

Summary
Advances in the understanding of the tumor microenvironment have led to development of immunotherapeutic strategies, such as chimeric antigen receptor T cells (CAR-Ts). However, despite success in blood malignancies, CAR-T therapies in solid tumors have been hampered by their restricted infiltration. Here, we used our understanding of early cytotoxic lymphocyte infiltration of human lymphocytes in solid tumors in vivo to investigate the receptors in normal, adjacent, and tumor tissues of primary non-small-cell lung cancer specimens. We found that CX3CL1-CX3CR1 reduction restricts cytotoxic cells from the solid-tumor bed, contributing to tumor escape. Based on this, we designed a CAR-T construct using the well-established natural killer group 2, member D (NKG2D) CAR-T expression together with overexpression of CX3CR1 to promote their infiltration. These CAR-Ts infiltrate tumors at higher rates than control-activated T cells or IL-15-overexpressing NKG2D CAR-Ts. This construct also had similar functionality in a liver-cancer model, demonstrating potential efficacy in other solid malignancies.
Subject areas: Oncology, Immunology, Cancer
Graphical abstract
Highlights
-
•
Tumor establishment prevents infiltration of TILs, regardless of their survival
-
•
CX3CR1/CX3CL1 axis plays a major role in TIL infiltration and activity in solid tumors
-
•
Addition of CX3CR1 to NKG2D CAR-T enhanced infiltration and killing efficiency in vivo
Oncology; Immunology; Cancer
Introduction
Over the past decade, significant advances have been made in the immunotherapy of hematologic malignancies, but the efficacy of immunotherapy in solid tumors remains limited.1 Through the advent of novel immunotherapies, it is well established that deficiency of tumor-infiltrating lymphocytes (TILs), including cytotoxic T cells and natural killer (NK) cells in solid tumors, is a factor in tumor progression and survival as well as an obstacle to overcome for reducing tumor burden.1,2,3 In particular, accumulation of lymphocytes near or into the tumor bed has been strongly correlated with a favorable prognosis in several solid malignancies.4 This suggests that circulation and infiltration of lymphocytes into the tumor are critical for antitumor immunity and perhaps for developing immunotherapeutic strategies, although the exact mechanisms involved remain to be elucidated. However, despite this, therapeutic targeting of solid malignancies remains a key issue for the effectiveness of most immunotherapies because of decreased lymphocyte infiltration into the tumor microenvironment (TME).5 Therefore, a better understanding of TIL-restriction mechanisms in the solid TME could play a key role in developing novel immunotherapies more efficient at overcoming this restriction.
Among the many chemokines relevant in tumor recruitment and infiltration is CX3CL1, whose receptor CX3CR1 is expressed in many immune cells, including lymphocytes.6,7,8,9,10 In addition, CX3CL1 has been previously shown to induce homing toward xenograft tumors when exogenously expressed in T cells,11 although its expression inside the tumor bed remains unknown or its relevance in lymphocyte restriction remains unknown. CX3CL1, or fractalkine, is a chemokine expressed in many tissues in response to inflammation; fractalkine has both a membrane-bound and a released form.12 The released form acts as a recruiting chemokine, forming a gradient between the releasing tissues and the periphery, whereas the membrane-bound form mediates adhesion and retention of CX3CR1+ cells in the target tissues.13,14,15 Importantly, increased levels of CX3CL1, which captures CX3CR1+ cytotoxic T cells, NK cells, and CD14+ monocytes in the tumor-bed cancers, such as colorectal, breast, gastric adenocarcinoma, and the majority of lung cancer cases, are a favorable prognostic marker in cancer through enhancing antitumor immunity.7,13,16,17 Importantly, other than for developing antibodies against this receptor-ligand pair for autoimmune disorders, such as rheumatoid arthritis, its use in developing immunotherapeutic strategies for cancer is rare. However, chimeric antigen receptor T (CAR-T) cells allow for retargeting T cells toward the tumor on a major histocompatibility complex––independent activation of recognition18,19,20; this retargeting is an excellent way to test the role of CX3CR1-CX3CL1 in inducing infiltration in solid malignancies. While CAR-Ts’ biggest success has been treating CD19+ B-cell lymphomas, the first clinical trials for this type of immunotherapy were conducted on solid-tumor malignancies.21 Because identifying targets for solid malignancies is rare, CAR-Ts have been developed to take advantage of the overexpression of NK group 2, member D (NKG2D), shown as effective in many preclinical and clinical studies.3,22,23,24 The rationale for using CAR-Ts is that engaging this receptor with upregulated NKG2D ligands in tumors triggers effector functions in cytotoxic cells, such as cellular proliferation, proinflammatory cytokine production, and granzyme-mediated cytolysis of target cells.25,26,27,28,29,30 However, clinically, NKG2D CAR-T alone has not been successful in solid tumors because of the lack of infiltration in the tumor.31
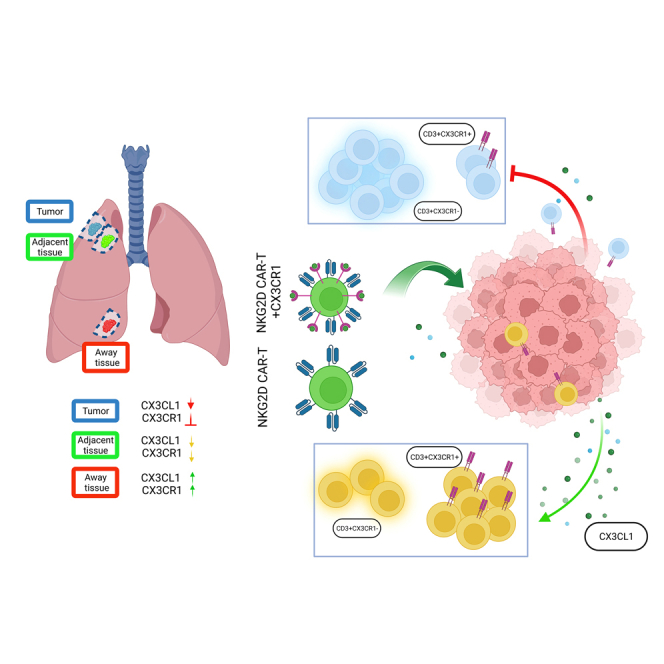
In the current study, we analyzed infiltrating lymphocytes from lung cancer primary tissues isolated from 3 separate areas: normal (N), adjacent (A), and tumor (T) tissues. We discovered a significant deficiency of CX3CR1+ TILs in the tumor bed and a reduced signal of lymphocytes from A compared to N tissues. A concomitant reduction of CX3CL1 in primary tumor tissues suggested that this receptor may recruit effector lymphocytes into the solid tumor. We took advantage of this discovery to include CX3CR1 receptor expression into the well-known NKG2D–CAR-T construct and tested it in a lung in vivo model. This combination reduced tumor burden in solid tumor-bearing mice and provides a unique alternative to overcome the reduced infiltration of CAR-T into the tumor.
Results
Early intratumoral restriction of cytotoxic cells in a lung-xenograft immunotherapy model
Our previous work focused on understanding the role of the TME in cytotoxic-cell suppression in lung cancer.32,33 Therefore, to understand how intratumoral lymphocyte restriction works, we tested whether engraftment of A549 tumors, a human non-small-cell lung cancer (NSCLC), into NOD (non-obese diabetic)-scid IL2Rγnull (NSG) mice34 can support immunotherapy of human lung cancer after establishment of the initial solid mass. Because NK cells are one of the early players for tumor immune surveillance and naturally express NKG2D, we created lung-tumor models surveyed by primary NK cells at different time points. As shown in Figure 1A, 3 × 105 luciferase-expressing human A549 cells could grow subcutaneously as a small, localized tumor in NSG mice that received a delayed administration (day 7) of 3 × 106 interleukin-2 (IL-2)-activated human NK cells while parallel administration at day 0 reduced or even prevented tumor growth (Figures 1A, 1B, and S1A), indicating that early activity of NK cells prevents the growth and establishment of solid tumors.
Figure 1.

Infiltration of NK cells is restricted by the growing tumor
(A) Representative images of early tumor-development model: NSG mice were inoculated s.c. with luciferase-expressing A549 tumor cells and administered NK cells at day 0 or day 7 after tumor establishment. Tumors were detected by luminescence.
(B) Quantification of luminescence from the experiment shown in A (n = 10 mice per group, and NK cells from 5 human donors). Mice with no luciferase tumors were measured to assess background. Groups were not normally distributed and analyzed with the Friedman test (p values shown in figure). Error bars represent the standard error of the mean of n.
(C) Established tumor-model schematics: luciferase-expressing A549 cells were inoculated into mice, and 2 weeks after (when tumor was palpable), isolated primary human NK cells were injected. Tumors were collected 5 days after injection, and mononuclear cells were analyzed by flow cytometry for the presence of human Cluster of Differentiation (hCD)45+ cells.
(D) Flow cytometric quantification of the percentage of hCD45+ NK-cell infiltration in tissues of A549 tumor-bearing mice treated as described in C (representative figure of n = 3). Inset shows the percentage of human CD45+ cells inside the gate shown.
(E) Flow cytometric quantification as in D of hCD45+ NK cells precultured in the absence (upper panel) or presence (lower panel) of 10 ng/mL IL-15 for 3 days before injection and injected in 10 μg/1 × 106 cells IL-15-supplemented vehicle (representative figure of n = 3). Inset shows the percentage of human NK cells in each tissue. Abbreviations: NK, Natural killer; NSG, NOD-scid gamma.
To understand if the effect on the TME is restricted to the tumor or caused by a general reduction of cell survival, we performed a delayed NK injection (2 weeks after tumor) to assess the infiltration of human (h)CD45+ NK cells in blood, liver, spleen, or tumor after injection in mice, as schematized in Figure 1C. As expected, we found no infiltration of primary activated NK cells into the tumor (Figure 1D) compared to other tissues. This was not cell line-specific because similar results were obtained with a different NSCLC model (Figure S1B). This suggests that the restriction of NK cells from the tumor is linked to survival and migration of NK cells. IL-15, a survival cytokine critical for cytotoxic-cell development, function, and proliferation,35 works like IL-2, albeit more effectively because its higher receptor affinity leads to cytotoxic-cell activation at lower doses.36 Because of its essential role in the survival and activation of cytotoxic cells,37,38,39,40 we tested whether increased NK-cell survival and activation with IL-15 could increase tumor infiltration. We tested the infiltration of primary NK cells cultured with the cytotoxic lymphocyte-activating cytokine IL-15 and supplemented with IL-15 during the intravenous administration in the same tumor model. Stimulating with IL-15, which, as expected, increased CD56bright NK population upon activation41 and improved survival of NK cells but not the amount of tumor infiltration in vivo, suggesting that mechanisms other than lymphocyte activation are relevant to infiltration (Figure 1E).
Restricted infiltration of functional cytotoxic cells in lung cancer
Based on the early xenograft model, as well as reports that lymphocytes are restricted from tumor tissues in many malignancies,42,43,44,45 we analyzed 16 fresh primary NSCLC specimens through phenotypic mapping of lymphocyte infiltration. This allowed systematic characterization of the lymphocytic presence and distribution in the solid mass and its surrounding tissues. Lymphocytes from primary lung-tissue specimens were isolated from N, A, or T tissues (Figures S2A and S2B) similar to a previous description46 and analyzed individually or by T-distributed stochastic neighbor embedding (tSNE) modeling for visualization of multidimensional data47 (Figure S2C). A gradient-like decrease was observed in the percentage of NK cells but with an increase of suppressive regulatory T cells (T regs), while the overall percentage of T cells (CD4 and CD8) was not significantly changed (Figures 2A and S3A). To determine if there were changes in subpopulations of T cells, we analyzed the isolated cells for changes in different functional markers. We detected that NK cells expressing the activation markers CD16, NKG2D, DNAM1, and Tbet were significantly downregulated in both the A and T tissues (Figure S3B). However, markers that represent an increase of exhaustion and immune suppression, NKp44, NKG2A, and Tim3,48 were increased. Similarly, CD8+ T cells had a decrease in DNAM1 and Tbet expression but not NKG2D, while only PD1 (but not Tim3, TIGIT, or CTLA4) was significantly upregulated in the cytotoxic T cells infiltrating the tumor (Figure S3C). Interestingly, we observed a decreased expression in the mobilization-linked cytokine receptor CX3CR1 in cytotoxic cells NK, NKT, and CD8+ (Figure 2B), which may help explain our earlier observations of TIL restriction in the TME. CX3CR1 is established—along with its ligand CX3CL1 (fractalkine)—as driving migration, increasing responsiveness to chemo-attractants, promoting interferon-gamma (IFNγ) expression, and activating cytotoxic T cells.49,50,51 Moreover, while CX3CR1+CD4+ cells were also reduced in the tumor, this reduction was not as striking as CX3CR1-expressing CD8+ T cells or NK cells and likely explained by the increased level of T regs present (Figures 2A and S3A). CX3CR1 levels also affect migration of myeloid-derived suppressor cells (MDSCs), and a reduction of this receptor in TILs enhances the phagocytic ability of MDSCs.52,53 MDSCs are well established as a main immunosuppressive cell type, playing a critical role in tumor immune escape by secreting suppressive cytokines, reducing antitumor adaptive responses, and increasing suppressive cells, including T regs.54,55 For that reason, we investigated not only the overall phenotypic MDSC levels but also the levels of the MDSC subsets: early stage (e)MDSC, defined as Lin−HLA-DRlo/−CD11b+CD33+; monocytic MDSC (M-MDSC), defined as CD11b+CD14+HLA-DRlo/−CD15−; and granulocytic MDSC (polymorphonuclear PMN-MDSC), defined as CD14−CD11b+CD15+.56 To confirm these are MDSCs, we performed a functional suppression assay with enriched CD11b+ cells, isolated from either N, A, or T tissues, with either autologous or allogeneic T cells (Figure S3D) confirming the suppressive nature of these cells. The total population of MDSCs was significantly upregulated in the tumor compared to either N or A tissues (Figure 2C). The most significant was the percentage of early immature eMDSC in the tumor, compared to either N or A tissues, whereas M-MDSCs were only significant compared to A tissues, and PMN-MDSCs were not significant, albeit trending toward increases in the tumor. This suggests that the increase in suppression in the TME contributes to the reduced immune infiltration and is associated with decreases of CX3CR1 in the tumor. To corroborate the architecture of CX3CR1 lymphocytic restriction, we used the immunohistochemistry of the same locations on the same primary lung cancer specimens. This confirmed the CX3CR1-gradient reduction of cytotoxic lymphocyte infiltration into the tumor (Figure 2D and lower magnification of the same images in Figure S4), which was paired with a similar gradient reduction of its ligand, CX3CL1 (Figure 2E and lower magnification of the same images in Figure S5). Importantly, CX3CR1-CX3CL1 was significantly reduced yet not abrogated because some expression in the tumor tissues remained.
Figure 2.

Primary non-small-cell lung cancer reduces cytotoxic cells and CX3CR1 levels in the tumor microenvironment
(A) Percentage of total live CD45+ lymphocytes isolated from primary lung tissues (NK cells, Lin−CD56+CD16+; T cell subsets CD3+CD4+ T helper cells, CD3+CD8+ cytotoxic cells, and CD3+CD25+Foxp3+ T regs). NK cells were not normally distributed and analyzed with the Friedman test, while the other groups were normally distributed and analyzed by one-way ANOVA.
(B) Percentage of CX3CR1+ NK cells, T helper cells, and NKT cells (defined as in A). CX3CR1+NK cells were normally distributed and analyzed by one-way ANOVA, while the other CX3CR1+ groups were not normally distributed and analyzed with the Friedman test.
(C) The percentage of total MDSC and MDSC subsets: eMDSC (Lin−HLA-DRlo/−CD11b+CD33+), M-MDSC (CD11b+CD14+HLA-DRlo/−CD15−), and PMN-MDSC (CD14−CD11b+CD15+). Except for PMN-MDSC, all data in C were normally distributed and analyzed by one-way ANOVA.
(D) Immunohistochemistry staining of primary lung-tissue specimens’ normal, adjacent, and tumor sections (2 separate specimens shown) showing the expression of CX3CR1 (magenta) and cytotoxic cells (CD8 T cells in green, CD56 NK in yellow). Zoomed-out high-resolution images of the same specimens are shown in Figure S4.
(E) Immunohistochemical staining of primary lung-tissue specimens’ normal, adjacent, and tumor sections (2 separate specimens shown) showing the expression of CX3CR1 (magenta) and CX3CL1 (yellow). Zoomed-out high-resolution images of the same specimens are shown in Figure S5. All these data come from up to a total of n = 16 specimens (depending on availability for each study). Abbreviation: MDSC, myeloid-derived suppressor cells; NK, Natural killer.
In all graphs error bars represent the standard error of the mean of n.
High CX3CR1 expression is linked to higher survival in lung cancer and other solid malignancies
Considering the data on CX3CR1-mediated restriction of cytotoxic lymphocytes in NSCLC tumors, the main question remains, what is the clinical impact of CX3CR1+ cells in the outcomes of patients with solid malignancies? Using gene-expression analysis from The Cancer Genome Atlas (TCGA) database, we confirmed reduced CX3CR1 and CX3CL1 gene expression in different lung cancers (Figures 3A and 3B, respectively), as well as in different lung cancer cell lines by flow cytometric analysis (Figure S6A). Using OncoLnc (oncolnc.org), which extracts gene-expression and survival data from TCGA, we performed a survival analysis using killer-cell lectin receptor subfamily K member 1 (KLRK1, the gene encoding NKG2D)57,58 and CX3CR1 expression in lung adenocarcinoma and liver hepatocellular carcinoma, as well as 2 other solid malignancies: bladder urothelial carcinoma and head-and-neck squamous-cell carcinoma. Because NKG2D is expressed in cytotoxic lymphocytes,59 this search checks the survival of patients based on CX3CR1 expression in TILs. Samples were categorized as high expressers if they had above-median expression for both genes and low expressers if they had below-median expression for both genes (Figure 3C). In these solid malignancies, the higher expression of KLRK1-CX3CR1 was linked with better survival, supporting the hypothesis that higher CX3CR1-expressing cytotoxic lymphocytes reduce tumors in solid malignancies and are associated with patient prognosis. Moreover, this suggests that reducing both CX3CR1 and CX3CL1 in the TME also reduces the infiltration of antitumor cytotoxic cells.
Figure 3.

CX3CR1 and CX3CL1 correlate with improved survival in solid malignancies
Gene expression of A) CX3CR1 and B) CX3CL1 in lung cancer (lung squamous-cell carcinoma and adenocarcinoma) and normal tissues denoted as FPKM.
(C) Survival analysis of KLRK1 and CX3CR1 expression in lung adenocarcinoma, liver hepatocellular carcinoma, bladder urothelial carcinoma, and head-and-neck squamous-cell carcinoma from The Cancer Genome Atlas database.
(D) Infiltration of primary human CD45+ NK cells into A549 tumors stably overexpressing either CX3CL1 or a scrambled control grafted onto NSG mice. The image represents paraffin-tissue slides immune stained with human CD45 (NK cells, blue cells) and counterstained pink. Spleens serve as positive infiltration control (representative figure).
(E) Quantification of hCD45+ NK cells of experiment as in C. Significance was estimated by the Wilcoxon rank-sum test. Error bar represent the standrd error of the mean of n.
(F) Flow cytometric analysis of circulating human NK cells in the peripheral blood of tumor-bearing mice as in C (representative figure). Isolated NK cells serve as positive control.
(G) Cytotoxicity assay of primary human NK cells against A549 cells with or without CX3CL1 overexpression. Unpaired t-test was used to compare the area under the curve of each group. Abbreviations: FPKM, fragments per kilobase million, NK, Natural killer.
To further establish the role of the CX3CR1-CX3CL1 axis in TIL restriction in solid malignancies, we repeated the A549 xenograft experiment as in Figure 1C with the addition of overexpressing CX3CL1 in the tumor cells and measured infiltration through quantification of human CD45 cells in the tumor bed. While A549, as well as 2 other lung cell lines (H1299 and H1355), had low endogenous CX3CL1 expression measured by flow cytometry (Figure S6A), overexpression of CX3CL1 in A549 cells increased cellular expression and secretion of CX3CL1 (Figures S6B and S6C). Compared to control-transfected A549, CX3CL1-overexpressing A549 cells, xenografted onto NSG mice, had an increased infiltration of hCD45+ NK cells measured by tissue immunostaining (Figures 3D and 3E) and validated by isolating and quantifying hCD45+–circulating cells in the peripheral blood of these mice by flow cytometry (Figure 3F). The CX3CR1-CX3CL1 axis has also been reported to be important in the cytotoxic activity of lymphocytes.60,61,62,63 Hence, we tested the ability of healthy donor NK cells to kill A459 cells overexpressing CX3CL1. We found that CX3CL1 overexpression resulted in superior killing, compared to control-transfected A549 cells, proving that the CX3CR1-CX3CL1 axis not only aids in infiltration of cytotoxic cells into solid malignancies but also increases the cytolytic activity of the recruited cells (Figure 3G). This novel finding points toward exploiting this axis for the therapeutic efficacy of immunotherapies using cytotoxic cells.
Novel design of NKG2D-CAR-T overexpressing CX3CR1
While we have demonstrated that overexpression-induced secretion of CX3CL1 by the tumor can restore infiltration and increase functionality of cytotoxic cells, modifying the tumor is a challenging approach to induce infiltration clinically. Moreover, previous work by Siddiqui et al. has shown that overexpression of CX3CR1 in T cells can aid TIL migration toward tumors in vivo: although their work demonstrates recruitment of TILs toward the tumor, it did not look for intratumoral infiltration.11 For this reason, we built upon the effectiveness of NKG2D CAR-T through the co-expression of CX3CR1 in the design as schematized in Figure S7A. Our CAR-T design, a second-generation CAR-T, focuses on using the extracellular domain of NKG2D, followed by a hinge and transmembrane region that leads to the signaling domains 4-1BB and CD3ζ to achieve a strong expansion and persistence of CAR-Ts.64,65 Because we have shown that IL-15 does not induce infiltration, our control co-expresses IL-15 to test the concept that CX3CR1 receptor co-expression on these cells can induce infiltration. Lastly, our design includes using epidermal growth factor receptor (EGFR) as a target for cell depletion to induce rapid T cell elimination after treatment to reduce adverse events.66 Empty vector (pCDCAR) T cells did not survive and proliferate in culture, but NKG2D CAR-Ts did. Overexpression of CX3CR1 enhanced the numbers of the CAR-Ts in culture (Figure 4A), although both CAR-T constructs (CX3CR1 or IL-15) had strong in vitro killing activity against either A549 or HepG2 cells (Figures 4B and S7B), likely because of saturation. However, the NKG2D-CX3CR1 cells retained higher levels of CFSE (CFDA-SE: carboxyfluorescein diacetate succinimidyl ester) compared to the IL-15 control cell, suggesting that they cycle slower and thus limits the rate at which they reach exhaustion (Figure S7C). Interestingly, CX3CR1 overexpression did allow for a stronger activation state than IL-15 overexpression, as indicated by the higher level of IFNγ activation (Figure 4C) and significantly increased mobilization (Figure S7D) when in co-culture with tumor cells.
Figure 4.

NKG2D-CAR-T co-expressing CX3CR1 has higher activation in vitro
(A) Representative flow cytometric analysis of CAR-T proportions (NKG2D+EGFR+) and sustainability after CAR-T viral transduction of primary isolated human T cells (n = 6). pCDCAR is the control vector, while NKG2D-IL-15 overexpresses IL-15 and NKG2D-CX3CR1 CAR-T overexpresses CX3CR1.
(B) CAR-T cytotoxic assay of A549 target cells. One-way ANOVA was used to compare the area under the curve of each group, and p values are shown in the graphs.
(C) IFNγ ELISA of supernatants from primary CAR-T cells co-cultured with target A549 cells at the ratio shown. D) tSNE analysis of expression of CD4, EGFR, PD1, Tim3, and LAG3 in CAR-T cells after co-incubation with target cells.
It is well accepted that CAR-T activation leads to increased parallel-checkpoint receptor expression. Co-expression of these receptors has been associated with prominent T cell activation (CD69/CD137), effector function (Granzyme-B), and proliferation (Ki-67) in NSCLC.67 Because of this, we tested the expression of PD1, Tim 3, and LAG3 on the NKG2D-IL-15 and NKG2D-CX3CR1 CAR-T cells and their distribution in a tSNE analysis before and after co-culture with tumor cells (Figures S8 and 4D, respectively). In both figures, the first column shows the distribution of cells in pseudocoloring, followed by the cells expressing CD4 (orange), with the green cells being CD8 T cells and where EGFR expression (CAR-T transduction) is present. We saw increased co-expression of PD-1, Tim-3, and LAG-3 receptors in both CAR-T-transduced T cells, after encounter with tumor cells (Figure 4D), consistent with the activation observed earlier but not before stimulation (Figure S8). These data suggest that CX3CR1 overexpression may have an advantage not only for inducing mobilization but also for enhancing the activation of these cells.
Testing of CX3CR1-overexpressing CAR-T in preclinical models of solid cancer
To validate the ability of CX3CR1 overexpression to enhance the infiltration of CAR-T into solid malignancies, we tested the ability of CX3CR1-overexpressing CAR-Ts to infiltrate a solid tumor in the A549 lung cancer model (schematic representation in Figure S9A). The tumors were established 2 weeks prior, as shown in Figures 1 and 3, to demonstrate a break in tumor-bed restriction with CX3CR1 overexpression in CAR-T. Testing CAR-Ts in NSG mice bearing human-tumor xenografts is the standard for testing the efficacy of CAR-T in vivo, including the support for clinical development of other CAR-Ts.68 While NKG2D-IL-15 CAR-Ts slowed the growth rate of A549 tumors, the presence of CX3CR1 in NKG2D CAR-T had the highest impact on tumor growth measured by luciferase expression (Figures 5A and 5B). In the NKG2D-CX3CR1 CAR-T mice, the tumors were barely palpable after 4 weeks, suggesting that the presence of CX3CR1 allowed these cells to effectively target a remote solid malignancy after intravenous injection. To understand the role of CX3CL1 ligation to CX3CR1+ CAR-T, we repeated the experiment using A549-CX3CL1-overexpression cells. Similar to our observations of increased TIL infiltration, there was a significantly reduced tumor burden when CX3CL1 was overexpressed in A549 cells, regardless of the CAR-T construct used, although CX3CL1-overexpressing tumors, treated with the CX3CR1-overexpressing CAR-Ts, completely reduced tumor luminescence by 3 weeks (Figure 5B). The highest level of circulating CAR-Ts was observed in the NKG2D-CX3CR1-treated cohort of mice (Figures 5C and 5D; Figure S9B), and this correlated with increased levels of IFNγ in the peripheral blood (Figure 5E). We confirmed that this increased activation is caused by higher intratumoral mobilization rather than from increased proliferation, by observing the infiltration of CAR-T injected peritumorally 72 h after injection (Figures S11A and S11C) with increased infiltration of pCDCAR in CX3CL1-overexpressing tumors (Figure S11D). The CAR-T phenotype was also analyzed from the blood collected at the end of the experiment, including memory/effector state and exhaustion profile. NKG2D-CX3CR1 CAR-T had a robust naive/central memory population (CD62L+CD127+) increase (Figures S12A and S12B) with the PD1 expression comparably lower to NKG2D-IL-15 CAR-T (Figure S12C). This experiment would indicate whether CX3CR1-overexpressing CAR-T cells may be better equipped to persist and have an effect in an intact TME with active suppression.
Figure 5.

NKG2D-CAR-T co-expressing CX3CR1 reduces tumor burden in solid tumor murine models
(A) Luminescence image of NSG mice carrying A549-luciferase tumors and treated with primary human CAR-T as shown (day 28).
(B) Luminoscore (pixels/second) of NSG mice carrying A549 tumors, stably transfected with either scrambled or CX3CL1 overexpression vectors and treated with primary human CAR-T cells (n = 5 per group). One-way ANOVA was used to compare the AUC of each group, and p values are shown in the figure graphs. StoS compares indicated A549 scramble tumor group to scramble group while StoOE denotes scramble group compared to CX3CL1-overexpression tumor group.
(C) Age of circulating human CD45+ CAR-T cells isolated after euthanasia in mice as in A (n = 10 per group). One-way ANOVA was used to compare the AUC of each group.
(D) Percentage of human CD45+EGFR+CX3CR1+ CAR-T cells in the blood of mice as in A. Statistical analysis used was Kruskal-Wallis test with multiple comparison analysis.
(E) IFNγ ELISA of peripheral blood plasma of mice as in A. Statistical analysis used was the Kruskal-Wallis test with multiple comparison analysis.
(F) Luminosity image of NSG mice carrying HepG2-luciferase tumors and treated with primary human CAR-T (day 28).
(G) Luminoscore (pixels per second) of NSG mice carrying HepG2-luciferase treated with primary human CAR-T cells (n = 6 mice per group). One-way ANOVA was used to compare the area under the curve (AUC) of each group). H) Kaplan-Meyer survival plot of HepG2 tumor mice as in E. In all graphs, error bars denote the SEM of the number of animals in the experiment (n = 6 mice per group). Abbreviation: AUC, area under the curve.
In all figures error bars denote the standard error of the mean of n.
To confirm the impact of CX3CR1 overexpression with CAR-T on solid malignancies beyond lung cancers, we repeated the experiment with HepG2 hepatocellular carcinoma cells and observed a similar decrease in tumor burden (Figures 5F and 5G). Interestingly, while the tumor cell lines used did not have, or induce, extra CX3CL1 production while on in vitro cytotoxic assays with naive CAR-T (Figures S11A and S11B), the level of CX3CL1 produced in co-cultures of T cells isolated from the blood of tumor-bearing mice showed a higher level of CX3CL1 secretion independent of the CAR-T used (Figures S11C–S11E). This suggests that NKG2D-CX3CR1 CAR-Ts are not exclusively activated through tonic signaling of the added chemokine receptor. Importantly, treatment with NKG2D-CX3CR1 CAR-T led to increased survival (Figure 5H) of the mice. These data confirm that this receptor enhances the activity of cytotoxic cells in solid malignancies by enhancing recruitment and providing preclinical rationale for using this CAR-T enhancement in the future for solid malignancies.
Discussion
While a myriad of dysfunctions have been linked to the failure of antitumor immunotherapies in solid malignancies, one of the critical challenges to overcome has been trafficking cytotoxic cells toward and into the tumor.3 This problem is best exemplified by reduced level of cytotoxic cells in the tumor bed as an independent predictor of progression-free survival,1,2 likely due to either reduced chemokine secretion by tumor cells and/or reduced receptors in cytotoxic cells.69 In the current study, to overcome cytotoxic-cell restriction in a murine xenograft model of lung cancer, we studied the immune cell receptor architecture in primary NSCLC, a solid tumor with a decreasing survival rate with increasing stage and low response to adjuvant chemotherapy.70,71 Our murine model demonstrates an early cytotoxic-cell restriction induced by the developing tumor that is not linked to the cell’s survival or activation as IL-15 did not restore their infiltration. Considering this restriction has been established in many solid malignancies,1,2,3 we investigated the proportion of immune cells and key receptor expression on those cells in 3 different zones: in the tumor, surrounding the tumor, and away from the tumor (healthy tissues). We observed that cytotoxic cells restricted from the tumor were linked to a reduction to their expression of CX3CR1 along with its ligand CX3CL1 in the tumor, and enhancement of either receptor or ligand was sufficient to restore migration of TILs into tumors. This is inconsistent with a recent study on a melanoma model that CX3CR1 expression can define subsets in the tumor: (a) CX3CR1– subset that represents less-differentiated CD8+ T cells with severely impaired effector function; (b) CX3CR1int subset that also expresses high levels of coinhibitory receptors but has relatively preserved cytokine production capacity in the tumor; and (c) the terminally differentiated CX3CR1hi subset that exhibits robust cytotoxicity.63 We showed that this chemokine and its receptor play a key role in survival of mice in preclinical models of several different solid malignancies. These results are in accordance with others demonstrating that cytotoxic lymphocytes are localized mainly in the neighboring tissues and surrounding the tumor in NSCLC72 and strongly reduced inside the tumor. Particularly, CX3CR1 expression in TILs (KLRK1+) was linked to survival in solid malignancies, indicating that trafficking of lymphocytes and CX3CR1-CX3CL1 is a major roadblock in CAR-T therapeutics in solid tumors. This is proven as CX3CR1-CX3CL1 can serve as prognostic biomarkers in several malignancies with higher levels linked with a good prognosis,4,16,73 including our own observations. Moreover, Siddiqui et al. previously demonstrated that overexpression of CX3CR1 in T cells allows homing of T cells toward the tumor in vivo, although it did not strongly reduce tumor burden.11 However, because they examined aggregated tumors, it was not clear if the receptor allowed infiltration into the tumor bed. This increased migration is likely caused by the consequences of the change in expression of CX3CR1 in cytotoxic cells because they represent a differentiated migrating effector-cell population.74 Moreover, the expression level of CX3CL1 also correlates with the recruitment of TILs.16,17,75
The CX3CR1-CX3CL1 axis can also drive the infiltration of myeloid cells, including MDSCs, which comprise a significant subset of the CX3CR1 population of infiltrating cells and migrate faster toward CX3CL1 than NK cells and CD8+ cells.74,75,76,77,78,79 This was confirmed in our work through the increased levels of MDSCs in the TME that correlated with the decreased levels of cytotoxic-cell populations. Importantly, MDSC can contribute to the modulation of other CX3CR1+ cells function, modulate the release of CX3CL1, and induce the accumulation of Tregs.46,80 Active expression of CX3CR1 in our CAR-T design can overcome the effects of MDSC by enhancing antitumor responses in the TME, even if surface CX3CL1 expression is reduced in tumor cells. Moreover, the naive/central memory population of NKG2D-CX3CR1 CAR-T cells had lower levels of PD1 expression in the circulation, suggesting a less-exhausted phenotype that can aid in longer lasting responses.
Our way to therapeutically achieve the goal of increasing antitumor responses, while evading the intratumor TIL restriction and increased immune suppressiveness of MDSC, is by increasing the expression of CX3CR1 in CAR-Ts. We selected the use of NKG2D CAR-Ts because it has been shown preclinically and clinically against several types of solid malignancies and since its ligands are increased in these tumors compared to surrounding healthy cells.24,81,82,83,84,85 Moreover, studies have shown that NKG2D CAR-Ts can survive longer than other CAR-Ts, offering an advantage that may allow extra time for the cells to migrate toward tumors, although the cells are still limited in infiltrating into the solid tumor.23,24,69,86,87 NKG2D CAR-Ts also have the advantage that they can reduce immunosuppressive cells through NKG2D-mediated activation, including MDSCs and T regs, leading to the resumption of effective antitumor immune responses.24,88 Our data demonstrate that the introduction of CX3CR1 overexpression improved trafficking of CAR-Ts into the tumor, consistent with previous reports that induction of chemokine receptors, such as CXCR2 or CXCR3, in engineered T cells can be beneficial.89,90,91,92 CX3CR1 has advantages compared to other chemokine receptors because it can send pro-survival signals after ligation, inhibit Fas-mediated apoptosis, increase responsiveness to other chemokines, promote activation through IFNγ production and cytotoxicity, and have CX3CL1-independent effects on tumor cells.50,51,93,94,95,96 Therefore, adding the CX3CR1 receptor can increase the infiltration of these cytotoxic cells into the tumor while simultaneously taking advantage of other nonchemotactic functions to enhance the activation of NKG2D CAR-Ts.
CAR-Ts can have several pitfalls that leave them clinically ineffective and affect our construct. Among the most common is how to further reduce the tumor-induced immune-suppressive microenvironment, apart from the effects met by CX3CR1 in our construct. To overcome that, future studies on therapeutic combinations of our novel CAR-T construct with other reagents that can help counteract some of those effects will be warranted, including combination with MDSC-depleting agents,97 TGFβ inhibition with antagonists,98,99,100 enhancing the activation of NKG2D with IL-15,1,38,57,101,102,103 or checkpoint inhibitors (anti-PD1/PD-L1 antibodies).18 Most of these approaches work by reducing the suppressiveness of the TME. This combined with the overexpression of CX3CR1 in the CAR-T (i) should enhance both the function of the CAR-T and the accumulation of antitumor TILs; (ii) increase production of IFNγ and prevent downregulation of NKG2D in these cells; and (iii) enhance NKG2D-ligand expression. In summary, based on our study in human primary NSCLC, we discovered that reducing expression of CX3CR1 in cytotoxic lymphocytes is a key mechanism that prevents lymphocyte trafficking into the TME. Co-expression of CX3CR1 with NKG2D in CAR-T cells can significantly increase the migration of cytotoxic lymphocytes into the tumor site. These CAR-T cells have increased cytotoxicity of target tumor cells and high levels of activation with increased levels of cytokine and activation marker expression. Our work further demonstrates that the combination with CX3CR1 could significantly reduce the tumor burden and prolong survival of tumor-bearing mice. The clinical efficacy will need to be tested in the future phase I and II trials.
Limitations of the study
In the present study, we took advantage of the isolation tumor, adjacent, and away tissues from 16 primary NSCLC specimens. The opportunity to observe TILs in this specific architecture showcased the restriction of CX3CR1+ cytotoxic cells from the tumor bed. Apart from the limited number of samples (16) for this assessment, another potential limitation is the impossibility to demonstrate whether the presence of the CX3CR1 receptor in cytotoxic cells is reduced in the cells or the proportion of CX3CR1+ cells is reduced rather than a change in receptor expression. Another limitation is the use of the NSG A549 murine model. This model uses human T cells injected after a short time which makes it difficult to assess the full changes induced by the TME on endogenous TILs. Moreover, this system is also devoid of other immune cells, such as myeloid cells, that are known to also play a critical role in immunosuppression.
Data and materials availability
Except for primary specimens and CAR-T designs, all the materials used are commercially available as described in STAR Methods. All data and materials used in the analysis will be available to researchers upon request for purposes of reproducing or extending the analysis. The CAR-T designs shown here have been patented and will be made available only for research purposes under a material transfer agreement through Moffitt Cancer Center for noncommercial purposes.
STAR★Methods
Key resources table
| REAGENTS or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD16 PerCP C5.5 | BD Biosciences | catalog number 560717, RRID:AB_1727434 |
| Tbet PE CF594 | BD Biosciences | catalog number 562467, RRID:AB_2737621 |
| NKG2D PE-Cy7 | BD Biosciences | catalog number 562365, RRID:AB_11153309 |
| NKp46 APC | BD Biosciences | catalog number 558051, RRID:AB_398653 |
| LAG3 APC-R700 | BD Biosciences | catalog number 565774, RRID:AB_2744329 |
| CX3CR1 BV510 | BD Biosciences | catalog number 744487, RRID:AB_2742267 |
| NKp30 BV605 | BD Biosciences | catalog number 563384, RRID:AB_2738170 |
| DNAM1 BV650 | BD Biosciences | catalog number 742496, RRID:AB_2740829 |
| CD3 BV711 | BD Biosciences | catalog number 563724, RRID:AB_2744392 |
| IL-10 BV786 | BD Biosciences | catalog number 564049, RRID:AB_2738563 |
| NKp44 BUV395 | BD Biosciences | catalog number 744305, RRID:AB_2742135 |
| CD56 BUV737 | BD Biosciences | catalog number 744305, RRID:AB_2742135 |
| CD8 BUV496 | BD Biosciences | catalog number 564804, RRID:AB_2744460 |
| CD45 BUV805 | BD Biosciences | catalog number 564914, RRID:AB_2744401 |
| CD11b AF488 | BD Biosciences | catalog number 557701, RRID:AB_2129268 |
| CD68 PE-CF594 | BD Biosciences | catalog number 564944, RRID:AB_2739021 |
| CD195 PE-Cy7 | BD Biosciences | catalog number 557752, RRID:AB_396858 |
| HLA-DR APC-H7 | BD Biosciences | catalog number 641393, RRID:AB_1645739 |
| CD33 BV650 | BD Biosciences | catalog number 744352, RRID:AB_2742171 |
| CD3 BV650 | BD Biosciences | catalog number 563851, RRID:AB_2744391 |
| CD15 BV786 | BD Biosciences | catalog number 563851, RRID:AB_2744391 |
| CD14 BUV395 | BD Biosciences | catalog number 563561, RRID:AB_2744288 |
| CD56 BUV737 | BD Biosciences | catalog number 564447, RRID:AB_2744432 |
| CD19 BUV496 | BD Biosciences | catalog number 564655, RRID:AB_2744311 |
| CD45 BUV805 | BD Biosciences | catalog number 564914, RRID:AB_2744401 |
| EGFR AF488 | R&D Systems | catalog number FAB9577G |
| CD4 PerCP-Cy5.5 | BD Biosciences | catalog number 560650, RRID:AB_1727476 |
| CX3CR1 APC | Thermo Fisher Scientific | catalog number 17-6099-42, RRID:AB_11149136 |
| CD28 APC-H7 | BD Biosciences | catalog number 561368, RRID:AB_11154032 |
| PD1 BV421 | BD Biosciences | catalog number 562516, RRID:AB_11153482 |
| CD127 BV605 | BD Biosciences | catalog number 562662, RRID:AB_2737706 |
| CD62L BV711 | BD Biosciences | catalog number 565040, RRID:AB_2869642 |
| Tim3 BV786 | BD Biosciences | catalog number 742857, RRID:AB_2741100 |
| CCR7 BUV395 | Thermo Fisher Scientific | catalog number 17-6099-42, RRID:AB_11149136 |
| TIGIT APC-Fire750 | BioLegend | catalog number 372708, RRID:AB_2632755 |
| CTLA-4 PE-CF594 | BD Biosciences | catalog number 562742, RRID:AB_2737761 |
| CD8 BUV496 | BD Biosciences | catalog number 564804, RRID:AB_2744460 |
| CD27 BUV 737 | BD Biosciences | catalog number 564301, RRID:AB_2744350 |
| CD3 BUV605 | BD Biosciences | catalog number 564712, RRID:AB_2738908 |
| LAG3 APC-R700 | BD Biosciences | catalog number 565774, RRID:AB_2744329 |
| CD95 PE-CF594 | BD Biosciences | catalog number 562395, RRID:AB_11153666 |
| CD45RA PE-Cy7 | BD Biosciences | catalog number 560675, RRID:AB_1727498 |
| EGFR AF488 | R&D systems | catalog number FAB9577G |
| CX3CR1 APC | Thermo Fisher Scientific | catalog number 17-6099-42, RRID:AB_11149136 |
| CD45 BUV805 | BD Biosciences | catalog number 612891, RRID:AB_2870179 |
| NKG2D PE-Cy7 | BD Biosciences | catalog number 562365, RRID:AB_11153309 |
| CD56 antibody (clone: 123C3.D5) | Cell Marque | catalog number 156M-84, RRID:AB_1158181 |
| CX3CR1 antibody | Abcam | catalog number ab8021, RRID:AB_306203 |
| CX3CL1 antibody | Abcam | catalog number ab9819, RRID:AB_2292428 |
| CD8 antibody (clone:C8/144B) | Abcam | catalog number ab17147, RRID:AB_443686 |
| Mouse anti-Human CD45 Antibody (Leukocyte marker) | Raybiotech | catalog number 188-10998-1 |
| Chemicals peptides and recombinant proteins | ||
| DMEM media | GIBCO | catalog number: 11995-065 |
| FBS | GemCell | catalog numbers: 100-500 |
| Opti-MEM medium | GIBCO | catalog number 11058021 |
| Tet-System Approved FBS | Thermo Fisher Scientific | catalog number A4736401 |
| X-VIVO 15 T-cell media | Lonza | Catalog number BE02-060Q |
| Recombinant Human IL-2 | Peprotech | Catalog Number 200-02 |
| Ficoll-Paque PLUS density gradient media | Cytiva | catalog number 17144003 |
| Critical Commercial Assays | ||
| Human CX3CL1/Fractalkine DuoSet ELISA | R&D Systems | Catalog number: DY365 |
| Human IFNγ DuoSet ELISA | R&D Systems | Catalog number: DY285 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Catalog number L3000001 |
| Lenti-X Concentrator | Takara Bio | catalog number 631232 |
| Pan T Cell Isolation Kit | Miltenyi Biotec | catalog number 130-096-535 |
| Dynabeads™ Human T-Activator CD3/CD28 for T Cell Expansion and Activation | GIBCO | Catalog number: 11131D |
| Opal-IHC system | Akoya/PerkinElmer | catalog Nos. FP1487001KT, FP1488001KT, and FP1497001KT |
| antifade with DAPI | Perkin Elmer | catalog number FP1490 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit, for 405 nm excitation | Thermo Fisher Scientific | Catalog number: L34957 |
| Experimental Models: Cell lines | ||
| A549 cells | ATCC | catalog number CCL-185, RRID:CVCL_0023 |
| A549-luc | ATCC | RRID: CVCL_J242 |
| Lenti-X 293T cells | Takara Bio | catalog number 632180, RRID:CVCL_4401 |
| HepG2 cells | ATCC | RRID:CVCL_JG47 |
| Experimental Models: Organisms strains | ||
| NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG) mice | Jackson Laboratory | catalog number 005557 |
| Recombinant DNA | ||
| Lentiviral (accession number: BC001163) overexpression vectors (pLenti-GIII-CMV-GFP-2A-Puro) | ABM | catalog number LV130375 |
| CAR constructs (designed by us) | CreativeBioLabs | N/A |
| pRV-Rev | Addgene | N/A |
| pMDLg/pRRE | Addgene | Plasmid# 12251 |
| pMD2.G | Addgene | Plasmid# 12259 |
| Software and Algorithms | ||
| FlowJo software | FlowJo | RRID:SCR_008520 |
| InForm | Perkin Elmer | N/A, Microscopy Core Equipment |
| HALO Image Analysis Platform | Indica Labs | RRID:SCR_018350 |
| NIH GDC Data Portal | GDC Data Portal | RRID:SCR_014514; portal.gdc.cancer.gov |
| GraphPad Prism Software Version 8 | GraphPad Prism | RRID:SCR_002798 |
| Other | ||
| Consumables | ||
| 96-well RTCA E-Plate | Agilent Biosciences | catalog number 300601010 |
| Nunc-Immuno plate | Millipore-Sigma | Catalog number: M9410-1CS |
| Equipment | ||
| BD FACSymphony | BD | N/A Custom |
| IVIS Lumina Series II | Perkin Elmer | N/A, SAIL Core Equipment |
| Bruker Biospec 7T MRI | Perkin Elmer | N/A, SAIL Core Equipment |
| Vectra 3 Automated Quantitative Pathology Imaging System | Akoya Biosciences | N/A, Microscopy Core Equipment |
| Core Facilities | ||
| Moffitt Tissue Core | Moffitt Cancer Center | RRID:SCR_012364 |
| Moffitt Cancer Center Small Animal Imaging Laboratory | Moffitt Cancer Center | RRID:SCR_012401 |
| Moffitt Flow Cytometry Core | Moffitt Cancer Center | N/A |
| Moffitt Analytical Microscopy Core | Moffitt Cancer Center | N/A |
Resource availability
Lead contact
Further information on resources and reagents should be directed to the lead contact, Dr. Erika A. Eksioglu (Erika.eksioglu@moffitt.org).
Materials availability
Materials generated in this study are available from the lead contact, Dr. Erika A. Eksioglu (Erika.eksioglu@moffitt.org).
Experimental model and subject details
Patient specimens
Patients provided informed consent, and tissue specimens were collected through Moffitt Cancer Center’s (MCC) Total Cancer Care Protocol (PMID:22157297). De-identified tissues were released for this study according to stage and grade (when available at time of collection) under the protocol MCC17350 in 2017. Age and sex of the patients from where the tissue was obtained was not provided to us. Staff pathologists in MCC’s Tissue Core (RRID:SCR_012364), provided separated sections of normal away, normal adjacent, and tumor resected from consented patients and de-identified the information save for tumor stage and grade. Of the 8 adenocarcinoma specimens, 3 were provided without stage information, 4 were stage IA and one IB. Of the 5 squamous carcinoma specimens, for one no stage was given, 2 were stage IA, and one each was stage IIA or IIB. There were also 2 neuroendocrine tumors stage IA and one metastatic to lung fibrosarcoma. The tissues were divided for flow cytometric analysis while part of the tissue was embedded, paraffinized, and put on slides for immunohistochemical staining.
Cell lines
Lentiviral Scrambled (Scr, sequence randomized by vendor) and CX3CL1 (accession number: BC001163) overexpression vectors were obtained from ABM (pLenti-GIII-CMV-GFP-2A-Puro, catalog number LV130375; Applied Biological Materials Inc. Richmond, BC, Canada) and the viral particles were transduced into A549 (RRID: CVCL_ZX73) and A549-luc (RRID: CVCL_J242) cells resulting in A549-CX3CL1OE, A549-luc-CX3CL1OE, A549-Scr, and A549-Luc-Scr. All cell lines were maintained in DMEM media (GIBCO catalog number: 11995-065) with 10% FBS (GemCell, catalog numbers: 100–500), 1% PSG, 1% sodium pyruvate, 1% nonessential amino acids, and 0.2% mycozap.
Murine lung and liver tumor models
NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG, The Jackson Laboratory catalog number 005557) mice were used for the xenograft experiments. Due to availability, all the mice used in this study were male between 6 and 8 weeks of age. The 105 A549-luc (RRID: CVCL_J242) or HepG2 cells expressing luciferase (RRID:CVCL_JG47) were transplanted subcutaneously at the back of each mouse. When tumors grow up at measurable size, normally after 3 weeks, mice were injected intravenously with 3 × 106 of either CAR-T control T cells. The mice were injected a total of 2 times with the CAR-T cells, 2 months apart. Each mouse also got 2 × 105 U of human IL-2 (PeproTech, Cranbury, NJ, USA) injected intraperitoneally biweekly (a standard procedure to maintain the viability of human T cells in NSG mice and similar to the protocol used in patients undergoing adoptive T cell therapy reviewed in.104 Tumor growth was measured weekly using an IVIS Lumina Series II and later a Bruker Biospec 7T MRI when the tumor size became too large to be imaged by luciferase at the Moffitt Cancer Center Small Animal Imaging Laboratory (RRID:SCR_012401). Once endpoint was reached, the liver, lungs, spleen, and tumor were harvested and fixed to be stained for human CD45 (hCD45).
Method details
Cytotoxicity assay
Cytotoxicity assay to test the efficiency of NK cells or CAR-T cells against either Scrambled (same nucleotides in random order, does not produce protein) or CX3CL1 overexpressing A549 target cells was measured using the xCelligence Real-Time Cell Analysis (RTCA) instrument (ACEA Biosciences, San Diego, CA, USA) according to the manufacturer’s instructions as done before by others.105 One day prior, target cells were added to each of a 96-well E-Plate (ACEA Biosciences, San Diego, CA, USA) at a concentration of 104 cells/well. Target cells were allowed to settle, and measurements of target cell growth (impedance) were collected for 24 h at 37°C, 5% CO2. Primary isolated NK or CAR-T cells were added after 48 h onto target cells at 100 000 cells per well at an E:T ratio of 10:1. Cell growth was monitored for 10 days.
Enzyme-linked immunosorbent assay (ELISAs)
Following the manufacturer’s recommendation for the DuoSet kits, 96-well plates (Nunc-Immuno Plate, Millipore-SIGMA, Darmstadt, Germany) were coated with purified monoclonal antibody against either human CX3CL1 or IFNγ (Duo set, R&D systems, Minesota, USA) in 1X PBS, pH 7.4 at room temperature overnight. The plates were then incubated in blocking buffer (Pierce-Endogen, Rockford, IL, USA) for 2 h at room temperature and washed in 0.05% Tween 20. Wells were seeded with serially diluted recombinant human (rh) cytokines (standard, in blocking buffer in duplicates, provided in kits) or with 50 μL of supernatants in triplicate followed by addition 1 h later of biotin-labelled anti-cytokine antibody. After incubation wells were washed 5 times and coated with strepavidin-horseradish peroxidase for 20 min, washed again 5 times and developed by adding TMB substrate (Pierce-Endogen, Rockford, IL, USA). The reaction was stopped by the addition of an equal volume of 0.18 M H2SO4. The absorbance was read at 450 nm with deletion of background at 650 nm.
CAR constructs
The CAR constructs were designed and purchased from CreativeBioLabs (creativebiolabs.net). The NKG2D extracellular domain, IL15, CX3CR1 were singly or co-integrated in a pCDCAR backbone plasmid. The cell surface truncated the form of human epidermal growth factor receptor (EGFRt), expressed separately on CAR constructs, was used as a tracking marker for CAR-expressing cells as well as a safety mechanism to facilitate elimination of transferred cells in the event of adverse effects.
CAR-T virus production/transfection
Lenti-X 293T cells (RRID:CVCL_4401) were seeded in DMEM medium with 10% TET FBS medium in 100-mm Petri dishes. The cells were cultured in 37 °C, 5% CO2, until 60%–70% confluency. Two hours before transfection, medium was replaced with fresh DMEM supplied with 10% TET FBS. In one tube, Lipofectamine 3000 was diluted in Opti-MEM medium at a 1:37 dilution and mixed well (41 μL of Lipofectamine 3000 diluted in 1.5 mL medium). In a separate tube, 20 μg expression plasmid, 5 μg pRV-Rev, 10 μg pMDLg/pRRE, and 6 μg pMD2.G were mixed in 1.5 mL Opti-MEM media. Thirty-five μL of P3000 enhancer reagent was added to the plasmid mix followed by addition of Lipofectamine 3000 following the manufacturer’s recommendations. Three mLs of the DNA-lipid complex were added dropwise to the 293 T cells and incubated for 72 h. At that time, the supernatant was collected and spun at 3000 × g for 15 min at 4 C. The conditioned medium was filtered using a 0.45 μM PES unit filter. Lenti-X Concentrator was added at a 1:3 dilution and then ultracentrifuged using an SW-32Ti swinging-bucket rotor at 21 000 RPM. The supernatant was discarded and the pellet was resuspended in IL X-VIVO 15 T cell media with 5% human AB serum, 1% PSG, and 300 Iu/mL of IL2 (Peprotech) at 4 C overnight. Solutions were stored in aliquots at −80 C and thawed before using.
CAR-T transduction
CAR T-cells were generated by lentiviral transduction. Human peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque centrifugation. T cells were isolated using Pan T Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany). T cells were activated with magnetic CD3−CD28 Dynabeads at a 1:1 ratio for 48 h and later transduced with PCDCAR, NKG2D-IL15, and NKG2D-CX3CR1. The lentiviral stock solution was added to a fibronectin-coated plate and spun at 2000 × g for 2 h at 32 °C. 0.5 × 106 of the activated T cells were added to the virus-bound well and spun at 1000 × g for 1 h at 32°C. The plate was then incubated at 37 °C, 5% CO2 incubator for 2 to 3 days. The cells were collected, centrifuged, and resuspended in T cell media.
Flow cytometry
Mononuclear cells isolated from human lung tissues were analyzed with the following antibodies: CD16 PerCP C5.5 (BD Biosciences catalog number 560717, RRID:AB_1727434), Tbet PE CF594 (BD Biosciences catalog number 562467, RRID:AB_2737621), NKG2D PE-Cy7 (BD Biosciences catalog number 562365, RRID:AB_11153309), NKp46 APC (BD Biosciences catalog number 558051, RRID:AB_398653), LAG3 APC-R700 (BD Biosciences catalog number 565774, RRID:AB_2744329), CX3CR1 BV510 (BD Biosciences catalog number 744487, RRID:AB_2742267), NKp30 BV605 (BD Biosciences catalog number 563384, RRID:AB_2738170), DNAM1 BV650 (BD Biosciences catalog number 742496, RRID:AB_2740829), CD3 BV711 (BD Biosciences catalog number 563724, RRID:AB_2744392), IL-10 BV786 (BD Biosciences catalog number 564049, RRID:AB_2738563), NKp44 BUV395 (BD Biosciences catalog number 744305, RRID:AB_2742135), CD56 BUV737 (BD Biosciences catalog number 744305, RRID:AB_2742135), CD8 BUV496 (BD Biosciences catalog number 564804, RRID:AB_2744460), CD45 BUV805 (BD Biosciences catalog number 564914, RRID:AB_2744401), CD11b AF488 (BD Biosciences catalog number 557701, RRID:AB_2129268), CD68 PE-CF594 (BD Biosciences catalog number 564944, RRID:AB_2739021), CD195 PE-Cy7 (BD Biosciences catalog number 557752, RRID:AB_396858), HLA-DR APC-H7 (BD Biosciences catalog number 641393, RRID:AB_1645739), CD33 BV650 (BD Biosciences catalog number 744352, RRID:AB_2742171), CD3 BV650 (BD Biosciences catalog number 563851, RRID:AB_2744391), CD15 BV786 (BD Biosciences catalog number 563851, RRID:AB_2744391), CD14 BUV395 (BD Biosciences catalog number 563561, RRID:AB_2744288), CD56 BUV737 (BD Biosciences catalog number 564447, RRID:AB_2744432), CD19 BUV496 (BD Biosciences catalog number 564655, RRID:AB_2744311), CD45 BUV805 (BD Biosciences catalog number 564914, RRID:AB_2744401).
CAR-T were characterized in a panel containing: EGFR AF488 (R&D Systems catalog number FAB9577G), CD4 PerCP-Cy5.5 (BD Biosciences catalog number 560650, RRID:AB_1727476), CX3CR1 APC (Thermo Fisher Scientific catalog number 17-6099-42, RRID:AB_11149136), CD28 APC-H7 (BD Biosciences catalog number 561368, RRID:AB_11154032), PD1 BV421 (BD Biosciences catalog number 562516, RRID:AB_11153482), CD127 BV605 (BD Biosciences catalog number 562662, RRID:AB_2737706), CD62L BV711 (BD Biosciences catalog number 565040, RRID:AB_2869642), Tim3 BV786 (BD Biosciences catalog number 742857, RRID:AB_2741100), CCR7 BUV395 (Thermo Fisher Scientific catalog number 17-6099-42, RRID:AB_11149136), TIGIT APC-Fire750 (BioLegend catalog number 372708, RRID:AB_2632755), CTLA-4 PE-CF594 (BD Biosciences catalog number 562742, RRID:AB_2737761), CD8 BUV496 (BD Biosciences catalog number 564804, RRID:AB_2744460), CD27 BUV 737 (BD Biosciences catalog number 564301, RRID:AB_2744350), CD3 BUV605 (BD Biosciences catalog number 564712, RRID:AB_2738908), LAG3 APC-R700 (BD Biosciences catalog number 565774, RRID:AB_2744329), CD95 PE-CF594 (BD Biosciences catalog number 562395, RRID:AB_11153666), and CD45RA PE-Cy7 (BD Biosciences catalog number 560675, RRID:AB_1727498).
Mouse blood samples were collected weekly and stained with the following anti-human antibodies: EGFR AF488 (R&D systems catalog number FAB9577G), CX3CR1 APC (Thermo Fisher Scientific catalog number 17-6099-42, RRID:AB_11149136), CD45 BUV805 (BD Biosciences catalog number 612891, RRID:AB_2870179), Live/Dead Fixable Aqua, and NKG2D PE-Cy7 (BD Biosciences catalog number 562365, RRID:AB_11153309).
Flow cytometry was performed on a BD FACSymphony at the MCC Flow Cytometry Core Facility and analyzed with FlowJo software (FlowJo, RRID:SCR_008520). tSNE plots were made with FlowJo by first normalizing down each sample to create each sample with the same number of events and then creating a concatenated group specimen. The concatenated files containing the average of all the specimens in each group were then compared analytically by running tSNE analysis in FlowJo. The expression characteristic of each marker in these files was then analyzed for the specific expression of markers.
Immunohistochemical staining
Formalin-fixed paraffin-embedded baked tissue sections (N, A, and T) were manually stained using an Opal-IHC system (Akoya/PerkinElmer catalog Nos. FP1487001KT, FP1488001KT, and FP1497001KT) following the manufacturer’s instructions. Briefly, slides were treated with the PerkinElmer blocking buffer for 10 min and incubated with the specific primary antibodies: CD56 (clone: 123C3.D5, Cell Marque catalog number 156M−84, RRID:AB_1158181), CX3CR1 (Abcam catalog number ab8021, RRID:AB_306203), CX3CL1 (Abcam catalog number ab9819, RRID:AB_2292428), CD8 (clone:C8/144B, Abcam catalog number ab17147, RRID:AB_443686) followed by Opal-HRP polymer and one Opal fluorophore contained in the kits named above. Individual antibody complexes were stripped after each round of detection and antifade with DAPI (Perkin Elmer catalog number FP1490) applied before the addition of a coverslip. Autofluorescence slides (negative control) included primary and secondary antibodies, omitting the Opal fluorophores. All multiplex slides were imaged with the Vectra 3 Automated Quantitative Pathology Imaging System (Akoya Biosciences, Marlborough, MA, USA). The 20× ROI were tiled across the tissue and sequentially scanned. InForm (PerkinElmer) was used to unmix and export the scanned images. The 20× multilayer TIFF images from each slide were imported into HALO Image Analysis Plat-form (Indica Labs, Albuquerque, NM, USA) to fuse the images into a WTS digital image. All the analysis was performed at MCC’s Microscopy Core facilities.
Survival analysis
Survival analysis of KLRK1 and CX3CR1 expression in lung adenocarcinoma, lung squamous cell carcinoma, and liver hepatocellular carcinoma after categorization of samples as high expressers (above median expression) and low expressers (below median expression). Data was used from Oncolnc, which pulls gene expression and survival data from TCGA.
Gene expression of CX3CR1 and CX3CL1 in lung cancer and normal tissues
We obtained RNA-seq data from TCGA-LUAD and TCGA-LUSC projects from the NIH GDC Data Portal (GDC Data Portal, RRID:SCR_014514; https://portal.gdc.cancer.gov). Fragment per kilobase per million mapped reads (FPKM) values from HTSeq-FPKM workflow were used to evaluate gene expression level for CX3CR1 and CX3CL1. Normal tissue and lung cancer cases were determined by based on GDC downloaded sample sheet. Analysis was performed at Moffitt’s Bioinformatics Core.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism Software Version 8 (GraphPad Prism, RRID:SCR_002798). Statistical significance to assess 2 treatment groups only were determined by Student t tests. In Figure 2, patient specimens were first tested for normal distribution and those found to not be normally distributed were analyzed with Friedman test, since samples are matched. Figure 3E samples were also not normally distributed, and we used Wilcoxon Rank-Sum Test. All other analyses of 2 variables or more that were normally distributed were analyzed using Ordinary one-way ANOVA with multiple comparison analysis. All analysis and graphics show standard error of the mean bars (SEM) and p values <0.05 were considered to be statistically significant.
Acknowledgments
Editorial assistance was provided by Moffitt Cancer Center’s Office of Scientific Publishing by Daley Drucker and Gerard Hebert; no compensation was given beyond their regular salaries. This work was supported by the Department of Defense grant W81XWH-15-1-0440 (SW), James and Esther King Discovery Science Award from the State of Florida (SW), NIH K01 Career Award CA187020-02 (EAE), T32 training grant CA115308 (NT), and NCI Cancer Center Support Grant P30-CA076292 that supports Moffitt Cancer Center’s core facilities, including the Flow Cytometry and Bioinformatics Cores.
Author contributions
Conceptualization: TLT, SW, and EAE; methodology: WAA, AC, NT, RD, TS, MW, GW, EK, SC, LC, AM, DG, NL, CMS, JN, WK, XC, and PC; validation: TLT, NT, XC, PC, GW, DC, and EAE; formal analysis: TLT, EAE, PC, LC, and SW; investigation: TLT, WAA, AC, NT, RD, TS, MW, GW, GW, DC, EK, SC, LC, AM, DG, NLB, CMS, JN, WK, XC, and PC; resources: NLB, CMS, JN, and MW; data curation: EAE, TLT, GW, and NT; writing—original draft preparation: EAE and TLT; writing—review and editing: EAE, TLT, JL, GW, DC, KLW, and SW; visualization: EAE; supervision: EAE, KLW, and SW; project administration: EAE, KLW, and SW. All authors have read and agreed to the published version of the manuscript.
Declaration of interests
The SW laboratory received unrelated support from Celgene and Genentech. SW has received unrelated support from Blackbird BioFinance, LLC. EAE and KLW have an unrelated project funding through Tolero Pharmaceuticals. All other authors have no conflict of interest to declare for this research. SW and Moffitt Cancer Center filed a patent on the CAR-T construct described here.
Published: March 21, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106443.
Supplemental information
Data and code availability
-
•
All data presented in this study will be shared upon reasonable request by the lead contact, Dr. Erika A. Eksioglu (Erika.eksioglu@moffitt.org).
-
•
This paper does not report any original code.
-
•
Additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Nayyar G., Chu Y., Cairo M.S. Overcoming resistance to natural killer cell based immunotherapies for solid tumors. Front. Oncol. 2019;9:51. doi: 10.3389/fonc.2019.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rusakiewicz S., Semeraro M., Sarabi M., Desbois M., Locher C., Mendez R., Vimond N., Concha A., Garrido F., Isambert N., et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013;73:3499–3510. doi: 10.1158/0008-5472.CAN-13-0371. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z., Chen W., Zhang X., Cai Z., Huang W. A long way to the battlefront: CAR T cell therapy against solid cancers. J. Cancer. 2019;10:3112–3123. doi: 10.7150/jca.30406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melero I., Rouzaut A., Motz G.T., Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014;4:522–526. doi: 10.1158/2159-8290.CD-13-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hendry S., Salgado R., Gevaert T., Russell P.A., John T., Thapa B., Christie M., van de Vijver K., Estrada M.V., Gonzalez-Ericsson P.I., et al. Assessing tumor-infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the international immuno-oncology biomarkers working group: Part 2: TILs in melanoma, gastrointestinal tract carcinomas, non-small cell lung carcinoma and mesothelioma, endometrial and ovarian carcinomas, squamous cell carcinoma of the head and neck, genitourinary carcinomas, and primary brain tumors. Adv. Anat. Pathol. 2017;24:311–335. doi: 10.1097/PAP.0000000000000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mantovani A. The chemokine system: redundancy for robust outputs. Immunol. Today. 1999;20:254–257. doi: 10.1016/s0167-5699(99)01469-3. [DOI] [PubMed] [Google Scholar]
- 7.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 8.Niess J.H., Brand S., Gu X., Landsman L., Jung S., McCormick B.A., Vyas J.M., Boes M., Ploegh H.L., Fox J.G., et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 9.Combadiere C., Salzwedel K., Smith E.D., Tiffany H.L., Berger E.A., Murphy P.M. Identification of CX3CR1. A chemotactic receptor for the human CX3C chemokine fractalkine and a fusion coreceptor for HIV-1. J. Biol. Chem. 1998;273:23799–23804. doi: 10.1074/jbc.273.37.23799. [DOI] [PubMed] [Google Scholar]
- 10.Combadiere C., Gao J., Tiffany H.L., Murphy P.M. Gene cloning, RNA distribution, and functional expression of mCX3CR1, a mouse chemotactic receptor for the CX3C chemokine fractalkine. Biochem. Biophys. Res. Commun. 1998;253:728–732. doi: 10.1006/bbrc.1998.9849. [DOI] [PubMed] [Google Scholar]
- 11.Siddiqui I., Erreni M., van Brakel M., Debets R., Allavena P. Enhanced recruitment of genetically modified CX3CR1-positive human T cells into Fractalkine/CX3CL1 expressing tumors: importance of the chemokine gradient. J. Immunother. Cancer. 2016;4:21. doi: 10.1186/s40425-016-0125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bazan J.F., Bacon K.B., Hardiman G., Wang W., Soo K., Rossi D., Greaves D.R., Zlotnik A., Schall T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 13.Su Y.C., Chang H., Sun S.J., Liao C.Y., Wang L.Y., Ko J.L., Chang J.T. Differential impact of CX3CL1 on lung cancer prognosis in smokers and non-smokers. Mol. Carcinog. 2018;57:629–639. doi: 10.1002/mc.22787. [DOI] [PubMed] [Google Scholar]
- 14.Fong A.M., Robinson L.A., Steeber D.A., Tedder T.F., Yoshie O., Imai T., Patel D.D. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J. Exp. Med. 1998;188:1413–1419. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imai T., Hieshima K., Haskell C., Baba M., Nagira M., Nishimura M., Kakizaki M., Takagi S., Nomiyama H., Schall T.J., Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 16.Hyakudomi M., Matsubara T., Hyakudomi R., Yamamoto T., Kinugasa S., Yamanoi A., Maruyama R., Tanaka T. Increased expression of fractalkine is correlated with a better prognosis and an increased number of both CD8+ T cells and natural killer cells in gastric adenocarcinoma. Ann. Surg Oncol. 2008;15:1775–1782. doi: 10.1245/s10434-008-9876-3. [DOI] [PubMed] [Google Scholar]
- 17.Park M.H., Lee J.S., Yoon J.H. High expression of CX3CL1 by tumor cells correlates with a good prognosis and increased tumor-infiltrating CD8+ T cells, natural killer cells, and dendritic cells in breast carcinoma. J. Surg. Oncol. 2012;106:386–392. doi: 10.1002/jso.23095. [DOI] [PubMed] [Google Scholar]
- 18.Lim W.A., June C.H. The principles of engineering immune cells to treat cancer. Cell. 2017;168:724–740. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gross G., Waks T., Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA. 1989;86:10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuwana Y., Asakura Y., Utsunomiya N., Nakanishi M., Arata Y., Itoh S., Nagase F., Kurosawa Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987;149:960–968. doi: 10.1016/0006-291x(87)90502-x. [DOI] [PubMed] [Google Scholar]
- 21.Kershaw M.H., Westwood J.A., Parker L.L., Wang G., Eshhar Z., Mavroukakis S.A., White D.E., Wunderlich J.R., Canevari S., Rogers-Freezer L., et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernández L., Metais J.Y., Escudero A., Vela M., Valentín J., Vallcorba I., Leivas A., Torres J., Valeri A., Patiño-García A., et al. Memory T cells expressing an NKG2D-CAR efficiently target osteosarcoma cells. Clin. Cancer Res. 2017;23:5824–5835. doi: 10.1158/1078-0432.CCR-17-0075. [DOI] [PubMed] [Google Scholar]
- 23.Weiss T., Weller M., Guckenberger M., Sentman C.L., Roth P. NKG2D-Based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. 2018;78:1031–1043. doi: 10.1158/0008-5472.CAN-17-1788. [DOI] [PubMed] [Google Scholar]
- 24.Yang D., Sun B., Dai H., Li W., Shi L., Zhang P., Li S., Zhao X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J. Immunother. Cancer. 2019;7:171. doi: 10.1186/s40425-019-0642-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markiewicz M.A., Carayannopoulos L.N., Naidenko O.V., Matsui K., Burack W.R., Wise E.L., Fremont D.H., Allen P.M., Yokoyama W.M., Colonna M., Shaw A.S. Costimulation through NKG2D enhances murine CD8+ CTL function: similarities and differences between NKG2D and CD28 costimulation. J. Immunol. 2005;175:2825–2833. doi: 10.4049/jimmunol.175.5.2825. [DOI] [PubMed] [Google Scholar]
- 26.Markiewicz M.A., Wise E.L., Buchwald Z.S., Pinto A.K., Zafirova B., Polic B., Shaw A.S. RAE1epsilon ligand expressed on pancreatic islets recruits NKG2D receptor-expressing cytotoxic T cells independent of T cell receptor recognition. Immunity. 2012;36:132–141. doi: 10.1016/j.immuni.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meresse B., Chen Z., Ciszewski C., Tretiakova M., Bhagat G., Krausz T.N., Raulet D.H., Lanier L.L., Groh V., Spies T., et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. 2004;21:357–366. doi: 10.1016/j.immuni.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 28.Verneris M.R., Karimi M., Baker J., Jayaswal A., Negrin R.S. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood. 2004;103:3065–3072. doi: 10.1182/blood-2003-06-2125. [DOI] [PubMed] [Google Scholar]
- 29.Nausch N., Cerwenka A. NKG2D ligands in tumor immunity. Oncogene. 2008;27:5944–5958. doi: 10.1038/onc.2008.272. [DOI] [PubMed] [Google Scholar]
- 30.Antonangeli F., Soriani A., Cerboni C., Sciumè G., Santoni A. How mucosal epithelia deal with stress: role of NKG2D/NKG2D ligands during inflammation. Front. Immunol. 2017;8:1583. doi: 10.3389/fimmu.2017.01583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parihar R., Rivas C., Huynh M., Omer B., Lapteva N., Metelitsa L.S., Gottschalk S.M., Rooney C.M. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol. Res. 2019;7:363–375. doi: 10.1158/2326-6066.CIR-18-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trinh T.L., Kandell W.M., Donatelli S.S., Tu N., Tejera M.M., Gilvary D.L., Eksioglu E.A., Burnette A., Adams W.A., Liu J., et al. Immune evasion by TGFbeta-induced miR-183 repression of MICA/B expression in human lung tumor cells. OncoImmunology. 2019;8:e1557372. doi: 10.1080/2162402X.2018.1557372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donatelli S.S., Zhou J.M., Gilvary D.L., Eksioglu E.A., Chen X., Cress W.D., Haura E.B., Schabath M.B., Coppola D., Wei S., Djeu J.Y. TGF-beta-inducible microRNA-183 silences tumor-associated natural killer cells. Proc. Natl. Acad. Sci. USA. 2014;111:4203–4208. doi: 10.1073/pnas.1319269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller J.S., Rooney C.M., Curtsinger J., McElmurry R., McCullar V., Verneris M.R., Lapteva N., McKenna D., Wagner J.E., Blazar B.R., Tolar J. Expansion and homing of adoptively transferred human natural killer cells in immunodeficient mice varies with product preparation and in vivo cytokine administration: implications for clinical therapy. Biology of blood and marrow transplantation. Biol. Blood Marrow Transplant. 2014;20:1252–1257. doi: 10.1016/j.bbmt.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanamachi D.M., Moniot D.C., Cado D., Liu S.D., Hsia J.K., Raulet D.H. Genomic Ly49A transgenes: basis of variegated Ly49A gene expression and identification of a critical regulatory element. J. Immunol. 2004;172:1074–1082. doi: 10.4049/jimmunol.172.2.1074. [DOI] [PubMed] [Google Scholar]
- 36.Mortier E., Woo T., Advincula R., Gozalo S., Ma A. IL-15Ralpha chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J. Exp. Med. 2008;205:1213–1225. doi: 10.1084/jem.20071913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kennedy M.K., Glaccum M., Brown S.N., Butz E.A., Viney J.L., Embers M., Matsuki N., Charrier K., Sedger L., Willis C.R., et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med. 2000;191:771–780. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans R., Fuller J.A., Christianson G., Krupke D.M., Troutt A.B. IL-15 mediates anti-tumor effects after cyclophosphamide injection of tumor-bearing mice and enhances adoptive immunotherapy: the potential role of NK cell subpopulations. Cell. Immunol. 1997;179:66–73. doi: 10.1006/cimm.1997.1132. [DOI] [PubMed] [Google Scholar]
- 39.Fehniger T.A., Cai S.F., Cao X., Bredemeyer A.J., Presti R.M., French A.R., Ley T.J. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity. 2007;26:798–811. doi: 10.1016/j.immuni.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 40.Mao Y., van Hoef V., Zhang X., Wennerberg E., Lorent J., Witt K., Masvidal L., Liang S., Murray S., Larsson O., et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood. 2016;128:1475–1489. doi: 10.1182/blood-2016-02-698027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poznanski S.M., Ashkar A.A. Shining light on the significance of NK cell CD56 brightness. Cell. Mol. Immunol. 2018;15:1071–1073. doi: 10.1038/s41423-018-0163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okamura T., Kodama Y., Kamegawa T., Sano C., Kumashiro R., Inokuchi K. Gastric carcinoma with lymphoid stroma: correlation to reactive hyperplasia in regional lymph nodes and prognosis. Jpn. J. Surg. 1983;13:177–183. doi: 10.1007/bf02469473. [DOI] [PubMed] [Google Scholar]
- 43.Songun I., van de Velde C.J., Hermans J., Pals S.T., Verspaget H.W., Vis A.N., Menon A.G., Litvinov S.V., van Krieken J.H. Expression of oncoproteins and the amount of eosinophilic and lymphocytic infiltrates can be used as prognostic factors in gastric cancer. Dutch Gastric Cancer Group (DGCG) Br. J. Cancer. 1996;74:1783–1788. doi: 10.1038/bjc.1996.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jass J.R., Atkin W.S., Cuzick J., Bussey H.J.R., Morson B.C., Northover J.M.A., Todd I.P. The grading of rectal cancer: historical perspectives and a multivariate analysis of 447 cases. Histopathology. 2002;41:59–81. [PubMed] [Google Scholar]
- 45.Usubütün A., Ayhan A., Uygur M.C., Ozen H., Toklu C., Ruacan S. Prognostic factors in renal cell carcinoma. J. Exp. Clin. Cancer Res. 1998;17:77–81. [PubMed] [Google Scholar]
- 46.Lavin Y., Kobayashi S., Leader A., Amir E.A.D., Elefant N., Bigenwald C., Remark R., Sweeney R., Becker C.D., Levine J.H., et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169:750–765.e17. doi: 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toghi Eshghi S., Au-Yeung A., Takahashi C., Bolen C.R., Nyachienga M.N., Lear S.P., Green C., Mathews W.R., O'Gorman W.E. Quantitative comparison of conventional and t-SNE-guided gating analyses. Front. Immunol. 2019;10:1194. doi: 10.3389/fimmu.2019.01194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bi J., Tian Z. NK cell exhaustion. Front. Immunol. 2017;8:760. doi: 10.3389/fimmu.2017.00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castriconi R., Dondero A., Bellora F., Moretta L., Castellano A., Locatelli F., Corrias M.V., Moretta A., Bottino C. Neuroblastoma-derived TGF-beta1 modulates the chemokine receptor repertoire of human resting NK cells. J. Immunol. 2013;190:5321–5328. doi: 10.4049/jimmunol.1202693. [DOI] [PubMed] [Google Scholar]
- 50.Nishimura M., Umehara H., Nakayama T., Yoneda O., Hieshima K., Kakizaki M., Dohmae N., Yoshie O., Imai T. Dual functions of fractalkine/CX3C ligand 1 in trafficking of perforin+/granzyme B+ cytotoxic effector lymphocytes that are defined by CX3CR1 expression. J. Immunol. 2002;168:6173–6180. doi: 10.4049/jimmunol.168.12.6173. [DOI] [PubMed] [Google Scholar]
- 51.Yoneda O., Imai T., Goda S., Inoue H., Yamauchi A., Okazaki T., Imai H., Yoshie O., Bloom E.T., Domae N., Umehara H. Fractalkine-mediated endothelial cell injury by NK cells. J. Immunol. 2000;164:4055–4062. doi: 10.4049/jimmunol.164.8.4055. [DOI] [PubMed] [Google Scholar]
- 52.Lee M., Lee Y., Song J., Lee J., Chang S.Y. Tissue-specific role of CX3CR1 expressing immune cells and their relationships with human disease. Immune Network. 2018;18:e5. doi: 10.4110/in.2018.18.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kittang A.O., Kordasti S., Sand K.E., Costantini B., Kramer A.M., Perezabellan P., Seidl T., Rye K.P., Hagen K.M., Kulasekararaj A., et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. OncoImmunology. 2016;5:e1062208. doi: 10.1080/2162402X.2015.1062208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gabrilovich D.I., Ostrand-Rosenberg S., Bronte V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Engblom C., Pfirschke C., Pittet M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer. 2016;16:447–462. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 56.Bronte V., Brandau S., Chen S.H., Colombo M.P., Frey A.B., Greten T.F., Mandruzzato S., Murray P.J., Ochoa A., Ostrand-Rosenberg S., et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016;7:12150. doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu H., Wang S., Xin J., Wang J., Yao C., Zhang Z. Role of NKG2D and its ligands in cancer immunotherapy. Am. J. Cancer Res. 2019;9:2064–2078. [PMC free article] [PubMed] [Google Scholar]
- 58.Obeidy P., Sharland A.F. NKG2D and its ligands. Int. J. Biochem. Cell Biol. 2009;41:2364–2367. doi: 10.1016/j.biocel.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 59.Wensveen F.M., Jelenčić V., Polić B. NKG2D: a master regulator of immune cell responsiveness. Front. Immunol. 2018;9:441. doi: 10.3389/fimmu.2018.00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Böttcher J.P., Beyer M., Meissner F., Abdullah Z., Sander J., Höchst B., Eickhoff S., Rieckmann J.C., Russo C., Bauer T., et al. Functional classification of memory CD8(+) T cells by CX3CR1 expression. Nat. Commun. 2015;6:8306. doi: 10.1038/ncomms9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gerlach C., Moseman E.A., Loughhead S.M., Alvarez D., Zwijnenburg A.J., Waanders L., Garg R., de la Torre J.C., von Andrian U.H. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity. 2016;45:1270–1284. doi: 10.1016/j.immuni.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferretti E., Pistoia V., Corcione A. Role of fractalkine/CX3CL1 and its receptor in the pathogenesis of inflammatory and malignant diseases with emphasis on B cell malignancies. Mediators Inflamm. 2014;2014:480941. doi: 10.1155/2014/480941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamauchi T., Hoki T., Oba T., Saito H., Attwood K., Sabel M.S., Chang A.E., Odunsi K., Ito F. CX3CR1-CD8+ T cells are critical in antitumor efficacy but functionally suppressed in the tumor microenvironment. JCI insight. 2020;5:e133920. doi: 10.1172/jci.insight.133920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hay K.A., Turtle C.J. Chimeric antigen receptor (CAR) T cells: lessons learned from targeting of CD19 in B-cell malignancies. Drugs. 2017;77:237–245. doi: 10.1007/s40265-017-0690-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Savoldo B., Ramos C.A., Liu E., Mims M.P., Keating M.J., Carrum G., Kamble R.T., Bollard C.M., Gee A.P., Mei Z., et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X., Chang W.C., Wong C.W., Colcher D., Sherman M., Ostberg J.R., Forman S.J., Riddell S.R., Jensen M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Datar I., Sanmamed M.F., Wang J., Henick B.S., Choi J., Badri T., Dong W., Mani N., Toki M., Mejías L.D., et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin. Cancer Res. 2019;25:4663–4673. doi: 10.1158/1078-0432.CCR-18-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mhaidly R., Verhoeyen E. Humanized mice are precious tools for preclinical evaluation of CAR T and CAR NK cell therapies. Cancers. 2020;12:1915. doi: 10.3390/cancers12071915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harlin H., Meng Y., Peterson A.C., Zha Y., Tretiakova M., Slingluff C., McKee M., Gajewski T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goldstraw P., Chansky K., Crowley J., Rami-Porta R., Asamura H., Eberhardt W.E.E., Nicholson A.G., Groome P., Mitchell A., Bolejack V., et al. The IASLC lung cancer staging project: proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J. Thorac. Oncol. 2016;11:39–51. doi: 10.1016/j.jtho.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 71.Pignon J.P., Tribodet H., Scagliotti G.V., Douillard J.Y., Shepherd F.A., Stephens R.J., Dunant A., Torri V., Rosell R., Seymour L., et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J. Clin. Oncol. 2008;26:3552–3559. doi: 10.1200/JCO.2007.13.9030. [DOI] [PubMed] [Google Scholar]
- 72.Platonova S., Cherfils-Vicini J., Damotte D., Crozet L., Vieillard V., Validire P., André P., Dieu-Nosjean M.C., Alifano M., Régnard J.F., et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011;71:5412–5422. doi: 10.1158/0008-5472.CAN-10-4179. [DOI] [PubMed] [Google Scholar]
- 73.Ohta M., Tanaka F., Yamaguchi H., Sadanaga N., Inoue H., Mori M. The high expression of Fractalkine results in a better prognosis for colorectal cancer patients. Int. J. Oncol. 2005;26:41–47. [PubMed] [Google Scholar]
- 74.McComb J.G., Ranganathan M., Liu X.H., Pilewski J.M., Ray P., Watkins S.C., Choi A.M.K., Lee J.S. CX3CL1 up-regulation is associated with recruitment of CX3CR1+ mononuclear phagocytes and T lymphocytes in the lungs during cigarette smoke-induced emphysema. Am. J. Pathol. 2008;173:949–961. doi: 10.2353/ajpath.2008.071034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamin R., Lecker L.S.M., Weisblum Y., Vitenshtein A., Le-Trilling V.T.K., Wolf D.G., Mandelboim O. HCMV vCXCL1 binds several chemokine receptors and preferentially attracts neutrophils over NK cells by interacting with CXCR2. Cell Rep. 2016;15:1542–1553. doi: 10.1016/j.celrep.2016.04.042. [DOI] [PubMed] [Google Scholar]
- 76.Jung S., Aliberti J., Graemmel P., Sunshine M.J., Kreutzberg G.W., Sher A., Littman D.R. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geissmann F., Jung S., Littman D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 78.Palframan R.T., Jung S., Cheng G., Weninger W., Luo Y., Dorf M., Littman D.R., Rollins B.J., Zweerink H., Rot A., von Andrian U.H. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J. Exp. Med. 2001;194:1361–1373. doi: 10.1084/jem.194.9.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., Sosman J.A., McDermott D.F., Powderly J.D., Gettinger S.N., et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mussai F., Egan S., Hunter S., Webber H., Fisher J., Wheat R., McConville C., Sbirkov Y., Wheeler K., Bendle G., et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. 2015;75:3043–3053. doi: 10.1158/0008-5472.CAN-14-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spear P., Wu M.R., Sentman M.L., Sentman C.L. NKG2D ligands as therapeutic targets. Cancer Immun. 2013;13:8. [PMC free article] [PubMed] [Google Scholar]
- 82.Hüe S., Mention J.J., Monteiro R.C., Zhang S., Cellier C., Schmitz J., Verkarre V., Fodil N., Bahram S., Cerf-Bensussan N., Caillat-Zucman S. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21:367–377. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 83.Lehner M., Götz G., Proff J., Schaft N., Dörrie J., Full F., Ensser A., Muller Y.A., Cerwenka A., Abken H., et al. Redirecting T cells to Ewing's sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS One. 2012;7:e31210. doi: 10.1371/journal.pone.0031210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Song D.G., Ye Q., Santoro S., Fang C., Best A., Powell D.J., Jr. Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum. Gene Ther. 2013;24:295–305. doi: 10.1089/hum.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y., Lin G., Guo Z.Q., Zhou Z.F., He Z.Y., Ye Y.B. Effects of MICA expression on the prognosis of advanced non-small cell lung cancer and the efficacy of CIK therapy. PLoS One. 2013;8:e69044. doi: 10.1371/journal.pone.0069044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bagley S.J., Desai A.S., Linette G.P., June C.H., O'Rourke D.M. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro Oncol. 2018;20:1429–1438. doi: 10.1093/neuonc/noy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chang Y.H., Connolly J., Shimasaki N., Mimura K., Kono K., Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73:1777–1786. doi: 10.1158/0008-5472.CAN-12-3558. [DOI] [PubMed] [Google Scholar]
- 88.Barber A., Rynda A., Sentman C.L. Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J. Immunol. 2009;183:6939–6947. doi: 10.4049/jimmunol.0902000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Di Stasi A., De Angelis B., Rooney C.M., Zhang L., Mahendravada A., Foster A.E., Heslop H.E., Brenner M.K., Dotti G., Savoldo B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392–6402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moon E.K., Carpenito C., Sun J., Wang L.C.S., Kapoor V., Predina J., Powell D.J., Jr., Riley J.L., June C.H., Albelda S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011;17:4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Craddock J.A., Lu A., Bear A., Pule M., Brenner M.K., Rooney C.M., Foster A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Newick K., O'Brien S., Sun J., Kapoor V., Maceyko S., Lo A., Puré E., Moon E., Albelda S.M. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol. Res. 2016;4:541–551. doi: 10.1158/2326-6066.CIR-15-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meucci O., Fatatis A., Simen A.A., Miller R.J. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. USA. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cardona A.E., Pioro E.P., Sasse M.E., Kostenko V., Cardona S.M., Dijkstra I.M., Huang D., Kidd G., Dombrowski S., Dutta R., et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 95.Boehme S.A., Lio F.M., Maciejewski-Lenoir D., Bacon K.B., Conlon P.J. The chemokine fractalkine inhibits Fas-mediated cell death of brain microglia. J. Immunol. 2000;165:397–403. doi: 10.4049/jimmunol.165.1.397. [DOI] [PubMed] [Google Scholar]
- 96.Yu Y.R.A., Fong A.M., Combadiere C., Gao J.L., Murphy P.M., Patel D.D. Defective antitumor responses in CX3CR1-deficient mice. Int. J. Cancer. 2007;121:316–322. doi: 10.1002/ijc.22660. [DOI] [PubMed] [Google Scholar]
- 97.Burga R.A., Thorn M., Point G.R., Guha P., Nguyen C.T., Licata L.A., DeMatteo R.P., Ayala A., Joseph Espat N., Junghans R.P., Katz S.C. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer immunology, immunotherapy. Cancer Immunol. Immunother. 2015;64:817–829. doi: 10.1007/s00262-015-1692-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bollard C.M., Rössig C., Calonge M.J., Huls M.H., Wagner H.J., Massague J., Brenner M.K., Heslop H.E., Rooney C.M. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179–3187. doi: 10.1182/blood.v99.9.3179. [DOI] [PubMed] [Google Scholar]
- 99.Kopp H.G., Placke T., Salih H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009;69:7775–7783. doi: 10.1158/0008-5472.CAN-09-2123. [DOI] [PubMed] [Google Scholar]
- 100.Laouar Y., Sutterwala F.S., Gorelik L., Flavell R.A. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat. Immunol. 2005;6:600–607. doi: 10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- 101.Cao S., Troutt A.B., Rustum Y.M. Interleukin 15 protects against toxicity and potentiates antitumor activity of 5-fluorouracil alone and in combination with leucovorin in rats bearing colorectal cancer. Cancer Res. 1998;58:1695–1699. [PubMed] [Google Scholar]
- 102.Zhu X., Marcus W.D., Xu W., Lee H.I., Han K., Egan J.O., Yovandich J.L., Rhode P.R., Wong H.C. Novel human interleukin-15 agonists. J. Immunol. 2009;183:3598–3607. doi: 10.4049/jimmunol.0901244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu W., Jones M., Liu B., Zhu X., Johnson C.B., Edwards A.C., Kong L., Jeng E.K., Han K., Marcus W.D., et al. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor alphaSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res. 2013;73:3075–3086. doi: 10.1158/0008-5472.CAN-12-2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guil-Luna S., Sedlik C., Piaggio E. Humanized mouse models to evaluate cancer immunotherapeutics. Annu. Rev. Cancer Biol. 2021;5:119–136. doi: 10.1146/annurev-cancerbio-050520-100526. [DOI] [Google Scholar]
- 105.Shrestha B., Zhang Y., Yu B., Li G., Boucher J.C., Beatty N.J., Tsai H.C., Wang X., Mishra A., Sweet K., et al. Generation of antitumor T cells for adoptive cell therapy with artificial antigen presenting cells. J. Immunother. 2020;43:79–88. doi: 10.1097/CJI.0000000000000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data presented in this study will be shared upon reasonable request by the lead contact, Dr. Erika A. Eksioglu (Erika.eksioglu@moffitt.org).
-
•
This paper does not report any original code.
-
•
Additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.