

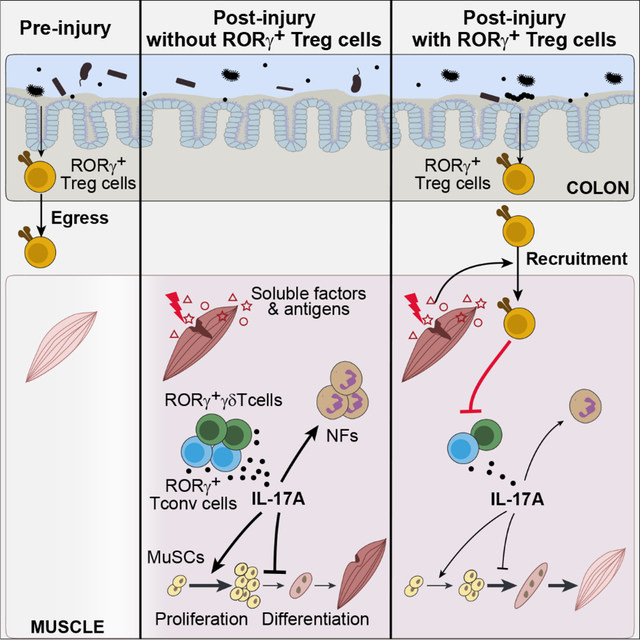
SUMMARY
Specific microbial signals induce the differentiation of a distinct pool of RORγ+ regulatory T (Treg) cells crucial for intestinal homeostasis. We discovered highly analogous populations of microbiota-dependent Treg cells that promote tissue regeneration at extra-gut sites, notably acutely injured skeletal muscle and fatty liver. Inflammatory meditators elicited by tissue damage combined with MHC-class-II-dependent T cell activation to drive the accumulation of gut-derived RORγ+ Treg cells in injured muscle, wherein they regulated the dynamics and tenor of early inflammation and helped balance the proliferation versus differentiation of local stem cells. Reining in IL-17A-producing T cells was a major mechanism underlying the rheostatic functions of RORγ+ Treg cells in compromised tissues. Our findings highlight the importance of gut-trained Treg cell emissaries in controlling the response to sterile injury of non-mucosal tissues.
Keywords: Muscle, Colon, Microbiota, Treg, RORγ, IL-17, Tissue regeneration, Stem cell
eTOC BLURB
Colonic RORγ+ Treg cells are a specialized microbiota-dependent population crucial for intestinal homeostasis, but their role in extra-gut tissues remains unexplored. Hanna et al. report that these cells seed non-mucosal tissues in response to injury wherein they can rein in IL-17-driven inflammation, regulate stem cell activities and promote tissue regeneration.
Graphical Abstract
INTRODUCTION
Foxp3-expressing Treg cells are a subset of CD4+ T lymphocytes key for maintaining immunological tolerance. So-called “tissue-Treg cells” are a specialized category of Treg cells, with distinct transcriptomes and T cell receptor (TCR) repertoires, that are located in nonlymphoid tissues – for example, visceral adipose tissue, skeletal muscle, skin, and the colonic lamina propria1. Powered by their ability to dynamically cross-talk with a wide array of immunological and non-immunological cells in their microenvironment, tissue-Treg cells exert diverse functions, and thereby guard tissue homeostasis in various contexts, such as regulating local and systemic metabolism, enforcing tolerance to the microbiota, and regulating tissue regeneration in response to injury1–10. This diverse functional palette can manifest even within the same organ, as highlighted by skeletal muscle7,11. Treg cells rapidly accumulate in response to sterile muscle injury and promote tissue regeneration via two major modes: 1) by controlling the activities of innate and adaptive immunocytes; and 2) by enhancing the regenerative capacity of muscle satellite cells (MuSCs), at least in part through secretion of the growth factor Areg.
While surveying Treg cell heterogeneity in nonlymphoid tissues, we noticed a population of RORγ+ Treg cells that accrued in regenerating skeletal muscle early after acute injury. Unlike the majority of Treg cells generated as such in the thymus, intestinal RORγ+ Treg cells are mostly locally induced in response to microbial or food antigens3–5,12,13. The function of RORγ+ Treg cells has so far been linked primarily to mucosal health as they play an indispensable role in maintaining intestinal tolerance, with their loss resulting in increased incidences of colitis, colon cancer and food allergies3,4,12,14–16.
In addition to its role in shaping the gut immune system, impacts of the microbiota on systemic immunity are increasingly evident. So far, such effects are best characterized for the effector arm of the adaptive immune system, where multiple effector T (Teff) cell subtypes generated or educated in the gut in response to microbial signals influence immunity at distal sites17–24. In contrast, whether RORγ+ Treg cells play a role beyond their local function in mucosal tissues remains essentially unexplored. Our observation of a population of RORγ+ Treg cells in regenerating skeletal muscle prompted us to investigate whether intestinal RORγ+ Treg cells can drive tissue homeostasis at distal sites, thereby constituting a mode of cross-tissue communication coordinated by microbiota:Treg cell interactions.
RESULTS
scRNA-seq identifies a population of RORγ+ Treg cells that accumulate in skeletal muscle at early times after acute injury
In response to the local damage provoked by intramuscular (i.m.) injection of cardiotoxin (CTX), Treg cells rapidly accumulate in regenerating muscle, peaking numerically at day 3–4 post-injury7. This timepoint marks transition of the tissue milieu from a pro- to an anti-inflammatory state, a step that is critical for efficient repair7,25. Considering the diverse cellular interactions in which muscle Treg cells participate in this complex, dynamic microenvironment7,11,26,27, we investigated their transcriptional heterogeneity three days after CTX-induced injury via single-cell RNA sequencing (scRNA-seq) [quality-control metrics in Table S1]. Clustering, visualized by Uniform Manifold Approximation and Projection (UMAP), revealed five distinct groupings (Figure 1A). According to the transcripts differentially expressed by these clusters, we identified them as “resting”, “early-activation”, “reparative”, “RORγ+”, and “interferon (IFN)-responding” Treg cells (Figure S1A–F). Most relevant here, expression of Rorc, encoding the RORγ transcription factor (TF), distinguished the RORγ+ cluster, which also preferentially expressed colonic RORγ+ Treg cell marker genes such as Maf (encoding c-Maf), and Rorc target loci such as Ltb4r1 and Furin (Figures 1B, S1A, E).
Figure 1: Accumulation of RORγ+ Treg cells in skeletal muscle early after acute injury.
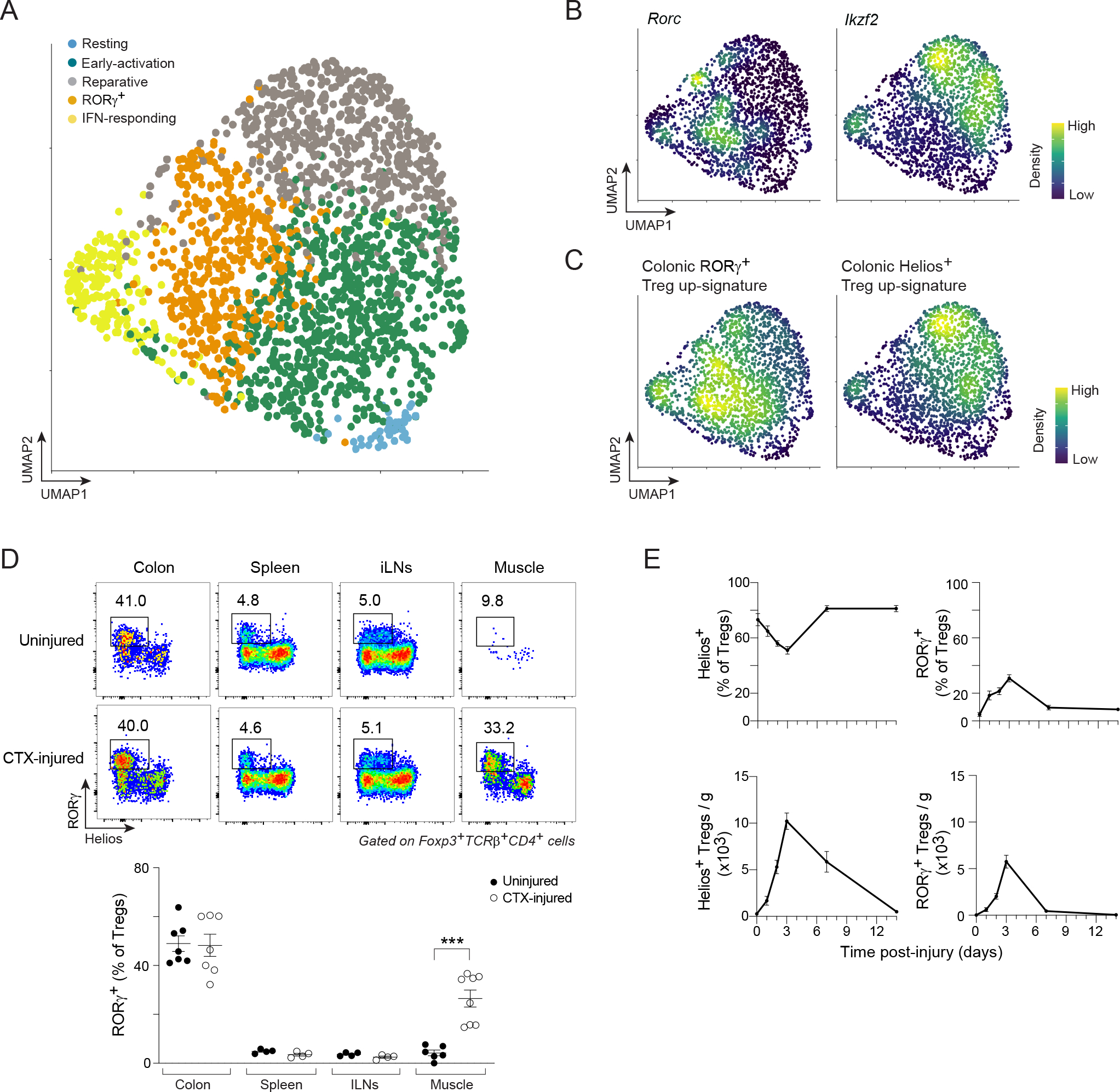
(A–C) scRNA-seq of Treg cells from hindlimb muscles of Foxp3GFP mice 3 days after CTX-induced injury. A) 2D UMAP plot. B, C) Density plots of the expression of the indicated genes and signatures. (D) Flow-cytometry of RORγ+ Treg cells at steady-state and 3 days after CTX-induced injury. Upper panel: representative dot-plots (2–3 independent experiments); lower panel: summary data. (E) Time course of fractions (top) and numbers (bottom) of Helios+ and RORγ+ Treg cells in CTX-injured muscles. Unpaired t-test (D). See also Figure S1.
Our attention was quickly drawn to the largely non-overlapping expression patterns of Rorc and Ikzf2, which encodes Helios (Figure 1B). Ikzf2 showed an expression gradient spanning the reparative and part of the early-activation clusters, while expression of Rorc was elsewhere and more circumspect. Since an essentially non-overlapping expression pattern of RORγ and Helios is characteristic of the intestinal Treg compartment3,4, we further addressed the identity of the seemingly analogous muscle Treg cell clusters by superimposing the transcriptional signatures distinguishing colonic RORγ+ and Helios+ Treg cells on the UMAP (STAR Methods for details). As observed for the driving TFs, expression of the two signatures was essentially bimodal: the colonic RORγ+ Treg cell up-signature was enriched in the muscle RORγ+ cluster whereas the colonic Helios+ up-signature was highest in the reparative and early-activation clusters (Figure 1C).
In search of clues to their function in muscle regeneration, we examined the transcriptome of muscle RORγ+ Treg cells vis-à-vis other local subtypes (Figure S1G). Several of the up-regulated transcripts in muscle RORγ+ Treg cells encoded proteins that promote immunocyte recruitment to inflammatory sites (e.g. Ccr2, Ccr4, Cxcr6, Ltb4r1) as well as cytoskeleton and cell adhesion proteins (e.g. Vim, Actg1, Rac2 and Cdc42). Additionally, this cluster preferentially made more transcripts encoding a distinct set of Treg cell effector molecules (e.g. Gzmb, Lgals1, Lgals3 and Furin) and proteins known to dampen the function of inflammatory cells (e.g. Il1r2, Anxa1, Cd47, Serpinb1a and Slamf6). Pathway analysis of genes preferentially up-regulated in the muscle RORγ+ cluster highlighted cell locomotion and extravasation pathways (Figure S1H).
We then examined the expression of RORγ and Helios in Treg cells from skeletal muscle as well as other relevant tissues at the protein level using flow cytometry (Figure 1D). As expected, colonic Treg cells included a much larger fraction of RORγ+Helios− Treg cells (referred to as RORγ+ Treg cells from here on) than did Treg cells from the spleen and muscle-draining inguinal lymph nodes (iLNs), of which RORγ−Helios+ Treg cells (referred to as Helios+ Treg cells from here on) were the predominant population. Uninjured muscle harbored a small population of RORγ+ Treg cells comprising 0–10% of the Treg compartment at steady-state. Three days after CTX-induced injury, a sizable population of RORγ+ Treg cells had accumulated in regenerating muscle, an increase not observed in other tissues.
Preferential expression of key RORγ+ Treg cell marker genes was also confirmed at the protein level: c-Maf and CXCR6, markers of RORγ+ Treg cells, were expressed in more muscle RORγ+ than Helios+ Treg cells (Figure S1I). Gata3 and ST2, markers of colonic Helios+ Treg cells, showed the opposite pattern of expression. RORγ+ Treg cells rapidly accumulated upon muscle injury, peaking at day 3 and declining thereafter, when Helios+ Treg cells became the major population (Figure 1E).
These data revealed an unanticipated heterogeneity of Treg cells in regenerating skeletal muscle. They also uncovered a distinct “early-responder” population of RORγ+ Treg cells rapidly accruing after acute skeletal muscle injury.
Muscle RORγ+ Treg cells have a colonic provenance
To address the possibility that muscle RORγ+ Treg cells arose either from Foxp3− CD4+ conventional T (Tconv) cells that transdifferentiated locally upon recruitment or from lymphoid-organ Treg cells that acquired RORγ expression in regenerating muscle, we transferred congenically marked, naïve, splenic Tconv (CD45.2+) and Treg (CD45.1+) cells into Treg-cell-depleted Foxp3DTR mice (CD45.1+2+), subsequently subjected to muscle injury (Figure S2A). The few donor-derived Foxp3+ cells detected in injured muscle lacked RORγ expression, suggesting that these cells were unlikely to be the main source of muscle RORγ+ Treg cells.
Thus, it seemed more plausible that muscle injury promoted the migration of colonic RORγ+ Treg cells to skeletal muscle, a possibility that we interrogated using multiple approaches. As a prelude, we used our recently published scRNA-seq data28 to perform a two-way comparison of the transcriptomes of muscle RORγ+ versus total spleen Treg cells on day 4 post-injury with those of colon RORγ+ versus total spleen Treg cells. There was an evident similarity between the transcriptional profiles of muscle and colon RORγ+ Treg cells (Figure 2A). In both cases, there was enrichment of the colon RORγ+ Treg cell up-signature, although more strongly so for the colon samples, as indicated by the off-diagonal cloud of transcripts on the fold-change/fold-change (FC/FC) plot. Both muscle and colon RORγ+ Treg cells exhibited enrichment of transcripts encoding Treg cell effector molecules (e.g. Gzmb, S100a4 and S100a6) and specific chemokine receptors (e.g. Ccr2, Ccr4 and Ccr5) while being relatively depleted of transcripts encoding naïve Treg cell markers (e.g. Ccr7 and Sell). However, some transcripts, such as Ccr9 (encoding a gut homing receptor), Lag3 and Il10 were preferentially enriched in the colon cells. Such transcriptional differences were confirmed via population-level RNA-seq of sorted muscle vs colon RORγ+ Treg cells (Figure S2B–D).
Figure 2: A colonic provenance for muscle RORγ+ Treg cells.
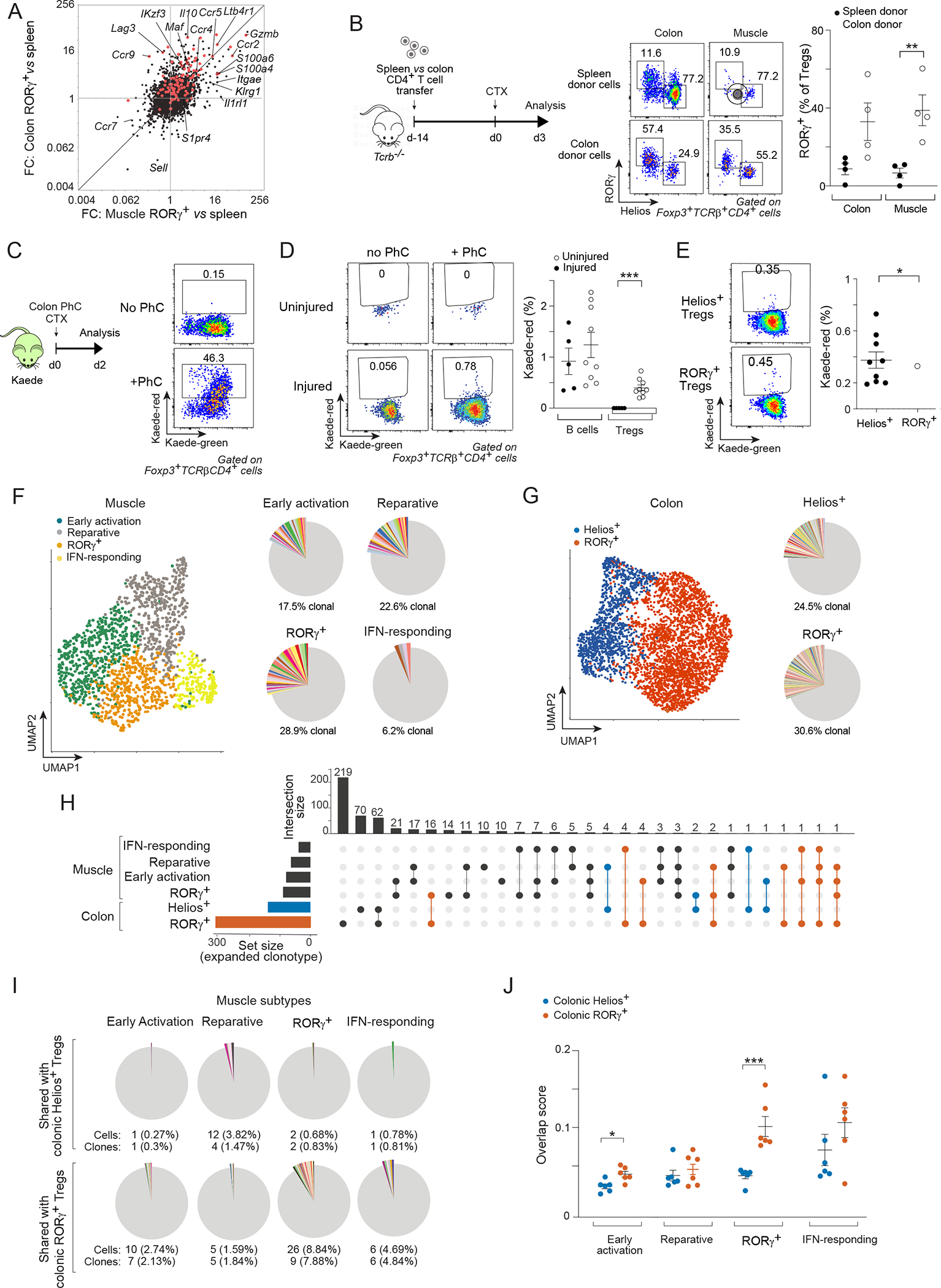
(A) Transcriptomic fold-change/fold-change (FC) plots comparing muscle RORγ+ vs spleen Treg cells at day 4 after CTX-induced injury vis-a-vis colon RORγ+ vs spleen Treg cells. Red: colonic RORγ+ Treg up-signature. (B) Spleen or colon CD4+ T cells were transferred into Tcrb−/− mice followed by CTX injection. Experimental design (left), representative dot-plots (middle), and summary data (right) of RORγ+ Treg cells in colon and muscle. (C-E) Colonic photoconversion (PhC) of Kaede mice ± CTX-induced injury. C) Experimental design (left) and representative dot-plots (right) of colon PhC efficiency at day 0. D, E) emigration of the indicated colonic cells to hindlimb muscles after 48 hr. Representative dot-plots (left) and summary data (right). (F-J) Paired scRNA-seq and scTcr-seq of muscle and colon Treg cells on day 3 after CTX injection. F) Left: UMAP of muscle data; right: pie-charts of scTcr-seq data showing the proportion of clonally expanded cells in each cluster. Different colors depict individual clones; non-expanded clones in gray. G) Same as panel F for colon Treg cells. H) Upset plot of intra-organ and inter-muscle-colon clonotype sharing. Set size indicates total number of expanded clones for each subtype. On top, the total number of shared clones is displayed. Incidences of clone sharing between any muscle Treg subtype and colonic RORγ+ (red) or Helios+ (blue) Treg cells are highlighted. I) Pie-charts depicting numbers and frequencies of clonotypes shared between muscle Treg cell subtypes and colonic RORγ+ or Helios+ Treg cells. Different colors depict individual clones; non-expanded clones in gray. J) Clonal overlap score between muscle Treg subtypes and colonic RORγ+ vs Helios+ Treg cells in individual mice. Representative dot-plots are from 2–3 independent experiments. Unpaired t-test (B, D), paired t-test (E), or two-way ANOVA (J). See also Figure S2.
Two strategies were employed to evaluate the ability of the colonic T cell compartment to engender muscle RORγ+ Treg cells. First, we transferred CD4+ T cells from the spleen versus colon into T-cell-deficient Tcrb−/− mice, followed by acute muscle injury (Figure 2B). We chose to transfer total CD4+ T cells rather than sorted Foxp3+ cells in order to avoid the previously documented unphysiological outgrowth of cells with unstable Foxp3 expression when Treg cells are transferred alone into lymphopenic mice29. Three days post-injury, the RORγ+ subtype constituted a much larger fraction of both the colon and muscle Treg compartments in mice transferred with colonic compared with splenic CD4+ T cells (Figure 2B). Second, we turned to the Kaede transgenic mouse system, wherein the photoconvertible fluorescent reporter, Kaede, is ubiquitously expressed30. Upon exposure to violet light, reporter fluorescence irreversibly converts from green to red. By briefly photoconverting part of the descending colon of Kaede mice using endoscopic laser exposure, one can conveniently tag a fraction of colonic leukocytes and subsequently track their trafficking from the gut to extra-gut tissues (Figure 2C). Our previous studies using this system revealed active migration of most leukocyte classes from the colon to extra-gut lymphoid organs even at steady-state31. Forty-eight hours after photoconversion, colonic B cells, which generally display the most active emigration amongst colonic lymphocytes31, migrated to skeletal muscle of both uninjured and injured mice (Figure 2D). Treg cells of colonic origin were not detectable in uninjured muscle, whereas CTX-induced injury consistently provoked their muscle accumulation (Figure 2D). The muscle RORγ+ and Helios+ Treg populations both contained cells of colonic origin, somewhat more in the RORγ+ compartment (Figure 2E).
We also exploited TCR sequence information as a cellular barcode to examine the ontogenic relationship between muscle and colonic Treg cells, in particular between the RORγ+ subtypes of these two organs. We performed scRNA-seq paired with scTcr-seq on matched muscle and colon Treg cells from six individual mice 3 days after CTX-induced injury. Clustering analysis of the muscle Treg cell scRNA-seq data largely recapitulated the major Treg cell subtypes we previously identified, the set of diagnostic transcripts of each cluster and, most importantly, the dichotomous expression pattern of Rorc and Ikzf2 (Figures 2F, S2E–K). For colonic Treg cells, 6 distinct clusters emerged (Figure S2L). Of relevance to this study, we split them broadly into RORγ+ and Helios+ cells based on Rorc vs Ikzf2 transcripts and their expression of the colon RORγ+ vs Helios+ Treg signatures (Figures 2G, S2N). Data integration confirmed a balanced distribution of cells from the two independent experiments across different clusters in both muscle and colon (Figure S2E, M).
We then used the TCR information to determine the degree of clonal expansion (≥2 cells sharing the exact same Tcra and Tcrb nucleotide sequences) in each subtype in each organ. In contrast to the polyclonal nature of most Treg cells in lymphoid organs7, both muscle and colon Treg cell subtypes displayed a less diverse TCR repertoire, with expansion of multiple clones (Figure 2F, G). There was a considerable degree of intra-muscle and intra-colon clonotype sharing (Figures 2H, S2O). Notably, we detected multiple incidences of sharing between muscle and colon Treg cells, mostly between the two RORγ+ subtypes (Figure 2H, I). A smaller fraction of muscle Treg cells, mostly restricted to the muscle reparative cluster, showed clonal overlap with the colonic Helios+ Treg compartment. To further highlight the preferential clonal overlap between muscle and colon RORγ+ Treg cells, we used the scRepertoire package32 to calculate, for each individual mouse, an overlap score between all muscle Treg cell subtypes and colon RORγ+ vs Helios+ Treg cells. The score was highest between muscle and colon RORγ+ Treg cells (Figure 2J).
In brief, multiple experimental approaches provided data arguing that muscle RORγ+ Treg cells were engendered in the colon. Muscle and colon RORγ+ Treg cells shared transcriptional particularities and TCR specificities. Both adoptive transfer and phototagging experiments evidenced colon to muscle cell flow.
RORγ+ Treg cell accumulation in regenerating muscle depends on induction of soluble factors and TCR:MHC-II interactions
In search of mediators of RORγ+ egress from the colon, we focused on sphingosine-1-phosphate receptors (S1PRs) since they are increasingly appreciated as regulators of lymphocyte egress from nonlymphoid tissues in addition to their classical role in regulating lymph node exit33–36. Kaede mice were treated with the drug FTY720, which provokes S1PR downregulation, followed by photoconversion of the distal colon. As expected31, a fraction of Tconv cells, RORγ+, and Helios+ Treg cells had egressed from the colon of vehicle-treated mice by 24 hours, evidenced by a drop in the Kaede-red pool retained in the colon (Figure 3A). Concomitantly, colon-derived immigrants were readily detectable in the spleen (Figure 3B). FTY720 treatment completely abrogated both egress of these cells from the colon and their appearance in the spleen (Figure 3A, B), indicating that S1PRs strictly regulated the egress of multiple types of CD4+ lymphocytes from the colon to extra-gut tissues at steady-state.
Figure 3: Mechanisms of RORγ+ Treg cell accumulation in regenerating muscle.
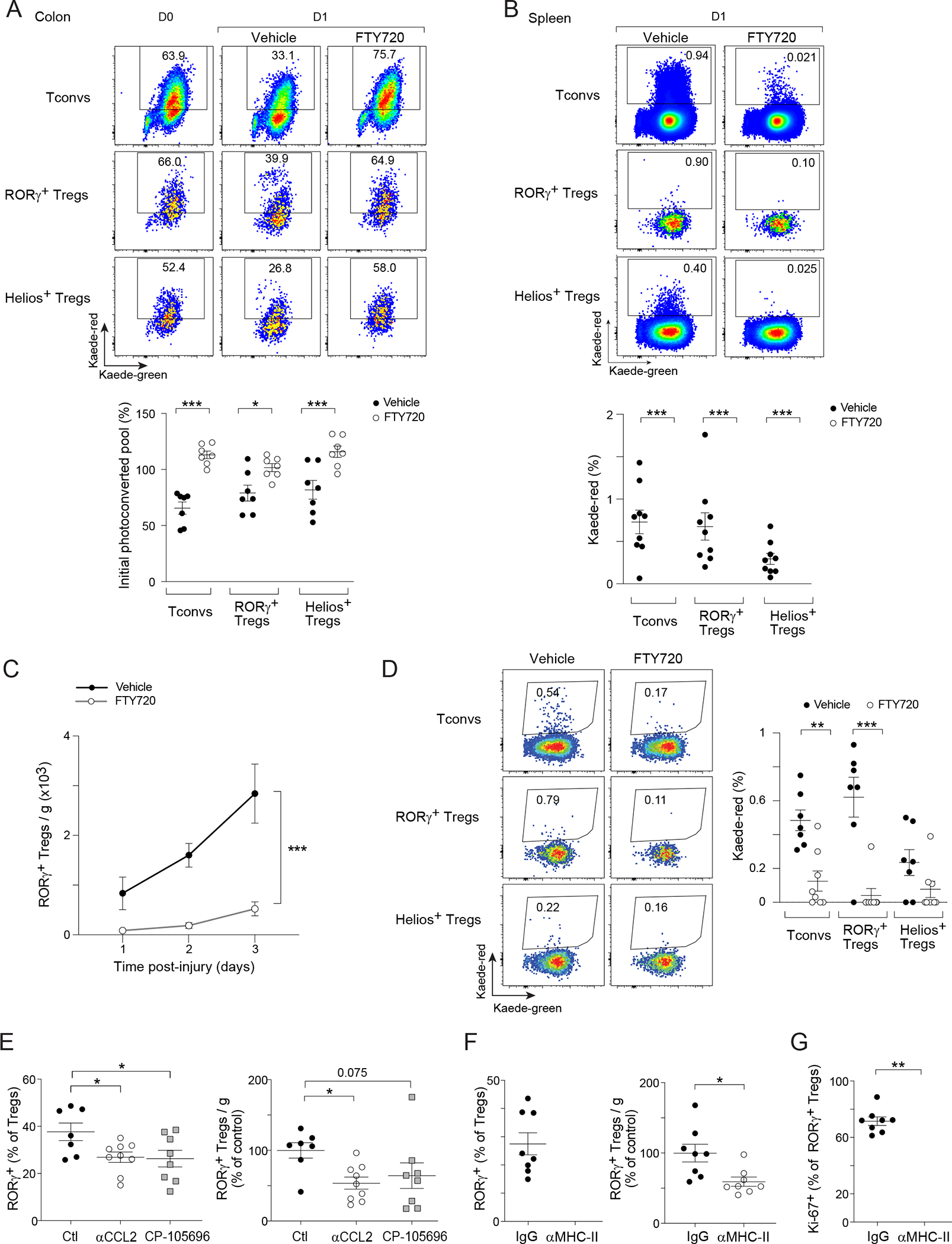
(A) Egress of diverse lymphocyte populations from the colon, and (B) their emigration to spleen after 24 hr of colonic PhC of Kaede mice. Top: representative dot-plots; bottom: summary data. (C) Muscle RORγ+ Treg cell numbers after CTX-induced injury in vehicle- or FTY720-treated mice. (D) Colon PhC coupled with CTX-induced injury as per Figure 2C. Representative dot-plots (left) and summary data (right) of emigration of colonic cells to hindlimb muscles after 48 hr. (E) RORγ+ Treg cell fraction (left) and number (right) in hindlimb muscles 2 days after CTX-induced injury in in control- (Ctl), αCCL2-, or CP-105696-treated mice. (F, G) Mice were treated with isotype (IgG) or αMHC-II antibody. F) Muscle RORγ+ Treg cell fraction (left) and number (right), and G) Ki-67 expression 3 days after CTX-induced injury. Representative dot-plots are from 2–3 independent experiments. Unpaired t-test (A, B, F, G), two-way ANOVA (C), or one-way ANOVA (E). See also Figure S3.
FTY720 treatment of B6 mice resulted in a large drop in the numbers of muscle RORγ+ Treg cells at various timepoints post-injury (Figure 3C). Since FTY720 can impair lymphocyte egress from lymphoid organs, we turned back to the Kaede system to specifically address whether S1PRs were required for colon-derived RORγ+ Treg cell emigration to regenerating muscle. Using the experimental scheme illustrated in Figure 2C, we detected tagged colonic Tconv cells, RORγ+, and Helios+ Treg cells in muscles of vehicle-treated mice 48 hours after injury. FTY720 almost completely abolished the migration of colonic RORγ+ Treg cells to regenerating muscle (Figure 3D). The drop in muscle RORγ+ Treg cell numbers in drug-treated mice was not due to increased cell death, either in the colon or in muscle, as indicated by comparable staining of cleaved caspase-3 (Figure S3A).
Several points argued against the possibility that muscle injury promoted RORγ+ Treg cell migration from the colon to injured muscle by more indirect or non-specific means. We did not observe any differences in the major immunocyte populations of the colonic lamina propria of control versus CTX-injured mice (Figures 1D, S3B). Nor did we detect a significant change in lymphocyte egress from the colon in injured compared with uninjured mice (Figure S3C). Consistently, CTX-induced injury did not result in increased intestinal permeability (Figure S3D), excluding the possibility that increased gut leakiness was the cause of muscle RORγ+ Treg cell accumulation.
As injury did not detectably influence colonic egress of RORγ+ Treg cells, we sought to identify inflammatory mediators whose receptors were expressed by RORγ+ Treg cells and whose expression was locally induced early after muscle injury. The scRNA-seq data introduced in Figure 1A revealed significant upregulation of transcripts encoding CCR2 and LTB4R1 (Leukotriene B4 receptor 1) in both colonic and muscle RORγ+ Treg cells (Figures 2A, S1E). Additionally, the ligands for both these receptors, CCL2 and LTB4, are well-established early inflammatory mediators, rapidly produced by invading myeloid cells and local stromal cells in response to injury37,38. We treated CTX-injured mice with a CCL2 neutralizing antibody or with an LTB4R1-specific antagonist, CP-105696. Administration of either agent reduced the representation of muscle RORγ+, but not Helios+, Treg cells 2 days after CTX injection (Figures 3E, S3E), suggesting that CCL2 and LTB4 preferentially attracted RORγ+ Treg cells to injured muscle.
We also explored whether local signals promoted proliferation and/or retention of the recruited RORγ+ Treg cells. Since muscle RORγ+ Treg cells displayed a restricted repertoire of TCRs (Figure 2F–H), we focused on TCR:MHC-II interactions, a pivotal axis in controlling tissue-Treg cell accumulation in multiple contexts1,39–41. We performed short-term antibody-mediated MHC-II blockade in CTX-injured mice, and analyzed the muscle Treg compartment 3 days post-injury. MHC-II blockade induced an ~40% drop in muscle RORγ+ Treg cell numbers and a decrease in their fraction of cycling cells (Figure 3F, G). As expected, the numbers and cycling of Helios+ muscle Treg cells were also affected by MHC-II blockade (Figure S3F, G). Reductions in RORγ+ Treg cell numbers and proliferation after short-term MHC-II blockade were not observed in the spleen, mesenteric lymph nodes (mLNs), or colon (Figure S3H, I), in line with reports that antigen presentation by specialized cells in the mLNs is required for the induction of colonic RORγ+ Treg cells early in life, but not for their maintenance in adulthood42.
Considering the results from this multi-pronged analysis, we favor the following scenario: Under steady-state conditions, RORγ+ (and other subtypes of) Treg cells generated in the colon constantly exit and redistribute through extra-gut lymphoid and non-lymphoid organs, being retained at sites where their TCR happens to recognize an antigen. Muscle injury induces the release of inflammatory stimuli such as CCL2 and LTB4, (perhaps coupled with increased antigen release), promoting recruitment of RORγ+ Treg cells, where those with TCRs that recognize muscle antigen(s) preferentially proliferate and are retained.
Muscle RORγ+ Treg cells are microbiota-dependent
One of the fundamental characteristics of intestinal RORγ+ Treg cells is their dependence on the microbiota3–5. Since our investigations pointed to colonic RORγ+ Treg cells as the original source of their muscle counterparts, we evaluated the microbiota dependence of muscle RORγ+ Treg cells by comparing their representation in germ-free (GF) vs specific-pathogen-free (SPF) mice and in antibiotic (Abx)- vs vehicle-treated animals. GF mice showed minimal RORγ+ Treg cell accumulation in muscle 3 days after CTX injection (Figures 4A, S4A). Similarly, treatment of SPF mice with a broad-spectrum Abx cocktail [ampicillin, neomycin, metronidazole and vancomycin; (VMNA)] drastically reduced the representation of muscle RORγ+ Treg cells (Figures 4B, S4B). RORγ+ Treg cell accumulation did not result from introduction of skin microbes during the injury process (Figure S4C, STAR Methods for details).
Figure 4: Microbiota-dependence of muscle RORγ+ Treg cells.
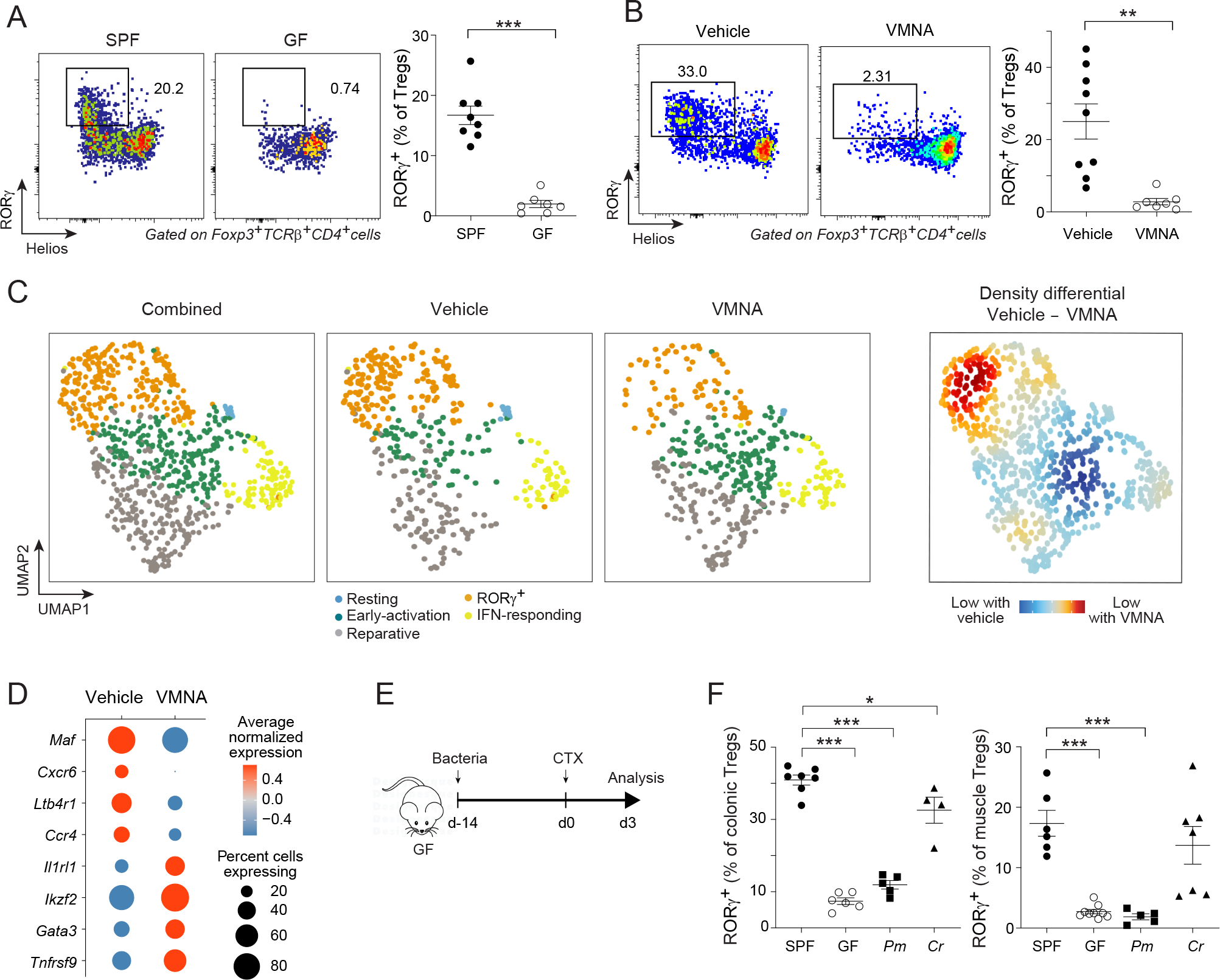
(A) Comparison of RORγ+ Treg cells from hindlimb muscles 3 days after CTX-induced injury of specific-pathogen-free (SPF) or germ-free (GF) mice. Left: representative dot-plots (3 independent experiments); right: summary data. (B) Same as panel A except SPF mice were treated or not with the Abx cocktail, VMNA (vancomycin, metronidazole, neomycin, ampicillin). (C, D) scRNA-seq comparison of muscle Treg cells from vehicle- and VMNA-treated Foxp3GFP mice 3 days after CTX-induced injury. C) 2D UMAP plot of the two conditions combined (left), each individual condition (middle) and their differential (right). (D) Bubble-plot of the average transcript expression of key marker genes in muscle Treg cells of vehicle- vs VMNA-treated mice. (E, F) Monocolonization. E) Experimental design: GF mice were orally gavaged with Peptostreptococus magnus (Pm) or Clostridium ramosum (Cr) before CTX-induced injury. F) RORγ+ Treg cells in colonic lamina propria (left) or muscles (right). Unpaired t-test (A, B) or one-way ANOVA (F). See also Figure S4.
We performed scRNA-seq on sorted Treg cells 3 days after CTX-induced injury in mice treated with VMNA or vehicle (Figures 4C, S4 D–I). The distribution of cells across the UMAP space differed between vehicle- and VMNA-treated animals, with the latter being relatively depleted of the RORγ+ cluster, while showing a proportional increase in the reparative and early-activation clusters (Figure 4C). Accordingly, muscle Treg cells from VMNA-treated animals had fewer transcripts preferentially expressed by RORγ+ Treg cells, such as Maf, Cxcr6 and Ltb4r1, while expressing more transcripts characteristic of reparative Treg cells, such as Ikzf2, Gata3 and Il1rl1 (Figure 4D), confirming the importance of microbial signals in shaping the muscle RORγ+ Treg compartment.
These results raised the question of whether the impact of the microbiota on muscle RORγ+ Treg cells was generic, or was specific to particular microbial strains. GF mice were monocolonized for 14 days with C. ramosum, which efficiently induces colonic RORγ+ Treg cells, or with P. magnus, which does not3, at which point hindlimb muscles were injected with CTX (Figure 4E). As expected, monocolonization with C. ramosum, but not P. magnus, restored colonic RORγ+ Treg cells to SPF levels. Three days after CTX injection, P. magnus-monocolonized mice showed no increase in their muscle RORγ+ Treg compartment above GF levels, whereas C. ramosum-monocolonized mice exhibited a specific expansion of the muscle RORγ+ Treg population to a degree comparable with that of SPF mice (Figure 4F).
In sum, these data indicate that the specific microbial signals that induce RORγ+ Treg cells in the colon predicate accumulation of the analogous RORγ+ Treg population in regenerating muscle. And they provide additional confirmation of the colonic provenance of muscle RORγ+ Treg cells.
Loss of muscle RORγ+ Treg cells induces more local IL-17A-producing cells and impairs muscle regeneration
Abx treatment severely impairs muscle regeneration43, probably due, at least in part, to the loss of microbiota-dependent RORγ+ Treg cells (Figure 4A–F). But other lymphocyte populations, namely IL-17A-producing γδT cells, are similarly shaped by the microbiota and were recently reported to promote muscle regeneration43. So we addressed the functional relevance specifically of RORγ+ Treg cells to muscle regeneration. Colonic RORγ+ Treg cell differentiation is dependent on the TF c-Maf; hence Foxp3cre × Maffl/fl (MafΔTreg) mice, which harbor a deletion of Maf specifically in Treg cells, are impoverished in colonic RORγ+ Treg cells14,15. Prior to injury, MafΔTreg mice had a similar fraction of muscle Treg cells and a distribution of the major immunocyte compartments similar to those of their Foxp3cre × Mafwt/wt (Mafwt) littermate controls (Figures 5A, S5A). Three days after CTX injection, MafΔTreg mice had a reduced fraction of muscle Treg cells (Figure 5A), with a diminished contribution of the RORγ+ subtype (Figure 5B). In contrast, neither the fraction nor numbers of Helios+ or Helios−RORγ− Treg cell subsets were reduced (Figure S5B). In addition, ST2 and Areg, two key markers of reparative Helios+ Treg cells, were similarly expressed in the two genotypes (Figure S5C). scRNA-seq of muscle Treg cells confirmed a reduced RORγ+ cluster in MafΔTreg mice, with proportional increases in the reparative and IFN-responding clusters (Figures 5C, S5E–J). Importantly, the reparative Treg cell cluster in MafΔTreg mice showed typical expression of diagnostic signature genes (Figure S5D), further confirming that Maf deletion primarily impacted the muscle RORγ+ Treg compartment, without obvious influences on other subtypes.
Figure 5: Impacts of RORγ+ Treg cell depletion on immunocytes and tissue repair.
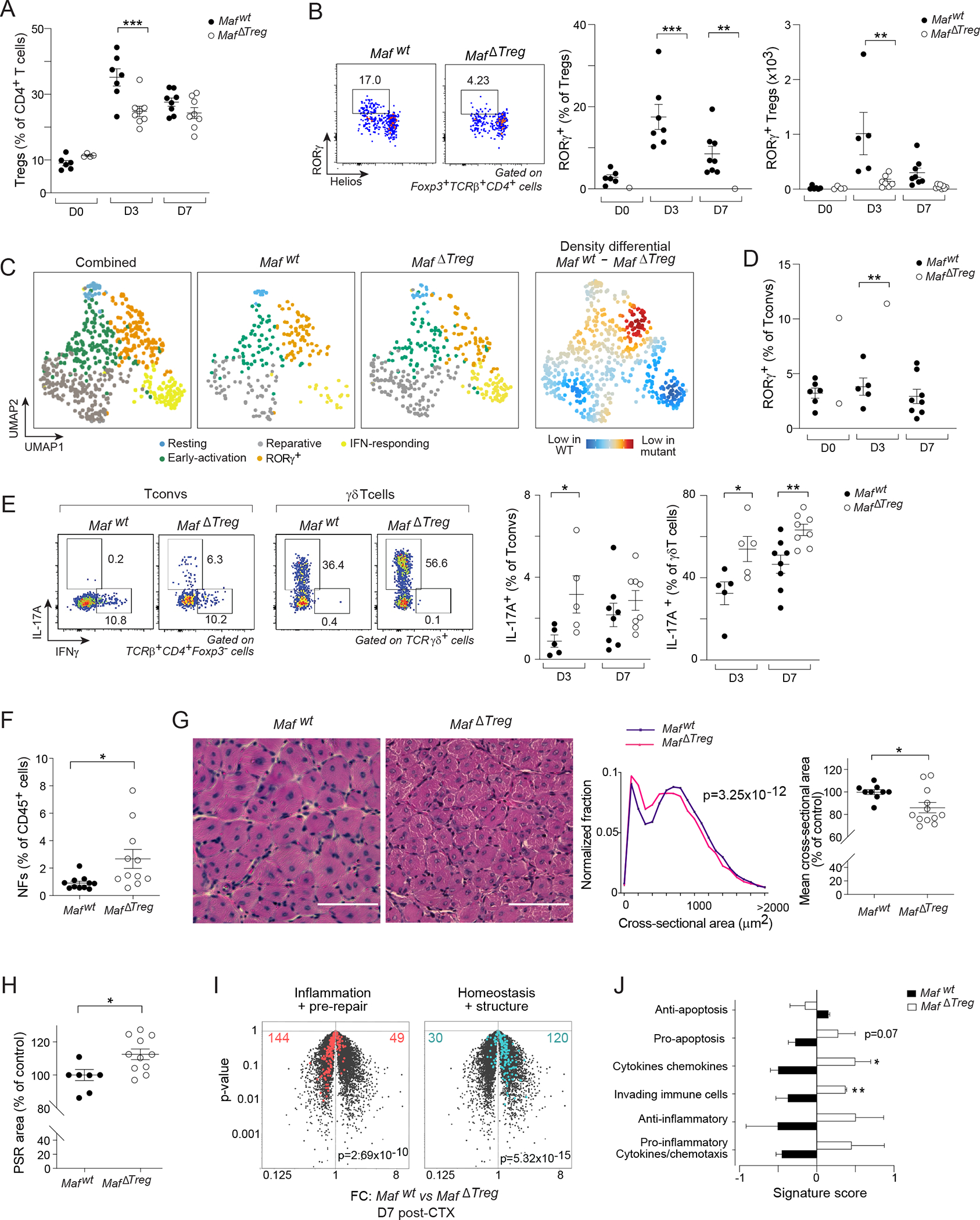
Comparisons of hindlimb muscles from Mafwt and MafΔTreg littermates 0, 3, or 7 days after CTX-induced injury. Quantification of (A) Total, and B) RORγ+ Treg cells. (C) scRNA-seq of muscle Treg cells 3 days after CTX-induced injury. 2D UMAP plot of combined data from the two genotypes (left); each individual condition (middle) and their differential (right). (D) Quantification of muscle RORγ+CD4+Foxp3− (Tconv) cells, and (E) production of IFNγ and IL-17A by Tconv and γδT cells. (F-J) Analysis of muscle repair efficiency 7 days after CTX-induced injury. F) Quantification of muscle neutrophils (NFs). G) Representative images of H&E staining (left); distribution of cross-sectional areas of individual centrally nucleated fibers (middle); average fiber areas for individual mice (right). H) Quantification of fibrotic areas via picrosirius red (PSR) staining. (I, J) Transcriptional analyses of whole muscle (n=3). I) Volcano plot illustrating FC differences between the two genotypes. Repair-related signatures43 are highlighted. J) Gene-signature scores of inflammatory gene sets44. Representative dot-plots and images are from 2–4 independent experiments. t-test of weighted sums (G middle), Chi-squared test (I), otherwise unpaired t-test. See also Figure S5.
Having established the validity of MafΔTreg mice as a model system, we probed the consequences of muscle RORγ+ Treg cell loss by analyzing expression of T-bet, Gata3, and RORγ (key TFs for Th1, Th2, and Th17 cell differentiation, respectively) in the Tconv population of hindlimb muscles from Mafwt and MafΔTreg littermates. Three days after CTX injection, comparable fractions of T-bet+ and Gata3+ Tconv cells were observed in the two genotypes, whereas there was an increase in RORγ+ Tconv cells in the mutant mice (Figures 5D, S5J). Accordingly, a higher percentage of Tconv cells from MafΔTreg than from Mafwt mice expressed IL-17A; but not IFNγ, IL-4, IL-5, or IL-13 (Figures 5E, S5K). In MafΔTreg mice, muscle γδT cells, the main cell-type producing IL-17A after injury43, had an increased proportion of IL-17A+ cells (Figure 5E). These data demonstrate that muscle RORγ+ Treg cells reined in a type-17 inflammatory response induced by muscle injury.
That RORγ+ Treg cells also regulated muscle regeneration was established by comparing Mafwt and MafΔTreg mice 7 days post-injury via several assays. First, the mutant mice had more muscle neutrophils (NFs) (Figure 5F), a sign of poor resolution of inflammation. Secondly, histological analysis of individual muscle fibers revealed that mice lacking RORγ+ Treg cells to have a significantly smaller distribution of fiber sizes and of the average cross-sectional area than control littermates (Figure 5G), similar in magnitude to the difference previously reported for mice lacking reparative (ST2+) Treg cells26, and indicative of less efficient regeneration in the mutant mice. Thirdly, muscles from MafΔTreg mice had greater collagen deposition, as measured by picrosirius red (PSR) staining (Figures S5L, 5H), signaling increased fibrosis, again a sign of poor muscle regeneration. Fourthly, as an orthogonal measure of tissue regeneration, we performed RNA-seq analysis on whole muscles from the two groups, and assessed expression of two gene modules previously demonstrated to dynamically change in response to injury: 1) an “inflammation + pre-repair” signature that is weakly expressed prior to injury, peaks at day 3–4, and is progressively turned off by days 7–14; and 2) a “homeostasis + structure” signature, comprised of genes involved in muscle filament structure and contractile function, which is turned down during active regeneration, and gradually turned back up by days 7–1443. MafΔTreg mice aberrantly maintained up-regulation of the inflammation + pre-repair signature and down-regulation of the homeostasis + structure signature at day 7 post-injury (Figure 5I). These observations were confirmed with an independent set of inflammatory gene signatures characteristic of muscle injury44 (Figure 5J).
In sum, muscle RORγ+ Treg cells are crucial for controlling local IL-17A production by multiple cell types. They also promote early resolution of inflammation and, ultimately, efficient tissue regeneration.
Failure to regulate IL-17A impairs MuSC differentiation and thereby muscle regeneration
Inflammatory cytokines such as tumor necrosis factor (TNF)α and IFNγ play key roles at early stages of muscle regeneration11,25,45–49. However, failure to down-regulate these same cytokines at subsequent stages is detrimental to the regenerative process, highlighting the need for dynamic regulation of inflammation. We recently reported that Tγδ17 and Th17 cells accumulate at early stages after muscle injury, which induces the proliferation of MuSCs and thereby promotes efficient regeneration43. Since depletion of RORγ+ Treg cells resulted in more cells making IL-17A (Figure 5E), failure to temporally regulate its production could paradoxically impede the process. Therefore, we compared the effects of administering rIL-17A early, late, or continuously after CTX-induced injury and evaluated muscle regeneration 7 days later (Figure 6A). These experiments were conducted on animals from Jackson Laboratory since they are known to have relatively low numbers of RORγ+ T cells and thus low basal IL-17A production43, which enabled us to supplement IL-17A at diverse stages of tissue regeneration, and thereby simulate the impact of IL-17A dysregulation subsequent to the loss of RORγ+ Treg cells. Seven days after CTX injection, the cross-sectional area of individual muscle fibers in mice treated with rIL-17A at only early times after injury was increased compared with that of vehicle-treated controls (Figure 6B). In contrast, mice with late or continuous rIL-17A treatment did not exhibit a regenerative boost (Figure 6B). Additionally, late rIL-17A treatment resulted in more muscle NFs and greater muscle fibrosis (Figure 6C, D). These results illustrate the necessity of temporally correct regulation of IL-17A production during muscle regeneration: pro-reparative when restricted to early stages; anti-reparative with prolonged exposure.
Figure 6: Impacts of IL-17A on muscle stem cells and repair.

(A-D) B6 mice were treated with vehicle or rIL-17A early, late or continuously (cont.) after CTX-induced injury. Muscles were analyzed 7 days post-injury. A) Treatment schematic. B) Distribution of cross-sectional areas of individual centrally nucleated fibers (left), and average fiber areas for individual mice (right). C) Quantification of muscle fibrotic areas via PSR staining. D) Quantification of muscle NFs. (E-G) RNA-seq analysis of sorted MuSCs from PBS- or rIL-17A-treated mice on days 0, 1 and 3 after CTX-induced injury. Treatment schedule as per panel A. E) Il17ra transcript quantification. F) k-means clustering of dynamically differential transcripts. Only select clusters are depicted. G) FC/FC plots for PBS- vs rIL-17A-treated mice on day 3 versus 1 after injury. MuSC differentiation signature genes54 highlighted in red. (H-J) Freshly isolated MuSCs were cultured in vitro with or without rIL-17A. Representative images (left, 2–3 independent experiments) and summary data (right) of H) EdU incorporation, I) myogenin (MyoG), and J) Myosin Heavy Chain (MyHC) expression after 2, 3 and 4 days of culture, respectively. Kruskal-Wallis test (B left), Chi-squared test of the number of signature genes falling on either side of the diagonal (G), Unpaired t-test (H-J), otherwise one-way ANOVA. See also Figure S6.
To explore the mechanisms underlying the detrimental effects of prolonged IL-17A exposure, we first identified cells capable of expressing both chains of the IL-17R using previously published scRNA-seq data from injured muscle50. Il17ra transcripts were abundant in multiple immunologic and non-immunologic cell types, while transcripts of Il17rc were restricted to MuSCs and mesenchymal stromal cells (Figure S6A, B). Because of their obvious relevance, we focused on MuSCs, a population of quiescent stem cells that become activated and proliferate for a certain number of cycles in response to muscle injury25,51,52. They must then exit the cell cycle to initiate the highly orchestrated differentiation program leading to myocytes, which eventually fuse with each other and with existing myofibers to regenerate healthy muscle tissue. Consequently, various inflammatory cytokines that promote MuSC proliferation at early stages of injury can prohibit their myogenic differentiation upon prolonged exposure as they do not exit the cell cycle and myogenic TFs remain suppressed48,49,53. Thus, controlling inflammation could be an important checkpoint for effective muscle regeneration. RNA-seq of flow-cytometrically sorted MuSCs (CD45−CD31−SCA-1−VCAM-1+, Figure S6C) on days 0, 1, and 3 after injury revealed dynamic expression of Il17ra by MuSCs, highest at steady-state and progressively reduced on days 1 and 3 after injury (Figure 6E).
Next, we performed RNA-seq on sorted MuSCs from rIL-17A- or vehicle-treated mice on days 0, 1 and 3 after CTX-induced injury (administration scheme as per Figure 6A). k-means clustering of the MuSC time-course highlighted 15 gene clusters coordinately modulated during the regenerative process. Focusing on clusters differentially expressed between rIL-17A- and vehicle-treated mice at day 3 after injury, we identified a set of differentiation-related gene modules pertaining to muscle structure, myogenic differentiation and metabolism (TCA cycle and oxidative phosphorylation) with reduced expression in rIL-17A-treated mice (Figure 6F). Relatedly, expression of a previously identified myogenic differentiation signature54 was down-regulated in MuSCs from rIL-17A-treated mice, as evidenced by the off-diagonal cloud on the FC/FC plot of Figure 6G. On the other hand, expression of a cell-division module was enriched in the rIL-17A-treated group (Figure 6F).
To determine whether this inhibitory effect reflected a direct impact of IL-17A on MuSCs, we sorted them from uninjured mice, and cultured them in vitro with or without rIL-17A under conditions that promote myogenic differentiation. In response to the isolation process and culturing with serum, MuSCs are activated and enter the cell cycle; subsequently, they commit to the myogenic fate and differentiate into myotubes55. After 2 days of culture, a greater fraction of rIL-17A-treated MuSCs were in cycle, as measured by EdU incorporation (Figure 6H). On day 3 and 4, we assessed MuSC differentiation by quantifying the expression of the myogenic commitment factor, Myogenin (MyoG), and myosin heavy chain (MyHC), a marker of differentiated myotubes. In line with the above-mentioned transcriptomic data (Figure 6 F, G), continuous rIl-17A treatment reduced the fraction of differentiated MyoG+ and MyHC+ cells (Figure 6I, J). These results agree with previous reports showing an inhibitory effect of IL-17A on MyoG expression and the consequent myogenic differentiation of myoblast cell lines56.
Thus, our data indicate a direct, time-dependent impact of IL-17A on MuSCs, initially promoting their proliferation, but impeding their differentiation upon prolonged exposure. Given the critical role of RORγ+ Treg cells in the temporal regulation of IL-17A production in regenerating muscle (Figure 5E), timely recruitment of RORγ+ Treg cells at early stages of regeneration appears to be crucial for shielding differentiating MuSCs from IL-17A, thereby promoting return to homeostasis.
RORγ+ Treg cells regulate IL-17A-driven inflammation in a model of non-alcoholic steatohepatitis (NASH) in a microbiota-dependent manner
Lastly, we probed the generality of RORγ+-Treg-cell-mediated inter-tissue communication. As a preliminary step, we examined RORγ expression in various tissue-Treg compartments at steady-state. A population of RORγ+ Treg cells was detected in multiple extra-gut tissues, including the liver, lung and kidney, usually comprising 5–10% of the Treg pool (Figures 7A, S7A). As in the colon and muscle, the RORγ+ Treg populations in these tissues were microbiota-dependent. Moreover, analysis of a previously published scRNA-seq dataset from CD4+ T cells isolated from cardiac muscle after myocardial infarction57 identified a distinct population of RORγ+ Treg cells with a marker profile similar to that of their colonic and skeletal muscle counterparts (Figures 7B–C, S7B–F).
Figure 7: Generality of RORγ+ Treg cells’ role in regulating tissue inflammation.
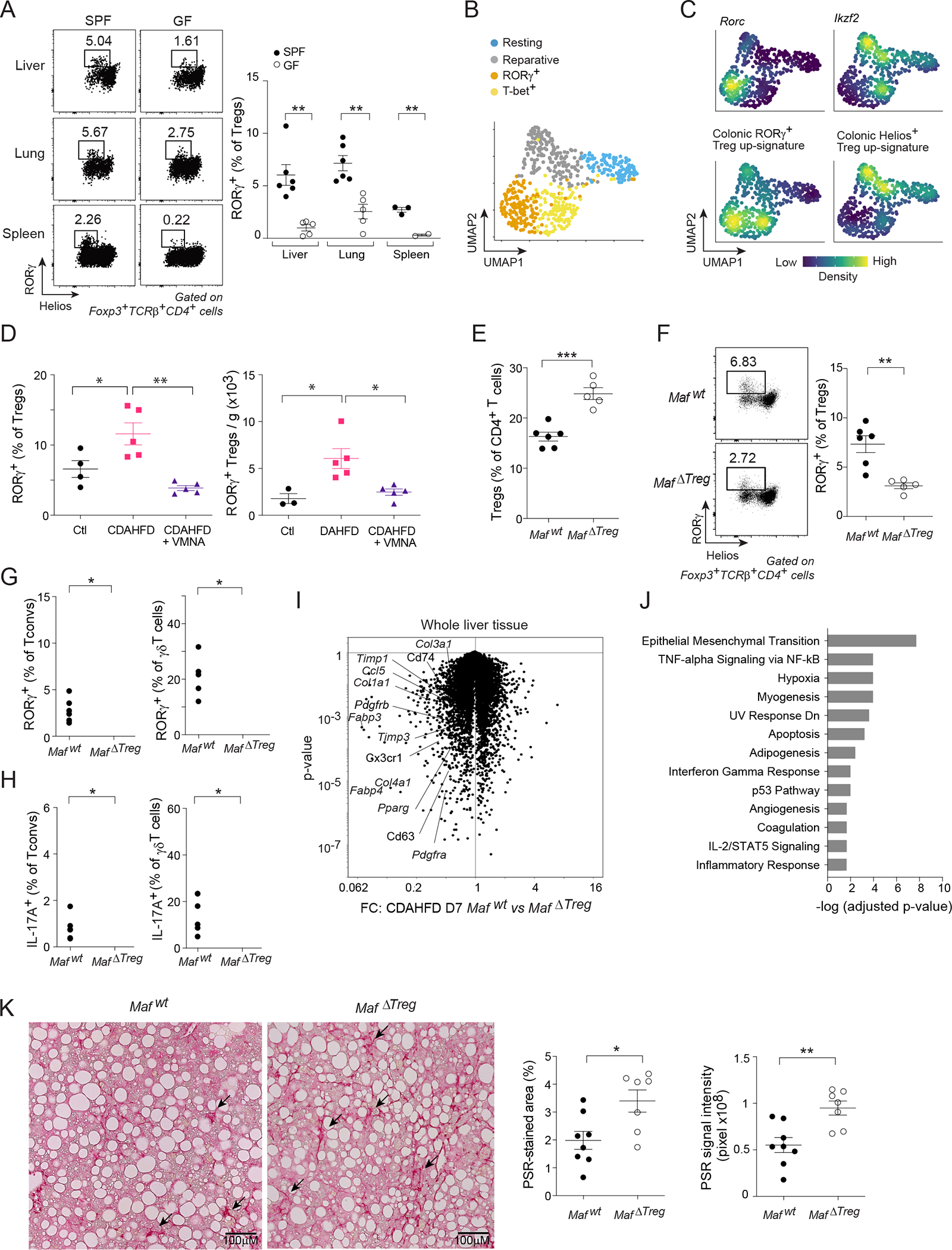
(A) RORγ+ Treg cell fractions in diverse tissues of SPF and GF mice. (B, C) scRNA-seq of cardiac muscle Treg cells after myocardial infarction57. B) 2D UMAP plot. C) Density plot of the indicated genes and signatures. (D) Quantification of liver RORγ+ Treg cells after one week of feeding with control diet (Ctl) or choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) ± VMNA treatment. (E-J) MafΔTreg or Mafwt mice were fed with CDAHFD for one week. Quantification of liver E) total Treg cells; F) RORγ+ Treg cells; G) RORγ+ Tconv (left) and γδT cells (right); and H) IL-17A expression. I) Volcano plot illustrating the transcriptional differences in livers from the two genotypes. J) Pathway analysis of transcripts significantly up-regulated ≥ 1.5-fold in MafΔTreg livers. (K) Mice were fed with CDAHFD for 8 weeks. Representative images (left) and summary data (right) of liver PSR staining. Representative dot-plots and images are from 2 independent experiments. Unpaired t-test (A, D-F, K), or paired t-test (G, H). See also Figure S7.
As the steady-state RORγ+ Treg populations in liver, lung and kidney were generally smaller than that of regenerating muscle, we wondered whether they would increase in response to sterile injury or inflammation. We chose a diet-induced NASH model to address this question because the gut:liver axis has been implicated in promoting multiple hepatic pathologies, and because IL-17A-mediated inflammation plays a central role in NASH progression58–60. Feeding mice a choline-deficient, amino-acid-defined, high-fat diet (CDAHFD) rapidly induces hepatocellular death and inflammation, with mice succumbing to tissue fibrosis at later disease stages61. We analyzed liver RORγ+ Treg cells after one week of CDAHFD feeding, an early time-point at which intestinal permeability and the colonic RORγ+ Treg compartment were not altered, and observed a fractional and numerical increase in liver RORγ+ Treg cells vis-a-vis mice fed control diet (Figures 7D, S7G–I). Mirroring our observations on regenerating skeletal muscle, the CDAHFD-induced increase in liver RORγ+ Treg cells was abolished in VMNA-treated animals (Figure 7D).
To assess the functional relevance of RORγ+ Treg cells in this context, we again turned to MafΔTreg mice. At steady-state, mutant mice had a normal representation of the major immunocyte populations (Figure S7J). But, one week after the introduction of CDAHFD, they showed an increase in total liver Treg cells (Figure 7E), though a substantially reduced population of RORγ+ Treg cells (Figure 7F). This reduction was accompanied by higher percentages of liver RORγ+ Tconv and γδT cells (Figure 7G). Accordingly, the fraction of cells producing IL-17A, but not those making IFNγ, IL-4, IL-5 or IL-13, in MafΔTreg mice was higher for both liver Tconv and γδT cells (Figures 7H, S7K, L).
IL-17A is a well-established driver of NASH pathology through its promotion of inflammation, metabolic dysregulation, and the fibrogenic machinery in hepatic stellate cells59,60,62. To explore impacts on these processes, we examined the effect of RORγ+ Treg cell loss on whole-liver gene expression profiles after a brief course of CDAHFD feeding. 387 and 204 genes were significantly up- or down-regulated, respectively, >1.5-fold in MafΔTreg compared with Mafwt mice (Figure 7I). Loci preferentially expressed in the mutant livers included metabolic genes implicated in NASH pathology (e.g Pparg and Fabp4), inflammatory genes (e.g. Ccl5, Cx3cr1 and Cd63) and multiple extracellular matrix genes (e.g. Col1a1, Col3a1, Col4a1 and Timp3). Consistently, pathway analysis pointed to epithelial-mesenchymal transition and immune-response modules as the top up-regulated pathways in the absence of RORγ+ Treg cells (Figure 7J).
We questioned whether the early changes in response to injury would translate into increased NASH-induced fibrosis at later stages by analyzing MafΔTreg mice after 8 weeks of CDAHFD feeding, a timepoint at which liver fibrosis has been established61. At this timepoint, the mutant mice showed a decreased fraction of RORγ+, but not Helios+, ST2+, or Areg+ Treg cells, as well as increased RORγ+ and IL-17A+ Tconv populations (Figure S7M–Q). In addition, they had an increased representation of inflammatory macrophages and NFs (Figure S7R). While the plasma concentrations of liver transaminases were comparable between the two genotypes (Figure S7S), MafΔTreg livers had significantly larger fibrotic scars, as measured by PSR staining (Figure 7K), indicating a protective role of RORγ+ Treg cells in inflammation-induced liver fibrosis.
These results serve to generalize our findings from regenerating muscle. The homeostatic function of microbiota-dependent RORγ+ Treg cells extends far beyond the intestine to multiple non-mucosal tissues.
DISCUSSION
Our temporal scRNA-seq studies uncovered a population of RORγ+ Treg cells accruing at early times after acute skeletal muscle injury. Explorations of these cells’ role in muscle regeneration yielded several important insights: 1) that the microbiota regulated muscle repair via RORγ+ Treg cells emanating from the gut; 2) that local inflammatory signals and TCR engagement drove their migration to and expansion within regenerating muscle; 3) that these Treg cells controlled the tenor and dynamics of injury-induced inflammation while also helping to balance the proliferative and differentiative activities of muscle stem cells; and 4) that RORγ+ Treg cell emissaries from the gut appeared to play a general role in the homeostasis of extra-gut tissues.
The original discovery of RORγ+ Treg cells as a microbiota-dependent subtype of colonic regulatory T cells emphasized their role as guardians of intestinal health through promotion of tolerance to food antigens and local microbes3,4,12,14–16. Here we demonstrated that the impact of colonic RORγ+ Treg cells extended far beyond their site of generation: they formed a specialized pool of gut-trained regulators that could be rapidly mobilized in response to tissue emergencies at non-mucosal sites. Equipped with a distinct set of TCRs, homing receptors, and effector molecules, these cells orchestrated effective tissue regeneration and return to homeostasis. Since RORγ+ Treg cells are thought to mainly react to commensal or food antigens, the TCR-mediated proliferation and retention of these cells in regenerating muscle raises the question of how local antigens promote this response. Cross-reactivity between microbial (including commensal) antigens and autoantigens is well-documented63–69. Thus, some form of epitope mimicry between microbial or food and muscle antigens might be at root, especially since that tissue damage and the associated inflammation could increase the strength and breadth of autoantigen presentation. It is by now well established that enteric microbes can drive effector T cell responses to a variety of challenges outside the gut17–21. Extending this concept to Treg cells raises the possibility of a self-contained system of gut-trained effector and regulatory T cell recruits, which could both coordinate rapid and efficient return to homeostasis and avoid over-shooting of microbiota-induced inflammatory responses.
Tissue regeneration in general, and muscle regeneration in particular, is a dynamic, multi-cellular process in which temporal regulation of pro-inflammatory mediators is fundamental to fidelity and efficiency25,45–49. While crucial for the early response to tissue injury, pro-inflammatory molecules can paradoxically impede tissue repair if not controlled properly. We have uncovered a spike in various classes of IL-17A-producing cells early after muscle injury (herein and43); to our knowledge, IL-17A had not previously been associated with muscle repair. One function of IL-17A is to recruit NFs, which have important early roles in secreting mediators and clearing debris, but must be reined in at later times to permit effective tissue regeneration43,70. Akin to its role in expanding stem cell pools in other contexts71–75, IL-17A also promoted the proliferation of MuSCs via two mechanisms: by directly stimulating their cell-cycle progression and, indirectly, by promoting the accumulation of NFs, which can also induce MuSC proliferation70. Our data highlight a rheostatic role for RORγ+ Treg cells in controlling the dynamics of IL-17A activity along the course of tissue regeneration. By limiting the activity of IL-17A to early stages of injury, they shielded MuSCs from prolonged exposure to IL-17A, which could impair myocyte differentiation, resulting in inferior repair and enhanced fibrosis. Similar phenomena wherein IL-17A inhibits tissue stem-cell differentiation directly or indirectly (in the CNS and skin) have been observed10,76.
Our data suggest that regulation of IL-17-mediated inflammation by microbiota-dependent RORγ+ Treg cells may be a general axis in play across multiple extra-gut sites. Thus, dysbiotic gut conditions capable of inhibiting the generation or export of RORγ+ Treg cells are likely to have systemic impacts on tissue homeostasis at multiple extra-gut sites. The potential for such detrimental outcomes advocates for judicious use of antibiotics after tissue injury. On the other hand, modulating RORγ+ Treg cells via diet or personalized probiotics77,78 could prove to be potent therapeutic add-ons in such contexts.
LIMITATIONS OF THE STUDY
Considering that a fraction of RORγ+ Treg cells constantly egresses from the colon to extra-gut sites at steady state, it is not clear to what extent the RORγ+ Treg population in injured muscle issued directly from the colon rather than first sojourning through one or more other tissues. Complex cell tracking experiments will be required to formally address this point. While injury-induced accumulation of RORγ+ Treg cells depended on the ligands of CCR2 and LTB4R1, it is theoretically possible that this dependency reflects indirect effects on non-Treg cells expressing these receptors.
STAR★Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Diane Mathis (dm@hms.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data newly reported in this paper were deposited in the Gene Expression Omnibus (GEO) database under accession no. GSE196338.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Foxp3IRES-GFP (Foxp3GFP) mice79 were obtained from V. Kuchroo (Brigham and Women’s Hospital, Boston, MA). C57BL/6J, Tcrb−/− and B6.129P2(Cg)-Rorctm2Litt/J (RorcGFP) mice80 were purchased from Jackson Laboratory. The Kaede transgenic mouse line (B6.Kaede)30, obtained from O. Kanagawa, and RorcGFP mice were crossed to Foxp3Thy1.1 mice81 (from A. Rudensky, Memorial Sloan Kettering Cancer Center, New York, NY). Foxp3DTR mice82 were obtained from A. Rudensky and crossed to B6.CD45.1 mice from Jackson Laboratory. To generate Treg-specific c-Maf-deficient mice, we crossed Foxp3Cre mice83 (from A. Rudensky) to Maffl mice84 (from C. Birchmaier; Max Delbrück Center, Berlin, Germany). Six- to 12-wk-old male and female mice were used for all experiments. Sexes were not mixed in transcriptomic experiments. All mice were housed in our SPF facility at Harvard Medical School. All experiments were conducted under protocols approved by Harvard Medical School’s Institutional Animal Care and Use Committee.
METHOD DETAILS
Mouse treatments and colonizations
To induce muscle injury, we anesthetized mice by intraperitoneal (i.p.) administration of ketamine:xylazine in combination (10 mg/kg: 2 mg/kg), and i.m.-injected them with 0.03 mL per muscle of CTX from Naja mossambica or Naja pallida (0.03 mg/mL; Sigma-Aldrich) in one or more hindlimb muscles (tibialis anterior (TA), gastrocnemius, quadriceps), as previously described7. To exclude the possibility that RORγ+ Treg cell accumulation resulted from introduction of skin microbes during our standard injury protocol, we employed a sterile surgical procedure: the skin was opened and injury was induced without the needle passing through skin. As shown in Figure S4C, this approach yielded a fraction of RORγ+ Treg cells similar to that induced by the standard injury protocol. To block egress of lymphocytes from peripheral organs, we i.p.-injected mice with 1 mg/kg of the S1P receptor agonist, FTY720 (Cayman Chemical), 4 hr prior to Kaede photoconversion or CTX-induced injury, and daily thereafter. For in vivo CCL2 and LTB4R1 blockade, mice were treated with anti-CCL2 neutralizing antibody (200 μg i.p.; clone 2H5) or with the LTB4R1 antagonist CP-105696 (20 mg/kg body weight by oral gavage; Sigma-Aldrich) 6 hr prior and 1 d after CTX injection. For in vivo MHC-II blockade, mice were i.p.-injected with 500 μg of anti-mouse I-A/I-E (clone M5/114) or rat IgG2b control (clone LTF-2) 6 hr prior to CTX-induced injury followed by 200 μg on each subsequent day. All in vivo antibodies were purchased from Bioxcell. For in vivo rIL-17A administration, 500 ng of rIL-17A (Peprotech) was i.p.-injected in sterile phosphate buffered saline (PBS) at various timepoints pre- or post-injury, as indicated in the schematic diagram in Figure 6A. To increase muscle specificity, rIL-17A (100 ng/muscle) was i.m.-injected on day 0 along with CTX.
For mono-colonization, GF mice were orally gavaged with single bacterial species at 4 weeks of age for 2 weeks. Stool samples were collected and plated at 2 weeks to verify colonization and rule out contamination from other species. For Abx treatment, mice were treated with 0.5 mg/ml vancomycin (RPI), 1 mg/ml metronidazole (Sigma-Aldrich), 1 mg/ml neomycin (Fisher Scientific), 1 mg/ml ampicillin (Sigma-Aldrich) dissolved in drinking water for three weeks prior to muscle injury.
For liver studies, male mice were fed a control diet containing 10 kcal% Fat (A08051501i) or CDAHFD (A06071309i), both purchased from Research Diets.
Isolation of leukocytes from lymphoid and nonlymphoid tissues
Skeletal muscle:
Isolation of skeletal muscle lymphocytes was done as previously described7. Briefly, hindlimb muscles were excised, minced and digested at 37°C for 30 min in a solution containing Dulbecco’s Modified Eagle’s Medium (DMEM, 2% fetal calf serum (FCS), collagenase II (2 mg/mL; Thermo Fisher Scientific) and DNase I (100 μg/mL; Sigma-Aldrich). Leukocytes were enriched by Percoll (GE Healthcare) density centrifugation (40%:80%, 25 min at 1,126 × g). The interphase containing leukocytes was recovered, washed, and stained for analysis or sorting by flow cytometry.
Colon:
Intestinal tissues were cleaned and treated with Roswell Park Memorial Institute (RPMI) medium containing 1 mM dithiothreitol, 20 mM ethylenediaminetetraacetic acid (EDTA) and 2% FCS at 37°C for 15 min to remove epithelial cells, minced and dissociated in collagenase solution (1.5 mg/ml collagenase II (Gibco), 0.5 mg/ml dispase and 1% FCS in RPMI) with constant stirring at 37°C for 30 min. Single-cell suspensions were filtered and washed with 10% FCS RPMI solution before staining and sorting by flow cytometry.
Liver, lung and kidney:
Mice were perfused with ice-cold PBS through the left ventricle of the heart. Tissues were excised and minced in DMEM containing collagenase IV (0.5 mg/ml; Gibco), DNase I (150 μg/ml; Sigma-Aldrich) and FCS (1%), and were incubated in a 37°C water bath with shaking for 30–45 min. Digested tissues were passed through a 70-μm cell strainer and washed in DMEM/FCS. Leukocytes from liver were enriched by Percoll density centrifugation (36%, 10 min at 800 × g) as previously described85.
Spleen and lymph nodes:
Lymphocytes were obtained by mechanical disruption, followed by red blood cell lysis, and filtered and washed with 10% RPMI solution.
Isolation and analysis of muscle stem cells
Isolation of muscle MuSCs was done as previously described86. Briefly, hindlimb muscles were minced and digested by collagenase II (800 U/ml) in Ham’s F10 with 10% horse serum at 37°C for 90 min, followed by a second digestion step using collagenase II (80 U/ml) and dispase (11 U/ml) for 30 min. Cell suspensions were passed through a 20-Gauge needle 10 times and then filtered through a 40-μm cell strainer, washed, and stained for analysis or sorting by flow cytometry.
Flow cytometry
The following antibodies were used for flow-cytometric staining: anti-CD4, -CD8a, -CD45, -CD45.1, -CD45.2, -Thy1.1, -CD44, -CXCR6, -CD11b, -CD3, -TCRγδ, -IL-17A, -IFN-γ, -IL-4, -IL-5, -CD31, -Sca-1, -VCAM-1, -Helios, -T-bet, all from Biolegend; and -Foxp3, -RORγt, -c-Maf, -Gata3, -ST2, -IL-13 from eBioscience; -TCRβ, -Ki-67 from BD; -Areg from R&D; and -cleaved caspase-3 from Cell Signaling. Surface staining was performed for 30 minutes at 4°C, and viability was assessed using Zombie UV™ Fixable Viability Dye (Biolegend) as per the manufacturer’s instructions. Intracellular staining was performed using eBioscience™ Foxp3/Transcription Factor Staining Buffer Set. After surface staining of cells from Kaede mice, cells were fixed with 2% ice-cold paraformaldehyde for 30–60 min at 4°C, washed twice with PBS before proceeding to intracellular staining using eBioscience™ Foxp3/Transcription Factor Staining Buffer Set. For ex vivo intracellular cytokine and Areg staining, single-cell suspensions were stimulated for 3.5 hours at 37°C with 50 ng/mL phorbol 12-myristate 13-acetate (PMA) and 1 μM ionomycin (both from Sigma-Aldrich) in the presence of protein transport inhibitor cocktail (eBioscience) in complete RPMI-1640 media supplemented with 10% fetal bovine serum (Thermo Fisher Scientific). Cells were acquired with a FACSymphony flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (Tree Star).
Cell transfers
Treg and Tconv cell transfers were performed as previously described87. Briefly, 105 Treg cells (CD4+TCRβ+Foxp3GFP+) or 106 naïve CD4+ Tconv cells (CD4+TCRβ+CD44loFoxp3GFP−) were sorted from the spleens of CD45.1+ Foxp3GFP or B6.Foxp3GFP mice, respectively using MoFlo (Beckman Coulter) and were i.v. injected into CD45.1+/CD45.2+ Foxp3DTR hosts. At 1d prior and 1d after transfer, host Treg cells were depleted via i.p. injection with diphtheria toxin (Sigma-Aldrich) at 20 ng/g of body weight. For cell transfer into Tcrb−/− mice, CD4+ T cells were sorted from the spleen or colon of B6 mice, and matched cell numbers (1–5 × 105 cells) were i.v. injected into recipient mice. All cell transfers were performed 2 weeks prior to CTX-induced injury.
Analysis of cell migration via the Kaede system
Kaede mice were anesthetized with ketamine:xylazine in combination (10 mg/kg:2 mg/kg i.p). Photoconversion of the descending colon was done as previously described31. A custom-built fiberoptic endoscope (ZIBRA Corporation) was coupled to the handheld 405-nm laser via an in-house, custom-made connection device (fixed mounts were purchased from ThorLabs). After cleansing the colon of fecal pellets with PBS, we inserted the fiberoptic endoscope through the anus into the descending colon to a depth of 2.5 cm. The laser was switched on, thereby exposing the inner colon to violet light (beam diameter was 3.5 mm). Subsequently, the endoscope was gently retracted, pausing at 2-mm increments for 30s light pulses at each interval (for a total of up to 10 min). The fractions of Kaede-red+ cells in various immunocyte populations were analyzed by flow cytometry at the indicated time-points.
Single-cell studies
For standard genome-wide scRNA-seq analyses, Treg cells from hindlimb muscles of Foxp3GFP or Foxp3Cre × Maffl/fl mice were sorted by flow cytometry and encapsulated using the Chromium Single Cell 3′ v3 platform. For coupled scRNA-seq and scTcr-seq, Treg or CD4+ T cells from muscles and colon of six individual Foxp3GFP mice were sorted and encapsulated using the Chromium Single Cell 5′ v1.1 and V(D)J platform (10x Genomics). In some experiments, individual samples were tagged (hashed) with DNA-coded anti-CD45/MHC-I antibodies (Biolegend). For standard scRNA-seq, data analysis was done as previously described88,89. Data were processed using the standard cellranger pipeline (10x Genomics). HTO counts were obtained using the CITE-seq-Count package. Single-cell data were analyzed using the Seurat package90. HTOs were assigned to cells using the HTODemux function, and doublets and negative cells were eliminated from analysis. Low quality cells and doublets were also excluded from the analysis according to the criteria in Table S1. Clusters encompassing Treg cells were determined based on the expression of Treg marker genes (e.g. Foxp3, Il2ra, Ctla4 and Ikzf2), while clusters encompassing non-Treg cells were removed from analysis using the SubsetData function. Data were normalized using the NormalizeData function and scaled using the ScaleData function. Variable genes were determined using the FindVariableGenes function. Principal components (PCs) were calculated using the RunPCA function, and top significant PCs were selected using JackStraw() and ElbowPlot() functions. Clustering was done on selected PCs using the FindNeighbors followed by the FindClusters functions, and UMAP dimensionality reduction was calculated using the RunUMAP function. Signature scoring was done with Seurat’s AddModuleScore function. Differentially expressed genes were calculated using the FindAllmarkers and FindMarkers functions. Colon Treg signatures were generated from scRNAseq data of colonic CD4+ T cells from SPF mice88 using the FindMarkers function in Seurat to identify differentially expressed genes (>0.25 logFC with p-value<0.05) between RORγ+ and Helios+ colonic Treg cells. Visualization of expression density of transcripts or signature scores was done suing the Nebulosa package91. Comparison of colon and muscle RORγ+ Treg transcriptomes was done using our previously published tissue-Treg scRNAseq dataset28. [Note: muscle Treg cells in this dataset were isolated at day 4 after injury. Since different batches of CTX induce muscle injury and repair with slightly different kinetics, the peak of inflammation can be on either day 3 or day 4. We routinely titer different batches of CTX to adjust the time course accordingly]. scRNA-seq of muscle, colon and matched total spleen Treg cells was analyzed using the standard Seurat pipeline, and the RORγ+ Treg cluster in each tissue was identified by high expression of the colonic RORγ+ Treg signature. Average expression of genes in each cluster was calculated using the AverageExpression function in Seurat, and FC/FC plots were generated by Multiplot Studio (GenePattern; Broad Institute) using matched spleen Treg samples as a comparator for RORγ+ Treg cells in each tissue. Integration of scRNA-seq data from different experimental batches was done using the Harmony pipeline92. Analysis of previously published scRNA-seq dataset from cardiac muscle CD4+ T cells after myocardial infarction57 was done as described above. Briefly, clusters encompassing Treg cells were determined based on high expression of Foxp3, Il2ra, Ctla4 and Ikzf2, and clusters with high expression of Teff markers (e.g. Ifng, Il17a, Il4) were removed from analysis using the SubsetData function.
The Tcr-seq library was processed with the cellranger vdj pipeline (v4.0), using default parameters. This pipeline was used for the assignment of V and J genes, CDR3 regions and sequences. Resulting data were analyzed using the scRepertoire package32. Over two independent experiments, we recovered 1529 and 3918 individual muscle and colon Treg cells, while retaining Tcra and Tcrb sequence information from 72 and 79% of the cells, respectively. Repeated clonotypes were defined using the “strict” criteria in scRepertoire as cells sharing the VDJC segments of both Tcra and Tcrb with identical Cdr3 sequences at the nucleotide level. Clonal overlap scores, defined by scRepertoire as the intersection size of clonotypes between two populations divided by the length of the smallest one, were calculated using the clonalOverlap() function.
RNA-seq library preparation, sequencing and data processing
Cells were double-sorted using MoFlo Astrios or BD Aria II into TCL buffer (Qiagen) containing 1% 2-mercaptoethanol (2-ME, Sigma-Aldrich). For whole-muscle RNA-seq, total RNAs were isolated from TA muscles using TRIzol (Invitrogen) according to the manufacturer’s instructions. Total RNAs were isolated from liver samples using Rneasy Lipid Tissue Mini Kit (Qiagen). Two nanograms of RNA were suspended in 5 μL of TCL+1% 2-ME. Libraries were constructed, sequenced, and the data was processed according to the Immgen protocol (https://www.immgen.org/img/Protocols/ImmGenULI_RNAseq_methods.pdf), and as previously described43. Subsequent normalization of raw read counts was performed in the Bioconductor package DESeq2 using the median of ratios method. Batch effects were removed using the removeBatchEffect() function in the limma package. Following normalization, GCT and CLS files were generated and used for downstream analysis. After normalization, reads were further filtered by minimal expression and coefficient of variation in Multiplot Studio (GenePattern; Broad Institute).
Analysis of bulk RNA-seq data
Gene expression dynamics in PBS- and rIL-17A-treated MuSCs time-series data were assessed using a two-step regression modeling strategy implemented with maSigPro93. An alpha of 0.05 was used to account for multiple hypothesis testing and a false discovery rate of 5% was applied to first identify differentially expressed transcripts. To identify co-expression clusters (or modules) during the time-series, silhouette analysis partitioned differential genes into 15 clusters (cluster correlation, r2>0.65). Clusters were visualized using Morpheus (Broad Institute). For gene signature scoring, the normalized gene-expression matrix was log-transformed and the z-score of each gene was calculated. This matrix was used to calculate normalized gene-expression scores for each module using the score_genes function in the scanpy package in python. The annotated list of gene modules was obtained from a previously published study that identified transcriptional changes during muscle injury44. Volcano and FC/FC plots were generated using Multiplot Studio (Broad Institute). Pathway analysis of differentially expressed genes between classes of samples was performed using Metascape and Enrichr.
Analysis of intestinal permeability
Mice were fasted for 4 hr before oral gavage with 150 μl of 25 mg/ml 4 kDa FITC-dextran (Chondrex). Blood samples were collected after 4 hr, allowed to clot at room temperature for 30 min, and serum was isolated by centrifugation at 2000 g for 15 min. FITC fluorescent signal in the serum was measured on a BioTek Synergy Mx Microplate Reader.
Histology
TA muscles and livers were collected and fixed in 10% formalin. Tissues were processed at the Rodent Histopathology Core at Harvard Medical School for hematoxylin and eosin (for muscle fiber size analysis) and picrosirius red (for fibrosis) staining. Four muscle sections, 200 μm apart, were recovered and stained for analysis. Images were acquired with a Nikon Ti inverted microscope. Efficiency of muscle repair was assessed histologically, as previously described26,43,94. In brief, we first identified the section from each sample with the largest number of regenerating muscle fibers (centrally nucleated fibers). For 4–5 representative regions in each of these sections, the cross-sectional area of individual muscle fibers was calculated by manually tracing the fiber circumference using ImageJ 1.53c95. Approximately 1,000 fibers were traced per sample. To assess fibrosis, the fraction of Picrosirius-Red-stained area in TA muscle and liver sections was determined using ImageJ 1.53c.
MuSC functional assays
MuSCs (CD31−CD45−Sca1−Vcam1+) were sorted from hindlimb muscle using a BD FACSAria II cell sorter. For the 5-Ethynyl-2′-deoxyuridine (EdU) incorporation assay, freshly sorted MuSCs were cultured in Ham’s F10 with 10% horse serum and 1% penicillin-streptomycin (P/S) supplemented with 100ng/ml rIL-17A (Peprotech) for 36 h followed by a 4 h pulse with 10uM EdU. For the differentiation assay, freshly sorted MuSCs were cultured in Ham’s F10 with 10% horse serum and 1% P/S supplemented with 100 ng/ml rIL-17A. The medium was changed every 2 days. Cultured cells were fixed in 4% paraformaldehyde for 5 min, washed with 0.1% PBS with Tween (PBST), permeabilized by 0.5% PBST for 20 min and then blocked with 4% IgG-free bovine serum albumin in PBST for 1 h. Samples were incubated with mouse anti-MyoG (clone F5D, Santa Cruz) or mouse anti-MyHC (Clone MF20, R&D) antibodies at 4°C overnight, washed 3 times, followed by incubation with Cy-3 donkey anti-Mouse secondary antibody (Jackson ImmunoResearch) for 1 h. The nuclei were counterstained with 100 ng/ml 4’,6-diamidino-2-phenylindole (DAPI). After 3 washes, the samples were mounted and observed under a fluorescence microscope. The EdU staining was performed using a Click-iT™ imaging kit (Thermo Fisher Scientific). Images were acquired with a Nikon Ti inverted microscope. Image analysis was done using ImageJ 1.53c.
Measurement of liver transaminases
Blood samples were collected in EDTA-coated tubes and plasma was isolated by centrifugation at 2000 g for 15 min. Concentrations of plasma alanine transaminase and aspartate transaminase were analyzed on the Siemens Chemistry XPT clinical analyzer.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism software. Data were routinely presented as mean ± SEM. Statistical significance was calculated by unpaired Student’s t-test (two-tailed), t-test of weighted sums, Kruskal-Wallis test, one-way or two-way ANOVA. P-values for gene-signature enrichment either up or down between a pairwise comparison in volcano and FC/FC plots were determined using the χ2 test. Statistical significance is denoted as the following: *p<0.05, **p<0.01, ***p<0.001.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Brilliant Violet 605™ anti-mouse CD45 (clone 30-F11) | Biolegend | Cat# 103140 |
| APC anti-mouse CD45 (clone 30-F11) | Biolegend | Cat# 103112 |
| PE anti-Mouse TCRβ (clone H57-597) | Biolegend | Cat# 109208 |
| BUV737 anti-Mouse TCRβ (clone H57-597) | BD | Cat# 612821 |
| Brilliant Violet 711™ anti-mouse CD4 (Clone RM4-5) | Biolegend | Cat# 100550 |
| PE-eFluor™ 610 anti-mouse CD4 (clone RM4-5) | eBioscience | Cat# 61-0042-82 |
| APC/Cyanine7 anti-mouse CD8a (clone 53-6.7) | Biolegend | Cat# 100714 |
| Brilliant Violet 605™ anti-mouse CD45 (clone 30-F11) | Biolegend | Cat# 103140 |
| PerCP/Cyanine5.5 anti-mouse CD45.1 (clone A20) | Biolegend | Cat# 110728 |
| APC/Cyanine7 anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109824 |
| PE/Cyanine7 anti-mouse CD90.1 (clone OX-7) | Biolegend | Cat# 202518 |
| PerCP/Cyanine5.5 anti-mouse CD44 (clone IM7) | Biolegend | Cat# 103032 |
| APC anti-mouse CXCR6 (clone SA051D1) | Biolegend | Cat# 151106 |
| Brilliant Violet 711™ anti-mouse CD25 (clone PC61) | Biolegend | Cat# 102049 |
| PerCP/Cyanine5.5 anti-mouse CD11b (clone M1/70) | Biolegend | Cat# 101228 |
| APC/Cyanine7 anti-mouse Ly6G (Clone 1A8) | Biolegend | Cat# 127624 |
| APC/Cyanine7 anti-mouse CD3ε (clone 145-2C11) | Biolegend | Cat# 100330 |
| PE/Cyanine7 anti-mouse TCRγδ (clome GL3) | Biolegend | Cat# 118124 |
| Pacific Blue™ anti-mouse CD31 (clone 390) | Biolegend | Cat# 102422 |
| FITC anti-mouse Ly-6A/E (Sca-1; clone E13-161.7) | Biolegend | Cat# 122505 |
| PE/Cyanine7 anti-mouse VCAM-1 (clone 429) | Biolegend | Cat# 105720 |
| PE-Cyanine7 anti-ST2 (clone RMST2-2) | eBioscience | Cat# 25-9335-82 |
| Pacific Blue™ anti-mouse Helios (clone 22F6) | Biolegend | Cat# 137220 |
| Brilliant Violet 785™ anti-T-bet (clone 4B10) | Biolegend | Cat# 644835 |
| APC anti-Foxp3 (clone FJK-16s) | eBioscience | Cat# 17-5773-82 |
| Alexa Fluor™ 488 anti-Foxp3 (clone FJK-16s) | eBioscience | Cat# 53-5773-82 |
| PE anti-RORγt (clone AFKJS-9) | eBioscience | Cat# 12-6988-82 |
| APC anti-RORγt (clone AFKJS-9) | eBioscience | Cat# 17-6988-82 |
| PE-Cyanine7 anti-c-Maf (clone sym0F1) | eBioscience | Cat# 25-9855-82 |
| Alexa Fluor™ 488 anti-Gata3 (clone TWAJ) | eBioscience | Cat# 53-9966-42 |
| Alexa Fluor® 488 anti-mouse IL-17A (clone TC11-18H10.1) | Biolegend | Cat# 506910 |
| Brilliant Violet 785™ anti-mouse IFN-γ (clone XMG1.2) | Biolegend | Cat# 505838 |
| PE anti-mouse IL-4 (clone 11B11) | Biolegend | Cat# 504104 |
| PE anti-mouse IL-5 (clone TRFK5) | Biolegend | Cat# 504304 |
| PE anti-IL-13 (clone eBio13A) | eBioscience | Cat# 12-7133-81 |
| Alexa Fluor® 700 anti-Ki-67 (clone B56) | BD | Cat# 561277 |
| Biotin anti-Areg (polyclonal) | R&D | Cat# BAF989 |
| Alexa Fluor® 647 anti-cleaved Caspase-3 (Asp175; clone D3E9) | Cell Signaling | Cat# 9602S |
| TotalSeq-A anti-mouse hashtags | Biolegend | Cat# 155811 Cat# 155813 Cat# 155815 Cat# 155817 Cat# 155819 Cat# 155821 Cat# 155823 |
| TotalSeq-C anti-mouse hashtags | Biolegend | Cat# 155861 Cat# 155863 Cat# 155865 Cat# 155867 Cat# 155869 Cat# 155871 Cat# 155873 Cat# 155875 Cat# 155877 |
| Anti-Myogenin (clone F5D) | Santa Cruz | Cat# sc-12732 |
| Anti-Myosin Heavy Chain (clone MF20) | R&D | Cat# MAB4470 |
| Cy™3 Donkey Anti-Mouse IgG | Jackson ImmunoResearch | Cat# 715-165-150 |
| InVivoMAb anti-mouse/human/rat CCL2 (clone 2H5) | BioXCell | Cat# BE0185 |
| InVivoMAb polyclonal Armenian hamster IgG | BioXCell | Cat# BE0091 |
| InVivoMAb anti-mouse MHC Class II (I-A/I-E) (clone M5/114) | BioXCell | Cat# BE0108 |
| InVivoMAb rat IgG2b control (clone LTF-2) | BioXCell | Cat# BE0090 |
| Bacterial and Virus Strains | ||
| Clostridium ramosum | Geva-Zatorsky et al.,201796 | NA |
| Peptostreptococcus magnus | Geva-Zatorsky et al.,201796 | NA |
| Chemicals, peptides, and recombinant proteins | ||
| Cardiotoxin from Naja mossambica | Sigma-Aldrich | Cat# C9759 |
| Cardiotoxin from Naja pallida | Sigma-Aldrich | Cat# 217503 |
| FTY720 | Cayman Chemical | Cat# 10006292 |
| CP-105696 | Sigma-Aldrich | Cat# PZ0363 |
| Diphtheria Toxin | Sigma-Aldrich | Cat# D0564 |
| Recombinant murine IL-17A | Peprotech | Cat# 210-17 |
| Metronidazole | Sigma-Aldrich | Cat# M1547 |
| Ampicillin | Sigma-Aldrich | Cat# A0166 |
| Neomycin | Fisher Scientific | Cat# BP2669-25 |
| Vancomycin | RPI | Cat# 1404-93-9 |
| FITC-Dextran, 4 kDa | Chondrex | Cat# 4013 |
| Collagenase type II (Gibco) | Thermo Fisher Scientific | Cat# 17101015 |
| Collagenase type IV (Gibco) | Thermo Fisher Scientific | Cat# 17104-019 |
| Dispase | Thermo Fisher scientific | Cat# 17105041 |
| DNase I from bovine pancreas | Sigma-Aldrich | Cat# DN25 |
| Phorbol 12-Myristate 13-acetate (PMA) | Sigma-Aldrich | Cat# P8139 |
| Ionomycin | Sigma-Aldrich | Cat# I0634 |
| Protein Transport Inhibitor Cocktail | eBioscience | Cat# 00-4980-03 |
| Percoll | GE Healthcare | Cat# 17089109 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M7522 |
| TCL RNA lysis buffer | Qiagen | Cat# 1031576 |
| TRIzol | Invitrogen | Cat# 15596026 |
| Critical commercial assays | ||
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| Click-iT™ EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 488 dye | Thermo Fisher Scientific | Cat# C10337 |
| Zombie UV™ Fixable Viability Kit | Biolegend | Cat# 423108 |
| RNeasy Lipid Tissue Mini Kit | Qiagen | Cat# 74804 |
| RNAClean XP beads | Beckman Coulter | Cat# A63987 |
| Nextera DNA Sample Prep Kit | Illumina | Cat# FC-121-1030 |
| Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 | 10x | Cat# PN-1000092 |
| Chromium Single Cell B Chip Kit | 10x | Cat# PN-1000074 |
| Chromium i7 Multiplex Kit | 10x | Cat# PN-120262 |
| Chromium Next GEM Single Cell 5’ Library & Gel Bead Kit v1.1 | 10x | Cat# PN-1000167 |
| Chromium Single Cell 5’ Library Construction Kit | 10x | Cat# PN-1000020 |
| Chromium Single Cell 5’ Feature Barcode Library Kit | 10x | Cat# PN-1000080 |
| Chromium Single Cell V(D)J Enrichment Kit, Mouse T Cell | 10x | Cat# PN-1000071 |
| Chromium Next GEM Chip G Single Cell Kit | 10x | Cat# PN-1000127 |
| Single Index Kit T Set A | 10x | Cat# PN-1000213 |
| Single Index Kit N Set A | 10x | Cat# PN-1000212 |
| 123count eBeads | Invitrogen | Cat# 01-1234-42 |
| Deposited data | ||
| scRNA-seq/scTcr-seq of muscle and colon Treg cells | This paper | GSE196337 |
| Bulk RNA-seq | This paper | GSE195966 |
| scRNA-seq of colon CD4+ T cells | Kiner et al., 202188 | GSE160053 |
| scRNA-seq of muscle Treg cells at day 4 after injury | DiSpirito et al., 201828 | GSE109742 |
| scRNA-seq of whole muscle | Oprescu et al., 202050 | GSE138826 |
| scRNA-seq of injured heart CD4+ T cells | Xia et al., 202057 | GSE155619 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | 000664 |
| Mouse: B6.CD45.1 | Jackson Laboratory | 002014 |
| Mouse: B6.129P2-Tcrbtm1Mom/J | Jackson Laboratory | 002118 |
| Mouse: B6.129P2(Cg)-Rorctm2Litt/J | Jackson Laboratory | 007572 |
| Mouse: B6.Foxp3GFP | Bettelli et al., 200679 | N/A |
| Mouse: B6.Foxp3Thy1.1 | Liston et al., 200881 | N/A |
| Mouse: B6.Foxp3DTR | Kim, et al., 200782 | N/A |
| Mouse: B6.Kaede | Tomura et al., 200830 | N/A |
| Mouse: B6.Foxp3Cre | Rubtsov et al., 200883 | N/A |
| Mouse: B6.Maffl/fl | Wende et al, 201284 | N/A |
| Germ Free C57BL/6J mice | GF C57BL/6 colony housed in the animal facility at Harvard Medical School. | N/A |
| Software and algorithms | ||
| Cell Ranger (v2.1.0, v4.0.0 and v6.0.0) | 10X Genomics | |
| CITE-seq-count v1.4.3 | doi:10.5281/zenodo.2590196 | https://hoohm.github.io/CITE-seq-Count/ |
| R v4.1.1 | The R Foundation | https://www.r-project.org |
| Seurat v4.0.3 | Hao et al., 202197 | https://satijalab.org/seurat/ |
| Harmony v0.1.0 | Korsunsky et al., 201992 | |
| Nebulosa v1.3.0 | Alquicira-Hernandez et al., 2021 91 | https://www.bioconductor.org/packages/release/bioc/html/Nebulosa.html |
| scRepertoire v1.4.0 | Borcherding et al., 2020 32 | https://github.com/ncborcherding/scRepertoire |
| Cuffquant version 2.2.1 | Trapnell et al., 201298 | http://cole-trapnell-lab.github.io/cufflinks/install/ |
| DESeq2 v1.28.1 | Love et al, 201499 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Limma | Ritchie et al, 2015 100 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| GenePattern software package | Broad Institute | http://software.broadinstitute.org/cancer/software/genepattern/ |
| maSigPro | Nueda et al., 201493 | https://www.bioconductor.org/packages/release/bioc/html/maSigPro.html |
| Scanpy | Wolf et al., 2018101 | https://github.com/scverse/scanpy |
| Metascape | Zhou et al., 2019 102 | https://metascape.org/ |
| Enrichr | Kuleshov et al., 2016103 | https://maayanlab.cloud/Enrichr/ |
| PRISM v9.2.0 | GraphPad | https://www.graphpad.com |
| FlowJo 10.6.2 | FlowJo, LLC | https://www.flowjo.com |
| ImageJ v2.1.0/1.53c | Schindelin et al., 201295 | https://imagej.net/software/fiji/ |
| Other | ||
| Control diet: L-Amino Acid Rodent Diet With 10 kcal% Fat (Amino Acid Complete) | Research Diets | Cat# A08051501i |
| CDAHFD: L-Amino Acid Diet With 45 kcal% Fat With 0.1% Methionine and No Added Choline | Research Diets | Cat#: A06071309i |
HIGHLIGHTS.
Muscle injury induces local accumulation of RORγ+ Treg cells emanating from the gut.
The microbiota regulates muscle repair via RORγ+ Treg cells.
Muscle RORγ+ Treg cells shield differentiating muscle stem cells from IL-17.
RORγ+ Treg cell emissaries play a general role in the homeostasis of extra-gut tissues.
ACKNOWLEDGEMENTS:
We thank K. Hattori, A. Ortiz-Lopez, L. Yang, O. Yaghi, P.K. Langston, D. Ramanan, T. Jayewickreme, Y. Zhu and X. Chi for experimental assistance; B. Vijaykumar for bioinformatics; C. Laplace for graphics; the Harvard Medical School (HMS) Rodent Histopathology Core; and the HMS Immunology Department Flow-Cytometry Core. This work was funded by grants from the NIH (R01 AR070334), the JPB Foundation, and Pfizer, Inc. to D.M. B.S.H. was partially supported by a Deutsche Forschungsgemeinschaft fellowship (HA 8510/1) and S.G.P. by an EMBO Long-term Fellowship (ACTF 547–2019).
Footnotes
COMPETING INTERESTS
D.M. is a co-founder and member of the Scientific Advisory Board of Abata Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Muñoz-Rojas AR and Mathis D (2021). Tissue regulatory T cells: regulatory chameleons. Nat Rev Immunol 21, 597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S et al. (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15, 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J et al. (2015). Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349, 993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S, Fedoseeva M et al. (2015). The microbiota regulates type 2 immunity through RORγ+ T cells. Science 349, 989–993. [DOI] [PubMed] [Google Scholar]
- 5.Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, Fohse L, Prinz I, Pezoldt J, Suerbaum S et al. (2016). Foxp3+ T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal. Immunol 9, 444–457. [DOI] [PubMed] [Google Scholar]
- 6.Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, and Rudensky AY (2015). A distinct function of regulatory T cells in tissue protection. Cell 162, 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C et al. (2013). A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, Gratz IK, Otto M, Moon JJ, Liese J et al. (2015). A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity 43, 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K, Ahn R, Corbin K, Lowe MM, Scharschmidt TC et al. (2017). Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell 169, 1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathur AN, Zirak B, Boothby IC, Tan M, Cohen JN, Mauro TM, Mehta P, Lowe MM, Abbas AK, Ali N et al. (2019). Treg-cell control of a CXCL5-IL-17 inflammatory axis promotes hair-follicle-stem-cell differentiation during skin-barrier repair. Immunity. 50, 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panduro M, Benoist C, and Mathis D (2018). Treg cells limit IFN-γ production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc Natl Acad Sci U S A 115, E2585–E2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdel-Gadir A, Stephen-Victor E, Gerber GK, Noval RM, Wang S, Harb H, Wang L, Li N, Crestani E, Spielman S et al. (2019). Microbiota therapy acts via a regulatory T cell MyD88/RORγt pathway to suppress food allergy. Nat Med 25, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim KS, Hong SW, Han D, Yi J, Jung J, Yang BG, Lee JY, Lee M, and Surh CD (2016). Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351, 858–863. [DOI] [PubMed] [Google Scholar]
- 14.Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ, Galan C, Belkaid Y, Bonneau R, and Littman DR (2018). c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumann C, Blume J, Roy U, Teh PP, Vasanthakumar A, Beller A, Liao Y, Heinrich F, Arenzana TL, Hackney JA et al. (2019). c-Maf-dependent Treg cell control of intestinal TH17 cells and IgA establishes host-microbiota homeostasis. Nat Immunol 20, 471–481. [DOI] [PubMed] [Google Scholar]
- 16.Al Nabhani Z, Dulauroy S, Marques R, Cousu C, Al BS, Dejardin F, Sparwasser T, Berard M, Cerf-Bensussan N, and Eberl G (2019). A weaning reaction to microbiota is required for resistance to immunopathologies in the adult. Immunity 50, 1276–1288. [DOI] [PubMed] [Google Scholar]
- 17.Mazmanian SK, Liu CH, Tzianabos AO, and Kasper DL (2005). An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118. [DOI] [PubMed] [Google Scholar]
- 18.Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, and Mathis D (2010). Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 32, 815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, and Iwasaki A (2011). Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A 108, 5354–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, Longman RS, Honda K, Littman DR, Choi GB et al. (2017). Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, Paik S, Stagg J, Groves RA, Gallo M et al. (2020). Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369, 1481–1489. [DOI] [PubMed] [Google Scholar]
- 22.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gauguet S, D’Ortona S, Ahnger-Pier K, Duan B, Surana NK, Lu R, Cywes-Bentley C, Gadjeva M, Shan Q, Priebe GP et al. (2015). Intestinal microbiota of mice influences resistance to staphylococcus aureus pneumonia. Infect. Immun. 83, 4003–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McAleer JP, Nguyen NL, Chen K, Kumar P, Ricks DM, Binnie M, Armentrout RA, Pociask DA, Hein A, Yu A et al. (2016). Pulmonary Th17 antifungal immunity is regulated by the gut microbiome. J Immunol 197, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tidball JG (2017). Regulation of muscle growth and regeneration by the immune system. Nat Rev Immunol 17, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, Benoist C, and Mathis D (2016). Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 44, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang K, Yaghi OK, Spallanzani RG, Chen X, Zemmour D, Lai N, Chiu IM, Benoist C, and Mathis D (2020). Neuronal, stromal, and T-regulatory cell crosstalk in murine skeletal muscle. Proc. Natl. Acad. Sci. U. S. A 117, 5402–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dispirito JR, Zemmour D, Ramanan D, Cho J, Zilionis R, Klein AM, Benoist C, and Mathis D (2018). Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci Immunol 3, eaat5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, and Hori S (2009). Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. U. S. A 106, 1903–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, and Kanagawa O (2008). Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc Natl Acad Sci U S A 105, 10871–10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morton AM, Sefik E, Upadhyay R, Weissleder R, Benoist C, and Mathis D (2014). Endoscopic photoconversion reveals unexpectedly broad leukocyte trafficking to and from the gut. Proc Natl Acad Sci U S A 111, 6696–6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borcherding N, Bormann NL, and Kraus G (2020). scRepertoire: an R-based toolkit for single-cell immune receptor analysis. F1000Res. 9, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fonseca R, Beura LK, Quarnstrom CF, Ghoneim HE, Fan Y, Zebley CC, Scott MC, Fares-Frederickson NJ, Wijeyesinghe S, Thompson EA et al. (2020). Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat Immunol 21, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laidlaw BJ, Gray EE, Zhang Y, Ramirez-Valle F, and Cyster JG (2019). Sphingosine-1-phosphate receptor 2 restrains egress of γδ T cells from the skin. J. Exp. Med 216, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ataide MA, Knopper K, Cruz de CP, Ugur M, Eickhoff S, Zou M, Shaikh H, Trivedi A, Grafen A, Yang T et al. (2022). Lymphatic migration of unconventional T cells promotes site-specific immunity in distinct lymph nodes. Immunity. [DOI] [PubMed] [Google Scholar]
- 36.Kunkel GT, Maceyka M, Milstien S, and Spiegel S (2013). Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 12, 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu H, Huang D, Ransohoff RM, and Zhou L (2011). Acute skeletal muscle injury: CCL2 expression by both monocytes and injured muscle is required for repair. FASEB J 25, 3344–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giannakis N, Sansbury BE, Patsalos A, Hays TT, Riley CO, Han X, Spite M, and Nagy L (2019). Publisher Correction: Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration. Nat Immunol 20, 765–767. [DOI] [PubMed] [Google Scholar]
- 39.Kolodin D, van PN, Li C, Magnuson AM, Cipolletta D, Miller CM, Wagers A, Germain RN, Benoist C, and Mathis D (2015). Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab 21, 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C, Dispirito JR, Zemmour D, Spallanzani RG, Kuswanto W, Benoist C, and Mathis D (2018). TCR transgenic mice reveal stepwise, multi-site acquisition of the distinctive fat-Treg phenotype. Cell 174, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho J, Kuswanto W, Benoist C, and Mathis D (2019). T cell receptor specificity drives accumulation of a reparative population of regulatory T cells within acutely injured skeletal muscle. Proc Natl Acad Sci U S A 116, 26727–26733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akagbosu B, Tayyebi Z, Shibu G, Paucar Iza YA, Deep D, Parisotto YF, Fisher L, Pasolli HA, Thevin V, Elmentaite R et al. (2022). Novel antigen presenting cell imparts Treg-dependent tolerance to gut microbiota. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mann AO, Hanna BS, Munoz-Rojas AR, Sandrock I, Prinz I, Benoist C, and Mathis D (2022). IL-17A-producing γδT cells promote muscle regeneration in a microbiota-dependent manner. J Exp Med 219, e20211504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aguilar CA, Shcherbina A, Ricke DO, Pop R, Carrigan CT, Gifford CA, Urso ML, Kottke MA, and Meissner A (2015). In vivo monitoring of transcriptional dynamics after lower-limb muscle injury enables quantitative classification of healing. Sci Rep 5, 13885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnold L, Henry A, Poron F, Baba-Amer Y, van RN, Plonquet A, Gherardi RK, and Chazaud B (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp. Med 204, 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng M, Nguyen MH, Fantuzzi G, and Koh TJ (2008). Endogenous interferon-gamma is required for efficient skeletal muscle regeneration. Am. J Physiol Cell Physiol 294, C1183–C1191. [DOI] [PubMed] [Google Scholar]
- 47.Chen SE, Jin B, and Li YP (2007). TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am. J Physiol Cell Physiol 292, C1660–C1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Londhe P and Davie JK (2011). Gamma interferon modulates myogenesis through the major histocompatibility complex class II transactivator, CIITA. Mol Cell Biol. 31, 2854–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Londhe P and Davie JK (2013). Interferon-gamma resets muscle cell fate by stimulating the sequential recruitment of JARID2 and PRC2 to promoters to repress myogenesis. Sci Signal. 6, ra107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oprescu SN, Yue F, Qiu J, Brito LF, and Kuang S (2020). Temporal dynamics and heterogeneity of cell populations during skeletal muscle regeneration. iScience. 23, 100993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sambasivan R, Yao R, Kissenpfennig A, Van WL, Paldi A, Gayraud-Morel B, Guenou H, Malissen B, Tajbakhsh S, and Galy A (2011). Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138, 3647–3656. [DOI] [PubMed] [Google Scholar]
- 52.Lepper C, Partridge TA, and Fan CM (2011). An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 138, 3639–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Girgenrath M, Weng S, Kostek CA, Browning B, Wang M, Brown SA, Winkles JA, Michaelson JS, Allaire N, Schneider P et al. (2006). TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J 25, 5826–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yartseva V, Goldstein LD, Rodman J, Kates L, Chen MZ, Chen YJ, Foreman O, Siebel CW, Modrusan Z, Peterson AS et al. (2020). Heterogeneity of satellite cells implicates DELTA1/NOTCH2 signaling in self-renewal. Cell Rep 30, 1491–1503. [DOI] [PubMed] [Google Scholar]
- 55.Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, Brunson C, Mastey N, Liu L, Tsai CR et al. (2014). mTORC1 controls the adaptive transition of quiescent stem cells from G0 to GAlert. Nature 510, 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kocic J, Santibanez JF, Krstic A, Mojsilovic S, Dordevic IO, Trivanovic D, Ilic V, and Bugarski D (2012). Interleukin 17 inhibits myogenic and promotes osteogenic differentiation of C2C12 myoblasts by activating ERK1,2. Biochim. Biophys. Acta 1823, 838–849. [DOI] [PubMed] [Google Scholar]
- 57.Xia N, Lu Y, Gu M, Li N, Liu M, Jiao J, Zhu Z, Li J, Li D, Tang T et al. (2020). A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation 142, 1956–1973. [DOI] [PubMed] [Google Scholar]
- 58.Wang R, Tang R, Li B, Ma X, Schnabl B, and Tilg H (2021). Gut microbiome, liver immunology, and liver diseases. Cell Mol. Immunol 18, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li F, Hao X, Chen Y, Bai L, Gao X, Lian Z, Wei H, Sun R, and Tian Z (2017). The microbiota maintain homeostasis of liver-resident γδT-17 cells in a lipid antigen/CD1d-dependent manner. Nat. Commun. 7, 13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gomes AL, Teijeiro A, Buren S, Tummala KS, Yilmaz M, Waisman A, Theurillat JP, Perna C, and Djouder N (2016). Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 30, 161–175. [DOI] [PubMed] [Google Scholar]
- 61.Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C, Ito T, Katsume A, and Sudoh M (2013). An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J Exp Pathol. 94, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M et al. (2012). Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 143, 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wucherpfennig KW and Strominger JL (1995). Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80, 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harkiolaki M, Holmes SL, Svendsen P, Gregersen JW, Jensen LT, McMahon R, Friese MA, van BG, Etzensperger R, Tzartos JS et al. (2009). T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity. 30, 348–357. [DOI] [PubMed] [Google Scholar]
- 65.Szymula A, Rosenthal J, Szczerba BM, Bagavant H, Fu SM, and Deshmukh US (2014). T cell epitope mimicry between Sjögren’s syndrome Antigen A (SSA)/Ro60 and oral, gut, skin and vaginal bacteria. Clin Immunol 152, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pianta A, Arvikar SL, Strle K, Drouin EE, Wang Q, Costello CE, and Steere AC (2017). Two rheumatoid arthritis-specific autoantigens correlate microbial immunity with autoimmune responses in joints. J Clin Invest 127, 2946–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hebbandi Nanjundappa R, Ronchi F, Wang J, Clemente-Casares X, Yamanouchi J, Sokke UC, Yang Y, Blanco J, Bassolas-Molina H, Salas A et al. (2017). A gut microbial mimic that hijacks diabetogenic autoreactivity to suppress colitis. Cell 171, 655–667. [DOI] [PubMed] [Google Scholar]
- 68.Carrasco Pro S, Lindestam Arlehamn CS, Dhanda SK, Carpenter C, Lindvall M, Faruqi AA, Santee CA, Renz H, Sidney J, Peters B et al. (2018). Microbiota epitope similarity either dampens or enhances the immunogenicity of disease-associated antigenic epitopes. PLoS One 13, e0196551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garabatos N and Santamaria P (2022). Gut microbial antigenic mimicry in autoimmunity. Front Immunol 13, 873607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seo BR, Payne CJ, McNamara SL, Freedman BR, Kwee BJ, Nam S, de L,I, Darnell M, Alvarez JT, Dellacherie MO et al. (2021). Skeletal muscle regeneration with robotic actuation-mediated clearance of neutrophils. Sci Transl. Med 13, eabe8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao J, Chen X, Herjan T, and Li X (2020). The role of interleukin-17 in tumor development and progression. J. Exp. Med 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen X, Cai G, Liu C, Zhao J, Gu C, Wu L, Hamilton TA, Zhang CJ, Ko J, Zhu L et al. (2019). IL-17R-EGFR axis links wound healing to tumorigenesis in Lrig1+ stem cells. J. Exp. Med 216, 195–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu L, Chen X, Zhao J, Martin B, Zepp JA, Ko JS, Gu C, Cai G, Ouyang W, Sen G et al. (2015). A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J. Exp. Med 212, 1571–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zepp JA, Zhao J, Liu C, Bulek K, Wu L, Chen X, Hao Y, Wang Z, Wang X, Ouyang W et al. (2017). IL-17A-induced PLET1 expression contributes to tissue repair and colon tumorigenesis. J. Immunol. 199, 3849–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang H, Kim HJ, Chang EJ, Lee ZH, Hwang SJ, Kim HM, Lee Y, and Kim HH (2009). IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death. Differ. 16, 1332–1343. [DOI] [PubMed] [Google Scholar]
- 76.Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, Chandrasekharan U, DiCorleto PE, Trapp BD, Ransohoff RM et al. (2013). Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat Neurosci. 16, 1401–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kolodziejczyk AA, Zheng D, and Elinav E (2019). Diet-microbiota interactions and personalized nutrition. Nat Rev Microbiol. 17, 742–753. [DOI] [PubMed] [Google Scholar]
- 78.Liwinski T and Elinav E (2020). Harnessing the microbiota for therapeutic purposes. Am. J Transplant. 20, 1482–1488. [DOI] [PubMed] [Google Scholar]
- 79.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, and Kuchroo VK (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238. [DOI] [PubMed] [Google Scholar]
- 80.Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, and Littman DR (2004). An essential function for the nuclear receptor RORγt in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 5, 64–73. [DOI] [PubMed] [Google Scholar]
- 81.Liston A, Nutsch KM, Farr AG, Lund JM, Rasmussen JP, Koni PA, and Rudensky AY (2008). Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc Natl. Acad Sci U S. A. 105, 11903–11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim JM, Rasmussen JP, and Rudensky AY (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol 8, 191–197. [DOI] [PubMed] [Google Scholar]
- 83.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr. et al. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558. [DOI] [PubMed] [Google Scholar]
- 84.Wende H, Lechner SG, Cheret C, Bourane S, Kolanczyk ME, Pattyn A, Reuter K, Munier FL, Carroll P, Lewin GR et al. (2012). The transcription factor c-Maf controls touch receptor development and function. Science 335, 1373–1376. [DOI] [PubMed] [Google Scholar]
- 85.Hardy CL, Bhathal PS, Snibson KJ, and Adams TE (1997). Comparison of intrahepatic lymphocytes from normal and growth hormone transgenic mice with chronic hepatitis and liver cancer. Immunology 90, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu L, Cheung TH, Charville GW, and Rando TA (2015). Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat. Protoc. 10, 1612–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pratama A, Schnell A, Mathis D, and Benoist C (2020). Developmental and cellular age direct conversion of CD4+ T cells into RORγ+ or Helios+ colon Treg cells. J Exp. Med 217, e20190428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kiner E, Willie E, Vijaykumar B, Chowdhary K, Schmutz H, Chandler J, Schnell A, Thakore PI, LeGros G, Mostafavi S et al. (2021). Gut CD4+ T cell phenotypes are a continuum molded by microbes, not by TH archetypes. Nat. Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li C, Muñoz-Rojas AR, Wang G, Mann AO, Benoist C, and Mathis D (2021). PPARγ marks splenic precursors of multiple nonlymphoid-tissue Treg compartments. Proc Natl Acad Sci U S A 118, e2025197118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alquicira-Hernandez J and Powell JE (2021). Nebulosa recovers single cell gene expression signals by kernel density estimation. Bioinformatics. [DOI] [PubMed] [Google Scholar]
- 92.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nueda MJ, Tarazona S, and Conesa A (2014). Next maSigPro: updating maSigPro bioconductor package for RNA-seq time series. Bioinformatics. 30, 2598–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Almada AE, Horwitz N, Price FD, Gonzalez AE, Ko M, Bolukbasi OV, Messemer KA, Chen S, Sinha M, Rubin LL et al. (2021). FOS licenses early events in stem cell activation driving skeletal muscle regeneration. Cell Rep 34, 108656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, Yanortsang TB, Yang L, Jupp R, Mathis D et al. (2017). Mining the human gut microbiota for immunomodulatory organisms. Cell 168, 928–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hao Y, Hao S, Andersen-Nissen E, Mauck WM III, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data newly reported in this paper were deposited in the Gene Expression Omnibus (GEO) database under accession no. GSE196338.