

Summary
ARID1A, encoding a subunit of the SWI/SNF complex, is mutated in ~50% of clear cell ovarian carcinoma (OCCC) cases. Here we show that inhibition of the mevalonate pathway synergizes with immune checkpoint blockade (ICB) by driving inflammasome-regulated immunomodulating pyroptosis in ARID1A-inactivated OCCCs. SWI/SNF inactivation downregulates the rate-limiting enzymes in the mevalonate pathway such as HMGCR and HMGCS1, which creates a dependence on the residual activity of the pathway in ARID1A-inactivated cells. Inhibitors of the mevalonate pathway such as simvastatin suppresses the growth of ARID1A mutant, but not wildtype, OCCCs. In addition, simvastatin synergizes with anti-PD-L1 antibody in a genetic OCCC mouse model driven by conditional Arid1a inactivation and in a humanized immunocompetent ARID1A mutant patient derived OCCC mouse model. Our data indicate that inhibition of the mevalonate pathway simultaneously suppresses tumor cell growth and boosts antitumor immunity by promoting pyroptosis, which synergizes with immune checkpoint blockade in suppressing ARID1A-mutated cancers.
Graphical Abstract
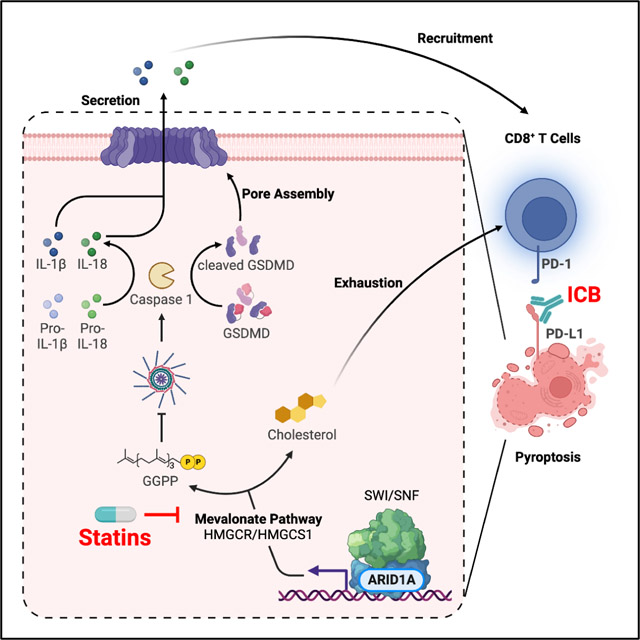
Modulating the tumor immune microenvironment in a molecularly defined manner remains a major challenge. Zhou et al report a molecular mechanism of ARID1A regulated immune-stimulatory cell death. The findings support that inhibition of the mevalonate pathway alone or in combination with immune checkpoint blockade represents an urgently needed therapeutic approach for ARID1A-mutated cancers.
Introduction
ARID1A encodes a subunit of the SWI/SNF chromatin-remodeling complex and functions as a tumor suppressor1. Notably, inactivating mutations in ARID1A occur frequently in ovarian clear cell carcinomas (OCCC; >50%) and ovarian endometrioid carcinomas (OEC; >30%)2,3. Over 90% of the ARID1A mutations observed in ovarian cancers are frameshift or nonsense mutations that result in the loss of ARID1A protein expression2–4. OCCC is generally refractory to standard agents used to treat ovarian cancers, and when diagnosed in advanced stages, OCCC carries the worst prognosis of all ovarian cancer subtypes5,6. Thus, there is an unmet clinical need for effective treatment modalities for OCCC.
The mevalonate pathway is required for the synthesis of a variety of isoprenoids derived from acetyl-CoA, including cholesterol and prenyl groups, which are required for multiple biological processes7. The mevalonate pathway is controlled by rate-limiting enzymes such as 3-hydroxy-3-methylglutaryl-CoA synthase 1 (HMGCS1) and HMG-CoA reductase (HMGCR). Previous implication of the mevalonate pathway in cancer has been its essential role in cell survival and proliferation7. Statins, a class of FDA-approved HMGCR inhibitors, are clinically safe and widely used in treating cardiovascular diseases8.
While there are reports that the SWI/SNF complex inactivation renders tumors sensitive to immune checkpoint blockade (ICB)9–13, others did not find a consistent association between SWI/SNF genomic alterations and improved clinical outcome to immune checkpoint inhibitors14. Notably, ARID1A mutation sensitizes ovarian cancers to ICB such as anti-PD-L113,15. Indeed, there was a trend toward improved response rate toward ICB in OCCC in clinical trials16. However, anti-PD-L1 treatment alone has a modest effect on improving the survival of mice bearing Arid1a-inactivated tumors13,15. This suggests that to achieve a complete eradication of ARID1A-mutated ovarian cancer, combinatorial therapeutic strategies are necessary.
Pyroptosis is an inflammatory form of cell death initiated by the formation of the inflammasome by NOD-like receptor (NLR) family proteins, such as NLRP3, through binding to an apoptosis-associated speck-like (ASC) protein containing a caspase activation and recruitment domain (CARD)17,18. ASC, in turn, interacts with the cysteine protease caspase 1 to trigger an inflammatory response as exemplified by the activation of caspase 1 and the maturation of its targets such as pro-IL-1β and pro-IL-18 into their bioactive forms19. In addition, caspase1 cleaves gasdermin protein family members such as GSDMD to form pores in the plasma membrane20. Notably, pyroptosis is associated with the release of danger-associated molecular patterns (DAMPs) including HMGB121. In contrast to immunologically silent apoptotic cell death, pyroptotic cell death induces the infiltration of immune cells such as CD8+ and CD4+ T cells through releasing inflammatory cytokines such as IL-1β and IL-18 and DAMPs such as HMGB122,23. Gain- and loss-of-function studies established IL-1β, IL-18, and HMGB1 released during pyroptosis of cancer cells mediate antitumor immune responses including in combination with immunotherapies such as ICB24–26. In addition, the infiltrated immune cells such as CD8+ T cells reciprocally induce pyroptosis in tumor cells23. Thus, pyroptosis in tumor cells establishes a positive tumor suppressive feedback loop with the associated immune microenvironment. Indeed, pyroptosis in less than 15% of tumor cells was sufficient to clear an entire tumor22. Altering the inflammatory state of the tumor immune microenvironment has implications for the response to immunotherapies such as ICB18,27. However, the role of ARID1A in regulating pyroptosis has not been investigated.
Here we report that inhibition of the mevalonate pathway in ARID1A-mutated ovarian cancer not only suppresses the growth of ARID1A-mutated cancer cells by inducing pyroptosis but also synergizes with ICB through modulating the tumor immune microenvironment.
Results
ARID1A inactivation sensitizes cells to pyroptosis induction
When culturing ARID1A knockout OVCA429 OCCC cells, we noticed that there was an increase in the percentage of cells with cytoplasmic DNA compared to the matched isogenic ARID1A wildtype controls (Fig. 1A and Fig. S1A–B). Indeed, in a panel of OCCC cell lines, the percentages of cells positive for cytoplasmic DNA were significantly higher in ARID1A mutant cells compared with wildtype cells (Fig. S1C–D). As a control, knockout of HNF1β did not cause an increase in cytoplasmic DNA, indicating that this did not a result from the process of gene knockout (Fig. S1E–G).
Figure 1. ARID1A inactivation sensitizes cells to pyroptosis induction.
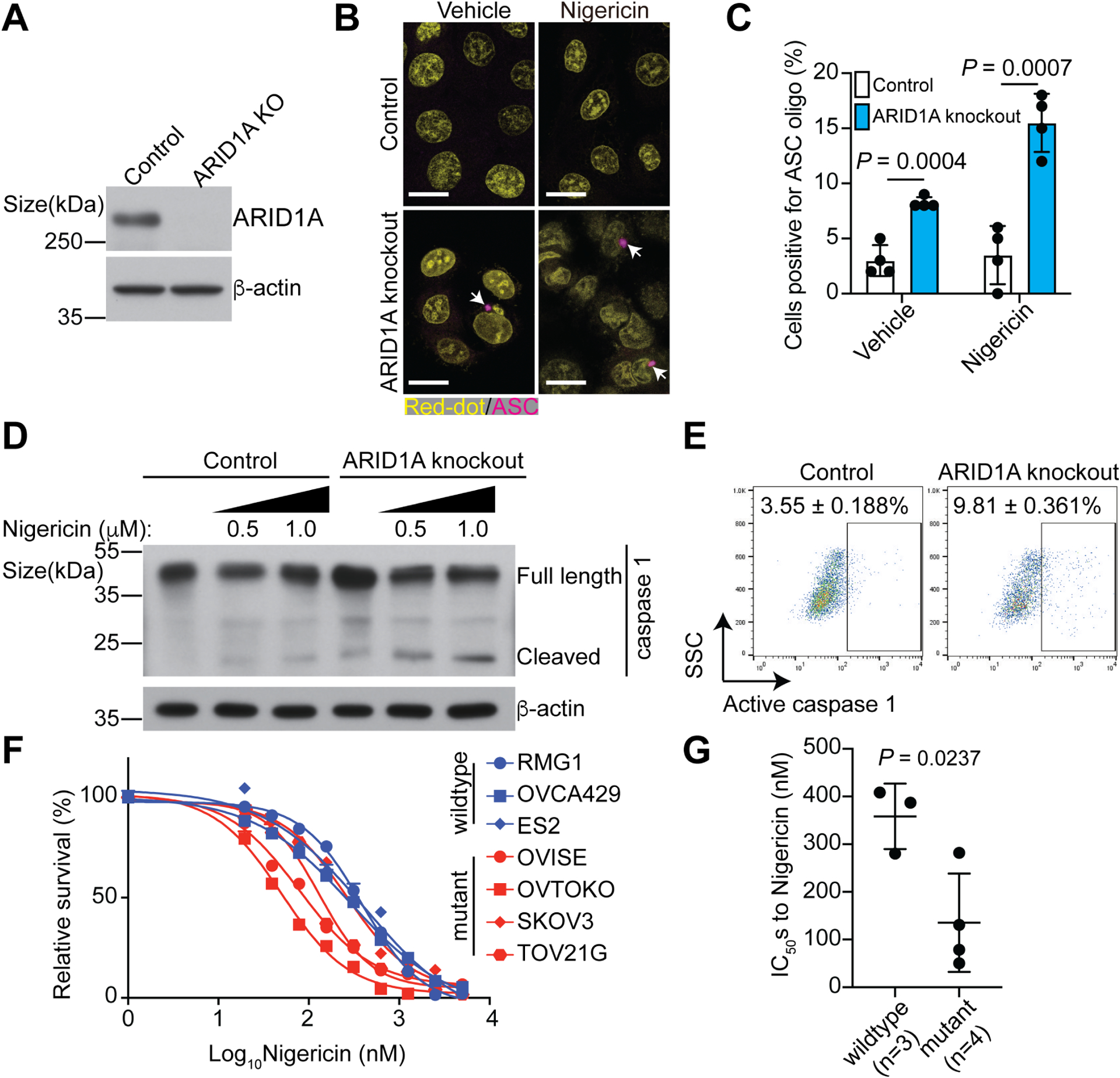
A, Validation of ARID1A knockout in ARID1A wildtype OVCA429 OCCC cells by immunoblot.
B, Representative immunofluorescence images of ASC-mCerulean staining in control and ARID1A knockout OVCA429 cells with or without Nigericin treatment (0.5 μM). Arrows point to examples of ASC oligo, a marker of inflammasome. Nuclei were visualized by RedDot staining. Scale bar = 20 μm.
C, Quantification of ASC positive cells in the indicated OVCA429 cells.
D, Expression of caspase 1 in the indicated OVCA429 cells treated with or without Nigericin was determined by immunoblot.
E. Activity of caspase 1 in the indicated OVCA429 cells treated with Nigericin (0.5 μM) was determined by flow cytometry. Percentages of caspase 1 positive cells were indicated based on three biological repeats.
F, Dose responsive curves of the indicated ARID1A wildtype or mutant OCCC cell lines to Nigericin were determined by the AlamarBlue assay.
G, IC50s of Nigericin are significantly higher in ARID1A wildtype (RMG1, OVCA429 and ES2) than mutant (OVISE, OVTOKO, SKOV3 and TOV21G) cell lines.
Error bars represent mean with SEM in C and G. P values were calculated using two-tailed Student t-test in C and G.
See also Figure S1.
Notably, cytoplasmic double stranded DNA triggers the activation of the cGAS pathway and/or inflammasome formation28. Thus, we first determined the status of the cGAS pathway by performing immunofluorescence staining for cGAS. We used etoposide treatment to induce cytoplasmic chromatin as a positive control29. Notably, cGAS was not localized to the cytoplasmic DNA in ARID1A knockout cells as compared to control etoposide-treated cells (Fig. S1A and S1H). As a positive control, a DNA damage marker γH2AX was positive in cytoplasmic DNA in both ARID1A knockout and etoposide-treated positive control cells (Fig. S1A and S1H). This was not due to downregulation of cGAS by ARID1A knockout (Fig. S1I). Consistently, ARID1A knockdown did not increase the levels of 2’3’-cGAMP, the enzymatic product of cGAS (Fig. S1J). These results suggest that cGAS is not activated by the cytoplasmic DNA observed in ARID1A knockout cells.
We next examined the inflammasome formation in control and ARID1A knockout cells. There was a significant increase in inflammasome formation in ARID1A knockout cells (Fig. 1B–C). Since inflammasomes are linked to the activation of pyroptosis17,18, we determined whether ARID1A knockout sensitizes cells to the pyroptosis inducer Nigericin30. Compared with controls, ARID1A knockout significantly increased the markers of pyroptosis such as inflammasome formation, and cleaved caspase 1 expression and activity (Fig. 1B–E). Similar findings were made in RMG1 OCCC cells (Fig. S1F and S1K–L), suggesting that this is not a cell line specific effect. Consistently, the IC50 of Nigericin was significantly lower in ARID1A-mutated compared with wildtype OCCC cell lines (Fig. 1F–G). Thus, we conclude that ARID1A inactivation induces inflammasome formation and sensitizes cells to pyroptosis induction.
ARID1A inactivation creates a dependence on the mevalonate pathway and sensitizes cells to pyroptosis induction by statins
We next determined the mechanism by which ARID1A regulates pyroptosis. Toward this goal, we performed pathway enrichment analyses based on proteins that are differentially expressed in control and ARID1A knockout OVCA429 cells31. The analyses revealed that the mevalonate/cholesterol pathway was among the top pathways that were suppressed by ARID1A knockout (Fig. 2A and Fig. S2A), which correlated with an enrichment of NLRP3 inflammasome signature (Fig. 2B). Conversely, wildtype ARID1A restoration in ARID1A-mutated OVISE cells enhanced the cholesterol pathway (Fig. S2B). We validated ARID1A knockout induced downregulation of the mevalonate pathway by confirming the reduction of endogenous cholesterol levels as determined by Amplex Red reaction and Filipin III staining (Fig. S2C–H). Likewise, ARID1A knockout decreased the levels of mevalonate (Fig. S2I). Notably, ectopic ARID1A expression rescued the downregulation of Filipin III induced by ARID1A knockout (Fig. S2F–H). However, ARID1A status does not affect expression of SREBP2, the master transcriptional regulator of the mevalonate pathway7 (Fig. S2J). Consistent with our finding that ARID1A knockout increased NLRP3 inflammasome signature and suppressed mevalonate pathway (Fig. 2A–B), there is evidence to suggest that a decrease in the mevalonate pathway triggers an inflammatory response32. Thus, we hypothesize that downregulation of the mevalonate pathway by ARID1A knockout may regulate inflammasome formation and pyroptosis.
Figure 2. Inhibition of the mevalonate pathway sensitizes ARID1A-inactivated cells to pyroptosis.
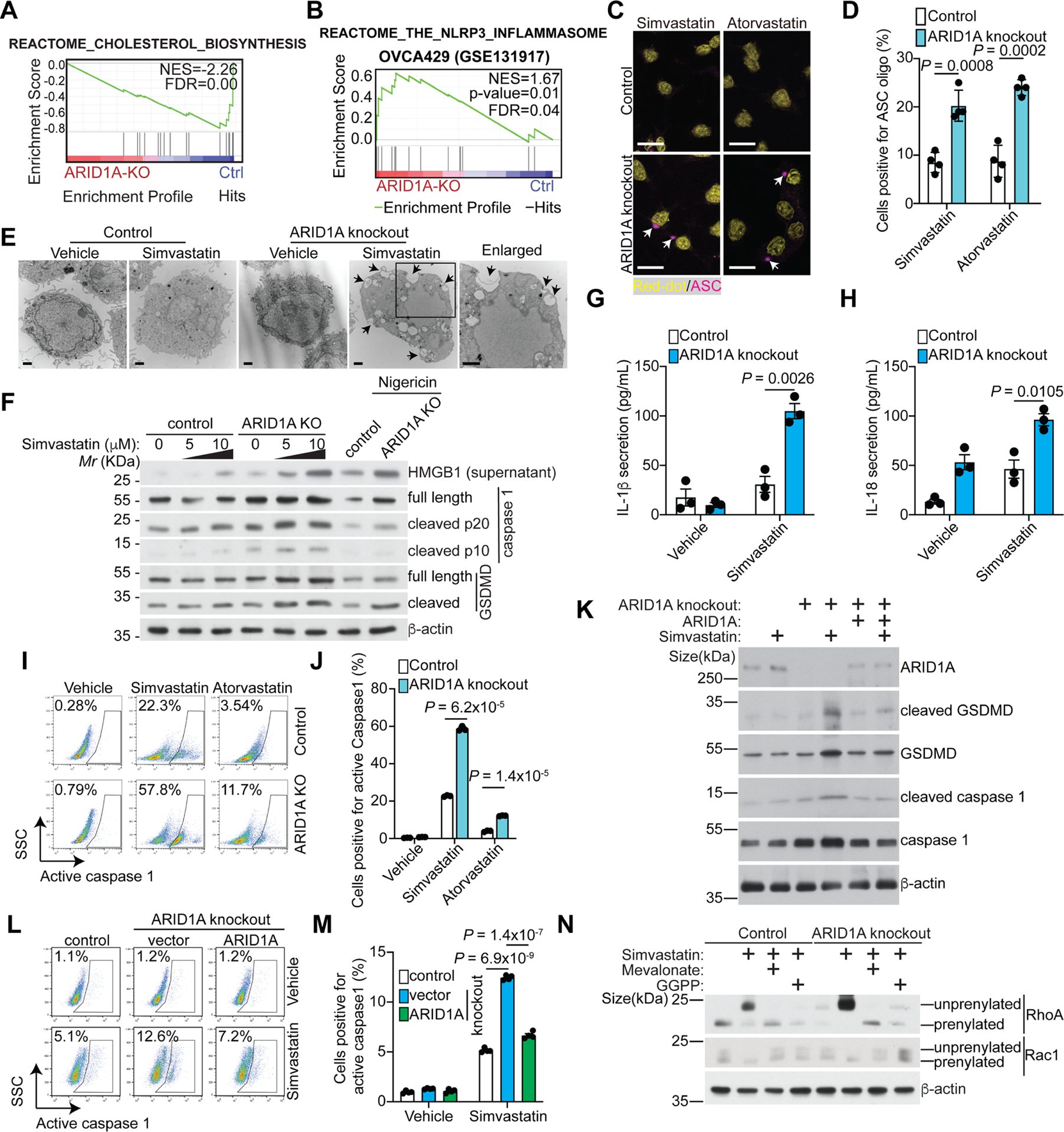
A, ARID1A knockout decreases the cholesterol biosynthesis pathway in OVCA429 cells based on GSEA enrichment analysis of differentially expressed proteins in a previously published proteomic analysis31.
B, ARID1A knockout enriches the NLRP3 inflammasome gene signature in OVCA429 cells based on GSEA analysis of differentially expressed genes in a previously published RNA-seq analysis (GEO: GSE131917)59.
C, Representative immunofluorescence images of ASC-mCerulean staining in the indicated OVCA429 cells treated with simvastatin (5 μM) or atorvastatin (10 μM). Arrows point to examples of ASC oligo, a marker of inflammasome. Nuclei were visualized by RedDot staining. Scale bar = 20 μm.
D, Quantification of ASC oligomerization positive cells in the indicated treatment groups.
E, Transmission electron microscope images of control and ARID1A knockout OVCA429 cells treated with or without simvastatin (10 μM for 48 hours). The cell surface exhibited disrupted regions and membrane pores (black arrows). Scale bar = 1 μm.
F, Expression of the indicated proteins in control and ARID1A knockout OVCA429 cells treated with or without the indicated concentration of simvastatin was determined by immunoblot. Nigericin treatment was used as a positive control.
G-H, Secretion of IL-1β (G) or IL-18 (H) in control and ARID1A knockout OVCA429 cells treated with or without simvastatin (10 μM for 48 hours) was examined by ELISA. n=3 independent experiments.
I-J, Activity of caspase 1 in control or ARID1A knockout OVCA429 cells treated with or without simvastatin (10 μM) and atorvastatin (10 μM) was examined by flow cytometry (I). And percentages of caspase 1 positive cells were quantified (J). n=3 independent experiments.
K, Expression of the indicated proteins in control and ARID1A knockout OVCA429 cells with or without wildtype ARID1A restoration treated with vehicle control or simvastatin (10 μM for 48 hours) was determined by immunoblot.
L-M, Activity of caspase 1 in control and ARID1A knockout OVCA429 cells with or without wildtype ARID1A restoration treated with vehicle control or simvastatin (10 μM for 48 hours) was examined by flow cytometry (L). And percentages of caspase 1 positive cells were quantified (M). n=3 independent experiments.
N, The prenylation of RhoA and Rac1 in control and ARID1A knockout OVCA429 cells treated with vehicle control, simvastatin (10 μM) with or without supplementation of mevalonate (100 μM) or GGPP (10 μM) for 48 hours were examined by immunoblot.
Error bars represent mean with SEM in D, G, H, J and M. P values were calculated using two-tailed Student t-test in D, G, H, J and M.
See also Figure S2.
To test our hypothesis, we treated control and ARID1A knockout cells with FDA-approved inhibitors of the mevalonate pathway, such as simvastatin and atorvastatin. Statins induced a significant increase in inflammasome formation in ARID1A knockout compared to control cells (Fig. 2C–D). Morphologically, transmission electron microscopic analysis revealed numerous plasma membrane disruptions, a characteristic of pyroptosis33, in ARID1A knockout cells treated with simvastatin (Fig. 2E). Consistently, other markers of pyroptosis such as cleaved caspase 1 and cleaved GSDMD were induced at a higher level in ARID1A knockout compared with control cells (Fig. 2F). Secretion of HMGB1 was increased by ARID1A knockout in response to simvastatin treatment (Fig. 2F). Likewise, secretion of IL-1β and IL-18 was increased by ARID1A knockout in response to simvastatin treatment (Fig. 2G–H). Further, the activity of caspase 1 was increased by ARID1A knockout in response to simvastatin or atorvastatin treatment for both percentage of positive cells (Fig. 2I–J and Fig. S2K–L) and the number of positive cells in ARID1A wildtype OCCC cells (Fig. 2I–J and Fig. S2M). Similar observations were also made in an endometriosis-derived 12Z cell line (Fig. S2N–Q). Notably, markers of pyroptosis such as caspase 1 activity and cleaved GSDMD and caspase 1 induced by simvastatin were rescued by ectopic wildtype ARID1A expression in ARID1A knockout cells (Fig. 2K–M). However, simvastatin did not increase the expression of the apoptosis specific cleaved PARP p8534 (Fig. S2R). In addition, apoptosis and pyroptosis share markers such as Annexin V positive staining34. To quantify the relative apoptosis vs. pyroptosis induced by simvastatin, we performed caspase 1 and Annexin V double staining. Our results showed that simvastatin induced a significantly higher levels of pyroptosis compared with non-pyrototic apoptosis in ARID1A inactivated cells (Fig. S2S–T).
GGPP, an isoprenoid product of the mevalonate pathway, plays an important role in protein prenylation35. Inhibition of prenylation of Rho GTPase family members such as RhoA triggers inflammasome assembly36,37. Consistently, RhoA prenylation was significantly reduced by inhibition of the mevalonate pathway using simvastatin in a mevalonate/GGPP dependent manner (Fig. 2N). As a control, simvastatin did not increase the unprenylated Rac1 (Fig. 2N). Thus, our results support that the mevalonate pathway represses pyroptosis by controlling RhoA prenylation in a mevalonate/GGPP dependent manner.
ARID1A knockout significantly reduced the IC50s of simvastatin and atorvastatin in ARID1A wildtype cells, which can be rescued by ectopic ARID1A expression (Fig. 3A–B and Fig. S3A–B). ARID1A knockout also decreased the IC50 of simvastatin in an endometriosis-derived 12Z cell line38 (Fig. 3C). Conversely, wildtype ARID1A restoration in ARID1A-mutated OCCC cell lines such as TOV21G and OVISE increased the IC50s of statins (Fig. 3D–E and Fig. S3C–E). These findings support the notion that the observed effects are not cell line specific effects.
Figure 3. ARID1A inactivation sensitizes cells to inhibition of the mevalonate pathway.
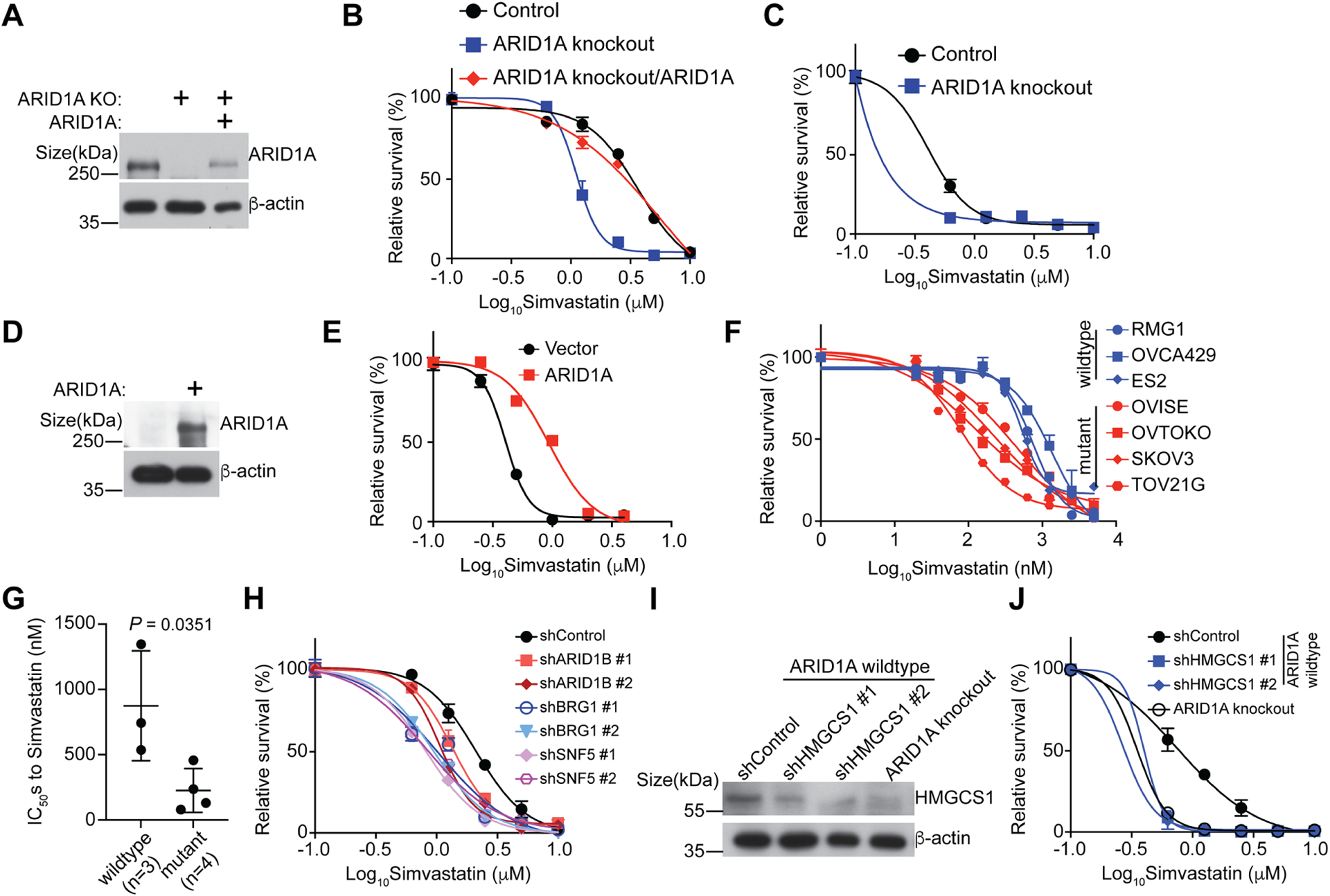
A, Expression of ARID1A in control and ARID1A knockout OVCA429 cells with or without wildtype ARID1A restoration was determined by immunoblot.
B, Dose response curves of the indicated control and ARID1A knockout OVCA429 cells with or without wildtype ARID1A restoration to simvastatin were determined by colony formation assay. n=4 independent experiments.
C, Dose response curves of the indicated control and ARID1A knockout endometriosis-derived 12Z cells to simvastatin were determined by colony formation assay. n=4 independent experiments.
D-E, Expression of ARID1A in control and ARID1A mutant TOV21G cells with or without wildtype ARID1A restoration was determined by immunoblot (D). Dose response curves of the indicated control and wildtype ARID1A restored ARID1A-mutated TOV21G OCCC cells to simvastatin were determined by colony formation assay (E). n=4 independent experiments.
F, Dose response curves of the indicated ARID1A wildtype or mutant OCCC cell lines to simvastatin were determined by the AlamarBlue assay. n=4 independent experiments.
G, IC50s of simvastatin in ARID1A wildtype (RMG1, OVCA429 and ES2) and (OVISE, OVTOKO, SKOV3 and TOV21G) cell lines.
H, Dose response curves of the indicated OVCA429 cells to simvastatin were determined by colony formation assay. n=4 independent experiments.
I-J, Expression of HMGCS1 in the indicated OVCA429 cells was determined by immunoblot (I). Dose response curves to simvastatin were determined by colony formation assay (J). n=4 independent experiments.
Error bars represent mean with SEM in B, C, E, F, G, H and J. P values were calculated using two-tailed Student t-test in G.
See also Figure S3.
In a panel of OCCC cell lines, the IC50s of simvastatin were significantly lower in ARID1A mutant compared with wildtype cell lines (Fig. 3F–G). In addition, knockdown of other subunits of the SWI/SNF complex such as ARID1B, BRG1 and SNF5 reduced the IC50s of simvastatin (Fig. 3H and Fig. S3F), suggesting that the observed effects are likely SWI/SNF complex dependent. Further, in the Project Achilles synthetic lethality database, HMGCR siRNA was more effective in suppressing the growth of cell lines with ARID1A mutation compared with wildtype cell lines (Fig. S3G)39. There was also a trend toward significance for increased sensitivity to HMGCS1 siRNA (Fig. S3G). For example, for uterine cancer cell lines in the database, siRNAs to HMGCR and HMGCS1 were significantly more effective against ARID1A mutant compared with wildtype cell lines (Fig. S3H)39. Our findings support that the residual mevalonate pathway activity is necessary for the survival of ARID1A inactivated cells.
To directly test this possibility, we knocked down HMGCS1, encoding a rate-limiting enzyme in the mevalonate pathway7, in ARID1A wildtype cells (Fig. 3I). HMGCS1 knockdown phenocopies ARID1A knockout in sensitizing ARID1A wildtype cells to simvastatin (Fig. 3J). Consistent with our results obtained using statins, ARID1A knockout cells were more sensitive to inhibition of the mevalonate pathway genetically by knocking down HMGCS1 (Fig. S3I–J). Thus, we conclude that ARID1A inactivation sensitizes cells to inhibition of the mevalonate pathway by inducing pyroptosis.
HMGCR and HMGCS1 are direct target genes of the ARID1A-containing SWI/SNF complex
We next determined the mechanism by which ARID1A regulates the mevalonate pathway. Toward this goal, we re-analyzed the previously published ARID1A ChIP-seq in ARID1A wildtype RMG1 cells40. We focused on genes encoding components of the mevalonate pathway. The analyses revealed that ARID1A is associated with the promoters of the HMGCR and HMGCS1 genes that encode the rate-limiting enzymes of the mevalonate pathway7 (Fig. 4A). Consistent with the notion that ARID1A promotes the expression of these genes, RNA-seq analysis revealed that HMGCR and HMGCS1 mRNAs are both downregulated by ARID1A knockout in OCCC cells (Fig. S4A). The cholesterol pathway showed a trend toward significance among genes regulated by ARID1A knockdown in other cancer types (e.g., Fig. S4B)41,42. Consistently, ARID1A knockout reduced the association of RNA polymerase II (Pol II) and active transcription epigenetic marker lysine 27 acetylation on histone H3 (H3K27ac) with the promoters of these genes based on ChIP-seq analysis (Fig. 4A). We validated the downregulation of both HMGCR and HMGCS1 at both mRNA and protein levels by ARID1A knockout in RMG1 and OVCA429 cells (Fig. 4B and Fig. S4C). Consistently, HMGCR activity was lower in ARID1A knockout cells, which was further reduced by simvastatin treatment (Fig. 4C). Similar downregulation of both HMGCR and HMGCS1 by ARID1A knockout was observed in ARID1A wildtype 12Z cells (Fig. 4D). Ectopic wildtype ARID1A restoration rescued the downregulation of HMGCR and HMGCS1 induced by ARID1A knockout (Fig. 4B and Fig. S4C). Likewise, ectopic wildtype ARID1A restoration in ARID1A-mutated OCCC cell lines such as TOV21G and OVISE upregulated both HMGCS1 and HMGCR (Fig. 4E and Fig. S4D). Similarly, restoration of SNF5 in SNF5 mutant rhabdoid tumor cell line G401 upregulated HMGCS1 and HMGCR (Fig. 4F). Consistent with the finding that knockdown of other subunits of the SWI/SNF complex such as ARID1B, BRG1 and SNF5 sensitized cells to simvastatin (Fig. 3H), the expression of HMGCS1 and HMGCR was reduced by their knockdown (Fig. S4E–G).
Figure 4. HMGCS1 and HMGCR are direct target genes of the ARID1A-containing SWI/SNF-complex.
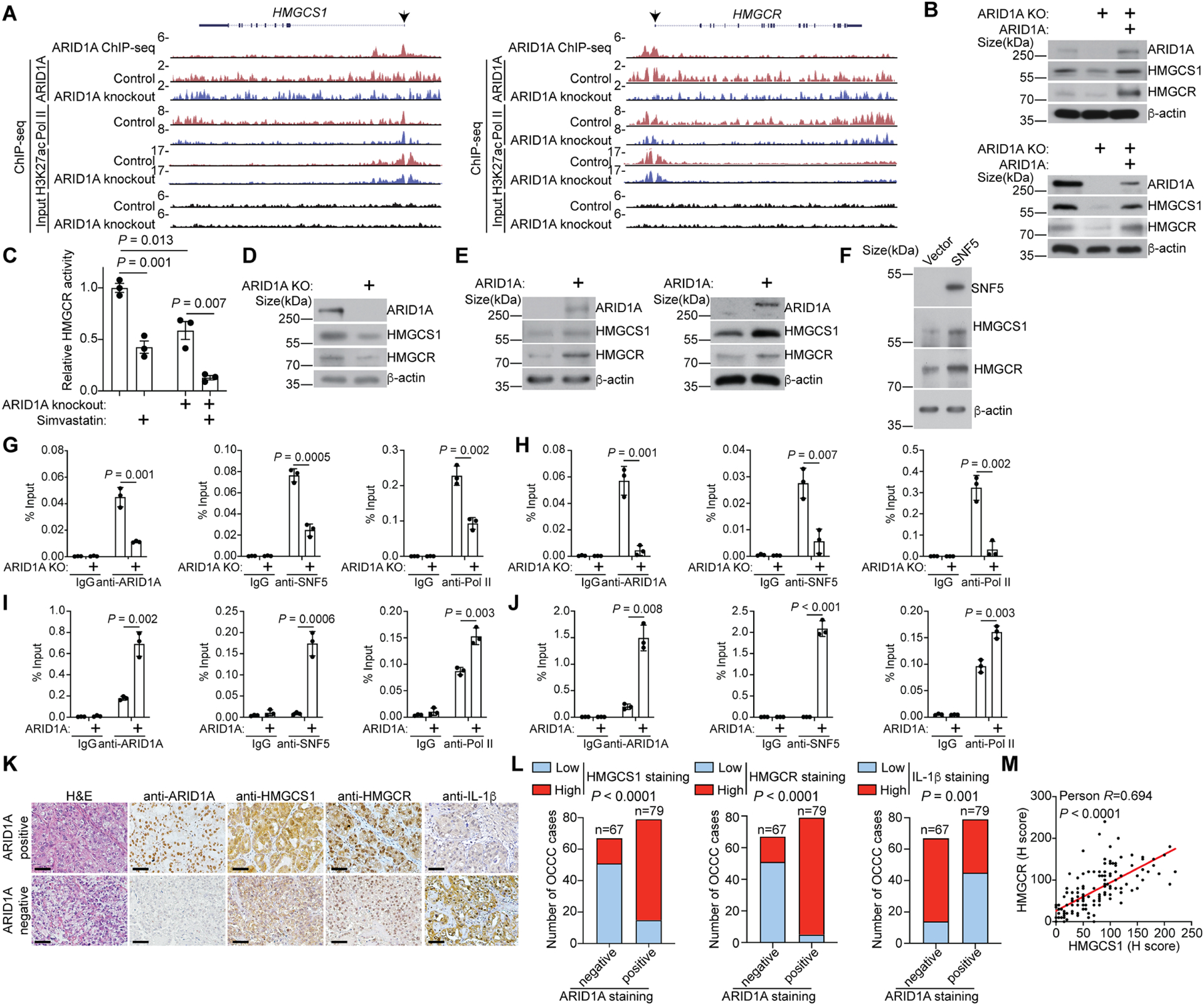
A, The indicated ChIP-seq and input tracks in the HMGCS1 and HMGCR gene loci in control and ARID1A knockout RMG1 cells from a previously published GEO dataset (GSE120060)40.
B, Expression of the indicated proteins in control, ARID1A knockout RMG1 (top) or OVCA429 (bottom) cells with or without wildtype ARID1A restoration was determined by immunoblot.
C, Relative enzyme activity of HMGCR in control or ARID1A knockout OVCA429 cells treated with or without simvastatin (10 μM) was examined. n=3 independent experiments.
D, Expression of the indicated proteins in control and ARID1A knockout 12Z cells was determined by immunoblot.
E, Expression of the indicated proteins in control and wildtype ARID1A restored TOV21G (left) or OVISE (right) cells were determined by immunoblot.
F, Expression of the indicated proteins in control and wildtype SNF5 restored SNF5 negative G401 rhabdoid tumor cells was determined by immunoblot.
G-J, The association of ARID1A, SNF5 and RNA Pol II with the HMGCS1 and HMGCR gene promoters in parental and ARID1A knockdown RMG1 cells (G and H) or control and wildtype ARID1A restored TOV21G cells (I and J) was determined by ChIP-qPCR analysis. An isotype matched IgG was used as a control. n=3 independent experiments.
K-M, Representative images of ARID1A, HMGCS1, HMGCR and IL-1β in ARID1A positive or negative cases from a tissue microarray with 146 OCCCs (K). Scale bar = 100 μm. Expression of ARID1A, HMGCS1, HMGCS1 and IL-1β were scored as high or low based on the median of histological scores (H. scores). HMGCR, HMGCS1 or IL-1β staining were quantified in the indicated ARID1A positive and negative OCCCs (L). The P value was calculated by Fisher’s exact test. In addition, correlation between HMGCR and HMGCS1 expression were examined by a two-sided Pearson analysis (M).
Error bars represent mean with SEM in C, G, H, I and J. P values were calculated using two-tailed Student t-test in C, G, H, I and J.
See also Figure S4.
We next validated the association of ARID1A and SNF5, a core subunit of the SWI/SNF complex1, with the promoters of the HMGCR and HMGCS1 genes (Fig. 4G–H and Fig. S4H–I). ARID1A knockout reduced the association of SNF5 and RNA Pol II with their promoters (Fig. 4G–H and Fig. S4H–I), which correlates with a reduction in the association of active transcription epigenetic markers such as H3K27ac and lysine 4 trimethylated histone H3 (H3K4me3) at their promoters (Fig. S4J–K). Conversely, wildtype ARID1A restoration in ARID1A-mutated TOV21G cells enhanced the association of both SNF5 and RNA Pol II with the promoters of the HMGCR and HMGCS1 genes (Fig. 4I–J), which correlated with their upregulation (Fig. 4E). This data supports that the ARID1A-containing SWI/SNF complex functions as an activator of HMGCR and HMGCS1 expression.
We next correlated the protein expression of ARID1A with HMGCR and HMGCS1 in a panel of 146 cases of human OCCCs. We observed that ARID1A positive OCCCs correlated with significantly higher levels of both HMGCR and HMGCS1 expression, while negatively correlates with IL-1β (Fig. 4K–L). In addition, there was a positive correlation between HMGCR and HMGCS1 in these OCCCs (Fig. 4M). Together, we conclude that HMGCR and HMGCS1 are direct target genes of the ARID1A-containing SWI/SNF complex and ARID1A inactivation suppresses the mevalonate pathway by downregulating HMGCR and HMGCS1.
Growth inhibition by statins depends on pyroptosis
We next determined whether the growth inhibition observed in ARID1A inactivated cells treated by statins are pyroptosis dependent. Toward this goal, we knocked down key pyroptosis regulators such as GSDMD or caspase 1 in ARID1A inactivated cells17,18. Indeed, knockdown of either GSDMD or caspase 1 rescued the observed decrease in simvastatin IC50s induced by ARID1A knockout (Fig. 5A–D). In addition, inhibition of caspase 1 activity by small molecule Ac-YVAD-CMK43 or Ac-YVAD-CHO44 rescued the observed decrease in simvastatin IC50s induced by ARID1A knockout (Fig. 5E). Similar results were also made using MCC950, a small molecule inhibitor of NLRP3 inflammasome45 (Fig. 5E). These findings support that the growth inhibition of ARID1A inactivated cells induced by inhibition of the mevalonate pathway is pyroptosis dependent.
Figure 5. Statin-induced growth inhibition is pyroptosis dependent.
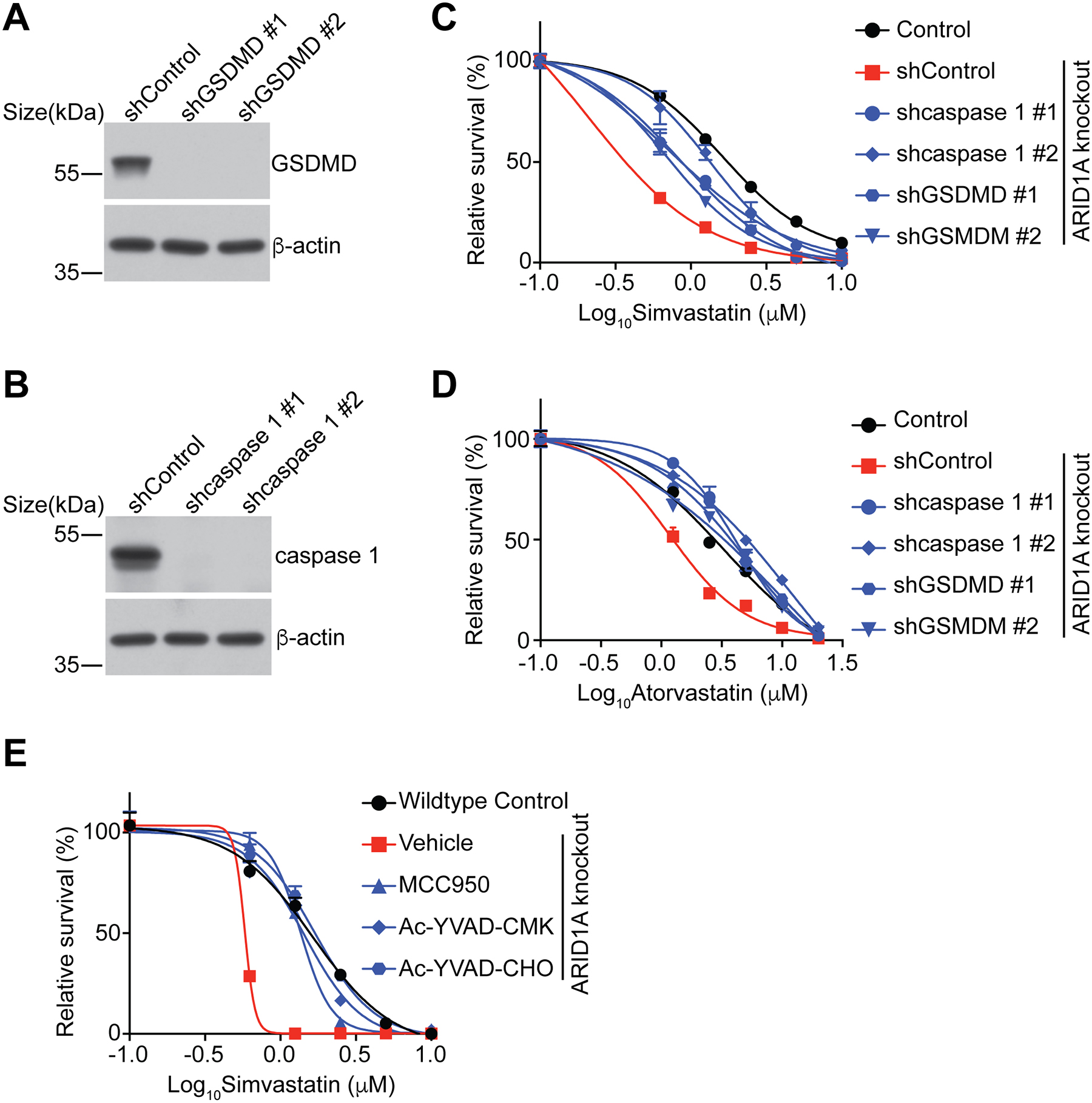
A, Expression of GSDMD in the indicated OVCA429 cells was determined by immunoblot.
B, Expression of caspase 1 in the indicated OVCA429 cells was determined by immunoblot.
C, Dose response curves of the indicated OVCA429 cells to simvastatin were determined by colony formation assay. n=4 independent experiments.
D, Dose response curves of the indicated OVCA429 cells to atorvastatin were determined by colony formation assay. n=4 independent experiments.
E, Dose response curves of the indicated OVCA429 cells treated with or without MCC950 (1 μM), Ac-YVAD-CMK (20 μM) and Ac-YVAD-CHO (20 μM) to simvastatin was determined by colony formation assay. n=4 independent experiments.
Error bars represent mean with SEM.
Pyroptosis induced by statins in ARID1A inactivated cells depends on the mevalonate pathway metabolite GGPP.
Statins inhibit the enzymatic activity of HMGCR, the rate-limiting enzyme in the mevalonate pathway7. Consequently, statins reduce the levels of downstream metabolites of the mevalonate pathway such as mevalonate, GGPP, and cholesterol7. To validate the on-target effects of statins in ARID1A inactivated cells, we performed the rescue experiments by supplementation of simvastatin or atorvastatin treated cells with mevalonate, GGPP, or cholesterol. Mevalonate and GGPP rescued the observed growth inhibition induced by statins in ARID1A knockout cells in a dose dependent manner (Fig. 6A–B). Similar observations were made in additional ARID1A knockout or mutant cell lines (Fig. S5A–C). In contrast, cholesterol further accentuated the growth inhibition induced by statins (Fig. 6A). Cholesterol suppresses the mevalonate pathway via a negative feedback loop7. Consistently, cholesterol sensitizes cells to low dose of simvastatin regardless of ARID1A status (Fig. S5D–E). Together, these findings support that suppression of the mevalonate pathway sensitizes cells to statins.
Figure 6. Pyroptosis induced by statins is mediated by mevalonate and GGPP metabolites.
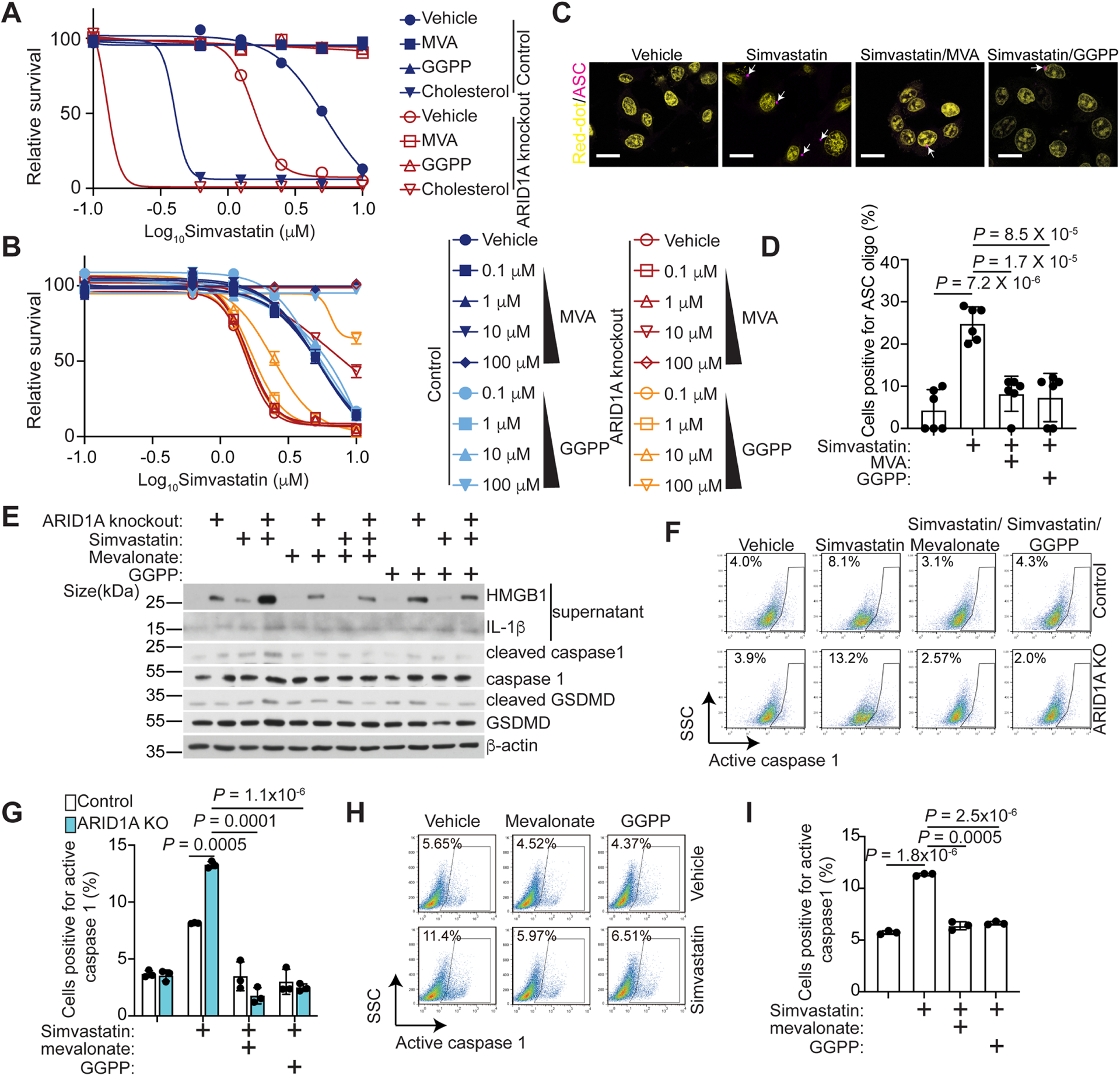
A, Dose response curves of control and ARID1A knockout RMG1 cells treated with vehicle control, mevalonate (100 μM), cholesterol (10 μM) or GGPP (10 μM) to simvastatin were determined by colony formation assay. n=4 independent experiments.
B, Dosage dependent rescue of simvastatin induced cell growth inhibition by mevalonate (0.1 μM, 1 μM, 10 μM and 100 μM) or GGPP (0.1 μM, 1 μM, 10 μM and 100 μM) in the indicated OVCA429 cells was determined by colony formation assay. n=4 independent experiments.
C. Representative immunofluorescence images of ASC-mCerulean staining in ARID1A knockout OVCA429 cells treated with vehicle control or simvastatin (5 μM) with or without MVA (100 μM) and GGPP (10 μM). Arrows point to examples of ASC oligo. Nuclei were visualized by Red-dot staining. Scale bar = 20 μm.
D. Quantification of ASC positive cells in the indicated treatment groups of the ARID1A knockout OVCA429 cells. n=6 independent experiments.
E, Expression of the indicated proteins in control or ARID1A knockout OVC429 treated with or without simvastatin together with or without mevalonate (100 μM) or GGPP (10 μM) was determined by immunoblot.
F, Caspase 1 activity in control and ARID1A knockout OVCA429 cells treated with simvastatin (10 μM) together with or without MVA (100 μM) or GGPP (10 μM) were examined by flow cytometry.
G, Quantification of caspase 1 positive cells in the indicated OVCA429 cells. n=3 independent experiments.
H, Caspase 1 activity in primary OCCC culture XVOA295 treated with simvastatin (10 μM) together with or without MVA (100 μM) or GGPP (10 μM) for 48 hours.
I, Quantification of percentages of caspase 1 positive XVOA295 cells in the indicated treatment groups. n=3 independent experiments.
Error bars represent mean with SEM in A, B, G and I. P values were calculated using two-tailed Student t-test in D, G and I.
See also Figure S5.
Given our findings that statins induce pyroptosis, we next determined the effects of mevalonate and GGPP on inflammasome formation and pyroptosis. Indeed, inflammasome formation induced by simvastatin was reduced by supplementation of either mevalonate or GGPP (Fig. 6C–D). Likewise, secretion of HMGB1 and IL-1β was rescued by mevalonate and GGPP (Fig. 6E). Further, simvastatin induced cleavage of caspase 1 and GSDMD was suppressed by mevalonate and GGPP (Fig. 6E). Finally, caspase 1 activity induced by simvastatin was reduced by mevalonate and GGPP (Fig. 6F–G and Fig. S5F–I). Similar observations were also made using primary ARID1A-mutated OCCC cultures (Fig. 6H–I). Thus, we conclude that pyroptosis induced by statins depends on the mevalonate pathway metabolite GGPP.
ARID1A inactivation sensitizes tumors to simvastatin in vivo.
We next determined whether ARID1A status affects the sensitivity to inhibition of the mevalonate pathway in vivo. Toward this goal, we used five different preclinical mouse models. First, we used orthotopic xenograft models of ovarian cancer formed by ARID1A wildtype RMG1 OCCC cells or the matched isogenic ARID1A knockout RMG1 cells. We orthotopically transplanted cells into the bursa-sac covering the ovary of immunocompromised nude mice to mimic the tumor microenvironment46,47. The injected cells were allowed to grow for one week to establish the orthotopic tumors (Table S1). Mice were then randomized and treated 3 times per week for 3 weeks with vehicle control or simvastatin (10 mg/kg), the same dose that was previously shown to be sufficient to suppress the mevalonate pathway in mouse models48. We used tumor weight as a surrogate for tumor burden. Simvastatin treatment significantly reduced the burden of orthotopic xenografts formed by ARID1A knockout RMG1 cells (Fig. 7A–B). In contrast, simvastatin did not significantly affect the growth of tumors formed by control RMG1 cells (Fig. 7A–B). Thus, the observed tumor suppressive effects by simvastatin treatment are ARID1A status dependent. Similar growth inhibition was also observed in orthotopic xenograft tumors formed by ARID1A-mutated TOV21G cells (Fig. 7C–D).
Figure 7. Simvastatin suppresses the growth of ARID1A inactivated OCCCs in vivo.
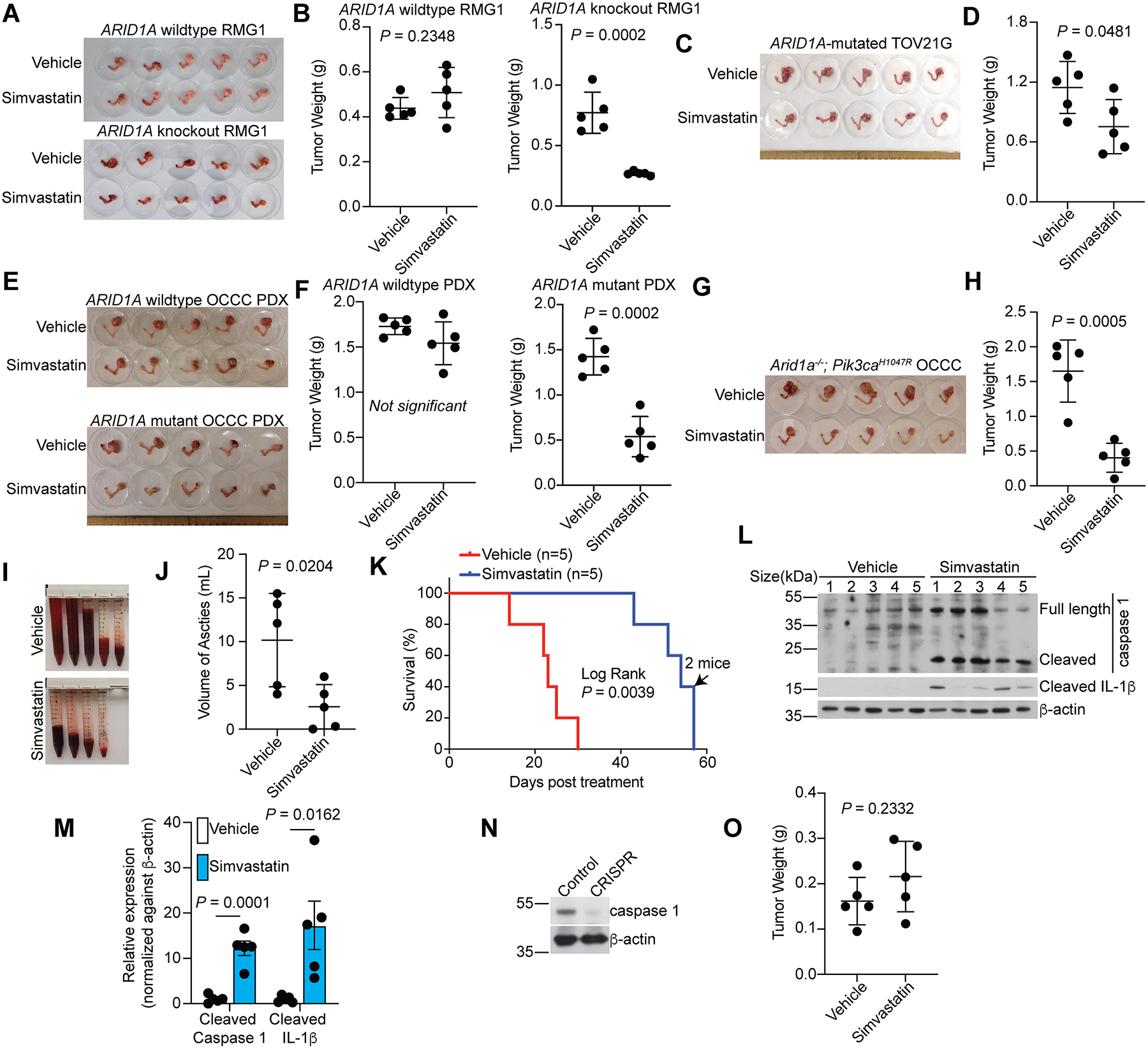
A-B, Orthotopic xenografts formed by control or ARID1A knockout RMG1 cells were treated with vehicle or simvastatin (n=5 mice/group). Shown are images of reproductive tracks with tumors from the indicated groups at the end of treatment (A). Tumor weights were measured as surrogates for tumor burden (B).
C-D, Same as A-B, but for orthoptic xenografts formed by ARID1A-mutated TOV21G cells.
E-F, Mice bearing wildtype or mutant ARID1A OCCC PDXs were treated with vehicle or simvastatin (n=5 mice/group). Shown are images of reproductive tracks with tumors from the indicated groups at the end of treatment (E). Tumor weights were measured as surrogates for tumor burden (F).
G-M, Mice bearing Arid1a−/−;Pik3caH1047R OCCCs were randomized into two indicated treatment groups (n=5 mice/group). Images of reproductive tracts with tumors from the indicated groups are showed (G). Tumor weight were measured as surrogate for tumor burden (H). Ascites produced in the indicated treatment groups (I) were quantified (J) at the end of treatment. After completing treatment, mice were followed for survival, and the Kaplan-Meier survival curves for each of the indicated groups are shown (K). The arrow indicates the time point when two mice met the criteria for euthanization. Expression of caspase 1 and IL-1β in tumors harvested at the end of experiments was determined by immunoblot (L), which was quantified by NIH ImageJ software (M).
N-O, Expression of caspase 1 and a loading control β-actin in Arid1a−/−;Pik3caH1047R tumors developed from mice injected with a lentivirus encoding both Cre-recombinase and a sgRNA to mouse Casp1 (N). Tumors developed from mice injected with only Cre-recombinase were used as a control for caspase 1 expression. The tumors developed in this model were randomized and treated with vehicle control or simvastatin the same as in (H) and tumor weight was measured as surrogate for tumor burden (O) (n=5 mice/group).
Error bars represent mean with SEM in B, D, F, H, J, M and O. P values were calculated using two-tailed Student t-test in B, D, F, H, J, M and O.
We next expanded these studies into OCCC patient-derived xenografts (PDX). Consistent with our mechanistic studies, compared with ARID1A wildtype OCCC PDX, HMGCR and HMGCS1 were downregulated in the OCCC PDX harboring a frameshift ARID1A mutation (Fig. S6). Indeed, simvastatin significantly reduced the tumor burden in ARID1A-mutated, but not ARID1A wildtype, OCCC PDXs (Fig. 7E–F). Finally, we examined whether simvastatin is effective in the Arid1a−/−;Pik3caH1047R immune competent OCCC genetic mouse model. Simvastatin significantly reduced the burden of established OCCCs and ascites produced in the Arid1a−/−;Pik3caH1047R genetic mouse model (Fig. 7G–J), which correlated with a significant improvement in the survival of tumor bearing mice (Fig. 7K).
We next determined whether simvastatin induces pyroptosis in vivo. Consistent with the notion that the observed reduction in tumor burden is due to the induction of pyroptosis in vivo, we observed an increase in both cleaved caspase 1 and IL-1β in tumors harvested from simvastatin treated mice compared with vehicle control treated mice (Fig. 7L–M). In addition, to directly determine whether the observed tumor suppressive effects induced by simvastatin are pyroptosis dependent, we knocked out caspase 1 in OCCCs developed from the Arid1a−/−;Pik3caH1047R genetic mouse model by injecting a lentivirus simultaneously encoding small guide RNA (sgRNA) to mouse Casp1 gene and a Cre-recombinase (Fig. 7N). Caspase 1 knockout abrogated the reduction in tumor burden induced by simvastatin treatment (Fig. 7O). However, simvastatin alone does not appear to be sufficient to eradicate established ARID1A-inactivated ovarian tumors (Fig. 7). Together, we conclude that inhibition of the mevalonate pathway suppresses the growth of ARID1A-inactivated ovarian cancer, but alone it is not sufficient to eradicate ARID1A-inactivated ovarian cancers.
Simvastatin synergizes with anti-PDL-1 in suppressing ARID1A-inactivated OCCCs
In contrast to immunologically silent apoptotic cell death, pyroptotic cell death induces the infiltration of immune cells such as CD8+ and CD4+ T cells by releasing IL-1β, IL-18, and HMGB122,23. Although the presence of intra-tumoral CD8+ T cells alone is not sufficient to predict the response to ICB, efficacy of ICB requires intra-tumoral CD8+ T cells. Consistently, it has been shown that pyroptosis induction synergizes with ICB in suppressing tumor growth18. In addition, ARID1A represses PD-L1 and anti-PD-L1 is effective against ARID1A-mutated ovarian cancer15,49. These findings raised the possibility that a combination of mevalonate pathway inhibition and the anti-PD-L1 antibody may be synergistic in suppressing ARID1A-inactivated ovarian cancers.
Accordingly, we examined whether simvastatin synergizes with anti-PD-L1 in the Arid1a−/−;Pik3caH1047R immune competent OCCC genetic mouse model. A combination of simvastatin and anti-PD-L1 was significantly more effective in reducing the ascites production and tumor burden compared with either of the individual treatments (Fig. 8A–D). Simvastatin increased the tumor infiltrating CD8+ and CD4+ T cells (Fig. 8E and Fig. S7A). Notably, simvastatin did not increase Ki67+ CD8+ T cells, indicating the observed increase was due to an increase in infiltration of these cells (Fig. S7B). However, single cell RNA-seq analysis did not reveal consistent changes in other immune cell populations (Fig. S7C), which was validated by FACS analysis for infiltration of immune cell populations such as M-MDSC, neutrophils, macrophage, tumor-associated immunosuppressive macrophages (Arg1+ or CD163+) by simvastatin treatment (Fig. S7D). Simvastatin induced the expression of cleaved caspase 3, a marker shared by apoptosis and pyroptosis34 (Fig. S7E–F). However, simvastatin upregulated caspase 1 activity in the treated tumors (Fig. 8F–G), which supports the notion that the observed increase in immune cell infiltration correlates with pyroptosis induction.
Figure 8. Simvastatin and anti-PD-L1 are synergistic in suppressing ARID1A inactivated OCCCs.
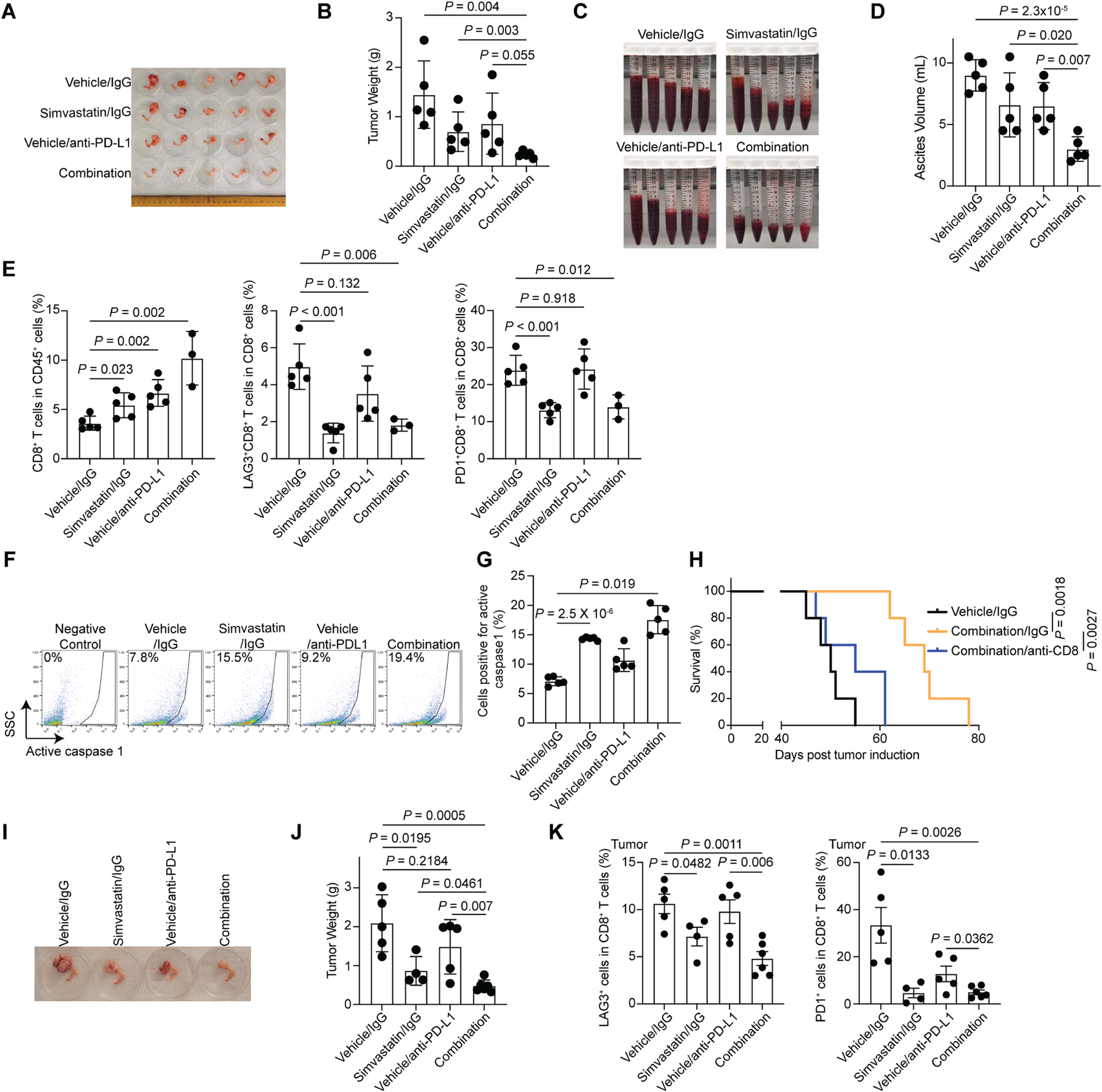
A-G, Mice bearing Arid1a−/−;Pik3caH1047R OCCCs were randomized into four indicated treatment groups (n=5 mice/group). Note that, for combination studies, we used 5 mg/kg simvastatin instead of 10 mg/kg in single treatment studies. Images of reproductive tracts with tumors from the indicated groups are showed at the end of treatment (A). Tumor weight were measured as surrogate for tumor burden (B). Statistical co-efficiency of drug interaction (CDI) analysis revealed that the CDI for the combination is 0.85 (<1, indicative of a synergistic effect)60. Ascites produced in the indicated treatment groups (C) were quantified (D) at the end of treatment. At end of treatment, percentages of tumor infiltrated CD8+ T cells, LAG3+ or PD-1+ CD8+ T cells were assessed by flow cytometry (E). In addition, caspase 1 activity in the tumors harvested from the indicated treatment groups was determined by flow cytometry (F) and percentages of caspase 1 positive cells were quantified (G).
H, Same as (A), but the mice were randomized into three indicated treatment groups. After completing treatment, mice were followed for survival, and the Kaplan-Meier survival curves for each of the indicated groups are shown (n=5 mice/group).
I-K, Humanized-BLT mice bearing ARID1A-mutated OCCC PDXs were randomized into four indicated treatment groups. Representative images of reproductive tracts with tumors from the indicated groups are showed at the end of treatment (I). Tumor weights were measured as surrogate for tumor burden (J). Statistical co-efficiency of drug interaction (CDI) analysis revealed that the CDI for the combination is 0.91 (<1, indicative of a synergistic effect)60. At end of treatment, percentages of tumor infiltrated human CD8+ T cells, LAG3+ or PD-1+ CD8+ T cells were assessed by flow cytometry (K).
Error bars represent mean with SEM in B, D, E, G, J and K. P values were calculated using two-tailed Student t-test in B, D, E, G, J and K.
See also Figures S7 and S8.
Reducing cholesterol levels in the tumor microenvironment prevents the functional exhaustion of CD8+ T cells50. Simvastatin reduced intratumoral cholesterol levels as determined Filipin III staining (Fig. S7E–F). Consistently, simvastatin treatment ameliorated CD8+ T cell exhaustion as evidenced by a decrease in LAG3+ or PD-1+ CD8+ T cells (Fig. 8E and Fig. S7A). These findings raised the possibility that the observed tumor suppressive effects induced by the combination is CD8+ T cell dependent. To test this possibility, we depleted the CD8+ T cells using an anti-CD8 antibody. Indeed, the observed improvement in the survival of tumor bearing mice by the combination was significantly reduced by anti-CD8 antibody treatment (Fig. 8H). Finally, we extended the combination study into a humanized patient derived ARID1A-mutated OCCC model (Fig. S7G). Similar synergy between simvastatin and anti-PD-L1 in reducing tumor burden was observed (Fig. 8I–J). Likewise, simvastatin reduced the markers of exhausted LAG3+ or PD-1+ CD8+ T cells (Fig. 8K). The combination at the doses used were well-tolerated. For example, the combination did not significantly affect the body weight of the treated tumor-bearing mice (Fig. S8A–B). In addition, the combination did not visibly change the histological morphology of liver, kidney and spleen (Fig. S8C). Together, we conclude that inhibition of the mevalonate pathway synergizes with immune checkpoint blockade in suppressing the growth of ARID1A-inactivated OCCCs.
Discussion
Here we report that ARID1A inactivation sensitizes cells to inhibition of the mevalonate pathway by reducing the expression of key mevalonate pathway regulators such as HMGCS1 and HMGCR. This is different from the typical therapeutic strategies that leverage the upregulation of tumor-promoting pathway induced by inactivation of tumor suppressors. Our findings are consistent with the essential role of the mevalonate pathway in cancer cell survival7. Knockdown of key mevalonate pathway regulators such as HMGCS1 in breast and lung cancer cell lines sensitizes them to statins51. Likewise, inhibiting the transcription of the HMGCS1 and HMGCR genes by dipyridamole synergizes with statins to suppress the growth of acute myelogenous leukemia patient samples52. These findings support that those genetic alterations reducing the mevalonate pathway may create a dependence on the residual activity of the mevalonate pathway and thus are synthetically lethal with inhibition of the pathway by statins.
Our results show that inhibition of the mevalonate pathway is effective in suppressing the growth of ARID1A inactivated cells by triggering pyroptosis. Using single-nucleotide polymorphisms in HMGCR as a proxy for therapeutic inhibition of HMCGR in a genome-wide association study, there were significantly lower odds in OECs and a trend toward lower odds in OCCCs53. Interestingly, in secondary analyses, there were no significant associations between circulating cholesterol and epithelial ovarian cancer in this study53. This is consistent with our findings that GGPP instead of cholesterol plays a key role in regulating statin-induced growth inhibition in ARID1A inactivated cells. However, the study did not consider ARID1A mutational status in their analysis. Thus, it will be interesting to examine whether the observed reduction in cancer risk associated with genetic proxy inhibition of HMGCR is primarily associated with ARID1A-mutated OCCCs and OECs. In addition, these findings raised the interesting possibility to explore FDA-approved statins as a precision prevention agent for women with ARID1A-mutated endometriosis, an early event during tumorigenesis2,54.
The injection of Cre-adenovirus and manipulation of the bursa could alter the immune microenvironment. However, mice in all the treatment groups received the same Cre-adenovirus injection and manipulation of the bursa. Thus, the observed effects induced by simvastatin treatment is independent of Cre-adenovirus injection and manipulation of the bursa.
Leveraging tumor cell intrinsic genetic alterations to drive immune infiltration is an attractive therapeutic strategy to boost the anti-tumor immune response. For example, pyroptosis induction in tumor cells will not only suppress the growth of tumor cells but also increase the infiltration of immune cells such as CD8+ T cells23. In addition, the infiltrated immune cells such as CD8+ T cells reciprocally induce pyroptosis in tumor cells to establish a positive tumor suppressive feedback loop with the associated immune microenvironment22,23. Further, our results show that simvastatin largely reduces the exhaustion of infiltrated CD8+ cells in the tumor bed50, which correlates with a decrease in cholesterol levels in the tumor microenvironment. Thus, inhibition of the mevalonate pathway suppresses ARID1A-mutated tumors through three different mechanisms: 1) induction of pyroptosis of tumor cells through cell intrinsic mechanism; 2) recruitment of immune cells such as CD8+ T cells to the tumor bed, which in turn induces pyroptosis in additional tumor cells; and 3) ameliorating the exhaustion of tumor infiltrating CD8+ T cells by decreasing cholesterol levels in the tumor microenvironment. Indeed, we observed a synergy between simvastatin and anti-PD-L1 in suppressing the growth of ARID1A-inactivated OCCCs. The improvement of survival of tumor bearing mice by the combination was significantly reduced by depleting CD8+ T cells, highlighting the importance of pyroptosis-induced antitumor immune response in the observed tumor suppression. Notably, in recurrent OCCC ICB durvalumab alone did not result in significant differences in progression-free survival or objective response rate in a phase II randomized trial55. This further highlights the advantage of the combinatorial therapeutic strategy reported here and it will be interesting to measure GGPP levels in these future studies.
Our findings support that a combination of statins and ICB is synergistic in suppressing ARID1A-mutated tumors because the combination leverages both the tumor intrinsic effects of inhibition of the mevalonate pathway and its effects on boosting the antitumor immunity in the tumor microenvironment. Both statins and ICB are FDA-approved. Thus, the combinatorial therapeutic strategy reported here is easily translatable to ARID1A-mutated cancer types such as OCCCs by repurposing these FDA-approved agents. Moreover, ARID1A is the most frequently mutated epigenetic regulator and SWI/SNF is altered in ~20% of all human cancers56–58. Thus, we expect our findings to have far-reaching implications in developing urgently needed therapeutic approaches for these cancers.
STAR METHODS
RESOURCE AVAILABILITY Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rugang Zhang (rzhang11@mdanderson.org)
Materials availability
This study did not generate new unique reagents
Data and code availability
The previously published ChIP-seq data that we reanalyzed here are available in the Gene Expression Omnibus (GEO) database under accession codes GSE12006040. Previously published RNA-seq data that we reanalyzed here are available under accession codes GSE13191759, GSE14847361 and GSE13113242. The previous published mass spectrometry proteomics data31 is available from ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) with dataset identifier PXD004570. Mutation status for cell lines and their genes inhibition dependency scores from Project Achilles were downloaded from DepMap portal (https://depmap.org/portal/download, dataset DepMap Public 20Q4 v2) and average dependency scores for cell lines from the same tissue origin were used for the analysis.
This paper does not report original code.
All other data supporting the findings of this study are available from the corresponding author on reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human ovarian clear cell carcinoma (OCCC) cell line RMG1 was cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C supplied with 5% CO2. OCCC cell lines OVCA429, TOV21G, OVISE, OVTOKO, SKOV3 and ES2 cells were cultured in RPMI-1640 with 10% FBS and 1% penicillin/streptomycin. Rhabdoid tumor cell line G401 was cultured in McCoy’s 5a medium with 10% FBS and 1% penicillin/streptomycin at 37°C supplied with 5% CO2. Immortalized human endometriotic cell line 12Z was cultured in DMEM with 10% FBS and 1% penicillin/streptomycin at 37°C supplied with 5% CO2. For lentivirus packing, human embryonic kidney cell line HEK-293T was cultured in DMEM with 10% FBS and 1% penicillin/streptomycin at 37°C supplied with 5% CO2. Primary cultures of human ovarian clear cell carcinoma XVOA295 was as described previously47. XVOA295 cells were cultured in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin. All the cell lines were authenticated using short tandem repeat DNA profiling. Mycoplasma was tested monthly using mycoplasma PCR detection kit (Sigma-Aldrich, Cat#: MP0035).
Human samples
PDX models were established by direct implantation of surgically removed human ovarian clear cell tumor tissues orthotopically in the bursal sac of the immunocompromised mice under a protocol approved by the IACUC of The Wistar Institute (No. 201205). Tumor tissue procurement was approved by the Institutionally Review Board at Christiana Care Health System and The Wistar Institute. The protocols were approved by the IACUC of The Wistar Institute.
Ovarian cancer tissue microarrays (TMA) which include 146 OCCC samples were kindly provided by Ronny Drapkin from The University of Pennsylvania.
Animal models
For mouse models, 2 different strains were used in this study. The transgenic mouse model of Arid1a−/−;Pik3caH1047R genetic clear cell ovarian tumor were generated by crossing Arid1aflox/flox mice with R26-PikcaH1047R as we previously published47. 6–8 weeks old female NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, RRID:IMSR_JAX:005557) from Jackson Laboratory were housed and maintained in individual microisolator cages in a rack system capable of managing air exchange with filters.
METHOD DETAILS
ARID1A CRISPR knockout
ARID1A knockout RMG1 and OVCA429 cells were generated as previously described40,47. For ARID1A knockout, pSpCas9(BB)-2A-GFP (Addgene, Cat#: 48138, RRID:Addgene_48138) and pFETCh_Donor (Addgene, Cat#: 63934, RRID:Addgene_63934) plasmids were purchased from Addgene. Guide RNA sequence (5’- TGTCCCACGGCTGTCATGAC -3’) targeting the terminal codon of ARID1A was inserted into pSpCas9(BB)-2A-GFP. About 500 base pairs of homologous arms at both sides of guide RNA targeting site were cloned and inserted into pFETCh-donor. ARID1A knockout clones were selected by puromycin (1 μg/ml) and validated by immunoblot.
Cell viability assay
3,000 cells were seeded into 96-well plated. For treatment, 5 repeats were performed for each concentration. Empty wells with same volume of complete medium were used as blank background. After 72 hours treatments, 10% AlamarBlue reagent (Fisher, Cat#: DAL1100) was added into each well and then incubated for 2 hours at 37°C. AlamarBlue fluorescence was then quantified at the respective excitation and emission wavelength of 540 and 595 nm using microplate reader (Perkin Elmer, 2104 EnVision).
Colony formation assay
3,000 to 5,000 cells were seeded into 24-well tissue culture plates. For treatment, cell medium was changed every three days with appropriate drug doses for a total of 10–14 days. Colonies were washed twice with PBS and fixed with 10% methanol. Fixed colonies were stained with 0.005% crystal violet in distilled water. Integrated density was measured using NIH ImageJ software (version 1.53a).
Immunoblotting
Cultured cells or tumor tissues were lysed with RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 1 mM EDTA, 1 mM dithiothreitol (DTT) and 1 mM PMSF) on ice for 30 min. Proteins were denatured using SDS loading buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 10% Glycerol, 0.1% Bromophenol blue and 100 mM DTT) at 100°C for 10 min. Proteins were then separated by SDS-PAGE gel and transferred to PVDF membrane (Millipore, IPVH00010). Membranes were blocked with 5% non-fat milk and incubated with following primary antibodies at 4°C overnight. Objective signals were amplified with HRP-conjugated secondary antibodies (Cell Signaling, Cat#: 7076 and Cat#: 7074) and detected by chemiluminescent substrate (Thermo Fisher, Cat#: 34094).
Protein prenylation assay
GTPases protein prenylation was reported previously62. The cells were harvested in ice-cold prenylation assay buffer (1% Nonidet P-40, 50 mM Tris-HCl pH 8.0, 5 mM EDTA, 1 mM PMSF and protease inhibitors cocktail). The lysate was centrifuged at 10,000× g to remove nuclei and the supernatants were used as cell lysates, mixed with SDS loading buffer and separated by 12% SDS-PAGE.
Cytoplasm and nucleus protein fractionation
Cells were resuspended in whole cell lysis buffer (10 mM HEPES pH 7.8, 0.34 M sucrose, 10% glycerol, 10 mM KCl, 1.5 mM MgCl2, 0.1% Triton X-100, 1 mM PMSF and protease inhibitors (Roche, Cat#: 11836170001)) and centrifuged for 5 min at 10,000× g. Supernatant were kept as cytoplasm fraction. Nucleus pellets were then lysed in nucleus lysis buffer (50 mM Tris-HCl pH 7.8, 420 mM NaCl, 0.34 M sucrose, 0.5% Nonidet P-40 and protease inhibitors) for 30 min on ice. Proteins were denatured using SDS loading buffer at 100°C for 10 min.
Chromatin immunoprecipitation (ChIP)
Cells were crosslinked with 1% formaldehyde for 10 min and then quenched by 0.125 M glycine for 5 min at room temperature. Fixed cells were lysed with ChIP lysis buffer 1 (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 1% Triton X-100, and 0.1% DOC) on ice and lysis buffer 2 (10 mM Tris pH 8.0, 200 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA) at room temperature. Chromatin was digested with micrococcal nuclease (MNase Cell Signaling, Cat#: 10010) in digestion buffer (10 mM Tris pH 8.0, 1 mM CaCl2, and 0.2% Triton X-100) at 37 °C for 15 min. Nucleus products were broken down by Bioruptor pulse at high frequency. The following antibodies were used for ChIP: rabbit anti-ARID1A antibody (Abcam, Cat#: ab182560, 5 μg per reaction), rabbit anti-SNF5 (Bethyl, Cat#: A301–087A, 5 μg per reaction), mouse anti-Pol II (Santa Cruz, Cat#: sc-47701, 5 μg per reaction), rabbit anti-Histone H3K4me3 (Active Motif, Cat#: 39159, 5 μg per reaction) and rabbit anti-Histone H3K27ac (Abcam, Cat#: ab4729, 5 μg per reaction). Isotype-matched IgGs were used as negative controls. ChIP DNA was purified by ChIP DNA clean and concentrator kit (Zymo Research, Cat#: D5205) and analyzed by qPCR. Primers targeting HMGCS1 or HMGCR promoters used for ChIP–qPCR are included in Table S2.
Reverse transcription and quantitative Real-Time PCR (qRT-PCR).
Total RNA was extracted using Trizol reagents (Thermo Fisher, Cat#: 15596026) according to the manufacturer’s protocol. RNA was reverse transcribed with the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher, Cat#: 4368813). qRT-PCR was performed using QuantStudio 3 Real-Time PCR System (Thermo Fisher). Primers used in this study are included in KEY RESOURCES TABLE.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-ARID1A | Abcam | Cat#: ab182560 |
| rabbit anti-ARID1B | Cell Signaling | Cat#: 92964; RRID:AB_2800195 |
| rabbit anti-BRG1 | Cell Signaling | Cat#: 49360; RRID:AB_2728743 |
| rabbit anti-SNF5 | Cell Signaling | Cat#: 91735; RRID:AB_2800172 |
| rabbit anti-HMGCS1 | Proteintech | Cat#: 17643–1-AP; RRID:AB_2248359 |
| mouse anti-HMGCR | Thermo Fisher | Cat#: CL0259; RRID:AB_2786972 |
| rabbit anti-MVD | Proteintech | Cat#: 15331–1-AP; RRID:AB_2147439 |
| mouse anti-FDPS | Proteintech | Cat#: 16129–1-AP; RRID:AB_2104749 |
| rabbit anti-SREBP2 | Proteintech | Cat#: 28212–1-AP; RRID:AB_2881091 |
| rabbit anti-HMGB1 | Abcam | Cat#: ab18256; RRID:AB_444360 |
| rabbit anti-cleaved IL-1β | Cell Signaling | Cat#: 83186; RRID:AB_2800010 |
| rabbit anti-GSDMD | Abcam | Cat#: ab210070; RRID:AB_2893325 |
| rabbit anti-caspase1 | Cell Signaling | Cat#: 3866; RRID:AB_2069051 |
| rabbit anti-cleaved caspase1 | Biovision | Cat#: A1004 |
| mouse anti-Histone H3 | Cell Signaling | Cat#: 14269; RRID:AB_2756816 |
| rabbit anti-α-tubulin | Cell Signaling | Cat#: 2125; RRID:AB_2619646 |
| mouse anti-β-actin | Sigma-Aldrich | Cat#: A5316; RRID:AB_476743 |
| rabbit anti-SNF5 | Bethyl | Cat#: A301–087A; RRID:AB_2191714 |
| mouse anti-Pol II | Santa Cruz | Cat#: sc-47701; RRID:AB_677353 |
| anti-mouse IgG, HRP-linked antibody | Cell Signaling | Cat#: 7076; RRID:AB_330924 |
| anti-rabbit IgG, HRP-linked antibody | Cell Signaling | Cat#: 7074; RRID:AB_2099233 |
| mouse anti-cGAS | Santa Cruz | Cat#: sc-515777; RRID:AB_2734736 |
| rabbit anti-γH2AX | Cell Signaling | Cat#: 9718; RRID:AB_2118009 |
| rabbit anti-cleaved PARP p85 | Cell Signaling | Cat#: 5625; RRID:AB_10699459 |
| mouse anti-RAC1 | Proteintech | Cat#: 66122–1-Ig; RRID:AB_2883264 |
| rabbit anti-RHOA | Proteintech | Cat#: 10749–1-AP; RRID:AB_2285104 |
| rabbit anti-HNF1β | Sigma-Aldrich | Cat#: HPA002083; RRID:AB_1080232 |
| rabbit anti-cleaved Caspase 3 | Cell Signaling | Cat#: 9661L; RRID:AB_2341188 |
| rabbit anti-Histone H3K4me3 | Active Motif | Cat#: 39159; RRID:AB_2615077 |
| rabbit anti-Histone H3K27ac | Abcam | Cat#: ab4729; RRID:AB_2118291 |
| IgG isotype control | Bio X Cell, | Cat#: BE0090; RRID:AB_1107780 |
| mouse PD-L1 antibody for in vivo | Bio X Cell | Cat#: BE0101; RRID:AB_10949073 |
| human PD-L1 antibody for in vivo | R&D Systems | Cat#: MAB10348 |
| Fc blocking anti-mouse CD16/CD32 | BD Bioscience | Cat#: 553142; RRID:AB_394657 |
| PE-Cy7 anti-mouse CD3e | BD Bioscience | Cat#: 552774; RRID:AB_394460 |
| BV711 anti-mouse CD45 | Biolegend | Cat#: 103147; RRID:AB_2564283 |
| PE/Cyanine7 anti-mouse PD-1 | Biolegend | Cat#: 109109; RRID:AB_572016 |
| BV421 anti-mouse LAG-3 | Biolegend | Cat#: 125221; RRID:AB_2572080 |
| Alexa Fluor 700 rat anti-mouse CD45 | BD Biosciences | Cat#: 560510; RRID:AB_1645208 |
| BB515 mouse anti-human CD45 | BD Biosciences | Cat#: 564586; RRID:AB_2869588 |
| BUV805 mouse anti-human CD3 | BD Biosciences | Cat#: 612895; RRID:AB_2870183 |
| BUV395 mouse anti-human CD4 | BD Biosciences | Cat#: 563550; RRID:AB_2738273 |
| PerCP-Cy5.5 mouse anti-human CD8 | BD Biosciences | Cat#: 565310; RRID:AB_2687497 |
| BV786 mouse anti-human PD-1 | BD Biosciences | Cat#: 563789; RRID:AB_2738425 |
| BV421 mouse anti-human LAG-3 | BD Biosciences | Cat#: 565720; RRID:AB_2744330 |
| PerCP/Cyanine5.5 anti-mouse CD19 | Biolegend | Cat#: 115534; RRID:AB_2072925 |
| APC/Fire750 anti-mouse CD8b.2 | Biolegend | Cat#: 140420; RRID:AB_2819885 |
| Alexa Fluor 647 anti-mouse Ki67 | Biolegend | Cat#: 652408; RRID:AB_2562139 |
| APC anti-mouse Arg1 | eBioscience | Cat#: 17369780; RRID:AB_2734834 |
| APC-Cyanine7 anti-mouse CD64 | Biolegend | Cat#: 139334; RRID:AB_2910296 |
| FITC anti-mouse Lyve1 | eBioscience | Cat#: 53-0443-82; RRID:AB_1633415 |
| BUV805 anti-mouse CD11b | BD Biosciences | Cat#: 741934; RRID:AB_2871246 |
| BV421 anti-mouse NK1.1 | Biolegend | Cat#: 108732; RRID:AB_2562218 |
| BV711 anti-mouse Ly6G | Biolegend | Cat#: 127643; RRID:AB_2565971 |
| BV785 anti-mouse Ly6C | Biolegend | Cat#: 128041; RRID:AB_2565852 |
| PE anti-mouse CD163 | Biolegend | Cat#: 156704; RRID:AB_2860724 |
| PE-Dazzle594 anti-mouse CD169 | Biolegend | Cat#: 142424; RRID:AB_2750059 |
| PE-Cyanine7 anti-mouse F4/80 | Biolegend | Cat#: 123114; RRID:AB_893478 |
| Bacterial and virus strains | ||
| DH5α competent E. coli | Thermo Fisher | Cat#: 18265017 |
| Stellar competent E. coli | Fisher | Cat#: DAL1100 |
| Chemicals, peptides, and recombinant proteins | ||
| simvastatin | Sigma-Aldrich | Cat#: S6196 |
| atorvastatin | Sigma-Aldrich | Cat#: PHR1422 |
| etoposide | Sigma-Aldrich | Cat#: E1383 |
| methyl-β-cyclodextrin | Cayman Chemical | Cat#: 21633 |
| Nigericin | InvivoGen | Cat#: tlrl-nig |
| mevalonate | Sigma-Aldrich | Cat#: 43987 |
| GGPP | Sigma-Aldrich | Cat#: G6025 |
| cholesterol | Sigma-Aldrich | Cat#: C4951 |
| lipofectamine 2000 | Thermo Fisher | Cat#: 11668027 |
| AlamarBlue reagent | Fisher | Cat#: DAL1100 |
| chemiluminescent substrate | Thermo Fisher | Cat#: 34094 |
| protease inhibitors cocktail | Roche | Cat#: 11836170001 |
| micrococcal nuclease | Cell Signaling | Cat#: 10010 |
| Trizol reagent | Thermo Fisher | Cat#: 15596026 |
| prolong antifade reagent | Thermo Fisher | Cat#: P36961 |
| RedDot nuclear stain | Biotium | Cat#: 40061 |
| Filipin III | Sigma-Aldrich | Cat#: F4767 |
| mammalian protein extraction reagent | Thermo Fisher | Cat#: 78501 |
| Cre recombinase expressing-adenovirus | Vector Viral Core | Cat#: VVC-U of Iowa-5 |
| red blood cell lysis buffer | Thermo Fisher | Cat#: 00-4333-57 |
| Fixable Viability Stain 510 | BD Biosciences | Cat#: 564406; RRID:AB_2869572 |
| Mayer’s Hematoxylin | Dako | Cat#: S3309 |
| Critical commercial assays | ||
| LookOut Mycoplasma PCR detection | Sigma-Aldrich | Cat#: MP0035 |
| ChIP DNA clean and concentrator kit | Zymo Research | Cat#: D5205 |
| Reverse Transcription kit | Thermo Fisher | Cat#: 4368813 |
| Cholesterol Assay Kit | Abcam | Cat#: ab133116 |
| Amplex Red Cholesterol Quantitation Kit | Thermo Fisher | Cat#: A12216 |
| 2’3’-cGAMP ELISA Kit | Cayman Chemical | Cat#: 501700 |
| FAM FLICA Caspase1 Kit | Bio-Rad | Cat#: ICT097 |
| Mouse Dissociation Kit | MiltenyiBiotec | Cat#: 130-096-730 |
| Viability staining Kit | Thermo Fisher | Cat#: L35957 |
| Dako EnVision+ system | Dako | Cat#: K4011 |
| HMG-CoA Reductase Assay Kit | Sigma-Aldrich | Cat#: CS1090 |
| Annexin V Conjugates for Apoptosis Detection | Thermo Fisher | Cat#: A35110 |
| Human IL-1β ELISA Kit | Abcam | Cat#: ab214025 |
| Human IL-18 ELISA Kit | Abcam | Cat#: ab215539 |
| True-Nuclea Transcription Factor Buffer Set | Biolegend | Cat#: 424401 |
| Experimental models: Cell lines | ||
| OVCA429 | Dr. Ie-Ming Shih, Johns Hopkins University | RRID:CVCL_3936 |
| RMG1 | JCRB | Cat# JCRB0172; RRID:CVCL_1662 |
| ES2 | ATCC | Cat#: CRL-1978; RRID: CVCL_3509 |
| SKOV3 | JCRB | Cat# NIHS0737; RRID:CVCL_4Y20 |
| OVISE | JCRB | Cat#: JCRB1043; RRID:CVCL_3116 |
| OVTOKO | JCRB | Cat#: NIHS0301; RRID:CVCL_3117 |
| TOV21G | ATCC | Cat#: CRL-11730; RRID:CVCL_3613 |
| HEK-293T | ATCC | Cat#: CRL-3216; RRID: CVCL_0063 |
| XVOA295 | PMID: 28737768 | N/A |
| G401 | PMID: 34085048 | N/A |
| 12Z | Dr. Ie-Ming Shih, Johns Hopkins University | RRID:CVCL_0Q73 |
| Experimental models: Organisms/strains | ||
| NSG mice | The Jackson Laboratory | Strain#: 005557; RRID:IMSR_JAX:005557 |
| Arid1a−/−;Pik3caH1047R transgenic mice | PMID: 28737768 | N/A |
| Oligonucleotides | ||
| Primers used in this study are listed in Table S2 | This paper | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP | Addgene | Cat#: 48138; RRID:Addgene_48138 |
| pFETCh_Donor | Addgene | Cat#: 63934; RRID:Addgene_63934 |
| pRP_ASC-LmCerulean | Addgene | Cat#: 41840; RRID:Addgene_41840 |
| plentiCRISPR v2 | Addgene | Cat#: 52961; RRID:Addgene_52961 |
| PlentiCRISPR-ARID1A | PMID: 30297712 | N/A |
| pLVX-M-puro | Addgene | Cat#: 125839; RRID:Addgene_125839 |
| pLVX-SNF5-puro | PMID: 34085048 | N/A |
| pCRISPR-Cre stuffer v4 | Addgene | Cat#: 158033; RRID:Addgene_158033 |
| pMD2.G | Addgene | Cat#: 12259; RRID:Addgene_12259 |
| psPAX2 | Addgene | Cat#: 12260; RRID:Addgene_12260 |
| pLKO.1-HMGCS1-shRNA1 | Molecular Screening Facility (Wistar) | TRCN0000045843 |
| pLKO.1-HMGCS1-shRNA2 | Molecular Screening Facility (Wistar) | TRCN0000045845 |
| pLKO.1-ARID1B-shRNA1 | Molecular Screening Facility (Wistar) | TRCN0000107361 |
| pLKO.1-ARID1B-shRNA2 | Molecular Screening Facility (Wistar) | TRCN0000107364 |
| pLKO.1-BRG1-shRNA1 | Molecular Screening Facility (Wistar) | TRCN0000015549 |
| pLKO.1- BRG1-shRNA2 | Molecular Screening Facility (Wistar) | TRCN0000015552 |
| pLKO.1-SNF5-shRNA1 | Molecular Screening Facility (Wistar) | TRCN0000039585 |
| pLKO.1-SNF5-shRNA2 | Molecular Screening Facility (Wistar) | TRCN0000039587 |
| pLKO.1-Caspase1-shRNA1 | Molecular Screening Facility (Wistar) | TRCN0000003502 |
| pLKO.1-Caspase1-shRNA2 | Molecular Screening Facility (Wistar) | TRCN0000003503 |
| pLKO.1-GSDMD-shRNA1 | Molecular Screening Facility (Wistar) | TRCN0000179394 |
| pLKO.1-GSDMD-shRNA2 | Molecular Screening Facility (Wistar) | TRCN0000180013 |
| Software and algorithms | ||
| GraphPad Prism version 8.0 | GraphPad Software Inc. | https://www.graphpad.com/ |
| FlowJo version 10.0 | FlowJo LLC | https://www.flowio.com/ |
| TraceFinder 4.1 | Thermo Fisher | https://www.thermofisher.com/order/catalog/product/OPTON-31001 |
| ImageJ version 1.53a | NIH | https://imagei.nih.gov/ii/ |
| GSEA version 4.1 | PMID: 16199517 | http://www.gsea-msigdb.org/gsea/index.isp |
Immunofluorescence staining
For ASC imaging45, OVCA429 cells were infected with lentivirus packaged pRP_ASC-LmCerulean plasmid (Addgene, Cat#: 41840, RRID:Addgene_41840). The ectopically expressed ASC-mCerulean oligo was imaged using a laser with 458 nm wavelength for excitation. Immunofluorescence staining was performed after indicated treatments by fixing cells in 4% paraformaldehyde and permeabilizing with 0.5% Triton-X 100. Samples were blocked with 5% FBS and incubated with following primary antibodies overnight at 4°C: mouse anti-cGAS (Santa Cruz, Cat#: sc-515777, RRID:AB_2734736), γH2AX (Cell Signaling, Cat#: 9718, RRID:AB_2118009). Cells were then incubated with highly cross absorbed secondary antibodies (Thermo Fisher, Cat#: A-11032 and A-11008, RRID:AB_2534091 and RRID:AB_143165) for 1 hour at room temperature and mounted with prolong antifade reagent (Thermo Fisher, Cat#: P36961). RedDot nuclear stain (Biotium, Cat#: 40061) was used to visualize the nucleus. Cells were imaged using scanning confocal microscope (Leica, TCS SP5 II).
Proteomic pathway enrichment
ARID1A wildtype and knockout OVCAR29 cells were utilized to perform the proteomic analysis to compare protein expression with or without ARID1A in isogenic OCCC cell lines31. Of the 4,952 protein groups identified with high confidence (two or more peptides, FDR < 1%), 264 proteins significantly changed in level with ARID1A knockout (|fold change| ≥ 2 and P value < 0.05), of which 95 increased and 169 decreased. The 169 proteins significantly decreased proteins (fold change ≤ 0.5 and P value < 0.05) in ARID1A knockout cells were used to perform a Wiki Pathway enrichment analysis on Enrichr website (https://maayanlab.cloud/Enrichr/). Next, we analyzed the effects of ARID1A overexpression in the ARID1A-mutated OCCC cell line OVISE31. A total of 5,760 protein groups were identified with high confidence, and 387 significantly changed with ARID1A expression (191 increased, 196 decreased). The entire protein expression dataset was utilized to perform gene set enrichment analysis on the Reactome Cholesterol Biosynthesis (Systematic name: M16227).
Gene set enrichment analysis (GSEA)
Integrated data of proteomics results or GEO datasets (GEO148473 and GEO131132) were utilized for the pathways enrichment analysis. GSEA was performed following the guidelines on the GSEA website of Broad Institute (http://www.broadinstitute.org/gsea/index.jsp) using gene sets including inflammasome pathway and steroid biosynthetic process.
Cholesterol detection assay
Cellular endogenous cholesterol was assayed by both immunofluorescence staining and flow cytometry. To examine the accumulation of cholesterol inside OVCA429 cells in response to ARID1A knockout, cells were seeded into 24-well plate at a density of 5 x 104 cells/well and cultured overnight. Cells were fixed with 4% paraformaldehyde and washed with cholesterol detection wash buffer (Abcam, Cat#: ab133116). Filipin III solution (Sigma-Aldrich, Cat#: 500 μg/mL) was added into each well for 40 min in the dark. After mounting, cholesterol staining was examined using Leica confocal microscope under excitation of 340–380 nm and emission of 385–470 nm. Red-Dot staining (Biotium, Cat#: 40061) was used to visualize the nucleus. To measure the cholesterol level by flow cytometry, cells were stained with 100 μg/mL Filipin III (Sigma-Aldrich, Cat#: F4767) for 40 min at room temperature. Cells were then washed twice and analyzed by flow cytometry (BD Biosciences, LSRII) for Filipin III at 355 nm laser.
Endogenous cholesterol quantification
Cellular endogenous cholesterol was extracted with 200 μL of chloroform: isopropanol: IGEPAL (7:11:0.1) in a micro homogenizer. Samples were centrifuged at 13,000× g for 10 min to remove insoluble material. The organic phase was transferred to a new tube and air dried to remove chloroform. And the pellet discarded. Residual organic solvent was removed under vacuum for 30 min. Dried lipids were dissolved in 200 μL of the cholesterol reaction buffer (Thermo Fisher, Cat#: A12216) and vortexed until the mixture was homogenous.
Both cholesterol standards and extracted lipids were set up to the Amplex Red reaction mix according to manufacturer’s instructions and seeded into 96-well plate. After 30 min incubation at 37 °C, plate was measured the fluorescence in a fluorescence microplate reader using excitation in the range of 560 nm and emission detection at 590 nm.
Quantification of mevalonate by LC-MS
Polar metabolites were extracted from control and ARID1A knockout OVCA429 cells with 80% ice-cold methanol and dried in a SpeedVac vacuum concentrator. Each sample was resuspended in 50 μl Buffer A (10 mM Ammonium Carbonate, 0.1% Ammonium Hydroxide in 60:40 ACN:H2O), and 4 μl was injected for LC-MS analysis on a Q Exactive HF-X mass spectrometer equipped with HESI II probe in-line with a Vanquish Horizon UHPLC system (Thermo Fisher). Samples were run in a pseudorandomized order. LC separation was performed on a ZIC-pHILIC column (Millipore, 2.1 × 150 mm ID) at 45 °C using an isocratic gradient of 100% Buffer A flowing at 0.1 mL/min, as described previously63. The mass spectrometer was operated in negative ionization mode, and full scans were acquired at 120,000 resolutions with a scan range of 120–460 m/z, automatic gain control target of 3E6, and maximum injection time of 100 ms. Mevalonate was quantified by the integrated area of the confirmed [M-H]-1 adduct peak using TraceFinder 4.1 (Thermo Fisher) followed by normalization to protein amount for each sample.
HMG-CoA reductase (HMGCR) enzymatic activity assay
HMGCR enzymatic activity was measured according to NADPH oxidation by HMGCR in the presence of the substrate HMG-CoA using manufacturer’s commercial kit (Sigma-Aldrich, Cat#: CS1090)64. 100 μg cell lysates from control and ARID1A knockout was mixed with NADPH and HMG-CoA (substrate), then incubated at 37°C for 5 minutes. Optical density at 340 nm was measured, and the decrease in OD340 represented the oxidation of NADPH by the catalytic subunit of HMGCR in the presence of the substrate HMG-CoA. HMGCR activity was normalized to OVCA429 control cells. Simvastatin treatment was applied as negative control.
2’3’-cGAMP ELISA detection
The endogenous level of 2’3’-cGAMP was examined by ELISA assay according to the manufacturer’s instructions (Cayman Chemical, Cat#: 501700). For cells quantification, 5×106 cells were harvested, washed with PBS, lysed by mammalian protein extraction reagent (Thermo Fisher, Cat#: 78501). Oncogenic Ras induced senescent IMR-90 fibroblast cells were used as positive control.
IL-1β and IL-18 ELISA detection
The endogenous level of IL-1β and IL-18 was examined by ELISA assay according to the manufacturer’s instructions (Abcam, Cat#: ab214025 and Cat#: ab215539). In brief, supernatant from control and ARID1A knockout OVCA429 cells were collected and centrifuged to remove cell debris. Supernatants, standards and all the reaction reagents were seeded into ELISA 96-plates. After 2 hours incubation, plates were washed and developed the signaling. Optical densities were recorded at 450 nm.
Caspase 1 activity assay
Activity of caspase 1 was determined with FAM FLICA caspase 1 Kit (Bio-Rad, Cat#: ICT097) followed by manufacturer’s instructions. In brief, cells with indicated treatments were resuspended in wash buffer and incubated with FAM labeled FLICA probe from 30 min at 37°C in the dark. After staining, cells were then washed twice and analyzed with flow cytometry (BD Biosciences, LSRII) at 488 nm laser.
Transmission electron microscopy
Transmission electron microscopy was performed on control and ARID1A knockout OVCA429 cells with or without simvastatin treatment. Samples were prepared by the Electron Microscopy Resource Laboratory of the University of Pennsylvania. Ultrathin sections from cell pellets were cut and mounted on the electron microscopy grids. Cell sections were then imaged using a transmission electron microscope (JEOL, JEM-1010). For pyroptosis analysis, imaging fields were taken in each group at ×10,000 or ×20,000 magnification.
OCCC orthotopic mouse models
The protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the Wistar Institute. Mice were maintained at 22–23°C with 40–60% humidity and 12 hours light/12 hours dark cycle. Briefly, 1×106 TOV21G or RMG1 cells were injected into the ovarian bursa sac of 6–8 weeks old female NSG mice. Tumor bearing mice were randomized into two groups (n=5 per group) one week after injection. The mice in each group were intraperitoneally injected with vehicle (10% DMSO in 100 μL PBS) or 10 mg/kg simvastatin 3 times per week for three weeks. Mice were then sacrificed, and tumor burden was examined using tumor weight as a surrogate in each treatment groups.
Human OCCC samples were obtained from Christiana Care. For the patients-derived xenograft (PDX) models of ovarian cancer, 5th passage of the previously described ARID1A wildtype and mutated PDXs65 were transplanted into ovarian bursa sac of 6–8 weeks old female NSG mice. Mice were randomized into 2 groups (n=5 per group) at three weeks after transplantation and treated with vehicle (10% DMSO in 100 μL PBS) or 10 mg/kg simvastatin 3 times per week for two weeks. Mice were then sacrificed, and tumor burden was examined using tumor weight as a surrogate in each treatment groups.
OCCC transgenic mouse model
For the conditional Arid1a−/−;Pik3caH1047R genetic OCCC mouse model47, Cre recombinase expressing-adenovirus (Ad-Cre, Vector Viral Core, Cat#: VVC-U of Iowa-5) was intrabursally injected to initiate tumorigenesis in 6–8 weeks old female mice. Mice were randomized at four weeks after Ad-Cre injection. For simvastatin (Sigma-Aldrich, Cat#: S6196) single treatment, the mice were randomized into the following 2 groups: vehicle (10% DMSO in 100 μL PBS) and simvastatin (10 mg/kg, 3 times per week, i.p.). After 3 weeks of treatment, mice were then sacrificed, and tumor burden was examined using tumor weight as a surrogate in each treatment groups. For survival experiments, the guideline of The Wistar Institute IACUC was used for endpoint assessment (e.g., tumor burden exceeds 10% of body weight).
For combination treatment, mice were randomized into the four treatment groups: vehicle plus IgG control (Bio X Cell, Cat#: BE0090, 5 mg/kg, 3 times per week, i.p.), simvastatin (5 mg/kg, 3 times per week, i.p.) plus IgG control, vehicle control plus anti-PD-L1 antibody (BioX Cell, Cat#: BE0101) (5 mg/kg, 3 times per week, i.p.), and combination of simvastatin and anti-PD-L1 antibody. After 3 weeks treatment, mice were then euthanized, and tumor burden was examined using tumor weight as a surrogate in each treatment groups.
For CD8+ T cell depletion, an anti-CD8 antibody (Bio X Cell, Cat#: BE0117, 10 mg/kg, twice per week) was used to deplete CD8+ T cells. An isotype-matched IgG (Bio X Cell, Cat#: BE0090, 10 mg/kg) was used as a negative control. For survival experiments, the guideline of The Wistar Institute IACUC was used for endpoint assessment (e.g., tumor burden exceeds 10% of body weight).
For caspase1 knockout in vivo, guide RNA (5’- GAGGGCAAGACGTGTACGAG -3’) targeting murine Casp1 gene was inserted into pCRISPR-Cre plasmid. After lentivirus packaging and concentration, lentivirus of Casp1-CRISPR-Cre was intrabursally injected into the ovarian sac of 6–8 weeks old Arid1a−/−;Pik3caH1047R transgenic mice. After four weeks, the OCCC tumors bearing mice were randomized for vehicle or simvastatin treatments (10 mg/kg, 3 times per week, i.p.).
Immune cell profiling
Immune cell profiling was previously described13,66. Tumors were chopped and digested with Mouse Dissociation Kit (Miltenyi Biotec, 130-096-730) according to the manufacturer’s instructions. Single cells were then harvested with 70 μm strainer and used for staining. Peritoneal cavity of mice was washed three times with 5 ml PBS and incubated in red blood cell lysis buffer (Thermo Fisher, Cat#: 00-4333-57). Live/dead cells were discriminated by viability staining Kit (Thermo Fisher, Cat#: L35957). Fc blocking (BD, Cat#: 553142) was followed by cell surface staining in FACS buffer (3% FBS in PBS buffer) using antibodies against CD3e (BD Biosciences, Cat#: 552774), CD45 (Biolegend, Cat#: 103147), CD4 (Biolegend, Cat#: 100516), CD8a (Biolegend, Cat#: 100708), CD69 (Biolegend, Cat#: 104510), PD1 (Biolegend, Cat#: 109109) and LAG3 (Biolegend, Cat#: 125221). Data was acquired using flow cytometry (BD Biosciences, LSRII) and analyzed using FlowJo software (version 10.0).
Single cell RNA sequencing (scRNA-seq) analysis
Pre-processing of the scRNA-seq data was performed using Cell Ranger Suite (pipeline v7.0.0, https://support.10xgenomics.com) with refdata-gex-mm10–2020-A transcriptome as a reference to map reads on mouse genome (mm10) using STAR67. Due to low overall expression, a rather lenient threshold was used for pre-filtering cells. Low-quality cells with less than 10 genes with reads and cells with over 10% mitochondrial content were filtered out. Cell clustering, marker identification and visualization were performed using Seurat v468. R package SingleR69 was used to determine cell types of the clusters using ImmGen data set as a reference for cell-specific gene signatures.
Generation of humanized-BLT mice
Hu-BLT mouse generation was conducted as we previously reported70,71 at the Wistar institute in accordance with IACUC-approved protocols. Briefly, 6 to 8 weeks old female NSG mice pretreated by busulfan at approximately 20 gram of body weight and then implanted with human fetal thymic tissue fragments and fetal liver tissue fragments under the murine renal capsule. Following the surgery, mice were injected via the tail vein with CD34+ hematopoietic stem cells isolated from human fetal liver tissues. Human fetal liver and thymus tissues were procured from Advanced Bioscience Resources.
Assessment of human immune cell reconstitution
After 12 weeks post-surgery, human immune cell reconstitution in peripheral blood was measured by flow cytometer (BD Biosciences, Symphony) using antibodies against mCD45 (BD Biosciences, Cat#: 560510), hCD45 (BD Biosciences, Cat#: 564586), hCD3 (BD Biosciences, Cat#: 612895), hCD4 (BD Biosciences, Cat#: 563550), hCD8 (BD Biosciences, Cat#: 565310) and Fixable Viability Stain (BD Biosciences, Cat#: 565310). Data were analyzed using FlowJo software (version 10.0).
Humanized OCCC PDX model
For humanized OCCC PDX model, 5th passage of a previously described ARID1A-mutated PDX (I832fs*)65 were transplanted to ovarian bursa sac of humanized-BLT mice. Three weeks after transplantation, mice were randomized into the following four treatment groups: vehicle plus IgG control (Bio X Cell, Cat#: BE0090, 5 mg/kg, 3 times per week, i.p.), simvastatin (5 mg/kg, 3 times per week, i.p.) plus IgG control, vehicle control plus anti-PD-L1 antibody (R&D Systems, Cat#: MAB10348, 5 mg/kg, 3 times per week, i.p.), and combination of simvastatin and anti-PD-L1 antibody. After 2 weeks treatment, mice were then euthanized, and tumor burden were compared using tumor weight as a surrogate in each of the treatment groups. Exhaustion of infiltrated human T cells in tumors were measured by flow cytometry (BD Biosciences, Symphony) using antibodies against mCD45 (BD Biosciences, Cat#: 560510), hCD45 (BD Biosciences, Cat#: 564586), hCD3 (BD Biosciences, Cat#: 612895), hCD4 (BD Biosciences, Cat#: 563550), hCD8 (BD Biosciences, Cat#: 565310), PD1 (BD Biosciences, Cat#: 563789), LAG3 (BD Biosciences, Cat#: 565720) and Fixable Viability Stain (BD Biosciences, Cat#: 564406). Data were analyzed with FlowJo software (version 10.0).
Immunohistochemistry (IHC) staining
Ovarian cancer tissue microarrays (TMA) were kindly provided by Ronny Drapkin from The University of Pennsylvania. For immunohistochemical staining72, TMA or mice tumor sections were incubated with primary following antibodies at 4°C overnight: anti-ARID1A (Abcam, Cat#: ab182560), anti-HMGCS1 (Proteintech, Cat#: 17643–1-AP), anti-HMGCR (Thermo Fisher, Cat#: CL0259), anti-cleaved IL-1β (Cell Signaling, Cat#: 83186) and anti-cleaved caspase 3 (Cell Signaling, Cat#: 9661L). The tissue sections were incubated with appropriate secondary antibodies (Dako, Cat#: K4011) for 1 hour at room temperature. Cell nuclei were stained using Mayer’s Hematoxylin (Dako, Cat#: S3309). Staining density was quantified using histologic score (H. Score)73.
Co-efficiency of drug interaction (CDI) analysis
CDI was calculated as follows: CDI = ((cA+cB)–cA×cB)/cAB, where cA represents the inhibitory rate of compound A, cB represents the inhibitory rate of compound B, and cAB represents the inhibitory rate of combination treatment of compound A and B74. Synergy is defined as CDI lower than 1.0, and antagonism is defined as CDI significantly greater than 1.0.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism software (version 8.0). Analysis of variance with Fisher’s least significant difference was used to identify significant differences in multiple comparisons. Spearman correlation analysis was used to examine the correlation between two factors. Log-rank test was used to compare the survival distributions among experimental groups. Experiments were repeated at least twice. Quantitative data are expressed as mean ± SEM unless otherwise stated. No statistical method was used to predetermine sample size. No data were excluded from the analyses. All analyses were performed blindly but not randomly. All mice for animal experiments were randomized.
Supplementary Material
Highlights.
ARID1A inactivation sensitizes cells to pyroptosis
ARID1A regulates HMGCR and HMGCS1, rate-limiting enzymes of the mevalonate pathway
Statins suppresses the growth of ARID1A-inactivated tumors via pyroptosis
Statins synergize with ICB in suppressing ARID1A-inactivated tumors
Acknowledgements
This work was supported by US National Institutes of Health grants (R01CA163377, R01CA202919, R01CA239128 and R01CA260661 to R. Z.; R01CA1665065 to L.J.M.; P50CA228991 to R. Z. and R.D.), US Department of Defense (OC180109 and OC190181, OC210124 to R. Z.; OC180192 to L.J.M.), and the Robert I. Jacobs Fund of The Philadelphia Foundation. Ovarian Cancer Research Alliance Ann and Sol Schreiber Mentored Investigator Award to W. Z. Support of Core Facilities was provided by Cancer Centre Support Grant (CCSG) CA010815 to The Wistar Institute and P30CA016672 to University of Texas MD Anderson Cancer Center.
Inclusion and Diversity Statement
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The Authors declare no competing interests.
References
- 1.Wu JN, and Roberts CW (2013). ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer discovery 3, 35–43. 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al. (2010). ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 363, 1532–1543. 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr., Vogelstein B, et al. (2010). Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330, 228–231. science.1196333 [pii] 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan B, Gao M, Wu CH, Wang TL, and Shih Ie M (2012). Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia 14, 986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan JK, Teoh D, Hu JM, Shin JY, Osann K, and Kapp DS (2008). Do clear cell ovarian carcinomas have poorer prognosis compared to other epithelial cell types? A study of 1411 clear cell ovarian cancers. Gynecologic oncology 109, 370–376. 10.1016/j.ygyno.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Mackay HJ, Brady MF, Oza AM, Reuss A, Pujade-Lauraine E, Swart AM, Siddiqui N, Colombo N, Bookman MA, Pfisterer J, and du Bois A (2010). Prognostic relevance of uncommon ovarian histology in women with stage III/IV epithelial ovarian cancer. International journal of gynecological cancer : official journal of the International Gynecological Cancer Society 20, 945–952. 10.1111/IGC.0b013e3181dd0110. [DOI] [PubMed] [Google Scholar]
- 7.Mullen PJ, Yu R, Longo J, Archer MC, and Penn LZ (2016). The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer 16, 718–731. 10.1038/nrc.2016.76. [DOI] [PubMed] [Google Scholar]
- 8.Gidding SS, Champagne MA, de Ferranti SD, Defesche J, Ito MK, Knowles JW, McCrindle B, Raal F, Rader D, Santos RD, et al. (2015). The Agenda for Familial Hypercholesterolemia: A Scientific Statement From the American Heart Association. Circulation 132, 2167–2192. 10.1161/CIR.0000000000000297. [DOI] [PubMed] [Google Scholar]
- 9.Jelinic P, Ricca J, Van Oudenhove E, Olvera N, Merghoub T, Levine DA, and Zamarin D (2018). Immune-Active Microenvironment in Small Cell Carcinoma of the Ovary, Hypercalcemic Type: Rationale for Immune Checkpoint Blockade. J Natl Cancer Inst. 10.1093/jnci/djx277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bosse D, Wankowicz SM, Cullen D, et al. (2018). Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806. 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. 10.1126/science.aao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anagnostou V, Niknafs N, Marrone K, Bruhm DC, White JR, Naidoo J, Hummelink K, Monkhorst K, Lalezari F, Lanis M, et al. (2020). Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nature Cancer 1, 99–111. 10.1038/s43018-019-0008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukumoto T, Fatkhutdinov N, Zundell JA, Tcyganov EN, Nacarelli T, Karakashev S, Wu S, Liu Q, Gabrilovich DI, and Zhang R (2019). HDAC6 inhibition synergizes with anti-PD-L1 therapy in ARID1A-inactivated ovarian cancer. Cancer Res. 10.1158/0008-5472.CAN-19-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abou Alaiwi S, Nassar AH, Xie W, Bakouny Z, Berchuck JE, Braun DA, Baca SC, Nuzzo PV, Flippot R, Mouhieddine TH, et al. (2020). Mammalian SWI/SNF Complex Genomic Alterations and Immune Checkpoint Blockade in Solid Tumors. Cancer Immunol Res. 10.1158/2326-6066.CIR-19-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, Nagel ZD, Zou J, Wang C, Kapoor P, et al. (2018). ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 10.1038/s41591-018-0012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, Kanai M, Mori Y, Matsumoto S, Chikuma S, et al. (2015). Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol 33, 4015–4022. 10.1200/JCO.2015.62.3397. [DOI] [PubMed] [Google Scholar]
- 17.Schroder K, and Tschopp J (2010). The inflammasomes. Cell 140, 821–832. 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 18.Rosenbaum SR, Wilski NA, and Aplin AE (2021). Fueling the Fire: Inflammatory Forms of Cell Death and Implications for Cancer Immunotherapy. Cancer discovery 11, 266–281. 10.1158/2159-8290.CD-20-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bedoui S, Herold MJ, and Strasser A (2020). Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol. 10.1038/s41580-020-0270-8. [DOI] [PubMed] [Google Scholar]
- 20.Kovacs SB, and Miao EA (2017). Gasdermins: Effectors of Pyroptosis. Trends Cell Biol 27, 673–684. 10.1016/j.tcb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, et al. (2012). Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674. 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, Huang H, Shao F, and Liu Z (2020). A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 579, 421–426. 10.1038/s41586-020-2079-1. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, et al. (2020). Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420. 10.1038/s41586-020-2071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karki R, and Kanneganti TD (2019). Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer 19, 197–214. 10.1038/s41568-019-0123-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou J, Hsu JM, and Hung MC (2021). Molecular mechanisms and functions of pyroptosis in inflammation and antitumor immunity. Mol Cell 81, 4579–4590. 10.1016/j.molcel.2021.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Jiang M, Qi L, Wu Y, Song D, Gan J, Li Y, and Bai Y (2021). Pyroptosis, a new bridge to tumor immunity. Cancer Sci 112, 3979–3994. 10.1111/cas.15059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erkes DA, Cai W, Sanchez IM, Purwin TJ, Rogers C, Field CO, Berger AC, Hartsough EJ, Rodeck U, Alnemri ES, and Aplin AE (2020). Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer discovery 10, 254–269. 10.1158/2159-8290.CD-19-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Motwani M, Pesiridis S, and Fitzgerald KA (2019). DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 20, 657–674. 10.1038/s41576-019-0151-1. [DOI] [PubMed] [Google Scholar]
- 29.Zhao B, Liu P, Fukumoto T, Nacarelli T, Fatkhutdinov N, Wu S, Lin J, Aird KM, Tang HY, Liu Q, et al. (2020). Topoisomerase 1 cleavage complex enables pattern recognition and inflammation during senescence. Nat Commun 11, 908. 10.1038/s41467-020-14652-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, and Nunez G (2013). K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldman AR, Bitler BG, Schug Z, Conejo-Garcia JR, Zhang R, and Speicher DW (2016). The Primary Effect on the Proteome of ARID1A-mutated Ovarian Clear Cell Carcinoma is Downregulation of the Mevalonate Pathway at the Post-transcriptional Level. Mol Cell Proteomics 15, 3348–3360. 10.1074/mcp.M116.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang XX, Ying P, Diao F, Wang Q, Ye D, Jiang C, Shen N, Xu N, Chen WB, Lai SS, et al. (2013). Altered protein prenylation in Sertoli cells is associated with adult infertility resulting from childhood mumps infection. J Exp Med 210, 1559–1574. 10.1084/jem.20121806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herr DR, Yam TYA, Tan WSD, Koh SS, Wong WSF, Ong WY, and Chayaburakul K (2020). Ultrastructural Characteristics of DHA-Induced Pyroptosis. Neuromolecular Med 22, 293–303. 10.1007/s12017-019-08586-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu P, Zhang X, Liu N, Tang L, Peng C, and Chen X (2021). Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther 6, 128. 10.1038/s41392-021-00507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waller DD, Park J, and Tsantrizos YS (2019). Inhibition of farnesyl pyrophosphate (FPP) and/or geranylgeranyl pyrophosphate (GGPP) biosynthesis and its implication in the treatment of cancers. Crit Rev Biochem Mol Biol 54, 41–60. 10.1080/10409238.2019.1568964. [DOI] [PubMed] [Google Scholar]
- 36.van der Burgh R, Pervolaraki K, Turkenburg M, Waterham HR, Frenkel J, and Boes M (2014). Unprenylated RhoA contributes to IL-1beta hypersecretion in mevalonate kinase deficiency model through stimulation of Rac1 activity. J Biol Chem 289, 27757–27765. 10.1074/jbc.M114.571810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skinner OP, Jurczyluk J, Baker PJ, Masters SL, Rios Wilks AG, Clearwater MS, Robertson AAB, Schroder K, Mehr S, Munoz MA, and Rogers MJ (2019). Lack of protein prenylation promotes NLRP3 inflammasome assembly in human monocytes. J Allergy Clin Immunol 143, 2315–2317 e2313. 10.1016/j.jaci.2019.02.013. [DOI] [PubMed] [Google Scholar]
- 38.Zeitvogel A, Baumann R, and Starzinski-Powitz A (2001). Identification of an invasive, N-cadherin-expressing epithelial cell type in endometriosis using a new cell culture model. Am J Pathol 159, 1839–1852. 10.1016/S0002-9440(10)63030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald ER 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, et al. (2017). Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 170, 577–592 e510. 10.1016/j.cell.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 40.Wu S, Fatkhutdinov N, Rosin L, Luppino JM, Iwasaki O, Tanizawa H, Tang HY, Kossenkov AV, Gardini A, Noma KI, et al. (2019). ARID1A spatially partitions interphase chromosomes. Sci Adv 5, eaaw5294. 10.1126/sciadv.aaw5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson MR, Reske JJ, Koeman J, Adams M, Joshi NR, Fazleabas AT, and Chandler RL (2022). SWI/SNF Antagonism of PRC2 Mediates Estrogen-Induced Progesterone Receptor Expression. Cells 11. 10.3390/cells11061000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo Q, Wu X, Chang W, Zhao P, Nan Y, Zhu X, Katz JP, Su D, and Liu Z (2020). ARID1A prevents squamous cell carcinoma initiation and chemoresistance by antagonizing pRb/E2F1/c-Myc-mediated cancer stemness. Cell Death Differ 27, 1981–1997. 10.1038/s41418-019-0475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schierle GS, Hansson O, Leist M, Nicotera P, Widner H, and Brundin P (1999). Caspase inhibition reduces apoptosis and increases survival of nigral transplants. Nat Med 5, 97–100. 10.1038/4785. [DOI] [PubMed] [Google Scholar]
- 44.Boost KA, Hoegl S, Hofstetter C, Flondor M, Stegewerth K, Platacis I, Pfeilschifter J, Muhl H, and Zwissler B (2007). Targeting caspase-1 by inhalation-therapy: effects of Ac-YVAD-CHO on IL-1 beta, IL-18 and downstream proinflammatory parameters as detected in rat endotoxaemia. Intensive Care Med 33, 863–871. 10.1007/s00134-007-0588-0. [DOI] [PubMed] [Google Scholar]
- 45.Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21, 248–255. 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih Ie M, Conejo-Garcia JR, et al. (2015). Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med 21, 231–238. 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, Zhai Y, Kossenkov AV, Vara-Ailor A, Rauscher FJ III, et al. (2017). ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol 19, 962–973. 10.1038/ncb3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Christensen M, Su AW, Snyder RW, Greco A, Lipschutz JH, and Madaio MP (2006). Simvastatin protection against acute immune-mediated glomerulonephritis in mice. Kidney Int 69, 457–463. 10.1038/sj.ki.5000086. [DOI] [PubMed] [Google Scholar]
- 49.Fukumoto T, Fatkhutdinov N, Zundell JA, Tcyganov EN, Nacarelli T, Karakashev S, Wu S, Liu Q, Gabrilovich DI, and Zhang R (2019). HDAC6 Inhibition Synergizes with Anti-PD-L1 Therapy in ARID1A-Inactivated Ovarian Cancer. Cancer Res 79, 5482–5489. 10.1158/0008-5472.CAN-19-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, Wang Q, Yang M, Kalady MF, Qian J, et al. (2019). Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab 30, 143–156 e145. 10.1016/j.cmet.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pandyra AA, Mullen PJ, Goard CA, Ericson E, Sharma P, Kalkat M, Yu R, Pong JT, Brown KR, Hart T, et al. (2015). Genome-wide RNAi analysis reveals that simultaneous inhibition of specific mevalonate pathway genes potentiates tumor cell death. Oncotarget 6, 26909–26921. 10.18632/oncotarget.4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pandyra A, Mullen PJ, Kalkat M, Yu R, Pong JT, Li Z, Trudel S, Lang KS, Minden MD, Schimmer AD, and Penn LZ (2014). Immediate utility of two approved agents to target both the metabolic mevalonate pathway and its restorative feedback loop. Cancer Res 74, 4772–4782. 10.1158/0008-5472.CAN-14-0130. [DOI] [PubMed] [Google Scholar]
- 53.Yarmolinsky J, Bull CJ, Vincent EE, Robinson J, Walther A, Smith GD, Lewis SJ, Relton CL, and Martin RM (2020). Association Between Genetically Proxied Inhibition of HMG-CoA Reductase and Epithelial Ovarian Cancer. JAMA 323, 646–655. 10.1001/jama.2020.0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noe M, Horlings HM, Lum A, Jones S, Senz J, Seckin T, et al. (2017). Cancer-Associated Mutations in Endometriosis without Cancer. N Engl J Med 376, 1835–1848. 10.1056/NEJMoa1614814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan DSP, Choi CH, Ngoi N, Sun H, Heong V, Ow SGW, Chay WY, Kim HS, Lim YW, Goss G, et al. (2022). A multicenter phase II randomized trial of durvalumab (D) versus physician’s choice chemotherapy (PCC) in patients (pts) with recurrent ovarian clear cell adenocarcinoma (MOCCA/APGOT-OV2/GCGS-OV3). Journal of Clinical Oncology 40, 5565–5565. 10.1200/JCO.2022.40.16_suppl.5565. [DOI] [Google Scholar]
- 56.Kadoch C, and Crabtree GR (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv 1, e1500447. 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385 e318. 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li J, Wang W, Zhang Y, Cieslik M, Guo J, Tan M, Green MD, Wang W, Lin H, Li W, et al. (2020). Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest 130, 2712–2726. 10.1172/JCI134402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cao SS, and Zhen YS (1989). Potentiation of antimetabolite antitumor activity in vivo by dipyridamole and amphotericin B. Cancer Chemother Pharmacol 24, 181–186. 10.1007/BF00300240. [DOI] [PubMed] [Google Scholar]
- 61.Wilson MR, Reske JJ, Holladay J, Neupane S, Ngo J, Cuthrell N, Wegener M, Rhodes M, Adams M, Sheridan R, et al. (2020). ARID1A Mutations Promote P300-Dependent Endometrial Invasion through Super-Enhancer Hyperacetylation. Cell Rep 33, 108366. 10.1016/j.celrep.2020.108366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aslam M, Troidl C, Tanislav C, Rohrbach S, Gunduz D, and Hamm CW (2019). Inhibition of Protein Prenylation of GTPases Alters Endothelial Barrier Function. Int J Mol Sci 21. 10.3390/ijms21010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baidoo EEK, Wang G, Joshua CJ, Benites VT, and Keasling JD (2019). Liquid Chromatography and Mass Spectrometry Analysis of Isoprenoid Intermediates in Escherichia coli. Methods Mol Biol 1859, 209–224. 10.1007/978-1-4939-8757-3_11. [DOI] [PubMed] [Google Scholar]
- 64.Wei TT, Lin YT, Chen WS, Luo P, Lin YC, Shun CT, Lin YH, Chen JB, Chen NW, Fang JM, et al. (2016). Dual Targeting of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase and Histone Deacetylase as a Therapy for Colorectal Cancer. EBioMedicine 10, 124–136. 10.1016/j.ebiom.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu S, Fukumoto T, Lin J, Nacarelli T, Wang Y, Ong D, Liu H, Fatkhutdinov N, Zundell JA, Karakashev S, et al. (2021). Targeting glutamine dependence through GLS1 inhibition suppresses ARID1A-inactivated clear cell ovarian carcinoma. Nat Cancer 2, 189–200. 10.1038/s43018-020-00160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin J, Guo D, Liu H, Zhou W, Wang C, Muller I, Kossenkov AV, Drapkin R, Bitler BG, Helin K, and Zhang R (2021). The SETDB1-TRIM28 Complex Suppresses Antitumor Immunity. Cancer Immunol Res 9, 1413–1424. 10.1158/2326-6066.CIR-21-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Du Y, Huang Q, Arisdakessian C, and Garmire LX (2020). Evaluation of STAR and Kallisto on Single Cell RNA-Seq Data Alignment. G3 (Bethesda) 10, 1775–1783. 10.1534/g3.120.401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20, 163–172. 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yuan Z, Kang G, Ma F, Lu W, Fan W, Fennessey CM, Keele BF, and Li Q (2016). Recapitulating Cross-Species Transmission of Simian Immunodeficiency Virus SIVcpz to Humans by Using Humanized BLT Mice. J Virol 90, 7728–7739. 10.1128/JVI.00860-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Somasundaram R, Connelly T, Choi R, Choi H, Samarkina A, Li L, Gregorio E, Chen Y, Thakur R, Abdel-Mohsen M, et al. (2021). Tumor-infiltrating mast cells are associated with resistance to anti-PD-1 therapy. Nat Commun 12, 346. 10.1038/s41467-020-20600-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin J, Liu H, Fukumoto T, Zundell J, Yan Q, Tang CA, Wu S, Zhou W, Guo D, Karakashev S, et al. (2021). Targeting the IRE1alpha/XBP1s pathway suppresses CARM1-expressing ovarian cancer. Nat Commun 12, 5321. 10.1038/s41467-021-25684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McCarty KS Jr., Szabo E, Flowers JL, Cox EB, Leight GS, Miller L, Konrath J, Soper JT, Budwit DA, Creasman WT, and et al. (1986). Use of a monoclonal anti-estrogen receptor antibody in the immunohistochemical evaluation of human tumors. Cancer Res 46, 4244s–4248s. [PubMed] [Google Scholar]
- 74.Huang Z, Zhou W, Li Y, Cao M, Wang T, Ma Y, Guo Q, Wang X, Zhang C, Zhang C, et al. (2018). Novel hybrid molecule overcomes the limited response of solid tumours to HDAC inhibitors via suppressing JAK1-STAT3-BCL2 signalling. Theranostics 8, 4995–5011. 10.7150/thno.26627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The previously published ChIP-seq data that we reanalyzed here are available in the Gene Expression Omnibus (GEO) database under accession codes GSE12006040. Previously published RNA-seq data that we reanalyzed here are available under accession codes GSE13191759, GSE14847361 and GSE13113242. The previous published mass spectrometry proteomics data31 is available from ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) with dataset identifier PXD004570. Mutation status for cell lines and their genes inhibition dependency scores from Project Achilles were downloaded from DepMap portal (https://depmap.org/portal/download, dataset DepMap Public 20Q4 v2) and average dependency scores for cell lines from the same tissue origin were used for the analysis.
This paper does not report original code.
All other data supporting the findings of this study are available from the corresponding author on reasonable request.