

Abstract
The diheme bacterial cytochrome c peroxidase (bCcP)/MauG superfamily is a diverse set of enzymes that remains largely uncharacterized. One recently discovered member, MbnH, converts a tryptophan residue in its substrate protein, MbnP, to kynurenine. Here we show that upon reaction with H2O2, MbnH forms a bis-Fe(IV) intermediate, a state previously detected in just two other enzymes, MauG and BthA. Using absorption, Mössbauer, and electron paramagnetic resonance (EPR) spectroscopies coupled with kinetic analysis, we characterized the bis-Fe(IV) state of MbnH and determined that this intermediate decays back to the diferric state in the absence of MbnP substrate. In the absence of MbnP substrate, MbnH can also detoxify H2O2 to prevent oxidative self damage, unlike MauG, which has long been viewed as the prototype for bis-Fe(IV) forming enzymes. MbnH performs a different reaction from MauG, while the role of BthA remains unclear. All three enzymes can form a bis-Fe(IV) intermediate but within distinct kinetic regimes. The study of MbnH significantly expands our knowledge of enzymes that form this species. Computational and structural analyses indicate that electron transfer between the two heme groups in MbnH and between MbnH and the target tryptophan in MbnP likely occurs via a hole-hopping mechanism involving intervening tryptophan residues. These findings set the stage for discovery of additional functional and mechanistic diversity within the bCcP/MauG superfamily.
Graphical Abstract
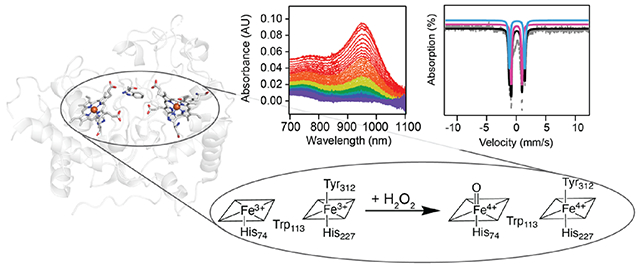
INTRODUCTION
Iron-dependent enzymes are widespread in nature and catalyze a range of biological redox reactions often involving potent high-valent oxoiron intermediates. To leverage these intermediates without causing oxidative damage, radical generation is frequently used in conjunction with long-range electron transfer (ET).1–5 In heme enzymes, two oxidizing equivalents can be harnessed by storing one on the heme iron and the second as an organic radical either on the porphyrin ring/axial ligand, denoted compound I (cmpd-I), or on a nearby amino acid residue, denoted compound ES (cmpd-ES). These two species have been characterized extensively in heme catalases, bacterial cytochrome c peroxidases (bCcPs), cytochrome P450s, cytochrome c oxidase, and NO synthase.6–10 A rarer example of a heme-enzyme state harboring two oxidizing equivalents has one on the usual oxoiron(IV) complex and the second on an additional second coordinate heme located 20 Å away (hereafter denoted bis-Fe(IV) state). The bis-Fe(IV) state has thus far been demonstrated only in two members of the broad diheme bCcP/MauG superfamily (PF03150), MauG and BthA.11,12
Whereas the physiological role of BthA is unknown, MauG uses its high-spin (HS), five-coordinate heme, heme 1, to capture hydrogen peroxide (H2O2) and form its bis-Fe(IV) state, which then oxidizes a pair of tryptophan residues in the methylamine dehydrogenase (MADH) precursor protein (preMADH) to the enzyme’s tryptophan tryptophylquinone cofactor.11 Although the bis-Fe(IV) state is presumed to be a potent oxidizing species,13 it is has an unusually long lifetime due to its unique environment within the protein scaffold.14 This long lifetime is thought to arise from electron transfer between the porphyrin ring of the HS, low-potential heme 1 and the low-spin (LS), high-potential, and six-coordinate heme 2, mediated by a tryptophan residue between the two hemes. Heme 2 is axially ligated by tyrosine and histidine, a coordination motif proposed to be critical to accessing its Fe(IV) oxidation state in the bis-Fe(IV) complex.14–16 Canonical bCcPs feature a Met–His ligation of heme 2 and are unable to form bis-Fe(IV) species. Instead, they use their diheme active site to reduce H2O2 to water by oxidizing a substrate protein.17 The MauG Tyr–His coordination motif results in Fe(III)/Fe(II) reduction potentials for the diheme active site of −159 mV and −244 mV (vs NHE) (Table S1),18 whereas the Met–His-coordinated LS heme 2 in bCcPs is tuned to a higher reduction potential (>+250 mV vs NHE), thus stabilizing the mixed-valent Feheme1(III)/Feheme2(II) state having the six-coordinate heme reduced.19–21
A recently discovered divergent member of the bCcP/MauG superfamily, MbnH,22 is proposed to act analogously to MauG in using its diheme active site (Figure 1) to generate a potent oxidizing intermediate that oxidizes a tryptophan residue in its substrate protein, MbnP, in this case to kynurenine.23 This reaction somehow accomplishes a transformation that, in catabolism of the free amino acid, requires the activities of two enzymes, tryptophan-2,3-dioxygenase and N-formyl kynurenine formamidase.24–26 The identity, reactivity, and mechanisms of formation and decay of the catalytic intermediate(s) effecting this complex transformation have not been established. Here, we show that Methylosinus trichosporium OB3b MbnH forms a bis-Fe(IV) state upon reaction with H2O2 and provide insight into how MbnH may stabilize and deploy this intermediate. In the absence of substrate, we propose that MbnH protects itself from oxidative damage by transferring oxidizing power away from the active site. Computational and structural modeling shed light on the mechanisms of ET between the two MbnH heme groups and between MbnH and its MbnP substrate. Finally, comparisons of MbnH to MauG and BthA set the stage for discovery of additional functional and mechanistic diversity within the bCcP/MauG superfamily.
Figure 1.
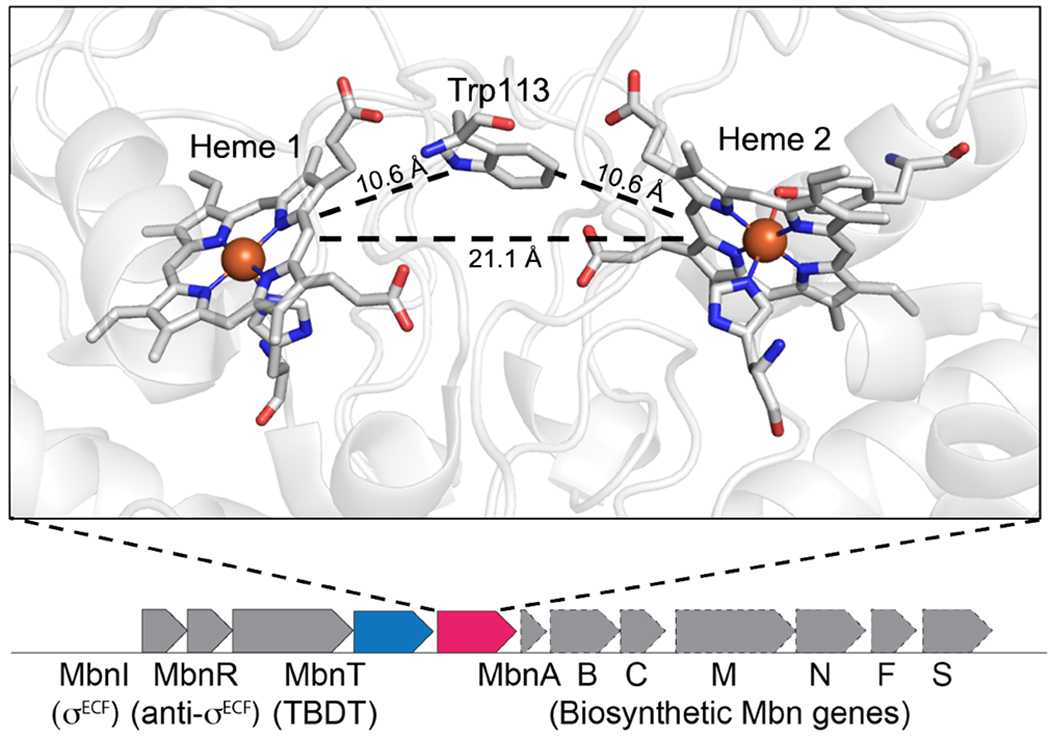
Crystal structure of MbnH. Active site of MbnH (PDB 6E1C) showing the five-coordinate, HS heme 1 (left) and the six-coordinate, LS heme 2 (right) (top). The mbnP (blue) and mbnH (pink) genes in M. trichosporium OB3b are found in an operon encoding the machinery to biosynthesize, regulate, and transport the copper chelator methanobactin (Mbn) (bottom).
RESULTS AND DISCUSSION
Decay of the High-Valent Intermediate in MbnH Occurs through H2O2 Detoxification.
Previous work demonstrated that upon reaction with H2O2, both the Soret peak and the α and β peaks in the visible Q-band region of MbnH shift and a broad feature forms in the near-infrared (nIR) region (~955 nm).22 This feature, which does not form in the absence of H2O2, is consistent with the formation of a bis-Fe(IV) state involving the two hemes and the intervening tryptophan.15,16 To further characterize this species, we monitored the decay of this nIR feature after the addition of varying quantities of H2O2. The addition of at least 3 equivalents of H2O2 is required for the development of maximum intensity, and the feature persists for tens of seconds at room temperature (Figure 2A). Singular value decomposition (SVD) analysis of the 800–1100 nm region as a function of time and H2O2 concentration reveals one distinct component that contributes to the spectra (Figure S1). While changes in temperature (5–25 °C) have no observable effect on the maximum absorption wavelength or the overall line shape (Figure S2), the feature decays more rapidly with increasing temperature (Figure 2B). Fitting the experimental nIR data using the Arrhenius equation yields an activation energy (Ea) of ~42 kJ/mol (~10 kcal/mol) for decay of the bis-Fe(IV) intermediate in the absence of substrate and a rate constant of 0.014 s−1 at 5 °C. Plotting the decay rate as a function of substrate concentration yields a hyperbolic shaped curve (Figure S3). This activation energy is significantly less than the ~76 kJ/mol (18.6 kcal/mol) reported for MauG,16 consistent with the 7-fold more rapid decay of the high-valent state in MbnH back to the diferric state than in MauG (0.002 s−1)16 and 60-fold more rapid decay of the bis-Fe(IV) state in BthA (0.014 min−1).27 Although all three proteins share similar cofactor structures, differences in the lifetimes of the bis-Fe(IV) state may arise from the differences in redox potentials across each system (Table S1). Recently, it has been suggested that the kinetics of electron transfer to the diheme cofactor of BthA may be fundamentally slower than that of MauG.12 By extension, it is possible that this ET process happens faster in MbnH, accounting for the shorter lifetime. Moreover, BthA lacks a coordinated calcium ion that is present in both MauG and MbnH which has been shown to impact the heme spin state and overall conformation and, by extension, may affect ET between the hemes. Last, all three proteins have different secondary coordination spheres, which may affect the stabilization of the bis-Fe(IV) state and/or ET kinetics. Notably, BthA features a serine residue positioned halfway between the hemes and likely employs a single-step tunneling mechanism involving the protein backbone.27 Conversely, MbnH and MauG feature a tryptophan residue halfway between the hemes,22,28 and MauG uses a hopping mechanism to form the bis-Fe(IV) cofactor. Additionally, the heme pocket of MauG features a threonine in position 67, whereas MbnH has an alanine residue in the analogous position. In MauG, this residue has been shown to be critical both in maintaining the hydrophobic environment of the high-spin heme and contributing the hydrogen bonding network. Mutation of this residue to an alanine to create the T67A mutant changes the kinetic properties of the bis-Fe(IV) state.29 Given the presence of an alanine instead of a threonine in MbnH, we believe that the hydrogen bonding environment may be perturbed.
Figure 2.
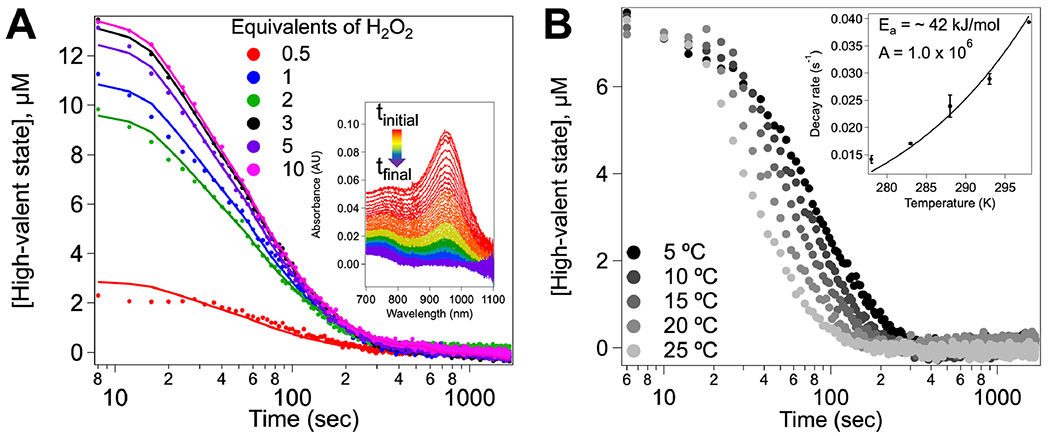
Decay kinetics of the high-valent state in MbnH. (A) Decay kinetics of the putative bis-Fe(IV) state monitored in the nIR region at room temperature upon the addition of 10–200 μM H2O2 to 20 μM MbnH. A global fit of the data to the proposed model for decay is shown. Inset shows the absorbance traces over time for the addition of 200 μM H2O2 to MbnH. (B) Decay of the nIR feature as a function of temperature upon mixing 200 μM H2O2 with 20 μM MbnH. The data were fit to a single exponential to extract kobs for the bis-Fe(IV) decay. The inset shows decay rates as a function of temperature with data fit to the Arrhenius equation.
In the absence of substrate, MauG oxidizes nearby methionine residues to methionine sulfoxides upon decay of its bis-Fe(IV) species.30,31 Protection from this oxidative damage occurs only in the presence of ascorbate.32 The addition of 4 equivalents of H2O2 inactivates MauG and prevents further formation of the bis-Fe(IV) species.31 By contrast, reaction of MbnH with 10 equivalents of H2O2 results in formation and decay of the nIR spectral feature attributable to the bis-Fe(IV) state, and addition of another 10 equivalents of H2O2 again yields the nIR feature. In one experiment, this addition of 10 equivalents H2O2 was repeated a total of 10 times, and MbnH repeatedly developed the nIR feature to the same intensity each time (Figure S4). We initially expected that in the absence of substrate, the high-valent state would decay via pathways accruing oxidative damage to the protein, similar to MauG. Instead, in the absence of MbnP, MbnH appears to avoid oxidative damage, enabling its catalytic decomposition of H2O2. Consistent with this hypothesis, fully heme-loaded MbnH was found to have peroxidase activity as measured by the o-dianisidine dye-linked assay (Figure S5A).22,33 Kinetic analysis yielded approximate KM and kcat values of 1.24 μM and 0.22 s−1, respectively (Figure S5B), and a specificity constant, kcat/KM, of 1.75 × 105 M−1 s−1 for peroxide decomposition (Table S1), approximately 10 times less than that of canonical bCcPs.27,34 Furthermore, using reduced myoglobin as a spectroscopic probe for oxygen production, we are able to show that oxygen is produced following the reaction of MbnH with H2O2 (Figure S6). Thus, the decay process of the high-valent species can be treated as a peroxidase/catalase-like reaction in which o-dianisidine or an additional molecule of H2O2 serves as the external electron donor to the diheme cofactor. The activation energy determined from the decay of the high-valent state is associated with this reaction.
MbnH High-Valent Intermediate Comprises Multiple States in a Rapid Equilibrium.
In addition to the appearance of the nIR feature upon treatment of MbnH with H2O2, we observed changes in the visible region of the spectrum. Included in these changes were a decrease in the intensity of the Soret band and a redshift from 404 to 411 nm (Figure 3A and Figure S7), consistent with perturbation of histidine-coordinated heme 1. Concomitant with the hypochromatic shift of the Soret band, features in the Q-band region attributed to the six-coordinate heme 2 (495, 538, 574, and 630 nm) shift due to oxidation of the six-coordinate heme 2 to an Fe(IV) species (Figure 3). Transient absorption features appear at ~523 and 526 nm (Figure 3B), and the 630 nm feature shifts to 660 nm. Cmpd-I like intermediates, which contain an Fe(IV) ion (SFe = 1) coupled to a radical (Srad = 1/2) on the porphyrin ring, typically exhibit absorption in the 660 nm region but lack a feature in the nIR region.6,35,15,36 Therefore, while not definitive, the presence of a transient absorption feature at 660 nm in MbnH is suggestive of the potential formation of a cmpd-I-like intermediate on the same time scale as the putative bis-Fe(IV) state.
Figure 3.
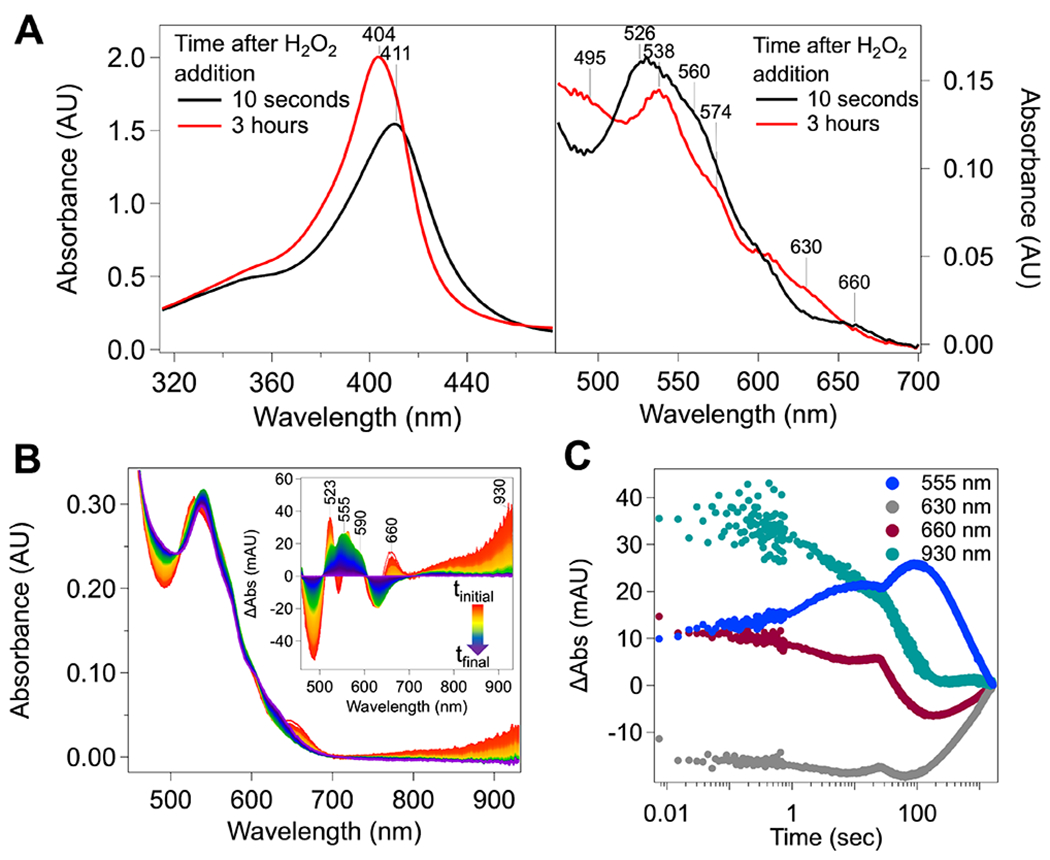
Reaction of MbnH with H2O2. (A) Changes in the MbnH visible region upon the addition of H2O2. Spectra were collected following the addition of 100 μM H2O2 to ~10 μM MbnH, and traces were collected every 30 s for 3 h in kinetic mode on a UV–vis spectrometer. (B) Baseline-corrected absorption spectra collected immediately following mixing of 200 μM H2O2 with 20 μM MbnH in a 1:1 volume ratio for 30 min. Inset shows the difference spectra at the heme α and β bands monitored at 5 °C by stopped-flow absorption spectroscopy. The final spectrum was subtracted from each spectrum to better visualize changes in the visible region. (C) Kinetic traces at wavelengths corresponding to bis-Fe(IV) (930 nm), 1e− oxidized cofactor intermediate (555 nm), 660 intermediate (660 nm), and the diferric MbnH product (630 nm) monitored at 5 °C using stopped-flow absorption spectroscopy. Spectra were collected immediately following mixing of 200 μM of peroxide with 20 μM MbnH in a 1:1 volume ratio for 30 min. The absorbance at 930 nm was used as a proxy for the 955 nm nIR feature.
SVD analysis of the Q-band region reveals four distinct components that contribute to the overall spectra (Figure S8). Deconvolution trying to incorporate five components resulted in a fifth component with a large amount of noise, a low significance value, and an autocorrelation value below 1 (Figure S9). For these reasons, modeling with four components was determined to be appropriate. In addition to the putative bis-Fe(IV) and 660 nm intermediates identified by the 955 and 660 nm features, respectively, a transient broad absorption between 500 and 600 nm (maximum, ~555 nm; blue line in Figure 3C) contributes. The kinetics of formation and decay of this spectral contribution also show a dependence on temperature (Figure S10). At 5 °C, the associated species forms only after the putative bis-Fe(IV) and 660 nm intermediates have begun to decay (Figure 3C). This spectral feature likely arises from a protein-based radical, possibly a tryptophan;37–40 the cationic forms of such a radical exhibits a distinct visible absorption spectra with maxima centered near 560 nm.41,42 At 20 °C, the formation and decay of the 660 nm intermediate and putative protein-based radical (555 nm) features are not observed (Figure S10). On the basis of these data, we posit that soon after reaction of H2O2 at the five-coordinate heme 1 to generate cmpd-I, reversible electron transfer from the heme 2 iron to the porphyrin cation-radical on heme 1 brings a cmpd-I-like and bis-Fe(IV) species into rapid equilibrium. This electron transfer proceeds by hole-hopping through the intervening Trp 113, thus introducing a third, cmpd-ES like species into the composite state (reactions 1–2 to 6, Scheme 1). The cmpd-ES state was not explicitly included in the model as it was not observed to build up to any appreciable amount. However, it is an implied state in the hole-hopping mechanism between cmpd-I and bis-Fe(IV) and therefore is visualized in the mechanism provided in Scheme 1. Given the ability of MbnH to detoxify peroxide, this 2e− oxidized equilibrium state likely decays through two pathways. In the first path, we imagine decay directly from the high-valent state to the diferric state via some catalase-like mechanism, resulting in the production of O2. This pathway would explain the peroxidase/catalase activity of MbnH, but it does not fully account for the protein-based radical observed at 555 nm. Therefore, we hypothesize there may also be a transfer of an electron from the protein surface to the high-valent state, thereby forming a mono-Fe(IV) intermediate observed at 555 nm (Figure 3A). There are several possible pathways composed of tryptophans and tyrosines (Figure S11) that could move an oxidizing equivalent to the protein surface, as well as a nearby cysteine residue (Figure S11) that could produce a radical species. Transfer of an oxidizing equivalent away from the diheme site may help explain how MbnH avoids oxidative damage in the absence of its substrate protein. Fitting the kinetic profiles for each monitored wavelength to the model (Figure S12) was used to extract rate constants for the formation and decay of each intermediate (Scheme 1).
Scheme 1. Proposed Model for Reaction of Diferric MbnH with Hydrogen Peroxidea.
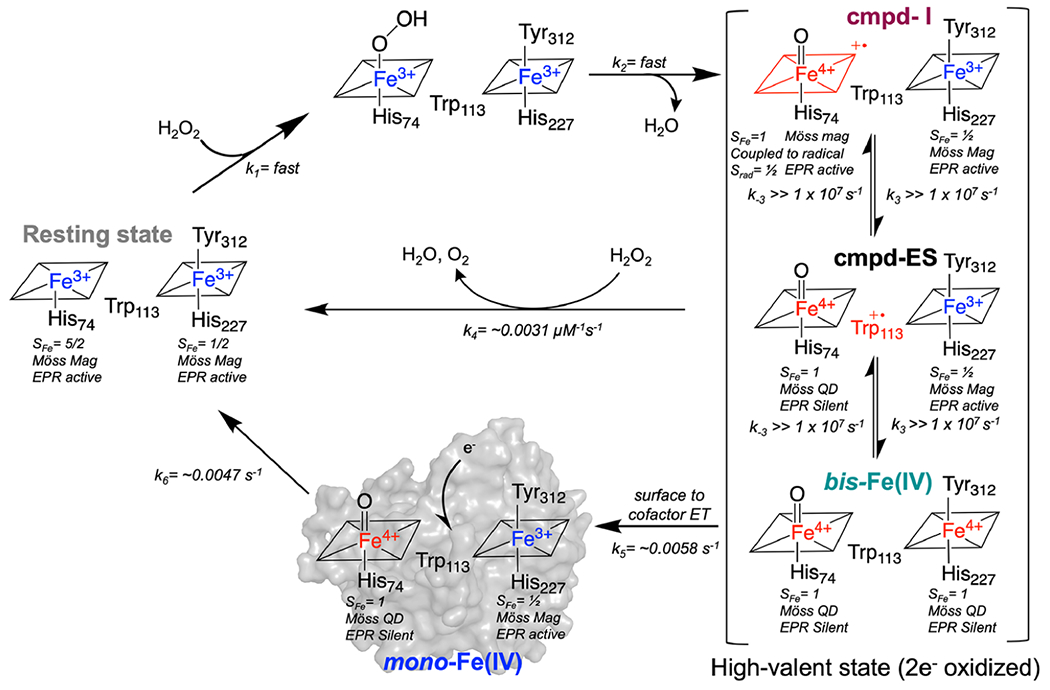
aRate constants were determined from fitting the proposed model to the kinetic profiles at 5 °C. Species that are less oxidized are shown in blue and species that are more oxidized are shown in red (Möss QD and Möss Mag indicate species that exhibit quadrupole doublet or magnetically split Mössbauer subspectra).
| (1–2) |
| (3) |
| (4) |
| (5) |
| (6) |
MbnH Forms a Stable High-Valent bis-Fe(IV) Species.
Exploiting the long-lived nature of the putative bis-Fe(IV) species, we prepared samples by the freeze–quench (FQ) method for further characterization by Mössbauer and electron paramagnetic resonance (EPR) spectroscopies. The Mössbauer spectrum of as-isolated 57Fe-enriched diferric MbnH in the absence of H2O2 reveals the presence of two distinct iron sites (Figure 4A and Figure S13). The Mössbauer parameters of these two species are consistent with a LS and a HS Fe(III) center in a relative abundance of approximately 1:1 (~55 and ~45%; Table S2). The nearly equal absorption area of the features is indicative of similar loading of the heme sites, consistent with the analysis by inductively coupled plasma mass spectrometry (ICP-MS) that showed two Fe atoms per MbnH protein monomer. Spin Hamiltonian simulations for the two ferric species were fit to the experimental spectrum using parameters derived from previous work (Figure S14).43,44 Two subspectra can also be seen in the X-band EPR spectrum of this diferric sample: a subspectrum with geff’⊥ = 5.61 and geff,∥ = 1.99, which is associated with the ground Kramers doublet of the S = 5/2 manifold of the HS heme with D >0 cm−1 and E/D ≈ 0, and a rhombic subspectrum with gx,y’z = 2.62, 2.23, 1.81, which is associated with the S = 1/2 ground state of the LS heme (Figure 4B and Table 1).
Figure 4.
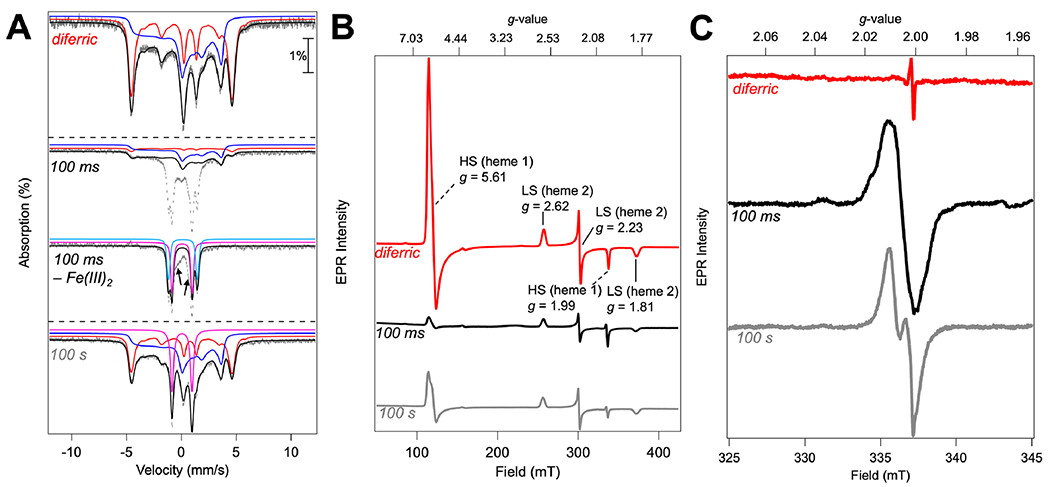
Freeze-quenched Mössbauer and EPR spectroscopy of WT MbnH reacted with H2O2. (A) Mössbauer spectra, recorded at 4.2 K in a 53 mT magnetic field applied parallel to the propagation of the γ-beam, of hand-quenched and rapid freeze-quenched samples from the reaction of diferric MbnH with H2O2. Spectrum of diferric resting-state MbnH (top) and MbnH reacted with 10 equivalents of H2O2 for 100 ms (middle) or 100 s (bottom). Both resting-state and 100 s samples were hand-quenched, while the 100 ms sample was frozen using rapid freeze-quenching. The experimental spectra are depicted by gray vertical bars reflecting the standard deviations of the absorption during spectral acquisition. The black solid lines depict the overall simulated spectra, and the colored lines are theoretical spectra illustrating the fractional contributions from HS Fe(III) heme 1 (red), LS Fe(III) heme 2 (blue), Fe(IV) intermediate from heme 1 (pink), and Fe(IV) intermediate from heme 2 (light blue) to the experimental spectra. Difference spectrum of 100 ms sample depicts the raw spectrum with the ferric components removed. Arrows indicate additional intermediate species not represented by the aforementioned Fe(III) or Fe(IV) components. Simulation parameters and relative areas of these spectra are provided in Table 1 and Table S2. (B) Full CW-EPR field sweep recorded at 10 K, microwave power of 1 mW, modulation amplitude of 1 mT, and frequency of 9.43684 GHz. (C) Field sweep in the radical region recorded at 70 K, microwave power of 1 mW, modulation amplitude of 1 mT, and frequency of 9.43684 GHz.
Table 1.
Simulation Parameters for Mössbauer Spectroscopy
| speciesb | δ (mm/s) | ΔEQ (mm/s) | linewidth (mm/s)a | S | g | D(cm−1) | (E/D) | AFe/gNβN (kG) | η |
|---|---|---|---|---|---|---|---|---|---|
| heme-1 Fe(III) | 0.42 | 1.68 | 0.40 | 5/2 | 2, 2, 2 | 5 | 0 | −182, −194, −190 | −0.1 |
| heme-2 Fe(III) | 0.30 | 2.65 | 0.35 | 1/2 | 1.811, 2.234, 2.620 | −452, 71, 288 | −2.3 | ||
| heme-1 Fe(IV) | 0.06 | 1.81 | −0.33 | ||||||
| heme-2 Fe(IV) | 0.13 | 2.63 | −0.28 |
Positive linewidth values correspond to a Lorentzian line shape and negative linewidth values correspond to a Voigt line shape.
Simulation parameters for the spectra shown in Figure 4A.
The Mössbauer spectrum of a sample prepared by rapid mixing of MbnH with 10 mM H2O2 and freeze-quenching after 100 ms exhibits diminished intensity of the two subspectra associated with the two ferric species (~5% HS and ~35% LS; Table S2) and development of several sharp absorption lines. Removal of the features of the two ferric subspectra reveals that the sharp lines are associated with two new quadrupole doublets. The first one has an isomer shift of δ1 = 0.06 mm/s and quadrupole splitting parameter of ΔEQ,1 = 1.81 mm/s and accounts for 26% of total intensity, while the second one has δ2 = 0.13 mm/s, ΔEQ,2 = 2.63 mm/s and accounts for 20% of total intensity (pink and light blue; Figure 4A, Figure S13, Table 1, and Table S2). These parameters indicate that the new features arise from two distinct Fe(IV) species. They match well with the previously reported parameters of the bis-Fe(IV) states in MauG and BthA (Table S1) and are assigned to the Fe(IV) states of hemes 1 and 2, respectively.14,27 In addition, there is residual, poorly resolved intensity in the center (~14% total intensity; see arrows in Figure 4A) that can be modeled with an asymmetric quadrupole doublet with δ ≈ 0.2 mm/s and ΔEQ ≈ 0.9 mm/s (Figure S15). While these features, in particular the broadened low-energy line, are reminiscent of those of cmpd-I of horseradish peroxidase (HRP-1), the unusually high isomer shift and lack of the expected (slight) field-orientation dependence (see Figure S15) are inconsistent with this assignment. The low relative amount of this feature (~14% of total intensity) and the fact that the signal overlaps with those of the other four components do not allow us to make a firm assignment. The EPR spectrum of a 100 ms FQ sample shows diminished intensity from the HS heme 1 signal and a more modest diminution of the LS heme 2 signal, consistent with the Mössbauer data showing the conversion of the ferric heme sites to integer spin Fe(IV) heme sites. Thus, like MauG and BthA, MbnH forms a significant amount (~40%) of a kinetically stable bis-Fe(IV) species.
The Mössbauer spectrum of a sample reacted for 100 s reveals marked changes compared to the spectrum of the 100 ms sample. The intensity of the magnetically split subspectra associated with ferric hemes increases, while that of quadrupole doublets associated with Fe(IV) hemes decreases. For heme 2, the quadrupole doublet associated with the Fe(IV) state has completely disappeared and the intensity of the LS Fe(III) site is (almost) back to the level observed for the reactant complex. For heme 1, the intensity of the quadrupole doublet is reduced to ~18% of total Fe and the intensity of the magnetically split subspectrum of HS Fe(III) increases to ~26% of total Fe, thus revealing that a significant fraction of heme 1 remains in the Fe(IV) state. Taken together, these results indicate that the bis-Fe(IV) state has completely decayed after 100 s and is reduced to a state with a ferryl heme 1 and LS ferric heme 2, which is likely generated by transfer of an exogenous electron from the surface to heme 2.
Furthermore, two different features at g ≈ 2.00 are observed in the EPR spectra of the 100 ms and 100s samples, consistent with the formation of two different protein-based radicals (Figure 4C). To further investigate these radical signals, we collected EPR spectra at varying temperatures (Figure S16) and varying powers (Figure S17). Organic radicals have differing magnetic and spin-relaxation properties depending on their location and ability to couple to nearby metal ions.45 While the signals for the two radicals are somewhat broad, they are also present at temperatures up to 70 K, consistent with the assignment to organic radicals. Given the broad EPR signals of exchange-coupled porphyrin cation radicals, these signals would not appear above temperatures of ~30 K and therefore likely do not arise from cmpd-I.46 A power saturation study of the sample at 100 ms revealed a P1/2 of 1.07 mW, consistent with a protein-based radical (Figure S17). At 100 s, the P1/2 of the radical species is 0.471 mW, again indicative of a protein radical behaving as a totally free radical located far away from the diheme site. As noted earlier, there is a broad absorption centered at 550 nm in the visible region of the absorption profile consistent with formation of a tryptophan radical. Advanced EPR methods on samples with specific isotopically labeled residues would be needed for further characterization and definitive assignment.
Mechanism of ET between the Two Hemes in the bis-Fe(IV) Species and to MbnP.
Having established that MbnH forms a bis-Fe(IV) species, we sought to understand its genesis. However, the appearance of the bis-Fe(IV) species upon addition of H2O2 is complete within the dead time of the stopped-flow instrument (~2 ms), preventing analysis of the kinetics of formation. Furthermore, it is not possible to measure the ET rate between the two hemes directly. Therefore, we estimated the ET rate and determined the most plausible ET mechanism by analyzing the MbnH crystal structure according to classical ET theory, applying the same theoretical analysis as Liu and coworkers used for MauG.47 Using the computational tool HARLEM,48 we examined the potential ET pathway between the two heme sites, considering the ET as occurring either in a single step by tunneling or as a two-step process with Trp113 as a hole-hopping mediator.15
For MbnH, the shortest distance (r) between the two hemes is ~21 Å, and the calculated decay constant (β) and electronic coupling (HAB) were determined to be 1.43 Å−1 and ~ 2.0 × 10−7, respectively. Given these values, the maximum possible predicted ET rate for single-step tunneling is ~7.8 s−1. For a two-step hopping reaction, an HAB value of 4.2 × 10−4 and a β of 1.09 Å−1 were calculated for hopping from the Fe of heme 2 to Trp113, and an HAB value of 4.5 × 10−4 and a β of 1.17 Å−1 were calculated for hopping from Trp113 to the Fe of heme 1. Each of the hopping segments has an HAB value that is 103-fold greater than that of the single-step tunneling reaction (Table 2). Because the rate of ET varies with HAB,2 the predicted ET rate for the hopping mechanism is ~106 times that for the single-step tunneling reaction, consistent with what was observed for MauG.15 Thus, the charge transfer from the porphyrin of one heme to the other heme during the formation and stabilization of the bis-Fe(IV) state occurs through a hole-hopping mechanism involving the intervening Trp113 residue. By contrast, BthA, which contains an intervening serine residue instead of a tryptophan, was predicted to use a single ET step through the backbone of histidine and arginine residues.27 Nevertheless, the delocalization over multiple redox centers in all three enzymes underscores the importance of charge and spin equilibrium for stabilization of the uncommon bis-Fe(IV) species.
Table 2.
HARLEM Analysis of ET Reactions in MbnH and MbnPH
| electron transfer in MbnH | donor | acceptor | H AB | r(Å) | β(Å−1) | max kET(s−1) |
|---|---|---|---|---|---|---|
| single-step tunneling | heme 2 Fe | heme 1 Fe | 2 × 10−7 | 21 | 1.43 | 7.8 |
| multistep hopping through Trp113 | heme 2 Fe | Trp113 | 4.2 × 10−4 | 11 | 1.09 | 4.0 × 108 |
| Trp113 | heme 1 Fe | 4.5 × 10−4 | 11 | 1.17 | 1.9 × 108 | |
| electron transfer from MbnP to MbnH | donor | acceptor | H AB | r(Å) | β(Å−1) | max kET(s−1) |
|
| ||||||
| single-step tunneling | Trp174 | heme 1 Fe | 6.7 × 10−8 | 19 | 1.86 | 3.9 × 10−2 |
| multistep hopping through Trp176 | Trp174 | Trp176 | 7.8 × 10−4 | 14 | 0.9 | 1.6 × 108 |
| Trp176 | heme 1 Fe | 1.9 × 10−5 | 16 | 1.24 | 2.7 × 108 | |
In the presence of the MbnP substrate during coexpression, MbnH oxidizes MbnP residue Trp174 to kynurenine.23 Therefore, we extended the analysis to predict the interprotein ET pathway. We first generated a model structure of the MbnPH complex using Alphafold2 (Figure S18).49 The shortest distance between MbnP Trp174 and the H2O2-binding heme 1 is ~19 Å, and the maximum possible predicted ET rate is ~3.9 × 10−2 s−1. The predicted pathway for oxidation includes MbnP Trp 176, a strictly conserved residue (Figure S18).23 For a multistep hopping pathway, HAB for the first segment from Trp 174 to Trp 176 has a calculated value of 7.8 × 10−4 with a β of 0.9 Å−1. The second segment from Trp 176 to heme 1 has a calculated HAB of 1.9 × 10−5 and a β of 1.24 Å−1. Thus, the predicted ET rate for the hopping mechanism is again ~106 times that for the single step tunneling reaction, suggesting that electron hopping is dominant in the MbnPH complex. Previous studies have shown that replacement of Trp176 with tyrosine does not affect the modification of Trp174,23 suggesting that tyrosine can also support this electron/hole hopping pathway.
Thus, MbnH uses tryptophan residues as hopping stations both between its two hemes and between heme 1 and the target of modification, Trp 174, in its substrate, MbnP. The Em value for the oxidation of tryptophan to kynurenine in MbnP is unknown, but reported Trp+•/Trp reduction potentials fall in the range of 0.64–1.08 V vs NHE,40,50 and the reported reduction potential of kynurenine to N-formylkynurenine in aqueous solution can be extrapolated as −1.1 V vs NHE.51,52 These values are very close to the range of 0.72–1.16 V vs NHE reported for other heme Fe(IV)/Fe(III) peroxidase systems, indicating that the ΔEm for the reaction will be close to 0.13 The repeated use of tryptophan along this relay is significant as hopping depends not only on the distance but also on ΔG°. Utilizing the same amino acid for each hopping station likely minimizes individual barrier heights due to the similar Em values. With ΔEm for the reaction near 0, the driving force for the oxidation of MbnP Trp 174 to kynurenine by the MbnH bis-Fe(IV) state is minimized. This is significant because following the initial hop from the diheme cofactor of MbnH, the energetic landscape of all other hopping steps is relatively flat, thereby ensuring that the hole moves to a specific site. This trend of breaking up long-range hopping into multiple energetically neutral steps is observed in other biological systems that deploy this mechanism for long-range ET.47,53–55
CONCLUSIONS
Our combined data indicate that the diheme enzyme MbnH forms a kinetically stable bis-Fe(IV) species. Charge transfer between the two heme groups occurs via a hole-hopping mechanism involving a conserved tryptophan residue, similar to the analogous process in MauG. However, the MbnH bis-Fe(IV) species decays more rapidly than that in MauG and by a mechanism that appears to be adaptive in that it serves to protect the protein from oxidation in the absence of MbnP by enabling the catalytic decomposition of H2O2. These distinctions, along with previously reported differences in redox potentials,22 are important given that MauG has been the paradigm for the bis-Fe(IV) forming enzymes within the MauG/bCcP superfamily for the past >15 years. Further studies of MbnH, BthA (which uses a different ET pathway for delocalization),27 and other family members should elucidate additional factors governing the formation and stabilization of bis-Fe(IV) intermediates. Moreover, MbnH is the only bis-Fe(IV)-forming enzyme besides MauG that has a confirmed oxidation target a distant tryptophan in MbnP23 and, as such, provides an excellent platform for investigating long-range ET linked to complex chemical transformations.
MATERIALS AND METHODS
Expression and Purification of MbnH.
Heterologous overexpression of MbnH from M. trichosporium OB3b (UniProt ID A0A2D2CY72) was accomplished as described previously.22 Briefly, a pCDFDuet-1 vector containing the gene for MbnP in MCS-1 and the gene for MbnH with an N-terminal PelB signal sequence and a C-terminal strep tag in MCS-2, was cotransformed with a pEC86 vector (containing the cytochrome c maturation or ccm genes) into BL21 DE3 cells. The ccm plasmid was necessary to improve incorporation of c-type hemes into MbnH. Transformants were first cultured in LB media at 37 °C supplemented with 70 μg/mL spectinomycin and 34 μg/mL chloramphenicol overnight and then diluted to 100 mL in LB media and grown for an additional 4 h at 37 °C. At this point, 10 mL of starter culture was transferred to ZYM-5052 autoinduction media. At an OD600 of 0.4–0.6, the temperature of the shaking incubator was lowered to 18 °C for 12–16 h for protein expression. MbnH was purified using a combination of affinity and ion size-exclusion chromatographies as described previously.22 The lysate containing MbnH was loaded onto a Strep-Tactin column (IBA), and protein was eluted using buffer containing 25 mM Tris, pH 7.4, 100 mM NaCl, and 2.5 mM desthiobiotin. Purification by size exclusion chromatography was performed using a Superdex 200 column (XK 16/100, GE Life Sciences), and protein was stored in 25 mM Tris, pH 7.4, 100 mM NaCl at 4 °C.
For expression and purification of 57Fe MbnH, ZYM-5052 autoinduction media lacking ferric citrate was prepared and supplemented with a final concentration of 50 μM 57FeSO4. 57FeSO4 was prepared by first dissolving ~25 mg of 57Fe0 (Isoflex USA) in 875 μL of 1 M H2SO4 anaerobically in a Coy chamber to produce a 500 mM 57FeSO4 solution. This solution was anaerobically heated at 60 °C for 4 h or until the 57Fe0 was mostly dissolved. The temperature was then lowered to 40 °C overnight to completely dissolve the remaining 57Fe0 anaerobically.
Determination of Protein Concentration and Metal Loading.
The purified MbnH protein concentration was determined using a standard DC assay (Bio-Rad) with equine cytochrome c (Sigma) as the standard curve protein. Metal loading was determined using ICP-MS at the Quantitative Bioelement Imaging Center at Northwestern University. Quantification of Fe was performed after digesting samples in concentrated trace nitric acid (> 69%, Thermo Fisher Scientific) and heating at 65 °C overnight for ~14 h. MilliQ H2O (18.2 MΩ·cm) was then added to produce a final solution of 3.0% nitric acid (v/v) in a 5 mL sample. A 100 μg/mL Fe element standard (Inorganic Ventures) was used to produce a 100 ng/mL 50 mL quantitative standard in 3.0% nitric acid (v/v). Samples were measured using a Thermo iCapQ ICP-MS (Thermo Fisher Scientific) controlled by the QTEGRA software in the same manner as described previously.23 Quantitative heme loading was achieved in the purified protein as determined by comparison of the Fe content to the protein concentration.
Absorption Spectroscopy.
Absorption spectra were measured on a Cary UV–vis compact Peltier (Agilent). MbnH samples were prepared in 25 mM Tris–HCl (pH 7.2) and 100 mM NaCl, and H2O2 (Fisher Scientific) solutions were prepared in the same solution. H2O2 was added to diferric MbnH protein samples, and data collection was started within 8 s of hand-mixing. Spectra were recorded over the 700–1100 nm range every 4 s for a total collection time of 30 min. Data were processed and analyzed using IgorPro 8 (Wavemetrics).
Stopped-flow absorption spectroscopy was performed using a Biologic Science Instruments (Seyssinet-Pariset, France) SFM-4000 stopped-flow spectrophotometer equipped with a broad-band UV–visible light source (Hamamatsu) and a TIDAS MCS UV/NIR 1910 photodiode array detector (J&M Analytik) housed in an MBraun anoxic chamber. The stopped-flow instrument was used in the single-mix configuration with a path length of 1 cm. For temperature-dependence studies, an external water chiller was used to maintain the temperature. MbnH at a concentration of 20 μM in 25 mM Tris–HCl (pH 7.2) and 100 mM NaCl was loaded into the sample syringe, and an H2O2 solution (varying concentration) prepared in the same buffer was loaded into the substrate syringe. Reactants were mixed in a 1:1 volume ratio to yield a final concentration of 10 μM MbnH with either 0.5, 1, 2, 3, 5, or 10 equivalents of H2O2. Reactions were monitored over the course of 30 min at temperatures of 5–25 °C. All absorption spectra were recorded over the 300–930 nm range, and data were processed and analyzed using Igor Pro 8 (Wavemetrics). The absorbance at 930 nm was used as a proxy for the 955 nm nIR feature because the weaker light emission of the source lamp in the IR region of the stopped-flow instrumentation prevented collection beyond 930 nm.
Kinetic Modeling.
SVD analysis and kinetic simulations were performed using KinTek software (version 7.2) with FitSpace Explorer and SpectraFit.56,57 Baseline-corrected absorption spectra truncated at wavelengths below 450 nm and above 850 nm due to saturation and noise were imported into KinTek, and SVD analysis was performed to deconvolute the spectra and isolate components with distinct spectral characteristics. The proposed kinetic model (reactions 1–2 to 6, Results and Discussion) was fit to the experimental data taken from the absorption profiles at select wavelengths to determine the best-fit rate constants at 5 °C. Rate constants for the decay of the bis-Fe(IV) and decay of the mono-Fe(IV) were constrained using experimentally determined values determined by fitting the decay of the absorption feature at 950 nm (for bis-Fe(IV)) and at 555 nm (for mono-Fe(IV)) at 5 and 25 °C, respectively. The best-fit rate constants are given in Scheme 1.
Mössbauer and EPR Spectroscopy.
Samples were prepared using hand-mixing (diferric resting state and 100 s) or by the FQ method (100 ms).42 The FQ instrument consists of an Update Instruments (Madison, WI) Ram Unit, a Model 705A computer controller, and a home-built, liquid nitrogen-cooled isopentane quench bath. After freeze quenching, Mössbauer samples were packed into custom cups for data collection. Mössbauer spectra were recorded on a spectrometer from SEECO (Edina, MN) equipped with a Janis SVT-400 variable-temperature cryostat. The reported isomer shifts are given relative to the centroid of the spectrum of α-iron metal at room temperature. External magnetic fields were applied parallel to the direction of propagation of the γ radiation. Simulations of the Mössbauer spectra were carried out using the WMOSS spectral analysis software from SEECO (www.wmoss.org, SEE Co., Edina, MN). Simulations of the spectral features of ferric hemes were carried using the spin Hamiltonian formalism and assuming the slow relaxation regime.
The hand-quenched EPR sample at 100 s was prepared using a different protein stock than for the FQ samples. EPR samples were transferred or packed into custom quartz X-band EPR tubes (Quartz Scientific, Inc., Fairport Harbor, Ohio), and continuous-wave X-band EPR spectra were recorded on a Magnettech MS5000X spectrometer equipped with an Oxford Instruments ESR-900 continuous-flow cryostat and an Oxford Instruments ITC-300 temperature controller. Data were collected at 10 K from 50 to 425 mT at a modulation amplitude of 1 mT and a microwave power of 1 mW. Power saturation experiments were performed to further analyze signals around g ≈ 2 at a modulation amplitude of 0.2 mT and across a power range from 0.001 to 100 mW at 75 K. The collected spectra were baseline corrected by subtraction of a fitted polynomial in Kazan Viewer58 and normalized by the square root of microwave power. The maximum intensity of the integrated spectra was extracted and fit to eq 7 below.
| (7) |
I represents the maximum normalized intensity at the appropriate magnetic field, P is the microwave power, and P1/2 is the power at half saturation.
Peroxidase/Catalase Activity Assays.
All UV–vis peroxidase assays were carried out on a Cary UV–vis compact Peltier (Agilent) in 25 mM Tris (pH 7.2) and 100 mM NaCl using o-dianisidine as the electron donor. A 3 mM stock of o-dianisidine was first prepared in ethanol as it is sparingly soluble in water. MbnH and o-dianisidine were added to buffer to final concentrations of 20 nM and 100 μM, respectively. Reactions were initiated by the addition of 1–100 μM H2O2, and the change in color of o-dianisidine upon oxidation was monitored at 460 nm. The concentration of oxidized o-dianisidine was calculated using the reported extinction coefficient of 11,300 M−1 cm−1.59 A control reaction lacking MbnH was performed to ensure that there was no background reaction from direct oxidation of the dye by H2O2. Initial velocities were determined using the method of initial slopes and plotted against the substrate concentration to determine the kinetic parameters KM, Vmax, and kcat. For reactions using myoglobin, first, myoglobin was reduced inside of an anaerobic Coy chamber using sodium dithionite (DT). Reduced myoglobin was then run through a self-packed PD-10 column to remove any excess DT and placed within a small vial containing a microstir bar. MbnH was placed in a separate vial with a microstir bar. Both vials were then placed within a septa-capped GC vial and sealed. Either 3 equivalents or 50 equivalents of H2O2 were added to only the MbnH vial, and the reaction was stirred at room temperature for 1 h. Following 1 h, a UV–vis spectra of myoglobin was collected to see if myoglobin had bound oxygen. A reaction lacking MbnH was used a negative control, and oxygen-sparged reduced myoglobin was used as positive control. Data were processed and analyzed using Igor Pro 8 (Wavemetrics).
Calculations of ET Pathways.
To evaluate ET pathways between the MbnH heme cofactors, the HARLEM program48 was used for predicting ET characteristics with PATHWAYS. This approach was also used for evaluating ET pathways between the MbnH heme cofactors and the substrate MbnP. Both the single-step tunneling pathway and an individual segment of a multistep process can be described by eq 8, in which h is Planck’s constant, R is the gas constant, T is temperature, ΔG° is the standard-state Gibbs free energy change, λ is the reorganization energy, and HAB is the electronic coupling matrix element. HAB depends on the ET distance as well as the medium, as shown in eq 9, in which the decay constant, β, quantifies how precipitously the ET rate decays with increasing distance (r – r0, where r is the distance between the donor and acceptor and r0 is the close contact distance) through a given medium.
| (8) |
| (9) |
The maximum possible ET rates under activationless conditions (i.e., when −ΔG° = λ) between the modified tryptophan of MbnP and either heme group of MbnH were estimated using the Alphafold249-predicted structure (as accessed through DeepMind’s Colab-fold) of the MbnP–MbnH complex and the direct distance approach of Dutton and co-workers60,61. The relevant ET parameters, including the decay rate (β) and electronic coupling element (HAB), were calculated for ET from the Trp174 of MbnP (donor) to the heme 1 of MbnH (acceptor 1) as well as to the heme 2 of MbnH (acceptor 2).
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to acknowledge Professor Alexey Silakov for use of his EPR spectrometer for EPR data collection.
Funding
This work was supported by NIH grants GM118035 (A.C.R.), GM127079 (C.K.), and F32 GM136156 (J.W.S.); a Simons Foundation Award through the Life Sciences Research Foundation (A.C.M.); and a Northwestern Summer Internship Grant (K.C.). The Northwestern University Quantitative Bioelement Imaging Center is supported by NASA Ames Research Center Grant NNA04CC36G.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.3c00021.
Supporting absorption, Mössbauer, and EPR spectroscopic data, Mössbauer simulation parameters, and structural models (PDF)
Accession Codes
M. trichosporium OB3b MbnH: UniProt ID A0A2D2CY72.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biochem.3c00021
The authors declare no competing financial interest.
Contributor Information
Anastasia C. Manesis, Departments of Molecular Biosciences and of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
Jeffrey W. Slater, Department of Chemistry and Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, Pennsylvania 16802, United States
Kenny Cantave, Departments of Molecular Biosciences and of Chemistry, Northwestern University, Evanston, Illinois 60208, United States.
J. Martin Bollinger, Jr., Department of Chemistry and Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, Pennsylvania 16802, United States
Carsten Krebs, Department of Chemistry and Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, Pennsylvania 16802, United States.
Amy C. Rosenzweig, Departments of Molecular Biosciences and of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
REFERENCES
- (1).Minnihan EC; Nocera DG; Stubbe J Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc. Chem. Res 2013, 46, 2524–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gray HB; Winkler JR Electron flow through metalloproteins. Biochim. Biophys. Acta, Bioenerg 2010, 1797, 1563–1572. [DOI] [PubMed] [Google Scholar]
- (3).Warren JJ; Ener ME; Viček A; Winkler JR; Gray HB Electron hopping through proteins. Coord. Chem. Rev 2012, 256, 2478–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Banerjee R; Jones JC; Lipscomb JD Soluble methane monooxygenase. Annu. Rev. Biochem 2019, 88, 409–431. [DOI] [PubMed] [Google Scholar]
- (5).Kang G; Taguchi AT; Stubbe J; Drennan CL Structure of a trapped radical transfer pathway within a ribonucleotide reductase holocomplex. Science 2020, 368, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rittle J; Green MT Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- (7).Millett ES; Efimov I; Basran J; Handa S; Mowat CG; Raven EL Heme-containing dioxygenases involved in tryptophan oxidation. Curr. Opin. Chem. Biol 2012, 16, 60–66. [DOI] [PubMed] [Google Scholar]
- (8).Yoshikawa S; Shimada A Reaction mechanism of cytochrome c oxidase. Chem. Rev 2015, 115, 1936–1989. [DOI] [PubMed] [Google Scholar]
- (9).Moody PCE; Raven EL The nature and reactivity of ferryl heme in compounds I and II. Acc. Chem. Res 2018, 51, 427–435. [DOI] [PubMed] [Google Scholar]
- (10).Lehnert N; Kim E; Dong HT; Harland JB; Hunt AP; Manickas EC; Oakley KM; Pham J; Reed GC; Alfaro VS The biologically relevant coordination chemistry of iron and nitric oxide: electronic structure and reactivity. Chem. Rev 2021, 121, 14682–14905. [DOI] [PubMed] [Google Scholar]
- (11).Geng J; Davis I; Liu F; Liu A Bis-Fe(IV): nature’s sniper for long-range oxidation. J. Biol. Inorg. Chem 2014, 19, 1057–1067. [DOI] [PubMed] [Google Scholar]
- (12).Rizzolo K; Weitz AC; Cohen SE; Drennan CL; Hendrich MP; Elliott SJ A stable ferryl porphyrin at the active site of Y463A BthA. J. Am. Chem. Soc 2020, 142, 11978–11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Battistuzzi G; Bellei M; Bortolotti CA; Sola M Redox properties of heme peroxidases. Arch. Biochem. Biophys 2010, 500, 21–36. [DOI] [PubMed] [Google Scholar]
- (14).Li X; Fu R; Lee S; Krebs C; Davidson VL; Liu A A catalytic di-heme bis-Fe(IV) intermediate, alternative to an Fe(IV)=O porphyrin radical. Proc. Natl. Acad. Sci 2008, 105, 8597–8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Geng J; Dornevil K; Davidson VL; Liu A Tryptophan-mediated charge-resonance stabilization in the bis-Fe(IV) redoxstate of MauG. Proc. Natl. Acad. Sci 2013, 110, 9639–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Geng J; Davis I; Liu A Probingbis-FeIV MauG: experimental evidence for the long-range charge-resonance model. Am. Ethnol 2015, 127, 3763–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Welinder KG Superfamily of plant, fungal and bacterial peroxidases. Curr. Opin. Struct. Biol 1992, 2, 388–393. [Google Scholar]
- (18).Li X; Feng M; Wang Y; Tachikawa H; Davidson VL Evidence for redox cooperativity between c-type hemes of MauG which is likely coupled to oxygen activation during tryptophan tryptophylquinone biosynthesis. Biochemistry 2006, 45, 821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gilmour R; Goodhew CF; Pettigrew GW; Prazeres S; Moura I; Moura JJG Spectroscopic characterization of cytochrome c peroxidase from Paracoccus denitrificans. Biochem. J 1993, 294, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Arciero DM; Hooper AB A di-heme cytochrome c peroxidase from Nitrosomonas europaea catalytically active in both the oxidized and half-reduced states. J. Biol. Chem 1994, 269, 11878–11886. [PubMed] [Google Scholar]
- (21).Wolf MW; Rizzolo K; Elliott SJ; Lehnert N Resonance Raman, electron paramagnetic resonance, and magnetic circular dichroism spectroscopic investigation of diheme cytochrome c peroxidases from Nitrosomonas europaea and Shewanella oneidensis. Biochemistry 2018, 57, 6416–6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kenney GE; Dassama LMK; Manesis AC; Chen S; Ross MO; Hoffman BM; Rosenzweig AC MbnH is a diheme MauG-like protein associated with microbial copper homeostasis. J. Biol. Chem 2019, 294, 16141–16151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Manesis AC; Jodts RJ; Hoffman BM; Rosenzweig AC Copper binding by a unique family of metalloproteins is dependent on kynurenine formation. Proc. Natl. Acad. Sci 2021, 118, No. e2100680118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).González Esquivel D; Ramírez-Ortega D; Pineda B; Castro N ; Ríos C; Pérez de la Cruz V Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 2017, 112, 331–345. [DOI] [PubMed] [Google Scholar]
- (25).Thackray SJ; Mowat CG; Chapman SK Exploring the mechanism of tryptophan 2,3-dioxygenase. Biochem. Soc. Trans 2008, 36, 1120–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Geng J; Weitz AC; Dornevil K; Hendrich MP; Liu A Kinetic and spectroscopic characterization of the catalytic ternary complex of tryptophan 2,3-dioxygenase. Biochemistry 2020, 59, 2813–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rizzolo K; Cohen SE; Weitz AC; Muñoz MML; Hendrich MP; Drennan CL; Elliott SJ A widely distributed diheme enzyme from Burkholderia that displays an atypically stable bis-Fe(IV) state. Nat. Commun 2019, 10, 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Jensen LMR; Sanishvili R; Davidson VL; Wilmot CM In crystallo posttranslational modification within a MauG/pre–methylamine dehydrogenase complex. Science 2010, 327, 1392–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shin S; Feng M; Li C; Williamson HR; Choi M; Wilmot CM; Davidson VL A T67A mutation in the proximal pocket of the high-spin heme of MauG stabilizes formation of a mixed-valent FeII/FeIII state and enhances charge resonance stabilization of the bis-FeIV state. Biochim. Biophys. Acta, Bioenerg 2015, 1847, 709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li X; Jones LH; Pearson AR; Wilmot CM; Davidson VL Mechanistic possibilities in MauG-dependent tryptophan tryptophylquinone biosynthesis. Biochemistry 2006, 45, 13276–13283. [DOI] [PubMed] [Google Scholar]
- (31).Yukl ET; Williamson HR; Higgins L; Davidson VL; Wilmot CM Oxidative damage in MauG: Implications for the control of high-valent iron species and radical propagation pathways. Biochemistry 2013, 52, 9447–9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ma Z; Davidson VL Ascorbate protects the diheme enzyme, MauG, against self-inflicted oxidative damage by an unusual antioxidant mechanism. Biochem. J 2017, 474, 2563–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Claiborne A; Fridovich I Purification of the o-dianisidine peroxidase from Escherichia coli B. Physicochemical characterization and analysis of its dual catalatic and peroxidatic activities. J. Biol. Chem 1979, 254, 4245–4252. [PubMed] [Google Scholar]
- (34).Bewley KD; Ellis KE; Firer-Sherwood MA; Elliott SJ Multi-heme proteins: nature’s electronic multi-purpose tool. Biochim. Biophys. Acta, Bioenerg 2013, 1827, 938–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Rittle J; Younker JM; Green MT Cytochrome P450: The active oxidant and its spectrum. Inorg. Chem 2010, 49, 3610–3617. [DOI] [PubMed] [Google Scholar]
- (36).Makris TM; von Koenig K; Schlichting I; Sligar SG The status of high-valent metal oxo complexes in the P450 cytochromes. J. Inorg. Biochem 2006, 100, 507–518. [DOI] [PubMed] [Google Scholar]
- (37).Huyett JE; Doan PE; Gurbiel R; Houseman ALP; Sivaraja M; Goodin DB; Hoffman BM Compound ES of cytochrome c peroxidase contains a Trp π-cation radical: characterization by continuous wave and pulsed Q-band external nuclear double resonance spectroscopy. J. Am. Chem. Soc 1995, 117, 9033–9041. [Google Scholar]
- (38).Benecky MJ; Frew JE; Scowen N; Jones P; Hoffman BM EPR and ENDOR detection of compound I from Micrococcus lysodeikticus catalase. Biochemistry 1993, 32, 11929–11933. [DOI] [PubMed] [Google Scholar]
- (39).Rigby SEJ; Jünemann S; Rich PR; Heathcote P Reaction of bovine cytochrome c oxidase with hydrogen peroxide produces a tryptophan cation radical and a porphyrin cation radical. Biochemistry 2000, 39, 5921–5928. [DOI] [PubMed] [Google Scholar]
- (40).Shafaat HS; Leigh BS; Tauber MJ; Kim JE Spectroscopic comparison of photogenerated tryptophan radicals in azurin: effects of local environment and structure. J. Am. Chem. Soc 2010, 132, 9030–9039. [DOI] [PubMed] [Google Scholar]
- (41).Baldwin J; Krebs C; Ley BA; Edmondson DE; Huynh BH; Bollinger JM Jr. Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 1. Evidence for a transient tryptophan radical. J. Am. Chem. Soc 2000, 122, 12195–12206. [Google Scholar]
- (42).Bollinger JM Jr.; Tong WH; Ravi N; Huynh BH; Edmondson DE; Stubbe J Mechanism of assembly of the tyrosyl radical-diiron(III) cofactor of E. coli ribonucleotide reductase. 3. Kinetics of the limiting Fe2+ reaction by optical, EPR, and Mössbauer spectroscopies. J. Am. Chem. Soc 1994, 116, 8024–8032. [Google Scholar]
- (43).Champion PM; Münck E; Debrunner PG; Hollenberg PF; Hager LP Mössbauer investigations of chloroperoxidase and its halide complexes. Biochemistry 1973, 12, 426–435. [DOI] [PubMed] [Google Scholar]
- (44).Debrunner PG Mössbauer spectroscopy of iron porphyrins. In EMR of Paramagnetic Molecules; Berliner LJ, Reuben J, Eds.; Biological Magnetic Resonance; Springer; US: Boston, MA, 1993; Vol. 13, pp. 139–234. DOI: 10.1007/978-1-4615-2892-0. [DOI] [Google Scholar]
- (45).Drago RS Chapter 13: Electron paramagnetic resonance spectra of transition metal ion complexes. In Physical Methods for Chemists; Surfside Scientific Publishers, 1992; pp. 559–603. [Google Scholar]
- (46).Colvin JT; Rutter R; Stapleton HJ; Hager LP Zero-field splitting of Fe3+ in horseradish peroxidase and of Fe4+ in horseradish peroxidase compound I from electron spin relaxation data. Biophys. J 1983, 41, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Tarboush NA; Jensen LMR; Yukl ET; Geng J; Liu A; Wilmot CM; Davidson VL Mutagenesis of tryptophan199 suggests that hopping is required for MauG-dependent tryptophan tryptophylquinone biosynthesis. Proc. Natl. Acad. Sci 2011, 108, 16956–16961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Kurnikov I HARLEM, 2000.
- (49).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).DeFelippis MR; Murthy CP; Faraggi M; Klapper MH Pulse radiolytic measurement of redox potentials: the tyrosine and tryptophan radicals. Biochemistry 1989, 28, 4847–4853. [DOI] [PubMed] [Google Scholar]
- (51).Bond AM; Tucker DJ; Qing Z; Rivett DE Electrochemical reduction of N-formylkynurenine at mercury electrodes in aqueous media. J. Electroanal. Chem. Interfacial Electrochem 1991, 317, 203–218. [Google Scholar]
- (52).Giles GI; Collins CA; Stone TW; Jacob C Electrochemical and in vitro evaluation of the redox-properties of kynurenine species. Biochem. Biophys. Res. Commun 2003, 300, 719–724. [DOI] [PubMed] [Google Scholar]
- (53).Stubbe J; Nocera DG; Yee CS; Chang MCY Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev 2003, 103, 2167–2202. [DOI] [PubMed] [Google Scholar]
- (54).Reece SY; Seyedsayamdost MR Long-range proton-coupled electron transfer in the Escherichia coli class Ia ribonucleotide reductase. Essays Biochem. 2017, 61, 281–292. [DOI] [PubMed] [Google Scholar]
- (55).Brettel K; Byrdin M Reaction mechanisms of DNA photolyase. Curr. Opin. Struct. Biol 2010, 20, 693–701. [DOI] [PubMed] [Google Scholar]
- (56).Johnson KA Chapter 23 Fitting enzyme kinetic data with kintek global kinetic explorer. In Methods in Enzymology; Elsevier, 2009; Vol. 467, pp. 601–626. DOI: 10.1016/S0076-6879(09)67023-3. [DOI] [PubMed] [Google Scholar]
- (57).Johnson KA; Simpson ZB; Blom T FitSpace Explorer: An algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem 2009, 387, 30–41. [DOI] [PubMed] [Google Scholar]
- (58).Silakov A Kazan Viewer.
- (59).Everse J; Johnson MC; Marini MA [36] Peroxidative activities of hemoglobin and hemoglobin derivatives. In Methods in Enzymology, Elsevier, 1994; Vol. 231, pp. 547–561. DOI: 10.1016/0076-6879(94)31038-6. [DOI] [PubMed] [Google Scholar]
- (60).Moser CC; Keske JM; Warncke K; Farid RS; Dutton PL Nature of biological electron transfer. Nature 1992, 355, 796–802. [DOI] [PubMed] [Google Scholar]
- (61).Page CC; Moser CC; Chen X; Dutton PL Natural engineering principles of electron tunnelling in biological oxidation–reduction. Nature 1999, 402, 47–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.