bodies in gene expression and disease. Biology (Basel). 2013;2(3):976–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lochs SJ, Kefalopoulou S, Kind J. Lamina associated domains and gene regulation in development and cancer. Cells. 2019;8(3):271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Van Steensel B, Belmont AS. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017;169(5):780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dechat T, Pfleghaar K, Sengupta K, et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes & Development. 2008;22(7):832–853. DOI: 10.1101/gad.1652708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vahabikashi A, Adam SA, Medalia O, et al. Nuclear lamins: structure and function in mechanobiology. APL Bioeng. 2022;6(1):011503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zullo JM, Demarco IA, Piqué-Regi R, et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell. 2012;149(7):1474–1487. DOI: 10.1016/j.cell.2012.04.035 [DOI] [PubMed] [Google Scholar]
- [10].Harr JC, Luperchio TR, Wong X, et al. Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. J Cell Biol. 2015;208(1):33–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Finlan LE, Sproul D, Thomson I, et al. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 2008;4(3):e1000039. DOI: 10.1371/journal.pgen.1000039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Robson MI, Jose I, Czapiewski R, et al. Tissue-specific gene repositioning by muscle nuclear membrane proteins enhances repression of critical developmental genes during myogenesis. Molecular Cell. 2016;62(6):834–847. DOI: 10.1016/j.molcel.2016.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rønningen T, Shah A, Oldenburg AR, et al. Prepatterning of differentiation-driven nuclear lamin A/C-associated chromatin domains by GlcNAcylated histone H2B. Genome Res. 2015;25(12):1825–1835. DOI: 10.1101/gr.193748.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brueckner L, Zhao PA, van Schaik T, et al. Local rewiring of genome–nuclear lamina interactions by transcription. Embo J. 2020;39(6):e103159. DOI: 10.15252/embj.2019103159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rao SS, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–1680. DOI: 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. DOI: 10.1126/science.1181369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].San Martin R, Das P, Reis Marques R D, et al. Chromosome compartmentalization alterations in prostate cancer cell lines model disease progression. J Cell Biol. 2022;221(2):221. DOI: 10.1083/jcb.202104108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cuomo AS, Seaton DD, McCarthy DJ, et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat Commun. 2020;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Grancharova T, Gerbin KA, Rosenberg AB, et al. A comprehensive analysis of gene expression changes in a high replicate and open-source dataset of differentiating hiPSC-derived cardiomyocytes. Sci Rep. 2021;11(1):1–21. DOI: 10.1038/s41598-021-94732-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shah PP, Keough KC, Gjoni K, et al. An atlas of lamina-associated chromatin across twelve human cell types reveals an intermediate chromatin subtype. Genome Biol. 2023;24:16. DOI: 10.1186/s13059-023-02849-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Peric-Hupkes D, Meuleman W, Pagie L, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Molecular Cell. 2010;38(4):603–613. DOI: 10.1016/j.molcel.2010.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dixon JR, Jung I, Selvaraj S, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518(7539):331–336. DOI: 10.1038/nature14222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bonev B, Cohen NM, Szabo Q, et al. Multiscale 3D genome rewiring during mouse neural development. Cell. 2017;171(3):557–72. e24. DOI: 10.1016/j.cell.2017.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Miura H, Takahashi S, Poonperm R, et al. Single-cell DNA replication profiling identifies spatiotemporal developmental dynamics of chromosome organization. Nature Genet. 2019;51(9):1356–1368. [DOI] [PubMed] [Google Scholar]
- [25].Leemans C, van der Zwalm MC, Brueckner L, et al. Promoter-intrinsic and local chromatin features determine gene repression in LADs. Cell. 2019;177(4):852–64. e14. DOI: 10.1016/j.cell.2019.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stik G, Vidal E, Barrero M, et al. CTCF is dispensable for immune cell transdifferentiation but facilitates an acute inflammatory response. Nature Genet. 2020;52(7):655–661. DOI: 10.1038/s41588-020-0643-0 [DOI] [PubMed] [Google Scholar]
- [27].Honda A, Hoeksema MA, Sakai M, et al. The lung microenvironment instructs gene transcription in neonatal and adult alveolar macrophages. J Immunol. 2022;208(8):ji2101192. DOI: 10.4049/jimmunol.2101192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kang YJ, Zhang W. Chapter 13 - stem cells and regenerative medicine. In: Goodman SR, editor. Goodman’s Medical Cell Biology, 4th edition. London, UK: Academic Press; 2021. pp. 361–380. DOI: 10.1016/B978-0-12-817927-7.00013-2. [DOI] [Google Scholar]
- [29].Keller G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Gene Dev. 2005;19(10):1129–1155. [DOI] [PubMed] [Google Scholar]
- [30].Caplan AI. Review: mesenchymal stem cells: cell–based reconstructive therapy in orthopedics. Tissue Eng. 2005;11(7–8):1198–1211. [DOI] [PubMed] [Google Scholar]
- [31].Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37(3):614–636. [DOI] [PubMed] [Google Scholar]
- [32].Nichols W, Murphy D, Cristofalo V, et al. Characterization of a new human diploid cell strain, IMR-90. Science. 1977;196(4285):60–63. [DOI] [PubMed] [Google Scholar]
- [33].Matsuo M, Kaji K, Utakoji T, et al. Ploidy of human embryonic fibroblasts during in vitro aging. J Gerontol. 1982;37(1):33–37. [DOI] [PubMed] [Google Scholar]
- [34].Brattain MG, Fine WD, Khaled FM, et al. Heterogeneity of malignant cells from a human colonic carcinoma. Cancer Res. 1981;41(5):1751–1756. [PubMed] [Google Scholar]
- [35].Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14(3):141–153. [DOI] [PubMed] [Google Scholar]
- [36].Yang S, Zhou J, Li D. Functions and diseases of the retinal pigment epithelium. Front Pharmacol. 2021;12. DOI: 10.3389/fphar.2021.727870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ponten J, Saksela E. Two established in vitro cell lines from human mesenchymal tumours. Int J Cancer. 1967;2(5):434–447. [DOI] [PubMed] [Google Scholar]
- [38].Lozzio CB, Lozzio BB. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood. 1975;45(3):321–334. [PubMed] [Google Scholar]
- [39].Kotecki M, Reddy PS, Cochran BH. Isolation and characterization of a near-haploid human cell line. Exp Cell Res. 1999;252(2):273–280. [DOI] [PubMed] [Google Scholar]
- [40].Schwenk H-U, Schneider U. Cell cycle dependency of a T-cell marker on lymphoblasts. Blut Zeitschrift für die Gesamte Blutforschung. 1975;31(5):299–306. [DOI] [PubMed] [Google Scholar]
- [41].Starnes AC, Huisingh C, Mcgwin G, et al. Multi-nucleate retinal pigment epithelium cells of the human macula exhibit a characteristic and highly specific distribution. Vis Neurosci. 2016;33. DOI: 10.1017/S0952523815000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pollreisz A, Neschi M, Sloan KR, et al. Atlas of human retinal pigment epithelium organelles significant for clinical imaging. Invest Ophthalmol Visual Sci. 2020;61(8):13. DOI: 10.1167/iovs.61.8.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Roy N, Hebrok M. Regulation of cellular identity in cancer. Dev Cell. 2015;35(6):674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kumaran RI, Spector DL. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J Cell Biol. 2008;180(1):51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hu H, Ji Q, Song M, et al. ZKSCAN3 counteracts cellular senescence by stabilizing heterochromatin. Nucleic Acids Res. 2020;48(11):6001–6018. DOI: 10.1093/nar/gkaa425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Falk M, Feodorova Y, Naumova N, et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature. 2019;570(7761):395–399. DOI: 10.1038/s41586-019-1275-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Smith CL, Lan Y, Jain R, et al. Global chromatin relabeling accompanies spatial inversion of chromatin in rod photoreceptors. Sci Adv. 2021;7(39):eabj3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Poleshko A, Smith CL, Nguyen SC, et al. H3k9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis. Elife. 2019;8:e49278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kourtis N, Moubarak RS, Aranda-Orgilles B, et al. FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat Cell Biol. 2015;17(3):322–332. DOI: 10.1038/ncb3121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Frank CL, Manandhar D, Gordân R, et al. HDAC inhibitors cause site-specific chromatin remodeling at PU. 1-bound enhancers in K562 cells. Epigenetics & Chromatin. 2016;9(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Basil P, Robertson MJ, Bingman WE, et al. Cistrome and transcriptome analysis identifies unique androgen receptor (AR) and AR-V7 splice variant chromatin binding and transcriptional activities. Sci Rep. 2022;12(1):1–18. DOI: 10.1038/s41598-022-09371-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Swift J, Ivanovska IL, Buxboim A, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341(6149):1240104. DOI: 10.1126/science.1240104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lammerding J, Fong LG, Ji JY, et al. Lamins a and C but not lamin B1 regulate nuclear mechanics. J Biol Chem. 2006;281(35):25768–25780. DOI: 10.1074/jbc.M513511200 [DOI] [PubMed] [Google Scholar]
- [54].Fischer T, Hayn A, Mierke CT. Effect of nuclear stiffness on cell mechanics and migration of human breast cancer cells. Front Cell Dev Biol. 2020;8:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Srivastava LK, Ju Z, Ghagre A, et al. Spatial distribution of lamin A/C determines nuclear stiffness and stress-mediated deformation. J Cell Sci. 2021;134(10):jcs248559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Stephens AD, Banigan EJ, Marko JF. Separate roles for chromatin and lamins in nuclear mechanics. Nucleus. 2018;9(1):119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cho S, Vashisth M, Abbas A, et al. Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Dev Cell. 2019;49(6):920–35. e5. DOI: 10.1016/j.devcel.2019.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Buchwalter A, Kaneshiro JM, Hetzer MW. Coaching from the sidelines: the nuclear periphery in genome regulation. Nat Rev Genet. 2019;20(1):39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Solovei I, Wang AS, Thanisch K, et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell. 2013;152(3):584–598. DOI: 10.1016/j.cell.2013.01.009 [DOI] [PubMed] [Google Scholar]
- [60].Amendola M, van Steensel B. Nuclear lamins are not required for lamina‐associated domain organization in mouse embryonic stem cells. EMBO Rep. 2015;16(5):610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Snyder MJ, Lau AC, Brouhard EA, et al. Anchoring of heterochromatin to the nuclear lamina reinforces dosage compensation-mediated gene repression. PLoS Genet. 2016;12(9):e1006341. DOI: 10.1371/journal.pgen.1006341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Robson MI, Jose I, Czapiewski R, et al. Constrained release of lamina-associated enhancers and genes from the nuclear envelope during T-cell activation facilitates their association in chromosome compartments. Genome Res. 2017;27(7):1126–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Forsberg F, Brunet A, Ali TML, et al. Interplay of lamin a and lamin B LADs on the radial positioning of chromatin. Nucleus. 2019;10(1):7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zuleger N, Boyle S, Kelly DA, et al. Specific nuclear envelope transmembrane proteins can promote the location of chromosomes to and from the nuclear periphery. Genome Bio. 2013;14(2):1–20. DOI: 10.1186/gb-2013-14-2-r14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Brunet A, Forsberg F, Fan Q, et al. Nuclear lamin B1 interactions with chromatin during the circadian cycle are uncoupled from periodic gene expression. Front Genet. 2019;10:917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Constantinescu D, Gray HL, Sammak PJ, et al. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells. 2006;24(1):177–185. [DOI] [PubMed] [Google Scholar]
- [67].Butler JT, Hall LL, Smith KP, et al. Changing nuclear landscape and unique PML structures during early epigenetic transitions of human embryonic stem cells. J Cell Biochem. 2009;107(4):609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kohwi M, Lupton Joshua R, Lai S-L, et al. Developmentally regulated subnuclear genome reorganization restricts neural progenitor competence in drosophila. Cell. 2013;152(1–2):97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chen H, Zheng X, Zheng Y. Age-associated loss of lamin-b leads to systemic inflammation and gut hyperplasia. Cell. 2014;159(4):829–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kohwi M, Lupton JR, Lai S-L, et al. Developmentally regulated subnuclear genome reorganization restricts neural progenitor competence in Drosophila. Cell. 2013;152(1–2):97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Clapier CR. Sophisticated conversations between chromatin and chromatin remodelers, and dissonances in cancer. Int J Mol Sci. 2021;22(11):5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mitchener MM, Muir TW. Oncohistones: exposing the nuances and vulnerabilities of epigenetic regulation. Molecular Cell. 2022;82(16):2925–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Dawson Mark A, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. [DOI] [PubMed] [Google Scholar]
- [74].Blanco E, González-Ramírez M, Alcaine-Colet A, et al. The bivalent genome: characterization, structure, and regulation. Trends Genet. 2020;36(2):118–131. [DOI] [PubMed] [Google Scholar]
- [75].Servant N, Varoquaux N, Lajoie BR, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Bio. 2015;16(1):1–11. DOI: 10.1186/s13059-015-0831-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bushnell B, Rood J, Singer E. Bbmerge–accurate paired shotgun read merging via overlap. PLoS ONE. 2017;12(10):e0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. DOI: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Anders S, Pyl PT, Huber W. Htseq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Zhang Y, Parmigiani G, Johnson WE. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom Bioinform. 2020;2(3):lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Bio. 2014;15(12):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Bio. 2008;9(9):1–9. DOI: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Landt SG, Marinov GK, Kundaje A, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–1831. DOI: 10.1101/gr.136184.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nature Biotechnol. 2008;26(12):1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Quinlan AR, Hall IM. Bedtools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ramírez F, Dündar F, Diehl S, et al. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42(W1):W187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets used for this project were obtained from NCBI GEO, EMBL ENA, 4DN Data Portal and ENCODE repositories and their accession IDs are given in Supplementary Table 1. Newly generated LaminB1 ChIP data for VCaP and LNCaP are available at GEO with accession number GSE213032 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE213032).