

Abstract
Extracellular vesicles (EVs) are an upcoming biomarker for disease. However, the measured concentrations of EVs by flow cytometry are incomparable due to analytical variables. This study aimed to investigate how the choice of fluorophore, and thereby brightness, affects the measured concentration of EVs. Four commonly used fluorophores allophycocyanin, Brilliant Violet‐421, fluorescein isothiocyanate, and phycoerythrin, all conjugated to CD61 antibodies, were used to label platelet‐derived extracellular vesicles (PEVs) in human plasma. PEVs were measured by flow cytometry. The concentration of EVs was obtained by manually set fluorescence gates, automatically determined fluorescence gates, and automatically determined fluorescence gates combined with specific size gates. Manually set fluorescence gates by five independent experts resulted in a variation coefficient (CV) of 41% between the measured PEV concentrations labeled with the four different fluorophores. A new algorithm for automatic determination of fluorescence gates was applied to reduce inter‐operator variability. Applying this algorithm resulted in a CV of 58%. However, when the algorithm was combined with a size gate to correct for differences in brightness between fluorophores, the CV reduced to 25%. In this study, we showed that different fluorophores can detect similar concentrations of EVs by (1) determining fluorescence gates automatically, and (2) by adding a size gate to correct for differences in brightness between fluorophores. Therefore, our research contributes to further standardization of EV concentration measurements by flow cytometry.
Keywords: automation, detection limit, extracellular vesicles, flow cytometry, fluorophores, immunostaining, inter‐operator variability, light scattering, sensitivity, standardization
1. INTRODUCTION
Extracellular vesicles (EVs) are cell‐derived particles enclosed by a phospholipid membrane that are present in biofluids [1]. Cells use EVs for intercellular communication and transporting biochemical information, including lipids, proteins, and RNA. Thus, the biochemical composition of EVs reflects both their cellular origin as well as the state of their parent cell [2]. Moreover, as the concentration of EVs changes with disease, EVs are potential biomarkers of disease [3].
The EV concentration can be measured by flow cytometry (FCM) [4]. At present, the measured EV concentrations are incomparable between instruments and institutes, due to differences in pre‐analytical, analytical, and post‐analytical variables [3, 5, 6, 7]. In a flow cytometry assay, the fluorophore is a pre‐analytical variable, because it affects the labeling process, and an analytical variable, because it affects the fluorescence brightness and detector used. The determination and application of gates are post‐analytical variables. In this manuscript, we show how the variation of measured concentrations of immunolabeled EVs is affected by the choice of fluorophore followed by applying [1] manually determined fluorescence gates, [2] automatically determined fluorescence gates, and [3] automatically determined fluorescence gates combined with a size gate.
We selected four commonly used fluorophores: allophycocyanin (APC), Brilliant Violet‐421 (BV421), fluorescein isothiocyanate (FITC), and phycoerythrin (PE), which were all conjugated to the same clone of CD61 (VI‐PL2). We selected the monoclonal antibody against CD61, because glycoprotein IIIa (CD61) is abundantly present on the membrane of platelet‐derived extracellular vesicles (PEVs) [8, 9], which in turn are abundantly present in human blood plasma. Moreover, the concentration of PEVs is widely studied, because it may be useful as an early biomarker of cardiovascular disease [10].
As APC, BV421, FITC, and PE differ in brightness, and only a part of all labeled EVs will exceed the detection limit of fluorescence detectors of the flow cytometer, we hypothesized that the choice of fluorophore may affect the measured concentrations of PEVs [11]. Based on the brightness of the fluorophores, which is defined by the extinction coefficient, F/P ratio, and quantum efficiency provided in Table 1, we expected that the highest PEV concentrations are measured by the brightest dyes, that is, BV421 and PE, followed by APC and FITC (Table S1.2). As manual gating to differentiate labeled EVs from background fluorescence adds to the variation in the measured PEV concentrations, we developed an algorithm to define these fluorescence gates automatically. We further hypothesized that an additional size gate, which only selects EVs exceeding the background fluorescence of each fluorophore, further reduces variation in the measured concentration of PEVs labeled with different fluorophores.
TABLE 1.
Specifications of the four used fluorophores.
| Fluorophore | Excitation source | QE | Extinction coefficient (M−1 cm−1) | F/P ratio | Brightness (M−1 cm−1) | Reference number |
|---|---|---|---|---|---|---|
| APC | Red | 0.68 | 700,000 | 1 | 4.8 × 105 | [15, 16, 17] |
| BV421 | Violet/blue | 0.69 | 2,500,000 | ‐ | 1.7 × 106 a | [17] |
| FITC | Green | 0.92 | 72,000 | 2.3 | 1.5 × 105 | [18] |
| PE | Blue‐green/yellow | 0.82 | 1,960,000 | 1 | 1.6 × 106 | [15, 17] |
Note: Excitation sources, quantum efficiency (QE), extinction coefficient, fluorophore to protein (F/P) ratio, and brightness of allophycocyanin (APC), Brilliant Violet 421 (BV421), fluorescein isothiocyanate (FITC), and phycoerythrin (PE). The F/P ratio and relative quantum yield for the four different fluorophores are either requested from the manufacturer or cited from literature.
Assuming that the F/P ratio is 1.
2. MATERIALS AND METHODS
2.1. Sample
Blood from 20 (10 male, 10 female) healthy donors was collected with a 21G needle in a 2.7 ml plastic citrate (3.2%) tube (no. 363083, from Becton Dickinson, United States). All donors gave informed consent. Plasma was prepared by centrifugation (Rotina 380R, Hettich Zentrifugen, Tuttlingen, Germany) at a room temperature (RT) of 20°C, 2500g (k‐factor = 11,585) for 15 min without brake. Plasma was collected to 10 mm above the buffy coat and the collected plasma was centrifuged using the same settings. The platelet‐depleted plasma was collected and pooled. Aliquots of pooled plasma (100 μl) were snap‐frozen in liquid nitrogen for 15 min and stored at −80°C. Before use, aliquots were thawed in a water bath for 1 min at 37°C, mixed and vortexed. Plasma dilution series were prepared in Dulbecco's phosphate‐buffered saline (DPBS) from 10‐fold to 316,500‐fold and the optimal dilution fold was determined for subsequent use.
2.2. Flow cytometry
2.2.1. Staining
Thawed plasma samples were analyzed for the presence of integrin β3 (CD61) using anti‐human CD61 clone VI‐PL2. Antibodies were conjugated to APC (eBioscience, Waltham, MA), BV421 (Becton, Dickinson and Company [BD], Franklin Lakes, NJ), FITC (BD), or PE (eBioscience). For BV421 and FITC, we also evaluated CD61+ clones RUU‐PL7F12 (BD) and Y2/51 (Dako, Glostrup, Denmark), respectively. To reduce background fluorescence by the presence of unbound reagents, antibodies were independently titrated by dilution in DPBS to find the optimal concentration of antibody that detects the highest PEV concentration with minimum background fluorescence. The manufacturers' reported antibody concentrations were used as starting concentrations for the titration series. Table 1 and Table S1.2 contain detailed specifications of the antibodies. To meet the optimal concentrations of the antibodies, dilutions were made and resulted in final concentrations of 8.3 μg/ml (APC, VI‐PL2), 6.3 μg/ml (BV421, VI‐PL2), 6.3 μg/ml (BV421, RUU‐PL7F12), 1.6 μg/ml (FITC, VI‐PL2), 12.5 μg/ml (FITC, Y2/51), and 3.0 μg/ml (PE, VI‐PL2). To remove antibody aggregates, these prediluted antibodies were centrifuged (Rotina 380R, Hettich Zentrifugen, Tuttlingen, Germany) for 5 min at RT at 18,890g (acceleration setting 9, deceleration setting 1, k‐factor = 5234). Subsequently, 10 μl supernatant was collected and transferred into a clean tube, from which 2.5 μl of the antibodies and thereafter 20 μl optimal diluted EV sample were added to a 96 well plate, shaken gently, and EVs were labeled in the dark for 2 h at RT. After incubation, 200 μl DPBS was added to dilute unbound labels. Next, samples were vortexed except for BV421, which was shaken gently, as recommended by the manufacturer. As controls, reagents in buffer control and plasma without reagents were included [6].
2.2.2. Flow cytometry
Labeled samples were measured in a 96‐well plate on an A60‐Micro (Apogee Flow Systems, Hemel Hempstead, UK). All samples were measured for 120 s at a flow rate of 3.0 μl/min using side scatter triggering with a wavelength of 405 nm. The trigger threshold was set at SSC 14 arbitrary units, corresponding to a side scattering cross section of 10 nm2 (Rosetta Calibration, Exometry, the Netherlands). The side scattering cross section is a hypothetical area of a particle that incoming light must impinge in order to be scattered toward the side scattering lens and is therefore independent of assumptions about the particle refractive index [12]. The side scattering cross section should be interpreted as a calibrated scale for light scattering signals that can be compared among flow cytometers with the same optical configuration. To avoid swarm detection, we made serial dilutions of plasma labeled with a single fluorophore at a time. The highest concentration of plasma without swarm detection was used for subsequent experiments and did not differ between fluorophores. Concentrations were determined by correcting the number of detected particles within defined gates for flow rate (3.0 μl/min), measurement time (120 s), and sample dilution. Standard deviations of the Poisson error and coefficients of variation (CV) were calculated for the concentrations of PEVs. In all measurements, the pulse height was analyzed, because for our flow cytometer [1] the pulse height results in substantially lower CVs then pulse area (Table S1.1) and [2] we estimated the overestimation of the registered signal introduced by pulse height analysis to be 13% at the thresholds of the fluorescence gates (Figure S1.2). All details of the FCM experiments can be found in the attached MIFlowCyt‐EV (Appendix S2).
2.2.3. Determine fluorescent gates manually
To investigate inter‐operator variability in setting fluorescence gates, five independent FCM experts set fluorescence gates to differentiate CD61+ labeled EVs from the background (FlowJo, 10.6.2, BD). Thereafter, the CV of the measured PEV concentrations for the fluorophores was calculated as follows:
2.2.4. Determine fluorescent gate automatically
We developed a script to automate fluorescence gating (S3). In short, a histogram of the fluorescence height parameter was created (MATLAB, R2018a, MathWorks, Natick, MA) on a logarithmic scale with 24 bins per decade (Figure 3). Values right from the peak and higher than 35% of the peak amplitude were linearly fitted. The intersection of the linear fit with the horizontal‐axis was multiplied with 1.7 and used as the automatically determined gate value. The gate values were separately determined for each fluorophore.
2.2.5. Relate scatter to diameter
To investigate whether the variation in the measured PEV concentrations between different fluorophores could be reduced by setting additional size gates, we related scatter to diameter. The scatter to diameter relationship was obtained by Rosetta Calibration [12], which involves three steps. First, a mixture of beads with known diameter and refractive index is measured. Second, the data are interpreted by Rosetta Calibration software, which automatically recognizes the beads and relates the arbitrary units of light scattering to the theoretical scattering cross section, thereby taking into account the optical configuration of the flow cytometer. Third, the theoretical scattering cross section is related to the diameter of EVs by assuming that EVs are core‐shell particles with a core refractive index of 1.38, a shell refractive index of 1.48, and a shell thickness of 6 nm. Detection ranges of the scatter and fluorescence detectors and data are reported according to MISEV and MIFlowCyt‐EV criteria [6, 13].
3. RESULTS
3.1. Optimal dilution to prevent swarm detection
To find the optimal dilution, that is, the highest count rate without swarm detection, we made serial sample dilutions. Figure 1A verifies that the total concentration of particles versus dilution is linear for all fluorophores within the white area. Dilutions of 104‐fold or more resulted in particle concentrations deviating from the linear fit due to insignificant data and a relatively high contribution of background counts. Dilutions of 100‐fold and less deviate from the linear fit due to swarm detection. To confirm swarm detection for dilutions of 100‐fold and less, Figure 1B,C shows the median scatter and fluorescence signals versus dilution. Because the median scattering cross‐section and median fluorescence increase for dilutions of 100‐fold and less, which is characteristic for swarm detection as shown in the right gray area, swarm detection is confirmed for these dilutions. Therefore, for our flow cytometer and this particular plasma sample, 316‐fold dilution is considered optimal, since with this dilution the highest number of particles is detected without swarm detection.
FIGURE 1.
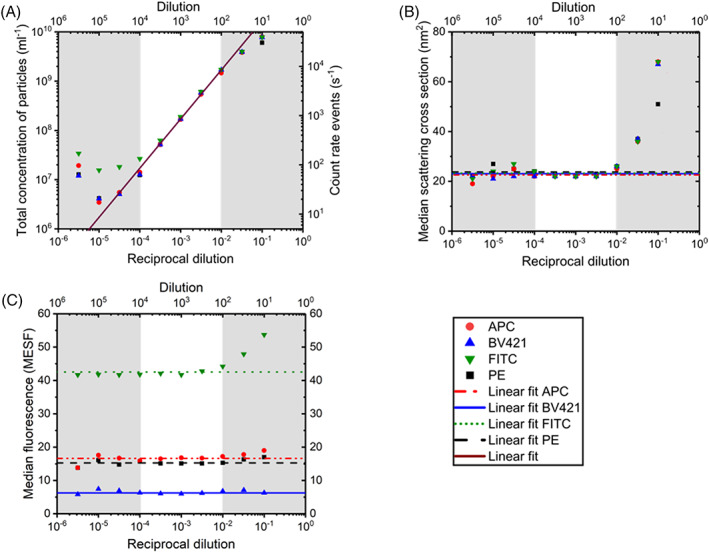
Selecting the optimal dilution of pooled human plasma to prevent swarm detection. (A) Total concentration of particles, (B) median scattering cross‐section, and (C) median fluorescence measured by flow cytometry versus reciprocal fold dilution of pooled human plasma labeled with either Allophycocyanin (APC, VI‐PL2), Brilliant Violet‐421 (BV421, RUUPL7F12), fluorescein isothiocyanate (FITC, Y2/51), or phycoerythrin (PE, VI‐PL2). Total concentration and reciprocal dilution axes have logarithmic scales. The right vertical axis of (A) shows the count rate of events. The median fluorescence in (C) is expressed in molecules of equivalent soluble fluorochrome (MESF) for all fluorophores. Data points within the white area follow a linear trend, as expected, so linear fits for these data points are given. Data points in the gray areas are deviating from these fits due to domination of particles in the reagents (left gray area) or due to swarm detection (right gray area). For the linear fit in (A) the R 2 is 1.00 and the slope is 1.00. For the linear fits in (B, C) a fixed slope of 0 is used. The error bars overlap with the symbols, meaning that the standard deviations are small and therefore omitted. The 316‐fold dilution is considered optimal since it detects the highest concentration of particles without swarm detection. [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Inter‐operator variability in setting gates
To investigate whether inter‐operator variability in setting fluorescence gates affects the measured concentrations of PEVs, Figure 2 shows the measured PEV concentrations in diluted plasma labeled with APC, BV421, FITC, or PE conjugated anti‐human CD61 (all VI‐PL2) gated by five different experts and independently from each other. Per fluorophore, the CV of the measured PEV concentration ranges from 12% for BV421 up to 17% for APC. Mean concentrations of PEVs differ up to 3‐fold between different fluorophores, resulting in an overall CV of 41% for manually gated data. As expected from the brightness of the fluorophores, BV421 resulted in detection of the highest concentrations of PEVs in plasma, followed by PE. APC and FITC resulted in detection of the lowest concentrations of PEVs.
FIGURE 2.
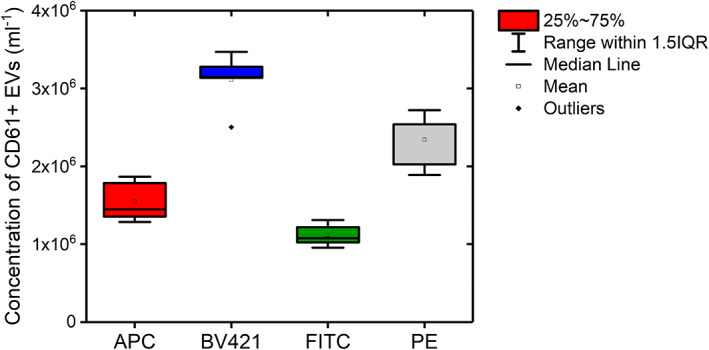
Concentration of CD61+ extracellular vesicles (EVs) measured by flow cytometry in 316‐fold diluted pooled human plasma labeled with Allophycocyanin (APC, VI‐PL2), Brilliant Violet‐421 (BV421, VI‐PL2), fluorescein isothiocyanate (FITC, VI‐PL2), or phycoerythrin (PE, VI‐PL2). The boxplot shows the CD61+ EV concentrations resulting from gates defined by five independent experts. Both the mean and median values of the concentration of CD61+ EVs gated by different experts are represented, as well as the interquartile range and outliers. Results do not only show variability in concentrations of CD61+ EVs detected by different fluorophores, but also between experts when analyzing exactly the same data. [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Automated determination of fluorescence gates
To automate the determination of fluorescence gates, an algorithm was developed. Figure 3A,C,E,G shows a histogram of the fluorescence height parameter of diluted plasma labeled with APC, BV421, FITC, or PE (all VI‐PL2), respectively. Based on the histograms and a fit of the background fluorescence (see Methods in Section 2), fluorescence gate values were automatically determined. Figure 3B,D,F,H shows the corresponding fluorescence intensity versus diameter plots, and, as a reference, the resulting automatically determined gate values (horizontal lines). The CV of the concentrations of PEVs labeled with different fluorophores and gated with an algorithm was 58%. As the same gating algorithm was applied to each fluorophore, the result shows that fluorophores are a source of variation to EV concentration measurements.
FIGURE 3.
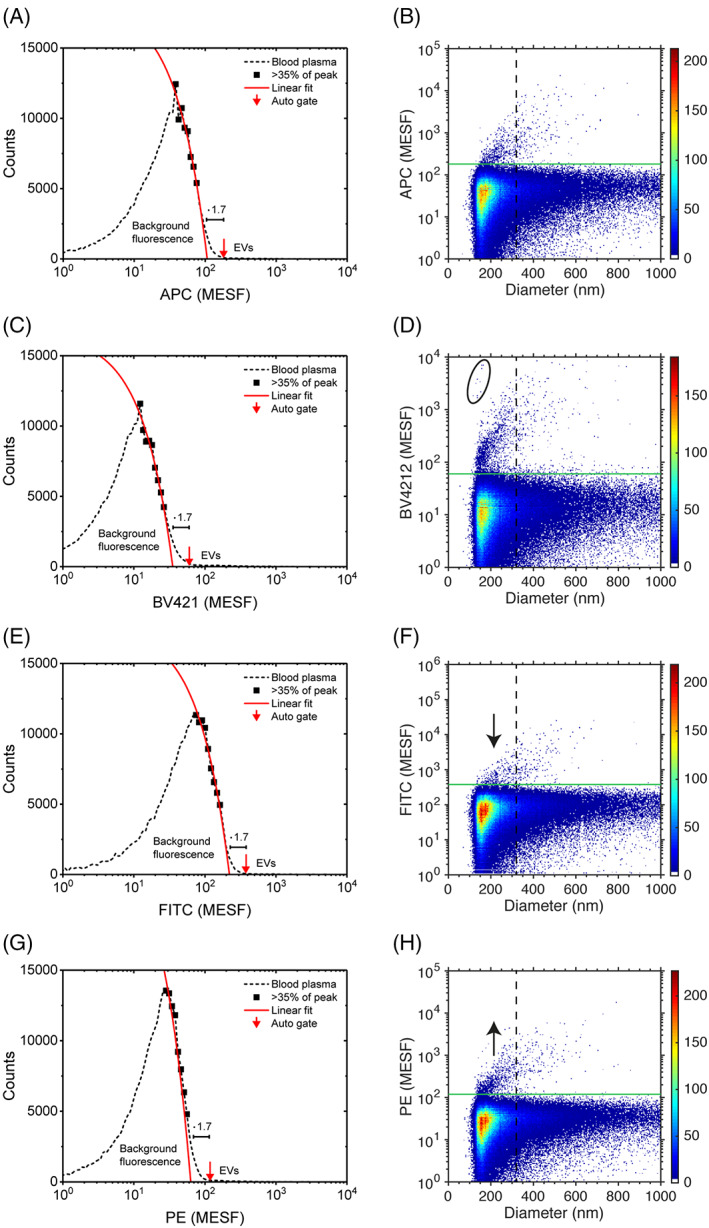
Automated determination of gates (left column) and fluorescence intensity versus diameter (with automatically determined gates; right column) for CD61+ extracellular vesicles (EVs) detected by flow cytometry in 316‐fold diluted human pooled plasma labeled with (A, B) Allophycocyanin (APC, VI‐PL2), (C, D) Brilliant Violet‐421 (BV421, VI‐PL2), (E, F) fluorescein isothiocyanate (FITC, VI‐PL2), or (G, H) Phycoerythrin (PE, VI‐PL2). To automate gate determination, a histogram of the fluorescence height parameter is created on a logarithmic scale with 24 bins per decade. Values right from the peak and higher than 35% of the peak amplitude are linearly fitted. The intersection of the linear fit with the horizontal‐axis multiplied with 1.7 is used as the automatically determined gate value. Fit variables are [1.7 × 102, 1.8 × 104], [4.7 × 102, 1.7 × 104], [0.8 × 102, 1.8 × 104], and [4.1 × 102, 2.6 × 104], resulting in autogate values of 1.8 × 102 MESF, 0.6 × 102 MESF, 3.8 × 102 MESF, and 1.2 × 102 MESF for APC, BV421, FITC, and PE, respectively (horizontal solid green lines). This results in median fluorescent intensities of positive particles above the gate threshold of 3.6 × 102, 2.1 × 102, 7.9 × 102, and 2.2 × 102 MESF, respectively. The vertical dashed black lines show size gates of 310 nm, which are used in Figure 4. Aggregates in BV421 (ellipse panel D) were excluded during further analyses, and a black arrow is displayed, showing a vertical translation of fluorescence for FITC and PE. Despite applying the same gating strategy, different fluorophores cause differences in the brightness of detected CD61+ EV concentrations. This results in vertical translations of data in the right columns (arrows), which particularly affects the CD61+ EV counts below 310 nm. MESF, molecules of equivalent soluble fluorochrome [Color figure can be viewed at wileyonlinelibrary.com]
Figure 3B,D,F,H shows further that the differences in brightness between fluorophores result in the different signal to background ratios. For example, FITC is a relatively dim fluorophore and therefore below a diameter of ~300 nm (vertical dashed lines), most PEVs labeled with FITC are below the fluorescence gate and do not exceed the background. In comparison to FITC, PE is a relatively bright fluorophore and PEVs labeled with PE do exceed the background, also for PEVs with a diameter below ~300 nm. Thus, for our flow cytometer, the brightness of the fluorophores particularly affects the PEV counts below ~300 nm. Therefore, we expect that the brightness of fluorophores has a negligible effect on concentration measurements of PEVs larger than ~300 nm in diameter.
3.4. Size gate reduces dependency of PEV concentrations on fluorophore
To investigate whether the variation in the measured PEV concentrations can be reduced by selecting a size range for which all CD61‐labeled EVs exceed the fluorescence background regardless of the used fluorophore, Figure 4 shows the measured PEV concentration versus the minimum diameter of an applied size gate. To exclude platelets, the maximum of the applied size gate is set at 1000 nm. Thus, Figure 4 shows the concentration of CD61+ EVs within a size range between the diameter indicated at the horizontal axis and 1000 nm. As expected, the larger the selected EVs are, the more the measured concentrations of PEVs labeled with the different fluorophores converge. The CV of the measured PEV concentrations reduces from 58% for a size gate of 0–1000 nm to 25% for a size gate of 310–1000 nm.
FIGURE 4.
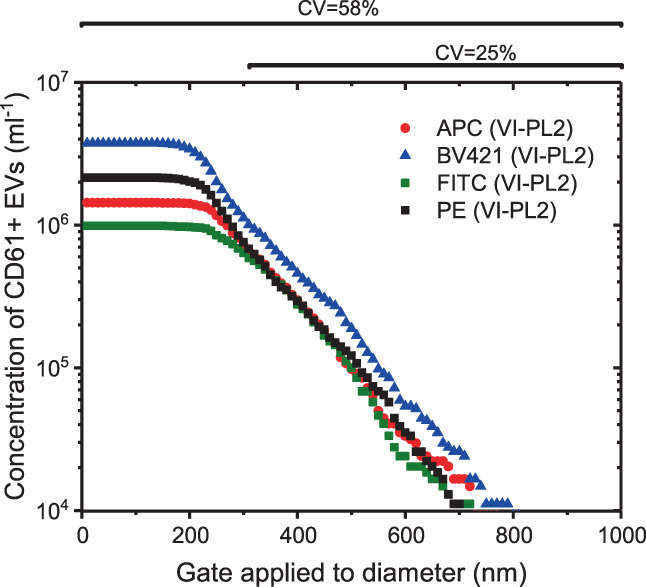
Concentration of CD61+ extracellular vesicles (EVs) measured by flow cytometry in 316‐fold diluted pooled human plasma labeled with Allophycocyanin (APC, VI‐PL2), Brilliant Violet‐421 (BV421, VIPL2), fluorescein isothiocyanate (FITC, VI‐PL2), or phycoerythrin (PE, VIPL2) versus minimum diameter of the selected EVs. In addition to a relevant fluorescence gate, a size gate was applied that ranges from the plotted minimum diameter to 1000 nm with subsequent steps of 10 nm. A size gate of 0–1000 nm results in up to 3.5‐fold differences in the measured CD61+ EV concentrations between fluorophores (CV = 58%). With increasing minimum diameter, the concentrations of CD61+ EVs labeled with different fluorophores converge. A minimum diameter of 310 nm provides more similar concentrations of CD61+ EVs for the different fluorophores (CV = 28%). [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
This study investigated the choice of commonly used fluorophores on the measured concentrations of immunolabeled EVs by flow cytometry. First, manual gating resulted in a CV of 41% for the measured concentrations of EVs labeled with different fluorophores. However, because the fluorescence gates were set by independent experts, inter‐operator variability of the measured EV concentrations ranged from 12% CV for BV421 up to 17% CV for APC. Second, to eliminate inter‐operator variability and obtain a more objective measure of the variation caused by the choice of fluorophore, we developed an algorithm to automatically define fluorescence gates. The automatically determined fluorescence gates resulted in a CV of 58% for the concentrations of EVs labeled with different fluorophores. Third, we applied the automatically determined fluorescence gate together with a size gate to select only EVs exceeding the detection limit of the fluorescence detector. The latter strategy reduced the CV to 25% for measured EV concentrations labeled with different fluorophores.
Due to the small size of EVs, immunolabeled EVs typically have orders of magnitude lower fluorescence signals than cells, and not all fluorescently labeled EVs exceed the background noise. Consequently, the fluorescence gate directly affects the measured EV concentration. In the research field of EVs, it is common practice to define the fluorescence gate manually. Here, we show that inter‐operator variability in setting the gates affects the obtained concentrations of immunolabeled EVs. The presence of inter‐operator variability in setting the gates does emphasize the need [1] to define gates on calibrated scales of fluorescence, and [2] to report the gates in order to make clinical research studies on EVs comparable [6]. To prevent variation in the measured concentrations of EVs between different experts, we recommend automating the determination of the fluorescence gates. Figure 3 describes one way to automate the determination of fluorescence gates.
The automatically determined fluorescent gates were 0.6 × 102 MESF for BV421, 1.2 × 102 MESF for PE, 1.8 × 102 MESF for APC, and 3.8 × 102 MESF for FITC. The reported MESF values of these gates are determined by extrapolating the MESF calibration (Figure S2.1) by one order of magnitude. Consequently, the uncertainty of the reported MESF values of these gates is unknown. An assessment of the uncertainty would require reference particles with a brightness <500 MESF, which are currently unavailable [14].
Following the expectations, brighter fluorophores resulted in lower gates, which in turn resulted in measuring higher concentrations of EVs. The EV concentration detected by BV421 was substantially higher than the concentration detected by PE, which we attribute to a higher F/P ratio than 1 for BV421. APC and FITC (FITC with several fluorophores attached to one CD61‐antibody) demonstrated, as expected, the lowest concentrations of EVs, since the brightness of these fluorophores is ~3 to 4‐fold dimmer compared to BV421 and PE. Previous research has proven that the reported concentrations of EVs, and particularly PEVs, are incomparable between instruments and institutes, partly due to differences in pre‐analytical and analytical variables [3, 5, 6, 7]. Our results agree with the finding that the measured PEV concentrations depend, among others, on the choice of fluorophore and the sensitivity of the fluorescence detectors. However, when we apply a size gate to select only EVs that exceed the fluorescence background, we obtain similar PEV concentrations (CV = 25%) while using different fluorophores. Compared to interlaboratory standardization studies, which report CVs of 28%, 37%, and 81% [5], a CV of 25% for the measured concentration of EVs labeled with different fluorophores is a good result. In addition, please note that the flow cytometer itself already accounts for a CV of 8%, which we based on unpublished repeatability experiments.
The current study was performed with anti‐CD61 antibody clone VI‐PL2. However, we performed preliminary experiments to investigate whether other anti‐CD61 antibody clones yield similar results. For example, by extending Figure 4 with the clones RUU‐PL7F12 (BV421) and Y2/51 (FITC), the CVs of the measured PEV concentrations were 50% without a size gate and 21% for a size gate of 310–1000 nm in diameter (Figure S1.3). Thus, regardless of the used CD61 antibody clone or fluorophore, the same number of antigens is stained at the surface of PEVs, which has two important implications. First, steric hindrance and quenching, which decrease the fluorescence intensity, are negligible for an abundantly expressed antigen like CD61, because if present these processes would have led to substantial differences between the measured concentrations of PEVs labeled with different fluorophores. Second, despite the small size of EVs compared to cells and fluorophores, staining of EVs is robust. Further investigation is required to confirm whether our findings hold true for other antibodies than CD61, other body fluids, and other flow cytometers.
In conclusion, with this study we showed that different fluorophores can detect similar concentrations of EVs by automating the determination of fluorescence gates and adding specific size ranges. Therefore, our research contributes to a new insight into the standardization of EV concentration measurements by flow cytometry.
CONFLICT OF INTEREST
E. van der Pol is co‐founder and stakeholder of Exometry B.V., Amsterdam, the Netherlands. All other authors report no conflicts of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/cyto.a.24665.
ETHICS STATEMENT
This article does not contain any studies involving human participants performed by any of the authors.
Supporting information
APPENDIX S1: Pulse height versus pulse area.
APPENDIX S2: MIFlowCyt‐EV of the research study “Automated fluorescence gating and size determination reduce variation in measured concentration of extracellular vesicles by flow.”
ACKNOWLEDGMENTS
The authors would like to acknowledge Dr. A. Gasecka, N. Hajji, and C.M. Hau (Laboratory of Experimental Clinical Chemistry, Amsterdam University Medical Centers, the Netherlands) for defining CD61+ EV gates in FlowJo. This work was supported by the Netherlands Organization for Scientific Research ‐ Domain Applied and Engineering Sciences (NWO‐TTW), research program VENI 15924 (E.v.d.P.). This project (18HLT01) has received funding from the EMPIR Program co‐financed by the Participating States and from the European Union‘s Horizon 2020 Research and Innovation Program.
Gankema AAF, Li B, Nieuwland R, Pol Evd. Automated fluorescence gating and size determination reduce variation in measured concentration of extracellular vesicles by flow cytometry. Cytometry. 2022;101(12):1049–1056. 10.1002/cyto.a.24665
Funding information European Union‘s Horizon 2020 Research and Innovation Program; Participating States; EMPIR Program
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in Figshare at http://doi.org/10.6084/m9.figshare.13584905.
REFERENCES
- 1. van der Pol E, Böing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012;64(3):676–705. [DOI] [PubMed] [Google Scholar]
- 2. Tao SC, Guo SC, Zhang CQ. Platelet‐derived extracellular vesicles: an emerging therapeutic approach. Int J Biol Sci. 2017;13(7):828–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tissot JD, Canellini G, Rubin O, Angelillo‐Scherrer A, Delobel J, Prudent M, et al. Blood microvesicles: from proteomics to physiology. Transl Proteom. 2013;1(1):38–52. [Google Scholar]
- 4. Gasecka A, Böing AN, Filipiak KJ, Nieuwland R. Platelet extracellular vesicles as biomarkers for arterial thrombosis. Platelets. 2017;28(3):228–34. [DOI] [PubMed] [Google Scholar]
- 5. Pol van der E, Sturk A, van Leeuwen T, Nieuwland R, Coumans F, Mobarrez F, et al. Standardization of extracellular vesicle measurements by flow cytometry through vesicle diameter approximation. J Thromb Haemost. 2018;16(6):1236–45. [DOI] [PubMed] [Google Scholar]
- 6. Welsh JA, Van Der Pol E, Arkesteijn GJA, Bremer M, Brisson A, Coumans F, et al. MIFlowCyt‐EV: a framework for standardized reporting of extracellular vesicle flow cytometry experiments. J Extracell Vesicles. 2020;9(1):1713526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rikkert LG, van der Pol E, van Leeuwen TG, Nieuwland R, Coumans FAW. Centrifugation affects the purity of liquid biopsy‐based tumor biomarkers. Cytometry A. 2018;93(12):1207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hagberg IA, Lyberg T. Blood platelet activation evaluated by flow cytometry: optimised methods for clinical studies. Platelets. 2000;11(3):137–50. [DOI] [PubMed] [Google Scholar]
- 9. Naeim F, Rao PN, Grody WW. Principles of immunophenotyping. In: Hematopathology. 2008:27–55. [Google Scholar]
- 10. Femminò S, Penna C, Margarita S, Comità S, Brizzi MF, Pagliaro P. Extracellular vesicles and cardiovascular system: biomarkers and cardioprotective effectors. Vasc Pharmacol. 2020;135(July):106790. 10.1016/j.vph.2020.106790 [DOI] [PubMed] [Google Scholar]
- 11. Steen HB. Noise, sensitivity, and resolution of flow cytometers. Cytometry. 1992;13(8):822–30. [DOI] [PubMed] [Google Scholar]
- 12. de Rond L, Coumans FA, Nieuwland R, van Leeuwen TG, van der Pol E. Deriving extracellular vesicle size from scatter intensities measured by flow cytometry. Curr Prot Cytom. 2018;86:e43. 10.1002/cpcy.43 [DOI] [PubMed] [Google Scholar]
- 13. Witwer KW, Soekmadji C, Hill AF, Wauben MH, Buzás EI, Di Vizio D, et al. Updating the MISEV minimal requirements for extracellular vesicle studies: building bridges to reproducibility. J Extracell Vesicles. 2017;6(1):1396823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuiper M, van de Nes A, Nieuwland R, Varga Z, van der Pol E. Reliable measurements of extracellular vesicles by clinical flow cytometry. Am J Reprod Immunol. 2021;85:e13350. 10.1111/aji.13350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKinnon KM. Flow cytometry: an overview. Curr Prot Immunol. 2018;20(18):5.1.1–5.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCartney LJ, Pickup JC, Rolinski OJ, Birch DJS. Near‐infrared fluorescence lifetime assay for serum glucose based on allophycocyanin‐labeled concanavalin A. Anal Biochem. 2001;292(2):216–21. [DOI] [PubMed] [Google Scholar]
- 17. Chattopadhyay PK, Gaylord B, Palmer A, Jiang N, Raven MA, Lewis G, et al. Brilliant violet fluorophores: a new class of ultrabright fluorescent compounds for immunofluorescence experiments. Cytometry Part A. 2012;81 A(6):456–66. [DOI] [PubMed] [Google Scholar]
- 18. Schwarze W, Bernhardt R, Jänig GR, Ruckpaul K. Fluorescent energy transfer measurements on fluorescein isothiocyanate modified cytochrome P‐450 LM2. Biochem Biophys Res Commun. 1983;113(1):353–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1: Pulse height versus pulse area.
APPENDIX S2: MIFlowCyt‐EV of the research study “Automated fluorescence gating and size determination reduce variation in measured concentration of extracellular vesicles by flow.”
Data Availability Statement
The data that support the findings of this study are openly available in Figshare at http://doi.org/10.6084/m9.figshare.13584905.