

Abstract
Riboswitches are 5’‐untranslated mRNA regions mostly found in bacteria. They are promising drug targets to overcome emerging bacterial resistance against commonly used antibiotics. The glmS riboswitch is unique among the family of riboswitches as it is a ribozyme that undergoes self‐cleavage upon binding to glucosamine‐6‐phosphate (GlcN6P). Previously, we showed that carba glucosamine‐6‐phosphate (carba‐GlcN6P) induces self‐cleavage of the riboswitch with a potency similar to that of GlcN6P. Here, we report a synthetic approach to a new class of carba‐GlcN6P derivatives with an alkoxy substituent in the carba position. Key features of the synthesis are a ring closing metathesis followed by a hydroboration. The strategy gives access to libraries of carba‐GlcN6P derivatives. Ribozyme cleavage assays unraveled new activators for the glmS riboswitch from Listeria monocytogenes and Clostridium difficile.
Keywords: carba-sugars, carbohydrate mimics, glmS riboswitch, ribozymes
A new class of carba‐sugars with an alkoxy substituent in the carba position has been synthesized. Key steps were a ring‐closing metathesis and a hydroboration. The compounds have been tested for their ability to induce self‐cleavage of the glmS ribozyme, which represents a potential drug target to fight bacterial resistance to antibiotics. Compound 32, with a methoxy substituent at the carba position, turned out to have the most promising properties.

Introduction
Antibiotic‐resistant bacteria are expected to cause around 10 million deaths yearly by 2050. [1] This dire prediction explains the urgent need for the development of new antibiotics. Ideally, these antibiotics use new modes of action unexploited by established drugs, thereby slowing down the development of multidrug‐resistant pathogens. [2] Riboswitches are promising drug targets. These 5’‐untranslated mRNA regions (5’‐UTRs) are found mostly in bacteria where they play an important role in gene regulation. [3] Targeting of these regulatory mechanisms could yield a fatal imbalance in the bacterial ability to control the expression of the corresponding gene. [4]
One riboswitch that is especially interesting in this aspect is the glmS riboswitch discovered in 2004 by Winkler et al. [5] It is found in a large variety of Gram‐positive bacteria as well as in a lower frequency in Gram‐negative bacteria. [6] It has also been predicted and recently characterized in Clostridium difficile and Listeria monocytogenes. [7] The glmS riboswitch is located in 5’‐UTR of the gene encoding for the enzyme glucosamine‐6‐phosphate‐synthase (GlmS) that catalyzes the reaction of glutamine and fructose‐6‐phosphate (Fru6P) to yield glutamate and glucosamine‐6‐phosphate (GlcN6P).[ 5 , 8 ] GlcN6P can in return bind to the glmS riboswitch and catalyze its self‐cleavage leading to the recruitment of RNase J1 and subsequent degradation of the downstream coding RNA. [9]
Artificial activation of the glmS riboswitch would impede the bacterial ability to produce GlcN6P which is needed for the biosynthesis of the bacterial cell wall. The design of artificial GlcN6P mimics is a possibility to inhibit bacterial growth by using a new and unexploited mode of action and, therefore, a promising contribution to the fight against antibiotic resistances.[ 4e , 5 , 10 ] Previous studies revealed the structural prerequisites of GlcN6P mimics to actively induce the self‐cleavage reaction of the glmS riboswitch (Figure 1).[ 5 , 10a , 10b ] It has been demonstrated that the cyclic structure, the equatorial amino group in the 2‐position, as well as the equatorial hydroxy group in the 4‐position are required. Removal of these functionalities or – in the case of the 4‐hydroxy group – inversion of the stereochemistry leads to a lack of activation.[ 10b , 10c , 10d ] The axial hydroxy group in position 1 greatly improves the efficacy of the activator, and a preferential binding of the alpha anomer of GlcN6P to the riboswitch has been demonstrated. [10c] The 1‐deoxy derivative of GlcN6P still activates the glmS riboswitch though with a 70‐fold lower activity for the rate constant k obs of the self‐cleavage reaction. [10b] The least critical position is the 3‐position; here an inversion of the stereochemistry decreases k obs by a factor of 3.5. [10b] In addition, phosphorylation of the 6‐OH group is required for efficient self‐cleavage of the riboswitch.[ 10a , 10b ] These experiments demonstrate that the design space for the exploration of new artificial activators of the glmS riboswitch is very limited.
Figure 1.

Structural requirements for GlcN6P derivatives acting as activators of the glmS riboswitch. Red: The cyclic structure and the equatorial substituents in the 2‐ and 4‐positions are essential. Green: Variation of the ring oxygen is tolerated. 1: Unsubstituted carba‐GlcN6P with similar activity to GlcN6P. 2, 3: Substituted carba‐GlcN6P derivatives (c.f. text).
Substitution of the ring oxygen with carbon is, however, tolerated. As we could show, carba‐sugar 1 (carba‐GlcN6P) has a similar potency to GlcN6P in modulating riboswitch activity. [11] Furthermore, it was demonstrated that carba‐GlcN treatment confers antibacterial activity. [10f] carba‐GlcN is taken up by the bacterial PTS and subsequently phosphorylated yielding carba‐GlcN6P. In this study it was shown that gene regulation through the glmS riboswitch occurs in carba‐GlcN‐treated bacteria. An X‐ray structure of the glmS riboswitch in complex with GlcN6P (PDB ID: 2Z75) [12] suggests that an equatorially oriented substituent in the carba position might be tolerated or even fill a potential binding pocket and thereby improve the activity of the mimic. Carba‐GlcN6P derivatives with substituents in the carba position, however, are not known except for derivative 2 with an axially oriented fluorine substituent. [13] Here, we present synthetic access to the new class of substances 3 with equatorial substituents in the carba position. We introduce an equatorial hydroxy group in the carba position that at a late stage of the synthesis can be alkylated with various groups thereby giving access to compound libraries for the study of structure‐activity relationships. Three compounds were tested in vitro for their activity to initiate the self‐cleavage reaction of the newly characterized [7] glmS riboswitches of C. difficile and L. monocytogenes. Among these compounds, the variant with a methoxy substituent at the carba position turned out to have the most promising properties.
The classical synthetic approach towards carba‐glucosamine uses the Ferrier rearrangement as a key step for the construction of the carbocycle followed by introduction of C‐6 [14] by olefination chemistry (Scheme 1, first line). [15] This synthetic route was also used by us [11] and others [16] for the preparation of carba‐GlcN(6P) and could be modified to introduce a fluorine substituent in the 5a position (Scheme 1, second line). [13] Initially, we tried to extend this strategy to carba‐sugars with an hydroxy or alkoxy substituent in the 5a position. These attempts, however, suffered from multiple drawbacks, including low yields. Furthermore, the desired substituent had to be introduced at an early stage of the synthesis rendering the preparation of a variety of compounds inefficient. In the synthesis of valienamine starting from glucose, the Kim group [17] and later also the Jung group [18] demonstrated the power of ring closing metathesis (RCM) for the construction of an tetrabenzylated cyclohexenol derivative (Scheme 1, third line) sharing some structural features with the anticipated carba‐GlcN6P derivative 3. Although it was questionable whether this route could be transferred to aminosugars, we envisioned the application of RCM as a suitable approach to the unsaturated core of carba‐glucosamine. The double bond would then be accessible for late‐stage introduction of alkoxy substituents in the 5a‐position by hydroboration and subsequent alkylation (Scheme 1 fourth line). In the following, we describe the successful realization of this approach.
Scheme 1.

Synthetic routes to carba‐sugars.
Results and Discussion
Synthesis
Retrosynthetically, the carba‐GlcN6P derivatives 31, 32, and 33 presented in this work were obtained through phosphorylation, protecting group manipulation, and derivatization of the common key intermediate 17 (Scheme 2). Key intermediate 17 was accessible by hydroboration of the unsaturated carba‐sugar derivative 15 that served as a suitable substrate for an anti‐Markovnikov introduction of a hydroxy group. [19] Cyclohexene 15 was obtained by RCM of acyclic diene 12 which was synthesized from 1,5‐diol 7. Diol 7 was envisioned to be accessible from commercially available N‐acetylglucosamine (GlcNAc) in several steps including a reduction of the hemiacetal functionality.
Scheme 2.

Retrosynthetic analysis of the carba‐GlcN6P derivatives 31, 32 and 33.
We started our synthesis with the preparation of pentabenzylated d‐glucosamine (GlcN) 6 (Scheme 3). Initially, we intended to obtain 6 by the cleavage of the acetal function of perbenzylated GlcN 8, which was obtained from GlcN⋅HCl according to Ye and co‐workers [16a] in one step. However, it turned out that acetal cleavage of 8 required harsh conditions that resulted in complete decomposition of the compound. The hindered acetal cleavage is readily explained by the presence of the positively charged ammonium group in the 2‐position under the acidic conditions that prevents further protonation of the acetal. A similar observation has been made by Yamamoto et al. during the cleavage of the methyl glycoside of GlcN. [20] Therefore, a new route to lactol 6 was established. GlcNAc was converted to the allyl glycoside by Fischer glycosidation and subsequently the acetamide was cleaved with Ba(OH)2 to give allyl glycoside 4 as reported. [21] Crude product 4 was perbenzylated with NaH and BnBr to provide compound 5 in 78 % yield over three steps. The allyl group of 5 was removed in two steps by IrI‐catalyzed rearrangement to the vinyl glycoside [22] followed by hydrolysis under mildly acidic conditions to give the desired pentabenzylated GlcN 6 in a yield of 73 %. Compared to the literature‐known route to 6 in 12 steps from GlcN⋅HCl, [23] our access represents a significant improvement. Reduction of lactol 6 with LiAlH4 gave the corresponding 1,5‐diol 7 in 95 %.
Scheme 3.

Synthesis of diol 7. TBAI=tetrabutylammonium iodide.
For the conversion of diol 7 into diene 12, the primary alcohol of 7 was selectively protected with TBDPSCl to give silyl ether 9 in 88 % yield (Scheme 4). Swern oxidation of the secondary alcohol of 9 to the corresponding ketone and subsequent Wittig reaction yielded olefin 10 in 80 % yield over 2 steps. A following treatment of 10 with TBAF recovered the primary alcohol 11 in a yield of 83 %. The primary alcohol 11 was oxidized under Swern conditions to the aldehyde followed by addition of vinylMgBr to give diene 12 as a mixture of diastereomers in a ratio of 1.2 : 1 according to HPLC analysis in a combined yield of 87 % yield over two steps. The subsequent ring closing metathesis (RCM) with second‐generation Grubbs catalyst [24] in toluene led to the unsaturated carba‐sugars 13 and 14 in a combined yield of 82 % preserving the ratio of diastereomers (13/14=1.2 : 1). It is known that amines are challenging substrates for the RCM due to their ability to coordinate to metal‐alkylidene complexes leading to decomposition of the catalyst. [25] Several attempts have been made to decelerate or even overcome the decomposition by decreasing the nucleophilicity of the amines through addition of organic acids or conversion of the amines to carbamates or amides, etc. [26] However, for the reaction of diene 12, addition of camphor sulfonic acid did not improve yields. In our case, best results were obtained by portion‐wise addition of the catalyst at equal time intervals over 2.5 h in toluene. [27] As the stereochemistry at C1 of key intermediate 17 corresponds to that of the minor RCM product 14, we developed an efficient way to convert 13 into 14 by Ley Griffith oxidation followed by a Luche reduction in 87 % over 2 steps.
Scheme 4.

Synthesis of cyclohexene 14. DMAP=4‐dimethylaminopyridine; NMO=N‐methylmorpholine N‐oxide; TBAF=tetrabutylammonium fluoride; TPAP=tetrapropylammonium perruthenate.
To convert cyclohexene 14 into key intermediate 17, 14 was benzylated to produce 15 in almost quantitative yield (Scheme 5A). Subsequently, a hydroboration was performed to introduce the hydroxy group at the carba‐position, yielding the diastereomeric cyclohexane derivatives 16 and 17 in a yield of 64 %. As expected, 16 was the major product of this reaction resulting from a syn addition of the borane to the double bond from the opposite side of the two benzyloxy substituents in the allylic positions. The stereochemistry at C5 and C‐5a, however, was easily inverted in a three‐step sequence consisting of Ley Griffith oxidation to 18, selective triethylamine‐induced isomerization of the 5‐position, and CBS reduction of ketone 19 to give the desired product 17 after a single purification at the end of the sequence in a yield of 74 % beside minor amounts of the 5a epimer with axial OH group (ratio of diastereoisomers 50 : 1). With this procedure, key intermediate 17 was readily available for further derivatization and phosphorylation.
Scheme 5.

Synthesis of key intermediate 17. BMS=borane dimethylsulfide complex.
To yield new potential activators of the glmS riboswitch, key intermediate 17 was treated with NaH and MeI to obtain methyl ether 20 in a yield of 66 % (Scheme 6). With allyl bromide instead, allyl ether 21 was obtained in a yield of 84 %. 20, 21, and alcohol 17 were hydrogenated to remove the benzyl protecting groups and, in case of 21, convert the allyl group into a propyl group in excellent yields. To achieve selective phosphorylation of the 6‐hydroxy group, we first reacted the TFA salts of 22, 23, and 24 with Cbz‐Cl leading to the carbamates 25, 26, and 27. Previously, the regioselective phosphorylation of primary hydroxy groups of nucleosides with phosphoryl chloride in the presence of water and pyridine has been reported. [28] We could show, however, that the application of this elegant method to hexoses with unprotected OH groups in the 4‐ and 6‐position is not possible due to the formation of cyclic phosphates. [11] Therefore, we investigated alternative methods for regioselective phosphorylation. Whereas the application of the phosphoramidite (BnO)2PN(iPr)2 and subsequent oxidation by meta‐chloroperbenzoic acid [29] resulted in only low yields, the phosphorus(V) species ClPO(OPh)2 in pyridine [30] successfully delivered the diphenyl phosphoric acid esters 28, 29, and 30. The attachment of the phosphate group to the 6‐position was confirmed by a significant downfield shift of the resonances of the protons in the 6‐position and by observation of indicative 3 J H,P and 4 J H,P coupling constants in a 1H,31P HMBC spectrum (see the Supporting Information). The final deprotection was carried out by firstly removing the Cbz group by catalytic hydrogenation with Pd‐charcoal followed by addition of HClaq to protonate the amine. Subsequently, the phenyl phosphoric acid esters were cleaved by catalytic hydrogenation using Adams’ catalyst to obtain the carba‐GlcN6P derivatives 31, 32, and 33 in quantitative yields. The protonation step before the second hydrogenation turned out to be crucial; [31] without this step, the reaction proceeded much slower and produced side products.
Scheme 6.

Final steps toward carba‐GlcN6P derivatives 31, 32 and 33.
Biological activity
We next investigated the glmS ribozyme‐activating properties of 31, 32, and 33 and of the corresponding non‐phosphorylated carba‐GlcN6P derivatives 22, 23, and 24. We employed the glmS ribozymes from L. monocytogenes and C. difficile [7] and the previously described metabolite‐induced cleavage assay. [11] The potential of the six compounds to induce ribozyme cleavage was tested at a concentration of 0.2 mM. These experiments revealed that hydroxy compound 31 and methoxy compound 32 are able to activate the glmS ribozyme from L. monocytogenes, whereas propoxy compound 33 is not (Figure 2a). As expected, the non‐phosphorylated derivatives 22, 23, and 24 did not activate the glmS ribozyme (Figure 2b). Activation of the glmS ribozyme from C. difficile by 31 and 32 was found to be less pronounced, whereas 33, and 22–24 did not activate ribozyme self‐cleavage (Figures 2c,d). Next, we determined concentration‐dependent activation of both glmS ribozymes by 31 and 32 (Figure 3). These experiments revealed an EC50 value of 0.080 mM for 32 for activation of the glmS ribozyme from L. monocytogenes whereas the value for the glmS ribozyme from C. difficile and those observed with 31 were significantly higher. Based on these results, we determined the time‐dependent cleavage of the glmS ribozyme from L. monocytogenes induced by the most potent compound (32) and determined a rate constant k obs of 0.041 (±0.01) min−1 at a concentration of 32 of 0.2 mM (Figure 4).
Figure 2.

Performance of GlcN6P and derivatives thereof at 0.2 mM in the metabolite‐dependent glmS riboswitch cleavage assay. Cleavage of the glmS riboswitch from L. monocytogenes induced by a) GlcN6P, GlcN, and compounds 31–33 and by b) GlcN6P, GlcN, and compounds 22–24. Cleavage of the glmS riboswitch from C. difficile induced by c) GlcN6P, GlcN, and compounds 31–33 and by d) GlcN6P, GlcN, and compounds 22–24. N=2, each N in duplicate.
Figure 3.

Determination of EC50 values for glmS riboswitch cleavage. Cleavage of the glmS riboswitch from L. monocytogenes induced by a) 32 and b) 31. Cleavage of the glmS riboswitch from C. difficile induced by c) 32 and d) 31. N=1 in duplicate.
Figure 4.
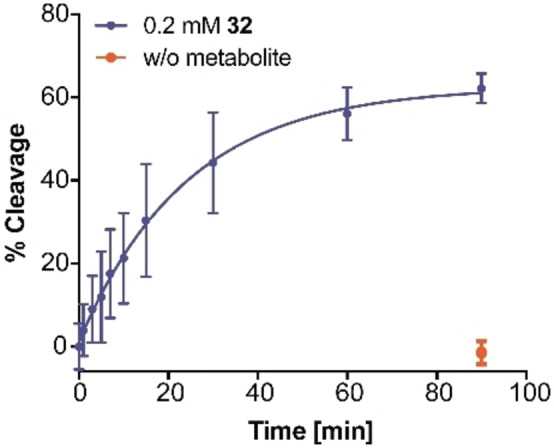
Time‐dependent cleavage of the glmS riboswitch from L. monocytogenes induced by 32 at a concentration of 0.2 mM (blue). In red: cleavage of the riboswitch after 90 min in the absence of a metabolite. N=1 in duplicate.
Conclusion
In conclusion, we have developed a synthetic access to a new class of carba‐sugars with an alkoxy substituent in the carba position. Such compounds were not accessible by the previously published routes to carba‐glucosamine derivatives. Ring closing metathesis followed by hydroboration of the double bond formed at a late stage of the synthesis gave access to key intermediate 17. This strategy allows access to a variety of carba‐GlcN6P derivatives with a substituent in the carba position, we demonstrated this by the synthesis of, for example, hydroxy compound 31, methoxy compound 32, and propoxy compound 33. Ribozyme cleavage assays revealed that carba‐GlcN6P derivatives with substituents in the carba position are able to activate the glmS ribozyme from L. monocytogenes and C. difficile. Methoxy‐substituted carba‐sugar 32 turned out to be the most active compound within this series. The observation that the larger methoxy substituent of 32 confers higher activity of the carba‐sugar than the smaller hydroxy group of 31 indicates that space is not a limiting factor for riboswitch activation. Although the propoxy substituent of 33 diminishes activity, it is quite possible that other substituents are beneficial for riboswitch activation possibly through intercalation between RNA nucleobases. The newly developed synthetic route opens the way for broad investigation in this direction, which we are currently undertaking.
Experimental Section
General methods: Anhydrous reactions were carried out under nitrogen or argon by using the Schlenk technique. Commercially available chemicals were used without further purification. Technical solvents were distilled prior to use. Thin‐layer chromatography (TLC) was performed using silica‐coated aluminum sheets (TLC Silica gel 60 F254) from Merck. Detection was carried out by excitation of the fluorescence at 254 nm or by dipping in one of the following staining solutions and subsequent gentle heating. Anisaldehyde reagent: ethanol (135 mL), conc. H2SO4 (5 mL), 4‐anisaldehyde (3.7 mL), glacial acetic acid (1.5 mL); vanillin reagent: ethanol (250 mL), conc. H2SO4 (2.5 mL), vanillin (6 g); potassium permanganate reagent: 0.1 % KMnO4 in 1 N NaOH. Preparative flash column chromatography (FC) was carried out on silica gel 60 (Geduran Si 60; 0.040‐0.063 mm particle size) from Merck. Solvent mixtures are given as volume ratio (v/v). Yields refer to chromatographically and spectroscopically pure compounds unless otherwise stated. NMR spectra were recorded on an Avance III 400 or Avance III 600 spectrometer from Bruker. Measurements were performed at room temperature. Chemical shifts are referenced to residual protic solvent signals (CDCl3: δ H=7.26 ppm, δ C=77.16; [D6]DMSO: δ H=2.50, δ C=39.52; D2O: δ H=4.79 CD3OD: δ H=3.35 ppm, δ C=49.3;). Signals were assigned by 2D NMR spectroscopy (COSY, HSQC, 1H,13C HMBC, 1H,31P HMBC & NOESY). As an analysis program, MestReNova v12.0 from Mestrelab Research S.L. was used. Preparative high‐performance liquid chromatography (HPLC) was performed on a LC‐20A instrument from Shimadzu containing the following components: degasser DGU‐20A3, auto sampler SIL‐20 A, pumps LC‐20AT, column oven CTO‐20AC, controller CMB‐20A, photodiode array detector SPD‐M20A, columns for separation and eluents are given in the synthesis procedures. Data analysis was performed with LCsolution v. 1.25 from Shimadzu. High‐resolution mass spectrometry (API+) was performed on a LTQ Orbitrap Velos instrument from Thermo Scientific. Samples were dissolved in water, acetonitrile, or methanol.
General procedure A: hydrogenation. The benzylated carba‐sugar derivative is dissolved in MeOH (6.7 mL mmol−1) and 10 % Pd−C catalyst (water wet, 35 % w/w of carba‐sugar) is added. After addition of TFA (10 equiv.) the reaction is placed in a laboratory autoclave and stirred under 15 bar hydrogen pressure until HPLC monitoring shows complete consumption of the starting material. Then, the catalyst is removed by filtration over a plug of celite followed by filtration through a regenerated cellulose syringe filter. The solvent is removed under reduced pressure to give the target compound as a colorless TFA salt.
General procedure B: Cbz protection of C‐2 amine. To a stirred solution of the CGlcN derivative in water (6.7 mL mmol−1), NaHCO3 (3 equiv.) and benzyl chloroformate (1.7 equiv.) are added at room temperature. The resulting solution is stirred overnight. The solvent is reduced under reduced pressure, and the crude product is purified as given in the individual procedures.
General procedure C: phosphorylation. To a stirred solution of the Cbz‐protected CGlcN derivative in pyridine (10.0 mL mmol−1), ClPO(OPh)2 (1.1 equiv.) is added at −40 °C in one portion upon the color of the solution turns from colorless to yellowish. The reaction mixture is stirred at room temperature overnight and quenched with water (5.0 mL/1.0 mmol). The solvent is removed under reduced pressure and toluene is evaporated from the residue. The crude product is purified as given in the individual procedures.
General procedure D: hydrogenation. The diphenyl phosphoric acid ester is dissolved in MeOH (8.0 mL mmol−1) and 10 % Pd−C catalyst (water wet, 10 % w/w of phosphoric acid ester) is added. The resulting suspension is stirred under hydrogen atmosphere (1 atm.) for 1 h. The catalyst is removed by centrifugation, and the supernatant is concentrated under reduced pressure. The residue is dissolved in MeOH (8.0 mL mmol−1), 1 m HClaq (1 equiv.) is added, and the reaction mixture is stirred for 2 h. Then, PtO2 (10 % w/w of phosphoric acid ester) is added and the suspension stirred under hydrogen atmosphere (1 atm) for 2 d. The catalyst is removed by filtration and the solvent is removed under reduced pressure.
Allyl 3,4,6‐tri‐O‐benzyl‐2‐(dibenzylamino)‐2‐deoxy‐d–glucopyranoside (5): Allyl glycoside 4 was synthesized from GlcNAc by a combination of two known procedures [21] for Fischer glycosidation and subsequent cleavage of the acetamide with slight modifications as follows. AcCl (15 mL, 77 mmol) was added dropwise to allylic alcohol (250 mL) under argon at 0 °C over a period of 5 min. At room temperature, GlcNAc (30 g, 135.6 mmol) was added, and the reaction mixture was stirred for 3 h at 70 °C. TLC analysis (CHCl2/MeOH 8 : 2) showed complete conversion of the starting material. Solid NaHCO3 was added to neutralize the solution to pH 7, and the suspension was filtered through celite and washed several times with MeOH. The solvent was removed under reduced pressure. The residue was used in the next step without further purification. To a stirred solution of the crude product in H2O (350 mL) was added Ba(OH)2 ⋅ 8H2O (150 g, 0.48 mol). The suspension was heated to reflux and stirred for 16 h. After cooling to room temperature, the precipitate was filtered off and the filtrate was carefully treated with dry ice to remove the barium salts. The solvent was removed by freeze drying. Crude product 4 was obtained as a colorless powder and was used in the next step without further purification. To a suspension of the crude product 4 in anhydrous DMF (340 mL) was added NaH (18.0 g, 60 % dispersion in mineral oil, 0.45 mmol) at 0 °C. The resulting suspension was stirred for 30 min at 0 °C. Then, benzyl bromide (38.7 mL, 0.33 mol) was added dropwise over 30 min at 0 °C. The solution was allowed to warm to room temperature and stirred for 1 h. The mixture was cooled to 0 °C, another portion of NaH (18.0 g, 60 % dispersion in mineral oil, 0.45 mmol) was added, and the suspension stirred for 30 min. at 0 °C. Benzyl bromide (38.7 mL, 0.33 mol) was added dropwise over 30 min at 0 °C. The solution was allowed to warm to room temperature and stirred for 1 h. The mixture was again cooled to 0 °C and an additional portion of NaH (18.0 g, 60 % dispersion in mineral oil, 0.45 mmol) was added and the suspension stirred for 30 min. at 0 °C followed by the addition of a third portion of benzyl bromide (38.7 mL, 0.33 mol) over 30 min. at 0 °C. The solution was allowed to warm to room temperature and stirred overnight. MeOH was added carefully to quench the excess of NaH. Then, the solvent was removed under reduced pressure, the residue was dissolved in CH2Cl2 (300 mL) and washed with water (60 mL) and brine (60 mL). The organic layer was dried over MgSO4 and concentrated under reduced pressure to give a brown oil. The crude product was purified by FC (petroleum ether/EtOAc=10 : 1) to give 5 as a yellow oil (66.3 g, 99.3 mmol, 78 % over 3 steps) as mixture of anomers (α/β=4 : 1). A small amount of the mixture was separated by FC to obtain analytical data for the individual isomers.
α‐Isomer: R f=0.39 (petroleum ether/EtOAc=10 : 1); 1H NMR (400 MHz, CDCl3): δ=7.56–7.10 (m, 25H, arenes), 5.98 (ddt, J=17.2 Hz, 10.5 Hz, 5.8 Hz, 1H, CH=CH2), 5.33 (dd, J=17.2 Hz, 1.6 Hz, 1H, CH=CHH’), 5.21 (dd, J=10.4 Hz, 1.5 Hz, 1H, CH=CHH’), 5.14 (d, 1H, J=11.5 Hz, O‐CHH’Ph), 5.04 (d, 1H, J=11.4 Hz, O−CHH'Ph), 4.92 (d, J=3.5 Hz, 1H, H‐1), 4.82 (d, J=10.7 Hz, 1H, O‐CHH'Ph), 4.69 (d, J=12.1 Hz, 1H, O‐CH’’H’’’Ph), 4.56 (m, 2H, O‐CHH’Ph, O‐CH’’H’’’Ph), 4.34 (dd, J=10.9 Hz, 8.5 Hz, 1H, H‐3), 4.27 (m, 1H, CHH’‐CH=CH2), 4.18 (d, J=13.6 Hz, 2H, N‐CH 2 Ph), 4.11 (ddt, J=12.7 Hz, 5.9 Hz, 1.5 Hz, 1H, CHH’‐CH=CH2), 3.90 (m, 3H, N‐CH 2 Ph, H‐5), 3.82 (dd, J=10.7 Hz, 3.7 Hz, 1H, H‐6a), 3.75 (dd,J=10.0 Hz, 8.5 Hz, 1H, H‐4), 3.70 (dd, J=10.6 Hz, 2.1 Hz, 1H, H‐6b), 3.08 ppm (dd, J=10.8 Hz, 3.5 Hz, 1H, H‐2); 13C NMR (CDCl3, 101 MHz): δ=141.0, 139.3, 138.3, 137.9, (5×Cquart.), 134.2 (CH=CH2), 128.8, 128.52, 128.50, 128.48, 128.24, 128.18 127.83, 127.79, 127.5, 127.4, 126.8, (arenes), 117.3 (CH=CH2), 100.8 (C‐1), 80.7 (C‐4), 80.2 (C‐3), 74.8, 73.7, 73.6 (3×O‐CH2Ph), 70.4 (C‐3), 68.8 (C‐6, CH2‐CH=CH2), 60.7 (C‐2), 56.1 ppm (2×N‐CH2Ph); HRMS (ESI) m/z calcd for C44H47NO5: 670.3527 [M+H]+ found: 670.3512.
β‐Isomer: R f=0.34 (petroleum ether/EtOAc=10 : 1); 1H NMR (400 MHz, CDCl3): δ=7.43–7.11 (m, 25H, arenes), 6.09 (m, 1H, CH=CH2), 5.43 (dd, J=17.2 Hz, 1.7 Hz, 1H, CH=CHH’), 5.33 (dd, J=10.4 Hz, 1.4 Hz, 1H, CH=CHH’), 5.04 (d, 1H, J=11.2 Hz, O‐CHH’Ph), 4.89 (d, 1H, J=11.1 Hz, O‐CHH’Ph), 4.75 (d, J=10.8 Hz, 1H, O‐CHH'Ph), 4.64 (d, J=7.7 Hz, 1H, H‐1), 4.60‐4.44 (m, 4H, O‐CHH’Ph, OCH2Ph, CHH’‐CH=CH2), 4.10 (m, 1H, CHH’‐CH=CH2), 4.03 (d, J=13.8 Hz, 2H, N‐CH 2 Ph), 3.92 (d, J=13.8 Hz, 2H, N‐CH 2 Ph), 3.81 (dd, J=9.9 Hz, 8.4 Hz, 1H, H‐3), 3.73 (dd, J=10.7 Hz, 2.2 Hz, 1H, H‐6a), 3.66 (dd, J=10.7 Hz, 5.1 Hz, 1H, H‐6b), 3.58 (dd, J=9.8 Hz, 8.4 Hz, 1H, H‐4), 3.45 (ddd, J=9.8 Hz, 5.3 Hz, 2.2 Hz, 1H, H‐5), 3.03 ppm (dd, J=9.9 Hz, 7.9 Hz, 1H, H‐2); 13C NMR (CDCl3, 101 MHz): δ=139.9, 139.1, 138.2, 138.1 (5×Cquart. arenes), 134.3 (CH=CH2), 128.9, 128.4, 128.3, 128.1, 127.89, 127.85, 127.7, 127.6, 127.3, 126.8 (arenes), 117.2 (CH=CH2), 101.2 (C‐1), 81.3 (C‐4), 79.4 (C‐3), 74.9 (C‐5), 74.8, 74.3, 73.5 (3×O‐CH2Ph), 69.7, 69.3 (C‐6, CH2‐CH=CH2), 63.5 (C‐2), 55.1 ppm (2×N‐CH2Ph); HRMS (ESI) m/z calcd for C44H47NO5: 670.3527 [M+H]+ found: 670.3514.
3,4,6‐Tri‐O‐benzyl‐2‐(dibenzylamino)‐2‐deoxy‐d–glucopyranose (6): Method A. [Ir(MePPh2)2(C8H12)]PF6 (44 μg, 0.05 mmol) was suspended in anhydrous THF (2 mL) under argon. The suspension was evacuated, diluted with H2 and stirred for 10 min at room temperature. After evacuation of excess H2, compound 5 (1.2 g, 1.75 mmol) dissolved in anhydrous THF (2.5 mL) was added dropwise over 15 min under argon atmosphere. The solution was stirred for further 10 min. TLC analysis (petroleum ether/EtOAc=15 : 1) showed complete conversion of the starting material. The solvent was removed under reduced pressure and the residue was purified by FC (petroleum ether/EtOAc=10 : 1) using a short column. The purified residue was dissolved in acetone/HClaq (1 N, 22 mL) and the solution was stirred for 1 h at 75 °C. TLC analysis (petroleum ether/EtOAc=3 : 1) showed complete conversion of the starting material into a slower moving compound. The solution was quenched with H2O (10 mL) and diluted with EtOAc (20 mL). The aqueous layer was extracted once more with EtOAc (20 mL) and the combined organic layers were washed with brine (5 mL), dried over MgSO4, and concentrated under reduced pressure. The residue was purified by FC (petroleum ether/EtOAc=10 : 1 to 3 : 1) to give 6 (800 mg, 1.30 mmol, 73 %) as a colorless syrup as a mixture of anomers (α/β=9 : 1).
Method B. Compound 5 (40.0 g, 59.7 mmol) was dissolved in degassed dry MeOH (320 mL). 1,3‐Dimethylbarbituric acid (23.3 g, 149.3 mmol) was added. Pd(PPh3)4 (3.5 g, 3 mmol) was added and the mixture heated to reflux overnight. The solvent was removed under reduced pressure and the residue dissolved in EtOAc (350 mL) and washed with saturated NaHCO3 (3x 150 mL) and brine (100 mL). The organic phase was concentrated under reduced pressure and purified by FC (petroleum ether/EtOAc=10 : 1→3 : 1) to give 6 (26.5 g, 42.1 mmol, 70 %) as a colorless syrup as a mixture of anomers (α/β=9 : 1). R f=0.6 (petroleum ether/EtOAc=3 : 1), vanillin; 1H NMR (CDCl3, 400 MHz): δ=7.51–7.14 (m, 25H, arenes), 5.25 (m, 0.34H, H‐1α), 5.07 (m, 2H, O‐CH 2 Ph), 4.79 (m, 1H, O‐CHH'Ph), 4.71 (d, J=7.9 Hz, 0.65H, H‐1β), 4.57 (m, 3H, O‐CHH’Ph, O‐CH 2 Ph), 4.31 (dd, J=10.8 Hz, 8.6 Hz, 0.34H, H‐3α), 4.11 (m, 1H, N‐CH2Ph, H‐5α), 3.97 (dd, J=10.3 Hz, 8.5 Hz, 0.66H, H‐3β), 3.89 (m, 2.69H, N‐CH 2 Ph), 3.73 (m, 2.35H, H‐6α/β, H‐4β), 3.64 (m, 0.66, H‐6α/β, H‐4α), 3.56 (br. S, 1H, OHβ), 3.48 (m, 1H, H‐5β), 3.02 (m, 1H, H‐2α), 2.87 (br. S, 1H, OHα), 2.75 ppm (dd, J=10.3 Hz, 7.9 Hz, 1H, H‐2β); 13C NMR (CDCl3, 101 MHz): δ=140.9, 139.4, 139.5, 138.7, 138.1, 138.0, 137.9, 137.7 (10×Cquart. arenes), 129.2, 128.7, 128.55, 128.54, 128.49, 128.47, 128.45, 128.2, 128.1, 127.86, 127.80, 127.78, 127.75, 127.72, 127.70, 127.5, 127.4, 127.3, 126.8 (arenes), 95.5 (C‐1α), 95.1 (C‐1β), 81.3 (C‐3β), 80.9 (C‐4α), 80.3 (C‐4β), 79.6 (C‐3α), 74.9 (C‐5β), 74.80, 74.75, 74.2, 73.7, 73.6, 73.5 (6×O‐CH2Ph), 70.3 (C‐5α), 69.0, 68.7 (2×C‐6), 64.3 (C‐2β), 60.8 (C‐2α), 55.9, 55.3 ppm (4×N‐CH2Ph); HRMS (ESI) m/z calcd for C41H43NO5: 630.3214 [M+H]+ found: 630.3198.
(2S,3R,4R,5R)‐3,4,6‐Tris(benzyloxy)‐2‐(dibenzylamino)hexane‐1,5‐diol (7): To a stirred solution of 6 (28 g, 44.5 mmol) in anhydrous THF (200 mL) was added LiAlH4 (3.7 mg, 97.8 mmol) at 0 °C under nitrogen atmosphere. After stirring for 4 h at room temperature, TLC analysis (petroleum ether/EtOAc=2 : 1) showed complete conversion of the starting material. The reaction mixture was carefully quenched with H2O, and afterwards HClaq (1 N, 400 mL) was added. The aqueous layer was extracted with EtOAc (3×150 mL) and the combined organic layers were washed with H2O and brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by FC (petroleum ether/EtOAc=2 : 1) to give 7 (26.6 g, 42.1 mmol, 95 %) as a colorless syrup. R f=0.5 (petroleum ether/EtOAc=2 : 1), vanillin; 1H NMR (CDCl3, 400 MHz): δ=7.43–7.27 (m, 25H, arenes), 4.87 (d, 11.2 Hz, O‐CHH'Ph), 4.63 (m, 4H, 2×O‐CH 2 Ph), 4.53 (d, 2H, J=11.9 Hz, O‐CHH'Ph), 3.97 (m, 4H, N‐CH 2 Ph, 2×CH), 3.83 (m, 3H, N‐CH 2 Ph, CH), 3.78 (dd, 1H, J=11.1 Hz, J=5.7 Hz, CHH’), 3.66 (m, 3H, CH2, CHH’), 3.46 (s, 2H, 2×OH), 3.30 ppm (td, 1H, J=7.8 Hz, 5.6 Hz, CH); 13C NMR (CDCl3, 101 MHz): δ=164.1, 139.4, 138.1, 138.0 (5×Cquart.), 129.4, 128.6, 128.48, 128.47, 28.40, 128.14, 128.07, 128.0, 127.89, 127.86, 127.8, 127.3 (arenes), 79.9, 77.3 (2×CH), 73.8, 73.7, 73.5 (3×O‐CH2Ph), 71.1 (CH2), 70.6 (CH), 59.5 (CH), 58.9 (CH2), 54.9 ppm (N‐CH2Ph); HRMS (ESI) m/z calcd for C41H45NO5: 632.3371 [M+H]+found: 632.3357; HPLC: t R=7.0 min (Eurosphere 100 C18, 16 mm, 9.0 mL min−1, 60–70 % MeCN in H2O in 20 min).
(2R,3R,4R,5S)‐1,3,4‐Tris(benzyloxy)‐6‐((tert‐butyldiphenylsilyl)oxy)‐5‐(dibenzylamino)hexan‐2‐ol (9): To a stirred solution of 7 (26.6 g, 42.1 mmol) in anhydrous CH2Cl2 (200 mL), tert‐butyldiphenylsilyl chloride (12.7 mL, 48.4 mmol), triethylamine (7.6 mL, 54.7 mmol) and N,N‐dimethylaminopyridine (205 mg, 1.7 mmol) were added at room temperature under nitrogen. After stirring for 20 h, TLC analysis (petroleum ether/EtOAc=8 : 1) showed complete conversion of the starting material. The reaction was stopped with H2O (100 mL) and the aqueous layer was extracted with CH2Cl2 (3×150 mL). The combined organic layers were washed with brine (150 mL), dried over MgSO4, and concentrated under reduced pressure. The residue was purified by FC (petroleum ether/EtOAc=8 : 1) to give 9 (32.4 g, 37.2 mmol, 88 %) as a colorless syrup. R f=0.29 (petroleum ether/EtOAc=8 : 1), vanillin; 1H NMR (CDCl3, 400 MHz): δ=7.71 (m, 4H, arenes), 7.53–7.19 (m, 31H, arenes), 4.81 (dd, 1H, J=11.6 Hz, J=2.3 Hz, O‐CHH’Ph), 4.68 (s, 2H, O‐CH 2 Ph), 4.56 (dd, 1H, J=11.6 Hz, J=2.3 Hz, O‐CHH'Ph), 4.40 (s, 2H, O‐CH 2 Ph), 4.16 (m, 5H, CH, 2×N‐CH 2 Ph), 3.97 (m, 1H, CH), 3.56 (m, 1H, CHH’), 3.49 (m, 2H, CH2), 3.42 (m, 2H, CH, CHH’), 3.10 (m, 1H, CH), 2.51 (s, 1H, OH), 1.10 ppm (m, 9H, 3×CH3); 13C NMR (CDCl3, 101 MHz): δ=140.2, 139.0, 138.8, 138.4 (5×Cquart., CH2 Ph), 135.8, 135.7 (arenes, SiPh), 133.41, 133.35 (2×Cquart., SiPh), 129.9, 129.8 (arenes, SiPh), 129.4, 128.3, 128.2, 127.83, 127.78, 127.6, 127.5, 127.41, 127.36, 127.2, 127.0 (arenes), 80.8, 80.3 (2×CH), 75.0, 74.4, 73.0 (3×O‐CH2Ph), 70.9 (CH2), 70.3 (CH), 59.3, 59.2, 55.8 (3×CH2), 27.1 (3×CH3), 19.1 ppm (Cquart., tBu); HRMS (ESI) m/z calcd for C57H63NO5Si: 870.4548 [M+H]+; found: 870.4528 ; HPLC: t r=8.90 min (Eurosphere 100 C18, 16 mm, 9.0 mL min−1, 80–100 % MeCN in H2O in 20 min).
(2S,3R,4R)‐N,N‐Dibenzyl‐3,4‐bis(benzyloxy)‐5‐((benzyloxy)methyl)‐1‐((tert‐butyldiphenylsilyl)oxy)hex‐5‐en‐2‐amine (10): To a stirred solution of oxalyl chloride (8.1 mL, 94.8 mmol) in anhydrous CH2Cl2 (250.0 mL) was added dimethyl sulfoxide (10.8 mL, 151.7 mmol) carefully at −78 °C under nitrogen and stirred for 30 min at the same temperature. The alcohol 9 (55.0 g, 63.2 mmol) was dissolved in anhydrous CH2Cl2 (150.0 mL) and was cooled to −78 °C and added to the reaction mixture. After stirring for 1 h at −78 °C, triethylamine (21.9 mL, 158.0 mmol) was added and stirred for 1 h at the same temperature. The cooling bath was removed, and the reaction mixture was allowed to warm up to room temperature and stirred for 30 min. The reaction was stopped by the addition of H2O (200 mL), and the aqueous layer was extracted with CH2Cl2 (3×200 mL). The organic layer was washed with brine (100 mL), dried over MgSO4 and concentrated under reduced pressure. Without further purification the residue was used in the next step. To a stirred solution of methyl triphenylphosphonium bromide (72.3 g, 202.2 mmol) dissolved in anhydrous THF (400.0 mL) was added NaHDMS (1 M in THF) (189.6 mL, 189.6 mmol) at 0 °C under nitrogen. The reaction mixture was stirred for 30 min. A solution of the ketone intermediate in anhydrous THF (200.0 mL) was added to the reaction mixture at 0 °C. The resulting mixture was stirred overnight at room temperature; afterwards the reaction was stopped by the addition of H2O (200 mL). The aqueous layer was extracted with EtOAc (3×200 mL) and the combined organic layers were washed with brine (100 mL), dried over MgSO4 and concentrated under reduced pressure. The residue was purified by FC (petroleum ether/EtOAc=7 : 1) to give 10 (43.9 mg, 50.6 mmol, 80 %) as a colorless oil. R f=0.34 (petroleum ether/EtOAc=8 : 1), vanillin; 1H NMR (CDCl3, 400 MHz): δ=7.68 (m, 4H, arenes), 7.46‐7.12 (m, 31H, arenes), 5.22 (m, 1H, C=CHH’), 4.87 (d, J=1.8 Hz, 1H, C=CHH’), 4.71 (d, 1H, J=11.6 Hz, O‐CHH'Ph), 4.55 (d, 1H, J=11.9 Hz, O‐CH’’H’’’Ph), 4.43 (m, 4H, O‐CH 2 Ph, O‐CHH’Ph, H‐4), 4.27 (d, 1H, J=11.9 Hz, O‐CH’’H’’’Ph), 3.97 (m, 7H, N‐CH 2 Ph, H‐1a/b, H‐6a/b, H‐3), 3.54 (d, 2H, J=13.6 Hz, N‐CH 2 Ph), 3.15 (m, 1H, H‐2), 1.11 ppm (s, 9H, 3×CH3); 13C NMR (CDCl3, 101 MHz): δ=142.2, 140.5, 139.2, 138.5, 138.4 (5×Cquart. arenes, Cquart. C‐5), 135.8, 135.7 (arenes, SiPh), 133.53, 133.50 (2×Cquart. SiPh), 129.74, 129.67, 129.4, 129.3, 128.43, 128.40, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.73, 127.70, 127.66, 127.55, 127.49, 127.47, 127.3, 127.0, 126.6 (arenes), 116.0 (C=CH2), 82.0 (C‐4), 80.5 (C‐3), 74.5, 72.4, 70.6 (3×O‐CH2Ph), 69.5 (C‐6), 60.4 (C‐1), 60.2 (C‐2), 55.8 (2×N‐CH2Ph), 27.1 (3×CH3), 19.2 ppm (Cquart. tBu); HRMS (ESI) m/z calcd for C58H63NO4Si: 866.4599 [M+H]+; found: 866.4581; HPLC: t R=9.20 min (Eurosphere 100 C18, 16 mm, 9.0 mL min−1, 80–100 % MeCN in 20 min).
(2S,3R,4R)‐3,4‐Bis(benzyloxy)‐5‐((benzyloxy)methyl)‐2‐(dibenzylamino)hex‐5‐en‐1‐ol (11): To a stirred solution of 7 (33.0 g, 38.1 mmol) in anhydrous THF (180.0 mL) was added tetra‐N‐butylammonium fluoride (1 N in THF; 64.8 mL, 64.8 mmol) under argon. After stirring for 24 h at room temperature, the reaction was stopped by addition of water (100 mL). The aqueous layer was extracted twice with EtOAc (2×150.0 mL) and the combined organic layers were washed with brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by FC (petroleum ether/EtOAc=3 : 1) to give 11 (20.2 g, 31.9 mmol, 84 %) as a colorless syrup. R f=0.3 (petroleum ether/EtOAc=3 : 1), vanillin; 1H NMR (CDCl3, 400 MHz): δ=7.41–7.20 (m, 25H, arenes), 5.37 (m, J=1.6 Hz, 1H, C=CHH’), 5.31 (m, 1H, C=CHH’), 4.80 (d, J=11.2 Hz, 1H, O‐CHH'Ph), 4.68 (d, J=11.9 Hz, 1H, OCH’’H’’’Ph), 4.56 (m, 3H, O‐CHH’Ph, O‐CH 2 Ph), 4.38 (d, J=11.9 Hz, OCH’’H’’’Ph), 4.33 (d, J=3.9 Hz, 1H, H‐4), 4.22 (m, 1H, H‐6a), 4.03 (m, 1H, H‐6b), 3.88 (dd, J=8.1 Hz, 3.9 Hz, 1H, H‐3) 3.82 (m, 4H, 2×N‐CH 2 Ph), 3.62 (dd, J=10.9 Hz, 5.4 Hz, 1H, H‐1a), 3.44 (dd, J=10.9 Hz, 8.7 Hz, 1H, H‐1b), 3.12 (ddd, J=8.7 Hz, 8.7 Hz, 5.4 Hz, 1H, H‐2), 2.95 ppm (br s, 1H, OH); 13C NMR (CDCl3, 101 MHz): δ=141.7, 139.9, 138.3, 138.2, 138.0 (5×Cquart. arenes, Cquart. C‐5), 129.3, 128.42, 128.39, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.70, 127.68, 127.66, 127.6, 127.0 (arenes), 116.0 (C=CH2) 81.3 (C‐3), 78.4 (C‐4), 73.3, 72.4 (2×O‐CH2Ph), 71.4 (C‐6), 71.3 (O‐CH2Ph), 59.6 (C‐2), 58.9 (C‐1), 54.8 ppm (2×N‐CH2Ph); HRMS (ESI) m/z calcd for C42H45NO4: 628.3421 [M+H]+; found: 628.3407; HPLC: t r=14.05 min (Eurosphere 100 C18, 16 mm, 9.0 mL min−1, 80–100 % MeCN in H2O in 20 min).
(4S,5R,6R)‐5,6‐Bis(benzyloxy)‐7‐((benzyloxy)methyl)‐4‐(dibenzylamino)octa‐1,7‐dien‐3‐ol (12): To a stirred solution of oxalyl chloride (4.1 mL, 47.8 mmol) in CH2Cl2 (200 mL) dimethyl sulfoxide (5.1 mL, 76.5 mmol) was carefully added at −78 °C under nitrogen atmosphere and stirred for 1 h at the same temperature. Alcohol 11 (20 g, 31.9 mmol) was dissolved in anhydrous CH2Cl2 (100 mL), cooled to −78 °C, and added to the reaction mixture. After stirring for 1 h at −78 °C, triethylamine (11.0 mL, 79.6 mmol) was added and the mixture stirred for 1 h −78 °C. The cooling bath was removed, and the reaction mixture was allowed to warm up to room temperature and stirred for 30 min. The reaction was stopped by the addition of H2O (100 mL) and the aqueous layer was extracted with CH2Cl2 (3×100 mL). The organic layer was washed with brine (50 mL), dried over MgSO4, and concentrated under reduced pressure. The obtained residue was used in the next step without further purification. To a stirred solution of the reaction intermediate in anhydrous THF (200 mL) was added vinylmagnesium bromide (1 N in THF) (69.9 mL, 69.9 mmol) carefully at 0 °C under nitrogen atmosphere. After stirring for 2 h at room temperature, the reaction was stopped by addition of saturated NH4Claq (100 mL) and extracted with CH2Cl2 (3×70 mL). The combined organic layers were washed with brine (40 mL), dried over MgSO4, and concentrated under reduced pressure. The residue was purified by FC (petroleum ether/EtOAc=5 : 1) to give 12 (18.1 g, 27.7 mmol, 87 %) as a colorless syrup as a mixture of diastereomers in a ratio of 1.2 : 1. The diastereomers could be separated by HPLC.
Isomer 1: R f=0.34 (petroleum ether/EtOAc=10 : 1), vanillin; 1H NMR (CDCl3, 400 MHz): δ=7.36–7.19 (m, 25H, arenes), 5.85 (ddd, J=17.1 Hz, 10.5 Hz, 4.6 Hz, 1H, H‐7), 5.32 (m, 2H, CH=CHH’, C=CH’’H’’’), 5.19 (m, 1H, C=CH’’H’’’), 5.06 (m, 1H, CH=HH’), 4.86 (d, J=11.1 Hz, 1H, O‐CHH'Ph), 4.60 (d, J=11.9 Hz, 1H, O‐CH’’H’’’), 4.47 (m, 4H, O‐CHH’Ph, O‐CH 2 Ph, H‐3), 4.32 (m, 2H, O‐CH’’H’’’, H‐6), 4.11 (m, 2H, H‐1a, H‐4), 3.95 (m, 3H, H‐1b, N‐CH 2 Ph), 3.78 (d, J=13.4 Hz, 2H, N‐CH 2 Ph), 3.62 (d, J=7.5 Hz, 1H, OH), 3.08 ppm (t, J=5.7 Hz, 1H, H‐5); 13C NMR (CDCl3, 101 MHz): δ=141.5, 140.2 (2×Cquart.), 139.7 (C‐7), 138.5, 138.3, 138.1 (3×Cquart.), 129.4, 128.53, 128.49, 128.4, 128.1, 128.0, 127.8, 127.1 (arenes), 117.5 (C=C‐2), 115.2 (C‐8), 81.7 (C‐4), 80.3 (C‐3), 74.2, 72.5, 71.1 (O‐CH2Ph), 70.8 (C‐1), 70.4 (C‐6), 60.8 (C‐5), 56.4 ppm (2×N‐CH2Ph); HRMS (ESI) m/z calcd for C44H47NO4: 654.3578 [M+H]+; found: 654.3560; HPLC: t R=12.2 min. (Luna Silica (2) 100 Å, 250×21.2 mm, 5–100 % EtOAc in hexane over 20 min, 10.0 mL min−1).
Isomer 2 (includes up to 20 % of isomer 1): R f=0.34 (petroleum ether/EtOAc=10 : 1), vanillin; 1H NMR (CDCl3, 400 MHz): δ=7.40–7.15 (m, 29.8H, arenes), 5.48 (ddd, J=17.2 Hz, 10.3 Hz, 7.0 Hz, 1H, H‐7), 5.37 (m, 1H, C=CHH’), 5.05 (m, 3H, C=CHH’, H‐8a, H‐8b), 4.86 (d, J=11.3 Hz, 1H, O‐CHH'Ph), 4.61 (m, 1H, O‐CH’’H’’’Ph), 4.51 (m, 3H, O‐CHH’Ph, O‐CH 2 Ph), 4.36 (d, J=12 Hz, 1H, O‐CH’’H’’’Ph), 4.29 (d, J=5.8 Hz, 1H, H‐3), 4.24 (t, J=7.4 Hz, 7.4 Hz, 1H, H‐6), 3.97 (m, 6H, 2×N‐CH 2 Ph, H‐1a, H‐1b), 3.88 (t, J=5.4 Hz, 1H, H‐4), 3.39 (s, 1H, OH), 2.87 ppm (dd, J=7.6 Hz, 5.2 Hz, 1H, H‐5); 13C NMR (CDCl3, 101 MHz): δ=142.1, 140.6 (Cquart.), 139.8 (C‐7), 139.0, 138.3, 138.2 (Cquart.), 129.5, 128.6, 128.5, 128.4, 128.3, 127.7, 127.5, 126.9 (arenes), 117.1 (C=C‐2), 116.9 (C‐8), 81.3 (C‐4), 81.2 (C‐3), 74.8, 72.8 (2×O‐CH2Ph), 72.5 (C‐6), 70.9 (O‐CH2Ph), 70.3 (C‐1), 62.9 (C‐5), 56.1 ppm (2×N‐CH2Ph); HRMS (ESI) m/z calcd for C44H47NO4: 654.3578 [M+H]+; found: 654.3566; HPLC: t R=12.7 min (Luna Silica (2) 100 Å, 250×21.2 mm, 5–100 % EtOAc in hexane over 20 min, 10.0 mL min−1).
(1R,4R,5R,6S)‐4,5‐Bis(benzyloxy)‐3‐((benzyloxy)methyl)‐6‐(dibenzylamino)cyclohex‐2‐en‐1‐ol (13) and (1S,4R,5R,6S)‐4,5‐Bis(benzyloxy)‐3‐((benzyloxy)methyl)‐6‐(dibenzylamino)cyclohex‐2‐en‐1‐ol (14): To a stirred solution of 12 (18 g, 27.53 mmol) dissolved in anhydrous degassed toluene (200 mL) was added Grubbs catalyst second‐generation (701 mg, 825 μmol) at room temperature under nitrogen. The reaction mixture was stirred for 50 min 90 °C while constantly exchanging the atmosphere above the reaction by a stream of N2. Afterwards, another portion of Grubbs catalyst second‐generation (701 mg, 825 μmol) was added After stirring for a further 50 min at 90 °C, the last portion of Grubbs catalyst second generation (701 mg, 825 μmol) was added and the reaction mixture was then stirred at 90 °C for further 50 min. The reaction mixture was cooled to room temperature, and the solvent was removed under reduced pressure. The obtained charcoal black residue was purified by column chromatography (petroleum ether/EtOAc=10 : 1→6 : 1) to afford 13 (7.81 g, 12.48 mmol, 45 %) and 14 (6.39 g, 10.21 mmol, 37 %) as a blackish oil as a separable mixture of diastereomers. 13: R f=0.35 (1R isomer, petroleum ether/EtOAc=6 : 1), vanillin; 1H NMR (CDCl3, 400 MHz): δ=7.54–7.25 (m, 25H, arenes), 5.87 (m, 1H, H‐5a), 5.18 (d, 1H, J=11.5 Hz, O‐CHH'Ph), 4.94 (d, 1H, J=11.5 Hz, O‐CHH’Ph), 4.85 (d, 1H, J=10.8 Hz, O‐CH’’H’’’Ph), 4.75 (d, 1H, J=10.9 Hz, O‐CH’’H’’’Ph), 4.51 (m, 2H, O‐CH 2 Ph), 4.45 (m, 1H, H‐4), 4.30 (m, 1H, H‐6a), 4.22 (m, 1H, H‐1), 4.10 (dd, 1H, J=10.9 Hz, J=7.0 Hz,H‐3), 3.91 (m, 5H, 2×N‐CH 2 Ph & H‐6b), 3.02 (s, 1H, OH), 2.90 ppm (dd, 1H, J=10.9 Hz, J=9.2 Hz, H‐2); 13C NMR (CDCl3, 101 MHz): δ=139.8 (2×Cquart. N‐CH2 Ph), 138.8, 138.3, 138.1 (3×Cquart. O‐CH2 Ph), 135.0 (Cquart. C‐5), 129.3, 129.2, 128.53, 128.50, 128.4, 127.8, 127.74, 127.70, 127.68, 127.66, 127.6, 127.3 (arenes), 83.0 (C‐4), 81.2 (C‐3), 73.8, 73.6, 72.3 (3×O‐CH2Ph), 70.3 (C‐6), 66.7 (C‐1), 64.7(C‐2), 55.1 ppm (2×N‐CH2Ph); HRMS (ESI) m/z calcd for C42H43NO4: 626.3265 [M+H]+ found: 626.3255. 14: R f=0.23 (1S isomer, petroleum ether/EtOAc=6 : 1), vanillin; 1H NMR (CDCl3, 400 MHz): δ=7.47–7.24 (m, 25H, arenes), 5.96 (m, 1H, H‐5a), 5.07 (d, 1H, J=11.6 Hz, O‐CHH'Ph), 4.85 (d, 1H, J=11.6 Hz, O‐CHH’Ph), 4.77 (m, 2H, O‐CH 2 Ph), 4.49 (d, 2H, J=2.4 Hz, O‐CH 2 Ph), 4.28 (m, 4H, H‐1, H‐3, H‐4, H‐6a), 4.16 (d, 2H, J=14.0 Hz, N‐CH 2 Ph), 3.97 (m, 3H, N‐CH 2 Ph, H‐6a), 3.01 (dd, 1H, J=9.2 Hz, J=4.5 Hz, H‐2), 2.50 ppm (m, 1H, OH); 13C NMR (CDCl3, 101 MHz): δ=140.5 (2×Cquart. N‐CH2 Ph), 139.0 (Cquart. O‐CH2 Ph), 138.6 (Cquart. C‐5), 138.3, 137.9 (Cquart. O‐CH2 Ph), 128.50, 128.45, 128.42, 128.39, 128.37, 127.9, 127.8, 127.70, 127.68, 127.6, 127.5, 127.4, 126.9 (arenes), 81.6 (C‐3/4), 78.1 (C‐3/4), 73.6, 72.8, 72.7 (3×O‐CH2Ph), 70.4 (C‐6), 68.7 (C‐1), 59.7 (C‐2), 56.8 ppm (2×N‐CH2Ph); HRMS (ESI) m/z calcd for C42H43NO4: 626.3265 [M+H]+; found: 626.3251.
Conversion of 13 to 14: Compound 13 (6.5 g, 10.4 mmol) was dissolved in anhydrous DCM (100 mL). TPAP (370 mg, 1.1 mmol) and NMO (2.4 g, 20.8 mmol) were added, and the solution was stirred for 1 h at room temperature. The reaction mixture was filtered over a short plug of silica which was washed with petroleum ether/EtOAc 1 : 1 (100 mL). The solvent was removed under reduced pressure, and the crude enone was dissolved in a 1 : 1 mixture of anhydrous THF and anhydrous MeOH (200 mL). The reaction mixture was cooled to −20 °C, and cerium chloride heptahydrate (4.7 g, 12.5 mmol) was added. The solution was stirred for 30 min, after which NaBH4 (590 mg, 15.6 mmol) was slowly added. The reaction mixture was stirred for 1 h, and the reaction stopped by the addition of saturated NH4Claq. (75 mL). The mixture was extracted with EtOAc (3×75 mL), and the combined organic phases were washed with brine (75 mL), dried over MgSO4, and concentrated under reduced pressure. The crude product was purified by FC (petroleum ether/EtOAc=10 : 1→6 : 1) to afford 14 (5.7 g, 9.1 mmol, 87 %).
(1S,2S,5R,6R)‐N,N‐Dibenzyl‐2,5,6‐tris(benzyloxy)‐4‐((benzyloxy)methyl)cyclohex‐3‐en‐1‐amine (15): To a stirred solution of 14 (8 g, 11.2 mmol) in anhydrous THF (100 mL) was added NaH (1.53 g, 60 % dispersion in mineral oil, 38.3 mmol) at 0 °C. Benzyl bromide (4.1 mL, 34.5 mmol) was added dropwise over 30 min. After stirring for 48 h, MeOH was added carefully. The solvent was removed under reduced pressure, and the residue was dissolved in CH2Cl2 (150 mL) and washed with water (20 mL) and brine (20 mL). The organic layer was dried over MgSO4 and concentrated under reduced pressure to give a brown oil. The crude product was purified by FC (petroleum ether/EtOAc=8 : 1) to give 15 as a yellow oil (9.2 g, 12.6 mmol, 98 %). R f=0.48 (petroleum ether/EtOAc=8 : 1), vanillin; 1H NMR (400 MHz, CDCl3): δ=7.48–7.28 (m, 30H, arenes), 6.11 (m, 1H, H‐5a), 5.28 (d, J=11.3 Hz, 1H, O‐CHH'Ph), 4.58–4.86 (m, 9H, H‐1, H‐3, 2×N‐CH 2 Ph, O‐CH 2 Ph, O‐CHH’Ph), 4.46 (s, 2H, O‐CH 2 Ph), 4.38 (d, J=6.8 Hz, 1H, H‐4), 4.25 (d, J=12.6 Hz, 1H, H‐6a), 4.04 (d, J=12.8 Hz, 2H, O‐CH 2 Ph), 3.95 (d, J=12.6 Hz, 1H, H‐6b), 3.36 ppm (dd, J=11.4 Hz, 3.4 Hz, 1H, H‐2); 13C NMR (CDCl3, 101 MHz): δ=139.6 (Cquart. C‐5), 138.0, 137.9, 137.8, 137.6, 131.0 (6×Cquart. arenes), 128.9, 128.65, 128.60, 128.5, 128.4, 128.3, 128.0, 127.92, 127.87, 128.8 (arenes), 125.2 (C‐7), 82.6 (C‐4), 76.4, 74.2 (C‐1/C‐3), 72.6 (2×N‐CH2Ph), 73.4, 71.9, 71.3 (3×O‐CH2Ph), 69.8 (C‐6), 59.4 (C‐2), 57.5 ppm (O‐CH2Ph); HRMS (ESI) m/z calcd for C49H49NO4: 716.3734 [M+H]+; found: 716.3714.
(1R,2R,3S,4R,5R,6R)‐2,4,5‐Tris(benzyloxy)‐6‐((benzyloxy)methyl)‐3‐(dibenzylamino)cyclohexan‐1‐ol (16) and (1S,2R,3S,4R,5R,6S)‐2,4,5‐tris(benzyloxy)‐6‐((benzyloxy)methyl)‐3‐(dibenzylamino)cyclohexan‐1‐ol (17): To a stirred solution of 15 (8.5 g, 11.9 mmol) in anhydrous THF (120 mL), BH3 ⋅SMe2 (28.5 mL, 142.5 mmol, 5 M in Et2O) was added dropwise, and the reaction mixture was stirred overnight at room temperature. The reaction was quenched carefully by addition of H2O (120 mL). Sodium perborate tetrahydrate (27.4 g, 178.1 mmol) was added carefully and the reaction stirred overnight. Then, the reaction was diluted with H2O (100 mL) and EtOAc (200 mL), and the organic layer was separated. The aqueous layer was extracted with EtOAc (2×100 mL), washed with brine (50 mL), dried over MgSO4, and concentrated under reduced pressure. The residue was purified by FC (petroleum ether/EtOAc=12 : 1→5 : 1) to give 16 (4.8 g, 6.54 mmol 55 %) and 17 (810 mg, 1.1 mmol, 9 %) as colorless syrups. 16: R f=0.23 (petroleum ether/EtOAc=5 : 1), vanillin; 1H NMR (600 MHz, CDCl3): δ=7.31–7.09 (30H, arenes), 4.81 (t, J=9.8 Hz, 1H, O‐CHH’Ph), 4.58 (d, J=11.1 Hz, 2H, O‐CH 2 Ph), 4.53 (d, J=10.9 Hz, 1H, O‐CHH'Ph), 4.45 (d, J=10.9 Hz, 2H, O‐CH 2 Ph), 4.31 (m, 2H, O‐CHH’Ph, H‐5a), 4.24 (d, J=11.8 Hz, 1H, O‐CHH'Ph), 4.06 (dd, J=9.3 Hz, 7.4 Hz, 1H, H‐3), 3.97 (d, J=14.0 Hz, 2H, N‐CH 2 Ph), 3.89 (d, J=13.9 Hz, 2H, N‐CH 2 Ph), 3.83 (m, 1H, H‐4), 3.80 (d, J=9.6 Hz, 1H, H‐6b), 3.73 (t, J=3.8 Hz, 1H, H‐1), 3.67 (dd, J=9.1 Hz, 5.3 Hz, 1H, H‐6a), 3.21 (dd, J=9.4 Hz, 3.7 Hz, 1H, H‐2), 2.41 ppm (m, 1H, H‐5); 13C NMR (CDCl3, 101 MHz): δ=141.0, 139.2, 138.34, 138.31, 138.0 (Cquart.), 128.5, 128.4, 128.28, 128.24, 128.15, 128.10, 128.0, 127.96, 127.73, 127.69, 127.44, 127.42, 127.39, 127.3, 127.2, 127.1, 127.0, (arenes), 84.2 (C‐1), 80.3 (C‐4), 77.4 (C‐3), 73.0, 72.8, 72.5, 72.0 (4×O‐CH2Ph), 68.1 (C‐6), 67.1 (C‐5a), 57.3 (C‐2), 56.5 (2×N‐CH2Ph), 43.9 ppm (C‐5); HRMS (ESI) m/z calcd for C49H51NO5: 734.3840 [M+H]+; found: 734.3835. 17: R f=0.56 (petroleum ether/EtOAc=5 : 1), vanillin; 1H NMR (600 MHz, CDCl3): δ=7.34–7.04 (m, 30H, arenes), 4.93 (t, J=17.3 Hz, 11.3 Hz, 2H, O‐CH 2 Ph), 4.82 (d, J=11.6 Hz, 1H, O‐CHH’Ph), 4.70 (d, J=18.9 Hz, J=10.9 Hz, 2H, O‐CH 2 Ph), 4.38 (m, 3H, O‐CH 2 Ph, O‐CHH'Ph), 4.17 (dd, J=11.0 Hz, 8.6 Hz, 1H, H‐3), 4.04 (t, J=2.3 Hz, 1H, H‐1), 3.98 (m, 4H, 2×N‐CH 2 ‐Ph), 3.73 (dd, J=9.1 Hz, 2.7 Hz, 1H, H‐6b), 3.55 (dd, J=9.1 Hz, 5.8 Hz, 1H, H‐6a), 3.46 (ddd, J=10.8 Hz, 5.3 Hz, 2.4 Hz, 1H, H‐5a), 3.28 (dd, J=11.0 Hz, 8.6 Hz, 1H, H‐4), 2.74 (d, J=5.3 Hz, 1H, OH), 2.64 (dd, J=11.1 Hz, 2.1 Hz, 1H, H‐2), 2.21 ppm (dddd, J=10.8 Hz, 10.8 Hz, 5.8 Hz, 2.7 Hz, 1H, H‐5); 13C NMR (CDCl3, 101 MHz): δ=139.6, 138.3, 138.8, 137.2, 136.7 (Cquart.), 127.5, 127.4, 127.3, 127.2, 127.1, 126.81, 126.80, 126.77, 126.60, 126.55, 126.4, 126.1, 125.9, 125.6 (arenes), 81.3 (C‐1), 80.0 (C‐4), 79.5 (C‐3), 74.2, 73.9, 72.3, 72.0 (4×O‐CH2Ph), 71.7 (C‐5a), 62.7 (C‐6), 57.7 (C‐2), 54.9 (2×N‐CH2Ph), 42.8 ppm (C‐5); HRMS (ESI) m/z calcd for C49H51NO5: 734.3840 [M+H]+; found: 724.3824.
Conversion of 16 to 17: Compound 16 (200 mg, 0.27 mmol) was dissolved in anhydrous DCM (5 mL). TPAP (10 mg, 27 μmol) and NMO (634 mg, 0.55 mmol) were added, and the solution was stirred for 1 h at room temperature. The reaction mixture was filtered over a short plug of silica which was washed with petroleum ether/EtOAc 1 : 1 (10 mL), and the solvent was removed under reduced pressure. The crude ketone 18 was dissolved in a 1 : 1 mixture of anhydrous DCM and anhydrous isopropanol (8 mL) to which NEt3 (376 μL, 2.7 mmol) was added. The reaction was stirred for 6 h, and the solvent was removed under reduced pressure. The crude isomerized ketone 19 was dissolved in anhydrous THF (5 mL) and (S)‐2‐methyl‐CBS‐oxazaborolidine catalyst (1 M in THF, 27 μL, 27 μM) was added. After stirring for 5 min, BH3 ⋅SMe2 (5 M in THF, 54 μL, 272 μmol) was added slowly. After stirring for 30 min, the reaction was quenched by the addition of water (2 mL) and extracted with DCM (3×4 mL). The combined organic phases were dried over MgSO4 and concentrated under reduced pressure. The crude product was purified by FC (petroleum ether/EtOAc=10 : 1→5 : 1) to yield 17 (148 mg, 202 μmol, 74 %). Although intermediate ketone 19 was immediately used for the CBS reduction, a small sample was purified by FC (petroleum ether/EtOAc=14 : 1→10 : 1) and characterized. 19: R f: 0.34 (petroleum ether/EtOAc=6 : 1); 1H NMR (500 MHz, CDCl3): δ=7.46–7.07 (m, 30H, arenes), 4.92 (d, 1H, J=11.3 Hz, O‐CHHPh), 4.84 (d, 1H, J=11.3 Hz, O‐CHHPh), 4.77 (d, 1H, J=10.7 Hz, O‐CHHPh), 4.53–4.35 (m, 5H, 2×O‐CH2Ph, H‐3), 4.14 (d, 1H, J=4.3 Hz, H‐1), 3.99–3.92 (m, 4H, 2×N‐ CH2Ph), 3.80–3.68 (m, 3H, H‐6a, H‐6b, H‐4), 3.07 (dd, 1H, J=9.2 Hz, 4.3 Hz, H‐2), 3.00 ppm (ddd, 1H, J=11.1 Hz, 4.4 Hz, 2.3 Hz, H‐5); 13C NMR (126 MHz, CDCl3): δ=207.0 (C‐5a), 140.1, 138.9, 138.2, 137.1 (arenes Cquart.), 128.7, 128.6, 128.6, 128.5, 128.5, 128.4, 128.4, 128.4, 128.3, 128.1, 128.0, 127.9, 127.9, 127.8, 127.7, 127.5, 127.3, 126.9 (arenes), 84.9 (C‐3), 81.6 (C‐1), 78.8 (C‐4), 75.2, 73.7, 73.4, 72.4 (4×O‐CH2Ph), 64.9 (C‐6), 59.5 (C‐2), 56.0 (2×N‐CH2Ph), 52.1 ppm (C‐5); HRMS (ESI) m/z calcd for C49H49NO5: 732.3684 [M+H]+; found: 732.3690.
(1S,2R,3R,4S,5S,6R)‐N,N‐Dibenzyl‐2,3,6‐tris(benzyloxy)‐4‐((benzyloxy)methyl)‐5‐methoxycyclohexan‐1‐amine (20): To a stirred solution of 17 (488 mg, 670 μmol) in dry THF (1.4 mL) under N2 atmosphere, NaH (53.2 mg, 1.33 mmol, 60 % dispersion in mineral oil) was added at 0 °C and stirred for 30 min at that temperature. Then, MeI (83.1 μL, 1.33 mmol) was added at 0 °C, and the reaction mixture was stirred overnight at room temperature. The reaction was quenched with MeOH (700 μL) and Et3N (420 μL). After stirring for 15 min at room temperature, H2O (2 mL) was added, and the aqueous layer was extracted with EtOAc (2×4 mL). The organic layer was dried over brine, MgSO4 and concentrated under reduced pressure. The obtained residue was purified by FC (petroleum ether/EtOAc=12 : 1). Product 20 (330 mg, 440 μmol, 66 %) was obtained as a colorless oil. 1H NMR (600 MHz, CDCl3): δ=7.63–7.34 (m, 30H, arenes), 5.19 (m, 2H, O‐CH 2 ‐Ph), 5.02 (m, 2H, O‐CH 2 ‐Ph), 4.77 (m, 2H, O‐CH 2 ‐Ph), 4.64 (m, 2H, O‐CH 2 ‐Ph), 4.46 (dd, J=2.3 Hz, 2.2 Hz, 1H, H‐1), 4.41 (dd, J=11.0 Hz, 8.7 Hz, 1H, H‐3), 4.25 (m, 4H, 2×N‐CH 2 ‐Ph), 3.93 (m, 2H, H‐6a, H‐6b), 3.82 (dd, J=11.0 Hz, 8.7 Hz, 1H, H‐4), 3.54 (s, 3H, CH3), 3.28 (dd, J=11.3, 1.9 Hz, 1H, H‐5a), 2.87 (dd, J=11.0, 1.9 Hz, 1H, H‐2), 2.46 ppm (m, 1H, H‐5); 13C NMR (151 MHz, CDCl3): δ=141.0, 139.8, 139.0, 138.9, 138.6 (Cquart. arenes), 128.53, 128.51, 128.44, 128.42, 128.40, 128.3, 128.1, 128.03, 127.95, 127.8, 127.7, 127.5, 127.3, 127.1, 126.8 (arenes), 80.9 (C‐3, C‐4), 79.9 (C‐7), 77.4 (C1), 75.3, 74.4, 73.4, 73.3 (4×O‐CH2Ph), 65.0 (C‐6), 59.2 (C‐2), 57.9 (CH3), 56.1 (2×N‐CH2‐Ph), 43.6 ppm (C‐5); HRMS (ESI) m/z calcd for C50H53NO5: 748.3997 [M+H]+; found: 748.3985.
(1S,2R,3S,4S,5R,6R)‐3‐(Allyloxy)‐N , N ‐dibenzyl‐2,5,6‐tris(benzyloxy)‐4‐((benzyloxy)methyl)cyclohexan‐1‐amine (21): To a stirred solution of 17 (11 mg, 15 μmol) in dry DMF (85 μL), NaH (1.8 mg, 45 μmol, 60 % dispersion in mineral oil) was added at 0 °C under N2 and stirred for 30 min at this temperature. Then, All‐Br (1.95 μL, 22.5 μmol) was added at 0 °C, and the reaction mixture was stirred overnight at room temperature. The reaction was carefully quenched with H2O (50 μL) and the aqueous layer was extracted with EtOAc (2×0.5 mL). The combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The obtained residue was purified by FC (petrol ether/EtOAc=10 : 1) to give product 21 (9.7 mg, 12.5 μmol, 84 %) as a colorless oil. R f=0.57 (petroleum ether/EtOAc 10 : 1); 1H NMR (800 MHz, CDCl3): δ=7.39–7.09 (m, 30H, arenes), 5.83 (dddd, J=17.2 Hz, 10.4 Hz, 5.6 Hz, 5.4 Hz, 1H, CH=CH2), 5.19 (dd, J=17.1 Hz, 1.7 Hz, 1H, CH=CHH’), 5.10 (dd, J=10.4 Hz, 1.7 Hz, 1H, CH=CHH’), 4.97 (m, 2H, O‐CHH'Ph, O‐CH’’H’’’Ph), 4.82 (d, J=11.5 Hz, 1H, O‐CHH’Ph, 4.73 (d, J=10.5 Hz, 1H, O‐CH’’’’H’’’’’Ph), 4.56 (d, J=10.3 Hz, 1H, O‐CH’’H’’’Ph), 4.50 (d, J=10.5 Hz, O‐CH’’’’H’’’’’Ph), 4.40 (d, J=2.3 Hz, 2H, O‐CH 2 Ph), 4.17 (m, 2H, H‐1 and H‐3), 4.05 (m, 1H, CHH’‐CH=CH2), 4.00 (m, 4H, 2×N‐CH 2 Ph), 3.89 (m, 1H, CHH’‐CH=CH2), 3.73 (m, 1H, H‐6a), 3.70 (m, 1H, H‐6b), 3.58 (dd, J=11.0 Hz, 8.7 Hz, 1H, H‐4), 3.20 (dd, J=11.3 Hz, 2.0 Hz, H‐5a), 2.62 (dd, J=11.0 Hz, 2.0 Hz, 1H, H‐2), 2.26 ppm (dd, J=11.1 Hz, 2.3 Hz, 1H, H‐5); 13C NMR (CDCl3, 151 MHz): δ=140.9, 139.6, 138.9, 138.7, 138.3 (Cquart.), 134.9 (CH=CH2), 128.4, 128.28, 128.27, 128.2, 128.14, 128.08, 127.90, 127.88, 127.7, 127.5, 127.3, 127.1, 126.9, 126.6 (arenes), 116.9 (CH=CH2), 80.70 (C‐4), 80.68 (C‐1 or C‐3), 78.3 (C‐1 or C‐3), 77.3 (C‐5a, overlapping with CDCl3), 75.2, 74.3, 73.3, 73.0 (4×O‐CH2Ph), 71.1 (CH2‐CH=CH2), 64.9 (C‐6), 58.9 (C‐2), 56.0 (2×N‐CH2Ph), 43.3 ppm (C‐5); HRMS (ESI) m/z calcd for C52H55NO5: 774.4153 [M+H]+; found: 774.4142.
(1R,2R,3S,4R,5S,6S)‐3‐Amino‐6‐(hydroxymethyl)cyclohexane‐1,2,4,5‐tetraol (22): Benzylated carba‐sugar 17 (27 mg, 37 μmol) was deprotected according to general procedure A to give the title compound as TFA salt 22×TFA (11 mg, 37 μmol, quant.) as a colorless solid with minor impurities. A small portion of the product (5 mg) was purified by HPLC to give the free base 22 as a colorless solid that was used for biological assays. 1H NMR (600 MHz, D2O): δ=3.82 (dd, J=4.2 Hz, 4.2 Hz, 1H, CH), 3.66 (m, 5H, 3×CH, CH2), 3.01 (br s, 1H, CH), 1.87 ppm (br s, 1H, CH); 13C NMR (151 MHz, CDCl3): δ=71.9, 71.1, 70.6, 68.0 (4×CH), 59.3 (C‐6), 54.9, 44.6 ppm (2×CH); HPLC: t R=6.7 min (Phenomenex® Luna 5 μm HILIC 200 Å, 250×21.2 mm, 40 % isocrat. MeCN in 15 mM TEAB buffer pH 7.0, 10.0 mL min−1, ELSD);HRMS (ESI) m/z calcd for C7H15NO5: 194.1023 [M+H]+; found: 194.1020.
(1R,2R,3R,4R,5S,6R)‐3‐Amino‐6‐(hydroxymethyl)‐5‐methoxycyclohexane‐1,2,4‐triol (23): Benzylated carba‐sugar 20 (300 mg, 400 μmol) was deprotected according to general procedure A to give the title compound as TFA salt 23×TFA (123 mg, 384 μmol, 96 %) as a colorless solid with minor impurities. A small portion of the product (13.5 mg) was purified by HPLC to give the free base 23 as a colorless solid that was used for biological assays. 1H NMR (400 MHz, D2O): δ=4.34 (dd, J=2.8 Hz, 2.6 Hz, 1H, H‐1), 3.93 (dd, J=11.5 Hz, 2.8 Hz, 1H, H‐6b), 3.82 (dd, J=11.5 Hz, 2.8 Hz, 1H, H‐6a), 3.52 (dd, J=10.4 Hz, 9.2 Hz, H‐3), 3.41 (m, 4H, H‐4, O‐CH3), 3.37 (dd, J=8.5 Hz, 2.8 Hz, 1H, H‐5a), 2.71 (dd, J=10.4 Hz, 2.7 Hz, 1H, H‐2), 1.82 ppm (ddt, J=11.1 Hz, 8.4 Hz, 2.8 Hz, 1H, H‐5); 13C NMR (101 MHz, D2O): δ=76.8 (C‐7), 73.8 (C‐3), 69.9 (C‐4), 66.6 (C‐1), 56.8 (C‐6), 56.4 (CH3), 53.1 (C‐2), 43.2 ppm (C‐5); HPLC: t R=7.9 min (Phenomenex® Luna 5 μm HILIC 200 Å, 250×21.2 mm, 20–30 % MeCN in 15 mM TEAB buffer pH 7.0 over 15 min, 10.0 mL min−1, ELSD); HRMS (ESI) m/z calcd for C8H17NO5: 208.1179 [M+H]+; found: 208.1177.
(1R,2R,3R,4R,5S,6R)‐3‐Amino‐6‐(hydroxymethyl)‐5‐propoxycyclohexane‐1,2,4‐triol (24): Benzylated carba‐sugar 21 (122 mg, 157.6 μmol) was deprotected according to general procedure A to give the title compound as TFA salt 24×TFA (54.2 mg, 157.6 μmol, quant.) as a colorless solid with minor impurities. A small portion of the product was purified by HPLC to give the free base 24 as a colorless solid that can be directly used in biological assays. 1H NMR (600 MHz, D2O): δ=4.34 (dd, J=2.7 Hz, 2.7 Hz, 1H, H‐1), 3.93 (dd, J=11.4 Hz, 2.6 Hz, 1H, H‐6a), 3.84 (dd, J=11.3 Hz, 2.6 Hz, 1H, H‐6b), 3.71 (m, 1H, OCHH'CH2CH3), 3.61 (dd, J=10.7 Hz, 9.7 Hz, 1H, H‐3), 3.43 (m, 3H, H‐4, H‐5a, OCHH’CH2CH3), 2.89 (dd, J=10.7 Hz, 2.7 Hz, 1H, H‐2), 1.83 (m, 1H, H‐5), 1.60 (m, 2H, OCH2CH 2 CH3), 0.92 ppm (t, J=7.4 Hz, 3H, CH3); 13C NMR (D2O, 151 MHz): δ=75.0 (C‐4 or C‐5a), 72.5 (C‐3), 71.5 (OCH2CH2CH3), 70.0 (C‐4 or C‐5a), 66.5 (C‐1), 56.6 (C‐6), 53.3 (C‐2), 43.0 (C‐5), 22.3 (OCH2 CH2CH3), 9.9 ppm (CH3); HPLC: t R=9.6 min (Phenomenex® Luna 5 μm HILIC 200 Å, 250×21.2 mm, 50 % MeCN in 40 mM TEAB buffer pH 6.4 over 15 min, 10.0 mL min−1, ELSD); HRMS (ESI) m/z calcd for C10H21NO5: 236.1492 [M+H]+; found: 236.1486.
Benzyl ((1S,2R,3R,4S,5S,6R)‐2,3,5,6‐tetrahydroxy‐4‐(hydroxymethyl)cyclohexyl)carbamate (25): TFA salt 22×TFA (80.4 mg, 262 μmol) was treated according to general procedure B. The crude product was purified by HPLC. Product 25 was obtained as a colorless solid (21 mg, 65 μmol, 25 %). 1H NMR (400 MHz, CD3OD): δ=7.41–7.29 (m, 5H, arenes), 5.12 (s, 2H, CH 2 Ph), 3.93 (m, 3H, H‐6a/b, CH), 3.60 (m, 2H, 2×CH), 3.41 (m, 2H, 2x CH), 1.87 ppm (m, 1H, H‐5); 13C NMR (CD3OD, 101 MHz): δ=160.1 (C=O), 139.7 (Cquart. arenes), 130.8, 130.34, 130.30 (arenes), 75.4, 75.0, 74.2, 71.2 (4×CH), 68.9 (CH2Ph), 61.8 (C‐6), 57.8 (CH), 46.8 ppm (C‐5); HPLC: t R=9.1 min (KINETEX® C18 5 μm 100 Å, 250×21.2 mm, 15–30 % MeCN in H2O in 20 min, 10.0 mL min−1, UV) HRMS (ESI) m/z calcd for C15H21NO7: 328.1391 [M+H]+; found: 328.1387.
Benzyl ((1R,2R,3R,4R,5S,6R)‐2,3,6‐trihydroxy‐4‐(hydroxymethyl)‐5‐methoxycyclohexyl)carbamate (26): TFA salt 23×TFA (109.8 mg, 342 μmol) was treated according to general procedure B. The crude product was purified by FC (DCM/MeOH=5 : 1) to give 26 as a colorless solid (76 mg, 220 μmol, 65 %). 1H NMR (400 MHz, CD3OD): δ=7.42–7.30 (m, 5H, arenes), 5.14 (s, 2H, CH 2 Ph), 4.25 (dd, J=2.6 Hz, 2.6 Hz, 1H, H‐1), 3.94 (dd, J=10.8 Hz, 2.7 Hz, 1H, H‐6b), 3.82 (dd, J=10.8 Hz, 3.3 Hz, 1H, H‐6a), 3.60 (dd, J=10.7 Hz, 8.9 Hz), 3.41 (m, 4H, CH3, H‐4), 3.38 (dd, J=10.7 Hz, 2.6 Hz, 1H, H‐2), 3.27 (dd, J=11.3 Hz, 2.6 Hz, 1H, H‐5a), 1.87 ppm (ddt, J=11.0 Hz, 10.9 Hz, 3.1 Hz, 1H, H‐5); 13C NMR (101 MHz, MeOD4,): δ=158.7 (C=O), 138.3 (Cquart.), 129.4, 128.98, 128.95 (arenes), 78.5 (C‐7), 74.0 (C‐3), 72.3 (C‐4), 68.2 (C‐1), 67.6 (CH2Ph), 58.9 (C‐6), 57.0 (CH3), 56.3 (C‐2), 44.8 ppm (C‐5); HRMS (ESI) m/z calcd for C16H23NO7: 342.1547 [M+H]+; found: 342.1545.
Benzyl ((1R,2R,3R,4R,5S,6R)‐2,3,6‐trihydroxy‐4‐(hydroxymethyl)‐5‐propoxycyclohexyl)carbamate (27): TFA salt 24×TFA (34 mg, 102 μmol) was treated according to general procedure B. The crude product was purified by FC (DCM/MeOH=10 : 1) to give 27 as a colorless solid (26 mg, 67 μmol, 65 %). R f=0.54 (DCM/MeOH 10 : 1); 1H NMR (400 MHz, MeOD4): δ=7.43–7.30 (m, 5H, arenes), 5.14 (s, 2H, CH2Ph), 4.21 (d, J=2.7 Hz, H‐1), 3.97 (dd, J=10.7 Hz, 2.9 Hz, 1H, H‐6a), 3.86 (dd, J=10.6 Hz, 3.1 Hz, 1H, H‐6b), 3.67 (m, 1H, O‐CHH'CH2CH3), 3.61 (m, 1H, H‐3), 3.42 (m, 4H, H‐2, H‐4, H‐5a, O‐CHH’CH2CH3), 1.91 (m, 1H, H‐5), 1.64 (m, 2H, CH2CH 2 CH3), 0.99 ppm (t, J=7.4 Hz, 3H, CH3); 13C NMR (CDCl3, 101 MHz): δ=157.3 (C=O), 136.9 (Cquart.), 128.1, 127.6, 127.5 (arenes), 75.9 (C‐4 or C‐5a), 72.5 (C‐3), 71.2 (C‐4 or C‐5a), 70.7 (CH2CH2CH3), 67.8 (C‐1), 66.2 (CH2Ph), 58.0 (C‐6), 55.0 (C‐2), 43.4 (C‐5), 22.8 (CH2 CH2CH3), 9.6 ppm (CH3); HRMS (ESI) m/z calcd for C18H27NO7: 392.1680 [M+Na]+; found: 392.1663.
Benzyl ((1S,2R,3R,4S,5S,6R)‐4‐(((diphenoxyphosphoryl)oxy)methyl)‐2,3,5,6‐tetrahydroxycyclohexyl)carbamate (28): Compound 25 (5.3 mg, 16.2 μmol) was treated according to general procedure C. The crude product was purified by FC (DCM/MeOH=10 : 1) to give 28 as a colorless syrup (4.6 mg, 8.2 μmol, 51 %). 1H NMR (600 MHz, CD3OD): δ=7.46–7.22 (m, 15H, arenes), 5.12 (s, 2H, CH 2 Ph), 4.72 (ddd, J=9.5 Hz, 4.9 Hz, 2.0 Hz, 1H, H‐6b), 4.64 (ddd, J=9.5 Hz, 4.9 Hz, 2.0 Hz, 1H, H‐6a), 3.93 (s, 1H, H‐1), 3.56 (m, 2H, H‐3, H‐5a), 3.36 (m, 2H, H‐2, H‐4), 1.99 ppm (m, 1H, H‐5); 13C NMR (151 MHz, CD3OD): δ=158.7 (Cbz‐C=O), 151.94, 151.89, 138.3 (Cquart. arenes), 131.1, 129.4, 129.0, 128.9, 126.9, 121.33, 121.32, 121.30, 121.29 (arenes), 74.0 (C‐7), 73.4 (C‐1), 71.0 (C‐4), 67.9 (CH2Ph), 67.5 (C‐3), 66.7 (d, J C‐P=6.2 Hz, C‐6), 56.4 (C‐2), 45.0 ppm (d, J C‐P=8.2 Hz, C‐5); 31P NMR (162 MHz, CD3OD): δ=−12.29 ppm (s, 1P); HPLC: t R=22.6 min (KINETEX® C18 5 μm 100 Å, 250×21.2 mm, 5–60 % MeCN in H2O in 20 min, 10.0 mL min−1, UV); HRMS (ESI) m/z calcd for C27H30NO10P: 560.1680 [M+H]+; found: 560.1674.
Benzyl ((1R,2R,3R,4R,5S,6R)‐4‐(((diphenoxyphosphoryl)oxy)methyl)‐2,3,6‐trihydroxy‐5‐methoxycyclohexyl)carbamate (29): Compound 26 (60 mg, 176 μmol) was treated according to general procedure C. The crude product was purified by FC (DCM/MeOH=10 : 1) to give 29 as a colorless syrup (64 mg, 110 μmol, 64 %). 1H NMR (400 MHz, CDCl3): δ=7.42–7.21 (m, 15H, arenes), 5.57 (m, 1H, NH), 5.16 (m, 2H, CH 2 Ph), 4.69 (ddd, J=10.4 Hz, 8.9 Hz, 1.8 Hz, 1H, H‐6b), 4.45 (m, 1H, H‐6a), 4.20 (m, 1H, H‐1), 3.65 (dd, J=9.8 Hz, 9.8 Hz, 1H, H‐3), 3.43 (m, 1H, H‐2), 3.30 (s, 3H, CH3), 3.26 (m, 1H, H‐4), 3.08 (m, 1H, H‐5a), 2.55 (br s, 3H, 3×OH), 1.94 ppm (m, 1H, H‐5); 13C NMR (101 MHz, CDCl3): δ=156.9 (C=O), 150.5, 150.4, 150.3 (Cquart.), 136.3, 130.0, 129.9, 128.6, 128.20, 128.17, 125.9, 125.6, 120.32, 120.27, 120.14, 120.09 (arenes), 75.9 (C‐7), 72.7 (C‐3), 69.2 (C‐4), 67.4 (C‐1), 67.1 (NH‐CH2‐C=O), 64.6 (C‐6), 57.4 (CH3), 53.9 (C‐2), 42.5 ppm (d, J P‐C=6.2 Hz, 1 C, C‐5); 31P NMR (162 MHz, CDCl3): δ=−10.6 ppm (s, 1P); HRMS (ESI) m/z calcd for C28H32NO10P: 574.1837 [M+H]+; found: 574.1829.
Benzyl ((1R,2R,3R,4R,5S,6R)‐4‐(((diphenoxyphosphoryl)oxy)methyl)‐2,3,6‐trihydroxy‐5‐propoxycyclohexyl)carbamate (30): Compound 27 (20.7 mg, 56.0 μmol) was treated according to general procedure C. The crude product was purified by FC (DCM/MeOH=12 : 1) to give 30 as a colorless syrup (22.6 mg, 37.6 μmol, 67 %). R f=0.48 (DCM/MeOH=12 : 1); 1H NMR (600 MHz, CD3OD): δ=7.44–7.25 (m, 15H, arenes), 5.12 (s, 2H, CH 2 Ph), 4.78 (m, 1H, H‐6a), 4.58 (m, 1H, H‐6b), 4.14 (dd, J=2.5 Hz, 2.5 Hz, 1H, H‐1), 3.58 (dd, J=10.7 Hz, 8.9 Hz, 1H, H‐3), 3.51 (m, 1H, O‐CHH'CH2CH3), 3.40(m, 1H, H‐4), 3.35 (dd, J=10.7 Hz, 2.5 Hz, 1H, H‐2), 3.18 (dd, J=11.2 Hz, 2.6 Hz, 1H, H‐5a), 3.10 (m, 1H, O‐CHH’‐CH2CH3), 2.04 (m, 1H, H‐5), 1.50 (m, 2H, CH2CH 2 CH3), 0.84 ppm (t, J=7.4 Hz, 3H, CH3); 13C NMR (151 MHz, CD3OD): δ=158.7 (C=O), 151.9 (d, J P‐C=7.5 Hz, Cquart.), 151.8 (d, J C‐P=7.4 Hz, Cquart.), 138.3 (Cquart.), 131.1, 129.4, 129.0, 128.9, 126.9, 121.31, 121.28 (arenes), 76.0 (C‐5a), 73.9 (C‐3), 72.0 (O‐CH2CH2CH3), 70.9 (C‐4), 68.7 (C‐1), 67.6 (CH2Ph), 66.4 (d, J=6.2 Hz, C‐6), 56.2 (C‐2), 44.0 (d, J=8.8 Hz, C‐5), 24.0 (O‐CH2 CH2CH3), 10.9 ppm (CH3); 31P NMR (162 MHz, MeOD4): δ=−12.73 ppm (s, 1P); HRMS (ESI) m/z calcd for C30H36NO10P 624.1969 [M+Na]+; found: 624.1949.
((1S,2R,3R,4S,5R,6S)‐4‐Amino‐2,3,5,6‐tetrahydroxycyclohexyl)methyl dihydrogen phosphate (31): Compound 28 (5.2 mg, 9.4 μmol) was deprotected according to general procedure D to give the title compound as HCl salt 31⋅HCl (2.9 mg, 9.4 μmol, quant.) as a colorless solid. 1H NMR (400 MHz, D2O): δ=4.23 (m, 2H, H‐6a, H‐1 or H‐3), 4.13 (m, 1H, H‐6b), 3.82 (m, 1H, H‐4 or H‐5a), 3.76 (m, 1H, H‐1 or H‐3), 3.55 (dd, J=10 Hz, 10 Hz, 1H, H‐4 or H‐5a), 3.27 (dd, J=10 Hz, 2.8 Hz, 1H, H‐2), 1.92 ppm (m, 1H, H‐5); 13C NMR (151 MHz, D2O): δ=70.6 (C‐1 or C‐3), 69.2 (C‐1 or C‐3), 68.8 (C‐4 or C‐5a), 66.1 (C‐4 or C‐5a), 60.0 (d, J C‐P=5.0 Hz, 1 C, C‐6), 53.4 (C‐2), 43.2 ppm (d, J C‐P=8.2 Hz, 1 C, C‐5); 31P NMR (162 MHz, D2O): δ=0.12 ppm (s, 1P); HRMS (ESI) m/z calcd for C7H16NO8P: 272.0541 [M‐H]−; found: 272.0543.
((1R,2R,3R,4R,5R,6S)‐4‐Amino‐2,3,5‐trihydroxy‐6‐methoxycyclohexyl)methyl dihydrogen phosphate (32): Compound 29 (17.5 mg, 30 μmol) was deprotected according to general procedure D to give the title compound as HCl salt 32⋅HCl (9.7 mg, 30 μmol, quant.) as a colorless solid with minor impurities. A small sample of the product was purified by HILIC to give the title compound as triethylamonium salt 32⋅2 Et3N. 1H NMR (400 MHz, D2O): δ=4.48 (dd, J=2.7 Hz, 2.7 Hz, 1H, H‐1), 4.12 (m, 1H, H‐6b), 4.00 (m, 1H, H‐6a), 3.77 (dd, J=10.1 Hz, 10.1 Hz, 1H, H‐3), 3.61 (dd, J=10.1 Hz, 10.1 Hz, 1H, H‐4), 3.46 (m, 4H, OCH3, H‐5a), 3.23 (m, 13H, CH2, H‐2), 1.86 (m, 1H, H‐5), 1.31 ppm (t, J=7.3 Hz, 18H, CH3); 13C NMR (101 MHz, D2O): δ=75.8 (C‐7), 70.3 (C‐3), 68.9 (C‐4), 64.8 (C‐1), 58.89 (d, J C‐P=4.8 Hz, 1 C, C‐6), 57.0 (OCH3), 53.8 (C‐2), 46.7 (6×CH2), 42.96 (d, J C‐P=7.5 Hz, 1 C, C‐5), 8.2 ppm (6×CH3), 31P NMR (162 MHz, D2O): δ=3.0 ppm (s, 1P); HPLC: t R=5.5 min (Phenomenex® Luna 5 μm HILIC 200 Å, 250×21.2 mm, 40 % isocrat. MeCN in 15 mM TEAB buffer pH 6.99, 10.0 mL min−1, ELSD); HRMS (ESI) m/z calcd for C8H18NO8P: 286.0697 [M‐H]−; found: 286.0698.
((1R,2R,3R,4R,5R,6S)‐4‐Amino‐2,3,5‐trihydroxy‐6‐propoxycyclohexyl)methyl dihydrogen phosphate (33): Compound 30 (20.5 mg, 34.0 μmol) was deprotected according to general procedure D to give the title compound as HCl salt 33⋅HCl (11.9 mg, 34.0 μmol, quant.) as a colorless solid. 1H NMR (400 MHz, D2O): δ=4.44 (dd, J=2.8 Hz, 2.8 Hz, 1H, H‐1), 4.23 (m, 1H, H‐6a), 4.14 (m, 1H, H‐6b), 3.74 (m, 2H, H‐3, O‐CHH'CH2CH3), 3.54 (m, 3H,H‐4, H‐5a, O‐CHH’CH2CH3), 3.23 (dd, J=10.9 Hz, 2.7 Hz, 1H, H‐2), 1.94 (m, 1H, H‐5), 1.64 (m, 2H, OCH2CH 2 CH3), 0.95 ppm (t, J=7.4 Hz); 13C NMR (101 MHz, D2O): δ=74.2, 72.0, 70.6, 68.9 (4×C), 60.1 (d, J C‐P=4.7 Hz, 1 C, C‐6), 53.6 (C), 42.4 (d, J C‐P=9.4 Hz, 1 C, C‐5), 22.3 (OCH2 CH2CH3), 9.9 ppm (OCH2CH2 CH3); 31P NMR (162 MHz, D2O): δ=0.20 ppm (s, 1P); HRMS (ESI) m/z calcd for C10H22NO8P: 314.1010 [M‐H]−; found: 314.1009.
Preparation of glmS ribozyme RNA: PCR amplification of C. difficile and L. monocytogenes glmS riboswitch encoding DNA sequences was accomplished by GoTaq DNA Polymerase (Promega) using 5’ primers which incorporate the T7 promotor (annealing temperature for C. difficile DNA: 58 °C, for L. monocytogenes DNA: 61 °C). PCR products were purified on Nucleospin columns according to the manufacturer's instructions (Macherey & Nagel) and subsequentially transcribed using T7 RNA polymerase at 37 °C o/n. Transcription products were treated with DNase and purified by denaturing polyacrylamide gel electrophoresis (PAGE). The RNAs were 5’‐dephosphorylated using calf intestine alkaline phosphatase (Promega) and radioactively labeled by 5’‐phosphorylation through incubation with T4 polynucleotide kinase (NEB) and γ‐32P‐ATP (10 mCi mL−1 BEBm Zaventem, Belgium) at 37 °C for 30 min. The labeled ribozymes were desalted with G25 columns (GE Healthcare) that had been equilibrated with DEPC‐treated water and again purified by polyacrylamide gel electrophoresis.
Metabolite‐dependent glmS riboswitch cleavage assay: The 32P‐labeled glmS RNA from C. difficile or L. monocytogenes was incubated at 37 °C with GlcN, GlcN6P, compounds 22, 23, 24, 31, 32, 33 or without GlcN derivative in cleavage buffer (10 mM MgCl2, 50 mM HEPES, 200 mM KCl, pH 7.5). The potential cleavage reaction was terminated by the addition of PAGE loading buffer (45 mM HEPES, 45 mM boric acid, 4 M urea, 10 % sucrose, 0.05 % SDS, 100 mM EDTA) followed by separation on a 17 % denaturing polyacrylamide gel for analysis. Cleavage products were detected by autoradiography on a phosphorimager FLA‐3000 (Fujifilm) and individual cleavage per sample was assessed using AIDA (Elysa‐Raytest) and Prism (GraphPad) software. To determine the rate constants k obs of ribozyme cleavage, trace amounts of 32P‐labeled ribozyme‐RNA were incubated at 37 °C in cleavage buffer with indicated concentrations of carba‐sugar 31 or 32. Aliquots were withdrawn from the reaction pool at various time points and the reaction was quenched by the addition of PAGE loading buffer. The cleavage products were separated by denaturing PAGE and k obs were determined by plotting the fraction cleaved as a function of time. Curves were fitted according to pseudo‐first order association kinetics.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) Project‐ID 398967434‐TRR261 and MA 3442/2‐2 to G.M. D.S. acknowledges a stipend according to the Landesgraduiertenförderungsgesetz. Open Access funding enabled and organized by Projekt DEAL.
Stängle D., Silkenath B., Gehle P., Esser A., Mayer G., Wittmann V., Chem. Eur. J. 2023, 29, e202202378.
Contributor Information
Prof. Dr. Günter Mayer, Email: gmayer@uni-bonn.de.
Prof. Dr. Valentin Wittmann, Email: mail@valentin-wittmann.de.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.J. O'Neill, Tackling Drug-Resistant Infections Globally: Final Report and Recommendations, The Review on Antimicrobial Resistance, http://amr-review.org, 2016.
- 2. Dougan G., Dowson C., Overington J., Next Generation Antibiotic Discovery Symposium Participants, Drug Discovery Today 2019, 24, 452–461. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Mandal M., Breaker R. R., Nat. Rev. Mol. Cell Biol. 2004, 5, 451–463; [DOI] [PubMed] [Google Scholar]
- 3b. Serganov A., Nudler E., Cell 2013, 152, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Sudarsan N., Wickiser J. K., Nakamura S., Ebert M. S., Breaker R. R., Genes Dev. 2003, 17, 2688–2697; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Sudarsan N., Cohen-Chalamish S., Nakamura S., Emilsson G. M., Breaker R. R., Chem. Biol. 2005, 12, 1325–1335; [DOI] [PubMed] [Google Scholar]
- 4c. Blount K. F., Breaker R. R., Nat. Biotechnol. 2006, 24, 1558–1564; [DOI] [PubMed] [Google Scholar]
- 4d. Blount K. F., Wang J. X., Lim J., Sudarsan N., Breaker R. R., Nat. Chem. Biol. 2007, 3, 44–49; [DOI] [PubMed] [Google Scholar]
- 4e. Lünse C. E., Schüller A., Mayer G., Int. J. Med. Microbiol. 2014, 304, 79–92; [DOI] [PubMed] [Google Scholar]
- 4f. Panchal V., Brenk R., Antibiotics 2021, 10, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winkler W. C., Nahvi A., Roth A., Collins J. A., Breaker R. R., Nature 2004, 428, 281–286. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Barrick J. E., Corbino K. A., Winkler W. C., Nahvi A., Mandal M., Collins J., Lee M., Roth A., Sudarsan N., Jona I., Wickiser J. K., Breaker R. R., Proc. Natl. Acad. Sci. USA 2004, 101, 6421–6426; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. McCown P. J., Roth A., Breaker R. R., RNA 2011, 17, 728–736; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. McCown P. J., Winkler W. C., Breaker R. R., Methods Mol. Biol. 2012, 848, 113–129; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Matzner D., Mayer G., J. Med. Chem. 2015, 58, 3275–3286; [DOI] [PubMed] [Google Scholar]
- 6e. McCown P. J., Corbino K. A., Stav S., Sherlock M. E., Breaker R. R., RNA 2017, 23, 995–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Esser A., Mayer G., Chem. Eur. J. 2022, 28, e202202376. [Google Scholar]
- 8. Ferre-D'Amare A. R., Q. Rev. Biophys. 2010, 43, 423–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Collins J. A., Irnov I., Baker S., Winkler W. C., Genes Dev. 2007, 21, 3356–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. McCarthy T. J., Plog M. A., Floy S. A., Jansen J. A., Soukup J. K., Soukup G. A., Chem. Biol. 2005, 12, 1221–1226; [DOI] [PubMed] [Google Scholar]
- 10b. Lim J., Grove B. C., Roth A., Breaker R. R., Angew. Chem. Int. Ed. 2006, 45, 6689–6693; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6841–6845; [Google Scholar]
- 10c. Davis J. H., Dunican B. F., Strobel S. A., Biochemistry 2011, 50, 7236–7242; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Viladoms J., Fedor M. J., J. Am. Chem. Soc. 2012, 134, 19043–19049; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10e. Fei X., Holmes T., Diddle J., Hintz L., Delaney D., Stock A., Renner D., McDevitt M., Berkowitz D. B., Soukup J. K., ACS Chem. Biol. 2014, 9, 2875–2882; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10f. Schüller A., Matzner D., Lünse C., Wittmann V., Schumacher C., Unsleber S., Brötz-Oesterhelt H., Mayer C., Bierbaum G., Mayer G., ChemBioChem 2017, 18, 435–440. [DOI] [PubMed] [Google Scholar]
- 11. Lünse C. E., Schmidt M. S., Wittmann V., Mayer G., ACS Chem. Biol. 2011, 6, 675–678. [DOI] [PubMed] [Google Scholar]
- 12. Klein D. J., Wilkinson S. R., Been M. D., Ferré-D'Amaré A. R., J. Mol. Biol. 2007, 373, 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matzner D., Schüller A., Seitz T., Wittmann V., Mayer G., Chem. Eur. J. 2017, 23, 12604–12612. [DOI] [PubMed] [Google Scholar]
- 14.Throughout the manuscript, we use the numbering of the parent GlcN derivative for all carba-sugars. The carbaposition is numbered 5a (cf. Scheme 1, first line).
- 15. Barton D. H. R., Augy-Dorey S., Camara J., Dalko P., Delaumény J. M., Géro S. D., Quiclet-Sire B., Stütz P., Tetrahedron 1990, 46, 215–230. [Google Scholar]
- 16.
- 16a. Wang G.-N., Lau P. S., Li Y., Ye X.-S., Tetrahedron 2012, 68, 9405–9412; [Google Scholar]
- 16b. Babczyk A., Wingen L. M., Menche D., Eur. J. Org. Chem. 2020, 2020, 6645–6648. [Google Scholar]
- 17. Chang Y. K., Lee B. Y., Kim D. J., Lee G. S., Jeon H. B., Kim K. S., J. Org. Chem. 2005, 70, 3299–3302. [DOI] [PubMed] [Google Scholar]
- 18. Li Q. R., Kim S. I., Park S. J., Yang H. R., Baek A. R., Kim I. S., Jung Y. H., Tetrahedron 2013, 69, 10384–10390. [Google Scholar]
- 19. Brown H. C., Tetrahedron 1961, 12, 117–138. [Google Scholar]
- 20. Yamamoto T., Nishiuchi Y., Teshima T., Matsuoka H., Yamada K., Tetrahedron Lett. 2008, 49, 6876–6878. [Google Scholar]
- 21.
- 21a. Arosio D., Vrasidas I., Valentini P., Liskamp R. M. J., Pieters R. J., Bernardi A., Org. Biomol. Chem. 2004, 2, 2113–2124; [DOI] [PubMed] [Google Scholar]
- 21b. Neumann J., Weingarten S., Thiem J., Eur. J. Org. Chem. 2007, 2007, 1130–1144. [Google Scholar]
- 22. Baudry D., Ephritikhine M., Felkin H., J. Chem. Soc. Chem. Commun. 1978, 694–695. [Google Scholar]
- 23.
- 23a. Gent P. A., Gigg R., J. Chem. Soc. Perkin Trans. 1 1974, 1446–1455; [Google Scholar]
- 23b. Gent P. A., Gigg R., Conant R., J. Chem. Soc. Perkin Trans. 1 1972, 1535–1542; [DOI] [PubMed] [Google Scholar]
- 23c. Gent P. A., Gigg R., Conant R., J. Chem. Soc. Perkin Trans. 1 1972, 277–278; [DOI] [PubMed] [Google Scholar]
- 23d. Gigg R., Warren C. D., J. Chem. Soc. C 1968, 1903–1911; [Google Scholar]
- 23e. Konstas S., Photaki I., Zervas L., Chem. Ber. 2006, 92, 1288–1293. [Google Scholar]
- 24. Scholl M., Ding S., Lee C. W., Grubbs R. H., Org. Lett. 1999, 1, 953–956. [DOI] [PubMed] [Google Scholar]
- 25. Lummiss J. A. M., Ireland B. J., Sommers J. M., Fogg D. E., ChemCatChem 2014, 6, 459–463. [Google Scholar]
- 26. Compain P., Adv. Synth. Catal. 2007, 349, 1829–1846. [Google Scholar]
- 27. Lahiri R., Kokatla H. P., Vankar Y. D., Tetrahedron Lett. 2011, 52, 781–786. [Google Scholar]
- 28. Sowa T., Ouchi S., Bull. Chem. Soc. Jpn. 1975, 48, 2084–2090. [Google Scholar]
- 29. Nikolaides N., Ganem B., Tetrahedron Lett. 1989, 30, 1461–1464. [Google Scholar]
- 30. Maley F., Lardy H. A., J. Am. Chem. Soc. 1956, 78, 1393–1397. [Google Scholar]
- 31. Joseph A. A., Dhurandhare V. M., Chang C.-W., Verma V. P., Mishra G. P., Ku C.-C., Lin C.-C., Wang C.-C., Chem. Commun. 2015, 51, 104–106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.