

Abstract
Aim
To use continuous glucose monitoring (CGM)‐based time‐in‐range (TIR) as a primary efficacy endpoint to compare the second‐generation basal insulin (BI) analogues insulin glargine 300 U/ml (Gla‐300) and insulin degludec 100 U/ml (IDeg‐100) in adults with type 1 diabetes (T1D).
Materials and Methods
InRange was a 12‐week, multicentre, randomized, active‐controlled, parallel‐group, open‐label study comparing glucose TIR and variability between Gla‐300 and IDeg‐100 using blinded 20‐day CGM profiles. The inclusion criteria consisted of adults with T1D treated with multiple daily injections, using BI once daily and rapid‐acting insulin analogues for at least 1 year, with an HbA1c of 7% or higher and of 10% or less at screening.
Results
Overall, 343 participants were randomized: 172 received Gla‐300 and 171 IDeg‐100. Non‐inferiority (10% relative margin) of Gla‐300 versus IDeg‐100 was shown for the primary endpoint (percentage TIR ≥ 70 to ≤ 180 mg/dl): least squares (LS) mean (95% confidence interval) 52.74% (51.06%, 54.42%) for Gla‐300 and 55.09% (53.34%, 56.84%) for IDeg‐100; LS mean difference (non‐inferiority): 3.16% (0.88%, 5.44%) (non‐inferiority P = .0067). Non‐inferiority was shown on glucose total coefficient of variation (main secondary endpoint): LS mean 39.91% (39.20%, 40.61%) and 41.22% (40.49%, 41.95%), respectively; LS mean difference (non‐inferiority) −5.44% (−6.50%, −4.38%) (non‐inferiority P < .0001). Superiority of Gla‐300 over IDeg‐100 was not shown on TIR. Occurrences of self‐measured and CGM‐derived hypoglycaemia were comparable between treatment groups. Safety profiles were consistent with known profiles, with no unexpected findings.
Conclusions
Using clinically relevant CGM metrics, InRange shows that Gla‐300 is non‐inferior to IDeg‐100 in people with T1D, with comparable hypoglycaemia and safety profiles.
Keywords: basal insulin, continuous glucose monitoring, glycaemic control, insulin analogues, randomized trial, type 1 diabetes
1. INTRODUCTION
Type 1 diabetes (T1D) affects approximately 10% of the overall diabetes population and its incidence is increasing globally. 1 Initial optimal control is crucial in T1D, and earlier implementation of intensive therapy is associated with greater reduction in the risk of macrovascular and microvascular complications. 2 However, intensive therapy increases the risk of hypoglycaemia, which may present a barrier to achieving appropriate glycaemic control and could be one reason why many people with T1D fail to achieve recommended HbA1c levels of less than 7% (<53 mmol/mol). 3 , 4 In a recent US study of health records from more than 30 000 people with T1D, 80% had suboptimal (≥ 7%) HbA1c levels. 5 Use of continuous glucose monitoring (CGM) in T1D management is becoming more common, 6 and is associated with improved metabolic control in adults and children/adolescents. 7 , 8 Despite the increasing use of diabetes technology including CGM, hypoglycaemia remains a significant issue in T1D management. 4
The second‐generation basal insulin (BI) analogues insulin glargine 300 U/ml (Gla‐300) and insulin degludec 100 U/ml (IDeg‐100) offer more stable and prolonged pharmacokinetic and pharmacodynamic (PK/PD) profiles versus the first‐generation analogue insulin glargine 100 U/ml (Gla‐100). 9 , 10 In randomized controlled trials (RCTs), these PK/PD profiles translated into similar glycaemic control versus Gla‐100, with less hypoglycaemia. 11 , 12 Previous direct PK/PD comparisons of Gla‐300 versus IDeg‐100 in people with T1D have reported somewhat conflicting results. In a study by Bailey et al., Gla‐300 provided more stable 24‐hour PD and more even PK profiles than IDeg‐100, 13 whereas a study by Heise et al. indicated lower day‐to‐day and within‐day variability with IDeg‐100 versus Gla‐300. 14 A more recent crossover PK/PD analysis of Gla‐300 and IDeg‐100 in people with T1D after 3 months of optimal insulin titration concluded that Gla‐300 and IDeg‐100 provided similar glycaemic control and equivalent PD profiles. 15 Direct comparisons of Gla‐300 and IDeg‐100 in people with T1D in more clinically relevant settings are limited. A crossover study of Gla‐300 and IDeg‐100 in 46 people with T1D reported that both treatments had similar glucose‐stabilizing effects with regards to day‐to‐day variability of fasting plasma glucose. 16 In the OneCARE study, an observational, retrospective, multicentre, cross‐sectional study using CGM including 199 people with T1D, switching from first‐generation BI analogues to either Gla‐300 or IDeg‐100 resulted in generally similar effectiveness and safety profiles, with more time in glucose range (TIR; ≥ 70 to ≤ 180 mg/dl; ≥ 3.9 to ≤ 10 mmol/L) during the night for Gla‐300. 17
The use of CGM has allowed the generation of clinical goals such as the percentage of time within the target glucose range (i.e. TIR), time above range (TAR) and time below range (TBR), and glucose variability metrics such as glucose coefficient of variation (CV), which add to and complement traditional metrics such as HbA1c. 18 , 19 Furthermore, standard blood glucose monitoring can underestimate hypoglycaemia, 20 , 21 , 22 with one study in type 2 diabetes reporting a 3‐8–fold increase in the frequency of events with CGM. 20 Such a differential is probable to be magnified at night when some events will be undetected by the person with diabetes. The utility of CGM is therefore becoming clear, and during 2013‐2019, CGM was used in 11% of clinical trials for BIs. 23 However, to date no RCTs have used TIR metrics as primary/secondary endpoints in head‐to‐head investigations of Gla‐300 and IDeg‐100.
InRange is the first large RCT designed to use CGM‐derived metrics to directly assess the efficacy and safety of Gla‐300 versus IDeg‐100 in people with T1D, with TIR (≥ 70 to ≤ 180 mg/dl; ≥ 3.9 to ≤ 10 mmol/L) as the primary endpoint.
2. MATERIALS AND METHODS
2.1. Study design
The study design of InRange (NCT04075513) has been described previously. 24 Briefly, this was a 12‐week, phase 4, multicentre (40 sites, seven countries), randomized, active‐controlled, parallel‐group, open‐label study to compare TIR and glucose variability between Gla‐300 and IDeg‐100 using blinded CGM. The study was conducted in accordance with the Declaration of Helsinki with all relevant amendments, the Council for International Organizations of Medical Sciences International Ethical Guidelines, the applicable International Conference on Harmonization Good Clinical Practice Guidelines, and all applicable laws and regulations of the countries in which the study was carried out.
2.2. Study population
The inclusion criteria were age 18‐70 years with T1D; an HbA1c of 7% or higher to 10% or less (≥ 53 to ≤ 86 mmol/mol) at screening; prior treatment with a BI analogue once daily and rapid‐acting insulin analogues for at least 1 year; and a stable basal/bolus insulin regimen for 30 days prior to screening. Further inclusion and exclusion criteria can be found in the published study design article. 24
2.3. Randomization and study intervention
The study length was approximately 18 weeks, including 1‐2 weeks screening; a 4‐week run‐in; a 12‐week treatment period; and a 2‐ to 4‐day follow‐up period (Figure S1). Five onsite visits were scheduled and there were weekly telephone contacts to monitor insulin titration between visits. During the run‐in, participants were trained on study‐related procedures, provided with a glucometer (Accu‐Chek Performa) and accessories and underwent stabilization of their current basal and mealtime insulin treatment (weeks −4 to −3). Current basal and mealtime insulins were titrated to stabilize fasting and 2‐hour postprandial self‐measured plasma glucose (SMPG) targets of 70 mg/dl or higher to less than 100 mg/dl (≥ 3.9 to < 5.6 mmol/L), and 130 mg/dl or higher to less than or equal to 180 mg/dl (≥ 7.2 to ≤ 10 mmol/L), respectively, while avoiding hypoglycaemia. Dose adjustment of mealtime insulin could be based on a pattern of postmeal SMPG data from the prior 3 days (simple titration) or the carbohydrate content of the meal.
Baseline CGM values were determined using the blinded CGM device (G6, Dexcom Inc, San Diego, CA) for 20 days at the end of the run‐in period after stabilization of current insulins. To be eligible for randomization, a minimum of 10 days (not necessarily consecutive) of useable CGM data generated during the run‐in period was required. Eligible participants were randomized 1:1 to receive a once‐daily subcutaneous injection of Gla‐300 or IDeg‐100 in the morning. The randomization scheme was provided by the study statistician to an interactive response technology (IRT) system, which generated the randomization list and allocated participants to treatment arms. At the screening visit, the investigator contacted the IRT centre to receive the participant number.
The randomized treatment period (weeks 0‐12) included an insulin titration period (expected to be up to week 8) and a CGM data collection period over 20 consecutive days during weeks 10‐12 (to ensure 10 days of evaluable data). Initiation doses of Gla‐300 or IDeg‐100 corresponded to the previous BI dose. During the titration period, doses of Gla‐300 or IDeg‐100 were titrated until participants achieved the target fasting SMPG of 70 mg/dl or higher to less than 100 mg/dl (≥ 3.9 to < 5.6 mmol/L), while avoiding hypoglycaemia. Dose adjustments for Gla‐300 and IDeg‐100 were based on median fasting SMPG values from the previous 3 days, including values recorded on the day of titration, as measured by the participants using glucometers. The mealtime insulin analogue was also actively titrated to achieve the 2‐hour postprandial SMPG target (≥ 130 to ≤ 180 mg/dl [≥ 7.2 to ≤ 10 mmol/L]), while avoiding hypoglycaemia. The dose was titrated at least weekly (but not more often than every 3 days). The best efforts were made to reach the targets by 8 weeks after randomization; thereafter, the dose was maintained until the end of the study. CGM data were blinded to both investigators and participants. Post‐treatment safety information was collected 2‐4 days after the last insulin dose.
2.4. Study objectives and endpoints
The primary objective of InRange was to show the non‐inferiority of Gla‐300 versus IDeg‐100 on glycaemic control, assessed by TIR and glycaemic variability. Secondary objectives were to evaluate glycaemic control and variability metrics using CGM, and to evaluate the safety of Gla‐300 in comparison with IDeg‐100.
The primary endpoint was percentage TIR ≥ 70 to ≤ 180 mg/dl (≥ 3.9 to ≤ 10 mmol/L) at week 12, assessed using blinded CGM. The main secondary efficacy endpoint was glucose total CV at week 12, and the additional secondary efficacy endpoints were the change from baseline to week 12 in HbA1c, a percentage TAR of more than 180 mg/dl (> 10 mmol/L) and a TBR of less than 70 mg/dl (< 3.9 mmol/L) at week 12, as well as within‐day and between‐day glucose CV at week 12. Safety endpoints were assessed in the safety population (all randomized participants who received at least one dose of study drug) and analysed according to the treatment received. These were the incidence and event rates of hypoglycaemia during the on‐treatment period reported by each participant and were based on SMPG readings from a glucometer (any, severe [Level 3, as defined by the American Diabetes Association {ADA} 25 ], documented < 70, < 70 and ≥ 54 mg/dl [Level 1], or < 54 mg/dl [Level 2], analysed as nocturnal [00:00 AM‐05:59 AM], diurnal [06:00 AM‐11:59 PM] or at any time [24 hours]) and the incidence of adverse events (AEs). As a post hoc analysis, the rates of hypoglycaemia were also analysed using CGM data from weeks 10‐12 (the number of periods with at least 15 minutes of sensor glucose values of < 70 mg/dl, < 70 mg/dl and ≥ 54 mg/dl, and < 54 mg/dl, respectively, with an event ending when glucose returned to above 70 mg/dl for at least 15 minutes; an event of < 70 and ≥ 54 mg/dl was defined as a period of < 70 mg/dl without a 15‐minute excursion of < 54 mg/dl before the end of the event) and prolonged hypoglycaemia (the number of periods with at least 120 minutes of sensor glucose of < 70 mg/dl). SMPG‐derived hypoglycaemia rates during the CGM data collection period, weeks 10‐12, were also analysed post hoc. Daily insulin dose was assessed as an exploratory endpoint in the safety population.
2.5. Statistical analysis
Non‐inferiority on the primary endpoint (percentage TIR ≥ 70 to ≤ 180 mg/dl [≥ 3.9 to ≤ 10 mmol/L]) was tested with a one‐sided type I error of 2.5%, and a relative non‐inferiority margin of 10% (note relative, not a difference in % units; e.g. a 10% relative margin for 50% TIR is equal to a 5% difference in TIR). A sample size of 169 randomized participants per treatment group was calculated to provide at least 90% power to show non‐inferiority, assuming a TIR of 56% in the IDeg‐100 arm, a common SD of 14.7% and a non‐evaluability rate of 22%. The percentage TIR at week 12 (using available data from the 12‐week randomized period) was analysed in the intention‐to‐treat (ITT) population (all randomized participants, irrespective of the treatment received), according to the treatment group allocated by randomization using an analysis of covariance (ANCOVA) model, including the fixed categorical effects of treatment group, randomization stratum of screening HbA1c (< 8.0% vs. ≥ 8.0% [< 64 vs. ≥ 64 mmol/mol]), as well as the continuous fixed covariate of baseline percentage TIR value. A multiple imputation method (1000 imputations) under a missing at random assumption was used to address missing values in the ITT population. This procedure provided baseline‐adjusted least‐squares (LS) means estimates at week 12 for both treatment groups, and the between‐group differences, with corresponding standard error (SE) and 95% confidence intervals (CIs). Non‐inferiority on the primary endpoint was shown if the lower bound of the two‐sided 95% CI of the adjusted difference estimate for m1 − 0.9*m0 (where m1 and m0 are the true means for the Gla‐300 and IDeg‐100 groups, respectively) was greater than 0. Sensitivity analyses were conducted for the primary endpoint in the per protocol (PP) population and in the ITT population using multiple imputation under the missing not at random (MNAR) assumption.
The total glucose CV was analysed using the same model described for the primary endpoint using the ITT population. Non‐inferiority was shown if the upper bound of the two‐sided 95% CI of the adjusted difference estimate of m1 − 1.1*m0 (m1 = true mean of Gla‐300 for the glucose total CV and m0 = true mean of IDeg‐100) at week 12 was less than 0.
To control the type I error, a hierarchical step‐down testing procedure was applied for the primary endpoint and the main secondary endpoints. Step 1 assessed non‐inferiority of Gla‐300 versus IDeg‐100 on the percentage TIR with a relative non‐inferiority margin of 10%. Step 2: only if non‐inferiority was shown in step 1, then non‐inferiority of Gla‐300 versus IDeg‐100 on the glucose total CV (main secondary endpoint) was tested. Step 3: only if non‐inferiority was shown in step 2, then superiority of Gla‐300 versus IDeg‐100 on the percentage TIR was tested.
For other endpoints, no multiplicity adjustments were made, and any 95% CI values are presented for descriptive purposes only. Changes in HbA1c were analysed using an ANCOVA model that included the fixed categorical effect of treatment group and the continuous fixed covariate of baseline HbA1c.
3. RESULTS
3.1. Study population
A total of 343 participants were randomized, 172 to treatment with Gla‐300 and 171 to treatment with IDeg‐100 (Figure S 2 ). At the end of the run‐in period, before randomization, 23 of the screened patients were excluded because of failure to obtain a minimum of 10 days of useable baseline CGM data. Before randomization, most participants (91.0%) used Gla‐100 as their BI. All participants randomized were treated as planned and were included in the ITT and safety populations. There were 153 and 159 participants in the PP populations for Gla‐300 and IDeg‐100, respectively. Most participants completed the 12‐week treatment period (95.3% Gla‐300, 97.7% IDeg‐100). A total of eight participants in the Gla‐300 group and four participants in the IDeg‐100 group discontinued the study treatment prematurely. No treatment discontinuation was related to COVID‐19, and no participant was prematurely withdrawn from treatment because of an AE. Demographic, baseline and disease characteristics were overall balanced between the two treatment groups (Table 1). The mean (SD) age was 42.8 (13.28) years and body mass index was 27.3 (4.77) kg/m2, with 39.2% of participants categorized as overweight and 25.7% obese. Mean (SD) time since diagnosis of T1D was 20.5 (12.78) years; time since first intake of BI analogue and mealtime insulin analogue was 8.6 (6.24) and 9.17 (7.27) years, respectively. At week 12, 96.9% and 93.2% of participants had at least 10 days of evaluable CGM data, for Gla‐300 and IDeg‐100, respectively, with a mean of 16 days' evaluable data in both groups.
TABLE 1.
Baseline characteristics
| Baseline characteristic | Gla‐300 N = 172 | IDeg‐100 N = 171 | All N = 343 |
|---|---|---|---|
| Age, y, mean (SD) | 42.9 (13.53) | 42.8 (13.05) | 42.8 (13.28) |
| Sex, female, n (%) | 86 (50) | 74 (43.3) | 160 (46.6) |
| Body weight, kg, mean (SD) | 80.5 (15.95) | 78.8 (14.57) | 79.6 (15.28) |
| BMI, kg/m2, mean (SD) | 27.6 (5.07) | 27.0 (4.44) | 27.3 (4.77) |
| Time since T1D diagnosis, y, mean (SD) | 20.7 (12.47) | 20.3 (13.12) | 20.5 (12.78) |
| Age at diagnosis, y, mean (SD) | 22.7 (13.22) | 23.1 (12.84) | 22.9 (13.01) |
| Time since first intake of BI analogue treatment, y, mean (SD) | 8.1 (6.20) | 9.1 (6.26) | 8.6 (6.24) |
| Time since first intake of mealtime insulin analogue treatment, y, mean (SD) | 8.6 (7.22) | 9.8 (7.28) | 9.2 (7.27) |
| Method of mealtime insulin titration, n (%) | |||
| Postprandial SMPG data from the last 3 d | 75 (43.6) | 82 (48.8) | 157 (46.2) |
| Carbohydrate content of meal | 97 (56.4) | 86 (51.2) | 183 (53.8) |
| At least one diabetes complication, n (%) | 60 (35.1) | 55 (32.5) | 115 (33.8) |
| Diabetic retinopathy | 30 (17.5) | 36 (21.3) | 66 (19.4) |
| Non‐proliferative diabetic retinopathy | 25 (14.6) | 31 (18.3) | 56 (16.5) |
| Proliferative diabetic retinopathy | 4 (2.3) | 5 (3.0) | 9 (2.6) |
| Diabetic neuropathy | 32 (18.7) | 32 (18.9) | 64 (18.8) |
| Diabetic nephropathy | 13 (7.6) | 8 (4.7) | 21 (6.2) |
| HbA1c ≥ 8%, n (%) | 106 (61.6) | 106 (62.0) | 212 (61.8) |
Abbreviations: BI, basal insulin; BMI, body mass index; Gla‐300, insulin glargine 300 U/ml; IDeg‐100, insulin degludec 100 U/ml; SD, standard deviation; SMPG, self‐measured plasma glucose; T1D, type 1 diabetes.
3.2. Primary and main secondary efficacy endpoints
Non‐inferiority of Gla‐300 with respect to IDeg‐100 was shown on the primary efficacy endpoint (percentage TIR ≥ 70 to ≤ 180 mg/dl [≥ 3.9 to ≤ 10 mmol/L]). The LS mean estimates (95% CI) for TIR at week 12 were 52.74% (51.06%, 54.42%) for Gla‐300 and 55.09% (53.34%, 56.84%) for IDeg‐100; LS mean difference (non‐inferiority) was 3.16% (0.88%, 5.44%) (non‐inferiority P = .0067; Table 2). Non‐inferiority was confirmed in sensitivity analyses of the PP population (P = .0127) and using multiple imputation under the MNAR assumption (P = .0068).
TABLE 2.
Efficacy endpoints
| Gla‐300 | IDeg‐100 | |
|---|---|---|
| Efficacy endpoint (ITT population) | (N = 172) | (N = 171) |
| Time in glucose range ≥ 70 to ≤ 180 mg/dl (≥ 3.9 to ≤ 10 mmol/L) (%) | ||
| Baseline (after optimization of previous insulin therapy), mean (SD) | 51.19 (12.38) | 52.37 (14.24) |
| Week 12 | ||
| LS mean [95% CI] | 52.74 [51.06, 54.42] | 55.09 [53.34, 56.84] |
| Non‐inferiority LS mean difference [95% CI] a | 3.16 [0.88, 5.44] | |
| Non‐inferiority P value | P = .0067 | |
| LS mean difference [95% CI] | −2.35 [−4.75, 0.05] | |
| Superiority P value b | P = .0548 | |
| Glucose total CV (%) | ||
| Baseline (after optimization of previous insulin therapy), mean (SD) | 41.05 (6.92) | 39.90 (6.39) |
| Week 12 | ||
| LS mean [95% CI] | 39.91 [39.20, 40.61] | 41.22 [40.49, 41.95] |
| Non‐inferiority LS mean difference [95% CI] c | −5.44 [−6.50, −4.38] | |
| Non‐inferiority P value | P < .0001 | |
| LS mean difference [95% CI] | −1.32 [−2.32, −0.31] | |
| HbA1c (%) | ||
| Baseline, mean (SD) | 8.29 (0.82) | 8.34 (0.80) |
| Week 12, mean (SD) | 7.51 (0.76) | 7.38 (0.83) |
| LS mean change [95% CI] | −0.75 [−0.87, −0.64] | −0.92 [−1.03, −0.81] |
| LS mean difference | 0.17 [0.01, 0.32] | |
| HbA1c (mmol/mol) | ||
| Baseline, mean (SD) | 67.12 (8.92) | 67.68 (8.74) |
| Week 12, mean (SD) | 58.61 (8.30) | 57.21 (9.06) |
| LS mean change [95% CI] | −8.23 [−9.48, ‐6.98] | −10.04 [−11.26, −8.81] |
| LS mean difference | 1.81 [0.12, 3.50] | |
Abbreviations: CI, confidence interval; CV, coefficient of variation; Gla‐300, insulin glargine 300 U/ml; IDeg‐100, insulin degludec 100 U/ml; ITT, intention to treat; LS, least squares; SD, standard deviation; TIR, time in range.
Primary endpoint; non‐inferiority was tested with a relative margin of 10% (i.e. non‐inferiority mean difference = Gla‐300 − 0.9*IDeg‐100). Non‐inferiority was shown if the upper bound of the two‐sided 95% CI of the adjusted difference was > 0.
Superiority was tested because non‐inferiority for TIR and CV were shown.
Non‐inferiority was tested with a relative margin of 10% (i.e. non‐inferiority mean difference = Gla‐300 − 1.1*IDeg‐100). Non‐inferiority was shown if the upper bound of the two‐sided 95% CI of the adjusted difference was < 0.
Non‐inferiority of Gla‐300 with respect to IDeg‐100 was shown on the main secondary endpoint, glucose total CV. LS mean at week 12 was 39.91% (39.20%, 40.61%) for Gla‐300 and 41.22% (40.49%, 41.95%) for IDeg‐100 (Table 2); LS mean difference (non‐inferiority) was −5.44% (−6.50%, −4.38%) (P < .0001).
Following the hierarchical testing procedure, the superiority of Gla‐300 over IDeg‐100 on the percentage TIR was tested. Superiority was not shown, with an LS mean difference of −2.35% (−4.75%, 0.05%) and P = .0548.
3.3. Other secondary endpoints
Mean ± SD HbA1c values at baseline were 8.29% ± 0.82% (67.12 ± 8.92 mmol/mol) in the Gla‐300 group, and 8.34% ± 0.80% (67.68 ± 8.74 mmol/mol) in the IDeg‐100 group. At week 12, HbA1c was 7.51% ± 0.76% (58.61 ± 8.30 mmol/mol) in the Gla‐300 group and 7.38% ± 0.83% (57.21 ± 9.06 mmol/mol) in the IDeg‐100 group (Table 2 and Figure S3). The LS means (95% CI) of HbA1c change from baseline to week 12 were −0.75% (−0.87% to −0.64%) (−8.23 [−9.48, −6.98] mmol/mol) for Gla‐300 and −0.92% (−1.03% to −0.81%) (−10.04 [−11.26, −8.81] mmol/mol) for IDeg‐100.
For percentage TAR of more than 180 mg/dl (> 10 mmol/L) at week 12, LS means (95% CI) were 41.52% (39.60%, 43.44%) and 38.31% (36.34%, 40.28%) for Gla‐300 and IDeg‐100, respectively; LS mean difference was 3.21% (0.49%, 5.93%). LS means for percentage TBR less than 70 mg/dl (< 3.9 mmol/L) at week 12 were 5.55% (4.85%, 6.24%) and 6.49% (5.78%, 7.21%), respectively; LS mean difference was −0.95 (−1.93, 0.04). 26 Glucose within‐day CV at week 12 was 33.48% (32.81%, 34.16%) and 34.37% (33.68%, 35.06%), respectively; LS mean difference was −0.89% (−1.84%, 0.07%). 27 Glucose between‐day CV was 17.23% (16.44%, 18.03%) and 18.08% (17.27%, 18.90%), respectively; LS mean difference was −0.85% (−1.98%, 0.28%). 27
3.4. Anytime (24 hours) self‐reported/SMPG‐derived hypoglycaemia during the on‐treatment period
Overall, 165 participants (95.9%) treated with Gla‐300 and 166 participants (97.1%) treated with IDeg‐100 reported experiencing at least one hypoglycaemia event during the on‐treatment period. The rate of hypoglycaemia of any category at any time was comparable between groups: 2.1 events per patient‐week of exposure in the Gla‐300 group and 2.2 events per patient‐week in the IDeg‐100 group. The incidence and event rates of hypoglycaemia during the on‐treatment period (any, severe [Level 3], documented < 70, < 70 and ≥ 54 mg/dl [Level 1], or < 54 mg/dl [Level 2]) are presented in Figure 1. Severe hypoglycaemia at any time occurred in eight participants (4.7%) treated with Gla‐300 and in 10 participants (5.8%) treated with IDeg‐100, with a rate of 0.004 and 0.006 events per patient‐week (0.2 and 0.3 events per patient‐year), respectively. The incidence and event rates for symptomatic and asymptomatic hypoglycaemia are presented in Table S1.
FIGURE 1.
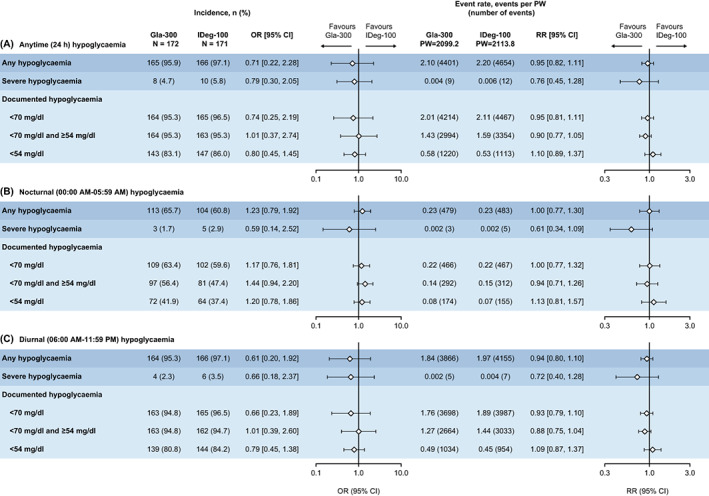
Self‐reported/SMPG‐derived hypoglycaemia incidence and event rate during the on‐treatment period for A) Anytime, B) Nocturnal, and C) Diurnal hypoglycaemia. CI, confidence interval; Gla‐300, insulin glargine 300 U/ml; IDeg‐100, insulin degludec 100 U/ml; OR, odds ratio; PW, patient‐week; RR, rate ratio; SMPG, self‐measured plasma glucose
3.5. Nocturnal/diurnal self‐reported/SMPG‐derived hypoglycaemia during the on‐treatment period
Comparable rates of any nocturnal (00:00 AM‐05:59 AM) hypoglycaemic event (0.23 events per patient‐week) were observed in both groups (Figure 1). Severe nocturnal hypoglycaemia occurred in three participants (1.7%) treated with Gla‐300 and in five participants (2.9%) treated with IDeg‐100, with a rate of 0.002 events per patient‐week (0.1 events per patient‐year) in both groups. The rate of any diurnal (06:00 AM‐11:59 PM) hypoglycaemic event was also similar between Gla‐300 and IDeg‐100 (1.84 and 1.97 events per patient‐week, respectively). Severe diurnal hypoglycaemia occurred in four participants (2.3%) treated with Gla‐300 and six participants (3.5%) treated with IDeg‐100, with a rate of 0.002 and 0.004 events per patient‐week (0.1 and 0.2 events per patient‐year), respectively. Incidence and event rates for nocturnal/diurnal symptomatic and asymptomatic hypoglycaemia are presented in Table S1.
3.6. CGM‐derived hypoglycaemia
Post hoc analysis of event rates for hypoglycaemia (documented < 70 mg/dl, < 70 mg/dl and ≥ 54 mg/dl [ADA Level 1], or < 54 mg/dl [ADA Level 2]) captured by CGM (weeks 10‐12) were comparable between treatment groups. However, CGM‐derived rates were between 2‐6–fold higher than SMPG‐derived rates during the CGM data collection period (Figure 2). The rate of prolonged hypoglycaemia captured by CGM was comparable between treatment groups.
FIGURE 2.

CGM‐derived and SMPG‐derived hypoglycaemia event rate during the on‐treatment CGM data collection period (weeks 10‐12), for A) Anytime, B) Nocturnal, and C) Diurnal hypoglycaemia. †CGM: number of periods with at least 15 minutes with sensor glucose < 70 mg/dl. Event ends when glucose returns above 70 mg/dl for at least 15 minutes. ‡CGM: number of periods with at least 15 minutes with sensor glucose < 70 mg/dl and ≥ 54 mg/dl. Event ends when glucose returns above 70 mg/dl for at least 15 minutes. §CGM: number of periods with at least 15 minutes with sensor glucose < 54 mg/dl. Event ends when glucose returns above 70 mg/dl for at least 15 minutes. ¶Number of periods with at least 120 minutes with sensor glucose < 70 mg/dl. Event ends when glucose returns above 70 mg/dl for at least 15 minutes. CGM, continuous glucose monitoring; CI, confidence interval; Gla‐300, insulin glargine 300 U/ml; IDeg‐100, insulin degludec 100 U/ml; PW, patient‐week; RR, rate ratio; SMPG, self‐measured plasma glucose
3.7. Safety
The proportion of participants with any treatment‐emergent adverse events (TEAEs) was 29.1% in the Gla‐300 group and 20.5% in the IDeg‐100 group. The proportions of participants with any serious TEAE were similar between treatment groups (4.1% in Gla‐300 vs. 4.7% in IDeg‐100). No unexpected TEAEs were reported. No participant had a TEAE leading to permanent treatment discontinuation, and none died from a TEAE (Table 3).
TABLE 3.
Safety
| Incidence, n (%) | Gla‐300 (N = 172) | IDeg‐100 (N = 171) |
|---|---|---|
| Any TEAE | 50 (29.1) | 35 (20.5) |
| Any treatment‐emergent SAE | 7 (4.1) | 8 (4.7) |
| Any TEAE leading to death | 0 | 0 |
| Any TEAE leading to permanent treatment discontinuation | 0 | 0 |
| Any treatment‐related TEAE | 4 (2.3) | 7 (4.1) |
Abbreviations: Gla‐300, insulin glargine 300 U/ml; IDeg‐100, insulin degludec 100 U/ml; SAE, serious adverse event; TEAE, treatment‐emergent adverse event.
3.8. Insulin dose and body weight
Daily insulin doses (including total daily insulin, basal insulin, mealtime insulin, ratio of basal to mealtime insulin dose) at baseline, week 8 and week 12 are presented in Table S2. Mean ± SD daily basal insulin dose at baseline was 30.4 ± 13.8 U (0.38 ± 0.15 U/kg) in the Gla‐300 group and 28.8 ± 14.0 U (0.36 ± 0.15 U/kg) in the IDeg‐100 group; daily basal insulin dose increased to 37.1 ± 15.3 U (0.46 ± 0.16 U/kg) and 33.5 ± 16.5 (0.41 ± 0.16 U/kg) at week 12, respectively. Mean changes from baseline to week 12 were 7.4 ± 9.2 U (0.09 ± 0.11 U/kg) in the Gla‐300 group and 4.4 ± 9.6 U (0.04 ± 0.12 U/kg) in the IDeg‐100 group. Mealtime insulin dose at baseline was 23.3 ± 15.8 U (0.29 ± 0.18 U/kg) for Gla‐300 and 21.8 ± 12.2 U (0.27 ± 0.14 IU/kg) for IDeg‐100 and changed very little over the 12‐week study period. Body weight change from baseline to week 12 (mean ± SD) was 0.97 ± 2.46 kg in the Gla‐300 group and 0.76 ± 2.82 kg in the IDeg‐100 group.
4. DISCUSSION
The primary objective of this study was achieved as the non‐inferiority of Gla‐300 to IDeg‐100 was shown for the primary endpoint (i.e. percentage TIR ≥ 70 to ≤ 180 mg/dl [≥ 3.9 to ≤ 10 mmol/L]). Superiority for the percentage TIR of 70 mg/dl or higher to 180 mg/dl or less (≥ 3.9 to ≤ 10 mmol/L) was not shown. Utilization of CGM TIR metrics in clinical trials has so far been limited, and InRange is the first RCT to use TIR as a primary endpoint to assess the two second‐generation BIs, Gla‐300 and IDeg‐100. With the increasing body of evidence regarding the advantages of using CGM to improve glycaemic control, CGM use in clinical practice is expanding, and it has been reported that each 5% improvement in TIR is associated with clinically significant benefits. 18 Considering the well‐documented clinical benefit of using CGM and the comprehensive data it provides, incorporation of CGM and TIR metrics into clinical trials is valuable and should be expanded further.
Assessing the wealth of data produced by CGM is particularly valuable as both TIR and glycaemic variability may be associated with an increased risk of diabetes complications such as retinopathy. 28 , 29 Furthermore, greater glycaemic variability may also be associated with lower quality of life and negative moods, 30 so treatment options that can reduce such variability may positively impact the lives of people with diabetes. InRange also showed non‐inferiority of Gla‐300 versus IDeg‐100 on the main secondary endpoint, glucose total CV (assessed prior to testing superiority on the primary endpoint). There were clinically relevant improvements in HbA1c in both treatment groups.
InRange results show that comparable incidences and event rates of hypoglycaemia were observed in the two treatment groups, for all categories assessed, and for all time periods (diurnal/nocturnal/anytime). The post hoc analysis of CGM‐derived hypoglycaemia data found that CGM captured approximately 2‐6–fold higher hypoglycaemia event rates than SMPG during the same period (weeks 10‐12), with the greatest disparity between nocturnal events, emphasizing the importance of using CGM in diabetes studies to adequately capture hypoglycaemia. CGM‐derived hypoglycaemia rates were comparable between treatment groups. Overall, the safety profile of Gla‐300 and IDeg‐100 was consistent with the known safety profile of each BI analogue, and no unexpected safety findings were identified.
The results of the InRange study support several other studies comparing the efficacy and safety of Gla‐300 and IDeg‐100. 15 , 16 , 17 It is particularly noteworthy that the findings of the non‐randomized OneCARE study, 17 which used CGM and TIR (≥ 70 to ≤ 180 mg/dl [≥ 3.9 to ≤ 10 mmol/L]) as an outcome, and which suggested that the efficacy and safety of Gla‐300 and IDeg‐100 are comparable in people with T1D insufficiently controlled with first‐generation BIs, have now been replicated in this randomized trial.
The strengths of the InRange study include its prospective, multicentre, randomized, active‐controlled, parallel‐group, comparative design, and the use of clinically relevant TIR metrics, as recommended by recent guidelines. CGM provides a comprehensive evaluation of glycaemic variability, allowing a thorough comparison of BI therapies. Compared with standard self‐monitoring of blood glucose, CGM has the potential to provide a more complete assessment of hypoglycaemia occurrence, particularly nocturnal events, which may be under‐reported. 31 In InRange, blinded CGM measurement was used and allowed a direct assessment of the properties of the insulins, rather than the effect of CGM. Furthermore, CGM data were blinded for both investigators and participants to avoid treatment decisions being made based on primary endpoint data.
However, the InRange study also has limitations. The study precludes conclusions for people with T1D using CGM in standard care, because the study was blinded and TIR was lower than the recommended general clinical target (> 70%). 18 Furthermore, the BIs were administered in the morning and therefore further study would be required to assess evening administration; however, based on the PK/PD profiles of Gla‐300 and IDeg‐100, a significant impact on the results would not be expected with different dosing times. The study also included only previous BI users, so further study would be needed to investigate insulin‐naïve people. In particular, it would be of interest to assess CGM during the initial titration period (when most insulin dose changes would be expected in insulin‐naïve people), because it was during this period that the BRIGHT study found lower rates of hypoglycaemia with Gla‐300 versus IDeg‐100, in insulin‐naïve people with type 2 diabetes. 32 The open‐label design was a limitation, as it was not possible to blind treatment because Gla‐300 and IDeg‐100 pens are distinguishable. However, the potential for bias because of the open‐label design was minimized by collecting objective glycaemic data. Finally, a treatment period of 12 weeks could be considered short compared with other clinical trials that evaluate changes in glycaemic control from baseline, however, the focus of the InRange study was the comparison of CGM‐derived TIR for Gla‐300 versus IDeg‐100, and the treatment duration was designed to allow dose optimization to occur before CGM data were collected. Although the CGM data collection period in InRange was of a length that has been shown to provide data that correlate with long‐term (3‐month) glycaemic outcomes, 18 , 29 , 33 longer term CGM data would provide more robust conclusions.
In summary, using two clinically relevant CGM metrics as primary and secondary endpoints for the first time, the InRange study of people with T1D shows that Gla‐300 is non‐inferior to IDeg‐100 in terms of glycaemic control as measured by TIR, and glycaemic variability as measured by glucose CV, with comparable occurrences of hypoglycaemia and safety profiles.
AUTHOR CONTRIBUTIONS
T.B., T.D. and E.R. participated in the conception and design of the study and interpretation of the data. S.V.E., P.C., R.M.B., J.W., B.M. and V.P. participated in the conception and design of the study, data acquisition and data analysis or interpretation. M.C. contributed to data acquisition and data analysis or interpretation. All authors participated in the writing, reviewing and editing of the manuscript, and had final responsibility for approving the published version. T.B. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
CONFLICT OF INTEREST
T.B. has received honoraria for participation on advisory boards for Novo Nordisk, Sanofi, Eli Lilly, Boehringer Ingelheim, Indigo and Medtronic, and as a speaker for AstraZeneca, Eli Lilly, Novo Nordisk, Medtronic, Pfizer, Sanofi, Dexcom, Abbott and Roche; owns stocks of DreaMed Diabetes; T.B.'s institution has received research grant support and travel expenses from Abbott Diabetes Care, Medtronic, Novo Nordisk, Sanofi, Sandoz, Novartis and Zealand Pharma. T.B. is funded in part by the Slovenian Research Agency grant # P3‐0343. T.D. declares speaker, advisory panel or research support from AstraZeneca, Bayer, Boehringer, Bristol‐Myers Squibb, Dexcom, Eli Lilly, Insulet, Medtronic, Novo Nordisk, Roche, Sanofi, Unomedical and Ypsomed and is a shareholder from DreaMed Diabetes, Ltd. S.V.E. is a board member of Senseonics and serves on the speakers' bureau and advisory panel for AstraZeneca, Dexcom, Eli Lilly, MannKind, Merck, Novo Nordisk and Sanofi. P.C. serves on advisory panels for Abbott, Cellnovo, Eli Lilly, Medtronic, Novo Nordisk, Roche and Sanofi; has received research support from Beta‐O2 and Medtronic; and serves on speakers bureaus for Abbott, Eli Lilly, Johnson & Johnson, Medtronic, Merck (MSD), Novo Nordisk, Roche and Sanofi. E.R. has received consultancy honoraria from Abbott, AstraZeneca, Boehringer Ingelheim, Dexcom Inc., Eli Lilly, Hillo, Insulet Inc., LifeScan, Medtronic, Medirio, Novo Nordisk, Roche and Sanofi, and research support from Dexcom Inc., Insulet Inc., Roche and Tandem. R.M.B. has received research support from, consulted for, or has been on a scientific advisory board for Abbott Diabetes Care, Dexcom, Eli Lilly, Johnson & Johnson, Medtronic, Novo Nordisk, Onduo, Roche, Sanofi and United HealthCare. His research is partly funded by the National Institute of Diabetes and Digestive and Kidney Diseases (National Institutes of Health grant DK108611). R.M.B.'s employer, the non‐profit HealthPartners Institute, contracts for his services and no personal income goes to R.M.B. J.W., B.M., V.P. and M.C. are employees and shareholders of Sanofi.
Supporting information
Figure S1. Study design
Figure S2. Study disposition
Figure S3. HbA1c change from baseline to Week 12 (ITT population; secondary endpoint, descriptive data)
Table S1. Symptomatic and asymptomatic SMPG‐derived hypoglycemia (safety population)
Table S2. Insulin dose change (ITT population)
ACKNOWLEDGEMENTS
The authors thank the study participants, trial staff and investigators for their participation (the list of investigators is provided in the Appendix). The authors thank Sirisha Pedapudi, MS, MSc (Sanofi), for coordinating the development, facilitating author discussions and critical review of this manuscript. Medical writing support and editorial assistance was provided by Lois Grant, PhD, of Fishawack Communications Ltd, part of Fishawack Health, and was funded by Sanofi.
APPENDIX A. List of Participating Principal Investigators
Brazil: Denise Franco, Joselita Maria França de Siqueira Bodart, Antônio Roberto Chacra, Miguel Nasser Hissa, Luis Henrique Canani, Joao Felício.
Germany: Thomas Schaum, Karsten Milek, Klaus Busch.
Hungary: László Könyves, László Deak, Zsuzsanna Papp, József Pauer, Gyöngyi Csécsei.
The Netherlands: Mazin AlHakim.
Turkey: Ilgin Yildirim Simsir, Oguzhan Deyneli.
UK: Melanie Davies, Jamali Shakeel, Shenaz Ramtoola.
United States: Jeannie Lucas, John Reed, Ola Odugbesan, Joanna Van, Bruce Bode, Paul Norwood, Nirmal Sunkara, Jeffrey Rothman, Samer Nakhle, Ronald Chochinov, Stephen Aronoff, David Klonoff, Michael Dempsey, Jean Dostou, Richard Bergenstal, Leon Fogelfeld, Julio Rosenstock, Jean Chen, Lubna Mirza, Timothy Bailey.
Battelino T, Danne T, Edelman SV, et al. Continuous glucose monitoring‐based time‐in‐range using insulin glargine 300 units/ml versus insulin degludec 100 units/ml in type 1 diabetes: The head‐to‐head randomized controlled InRange trial. Diabetes Obes Metab. 2023;25(2):545‐555. doi: 10.1111/dom.14898
Funding information Sanofi
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to patient‐level data and related documents. Patient‐level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data sharing criteria, eligible studies, and process for requesting access can be found at https://www.vivli.org.
REFERENCES
- 1. Mobasseri M, Shirmohammadi M, Amiri T, Vahed N, Hosseini Fard H, Ghojazadeh M. Prevalence and incidence of type 1 diabetes in the world: a systematic review and meta‐analysis. Health Promot Perspect. 2020;10:98‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lachin JM, Bebu I, Nathan DM, for the DCCT/EDIC Research Group . The beneficial effects of earlier versus later implementation of intensive therapy in type 1 diabetes. Diabetes Care. 2021;44:2225‐2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderbro T, Amsberg S, Adamson U, et al. Fear of hypoglycaemia in adults with type 1 diabetes. Diabet Med. 2010;27:1151‐1158. [DOI] [PubMed] [Google Scholar]
- 4. Holt RIG, DeVries JH, Hess‐Fischl A, et al. The management of type 1 diabetes in adults. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2021;44:2589‐2625. [DOI] [PubMed] [Google Scholar]
- 5. Pettus JH, Zhou FL, Shepherd L, et al. Differences between patients with type 1 diabetes with optimal and suboptimal glycaemic control: a real‐world study of more than 30 000 patients in a US electronic health record database. Diabetes Obes Metab. 2020;22:622‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foster NC, Beck RW, Miller KM, et al. State of type 1 diabetes management and outcomes from the T1D exchange in 2016‐2018. Diabetes Technol Ther. 2019;21:66‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beck RW, Riddlesworth T, Ruedy K, et al. Effect of continuous glucose monitoring on glycemic control in adults with type 1 diabetes using insulin injections: the DIAMOND randomized clinical trial. JAMA. 2017;317:371‐378. [DOI] [PubMed] [Google Scholar]
- 8. Tauschmann M, Hermann JM, Freiberg C, et al. Reduction in diabetic ketoacidosis and severe hypoglycemia in pediatric type 1 diabetes during the first year of continuous glucose monitoring: a multicenter analysis of 3,553 subjects from the DPV registry. Diabetes Care. 2020;43:e40‐e42. [DOI] [PubMed] [Google Scholar]
- 9. Heise T, Hovelmann U, Nosek L, Hermanski L, Bottcher SG, Haahr H. Comparison of the pharmacokinetic and pharmacodynamic profiles of insulin degludec and insulin glargine. Expert Opin Drug Metab Toxicol. 2015;11:1193‐1201. [DOI] [PubMed] [Google Scholar]
- 10. Becker RH, Dahmen R, Bergmann K, Lehmann A, Jax T, Heise T. New insulin glargine 300 units mL−1 provides a more even activity profile and prolonged glycemic control at steady state compared with insulin glargine 100 units mL−1 . Diabetes Care. 2015;38:637‐643. [DOI] [PubMed] [Google Scholar]
- 11. Bode BW, Buse JB, Fisher M, et al. Insulin degludec improves glycaemic control with lower nocturnal hypoglycaemia risk than insulin glargine in basal‐bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN([R]) basal‐bolus type 1): 2‐year results of a randomized clinical trial. Diabet Med. 2013;30:1293‐1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Danne T, Matsuhisa M, Sussebach C, et al. Lower risk of severe hypoglycaemia with insulin glargine 300 U/mL versus glargine 100 U/mL in participants with type 1 diabetes: a meta‐analysis of 6‐month phase 3 clinical trials. Diabetes Obes Metab. 2020;22:1880‐1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bailey TS, Pettus J, Roussel R, et al. Morning administration of 0.4U/kg/day insulin glargine 300U/mL provides less fluctuating 24‐hour pharmacodynamics and more even pharmacokinetic profiles compared with insulin degludec 100U/mL in type 1 diabetes. Diabetes Metab. 2018;44:15‐21. [DOI] [PubMed] [Google Scholar]
- 14. Heise T, Nørskov M, Nosek L, Kaplan K, Famulla S, Haahr HL. Insulin degludec: lower day‐to‐day and within‐day variability in pharmacodynamic response compared with insulin glargine 300 U/mL in type 1 diabetes. Diabetes Obes Metab. 2017;19:1032‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lucidi P, Candeloro P, Cioli P, et al. Pharmacokinetic and pharmacodynamic head‐to‐head comparison of clinical, equivalent doses of insulin glargine 300 units. mL−1 and insulin degludec 100 units. mL−1 in type 1 diabetes. Diabetes Care. 2021;44:125‐132. [DOI] [PubMed] [Google Scholar]
- 16. Miura H, Sakaguchi K, Otowa‐Suematsu N, et al. Effects of insulin degludec and insulin glargine U300 on glycaemic stability in individuals with type 1 diabetes: a multicentre, randomized controlled crossover study. Diabetes Obes Metab. 2020;22:2356‐2363. [DOI] [PubMed] [Google Scholar]
- 17. Conget I, Mangas MA, Morales C, et al. Effectiveness and safety of insulin glargine 300 u/ml in comparison with insulin degludec 100 u/ml evaluated with continuous glucose monitoring in adults with type 1 diabetes and suboptimal glycemic control in routine clinical practice: the OneCARE study. Diabetes Ther. 2021;12:2993‐3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Battelino T, Danne T, Bergenstal RM, et al. Clinical targets for continuous glucose monitoring data interpretation: recommendations from the international consensus on time in range. Diabetes Care. 2019;42:1593‐1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Battelino T, Edelman SV, Nishimura R, Bergenstal RM. Comparison of second‐generation basal insulin analogs: a review of the evidence from continuous glucose monitoring. Diabetes Technol Ther. 2021;23:20‐30. [DOI] [PubMed] [Google Scholar]
- 20. Levy JC, Davies MJ, Holman RR, Group TS . Continuous glucose monitoring detected hypoglycaemia in the treating to target in type 2 diabetes trial (4‐T). Diabetes Res Clin Pract. 2017;131:161‐168. [DOI] [PubMed] [Google Scholar]
- 21. Mangrola D, Cox C, Furman AS, Krishnan S, Karakas SE. Self blood glucose monitoring underestimates hyperglycemia and hypoglycemia as compared to continuous glucose monitoring in type 1 and type 2 diabetes. Endocr Pract. 2018;24:47‐52. [DOI] [PubMed] [Google Scholar]
- 22. Zick R, Petersen B, Richter M, Haug C, Group SS . Comparison of continuous blood glucose measurement with conventional documentation of hypoglycemia in patients with type 2 diabetes on multiple daily insulin injection therapy. Diabetes Technol Ther. 2007;9:483‐492. [DOI] [PubMed] [Google Scholar]
- 23. Fox BQ, Benjamin PF, Aqeel A, et al. Continuous glucose monitoring use in clinical trials for on‐market diabetes drugs. Clin Diabetes. 2021;39:160‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Battelino T, Bosnyak Z, Danne T, et al. InRange: comparison of the second‐generation basal insulin analogues glargine 300 u/ml and degludec 100 u/ml in persons with type 1 diabetes using continuous glucose monitoring‐study design. Diabetes Ther. 2020;11:1017‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. American Diabetes Association . 6. Glycemic targets: standards of medical care in diabetes—2021. Diabetes Care. 2020;44:S73‐S84. [DOI] [PubMed] [Google Scholar]
- 26. Battelino T, Danne T, Edelman S, et al. Parallel Session ‐ RCT evidence on time‐in‐range in type 1 diabetes [Invited speaker oral presentation]. The official journal of ATTD advanced technologies & treatments for diabetes conference 27‐30 April 2022 I Barcelona & Online. Diabetes Technol Ther. 2022;24(S1):A‐1‐A‐237: #951. [Google Scholar]
- 27. Bergenstal RM, Edelman S, Choudhary P, et al. 105‐LB: glucose variability with second‐generation basal insulin analogs glargine 300 U/mL and degludec 100 U/mL, evaluated by CGM in people with T1D—the In Range randomized controlled trial. Diabetes. 2022;71(Suppl 1):105‐LB. [Google Scholar]
- 28. Lu J, Ma X, Zhou J, et al. Association of time in range, as assessed by continuous glucose monitoring, with diabetic retinopathy in type 2 diabetes. Diabetes Care. 2018;41:2370‐2376. [DOI] [PubMed] [Google Scholar]
- 29. Beck RW, Bergenstal RM, Riddlesworth TD, et al. Validation of time in range as an outcome measure for diabetes clinical trials. Diabetes Care. 2018;42:400‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Penckofer S, Quinn L, Byrn M, Ferrans C, Miller M, Strange P. Does glycemic variability impact mood and quality of life? Diabetes Technol Ther. 2012;14:303‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Janapala RN, Jayaraj JS, Fathima N, et al. Continuous glucose monitoring versus self‐monitoring of blood glucose in type 2 diabetes mellitus: a systematic review with meta‐analysis. Cureus. 2019;11:e5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosenstock J, Cheng A, Ritzel R, et al. More similarities than differences testing insulin glargine 300 units/mL versus insulin Degludec 100 units/mL in insulin‐naive type 2 diabetes: the randomized head‐to‐head BRIGHT trial. Diabetes Care. 2018;41:2147‐2154. [DOI] [PubMed] [Google Scholar]
- 33. Riddlesworth TD, Beck RW, Gal RL, et al. Optimal sampling duration for continuous glucose monitoring to determine long‐term glycemic control. Diabetes Technol Ther. 2018;20:314‐316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Study design
Figure S2. Study disposition
Figure S3. HbA1c change from baseline to Week 12 (ITT population; secondary endpoint, descriptive data)
Table S1. Symptomatic and asymptomatic SMPG‐derived hypoglycemia (safety population)
Table S2. Insulin dose change (ITT population)
Data Availability Statement
Qualified researchers may request access to patient‐level data and related documents. Patient‐level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data sharing criteria, eligible studies, and process for requesting access can be found at https://www.vivli.org.