

Abstract
The number of FDA-approved protein drugs (biologics), such as antibodies, antibody–drug conjugates, hormones, and enzymes, continues to grow at a rapid rate; most of these drugs are used to treat diseases of the peripheral body. Unfortunately, most of these biologics cannot be used to treat brain diseases such as Alzheimer’s disease (AD), multiple sclerosis (MS), and brain tumors in a noninvasive manner due to their inability to permeate the blood–brain barrier (BBB). Therefore, there is a need to develop an effective method to deliver protein drugs into the brain. Here, we report a proof of concept to deliver a recombinant brain-derived neurotrophic factor (BDNF) to the brains of healthy and experimental autoimmune encephalomyelitis (EAE) mice via intravenous (iv) injections by co-administering BDNF with a BBB modulator (BBBM) peptide ADTC5. Western blot evaluations indicated that ADTC5 enhanced the brain delivery of BDNF in healthy SJL/elite mice compared to BDNF alone and triggered the phosphorylation of TrkB receptors in the brain. The EAE mice treated with BDNF + ADTC5 suppressed EAE relapse compared to those treated with BDNF alone, ADTC5 alone, or vehicle. We further demonstrated that brain delivery of BDNF induced neuroregeneration via visible activation of oligodendrocytes, remyelination, and ARC and EGR1 mRNA transcript upregulation. In summary, we have demonstrated that ADTC5 peptide modulates the BBB to permit noninvasive delivery of BDNF to exert its neuroregeneration activity in the brains of EAE mice.
Keywords: BDNF, blood–brain barrier, EAE, neuroregeneration, ADTC5, BBB modulator (BBBM), cadherin peptide
Graphical Abstract
INTRODUCTION
Brain diseases are difficult to diagnose and treat; thus, thousands of individuals suffer from brain diseases annually, including Alzheimer’s disease (AD), multiple sclerosis (MS), and brain tumors (e.g., glioblastoma, medulloblastoma). One of the primary reasons for this difficulty is that the blood–brain barrier (BBB) prevents functional molecules (e.g., drugs) and harmful toxins that are introduced into the bloodstream from entering the brain.1 To cross the BBB, a molecule must possess appropriate physicochemical properties. Furthermore, efflux pumps and enzymes expel and metabolize molecules, respectively, preventing certain molecular species from crossing the BBB. Because of the protective nature of the BBB, its selectivity imposes challenges for scientists to develop diagnostic and therapeutic agents for patients with brain diseases. This is especially apparent for modern biological drugs, such as monoclonal antibodies (mAbs), enzymes, and hormones. The physicochemical properties (i.e., size, charges, high hydrogen-bonding potentials, and hydrophilicity) of biologics prevent them from partitioning into the cell membranes of the BBB vascular endothelial cells and crossing through the cells (i.e., transcellularly).1 Due to their size, these molecules also cannot penetrate between the endothelial cells of the BBB (i.e., paracellularly) because the paracellular pathway’s tight junctions restrict the permeation of molecules with hydrodynamic radius larger than 11 Å.
Most available drugs for treatment of multiple sclerosis (MS) suppress only the immune response to halt the disease; for example, natalizumab (Tysabri) is a monoclonal antibody (mAb) drug that prevents brain infiltration of activated immune cells that could damage the axon myelin sheath.2 Another widely prescribed treatment, glatiramer acetate (Copaxone), has a mechanism of action that is not entirely understood, but it is believed to ameliorate the disease by modulating Th1 to Th2 cells responses.3 Although these current drugs may halt MS disease progression, they do not reverse neuronal damage in the central nervous system (CNS). Although many researchers are currently investigating potential drugs for remyelination, no available drugs on the market can reverse damage of the myelin sheaths of neuronal axons. It has been shown previously that the extent of MS patient’s disability can be correlated to the levels of the axonal damage in the CNS;4 therefore, a means to reverse axon demyelination could greatly improve MS patients’ quality of life.
One way to reverse MS is to deliver molecules that can repair demyelination and/or neuronal damage to the CNS. Monoclonal antibodies (mAb) such as anti-Nogo-A,5 anti-LINGO-1,6 sHIgM22,7 and VX15/25038 have been developed for inducing remyelination. Unfortunately, clinical trials for several of these mAbs, including anti-Nogo-A and anti-LINGO-1, have been terminated—anti-LINGO-1 mAb for lack of significant therapeutic efficacy, and anti-Nogo-A for reasons that have not been released. Alternatively, brain delivery of brain-derived neurotrophic factor (BDNF),9,10 nerve growth factor (NGF),11 and insulin-like growth factor 1 (IGF-1)12 has a potential benefit in reversing neurodegenerative diseases. BDNF stimulates neuron growth and remyelination in cell-culture systems and ex vivo brain slices and in vivo.9,10 Direct delivery of BDNF via intracerebroventricular (ICV) injection to the lateral ventricle of brains of adult rats generates new neurons in the olfactory bulb, thalamus, hypothalamus, and parenchyma-striatum.13 Remyelination in the brains of diseased animals allows the recovery of physical function, demonstrating a correlation between remyelination and recovery from disease symptoms in the EAE animal model.14,15 While intravenous (iv) administration of neuroregenerative proteins (e.g., BDNF, NGF) would be more practical, many attempts to deliver these proteins directly across the BBB via the systemic circulation have not proven successful in inducing neuroregeneration/repair because they cannot cross the BBB.16 Therefore, there is an urgent need to develop effective and noninvasive trans-BBB delivery methods for supplying remyelinating neurotrophic factors into the brain.
One alternative strategy to deliver drugs including proteins into the brain is via the paracellular pathway (i.e., intercellular junctions) of the BBB. In this case, modulation of intercellular junctions of the BBB can increase porosity of the paracellular pathways of the BBB. The osmotic method using a hypertonic mannitol solution has been successful in modulating intercellular junctions.17 The hypertonic solution shrinks the BBB endothelial cells to increase the porosity of the paracellular pathway and allows anticancer drugs, including mAbs, to cross the BBB to treat brain tumors.17 Alternatively, our group has investigated a new approach to modulating the BBB using cadherin peptides. In this case, cadherin peptides were designed to inhibit cadherin–cadherin interactions in an equilibrium and dynamic fashion to increase the porosity of the paracellular pathway.1,18 In healthy animals, in vivo cadherin peptides (e.g., HAV6: Ac-SHAVSS-NH2 and ADTC5: Cyclo-(1,7)Ac-CDTPPVC-NH2) have been shown to deliver small molecules, peptides, and proteins (e.g., galbumin) to the brain by modulating the BBB for a very short period of time.19–22 Recently, HAV6 and ADTC5 peptides have been shown to deliver various sizes of protein (i.e., 15 kDa lysozyme, 65 kDa albumin, 150 kDa IgG mAb) into the brain of healthy C57BL/6 mice when both BBB modulator and protein were delivered via iv administration.23 Second, novel cyclic HAV and ADT peptides have been designed to improve the BBB modulatory activity to deliver IgG mAb into the brain of C57BL/6 mice.24 Thirdly, a combination of HAV6 and an anticancer drug, adenanthin, was effective in suppressing medulloblastoma brain tumor growth and increasing the survival rate of the mice.25 Finally, HAV6 peptide improved oral absorption and brain delivery of eflornithine by improving its paracellular permeation.26
In this study, BDNF (13 kDa monomer) was delivered to the brains of relapsing-remitting experimental autoimmune encephalomyelitis (RR-EAE) mice using ADTC5 peptide via iv administrations to induce remyelination and neurorepair as a less invasive method compared to ICV.13 Four different groups of EAE mice were treated eight times with BDNF + ADTC5, BDNF alone, ADTC5 alone, or vehicle during the remission period of EAE. Therapeutic effects of delivering BDNF in vivo were evaluated by observing the amelioration of EAE relapse and comparing clinical body scores across treatment groups. Finally, the effects of BDNF in the brains of EAE mice were evaluated using several ex vivo analyses to indicate remyelination and the degree of NG2-glia activity as well as by probing mRNA transcript upregulation of proteins affected by BDNF.
MATERIALS AND METHODS
Animals.
The protocols to use live mice have been approved by the Institutional Animal Care and Use Committee (IACUC) at The University of Kansas. SJL/elite mice were purchased from Charles River Laboratories, Inc. (Wilmington, MA). All mice were housed under specific pathogen-free conditions at the animal facility at The University of Kansas approved by the university Animal Care Unit (ACU). The animals were maintained in the Animal Care Unit with free access to food, water, and rotating stimuli.
Cadherin Peptide Synthesis and Purification.
The syntheses of the ADTC5 and PLP139–151 peptides were accomplished using a solid-phase peptide synthesizer (Gyros Protein Technologies, Tucson, AZ). After peptide cleavage from the resin using TFA, the crude peptides were precipitated overnight in cold diethyl ether. In most cases, the crude precipitate showed high concentrations of the desired peptide. The formation of a disulfide bond in the cyclic peptide (i.e., ADTC5) was accomplished by vigorously stirring the precursor linear peptide in bicarbonate buffer solution under air oxidation at pH 9.0 in high dilution. The cyclization reaction produced primarily the desired monomer with minor oligomer side products; the monomer peptide was isolated from the mixture using a semipreparative HPLC X-bridge C18 column (Waters, Milford, MA). After purification with semipreparative HPLC, the isolated peptides had high purity (>95%) as determined by analytical HPLC. The exact mass of each peptide was determined by mass spectrometry.
EAE Mouse Model.
EAE disease in animals (5- to 8-week-old SJL/elite female mice, Charles River) was stimulated by injecting 200 μg of PLP139–151 peptide in a 0.2 mL emulsion containing equal volumes of phosphate-buffered saline (PBS) and complete Freund’s adjuvant (CFA) with killed mycobacterium tuberculosis strain H37RA (Difco, Detroit, MI; final concentration, 4 mg/mL), as described previously.27,28 Briefly, 50 μL of PLP/CFA emulsion was administered to four different regions above the shoulders and the flanks on day 0 followed by an intraperitoneal injection of 200 ng of pertussis toxin (List Biological Laboratories, Campbell, CA) on days 0 and 2. Clinical scores that reflect the disease progression were determined using an 11-point scale with 0.5 increments ranging from 0 to 5; 0 being no apparent disease and 5 being moribund. On day 21, the mice were randomly separated into four treatment groups: (i) BDNF (5.7 nmol/kg) + ADTC5 (10 μmol/kg; n = 7), (ii) BDNF alone (5.71 nmol/kg, n = 6), (iii) ADTC5 alone (10 μmol/kg; n = 5), and (iv) vehicle (n = 5). All of the mice received eight intravenous injections every 4 days beginning on day 21. The mice were euthanized via CO2 inhalation on day 55. Area under the curve (AUC) calculations were used to compare clinical scores across groups; AUC calculations were performed using the trapezoid rule from days 21 to 55.
Euthanasia, Brain Perfusion, and Extraction.
All of the mice were euthanized via a CO2 chamber. Immediately following euthanasia, the mice underwent cervical dislocation and were transcardially perfused with PBS + 0.2% Tween 20, followed by perfusion fixation with a 4% paraformaldehyde and 30% sucrose PBS solution. Following the fixation, the brains were extracted and postfixed overnight in the perfusion-fixation solution.
Immunohistochemistry.
Fixed brain samples were submitted to IHC World (Ellicott City, MD) for paraffin embedding, tissue sectioning (5 μm), anti-NG2 (Abcam, Cambridge, U.K.) staining via DAB, and Luxol fast blue staining. Staining protocols described on the IHC World website for Luxol fast blue and immunohistochemistry enzyme HRP were performed. For both procedures, brains were cut into 5 μm sections and then deparaffinized and rehydrated using xylenes and an ethanol–water gradient. For Luxol staining, the sections were incubated in Luxol fast blue solution at 56 °C overnight and subsequently rinsed with 95% ethyl alcohol followed by distilled water. For anti-NG2 mAb staining, the sections underwent antigen retrieval, followed by rinsing with PBS-Tween 20 for 2 × 2 min2. The sections were incubated with normal serum block, followed by primary antibody incubation with anti-NG2 mAb at 4 °C overnight and subsequently rinsed with PBS-Tween 20. The sections were then blocked using a peroxidase blocking solution for 10 min at room temperature (RT). Next, the samples were incubated with a biotinylated secondary antibody at 1–10 000 dilution in PBS for 30 min at RT. The sections were then incubated in streptavidin-HRP in PBS for 30 min at RT followed by incubation in DAB solution for 1–3 min. The sections were dehydrated through 95% ethanol for 2 min, 100% ethanol for 2 × 3 min2, and cleared with xylene. The sections were mounted using aqueous mounting media and coverslipped using 1.5 coverslips.
Luxol fast blue and anti-NG2 mAb images were taken under identical conditions on a Zeiss Axioplan 2 microscope (Oberkochen, Germany) equipped with a mercury lamp excitation source, and 40× (Luxol) and 20× (anti-NG2) air objective lenses. Grayscale images for quantification were taken using a 1344 × 1024 Orca ER CCD camera (Hamamatsu Photonics, Japan), and color images for qualitative purposes were taken using a 1.3 MP Spot Color camera (Spot Imaging, Sterling Heights, MI). To determine the degree of demyelination (i.e., breakages in the myelin sheath), five grayscale images from each group were randomly selected and converted to binary, and regions of interest (ROIs) were manually selected within the lateral corpus callosum using ImageJ (National Institute of Health, Bethesda, MD). A binary value of “1” (i.e., white signal) implied a lack of myelin, whereas a binary value of “0” (i.e., black signal) implied myelin. The mean value of each ROI from each image was recorded. To determine the degree of anti-NG2 staining, densitometry analysis was performed on DAB-stained sections; grayscale images were taken under equal exposure times, and five images per group were randomly selected and used for analysis. ROIs of identical size were selected within the medial corpus callosum. The integrated mean gray value for each ROI from each image was recorded. Staining background was controlled for by subtracting an aggregate of mean gray values from five ROIs of negative controls from each group.
Fluorescent in Situ Hybridization.
Coronal brain sections (5 μm thickness) from mid- and hindbrain were sectioned and washed three times in PBS before mounting on gelatin-coated glass slides (Superfrost Plus, Thermo Fisher Scientific). Tissue was allowed to dry at RT and then stored at −20 °C until use. Fluorescent in situ hybridization (FISH) was performed using RNAscope Technology 2.0, Advanced Cell Diagnostics (ACD), (Hayward, CA) Multiplex Reagent Kit V2.29–31 In short, mounted tissue sections were deparaffinized using xylene and serially dehydrated in 50, 70, 95, and 100% ethanol for 5 min each. In between all pretreatment steps, tissue sections were briefly washed with nanopure water. Pretreatment solution 1 (hydrogen peroxide reagent) was applied for 10 min at RT and then the tissue sections were boiled in pretreatment solution 2 (target retrieval reagent) for 15 min. Mounted slices were pretreated with solution 3 (protease reagent) for 30 min at 40 °C in the HybEz hybridization system (ACD). Following tissue pretreatment, the following transcript probes were applied to all sections: Mm-EGR1-C1 (Cat. # 423371), Mm-NOS1-C2 (Cat. # 437651-C2), and Mm-ARC-C3 (Cat. # 316911-C3), which correspond to early growth response 1 (EGR1), nitric oxide synthase 1 (NOS1), and activity-related cytoskeleton-associated protein (ARC). Probes were hybridized to sections for 2 h at 40 °C and then subsequently washed for 2 min at room temperature. Following hybridization, hybridize AMP 1 was applied to each slide, which was then incubated for 30 min at 40 °C. The same process was repeated for hybridize AMP 2 and 3. For HRP-C1 signal development (EGR1), HRP-C1 was applied to each slide, which was incubated for 15 min at 40 °C and then washed. For C1, TSA Plus fluorescein (PerkinElmer, Akron, OH) was applied and incubated for 30 min at 40 °C and then washed. Following the wash, a HRP blocker was applied to each slide, which was incubated for 15 min at 40 °C and then washed. This process was repeated for C2 (NOS1) and C3 (EGR1) using TSA Plus Cy3 and Cy5, respectively. The resulting transcript-fluorophore labeling is as follows: EGR1-fluorescein, NOS1-Cy3, ARC-Cy5. All sections were counterstained by incubating DAPI for 30 s at RT followed by rinsing. Slides were then covered using ProLong Gold Antifade Mountant and 1.5 coverslips. The slides were allowed to dry in the dark overnight at 4 °C. All sections were imaged within 2 weeks.
Fluorescent images were taken using an Olympus Inverted Epifluorescence Microscope XI81 (Olympus Life Solutions, Waltham, MA) running SlideBook Version 5.5 (3i, Ringsby, CT) equipped with a digital CMOS camera (2000 × 2000), automatic XYZ stage position, ZDC autofocus, and a xenon lamp excitation source. Images were taken using a 20× objective and appropriate filter sets for each fluorophore (i.e., DAPI, FITC, Cy3, C5). To determine the degree of mRNA transcript expression, five images of analogous regions of the cerebral cortex were randomly selected from mouse samples of each group, and the total number of cells expressing each mRNA transcript were counted using ImageJ. The number of cells expressing each mRNA transcript was normalized against the total number of cells (as determined by DAPI) to ensure that analyzed areas had equal cell density. For display purposes, images were pseudo-colored using ImageJ; green was assigned to fluorescein (EGR1), magenta was assigned to Cy5 (ARC), and blue to DAPI. NOS images were not incorporated due to virtually no signal detection.
Western Blots.
Female SJL/elite mice (5 weeks of age, Charles River) were initially intravenously injected via the lateral tail vein with 5.71 nmol/kg BDNF (Peprotech, Rocky Hill, NJ) with (n = 3) or without (n = 3) 10 μmol/kg ADTC5. BDNF was allowed to circulate for 20–30 min prior to euthanasia via CO2. Immediately following euthanasia, the mice were transcardially perfused with protease inhibitor infused Tris buffer (pH 7.4). The brains of the mice were extracted and placed in the perfusate buffer on ice. For Western blotting, 100–150 mg of brain tissue was sectioned from the most ventro-posterior portion of the brain and placed in 200–250 μL of solution mixture containing 66% tissue protein extraction reagent (TPER; Thermo Fisher, Waltham, WA) and 50 μL of 33% neural protein extraction reagent (NPER; Thermo Fisher) with protease and phosphatase inhibitors (Thermo Fisher). The tissue samples were lysed via sonication using a Sonic Disembrator 500 (Thermo Fisher) at an amplitude level of 15 Hz for a maximum of 10 s. Following sonication, the samples were vortexed for 1 min and then centrifuged at 4 °C and 13 000 rpm for 30 min. The sonication, vortexing, and centrifugation were repeated two times. Following lysis and centrifugation, NuPAGE 4–12% Bis-Tris Protein Gels (1.5 mm, 10-well, Thermo Fisher) were loaded with 60 μg of protein and Licor (Lincoln, NE) loading buffer. A BDNF standard of less than 1.0 μg was also loaded for positive control. The gel was run at 100 V for 2 h. Following the gel, the protein bands were transferred to a nitrocellulose membrane (Licor) at 36 V overnight. Following the transfer, the membrane was stained with REVERT (Licor) for 3 min and then washed using the REVERT Wash Solution for 2 min, followed immediately by scanning using a Licor Odyssey at 700 nm. Next, the membrane was washed using the REVERT Reversal Solution (Licor) and subsequently blocked for 2 h at 4 °C using Licor TBS blocking reagent. The membrane was then incubated with the primary antibody, anti-BDNF (Abcam), at a 1:1000 ratio in TBS + 0.1% Tween 20 for 36 h at 4 °C. Following primary antibody, the membrane was rinsed and incubated with the IR800-conjugated secondary antibody (Licor) for 1.5 h at room temperature in the dark. The membrane was then immediately scanned using a Licor Odyssey CLX at a wavelength of 800 nm. Following imaging of BDNF bands on the membrane, the membrane was stripped using stripping buffer to be reprobed for the phosphorylated-TrkB (pTrkB) receptor with anti-phospho-TrkB (EMD Millipore, Burlington, MA) at a 1:1000 dilution in TBS + 0.1% Tween 20 for 24 h at 4 °C. Following primary antibody incubation, the membrane was rinsed and incubated with the IR800-conjugated secondary antibody for 1.5 h at room temperature in the dark. The membrane was then immediately scanned using the same parameters as for the BDNF imaging. These bands were not densiometrically analyzed due to high background signal; however, they are shown for qualitative analysis.
To improve the level of detection of BDNF and pTrkB bands via Western blot, the above process was repeated with an increase in dosages of BDNF. The dosages of ADTC5 remained constant; the mice received 57.1 nmol/kg BDNF (10-fold increase) + 10 μmol/kg ADTC5 (n = 2), 28.6 nmol/kg BDNF (5-fold increase) + 10 μmol/kg ADTC5 (n = 1), or 28.6 nmol/kg BDNF alone (5-fold increase; n = 3). These images were not quantified due to the variation in dosing regiments; however, they are provided for qualitative analysis of BDNF brain depositions.
Statistics.
All statistics were performed using GraphPad Prism (San Diego, CA). Analysis of variance (ANOVA) and Student’s t-test were performed when appropriate, both operating at 95% confidence intervals with a p-value of less than 0.05 used as the criterion for statistical significance unless otherwise stated.
RESULTS
Effect of BDNF Brain Delivery by ADTC5 on Suppression of EAE Relapse.
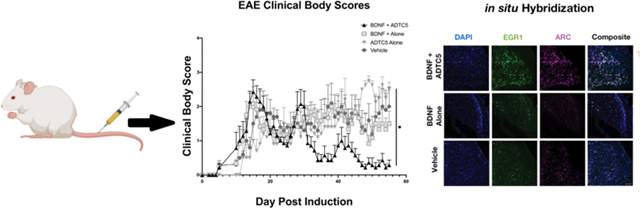
The ability of ADTC5 to deliver BDNF into the brains of mice after iv administrations was assessed by determining the effects of BDNF in suppressing disease relapse in the relapsing-remitting EAE animal model. The efficacy of BDNF (5.71 nmol/kg) + ADTC5 (10 μmol/kg; n = 7) was compared to that of BDNF alone (5.71 nmol/kg; n = 6), ADTC5 alone (10 μmol/kg; n = 5), and vehicle (n = 5). Intravenous injections were performed every 4 days up to eight injections starting from day 21 during the time of disease remission and relapse. EAE clinical scores were monitored daily from the beginning to the end of the study. The EAE mice that received injections of BDNF + ADTC5 had clinical body scores significantly lower over time compared to the mice that received BDNF alone, ADTC5 alone, or vehicle (Figure 1A). The mice that received injections of BDNF + ADTC5 showed normal locomotion on all four limbs, with some residual tail paralysis. In contrast, the mice that received BDNF alone, ADTC5 alone, or vehicle showed partial or full hind leg paralysis and full tail paralysis.
Figure 1.
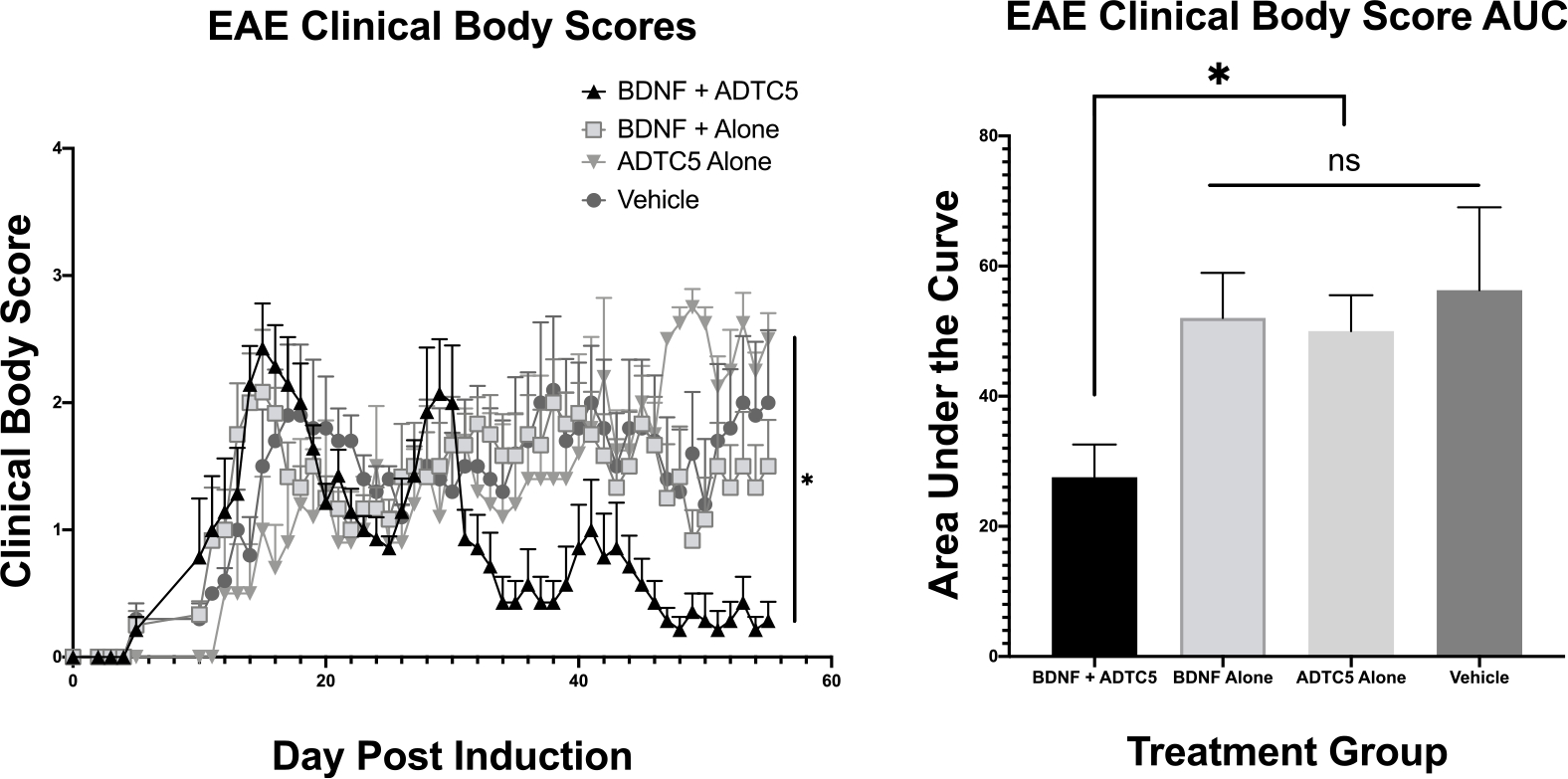
Effect of treatment of SJL/elite EAE mice, an animal model for MS, with BDNF (5.71 nmol/kg) + ADTC5 (10 μmol/kg; n = 7), BDNF alone (5.71 nmol/kg; n = 6), ADTC5 alone (10 μmol/kg; n = 5), or vehicle (n = 5) during remission on days 21, 25, 29, 33, 37, 41, 45, and 48. (A) Clinical disease score vs time of mice treated eight times with BDNF + ADTC5, BDNF alone, ADTC5 alone, or vehicle. (B) Comparison of area under the curve (AUC) of the disease scores from days 21 to 55 from EAE mice treated with BDNF + ADTC5, BDNF alone, ADTC5 alone, or vehicle. *p ≤ 0.05; one-way ANOVA (95% confidence).
The differences in clinical body scores were distinguished by generation of the areas under the curve (AUC) disease scores of all four groups from day 21 to day 55, after the peak of the disease. It was found that the mice that received injections of BDNF + ADTC5 had significantly lower ACU disease scores compared to those that received BDNF alone, ADTC5 alone, or vehicle (F(3,9) = 3.180; p ≤ 0.05; Figure 1B). There was no significant difference in the clinical scores between treatments with BDNF alone, ADTC5 alone, and PBS (F(2,13) = 0.128; p = 0.881). The results suggest that ADTC5 helps BDNF to penetrate the BBB to exert its biological activity in the brain while BDNF alone did not have efficacy due to its inability to penetrate the BBB. Further evaluation of the therapeutic efficacy of systemically delivered BDNF using ADTC5 peptide was assessed using histological, immunohistochemical, and hybridization methods.
Effect of BDNF on Remyelination.
The ability of BDNF to induce remyelination has been previously demonstrated using BDNF knockout mice in which myelin loss was shown to be sensitive to a lack of BDNF expression.32 Additionally, BDNF has been shown to improve remyelination and regeneration of nerve fibers after C7 ventral root avulsion and replantation.33 Thus, we probed myelin levels in the brains of mice as an indication that BDNF is successfully entering the CNS and exerting an effect. Myelin levels in the brain were imaged using Luxol fast blue chromogen staining. Figure 2A shows a notably more dense myelin staining in the lateral corpus callosum in mice that received BDNF + ADTC5 (n = 5) compared to those that received BDNF alone (n = 5) or vehicle (n = 5). The mice that received BDNF alone or vehicle showed myelin discontinuity (white spaces) in the corpus callosum. Quantification via densitometry using binary images of myelin staining showed a statistically significant increase in myelin density (decrease in white spaces) in the lateral corpus for the mice that received BDNF + ADTC5 compared to those that received BDNF alone or vehicle (F(2,12) = 21.72; p ≤ 0.001, Figure 2B). This result supports the idea that BDNF successfully entered the brain with the help of ADTC5 and induced remyelination in the corpus callosum.
Figure 2.
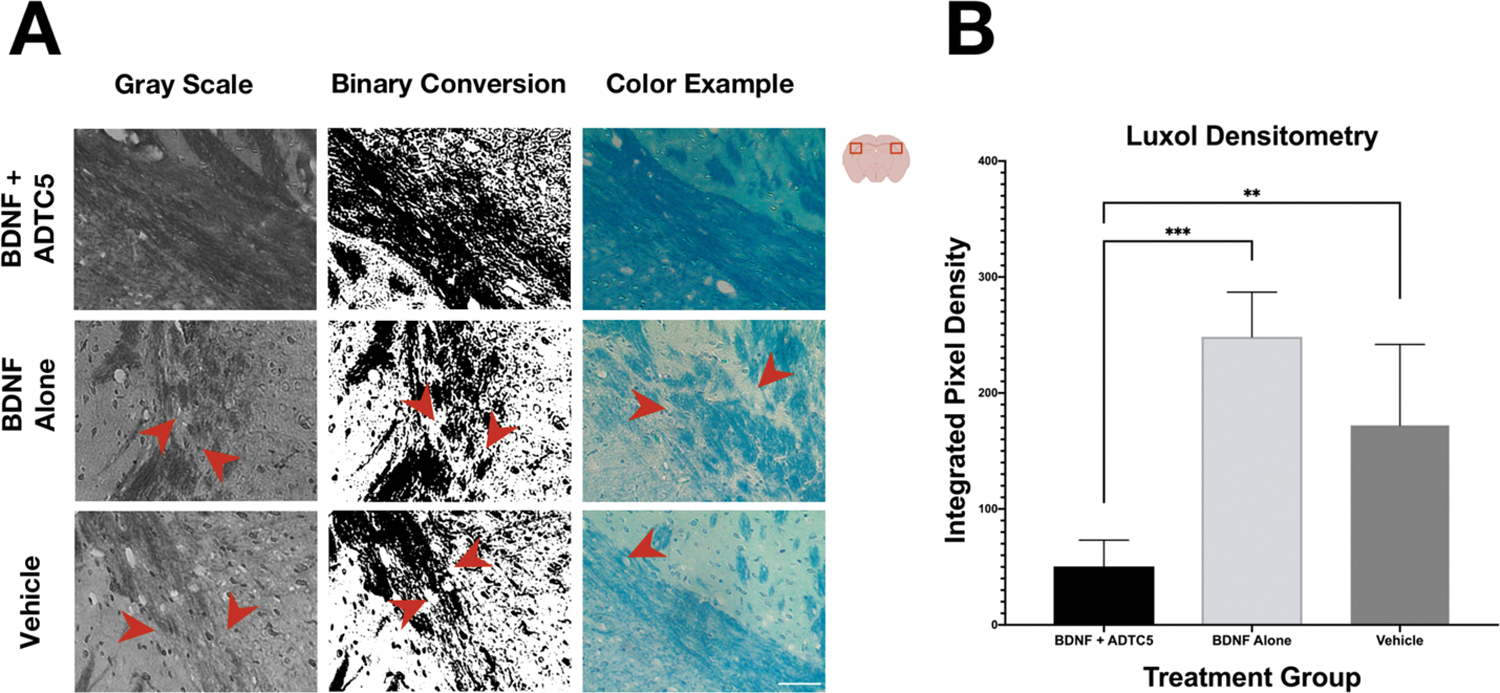
Effects of BDNF (5.71 nmol/kg) + ADTC5 (10 μmol/kg), BDNF alone (5.71 nmol/kg), or vehicle treatments on remyelination in the lateral corpus callosum and surrounding cortex of the brains of SJL/elite EAE mice as stained by Luxol fast blue. (A) Grayscale, the binary conversion, and color photomicrograph of myelin images taken under identical exposure of the lateral corpus callosum of EAE mice treated with BDNF + ADTC5, BDNF alone, or vehicle; the red arrows indicate breakages in the myelin. (B) Quantitative myelin densiometric comparison of white spaces (demyelination) in the brain of BDNF + ADTC5, BDNF alone, and vehicle-treated EAE mice; scale bar = 50 μm; **p ≤ 0.01 ***p ≤ 0.001; one-way ANOVA (95% confidence; n = 5).
Effect of BDNF on NG2-Glia.
The NG2 receptors have previously been shown to facilitate the maturation of oligodendrocyte precursor cells and have been demonstrated to be distinctly upregulated in the BDNF+/+ mice following the development of cuprizone-induced lesions.32,34 We further probed NG2 receptor presence as an additional indicator that BDNF is indeed entering the CNS and exerting a therapeutic effect. NG2 receptor levels were quantified using anti-NG2 immunohistochemistry staining. A higher degree of NG2 staining in the medial corpus callosum of the mice was found in animals that received BDNF + ADTC5 (n = 5) compared to those that received BDNF alone (n = 5) or vehicle (n = 5; Figure 3A). Quantification of the degree of NG2 staining was determined using mean gray values. The mice that received BDNF + ADTC5 showed a significantly increased level of anti-NG2 staining compared to those that received BDNF alone or vehicle (F(2,12) = 10.44, p ≤ 0.01; Figure 3B). These results are evidence that BDNF is inducing oligodendrocyte maturation and, in turn, remyelination.
Figure 3.
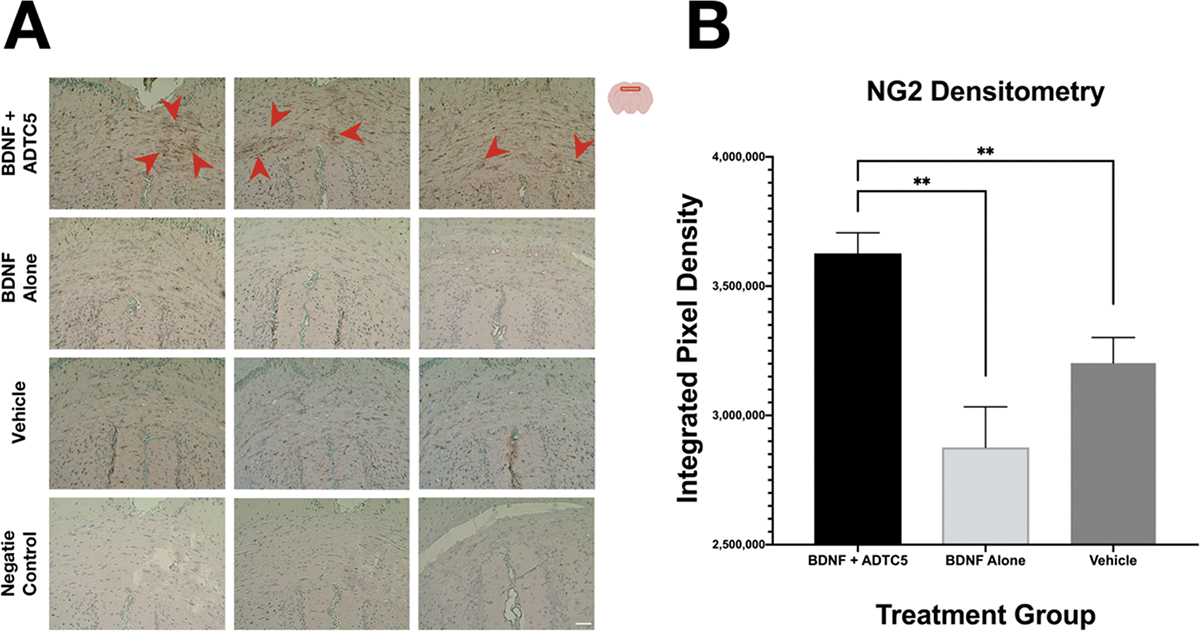
Effects of BDNF (5.71 nmol/kg) + ADTC5 (10 μmol/kg), BDNF alone (5.71 nmol/kg), or vehicle treatments in the presence of NG2 receptor in the medial corpus callosum of brains of SJL/elite EAE mice as stained by DAB. (A) Color photomicrograph of anti-NG2 staining (brown) taken under identical conditions from the medial corpus callosum for mice treated with BDNF + ADTC5, BDNF alone, vehicle; the red arrows point to dense regions of activated NG2-glia. (B) Quantitative NG2 density comparison among the EAE mice treated with BDNF + ADTC5, BDNF alone, and vehicle; scale bar = 50 μm; **p ≤ 0.01; one-way ANOVA (95% confidence; n = 5).
Effects of BDNF on EGR1, ARC, and NOS1 mRNA Transcript Expression.
BDNF exposure is well known to affect downstream transcription factors including c-fos, cAMP response element binding protein (CREB), early growth response 1 (EGR1), and EGR3.35,36 Furthermore, EGR1 has been demonstrated to target the activity-regulated ARC gene, and EGR1 is also upregulated by BDNF exposure.37,38 In addition, BDNF has not only been shown to upregulate specific downstream transcripts, but has also been shown to inhibit the expression of nitric oxide synthase 1 (NOS1).39 Therefore, we probed three mRNA transcripts, EGR1, ARC, and NOS1 for evidence that BDNF is entering the brain and exhibiting effects. The mRNA expression levels of EGR1, ARC, and NOS1 mRNA were quantified using fluorescent in situ hybridization (FISH). Figure 4A,B shows brain sections from the mid and hindbrain, respectively; the mice that received BDNF + ADTC5 have a notable upregulation of EGR1 and ARC mRNA transcripts compared to the mice that received BDNF alone or vehicle. However, images for the NOS1 mRNA expressions are not shown due to low level of detectability. The mRNA expression levels were quantified using cell counting that was normalized against the number of cell nuclei to ensure that analyzed areas were of equal cell density. Composite images of all fluorescent channels showed a pronounced increase in mRNA transcripts that can be seen for the mice that received BDNF + ADTC5 (n = 5) compared to the mice that received BDNF alone (n = 5) or vehicle (n = 5; Figure 4A,B). Figure 4C shows a significant increase in EGR1 (F(2,12) = 47.10; p ≤ 0.001) and ARC (F(2,12) = 33.43; p ≤ 0.001) expression levels for the mice that received BDNF + ADTC5 compared to those of the mice that received BDNF alone or vehicle. In contrast, there was no significant difference in NOS (F(2,12) = 1.826; p = 0.203) or DAPI (F(2,12) = 0.504; p = 0.617) staining across the three groups (Figure 4D).
Figure 4.
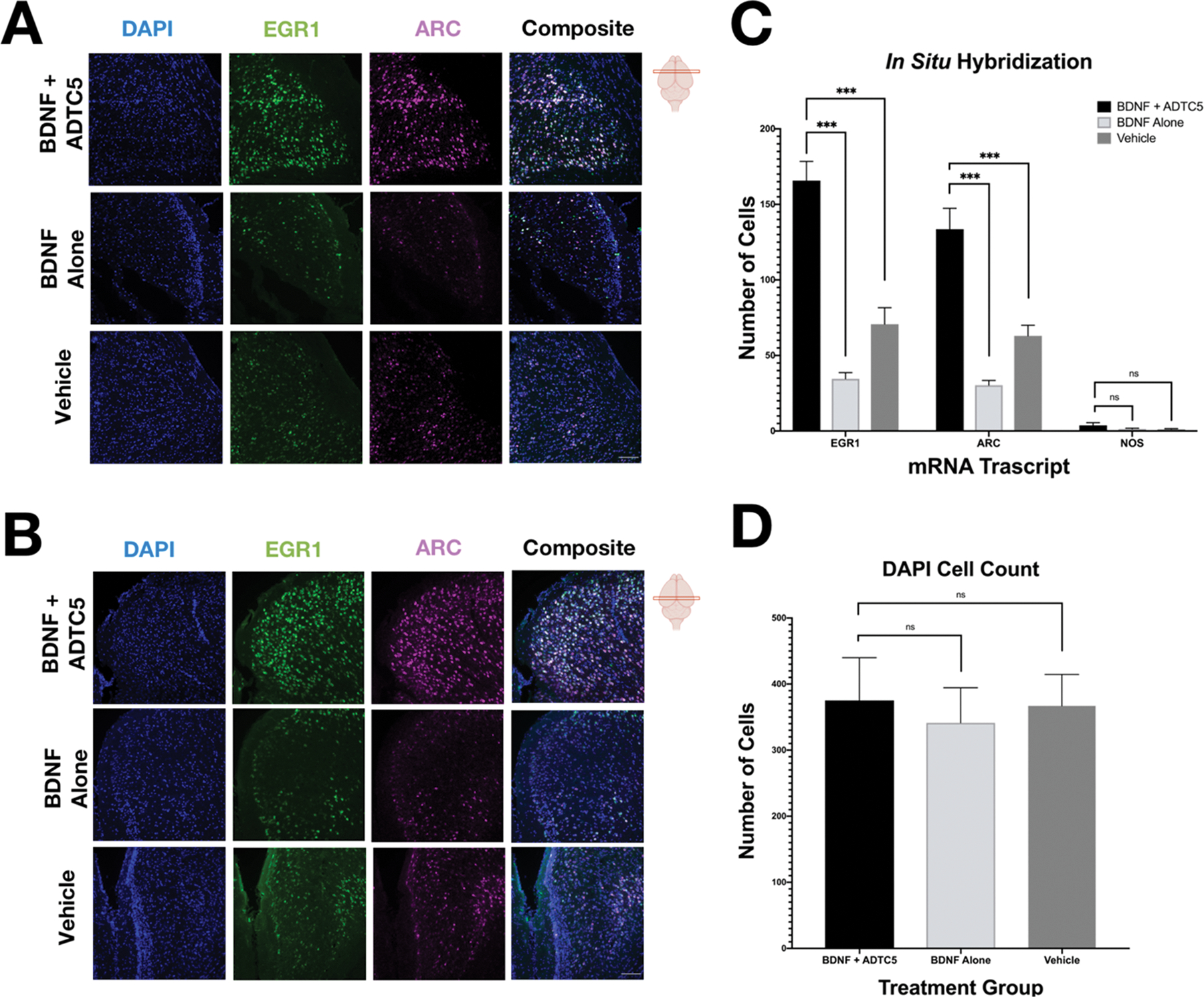
Effects of BDNF (5.71 nmol/kg) + ADTC5 (10 μmol/kg), BDNF alone (5.71 nmol/kg), or vehicle treatments on mRNA expression of EGR1 and ARC in the cortex of the brains of SJL/elite EAE mice. (A, B) Photomicrograph of DAPI (blue), EGR1 (green), ARC (magenta), and composite images taken of the cortex of the midbrain (A) and hindbrain (B) of EAE mice treated with BDNF + ADTC5, BDNF alone, or vehicle. (C) Quantitative comparison of EGR, ARC, and NOS1 mRNA transcript expression, as determined by cell count, for mice treated with BDNF + ADTC5, BDNF alone, or vehicle. (D) Quantitative comparison of DAPI cell count; scale bar = 50 μm; ***p ≤ 0.001; one-way ANOVA (99% confidence; n = 5). Contrast and brightness of images were adjusted only for display purposes.
Detection of BDNF in the Brain Using Western Blots.
The ability of ADTC5 to deliver BDNF into the brain was confirmed by Western blot analysis of the brain homogenates. To determine if BDNF entered the brain using the ADTC5 peptide, the mice were initially given a 5.71 nmol/kg BDNF injection with (n = 3) or without (n = 3) 10 μmol/kg ADTC5 and were sacrificed after 20 min to allow for sufficient circulation and activation of the pTrkb pathway. Figure 5A shows a notable increase in detection of BDNF bands in the brains of mice that received injections of BDNF + ADTC5 compared to those that received BDNF alone, where delivered BDNF was undetected. Because of high background, pTrkB could not be detected with confidence using this Western blot.
Figure 5.
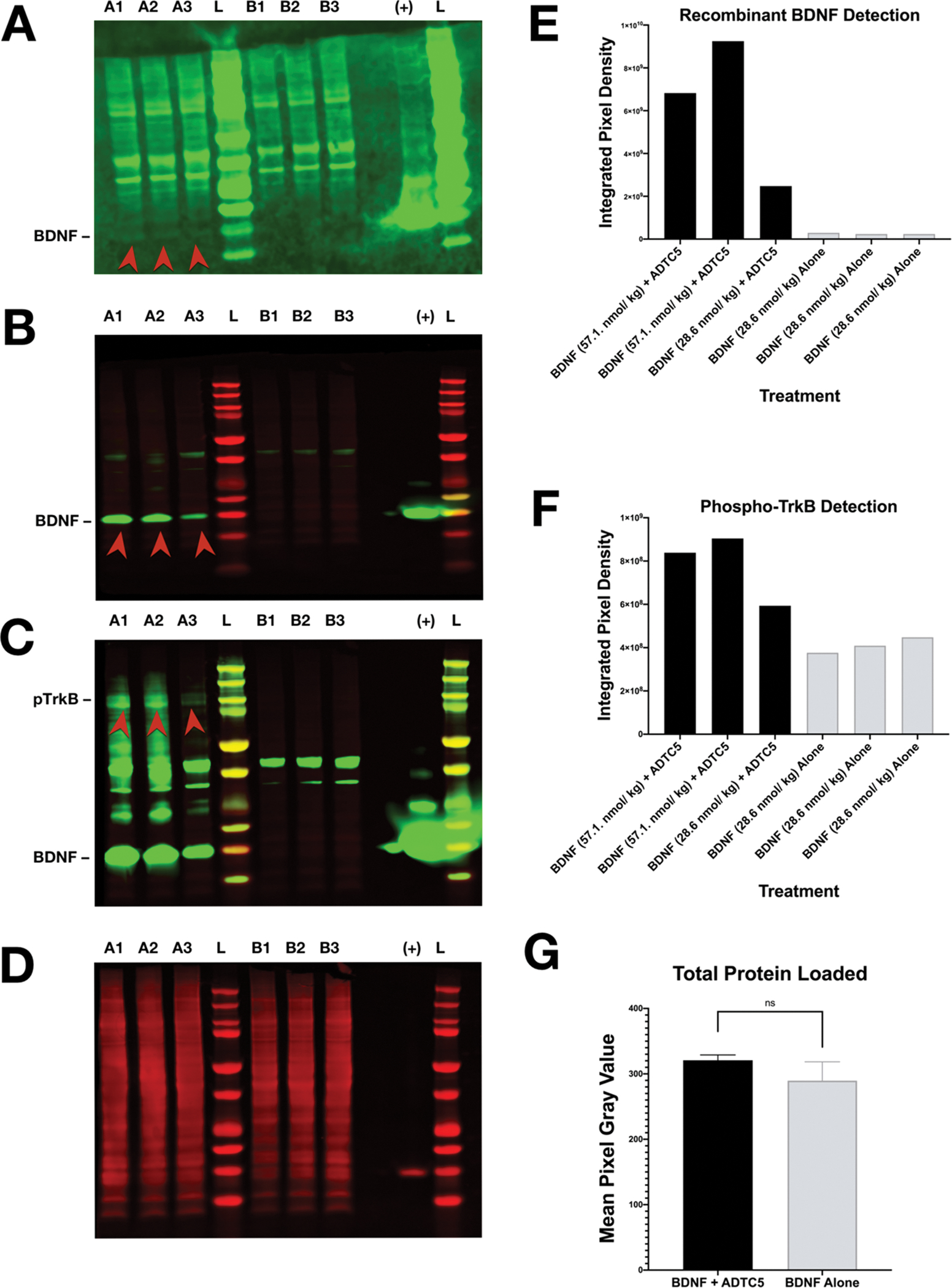
Western blot detection of recombinant BDNF and pTrkB from mice treated with either BDNF + ADTC5 or BDNF alone. (A) Western blot probing for recombinant BDNF in the brains of mice that received BDNF (5.71 nmol/kg) + ADTC5 (10 μmol/kg; A1, A2, A3) or BDNF alone (5.71 nmol/kg, B1, B2, B3); “L” represents molecular weight ladder; “+” represents the positive control of recombinant BDNF; the red arrows highlight increased recombinant BDNF detection. (B) Western blot probing for recombinant BDNF after dosage increase in healthy mice that received BDNF (57.1 nmol/kg) + ADTC5 (10 μmol/kg; A1, A2), BDNF (28.6 nmol/kg) + ADTC5 (10 μmol/kg; A3), or BDNF alone (28.6 nmol/kg; B1, B2, B3); the red arrows highlight increased recombinant BDNF detection. (C) Western Blot probing for pTrkB after dosage increase of healthy mice that received BDNF (57.1 nmol/kg) + ADTC5 (10 μmol/kg; A1, A2), BDNF (28.6 nmol/kg) + ADTC5 (10 μmol/kg; A3), or BDNF alone (28.6 nmol/kg; B1, B2, B3); the red arrows highlight increased pTrkB detection. (D) Total protein stain (loading control) for samples treated with BDNF 57.1 nmol/kg or 28.6 nmol/kg in (B) and (C). (E) Graphical representation of recombinant BDNF detection level in mice that received BDNF (57.1 nmol/kg) + ADTC5 (10 μmol/kg; A1, A2), BDNF (28.6 nmol/kg) + ADTC5 (10 μmol/kg; A3), or BDNF alone (28.6 nmol/kg; B1, B2, B3). (F) Graphical representation of pTrkB detection level for mice that received BDNF (57.1 nmol/kg) + ADTC5 (10 μmol/kg; A1, A2), BDNF (28.6 nmol/kg) + ADTC5 (10 μmol/kg; A3), or BDNF alone (28.6 nmol/kg; B1, B2, B3). (G) Graphical representation of total protein loaded among all groups. Contrast and brightness of images were adjusted only for display purposes.
Due to suboptimal detection of pTrkB using 5.71 nmol/kg BDNF injections, the above process was repeated with increases in dosage of BDNF to 57.1 nmol/kg but with the dosages of ADTC5 remaining constant. The mice received 57.1 μmol/kg BDNF (10-fold increase) + 10 μmol/kg ADTC5 (n = 2), 28.6 nmol/kg BDNF (5-fold increase) + 10 μmol/kg ADTC5 (n = 1), or 28.6 nmol/kg BDNF alone (5-fold increase; n = 3). Figure 5B,C more clearly shows an increase in detection of BDNF and pTrkB bands for the mice that were treated with BDNF + ADTC5 compared to the mice that were treated with BDNF alone. Additionally, to ensure that total protein loaded into each well across all groups was consistent, a total protein stain was performed (Figure 5D); this serves as a more reliable and accurate loading control in comparison to detecting a ubiquitous protein such as actin. There was no significant difference in the total protein loading across each group (t(4) = 1.808; p = 0.145). Due to the variation of dosages of BDNF administered, the densiometric BDNF and pTrkB bands cannot be statistically compared to confidence; however, the relative intensities are shown in Figure 5E,F. The aggregate results of these two Western blots indicate that BDNF is successfully entering the CNS and inducing an immediate effect on upregulation of pTrkB.
DISCUSSION
Here, we have demonstrated that multiple injections of BDNF + ADTC5 peptide during the remission period of the relapsing-remitting EAE mice suppressed the disease relapse compared to treatment with BDNF alone, ADTC5 alone, or vehicle. BDNF was selected in this study because it is an endogenous molecule; thus, its delivery may not cause adverse side effects in the EAE mice. There is a significant improvement of the disease clinical scores during remission when the EAE mice were treated with BDNF + ADTC5 compared to those treated with BDNF alone, ADTC5 alone, and vehicle (Figure 1A,B). A combination of BDNF + ADTC5 induced remyelination of the axons to reverse the neuronal damage caused by the immune cells (Figure 2A,B). As expected, administering BDNF alone, ADTC5 alone, or vehicle did not have any effect on suppressing the disease relapse. BDNF alone did not induce remyelination because BDNF alone could not penetrate the BBB. In addition, ADTC5 alone has no inherent neuroregenerative properties. The suppression of disease symptoms in the treatment group with BDNF + ADTC5 indirectly suggests that ADTC5 helps to deliver BDNF into the brain (Figure 1). This is also supported by the detection of BDNF and the upregulation of pTrkB receptors after its delivery with ADTC5 (Figure 5).
Our previous study showed that the effect of ADTC5 alone on the integrity the BBB intercellular junctions in vivo was reversible in healthy mice. Using transmission electron microscopy (TEM), evaluation of the BBB endothelial microvessels 2 h after treatment with ADTC5 showed that the morphology of the endothelial cells was similar to that in the vehicle-treated mice.20 ADTC5-treated mice have no change in appearance of the brain capillaries, and their tight junctions have normal ultrastructural characteristics. The vehicular activity in the vascular endothelial cells appears normal 2 h after ADTC5 treatment. In a parallel study, in vivo modulation of the BBB using a cadherin peptide, HAV6 peptide, in the mice did not upregulate ionized calcium binding adapter molecule 1 (Iba1), which is a marker for microglia activation and glial fibrillary acidic protein (GFAP), which is a marker of astrogliosis.25 This suggests that cadherin peptides may not induce neuroinflammation. This is different from the effects of osmotic BBB modulation, which shows altered morphology and disruption of the tight junction ultrastructure as well as potential astrogliosis.40–42 In addition, magnetic resonance imaging (MRI) studies showed that HAV6 did not influence blood flow in the brain; thus, the activity of HAV6 to enhance delivery of molecules into the brain was not due to the change in blood flow into the brain.21
Each BBBM (i.e., HAV6 and ADTC5) modulates the BBB with a certain duration of time of opening for a certain size of molecules.20–22 In this case, the durations of BBB opening created by HAV6 and ADTC5 for small molecules such as Gd-DTPA are less than 1 and 4 h, respectively.21,22 When administered together, HAV6 enhanced the brain delivery of 65 kDa galbumin when it was administered at a high dose (600 nmol/kg) as detected by MRI; however, a 10 min delay between the administration of HAV6 and galbumin did not produce any enhancement in brain deposition of galbumin.22 This indicates that the increase in the size of pores that allow galbumin to cross the BBB was gone in less than 10 min. Similarly, ADTC5 enhanced the brain delivery of galbumin when they were administered together. A 10 min delay between injections of ADTC5 and galbumin still improved brain deposition of galbumin.22 However, when there was a 40 min delay between their administrations, no brain delivery enhancement of galbumin was observed. Recently, we have shown that a 20 or 40 min delay between administration of ADTC5 and IgG mAb did not cause an increase in IgG mAb brain deposition. In summary, pore openings created by the BBBM peptides depend on (a) the structure of BBBM peptide (e.g., HAV6 and ADTC5), (b) time between administration of BBBM and the delivered molecules, and (c) the size of the molecule being delivered.
Using the data from various results, the mechanism of BBBM peptides to improve brain delivery of various sizes of molecules can be proposed. The hypothesis is that BBBM peptide modulates the BBB by binding to the EC1 domain of cadherins and blocking cadherin–cadherin interactions to create various subpopulations of pore sizes in the BBB intercellular junctions. For simplicity of discussion, the created pores have large, medium, and small sizes. It is hypothesized that each pore size has different time-dependent stability. The large pores rapidly collapse to medium-size and small-size pores in a time-dependent manner, followed by the collapse of medium-size pores to small pores. Finally, the small pores revert to the original pore size under normal conditions with resealed BBB intercellular junctions. Thus, the effects of time and molecular size on brain depositions of molecules can be explained by the time-dependent collapse of large pores (fast), medium-size pores (moderate), and small pores (slow). This mechanism could explain why large molecules have a shorter window of delivery than do small molecules. In the future, we will investigate this proposed mechanism.
As direct evidence that ADTC5 can deliver BDNF into the brain, unlabeled BDNF was delivered with and without ADTC5. After delivery, Western blots showed that recombinant BDNF was detected in brain homogenates from the mice treated with BDNF + ADTC5 compared to no detection of recombinant BDNF for the mice that received BDNF without ADTC5 (Figure 5B,C). Qualitatively, the increase in BDNF doses in the BDNF + ADTC5-treated mice shows increases in the amounts of detected BDNF in the brain homogenates in Western blots. In addition, the increase in BDNF doses upregulates the pTrkB receptor expression (Figure 5C).43,44 This is a first proof of concept for preclinical demonstration that ADTC5 can deliver BDNF into the brain after iv administrations to suppress EAE disease relapse in the mouse model.
Demyelination is a hallmark trait of multiple sclerosis as well as in cuprizone and EAE mouse models. Reduced myelin levels in the corpus callosum in humans and both animal models are commonly observed.45,46 In our case, the mice treated with BDNF + ADTC5 showed a significant increase in both myelin density, as determined by Luxol fast blue (Figure 2A,B) and NG2 presence in the corpus callosum, as indicated by immunohistochemistry staining (Figure 3A,B) compared to the mice that received BDNF alone or vehicle. These data are consistent with those from previous studies indicating that BDNF plays an integral role in remyelination and NG2 upregulation.34,47,48
Neurotrophins such as NGF, BDNF, neurotrophin-3,4/5 (NT3, NT4/5), and IGF1 have been known to regulate the viability, development, and function of neurons. However, BDNF has been by far the most studied to reveal its role in brain health.49 A decrease in BDNF levels in vivo in the BDNF+/− mice reduces the number of NG2+ cells and myelin levels throughout the development, suggesting a correlation between NG2 cells and remyelination.34,47,48 In the cuprizone animal model used for studying remyelination, the demyelination in the corpus callosum is correlated with the decrease in BDNF, suggesting a relationship between BDNF levels in the brain and demyelination. A cuprizone mouse model shows demyelination after 4- and 5-week treatments with cuprizone. Administration of cuprizone to BDNF+/+ mice exhibited a counter response by increasing remyelination as well as upregulation of NG2 receptors. However, this increase in NG2 receptors in the cuprizone-treated BDNF+/+ mice was significantly lower than those in untreated of BDNF+/+ mice. The results indicate that BDNF has an integral role in the proliferation of NG2 cells and the remyelination process.32,34
To further evaluate the effects of BDNF brain delivery in the EAE mice, upregulations of several BDNF-stimulated mRNAs were determined for those that delivered the information from the gene to protein expressions. BDNF is known to activate signaling pathways for rapidly modifying the function of local targets (e.g., phosphorylating TrkB); it also has a long-term effect on gene transcription (e.g., CREB, EGR1 upregulation, ARC synthesis).35 In this study, BDNF has been shown to stimulate phosphorylation of TrkB in normal mice (Figure 5C). The brain slices from the BDNF + ADTC5-treated EAE mice exhibited a significant increase in activity-regulated cytoskeletal-related (ARC) and early growth response 1 (EGR1) transcripts compared to those of controls treated with BDNF alone or vehicle (Figure 4). Thus, this indicates that BDNF enters the brain with the help of ADTC5 to exude its biological activity to stimulate ARC and EGR1 mRNA upregulations.36,50,51 BDNF has been demonstrated to upregulate ARC in cell cultures, and transcription of the ARC gene is essential for late-phase, long-term potentiation in the cortex.36,51 In addition, EGR1 and EGR3 have been shown to directly regulate ARC synthesis.37 The EGR1- or EGR3-deficient mice lack ARC protein in some neurons; however, when the mice are deficient in both EGR1 and EGR3, all of the neurons lack ARC protein. In this study, upregulation of EGR1 and ARC mRNAs was presumably due to delivered BDNF in the brain (Figure 4A–C).
Nitric oxide synthase 1 (NOS1) mRNA was also probed; however, no significant change in the NOS1 mRNA was observed by comparing brain slices for all three groups, indicating no effect of BDNF on NOS1 mRNA. It has been shown that the increase in BDNF mRNA is dependent on the increase in NOS1 mRNA upon exercise because NOS1 mRNA stimulates BDNF increase in the hippocampus during exercise as well as in the mouse brain after stroke.52 The lack of NOS1 notable expression may be because BDNF does not use the nitric oxide pathway as other neurotrophins (e.g., NGF, IGF1) do.53 Additionally, BDNF has been shown to block NOS expression in rats to achieve BDNF homeostasis levels in the brain.39
Many researchers have also investigated different ways for noninvasive brain delivery of BDNF. One of these is via transcytosis across the BBB using receptor-mediated transport; in this case, BDNF is conjugated to OX26 monoclonal antibody (mAb), which is a transferrin receptor (TfR) mAb.54,55 This method has been referred to as the “Trojan horse” method, and the conjugate has been shown to cross the BBB in an animal model. Unfortunately, this transcytosis process may not be very efficient for carrying sufficient amounts of BDNF into the brain. There are several potential reasons for the inefficiency of this transcytosis method. First, due to the tight binding of mAb to TfR, a higher percentage of the conjugate is degraded in lysosomes of the BBB microvessel endothelial cells; thus, a lower amount of the delivered conjugate undergoes transcytosis into the brain side of the microvessels. Second, when the conjugate is transported into the brain side of the vascular endothelium, the conjugate cannot be released from TfR into the brain due to the very high affinity of the mAb to the TfR. As a result, the conjugate cannot effectively diffuse into brain tissues where BDNF is needed. Finally, conjugation to the mAb moiety could lower the BDNF binding affinity to the BDNF receptor, resulting in lower in vivo efficacy. Another BDNF delivery method across the BBB utilizes ultrasound along with microbubbles; however, this method has not been applied to deliver BDNF in animal models of disease.56–58 Ultrasound methods have been regarded as safe and reversible based on a short-term histological assessment; however, the risks associated with recurrent and frequent uses of ultrasound have not been fully determined.59,60
Our previous studies have shown that ADTC5 enhanced brain delivery of 65 kDa galbumin into the brains of living mice as detected using magnetic resonance imaging (MRI) imaging.22 Galbumin distributions were found throughout the posterior, midbrain, and anterior regions of the brain with the highest deposition at the posterior followed by midbrain and finally the lowest deposition in the anterior region. Recently, ADTC5 has been shown to deliver various-size proteins into the brain of healthy mice, including IRdye800CW-labeled 15 kDa lysozyme, 65 kDa albumin, and 150 kDa IgG mAb.23 As in galbumin, with NIR, the protein was distributed throughout different brain regions. The quantitative amounts of each protein in the brain homogenates (picomol/g brain) were determined using a newly developed near-IR fluorescence (NIRF) imaging method.23 With the same condition, the amount of albumin is higher than IgG mAb, suggesting that the delivery of a larger molecule such as IgG mAb was more difficult than that of a smaller albumin.23 Using the same condition, the study found that ADTC5 did not enhance the delivery of 220 kDa fibronectin, suggesting that a potential cutoff size that can be delivered is 220 kDa.23
Recently, we have developed novel cyclic peptides with N- to C-terminal cyclization derived from the HAV6 and ADTC5 peptide to improve conformational rigidity, target selectivity for cadherin, and plasma stability.24 It was shown that cyclic peptides HAVN1 (Cyclo(1,6)SHAVSS) and HAVN2 (Cyclo(1,5)SHAVS) significantly enhanced the brain delivery of 150 kDa IgG mAb compared to control while linear HAV6 did not increase the brain delivery of IgG mAb.24 Finally, a cyclic ADTHAV (Cyclo(1,8)TPPVSHAV) with a sequence combination from ADTC5 and HAV6 has a better BBB modulatory activity than its linear counterpart.24 The results suggest that N- to C-terminal cyclization could improve the selectivity and BBB modulatory activity of cadherin peptides.24
HAV6 has also been shown to deliver the anticancer drug adenanthin into the brain of mice with medulloblastoma brain tumor.25 Adenanthin is a substrate for the efflux pump, P-glycoprotein (Pgp); thus, adenanthin alone cannot effectively penetrate the BBB. In this study, multiple treatments with HAV6 + adenanthin and adenanthin alone were compared to no-treatment in mice with medulloblastoma brain tumor (i.e., D425-Med-Luc tumors). The group treated with HAV6 + adenanthin showed a significant suppression of brain tumors compared to adenanthin-treated and nontreated groups.25 In addition, the HAV6 + adenanthin group had a median survival of 30 days post tumor cell injection. In contrast, the median day of survival after tumor cell injection for Ade-alone was 20 days, which was similar to that of nontreated groups (19 days).25 About 50% of the HAV6 + adenathin-treated group was able to complete a five cycle treatment, which resulted in 45 days of survival post tumor cell injection. These mice showed a complete elimination of brain tumor as detected by bioluminescence imaging.25 These results support the potential applicability of HAV6 peptide and other cadherin peptides (i.e., ADTC5, ADTHAV, HAVN1, HAVN2) for delivering therapeutic agents in models of brain diseases.
Throughout the course of our studies, we found that the in vivo iv administration of small and large molecules in mice was a better and more sensitive method than the in situ rat brain perfusion method. Previously, ADTC5 has been used to deliver 3H-PEG-1500 and 14C-PEG-40,000 into the brain using the in situ rat brain perfusion method.20 However, the results gave a trend of enhanced brain delivery for both poly(ethylene glycol) (PEG) molecules when co-delivered with ADTC5, but these enhancements were not statistically significant compared to control.20 In that study, the delivered radioactive PEG molecules were diluted with the nonradioactive PEG; thus, the detected radioactivity was a fraction of delivered molecules in the brain. In contrast, the brain deposition of IRdye800CW-labeled 25 kDa PEG was significantly higher when administered with a cadherin peptide (HAV6) compared to that without the peptide using in vivo iv administration with a 15 min circulation time.21 The differences in results in brain delivery of molecules (i.e., PEGs or proteins) in the in situ rat brain and in vivo delivery method in the mice could be attributed to the differences in experimental conditions in these two methods. In the in situ rat brain perfusion method, the BBB exposure time to ADTC5 and PEG molecules was only 2 min with one pass perfusion through the BBB vasculature before detecting deposition of delivered molecules in the brain. In contrast, in vivo delivery via iv administration of IRdye800CW-labeled molecules with cadherin peptides (i.e., ADTC5 or HAV6) has a 15 min circulation time; thus, the BBB exposure to the administered molecule was longer than those in the in situ rat brain perfusion studies. We also found that the dose of delivered molecules and the label used to detect the molecule could help the quantitation of the delivered molecules in the brain.
One of the potential disadvantages of BBBM peptides (e.g., HAV6 and ADTC5) to improve the brain delivery of molecules is that there could also be modulated cadherin–cadherin interactions in other organs such as liver, kidney, spleen, lung, and heart. In other words, if BBBM peptides have low selectivity toward the BBB compared to vasculature in other organs (i.e., liver, kidney, spleen, lung, and heart), then BBBM peptides may induce side effects due to off-target depositions of delivered molecules in other organs. To test this hypothesis, the effects of HAV6 or ADTC5 on depositions of delivered proteins were evaluated in the liver, kidney, spleen, lung, and heart.23,24 It was found that ADTC5 can enhance the brain delivery of 15 kDa lysozyme, 65 kDa albumin, and 150 kDa IgG mAb; however, HAV6 can only increase the delivery of lysozyme but not albumin and IgG mAb. It should be noted that HAV6 successfully increased brain depositions galbumin when administered in high dose (600 nmol/kg or 27×).22 During administration of lysozyme + ADTC5 or HAV6 peptide, the highest deposition of lysozyme other than in the brain was in the kidney; this is due to the glomerular filtration process in the kidney. In addition, ADTC5 but not HAV6 significantly enhanced the deposition of lysozyme in the kidney compared to control (lysozyme alone), suggesting that HAV6 did not have any effect in the kidney. It is interesting to find that both ADTC5 and HAV6 did not enhance the depositions of lysozyme in the liver, spleen, lung, and heart. For albumin and IgG mAb, most of the depositions of both proteins were in the liver with very low depositions in the kidney, spleen, lung, and heart. Both ADTC5 and HAV6 did not significantly increase the depositions of albumin and IgG mAb in the liver, kidney, spleen, lung, and heart. These results suggest that there is a reasonable selectivity of BBBM (i.e., ADTC5 and HAV6) for the brain compared to other organs, and their selectivity depends on the BBBM peptide used and the size of the delivered molecules.
A common concern of our method is the extended duration of BBB modulation that allows unwanted molecules to cross the BBB into the brain. A more important question is that what is the effect of repeated treatments with ADTC5 on the BBB, animal behavior, and toxicities to the brain and other organs in healthy and diseased animals. This study is currently planned to evaluate the potential side effects of BBBM peptides. As mentioned above, the BBB opening created by BBBM peptides was short especially for large proteins such as albumin and IgG mAb. The size of opening created by ADTC5 was limited because it could not enhance the delivery of 220 kDa fibronectin into the brain. The BBB modulation by HAV6 and ADTC5 is considered to have a short duration compared to the “osmotic delivery method” using a hypertonic mannitol solution, which disrupts the BBB for more than several hours. In the present study, multiple iv injections of ADTC5 in the presence of BDNF up to eight times did not show any toxicity to the mice. These findings in aggregate suggest that BBB modulation using ADTC5 is reversible, nontoxic, and does not induce long-term effects on the BBB junctions.
It is proposed that modulation of the BBB by cadherin peptides (i.e., ADTC5 and HAV6) to increase paracellular porosity is due to their binding to cadherins and inhibiting cadherin–cadherin interactions in a reversible and dynamic fashion. Cadherin–cadherin interactions are part of cell–cell adhesion and act as “Velcro” in the adherens junctions of the BBB, and ADTC5 and HAV6 peptides have been shown to enhance the penetration of molecules through the paracellular pathway.1,20 NMR binding studies between ADTC5 or HAV6 peptide to the EC1 domain of E-cadherin have indicated that each peptide binds to a different region of the EC1 domain. From these binding studies, ADTC5 is proposed to bind to the EC1–EC1 domain swapping region by blocking the EC1–EC1 trans-cadherin interactions to increase paracellular porosity. In contrast, HAV6 peptide is proposed to interact with the EC1 domain to block cis EC1–EC2 cadherin interactions. Although both peptides bind to different sites on the EC1 domain of E-cadherin, they are able to modulate cadherin interactions in the BBB.19,20 E-cadherin is primarily found in epithelial cells such as intestinal mucosa epithelium, and VE-cadherin (cadherin-5) is expressed in peripheral endothelium. However, it is still not clear whether the BBB has only VE-cadherin or a combination of E- and VE-cadherins. In Western blots analysis, anti-E-cadherin antibody but not anti-VE-cadherin (anti-cadherin-5) can detect cadherin in the homogenates of bovine brain microvessel endothelial cells (BBMEC).61 It has been suggested that cadherin in the intercellular junction of the BBB has E-cadherin-like properties and that the VE-cadherin might be contributing to the BBB function.62,63 Thus, there is still a need to study the selectivity of cadherin peptides to cadherins in the BBB versus cadherins in other body parts (e.g., intestinal mucosa, kidney, and lung).
CONCLUSIONS
The present study has demonstrated that BDNF can be delivered to the brains of mice via systemic administration using ADTC5 as a BBB modulator. To the best of our knowledge, this is the first demonstration of noninvasive delivery of BDNF that suppresses disease relapse in the EAE mice, an animal model for multiple sclerosis. Multiple iv administrations of BDNF + ADTC5 significantly improve the clinical body scores of the EAE mice compared to the mice that received BDNF alone, ADTC5 alone, or vehicle. BDNF can permeate the BBB and exert an immediate effect to upregulate pTrkB receptors. Additionally, delivered BDNF was shown to induce remyelination and increase the presence of NG2-glia cells as well as stimulate downstream EGR1 and ARC mRNA transcripts. These results demonstrate that ADTC5 could be used to modulate the BBB to improve noninvasive brain delivery of BDNF or other proteins to treat brain diseases. Further studies are being conducted for brain delivery of various-sized proteins in healthy and brain disease animal models.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (NIH), including R01-NS075374 from the National Institute of Neurological Disorders and Stroke (NINDS) and P30-AG035982 from the KU Alzheimer’s Disease Center-National Institute of Aging (NIA). The authors acknowledge support for B.M.K. from the NIH T32 Predoctoral Training Program on Pharmaceutical Aspects of Biotechnology (T32-GM008359). They also thank Nancy Harmony for proofreading this manuscript.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Laksitorini M; Prasasty VD; Kiptoo PK; Siahaan TJ Pathways and progress in improving drug delivery through the intestinal mucosa and blood-brain barriers. Ther. Delivery 2014, 5, 1143–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Steinman L Blocking adhesion molecules as therapy for multiple sclerosis: natalizumab. Nat. Rev. Drug Discovery 2005, 4, 510–518. [DOI] [PubMed] [Google Scholar]
- (3).Duda PW; Schmied MC; Cook SL; Krieger JI; Hafler DA Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J. Clin. Invest. 2000, 105, 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Trapp BD; Ransohoff R; Rudick R Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr. Opin. Neurol. 1999, 12, 295–302. [DOI] [PubMed] [Google Scholar]
- (5).Ineichen BV; Plattner PS; Good N; Martin R; Linnebank M; Schwab ME Nogo-A Antibodies for Progressive Multiple Sclerosis. CNS Drugs 2017, 31, 187–198. [DOI] [PubMed] [Google Scholar]
- (6).Ruggieri S; Tortorella C; Gasperini C Anti lingo 1 (opicinumab) a new monoclonal antibody tested in relapsing remitting multiple sclerosis. Expert Rev. Neurother. 2017, 17, 1081–1089. [DOI] [PubMed] [Google Scholar]
- (7).Ciric B; Howe CL; Paz Soldan M; Warrington AE; Bieber AJ; Van Keulen V; Rodriguez M; Pease LR Human monoclonal IgM antibody promotes CNS myelin repair independent of Fc function. Brain Pathol. 2003, 13, 608–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fisher TL; Reilly CA; Winter LA; Pandina T; Jonason A; Scrivens M; Balch L; Bussler H; Torno S; Seils J; Mueller L; Huang H; Klimatcheva E; Howell A; Kirk R; Evans E; Paris M; Leonard JE; Smith ES; Zauderer M Generation and preclinical characterization of an antibody specific for SEMA4D. mAbs 2016, 8, 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Crozier RA; Bi C; Han YR; Plummer MR BDNF modulation of NMDA receptors is activity dependent. J. Neurophysiol. 2008, 100, 3264–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Obermeyer JM; Tuladhar A; Payne SL; Ho E; Morshead CM; Shoichet MS Local Delivery of Brain-Derived Neurotrophic Factor Enables Behavioral Recovery and Tissue Repair in Stroke-Injured Rats. Tissue Eng., Part A 2019, 1175–1187. [DOI] [PubMed] [Google Scholar]
- (11).Nagahara AH; Bernot T; Moseanko R; Brignolo L; Blesch A; Conner JM; Ramirez A; Gasmi M; Tuszynski MH Long-term reversal of cholinergic neuronal decline in aged nonhuman primates by lentiviral NGF gene delivery. Exp. Neurol. 2009, 215, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kaspar BK; Llado J; Sherkat N; Rothstein JD; Gage FH Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 2003, 301, 839–842. [DOI] [PubMed] [Google Scholar]
- (13).Givalois L; Naert G; Rage F; Ixart G; Arancibia S; Tapia-Arancibia L A single brain-derived neurotrophic factor injection modifies hypothalamo-pituitary-adrenocortical axis activity in adult male rats. Mol. Cell. Neurosci. 2004, 27, 280–295. [DOI] [PubMed] [Google Scholar]
- (14).Crawford DK; Mangiardi M; Song B; Patel R; Du S; Sofroniew MV; Voskuhl RR; Tiwari-Woodruff SK Oestrogen receptor beta ligand: a novel treatment to enhance endogenous functional remyelination. Brain 2010, 133, 2999–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Steelman AJ; Thompson JP; Li J Demyelination and remyelination in anatomically distinct regions of the corpus callosum following cuprizone intoxication. Neurosci. Res. 2012, 72, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Oldendorf WH; Hyman S; Braun L; Oldendorf SZ Blood-brain barrier: penetration of morphine, codeine, heroin, and methadone after carotid injection. Science 1972, 178, 984–986. [DOI] [PubMed] [Google Scholar]
- (17).Kroll RA; Neuwelt EA Outwitting the blood-brain barrier for therapeutic purposes: Osmotic opening and other means. Neurosurgery 1998, 42, 1083–1099. [DOI] [PubMed] [Google Scholar]
- (18).Ulapane KR; Kopec BM; Moral MEG; Siahaan TJ Peptides and Drug Delivery. Adv. Exp. Med. Biol. 2017, 1030, 167–184. [DOI] [PubMed] [Google Scholar]
- (19).Alaofi A; On N; Kiptoo P; Williams TD; Miller DW; Siahaan TJ Comparison of Linear and Cyclic His-Ala-Val Peptides in Modulating the Blood-Brain Barrier Permeability: Impact on Delivery of Molecules to the Brain. J. Pharm. Sci. 2016, 105, 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Laksitorini MD; Kiptoo PK; On NH; Thliveris JA; Miller DW; Siahaan TJ Modulation of intercellular junctions by cyclic-ADT peptides as a method to reversibly increase blood-brain barrier permeability. J. Pharm. Sci. 2015, 104, 1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).On NH; Kiptoo P; Siahaan TJ; Miller DW Modulation of blood-brain barrier permeability in mice using synthetic E-cadherin peptide. Mol. Pharm. 2014, 11, 974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ulapane KR; On N; Kiptoo P; Williams TD; Miller DW; Siahaan TJ Improving Brain Delivery of Biomolecules via BBB Modulation in Mouse and Rat: Detection using MRI, NIRF, and Mass Spectrometry. Nanotheranostics 2017, 1, 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ulapane KR; Kopec BM; Siahaan TJ In Vivo Brain Delivery and Brain Deposition of Proteins with Various Sizes. Mol. Pharm. 2019, DOI: 10.1021/acs.molpharmaceut.9b00763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ulapane KR; Kopec BM; Siahaan TJ Improving In Vivo Brain Delivery of Monoclonal Antibody Using Novel Cyclic Peptides. Pharmaceutics 2019, 11, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sajesh BV; On NH; Omar R; Alrushaid S; Kopec BM; Wang W-G; Sun H-D; Lillico R; Lakowski TM; Siahaan TJ; Davies NM; Puno P-T; Vanan MI; Miller DW Validation of Cadherin HAV6 Peptide in the Transient Modulation of the Blood-Brain Barrier for the Treatment of Brain Tumors. Pharmaceutics 2019, 11, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yang S; Chen Y; Feng M; Rodriguez L; Wu JQ; Wang MZ Improving eflornithine oral bioavailability and brain uptake by modulating intercellular junctions with an E-cadherin peptide. J. Pharm. Sci. 2019, 3870–3878. [DOI] [PubMed] [Google Scholar]
- (27).Kobayashi N; Kiptoo P; Kobayashi H; Ridwan R; Brocke S; Siahaan TJ Prophylactic and therapeutic suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. Clin. Immunol. 2008, 129, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kobayashi N; Kobayashi H; Gu L; Malefyt T; Siahaan TJ Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. J. Pharmacol. Exp. Ther. 2007, 322, 879–886. [DOI] [PubMed] [Google Scholar]
- (29).Vasquez JJ; Hussien R; Aguilar-Rodriguez B; Junger H; Dobi D; Henrich TJ; Thanh C; Gibson E; Hogan LE; McCune J; Hunt PW; Stoddart CA; Laszik ZG Elucidating the Burden of HIV in Tissues Using Multiplexed Immunofluorescence and In Situ Hybridization: Methods for the Single-Cell Phenotypic Characterization of Cells Harboring HIV In Situ. J. Histochem. Cytochem. 2018, 66, 427–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gershon TR; Crowther AJ; Liu H; Miller CR; Deshmukh M Cerebellar granule neuron progenitors are the source of Hk2 in the postnatal cerebellum. Cancer Metab. 2013, 1, No. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Smith PA; Schmid C; Zurbruegg S; Jivkov M; Doelemeyer A; Theil D; Dubost V; Beckmann N Fingolimod inhibits brain atrophy and promotes brain-derived neurotrophic factor in an animal model of multiple sclerosis. J. Neuroimmunol. 2018, 318, 103–113. [DOI] [PubMed] [Google Scholar]
- (32).Vondran MW; Clinton-Luke P; Honeywell JZ; Dreyfus CF BDNF+/− mice /-exhibit deficits in oligodendrocyte lineage cells of the basal forebrain. Glia 2010, 58, 848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lang EM; Schlegel N; Reiners K; Hofmann GO; Sendtner M; Asan E Single-dose application of CNTF and BDNF improves remyelination of regenerating nerve fibers after C7 ventral root avulsion and replantation. J. Neurotrauma 2008, 25, 384–400. [DOI] [PubMed] [Google Scholar]
- (34).VonDran MW; Singh H; Honeywell JZ; Dreyfus CF Levels of BDNF impact oligodendrocyte lineage cells following a cuprizone lesion. J. Neurosci. 2011, 31, 14182–14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hu Y; Russek SJ BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J. Neurochem. 2008, 105, 1–17. [DOI] [PubMed] [Google Scholar]
- (36).Alder J; Thakker-Varia S; Bangasser DA; Kuroiwa M; Plummer MR; Shors TJ; Black IB Brain-derived neurotrophic factor-induced gene expression reveals novel actions of VGF in hippocampal synaptic plasticity. J. Neurosci. 2003, 23, 10800–10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Li L; Carter J; Gao X; Whitehead J; Tourtellotte WG The neuroplasticity-associated arc gene is a direct transcriptional target of early growth response (Egr) transcription factors. Mol. Cell. Biol. 2005, 25, 10286–10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yin Y; Edelman GM; Vanderklish PW The brain-derived neurotrophic factor enhances synthesis of Arc in synaptoneurosomes. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2368–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Novikov L; Novikova L; Kellerth JO Brain-derived neurotrophic factor promotes survival and blocks nitric oxide synthase expression in adult rat spinal motoneurons after ventral root avulsion. Neurosci. Lett. 1995, 200, 45–48. [DOI] [PubMed] [Google Scholar]
- (40).Engelhardt B; Liebner S Novel insights into the development and maintenance of the blood-brain barrier. Cell Tissue Res. 2014, 355, 687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cosolo WC; Martinello P; Louis WJ; Christophidis N Blood-brain barrier disruption using mannitol: time course and electron microscopy studies. Am. J. Physiol. 1989, 256, R443–R447. [DOI] [PubMed] [Google Scholar]
- (42).Farrell CL; Shivers RR Capillary junctions of the rat are not affected by osmotic opening of the blood-brain barrier. Acta Neuropathol. 1984, 63, 179–189. [DOI] [PubMed] [Google Scholar]
- (43).Yoshii A; Constantine-Paton M Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 2010, 70, 304–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Numakawa T; Suzuki S; Kumamaru E; Adachi N; Richards M; Kunugi H BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [DOI] [PubMed] [Google Scholar]
- (45).Reynolds R; Dawson M; Papadopoulos D; Polito A; Di Bello IC; Pham-Dinh D; Levine J The response of NG2-expressing oligodendrocyte progenitors to demyelination in MOG-EAE and MS. J. Neurocytol. 2002, 31, 523–536. [DOI] [PubMed] [Google Scholar]
- (46).Gold R; Hartung HP; Toyka KV Animal models for autoimmune demyelinating disorders of the nervous system. Mol. Med. Today 2000, 6, 88–91. [DOI] [PubMed] [Google Scholar]
- (47).Du Y; Lercher LD; Zhou R; Dreyfus CF Mitogen-activated protein kinase pathway mediates effects of brain-derived neurotrophic factor on differentiation of basal forebrain oligodendrocytes. J. Neurosci. Res. 2006, 84, 1692–1702. [DOI] [PubMed] [Google Scholar]
- (48).Tsiperson V; Huang Y; Bagayogo I; Song Y; VonDran MW; DiCicco-Bloom E; Dreyfus CF Brain-derived neurotrophic factor deficiency restricts proliferation of oligodendrocyte progenitors following cuprizone-induced demyelination ASN Neuro 2015, 71, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Habtemariam S The brain-derived neurotrophic factor in neuronal plasticity and neuroregeneration: new pharmacological concepts for old and new drugs. Neural Regener. Res. 2018, 13, 983–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Roberts DS; Hu Y; Lund IV; Brooks-Kayal AR; Russek SJ Brain-derived neurotrophic factor (BDNF)-induced synthesis of early growth response factor 3 (Egr3) controls the levels of type A GABA receptor alpha 4 subunits in hippocampal neurons. J. Biol. Chem. 2006, 281, 29431–29435. [DOI] [PubMed] [Google Scholar]
- (51).Ying SW; Futter M; Rosenblum K; Webber MJ; Hunt SP; Bliss TV; Bramham CR Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J. Neurosci. 2002, 22, 1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Chen MJ; Ivy AS; Russo-Neustadt AA Nitric oxide synthesis is required for exercise-induced increases in hippocampal BDNF and phosphatidylinositol 3′ kinase expression. Brain Res. Bull. 2006, 68, 257–268. [DOI] [PubMed] [Google Scholar]
- (53).Bonthius DJ; Karacay B; Dai D; Pantazis NJ FGF-2, NGF and IGF-1, but not BDNF, utilize a nitric oxide pathway to signal neurotrophic and neuroprotective effects against alcohol toxicity in cerebellar granule cell cultures. Dev. Brain Res. 2003, 140, 15–28. [DOI] [PubMed] [Google Scholar]
- (54).Wu D; Pardridge WM Neuroprotection with noninvasive neurotrophin delivery to the brain. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 254–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zhang Y; Pardridge WM Conjugation of brain-derived neurotrophic factor to a blood-brain barrier drug targeting system enables neuroprotection in regional brain ischemia following intravenous injection of the neurotrophin. Brain Res. 2001, 889, 49–56. [DOI] [PubMed] [Google Scholar]
- (56).Baseri B; Choi JJ; Deffieux T; Samiotaki G; Tung YS; Olumolade O; Small SA; Morrison B; Konofagou EE Activation of signaling pathways following localized delivery of systemically administered neurotrophic factors across the blood-brain barrier using focused ultrasound and microbubbles. Phys. Med. Biol. 2012, 57, N65–N81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Kinoshita M; McDannold N; Jolesz FA; Hynynen K Noninvasive localized delivery of Herceptin to the mouse brain by MRI-guided focused ultrasound-induced blood-brain barrier disruption. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 11719–11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Rodríguez-Frutos B; Otero-Ortega L; Ramos-Cejudo J; Martinez-Sanchez P; Barahona-Sanz I; Navarro-Hernanz T; Gomez-de Frutos Mdel C; Diez-Tejedor E; Gutierrez-Fernandez M Enhanced brain-derived neurotrophic factor delivery by ultrasound and microbubbles promotes white matter repair after stroke. Biomaterials 2016, 100, 41–52. [DOI] [PubMed] [Google Scholar]
- (59).Baseri B; Choi JJ; Tung YS; Konofagou EE Multi-modality safety assessment of blood-brain barrier opening using focused ultrasound and definity microbubbles: a short-term study. Ultrasound Med. Biol. 2010, 36, 1445–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Samiotaki G; Vlachos F; Tung Y-S; Konofagou EE A quantitative pressure and microbubble-size dependence study of focused ultrasound-induced blood-brain barrier opening reversibility in vivo using MRI. Magn. Reson. Med. 2012, 67, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Pal D; Audus KL; Siahaan TJ Modulation of cellular adhesion in bovine brain microvessel endothelial cells by a decapeptide. Brain Res. 1997, 747, 103–113. [DOI] [PubMed] [Google Scholar]
- (62).Vorbrodt AW; Dobrogowska DH Molecular anatomy of interendothelial junctions in human blood-brain barrier microvessels. Folia Histochem. Cytobiol. 2004, 42, 67–75. [PubMed] [Google Scholar]
- (63).Rubin LL; Staddon JM The cell biology of the blood-brain barrier. Annu. Rev. Neurosci. 1999, 22, 11–28. [DOI] [PubMed] [Google Scholar]