

SUMMARY
The human fallopian tube harbors the cell of origin for the majority of high-grade serous “ovarian” cancers (HGSCs), but its cellular composition, particularly the epithelial component, is poorly characterized. We perform single-cell transcriptomic profiling of around 53,000 individual cells from 12 primary fallopian specimens to map their major cell types. We identify 10 epithelial subpopulations with diverse transcriptional programs. Based on transcriptional signatures, we reconstruct a trajectory whereby secretory cells differentiate into ciliated cells via a RUNX3high intermediate. Computational deconvolution of advanced HGSCs identifies the “early secretory” population as a likely precursor state for the majority of HGSCs. Its signature comprises both epithelial and mesenchymal features and is enriched in mesenchymal-type HGSCs (p = 6.7 × 10−27), a group known to have particularly poor prognoses. This cellular and molecular compendium of the human fallopian tube in cancer-free women is expected to advance our understanding of the earliest stages of fallopian epithelial neoplasia.
Graphical abstract
In brief
Using single-cell transcriptomic profiling of around 53,000 cells from human fallopian tubes, Dinh et al. identify SOX17 as a critical factor in fallopian epithelia and construct a differentiation trajectory involving a RUNX3+ intermediate. Early secretory epithelial cells are revealed to be a likely cell of origin for high-grade serous “ovarian” carcinomas.
INTRODUCTION
The fallopian tube (FT) serves as a conduit between the ovary and the uterus to allow the transport of the fertilized egg for implantation in the endometrium. It is derived from the Müllerian ducts, which also give rise to the uterus, cervix, and upper vagina (Healey, 2012). The FT is a dynamic organ that undergoes structural modifications in response to the menstrual cycle, menopause, and tubal ligation (Crow et al., 1994; Donnez et al., 1985). The FT is composed of three layers: the mucosa, a seromuscular layer, and the serosa. The mucosa consists of the lamina propria and the epithelium. Within the lamina propria resides a network of fibroblast-mesenchymal cells (Wang et al., 2015a) and immune cells. T cells represent the dominant population of lymphoid cells found in the FT, with CD8+ T cells being the prevalent cell type (Ardighieri et al., 2014). Neutrophils are the second most prevalent population of immune cells and are relatively more numerous in the FT compared to the rest of the female reproductive tract (Givan et al., 1997; Smith et al., 2006).
Three histologic cell types comprise the epithelium: ciliated; secretory; and intercalary cells. Ciliated cells are responsible for oocyte pickup, embryo transport, and tubal fluid flow. The intercalary cells are thought to be a population of epithelial stem-like cells, which may play a role in the initiation of serous tumors (Paik et al., 2012). The secretory cells comprise approximately 60% of the epithelial cells found in the FT and secrete growth factors, nutrients, antimicrobial agents, and anti-cellular stress agents into the tubal fluid. FT secretory epithelial cells (FTSECs) are of particular interest, as there is now evidence that the majority of high-grade serous “ovarian” cancers (HGSCs) originate from the secretory cells of the distal FT, with the recent development of a stepwise progression model starting from a p53 signature that progresses into serous tubal intraepithelial carcinoma (STIC) and finally to HGSC (Lee et al., 2007; Reade et al., 2014; Wang et al., 2015b). Secretory cell expansion and loss of ciliated cells are thought to be early events in the development of serous cancers (Wang et al., 2015b), but molecular drivers of these phenomena are unknown, as the differentiation path for tubal epithelia is poorly understood. Here, we employed droplet-based single-cell RNA sequencing (scRNA-seq) to map the cellular composition of the human FT. We identify epithelial subpopulations, including early and transitioning populations, and the critical transcriptional networks that define cellular populations in this organ. We then leverage the signatures of epithelial subsets to identify putative cells of origin for HGSCs.
RESULTS
Single-cell transcriptomic analyses of normal human FTs
We performed scRNA-seq of 12 human FT tissue specimens from 8 cancer-free individuals (Figures 1A and 1B). The phenotypic and clinical characteristics are summarized in Table 1. Three specimens were from the distal, fimbriated region, where the tube is in closest proximity to the ovary; the remaining nine specimens were from mid-portion of the organ. In one patient, we separately profiled matched ampulla, infundibulum, and fimbrial portions of the FT. Patient age ranged from 31 to 62 years; all patients, except patient 8, were premenopausal. The interval between onset of last menstrual period to surgery ranged from 3 to 31 days. All patients in the cohort were undergoing gynecological surgeries for benign conditions, including leiomyoma (7 patients), adenomyosis (3 patients), and endometriosis (2 patients). Two patients were parous (each with two live births). None of the patients were known or suspected to be carriers of any inherited germline genetic abnormalities associated with hereditary cancer predisposition syndromes. In total, we profiled 64,042 single cells isolated from the 12 specimens. We then excluded cells with ≤200 genes expressed and/or high mitochondrial RNA content (mtRNA ≥ 10%). Potential doublets were removed on a sample-wise basis based on the number of captured cells. After this filtering, a total of 52,908 cells remained for analysis. The average number of genes surveyed was 1,083 per cell (range: 709–1,636), and the average number of reads per cell was 96,166 (range: 31,578–259,280).
Figure 1. A cellular gene expression atlas of human fallopian tubes in cancer-free women.

(A) Schematic showing overall study design.
(B and C) UMAP plot showing the major cellular clusters (B) by patient and (C) by cell type.
(D) Scaled expression level of cell-type-defining markers and percentage of positive cells in each cluster.
(E) UMAP plots overlaying cluster-specific expression patterns of representative cell-type-defining markers.
(F) Frequency of cells in each cluster, by patient.
(G) UMAP plots for 2 specimens before and after cryopreservation.
(H) Violin plots of representative marker expression in 2 specimens, before and after cryopreservation. UMAP, uniform manifold approximation and projection.
Table 1.
Sample cohort
| Additional annotation | Age at time of surgery | Reason for surgery | Gravida | Para | Days since LMPa | No. cells analyzed | Mean reads per cell | Median genes per cell |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Patient 1 | ||||||||
|
| ||||||||
| mid-portion | 47 | symptomatic fibroids, adenomyosis, endometriosis | 1 | 0 | 3 | 1,614 | 259,280 | 1,636 |
|
| ||||||||
| Patient 2 | ||||||||
|
| ||||||||
| mid-portion | 35 | symptomatic fibroids | 0 | 0 | 18 | 3,229 | 153,799 | 1,313 |
|
| ||||||||
| Patient 3 | ||||||||
|
| ||||||||
| ampulla | 47 | symptomatic fibroids | 3 | 2 | 17 | 2,873 | 203,866 | 1,032 |
|
| ||||||||
| Patient 4 | ||||||||
|
| ||||||||
| mid-portion | 41 | symptomatic fibroids, adenomyosis, endometriosis | 2 | 0 | 7 | 2,448 | 62,427 | 709 |
|
| ||||||||
| Patient 5 | ||||||||
|
| ||||||||
| mid-portion (left) | 33 | symptomatic fibroids | 0 | 0 | 31b | 3,063 | 81,873 | 882 |
| mid-portion (right) | 33 | symptomatic fibroids | 0 | 0 | 31b | 3,129 | 44,293 | 784 |
|
| ||||||||
| Patient 6 | ||||||||
|
| ||||||||
| ampulla | 31 | uterine didelphys | 0 | 0 | 13 | 10,764 | 36,015 | 967 |
| infundibulum | 31 | uterine didelphys | 0 | 0 | 13 | 11,572 | 31,578 | 936 |
| fimbriae | 31 | uterine didelphys | 0 | 0 | 13 | 8,343 | 54,001 | 1,596 |
|
| ||||||||
| Patient 7 | ||||||||
|
| ||||||||
| mid-portion | 46 | symptomatic fibroids | 3 | 2 | 23 | 4,498 | 84,674 | 1,048 |
| fimbriae | 46 | symptomatic fibroids | 3 | 2 | 23 | 7,173 | 50,127 | 967 |
|
| ||||||||
| Patient 8 | ||||||||
|
| ||||||||
| fimbriae | 62 | symptomatic fibroids, endometrial polyps | 2 | 0 | NAc | 5,336 | 92,059 | 1,124 |
Interval between start of last menstrual period (LMP) and date of surgery
Patient-reported regular 35-day cycles
Not applicable, patient is post-menopausal
These data were integrated and batch corrected using canonical correlation-based alignment analysis implemented in the Seurat package (Stuart et al., 2019). Using a Louvain-based clustering method, we detected 13 major cellular populations based on well-defined markers (STAR Methods; Figures 1D and 1E; Table S1). In the immune microenvironment of the tube, the major populations were lymphoid T cells, including CD4+ and CD8+ T cells, and natural killer cells. Myeloid cells (monocytes, macrophages, dendritic cells, and mast cells), B cells, and plasma cells were detected at lower frequencies. We also detected a large population of fibroblasts/smooth muscle cells and a small population of endothelial cells. Epithelial cells formed two distinct clusters: one composed of PAX8/OVGP1-expressing secretory epithelial cells and the other consisting of FOXJ1+ ciliated epithelia (Figures 1C–1E). All major clusters were detected in each of the specimens analyzed (albeit at different frequencies; Figure 1F). To determine the impact of cryopreservation on FT tissues, cells from two specimens from patients 6 and 7 were viably frozen in 10% DMSO/90% fetal bovine serum and then thawed around 6 months later and subjected to a second round of capture and sequencing. Gene signatures from the 13 major cell types present in the fresh tissue profiles were used as a reference to predict the cell type labels in the two frozen samples. Even though lower cell numbers were captured in these specimens compared to the fresh primary tissues, we were able to recover all the major cell types after cryopreservation (Figures 1G and S1). All the major immune cellular populations were present in the repeat sample, T cells were the most conserved, and myeloid cells (monocytes, macrophages, and dendritic cells) were depleted from cryopreserved samples. Epithelial cells and fibroblasts, although present, were also depleted following cryopreservation (Figure S1A), although, reassuringly, expression of genes in fresh and frozen epithelial cells was highly positively correlated (r = 0.94; p < 2.2 × 10−16; Pearson correlation test; Figure S1B), although we note that only 90 epithelial cells were present in the post-cryopreservation samples. Expression of key cell-type-defining markers was stable following thawing (Figure 1H). Taken together, these results indicate that freeze-thaw cycles do not significantly impact the expression profile of FT cells, albeit changing the cell-type frequencies, and that lymphocytes within dissociated FT tissues are particularly resilient to cryopreservation.
We asked whether scRNA-seq could reveal transcription factors (TFs) associated with major cell types in the FT by implementing the SCENIC pipeline (Aibar et al., 2017). SCENIC integrates co-expression network analysis and motif-based analyses to identify TFs and downstream genes (called regulons) that characterize the distinct transcriptional programs that define the major cell clusters (Figures 2A and S2). Transcriptional signatures of fibroblasts were enriched for multiple regulons, including KLF4, EGR1, GATA2, and TCF4. Epithelial factors included SOX17, BARX2, and BHLHE41 in secretory epithelial cells, plus KLF5, TTF3, and RFX3 in ciliated epithelial cells. We recently identified SOX17 as a master regulator in HGSCs (Reddy et al., 2019), but SOX17 is not known to be a marker in physiologically normal FT epithelial cells. To validate this finding, we performed immunohistochemistry for SOX17 on 14 FTs from cancer-free women; strong SOX17 staining was observed in all specimens and was independent of age, menopausal status, and BRCA1/2 mutation status (Figure 2B; Table S2). Evaluation of cytologic features, including cell shape, amount of cytoplasm, nuclear morphology, and the presence or absence of cilia, were used to determine differentiation state of the epithelia. Ciliated cells were identified by their polygonal shape, abundant cytoplasm, round nuclei, and the presence of apical cilia. Secretory cells were recognized by their columnar shape and scant cytoplasm, oval nuclei, and absence of apical cilia. All 14 FTs examined showed heterogeneous staining suggestive of weaker SOX17 nuclear staining in ciliated cells compared to secretory epithelial cells in at least some portion of the tissue. In 6 of the 14 cases examined, SOX17 staining intensity was greater in secretory cells compared to ciliated cells in the majority of the tubal epithelium (Figure 2B), consistent with the greater activation of the SOX17 regulon observed in secretory cells compared to ciliated cells in the SCENIC analyses (Figure 2A). The extent to which SOX17 was differentially expressed between ciliated and secretory cells did not correlate with age, menopausal status, or germline mutation status; however, we were underpowered to detect anything other than very strong associations. Additional analyses in larger cohorts, with tissues co-stained for secretory and ciliated cell markers, will be required to comprehensively describe the heterogeneous patterns of SOX17 staining in the human FT.
Figure 2. Transcription factor analyses in human FT cell populations.

(A) Identification of TF regulons enriched in specific cell types using SCENIC (t value from a linear model test of regulon activity score of one subset versus the rest; see STAR Methods). Numbers in brackets denote the number of genes contained in the regulon; “extended” means the regulons include motifs that have been linked to the TF using motif similarity from SCENIC pipeline.
(B) Validation of SOX17 expression in human FT tissues using immunohistochemistry. In all specimens examined, staining was suggestive of stronger staining in secretory cells, distinguished here by their oval nuclei compared to the rounded nuclear morphology of ciliated cells.
Epithelial components of the FT
As FT epithelial cells, particularly the secretory epithelial cells, are the likely precursors of most HGSCs (Karst et al., 2011; Lee et al., 2007; Perets et al., 2013; Reade et al., 2014; Wang et al., 2015b), we focused on defining the epithelial landscape of the FT. Epithelial cell populations were identified by expression of well-defined markers epithelial cell adhesion molecule (EPCAM), keratin genes (KRT7, KRT8, and KRT18), OVGP1, and FOXJ1; cells expressing at least one of these markers were annotated as epithelial. Clustering was performed on 4,592 epithelial cells (8.7% of the total cell population) using PAX8/OVGP1 and FOXJ1 as defining markers of secretory and ciliated epithelial cells, respectively. Integration analysis stringently required each epithelial subset to represent at least 2% of cells in at least 3 samples. Using these criteria, we identified a total of 10 epithelial cell clusters, which we categorized as PAX8high and OVGP1high secretory epithelial cells (3 clusters; secretory 1–3); FOXJ1high ciliated epithelial cells (4 clusters; ciliated 1–4); and three clusters that lowly expressed PAX8, OVGP1, and FOXJ1 and could not therefore be classified as secretory or ciliated (unclassified 1–3; Figure 3A). Across the 10 epithelial subpopulations, there was extensive heterogeneity in the expression of known FT epithelial markers. EPCAM was highly expressed in secretory cluster 2 and ciliated clusters 1, 2, and 4 but was low/absent in the remaining clusters. Secretory clusters 2 and 3 and ciliated cluster 2 expressed high levels of low-molecular-weight keratins 7, 8, and 18 (Figure 3B). Unclassified cluster 3, secretory clusters 2 and 3, and ciliated 4 expressed KRT10; KRT17 was expressed in unclassified 3, secretory cluster 2, and ciliated cluster 2. Overall, ciliated epithelial cells were more abundant than secretory cells (47.3%, range 11.4%–83.6% for ciliated epithelial cells, versus 38.4%, range 3.4%–84.2% for secretory epithelial cells), although this difference was not significant (p = 0.31; two-tailed paired t test; Figure 3C). Secretory clusters 2 and 3 and ciliated clusters 1, 2, and 4 each comprised ~10% of the epithelial cells captured. Cells from all samples contributed to each cluster (Figure S3).
Figure 3. Epithelial cell subpopulations in the human fallopian tube.

(A) UMAP representation of 10 distinct epithelial clusters revealed by graph-based Louvain clustering implemented in the Seurat single-cell data analysis package.
(B) Dot plot presentation of scaled expression of epithelial, secretory, and ciliated cell markers.
(C) Frequency histogram for the 10 epithelial clusters.
(D) Pathway analyses performed using the ReactomePA R package. All pathways with p < 0.05 are shown. Secretory cluster 2 has zero enriched pathways reaching p < 0.05.
(E–G) Expression of (E) dynein protein-encoding genes, (F) ATP synthase 5 subunit genes, and (G) genes encoding cilia proteins.
We performed differential gene expression analyses within each epithelial cell subtype to identify differentially expressed genes across the subpopulations of fallopian epithelial cells (average log2 fold change [log2FC] > 0.5; false discovery rate [FDR] adjusted p < 0.01; 25% of cells within the cluster expressing the gene of interest; Table S3). FTSEC clusters were defined by higher expression of PAX8 and OVGP1 compared to ciliated epithelial cells (Figure 3B). When we surveyed the expression of known biomarkers, secretory clusters 2 and 3 exhibited the highest expression of PAX8, KRT7, and ESR1, and clusters 1 and 2 expressed relatively higher levels of OVGP1 and WFDC2 (which encodes ovarian cancer biomarker HE4) compared to cluster 3. FTSECs expressed high levels of less-well-known markers, including cysteine-rich secretory protein 3 (CRISP3)—a protein that has been implicated in epithelial cell proliferation and adhesion in the endometrium (Evans et al., 2015), but not in the FT; secretory leukocyte protease inhibitor (SLPI), expressed by HGSCs (Cancer Genome Atlas Research Network, 2011); mesothelin (MSLN), a proposed target for immunotherapy in ovarian cancer (Haas et al., 2019); and secreted chaperone clusterin (CLU) (Figure 3B). Secretory cluster 1 was the most abundantly represented epithelial population in our dataset, with, on average, 23.2% ± 19.0% of the epithelial cells in each specimen assigned to this cluster (range 1.1%–56.5%). This cluster was defined by 129 genes that were strongly enriched in myriad pathways associated with translation elongation (adjusted p = 3.8 × 10−155; Table S4), reflecting the high secretory activity of these cells (Figure 3D). Similar pathways were enriched in the genes that defined secretory cluster 3 (on average 2.8% of cells per sample), albeit with more modest p values. This cluster expressed KRT17, which was absent in clusters 1 and 2 and likely corresponds to the KRT17+ secretory cluster (C4) recently described in tubal epithelium from women with gynecologic cancers (Hu et al., 2020).
Ciliated clusters 1, 2, and 4 consisted of differentiated ciliated cells (Figure 3D; Table S4). Cluster 3 highly expressed genes encoding dynein components (DYNLT1, DNALI1, and DYNLL1; Figure 3E) and genes associated with energy production, with six subunits of mitochondrial ATP synthase 5 overexpressed (Figure 3F). Cluster 3 expressed some genes encoding ciliaand flagella-associated proteins (CFAP53 and CFAP126), but other genes encoding cilia- and flagella-associated proteins were absent/lowly expressed (CFAP157, CFAP73, CFAP52, and CFAP126; Figure 3G), suggesting the cells in this cluster are not yet fully differentiated into a ciliated phenotype. In contrast, ciliated cluster 4 represented differentiated ciliated epithelial cells and expressed high levels of genes encoding dynein components in addition to genes encoding cilia-associated proteins (Figure 3G). Pathways preferentially enriched in ciliated cluster 2 included cellular responses to stress (adjusted p = 7.02 × 10−5), senescence (adjusted p = 0.008), and p53 regulated cell death (adjusted p = 0.04; Figure 3E; Table S4). In this cluster, suppressor of cytokine signaling 3 (SOCS3) was specifically expressed at high levels; this is a cytokine-inducible gene that negatively regulates cytokine signaling (Figure 3H) that may be involved in senescence of terminally differentiated ciliated cells (Mallette et al., 2010).
Reconstructing differentiation trajectories in the human FT
Pseudotime analysis was performed using Monocle2 to order epithelial cells based on expression patterns (Qiu et al., 2017). This analysis constructed a cellular trajectory whereby secretory cells differentiate into ciliated cells via unclassified clusters 2 and 3 (Figures 4A and S4A). Pseudotime estimations obtained using Monocle2 were positively correlated with those from an alternative tool, Phenopath (Pearson correlation coefficient r = 0.35; p < 2.2 3 10−16; Figure S4B; Campbell and Yau, 2018). Unclassified cluster 1 and secretory cluster 3 were enriched at the earliest time points and may represent putative progenitor cells and/or committed cells early in the differentiation process, before progressing into secretory cluster 2 and then 1. Unclassified cluster 1 exhibited low expression of all of the genes that define ciliated and secretory epithelial cells (Figure 3B) and expressed CD44 and EPCAM (Figure 4B), known features of tissue stem cells in many tissue types, including the FT (Paik et al., 2012). This cluster was defined by a set of 186 genes that were enriched in pathways associated with the extracellular matrix and responses to growth factors (Figure 3E). We searched for cell surface markers unique to this population and identified THY1, which encodes CD90 (Figure 4B). We validated the EPCAMhi/CD44hi/THY1hi population in an independent fallopian tissue specimen using flow cytometry. Single cells were isolated using the sample protocol implemented for scRNA-seq. CD45+ cells were depleted before performing flow cytometry with fluorescently tagged antibodies targeting EPCAM, CD44, or THY1. A triple-positive population comprised 4.7% of the EPCAM+ cells in this specimen (Figure 4C). In the scRNA-seq, this cluster also expressed high levels of IGFBP5, LGALS1 (which encodes galectin-1), and ACTA2 (which encodes smooth muscle actin; Figure 4B) and was the cluster with the highest expression of ESR1 (encoding the estrogen receptor; Figure 3B).
Figure 4. A differentiation trajectory for fallopian tube epithelia.

(A) Pseudotime analyses performed using discriminative dimensionality reduction with trees (DDRTree) implemented in the Monocle2 package. The upper portion of the panel shows inferred pseudotime, divided into 100 bins, from early (left) to late (right). Proportion of cells from each cluster assigned to each bin are shown below. The lower panel shows expression of known and newly identified biomarkers across the pseudotime trajectory identified using a differential expression test (linear model of pseudotime) in Monocle2.
(B) Expression of CD44, EPCAM, and markers enriched in unclassified cluster 1.
(C) Validation of the presence of unclassified cluster 1 in a fallopian tube tissue using flow cytometry.
(D) Transcription factor regulon analyses performed using SCENIC.
(E) Correlation of t values from linear model test contrasting 1 subset versus the other 9 subsets. Color of dot denotes direction of correlation (Spearman correlation test); size denotes correlation coefficient.
(F) UMAP plots showing expression of EPCAM and secretory/ciliated cell genes. Inset panels show a gradient of PAX8, OVGP1m and FOXJ1 expression in unclassified clusters 2 and 3.
(G) RUNX3 expression is restricted to unclassified clusters 2 and 3.
(H) Violin plots of RUNX3 and CAPS expression.
(I) RUNX3 expression in a benign human fallopian tube. Tissues were co-stained with anti-RUNX3 (brown chromogen) and anti-CD45 (green chromogen). Dual-positive intraepithelial lymphocytes with rounded nuclei (left inset panel) were detected, in addition to RUNX3+ epithelial cells (arrows and right inset panel).
(J) Floating boxplots showing RUNX3 expression after PAX8, SOX17, or WT1 knockdown. Upper and lower boundaries of the box denote maximum and minimum expression values; horizontal line denotes mean. TPM, transcripts per million; normalized RNA-seq reads. n = 2 independent small interfering RNA (siRNA) transfections. * |log FC| > 1; p = 0.02; negative binomial regression performed using DESeq2.
Transcriptional regulation network analyses, again performed using the SCENIC pipeline, highlighted the NR2F2, EMX2, and MYC regulons as enriched in the early secretory unclassified 1 subset. Each of these factors has been previously implicated in stem/progenitor cell biology (Figure 4D; Cheng et al., 2010; Kozak et al., 2020; Takahashi and Yamanaka, 2006). These regulons were highly specific to this early, partially differentiated state. We observed a strong inverse correlation between TF regulons enriched in differentiated ciliated cells (ciliated cluster 2) and differentiated secretory cells (secretory cluster 1; Spearman’s correlation coefficient = −0.87; Figure 4E) and across the secretory and ciliated subsets, a pattern of TF regulon activity that mirrored the pseudotime trajectory. Unclassified cluster 2, which was located at the interface of secretory and ciliated cell phenotypes, showed high activity of the RUNX3 cistrome (Figure 4D). The RUNX3 regulon was also activated, albeit to a lesser extent, in unclassified cluster 3 and ciliated cluster 3. Unclassified cluster 2 expressed a gradient of decreasing OVGP1 and increasing FOXJ1 along the pseudotime trajectory and likely represents a population of cells undergoing the transition from a secretory to a ciliated phenotype (Figure 4F). On average, 8.0% of epithelial cells were assigned to this cluster (standard deviation [SD] = 5.3%; range: 0.0%–15.4%). Unclassified cluster 3 comprised rare cells representing, on average, 1.8% of all epithelial cells in each specimen (SD = 1.5; range: 0.2%–5.2%). This cluster was characterized by decreasing expression of OVGP1 with mitotic pathways and genes strongly enriched (Figures 3E and S4C), even though cell cycle phase scores were regressed out during pre-processing to minimize the impact of cell cycle heterogeneity on cluster calls. This cluster likely corresponds to cluster C9 from Hu et al. (2020). Cells in this cluster appeared to represent an early transitioning cell population at the point of losing OVGP1 expression. Unclassified cluster 2 has begun to acquire some features of ciliated cells, including CAPS expression (Figure 4G) and then the pathway for ciliated differentiation progresses from ciliated 3 (which expresses dynein genes but few cilia genes) into ciliated clusters 1 and 4—two clusters of fully differentiated ciliated cells. Ciliated cluster 2, which comprises differentiated ciliated cells undergoing cell death and senescence, represents the endpoint in the differentiation trajectory (Figures 3D and 4A; Table S4).
Consistent with the RUNX3 regulon being specifically enriched in transitioning unclassified clusters 2 and 3, RUNX3 gene expression was unique to these clusters (Figures 4D and 4F). Immunohistochemistry was performed on eight FT specimens from six women to validate the presence of RUNX3+ epithelial cells, with dual staining for CD45 included with RUNX3 to enable exclusion of lymphocytes and to serve as an internal control. In 2 out of 8 specimens, RUNX3 epithelial cells were observed, albeit at a low frequency (Table S5). Discrete RUNX3+ epithelial cells were located in close proximity to the basement membrane (Figure 4H). TFs that characterize FT secretory epithelial cells are PAX8 (Levanon et al., 2010; Ozcan et al., 2011), SOX17 (this study; Reddy et al., 2019), and WT1, a clinically used biomarker of HGSC (Köbel et al., 2008; Shimizu et al., 2000). To understand the relationship of RUNX3 to these major secretory cell TFs, we transiently knocked down PAX8, SOX17, and WT1 in normal FT epithelial cells (FT282) and a HGSOC cell line (OVCAR4). RUNX3 expression was almost undetectable in the FT282 cells, but following depletion of PAX8, RUNX3 expression increased 3.0-fold (p = 0.02; negative binomial regression performed using DE-Seq2; Figures 4I and S4D–S4F). RUNX3 expression was not significantly altered following depletion of SOX17 or WT1 (log2 FC = 0.78, p = 0.67; FC = 2.3, p = 0.14). Knockdown of PAX8 and SOX17 in the OVCAR4 cell line modestly downregulated RUNX3 expression (log2 FC = −0.66, p = 8.50 3 10−7; log2 FC = −0.73, p = 1.29 3 10−7), whereas WT1 depletion led to a moderate increase in RUNX3 expression (log2 FC = 0.46; p = 1.6 3 10−4; Figure 4H). We note that the expression changes observed in the OVCAR4 tumor model did not reach the threshold of absolute log2 FC >1 typically applied to identified differentially expressed genes in RNA-seq data. This suggests that, in the normal physiological state, PAX8 inhibits RUNX3 expression but that this regulation is attenuated during tumorigenesis.
Early secretory cell signatures are enriched in ex vivo FT cultures and in HGSCs
We derived signatures of the 10 epithelial clusters, combining secretory clusters 2 and 3 and ciliated clusters 1, 3, and 4 due to overlap in the signature genes (Figure 5A). Informed by the characteristics described above, we re-annotated “unclassified cluster 1” as “early secretory,” and unclassified clusters 2 and 3 were denoted as “transitioning” clusters. Using the MuSiC method (Wang et al., 2019) with our combined signatures (see STAR Methods), 6 epithelial signatures were used to deconvolute bulk RNA-seq profiles of 68 FTSEC primary cultures (Lawrenson et al., 2019) to infer the cell subpopulations that preferentially grow in vitro. In 25 out of 68 cultures (39.4%), the early secretory signature was strongly enriched (at least top 25% percentile of estimated frequency, i.e., frequency ≥ 0.7). A subset of 21 samples with higher expression of OVGP1 were most enriched for the differentiated secretory cell signature (top 25% percentile; i.e., frequency > 0.34; Figure 5B). Signatures of secretory cluster 1 and ciliated cells were poorly enriched overall. To see whether this pattern is maintained after cellular immortalization with ectopic expression of TERT and mutant TP53, we applied cell type prediction via the label transfer function implemented in Seurat to map cellular identities from all 10 epithelial subsets in scRNA-seq profiles for the FT282 cell line (Karst et al., 2014; Lawrenson et al., 2019). Consistent with the results seen for the ex vivo cultures, in this immortalized cell line, the majority of cells were labeled as early secretory (705 out of 1,690 total cells; 41.7%; Figure S5). These data indicate that early secretory cells preferentially grow in vitro or that in vitro culture results in a partial dedifferentiation of secretory epithelial cells that is maintained following immortalization. This is consistent with previous observations that ciliated cells are lost in in vitro cultures of fallopian epithelia (Levanon et al., 2010; Lawrenson et al., 2019) and that, in general, cells often dedifferentiate during adaptation to the stresses of in vitro culture (Vilas-Boas et al., 2019).
Figure 5. Signatures of early secretory epithelia are enriched in fallopian epithelial cultures and HGSCs.

(A) Development of epithelial subset signatures informed by pseudotime analyses; subsets with similar profiles of marker genes were combined for computational deconvolution analysis.
(B) Deconvolution of bulk RNA-seq profiles of 68 ex vivo FTSEC cultures: heatmap of the composition of 6 subsets in each sample. A subset of patients had diagnoses of high-grade adenocarcinomas of the ovary, endometrium, or cervix.
(C) Deconvolution of 394 HGSCs from The Cancer Genome Atlas (TCGA).
(D) The NR2F2 regulon is dysregulated in HGSCs compared to FTSECs. Top differentially expressed genes were identified by calculating |(mean HGSC expression – mean FTSEC expression)| > 2. On all heatmaps, rows and columns are ordered using unsupervised hierarchical clustering based on Euclidian distances.
The six signatures were then applied to 394 HGSC RNA-seq profiles generated by The Cancer Genome Atlas (TCGA). Unsupervised clustering of tumors based on the deconvoluted cell-type composition identified five clusters of tumors with similar composition (Figure 5C). As with the normal FTSEC cultures, the early secretory signature was the most strongly enriched in the majority of tumors (cluster 2; 204 out of 394 cases) and was particularly enriched in tumors of the mesenchymal molecular subgroup (89/92 mesenchymal samples were in cluster 2; p = 6.7 3 10−27; hypergeometric test; Table S6; Cancer Genome Atlas Research Network, 2011; Tothill et al., 2008). Clusters 1 and 3 were characterized by tumors exhibiting the differentiated secretory 2+3 signature, respectively; both were enriched with the differentiated molecular subgroup (p = 1.4 3 10−7 and p = 1.2 3 10−5, respectively; hypergeometric test). Tumors in cluster 4 were enriched for the transitioning cell signature, and the immunoreactive TCGA subtype was overrepresented in this cluster (p = 1.6 3 10−18; hypergeometric test). Cluster 5 was composed of tumors with signatures of secretory cluster 1 and was enriched in the proliferative molecular subtype (p = 0.01). In sum, the gene signatures of epithelial subsets were enriched within each TCGA molecular subtypes, and the computational deconvolution highlights the association of those gene signatures with established molecular subtypes.
Finally, because the NR2F2 regulon was activated in the early secretory cluster (Figure 5D), we tested whether this regulon was dysregulated during tumorigenesis using our dataset of 68 normal FTSEC ex vivo cultures integrated with 394 HGSCs from TCGA (Lawrenson et al., 2019). Ninety-three genes from the NR2F2 regulon were expressed in our dataset (Figure S6). Unsupervised clustering based on expression of these genes clearly separated tumor cells from normal cells, with dysregulation of genes involved in negative regulation of cell proliferation (q = 0.001), such as AR, EPAS1, and SPARCL1, and regulation of cell-substrate adhesion, including LAMA4, CCL21, and BCAM (Figures 5D and S6).
DISCUSSION
Single-cell gene expression analyses bridge the interface of cell biology, pathology, and gene regulation. These analyses are transforming our understanding of intra-tissue heterogeneity in development, normal tissue homeostasis, and disease alike (Cao et al., 2019; Wang et al., 2020). Using scRNA-seq performed with the 10X Genomics platform, we generated a cellular atlas of the human FT in cancer-free women and identified the transcriptional networks driving the expression programs of 13 major cell types. Although the FT is located internally, the genital tract is exposed to the external environment and thus a complex network of lymphocytes, macrophages, and dendritic cells to control invading pathogens. The immune landscape of this organ was dominated by T cells, which were largely composed of CD8+ T cells, consistent with previous measurements by immunohistochemistry (IHC) (Ardighieri et al., 2014). Reproductive tract T cells have variable cytolytic capacity throughout the menstrual cycle, suggesting their function is hormonally regulated (White et al., 1997). The phenotype and function of FT T cells, particularly during tumorigenesis, will be of great interest for designing effective immunotherapies. Although previous work using IHC found CD68+ macrophages to be as abundant as CD8+ T cells in 10 non-cancerous FT samples, we measured a very low frequency (1.4% of leukocytes) of macrophages using scRNA-seq. This difference could be attributed to challenges in extracting macrophages from the extracellular matrix during sample preparation (Givan et al., 1997). Prior work using flow cytometry also identified neutrophils as one of the predominant leukocyte subtypes in healthy FTs (Givan et al., 1997; Smith et al., 2006), but IHC was unable to detect substantial numbers of extravascular neutrophils (Ardighieri et al., 2014). Similarly, in our analysis, we did not detect neutrophils, although they are generally challenging to capture in scRNA-seq data, probably due to their tendency to rapidly undergo apoptosis ex vivo and low RNA yield.
We identified 10 epithelial subpopulations present in the FT and projected a cellular trajectory based on the transcriptional signatures of each cluster (Figure 6). The model for differentiation pathway for tubal epithelia was challenged in recent years with the demonstration that, rather than secretory and ciliated cells deriving from a bipotent progenitor, in murine models, all FT epithelia derive from a unipotent PAX8+ precursor, with secretory cells giving rise to ciliated cells (Ghosh et al., 2017). Our data provide evidence that this model holds true in human tissues. We identified two clusters that were enriched early in the epithelial cellular trajectory and appear to correspond to partially differentiated, late progenitor populations (Snegovskikh et al., 2014). The first was KRT17+ secretory cluster 3 and the second the early secretory cluster (former unclassified cluster 1), which cluster was characterized by EPCAMhi, CD44hi, and THY1hi expression. The early secretory cluster expressed both epithelial (EPCAM and KRT19) and mesenchymal (THY1 and DCN) characteristics as well as smooth muscle actin and estrogen receptor and thus bore some similarities to myoepithelial cells of the uterus. This population expresses IGFBP5 and LGALS1, genes implicated in homeostasis of stem and/or progenitor populations in normal and diseased tissues (Byrd et al., 2019; Garcés et al., 2020; Tumbar et al., 2004; You et al., 2018), and MEG3, an imprinted long noncoding RNA. Previous reports have identified an enrichment of stem cells in the fimbrial portion of the tube (Paik et al., 2012). In our one patient with matched ampulla, infundibulum, and fimbrial tissue, the proportion of THY1+ cluster cells was highest in the fimbrial portion of the tissue (10% versus <3% in the mid- and proximal regions). The specimen with the highest proportion of THY1 early secretory cluster came from an individual with a diagnosis of endometriosis. Endometriosis was not observed in the portion of the tissue that was subjected to pathology review, although we cannot rule out the presence of microscopic foci of endometriosis in the segment of tissue subjected to scRNA-seq. Nonetheless, it is plausible that the altered pelvic microenvironment associated with endometriosis may impact epithelial differentiation in the FT. Larger studies will be required to test these hypotheses.
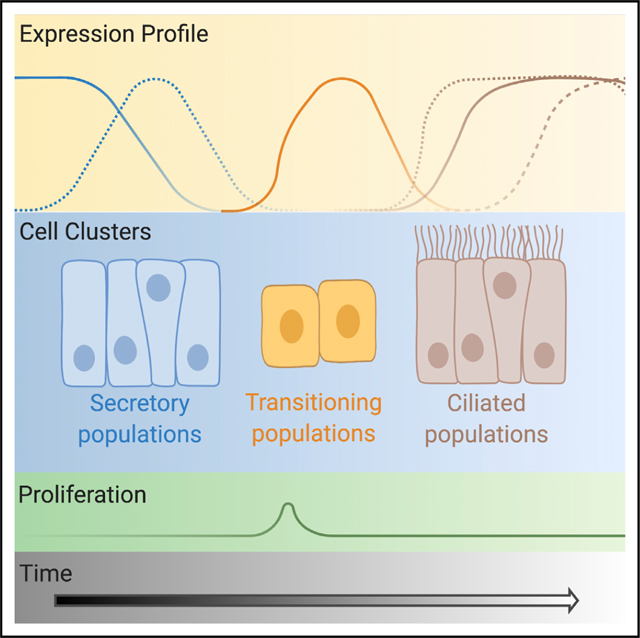
Figure 6. Diagram of the cellular differentiation path for fallopian tube epithelia.

Expression profiles of key markers and transcription factors are shown (not to scale). The “early secretory” cluster corresponds to the former “unclassified cluster 1,” the “early transitioning” cluster corresponds to “unclassified cluster 3,” and the “transitioning” cluster corresponds to “unclassified cluster 2.” Created with BioRender.com.
Epithelial cell dynamics in the FT have not been extensively studied, but in the last two decades, interest in this organ has been piqued by strong evidence to support a FT origin for the majority of high-grade serous ovarian carcinomas (Callahan et al., 2007; Chen et al., 2017; Lee et al., 2007; Medeiros et al., 2006; Meserve et al., 2017). The pseudostratified epithelium of the FT consists of two major cellular populations. A secretory population that expresses the PAX8 transcription factor is thought to be the predominant cell of origin for HGSC (Callahan et al., 2007; Chen et al., 2017; Ducie et al., 2017; Lawrenson et al., 2019; Lee et al., 2007; Medeiros et al., 2006; Meserve et al., 2017). Fallopian secretory epithelia are more proliferative in vitro and in vivo and less competent at repairing DNA damage compared to the FOXJ1+ ciliated population (Levanon et al., 2010). Our results suggest that an early secretory cell subset preferentially adapts to culture and is characterized by a molecular signature that is maintained in advanced tumors. Whether this specific cellular population represents a precursor of HGSC or whether a more-differentiated cell dedifferentiates during neoplastic transformation has yet to be determined. Because the development of long stretches of secretory cells, secretory cell outgrowths (SCOUTs), is thought to be an early step in the development of HGSC (Meserve et al., 2017), it is likely that a perturbation in the differentiation path for tubal epithelia is an early event in high-grade serous ovarian carcinogenesis. We identified RUNX3 as a putative regulator of a transitional state representing an intermediate between secretory and ciliated phenotypes. RUNX3 reportedly promotes proliferation and chemoresistance of ovarian cancer cells in vitro (Barghout et al., 2015; Lee et al., 2011; Nevadunsky et al., 2009). In our analyses, RUNX3 was overexpressed in tumor cells compared to FT cells, but repression by Müllerian TFs was context specific, suggesting regulation of RUNX3 is perturbed during tumorigenesis. Because an increased ratio of secretory to ciliated cells represents a more cancer-prone state, this suggests that loss of the normal transition of secretory to ciliated phenotype is an early step in neoplastic transformation; more work is needed to elucidate whether dysregulated RUNX3 plays a key role in the development of SCOUTs and STICs, as our data suggest.
One of the greatest insights emerging from this work is the identification of transcription factors regulating the different stages of tubal epithelial cell differentiation. SOX17 was enriched in secretory cells; this factor had not been implicated in HGSC or fallopian biology until recently when this protein was described as a master transcription factor for HGSC and both a regulator of and binding partner for PAX8 (Chaves-Moreira et al., 2020; Reddy et al., 2019; Shi et al., 2019). NR2F2 and EGR1 regulons were enriched in early secretory and secretory 1 clusters. We also observed a striking inverse correlation in the regulons enriched in differentiated endpoint for ciliated cells (ciliated cluster 2) and the most differentiated secretory cell cluster (secretory 1). Across the other clusters, activation state of these regulons mirrored the cellular trajectory inferred by pseudotime analyses. This suggests that TF networks required for differentiation of ciliated cells are largely suppressed in the differentiated secretory cell state. Master regulators underlying this negative feedback likely include PAX8, RUNX3, and FOXJ1 or interactions with these factors. Perturbation of this fine-tuned transcriptional regulation is likely an early step in tumor development.
We compared our epithelial subsets with the recently published work using SMART-Seq2 of EPCAM+ FT cells (Hu et al., 2020). We were able to match all of the subsets they identified using the gene signatures from their work. Our secretory cluster 3 had features of both the KRT17+ and inflammatory clusters, our unclassified cluster 2 likely corresponds to their “cell cycle” cluster, and our ciliated cluster expressed genes that corresponded to the “stress” gene set defined in this prior study. Our early secretory cluster expressed both genes from the differentiated subset (DPP4/LTBP4/SLC25A25) and epithelial-to-mesenchymal transition (EMT) subset (ACTA2/TIMP3), although we note that the Hu et al. study did not attempt to describe putative progenitor cells in the tube. The overall consistency between studies indicates that the epithelial subpopulations described are stable in women of different ages and diagnoses, independent of the technology used. Some of the variation between studies could be due to the limitations of the 10X platform, including reduced coverage of the transcriptome. There were also some unexpected findings, including co-expression of epithelial immune genes in unclassified (transitioning) cluster 2, even after doublet removal. Larger studies profiling more cells across more individuals will be required to validate all of the populations we have described. In addition, population-based scRNA-seq studies would enable the identification of significant perturbations in epithelial subpopulation frequencies and transcriptional signatures associated with age and lifetime ovulatory cycles, both major risk factors for HSGC.
Our study has some limitations. Although it is known that FT fimbriae likely harbor the cell of origin for the majority of HGSCs, the small sample size of our study precluded in-depth analyses of features specific to the fimbrial portion of the organ, to gain insight into why these cells are more prone to neoplastic transformation. Although the patients included in the study were not known to carry germline mutations associated with ovarian cancer risk, it is plausible that some of the cells profiled contained somatic mutations that could impact the expression signature of a cell or clone, although our analytic pipeline focused on cell populations reproducibly detected across multiple individuals to reduce the impact of private somatic alterations on the clusters we characterized. In addition, analyses of cells collected from women at high risk of ovarian cancer, such as women with germline mutations in the BRCA1/2 genes, are expected to be the most informative for discovery of biomarkers in early stages of pelvic serous carcinoma but were not included in the current study, as the extensive pathological examination of tubes prophylactically removed from BRCA1/2 mutation carriers (Medeiros et al., 2006) currently limits the application of scRNA-seq to these specimens. Finally, we were underpowered to explore the impact of the fluctuating steroid hormones due to phases in the menstrual cycle or menopause.
The data presented here will be valuable for the study of infertility and benign conditions, such as pelvic inflammatory disorder and ectopic pregnancy, but the greatest translational impact is likely to come from insights into origins and early development of HGSC. We observed that the fallopian epithelial cells that grow ex vivo and immortalized cultures represent an early differentiated FTSEC type and are therefore highly relevant for cancer studies, as the signature of these THY1+ early secretory cells was enriched in a majority of HGSCs. This signature was the most enriched in mesenchymal-type tumors, perhaps unsurprisingly, given the mesenchymal properties of this population, and suggests that further studies are warranted to characterize this population in the high-risk setting. The majority of patients with HGSC are diagnosed with advanced stage disease and have an overall 5-year survival rate of less than 40%, and there is a pressing need to study normal tubal tissues to understand the earliest stages of disease to develop more effective strategies for early detection and prevention. We expect this atlas of cells, transcriptomes, and TF networks in disease-free tissues will lead to translational insights by laying the foundation for identifying subtle alterations in the composition and functioning of the FT in the context of disease or elevated cancer risk.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kate Lawrenson (kate.lawrenson@cshs.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The RNA-Seq data generated during this study are available at NCBI GEO under the following accession numbers: GSE151214, GSE150443 and GSE151316.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell Lines
FT282 and OVCAR4 cells were obtained from Dr. Ronny Drapkin and Dr. William C. Hahn, respectively. OVCAR4 cells were cultured in RPMI-1640 supplemented with 10% FBS, 1 × NEAA, 11.4 μg/ml of insulin and 1 × penicillin/streptomycin. FT282 cells were cultured in DMEM/F12 supplemented with 10% FBS and 1 × penicillin/streptomycin. Cultures maintained at 37°C with 5% CO2. Cells were routinely passaged with 0.05% trypsin using standard cell culture procedures. Both cell lines were negative for Mycoplasma infections and were authenticated by profiling of short tandem repeats using the Promega Powerplex 16HS assay, performed at the University of Arizona Genomics Core (Table S7).
Patient Specimens
This project was performed with approval of the Institutional Review Board at Cedars-Sinai Medical Center (CSMC). All patients provided informed consent. Fresh or formalin fixed, benign human fallopian tubes were collected from women undergoing surgery at Cedars-Sinai Medical Center. Only females were included in this study as only females have fallopian tubes. For the single cell RNA-seq specimens, patient age ranged from 31–62 years. The phenotypic and clinical characteristics of the scRNA-seq cohort are summarized in Table 1. Clinical characteristics of the FFPE cohort are shown in Table S5.
METHOD DETAILS
Tissue processing
Human fallopian tube tissues were placed in sterile serum-free MEM at 4°C and transferred to the tissue culture laboratory. Pathology review was performed to confirm the absence of malignancy. Portions of primary fallopian tubes were transferred into a 10 cm cell-culture dish and the media carefully aspirated. The anatomic origin of each specimen was annotated where possible. Tissues were minced into ~1–2 mm pieces and digested with 1 × Collagenase/Hyaluronidase (STEMCELL Technologies) and 100 μg/mL DNase I (Sigma-Aldrich) in 6.33 mL serum-free MEM. The sample was incubated at 37°C with constant rotation for 90 mins. The supernatant was then harvested, and the remaining tissue flushed with serum free media to collect any remaining cells. The cell suspension was spun at 300 g for 10 mins at 4°C. To lyse red blood cells (RBCs), the resulting pellet was resuspended in an RBC lysis buffer (0.8% NH4Cl, 0.1% KHCO3, pH = 7.2) and incubated for 10 mins at room temperature. Cell suspensions were spun again at 300 g for 10 mins at 4°C and the cell pellet was resuspended in phosphate buffered saline (PBS), or, if > 5% dead cells were observed by trypan blue staining, cells were resuspended in dead cell removal buffer and dead cell removal was performed according to manufacturer’s instructions.
Remaining cells were frozen in 90% fetal bovine serum with 10% DMSO in a Mr. Frosty container placed at −80°C. Frozen cell vials were stored in LN2. Cells were thawed and transferred into a conical tube with 7 mL serum free media and spun at 300 g for 10 mins at 4°C. The cell pellet was resuspended in 100 μL PBS. Cells were counted using a hemocytometer and the sample volume adjusted to achieve a cell concentration between 100/μL to 2,000/μL. Due to the low numbers of cells available for the repeat analyses, lower numbers of cells were targeted for capture. For both patients, 10,000 cells were targeted in the fresh capture, but 5,000 cells were targeted in the thawed specimen for patient 6 and 2,000 were targeted in the thawed specimen for patient 7.
Single cell GEM and barcoding, scRNA-Seq library preparation and next-generation sequencing
Single cells are captured and barcoded using 10X Chromium platform (10X Genomics). scRNA-Seq libraries were prepared following the instructions from Chromium Single Cell 3′ Reagent Kits User Guide (v2 or v3). Briefly, Gel Bead-In EMulsions (GEMs) are generated using single cell preparations. After GEM-RT and cleanup, the cDNA from barcoded single cell RNAs were amplified before quantification using Agilent Bioanalyzer High Sensitivity DNA chips. The single cell 3′ gene expression libraries were constructed and cDNA corresponding to an insertion size of around 350 bp selected. Libraries were quantified using Agilent Bioanalyzer High Sensitivity DNA chip and pooled together to get similar numbers of reads from each single cell before sequencing on a NovaSeq S4 lane (Novogene). For three samples (patients 1–3), capture and NGS were performed by the Cedars-Sinai Applied Genomics, Computation and Translational Genomics Core; for the remaining samples capture and library preparation were performed in-house.
Immunohistochemistry
SOX17 immunohistochemistry was performed by the Cedars-Sinai Cancer Biobank and Translational Research Core. Immunohistochemistry was conducted on the Ventan Discovery Ultra Instrument (Roche Ventana). Paraffinized at 72°C with EZ solution (Roche Ventana). Antigen for SOX17 was retrieved via CC1 protocol with prediluted Tris solution, pH 8.0 (Roche Ventana) for 64 minutes at 95°C. This was followed with primary antibody incubation performed with 1:1000 goat anti-SOX17 (R&D) diluted with Antibody Dilution Buffer (Roche Ventana) for 60 minutes at 37°C. Detection was conducted with DISCOVERY UltraMap anti-Goat multimer RUO (Roche Ventana) for 12 minutes at 37°C, and finally with Chromogen DAB CM (Roche Ventana) for 12 minutes at room temperature. For evaluation of SOX17 staining, the following scheme was used: Distribution scoring: 1 = 0%–24%, 2 = 25%–49%, 3 = 50%–74%, 4 = 75%–100%; Staining intensity scoring: 0 = absent, 1 = weak, 2 = moderate, 3 = strong. Intensity score reflects the average highest intensity on the slide.
Validation of progenitor cell populations using flow cytometry
Fallopian tube tissue was obtained from a 48-year-old woman (gravida: 4, para: 2) with a large simple appearing cyst on her left ovary who underwent laparoscopic left salpingo-oophorectomy and right salpingectomy. Bilateral fallopian tubes (fimbria, ampulla, isthmus and infundibulum) were processed into single cells as described above. Dead cell removal was performed and CD45 microbeads used to deplete immune cells. Remaining cells were enumerated and resuspended in PBS containing 2% FBS at a concentration of 4 × 107 cells/ml. Cells were blocked for 15 minutes using an FcR blocking reagent and stained for 30 minutes in a 96-well round bottom plate using EPCAM-APC (1:50 dilution), CD44-PE (1:50 dilution), CD-90-FITC (1:50 dilution) and a live/dead stain. Singly labeled cells were included for compensation, and ‘fluorescence-minus one’ controls included for all stains to enable us to take fluorescence spread into account when placing the gates. Stained cells were washed, fixed with 1.5% formaldehyde, and maintained at 4°C. Acquisition was performed on BD Fortessa cell analyzer cytometer at the Cedars-Sinai Flow Cytometry Core.
PAX8, SOX17 and WT1 knockdown
OVCAR4 and FT282 cell line models were selected as they most closely recapitulate the cellular and molecular features of human HGSOC (Domcke et al., 2013) and FTSECs, respectively. Cells were reverse transfected using two different non-targeting Control siRNA pools (NT1 and NT2) or pooled oligonucleotides targeting PAX8, SOX17, or WT1 (Horizon Discovery Ltd.) 120 nM of each siRNA pool was diluted in Opti-MEM I (Thermo Fisher Scientific) for 5 minutes, which was then combined with a mix of Opti-MEM I and lipofectamine RNAiMAX (Thermo Fisher Scientific) and incubated for 20 minutes at room temperature. The transfection reagent mix was then combined with 300,000 cells and seeded in a 60mm dish. Media was replenished after 24 hours, and RNA harvested 48 hours later. Cells were washed with cold PBS, collected by scraping and RNA extraction performed using the Nucleospin RNA Plus kit (Macherey-Nagel) according to the manufacturer’s protocol.
Quantitative PCR
Total RNA was reverse transcribed to cDNA using random primers (Promega) and M-MLV Reverse Transcriptase RNase H (Promega). cDNA was amplified using Taqman Universal Master Mix with UNG (Thermo Fisher Scientific) along with probes for TUBB, HPRT1 (housekeeping genes) PAX8, SOX17, or WT1 (Thermo Fisher Scientific). Data were captured using the QuantStudio 12K Flex Real-Time PCR system (Thermo Fisher Scientific).
QUANTIFICATION AND STATISTICAL ANALYSIS
Single cell RNA-Seq data analysis
Preprocessing and integration
Raw reads were aligned to the hg38 reference genome, UMI (unique molecular identifier) counting was performed using Cell Ranger v.2.1.1 (10X Genomics) pipeline with default parameters. We use DoubletFinder (McGinnis et al., 2019) to remove potential doublets based on the expression proximity of each cell to artificial doublets. Only cells with low mitochondrial content (≤10%) were retained for further analysis. To integrate 12 FT scRNA-Seq samples accounting for potential technical variants, we used the anchoring integration method Seurat v3 (Butler et al., 2018; Stuart et al., 2019) which based on canonical correlation (CC) analysis. First, we used FindIntegrationAnchors function to find alignment anchors using default 2,000 high variable genes and 30 CC dimensions for defining neighbor search space. Then the anchors were determined and scored for all of 66 possible sample pairs for the integration process using the function IntegrateData to account for technical and individual sample variations. Cell type prediction of frozen and cell line single cell data using fresh data as the single cell reference was done using TransferData function in Seurat integration pipeline based on defining anchors between two datasets.
Identification of major cell types and epithelial subgroups
First, we used a standard deviation saturation plot to determine the optimal number of principal components from the integrated dataset. Then, we applied the Louvain community detection-based method which was performed on integrated expression values on shared-nearest-neighbor (SNN) graph as implemented in the FindCluster function from the Seurat package for clustering single cell data. Robustness and further clustering analysis were evaluated at the different resolution from 0.1 to 3 with different representative markers to identify the optimal cluster number so that a cluster was called are present at least 2% in at least 3 samples and two close clusters on the cluster tree are merged if there are no differentially expressed gene were detected at cutoff avg_logFC of 0.25 and at least 25% of cells expressed the markers (Differentially expression was performed using MAST (Finak et al., 2015), as implemented in the FindAllMarker and FindMarker functions in Seurat). The following marker genes were used for global annotation of cell types: Epithelial: EPCAM, Keratin genes (KRT7, KRT8, KRT18, PAX8, FOXJ1, OVGP1); T cells: CD3E, CD3D, TRBC1/2, TRAC; Myeloid cells: LYZ, CD86, CD68, FCGR3A; B cells and plasma cells: CD79A/B, JCHAIN, IGKC, IGHG3; Endothelial cells: CLDN5; Fibroblasts: DCN, C1R, COL1A1, ACTA2; Mast cells: TPSAB1. UMAP (uniform manifold approximation and projection) (Becht et al., 2018) was used for visualizing cell-types and clusters with representative markers.
Identification of markers of epithelial subgroups
Differential expression tests were performed using MAST (Finak et al., 2015), one of the top differential expression analysis methods according to a benchmark study (Soneson and Robinson, 2018) were used to identify markers for each of 10 identified epithelial subsets with average logFC 0.25, expressed in at least 25% of cells in a subset. Pathway analyses were performed using Reactome (Yu and He, 2016).
Regulatory network inference using SCENIC
We used Single-Cell Regulatory Network Inference And Clustering method, SCENIC (Aibar et al., 2017) to infer regulatory networks, namely regulons, from co-expression of transcription factor and candidate target genes (from RcisTarget database, https://aertslab.org/#scenic). We ran SCENIC for two scenarios: all major cell-types, and within 10 epithelial subsets (the input matrix was normalized expression matrix from Seurat, filtering genes that expressed in at least 10% of samples with minimal count per gene as 3% of the cells, as default in SCENIC). AUC scores for each regulon were used to compare a cluster with the rest to identify the specific regulon and associated transcription factor using a generalized linear model with FDR-corrected p values < 0.05.
Pseudotime trajectory analysis using monocle2
We used monocle2 with the DDR tree (Qiu et al., 2017) to infer the potential trajectory of epithelial cells. Epithelial cells were selected to perform pseudo-time ordering based on top 2,000 variable genes after integration (from the VariableFeature function in Seurat). Pseudo-time and cell states were inferred using orderCells function in monocle2, overlaying the cluster identification from Seurat for consistency. Cell trajectory was plotted using plot_cell_trajectory in monocle2 package. Then, we inferred genes associated with the pseudo-time (from early to late) using generalized linear models implemented in function differentialGeneTest with parameter “~sm.ns (Pseudotime)” using all the genes and RNA expression value (before integration). Hierarchical clustering was then used to infer 4 subset of genes associated with each stage of cellular pseudotime lineage.
Deconvolution of HGSCs using epithelial subsets
To estimate the potential contribution of epithelial subsets inferred by scRNA-Seq in the bulk sample profiled by RNA-Seq, we used computational deconvolution. First, we defined the subset-specific gene signatures for each of the subset from their differentially expressed genes compared to the rest with the stringent cutoff avg_logFC 1 and adjusted p value cutoff 0.05. The subsets which shared similar gene signatures after this step were merged leading to the combination of epithelial subsets 2 and 3; as well as the ciliated subsets 1,2,3. Then we used the recent method MuSiC (Wang et al., 2019), accounting for cross-subject gene weighting, to estimate the composition of 6 subsets for normal fallopian tube epithelial ex vivo cultures (Lawrenson et al., 2019) and HGSOC samples (Cancer Genome Atlas Research Network, 2011). Enrichment with TCGA molecular subtypes was evaluated by hypergeometric test phyper in R.
RNA-Seq data generation and analysis
Poly-A non-stranded libraries were built from each RNA sample and subjected to 150 bp paired-end sequencing to obtain ~40 million reads/sample. Each knockdown was sequenced in duplicate, using independent knockdown experiments performed at different passages. RNA-Seq was performed by BGI using the DNB-seq next generation sequencing platform. Data were filtered and aligned using hisat2–2.1.0 (reference genome: hg38, gencodev26 + ERCC92). A gene-level read count matrix generated using htseq-count (v0.5.4.p3) (Anders et al., 2015). ERCC read counts were removed, and differential gene expression analyses performed using the R package DESeq2 (version 1.24.0). Data for RUNX3 were extracted (Table S8) and DESeq2 statistics retrieved.
Supplementary Material
KEY RESOURCES TABLE
| Reagent or Resource | Source | Identifier |
|---|---|---|
|
| ||
| Antibodies | ||
| Anti-SOX17 | R&D Systems | AF1924; RRID: AB_355060 |
| CD326(EpCAM)-APC | Milteny Biotec | 130–111-000; RRID: AB_2657497 |
| CD90-FITC | Milteny Biotec | 130–117-684; RRID: AB_2784296 |
| CD44-PE | Milteny Biotec | 130–113-342; RRID: AB_2726118 |
|
| ||
| Biological samples | ||
|
| ||
| Human fallopian tube specimens (fresh) | Cedar-Sinai Medical Center - Center for Minimally Invasive Gynecologic Surgery (CMIGS) and Division of Gynecologic Oncology | GEO: GSE151214 |
| Human fallopian tube specimens (formalin fixed) | Cedar-Sinai Medical Center - Women’s Cancer Program, Division of Gynecologic Oncology and Department of Pathology and Laboratory Medicine | this study |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 10X Collagenase/Hyaluronidase in DMEM | STEMCELL Technologies | 07912 |
| Minimal Essential Medium (MEM) | Corning | 10–010-CV |
| Deoxyribonuclease I | Sigma-Aldrich | DN25–1G |
| Fetal bovine serum | Sigma-Aldrich | F0926–500ML |
| Formaldehyde | Avantor | 2106–01 |
| FcR blocking reagent | Milteny Biotec | 130–059-901 |
| Live/Dead Cell stain | Tonbo Biosciences | 13–0868-T100 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D2650–5X10ml |
| ON-TARGETplus Non-targeting Control Pool [NT1] |
Dharmacon Research/GE | D-001810–01-20 |
| ON-TARGETplus Non-targeting Control Pool [NT2] | Horizon Discovery Ltd | D-0012–03, D-001210–04, D-001210–05 (combined) |
| ON-TARGETplus PAX8 siRNA pool | Horizon Discovery Ltd | L-003778–00-0005 |
| ON-TARGETplus SOX17 siRNA pool | Horizon Discovery Ltd | L-013028–01-0010 |
| ON-TARGETplus WT1 siRNA pool | Horizon Discovery Ltd | L-009101–00-0005 |
| Opti-MEM I reduced serum medium | Thermo Fisher Scientific | 31–985-070 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | 13–778-150 |
| M-MLV reverse transcriptase RNase H | Promega | M5301 |
| Random primers | Promega | C1181 |
| Taqman Universal Master Mix with UNG | Thermo Fisher Scientific | 4440044 |
| HPRT1 probe | Thermo Fisher Scientific | Hs02800695_m1 |
| TUBB probe | Thermo Fisher Scientific | Hs00742828_s1 |
| PAX8 probe | Thermo Fisher Scientific | Hs00247586_m1 |
| SOX17 probe | Thermo Fisher Scientific | Hs00751752_s1 |
| WT1 probe | Thermo Fisher Scientific | Hs01103751_m1 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Dead Cell Removal Kit | Milteny Biotec | 130–090-101 |
| CD45 MicroBeads | Milteny Biotec | 130–045-801; RRID: AB_2783001 |
| Chromium Single Cell 3′ Library & Gel Bead Kit v2 | 10x Genomics | 120237 |
| Chromium Single Cell A Chip Kits | 10x Genomics | 120236 |
| Chromium Single Cell 3′ GEM, Library & Gel Bead Kit v3 | 10x Genomics | 1000075 |
| Chromium Chip B Single Cell Kit | 10x Genomics | 1000153 |
| Nucleospin RNA Plus kit | Macherey-Nagel | 740984.250 |
| High Sensitivity DNA | Agilent | 5067–4626 |
|
| ||
| Deposited data | ||
|
| ||
| Fallopian tube single cell RNA-Seq | This study | GEO: GSE151214 |
| RNA-Seq profiling of FT282 cells with PAX8, SOX17 or WT1 knocked down |
This study | GEO: GSE151316 |
| RNA-Seq profiling of OVCAR4 cells with PAX8, SOX17 or WT1 knocked down | This study | GEO: GSE150443 (raw data); GEO: GSE151316 (reprocessed analyzed data) |
| RNA-Seq dataset containing ex vivo fallopian tube epithelial cell cultures integrated with TCGA HGSOCs | Lawrenson et al., 2019 | https://github.com/lawrenson-lab/OvarianRNASeq |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| FT282 | Karst and Drapkin, 2012 | N/A |
| OVCAR4 | Hahn laboratory | RRID: CVCL_1627 |
|
| ||
| Software and algorithms | ||
|
| ||
| Cell Ranger | 10x Genomics; https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome | V.2.1.1; RRID: SCR_017344 |
| R package Seurat | https://satijalab.org/seurat/; Stuart et al., 2019 | V3; RRID: SCR_016341 |
| R package DoubletFinder | https://github.com/chris-mcginnis-ucsf/DoubletFinder; McGinnis et al., 2019 | N/A |
| R package MAST | https://github.com/RGLab/MAST; Finak et al., 2015 | N/A |
| R package Monocle2 | https://github.com/cole-trapnell-lab/monocle-release; Qiu et al., 2017 | N/A |
| R package SCENIC | https://github.com/aertslab/SCENIC; Aibar et al., 2017 | RRID: SCR_017247 |
| R package - MUSIC | https://github.com/xuranw/MuSiC; Wang et al., 2019 | RRID: SCR_008792 |
| HISAT | Kim et al., 2015 | 2–2.1.0; RRID: SCR_015530 |
| HTSeq | Anders et al., 2015 | V0.5.4.p; RRID: SCR_005514 |
| DEseq2 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | version 1.24.0; RRID: SCR_015687 |
| R package ReactomePA | http://bioconductor.org/packages/release/bioc/html/ReactomePA.html; Yu and He, 2016 | N/A |
| FlowJo | https://www.flowjo.com/ | v10 |
Highlights.
Single-cell transcriptomics create a cellular atlas for benign fallopian tubes
A secretory-intermediate-ciliated path is projected for epithelial differentiation
PAX8, SOX17, and RUNX3 potentially drive fallopian epithelial differentiation
“Early secretory” epithelial cells are likely precursors for the majority of HGSCs
ACKNOWLEDGMENTS
This project was supported by an Ovarian Cancer Research Alliance Liz Tilberis Early Career Award (599175; K.L.); Ovarian Cancer Research Alliance Program Project Development (373356; B.Y.K. and K.L.); a Southern California Clinical and Translational Science Institute Core Voucher (V132; K.L.); and a Leon Fine Translational Research award from the Cedars-Sinai Medical Center (K.L., M.T.S., and K.N.W.). K.L. is also supported by a Research Scholar’s Grant from the American Society (134005). This work was also supported in part by NIH R01 (5R01CA211707; K.L. and S.A.G.). R.N. is supported in part by a Ruth L. Kirschstein Institutional National Research Service Award (T32) from the NIH (grant number 5 T32 GM 118288–2). The research described was supported in part by National Center for Advancing Translational Sciences (NCATS) UCLA CTSI grant number UL1TR001881 and in part by Cedars-Sinai Cancer. This research used the following Cedars-Sinai Cores—the Flow Cytometry Core, the Applied Genomics, Computation and Translational (Genomics) Core, and the Cancer Biobank and Translational Research Core. We thank the core staff for their support of our research. We thank Dr. Sandra Orsulic and her laboratory for helpful discussions plus Jenny Lester and study staff from the Women’s Cancer Program biorepository and the Biologic and Epidemiologic Markers of Endometriosis (BEME) Study that provided the specimens used in this study. We also kindly thank the patients that generously donated the specimens used in our research. H.Q.D. thanks Dr. Lynn Hedrick for the support working on this project at LJI.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108978.
REFERENCES
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardighieri L, Lonardi S, Moratto D, Facchetti F, Shih, IeM., Vermi W, and Kurman RJ (2014). Characterization of the immune cell repertoire in the normal fallopian tube. Int. J. Gynecol. Pathol 33, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghout SH, Zepeda N, Vincent K, Azad AK, Xu Z, Yang C, Steed H, Postovit L-M, and Fu Y (2015). RUNX3 contributes to carboplatin resistance in epithelial ovarian cancer cells. Gynecol. Oncol 138, 647–655. [DOI] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol Published online December 3, 2018. 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd KM, Piehl NC, Patel JH, Huh WJ, Sequeira I, Lough KJ, Wagner BL, Marangoni P, Watt FM, Klein OD, et al. (2019). Heterogeneity within stratified epithelial stem cell populations maintains the oral mucosa in response to physiological stress. Cell Stem Cell 25, 814–829.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MJ, Crum CP, Medeiros F, Kindelberger DW, Elvin JA, Garber JE, Feltmate CM, Berkowitz RS, and Muto MG (2007). Primary fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. J. Clin. Oncol 25, 3985–3990. [DOI] [PubMed] [Google Scholar]
- Campbell KR, and Yau C (2018). Uncovering pseudotemporal trajectories with covariates from single cell and bulk expression data. Nat. Commun 9, 2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaves-Moreira D, Mitchell MA, Arruzza C, Rawat P, Sidoli S, Nameki R, Reddy J, Corona RI, Ma S, Winterhoff B, et al. (2020). PAX8 orchestrates an angiogenic program through interaction with SOX17. bioRxiv. 10.1101/2020.09.09.290387. [DOI] [Google Scholar]
- Chen F, Gaitskell K, Garcia MJ, Albukhari A, Tsaltas J, and Ahmed AA (2017). Serous tubal intraepithelial carcinomas associated with high-grade serous ovarian carcinomas: a systematic review. BJOG 124, 872–878. [DOI] [PubMed] [Google Scholar]
- Cheng L, Lu W, Kulkarni B, Pejovic T, Yan X, Chiang J-H, Hood L, Odunsi K, and Lin B (2010). Analysis of chemotherapy response programs in ovarian cancers by the next-generation sequencing technologies. Gynecol. Oncol 117, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow J, Amso NN, Lewin J, and Shaw RW (1994). Morphology and ultrastructure of fallopian tube epithelium at different stages of the menstrual cycle and menopause. Hum. Reprod 9, 2224–2233. [DOI] [PubMed] [Google Scholar]
- Domcke S, Sinha R, Levine DA, Sander C, and Schultz N (2013). Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun 4, 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnez J, Casanas-Roux F, Caprasse J, Ferin J, and Thomas K (1985). Cyclic changes in ciliation, cell height, and mitotic activity in human tubal epithelium during reproductive life. Fertil. Steril 43, 554–559. [DOI] [PubMed] [Google Scholar]
- Ducie J, Dao F, Considine M, Olvera N, Shaw PA, Kurman RJ, Shih I-M, Soslow RA, Cope L, and Levine DA (2017). Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun 8, 990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J, D’Sylva R, Volpert M, Jamsai D, Merriner DJ, Nie G, Salamonsen LA, and O’Bryan MK (2015). Endometrial CRISP3 is regulated throughout the mouse estrous and human menstrual cycle and facilitates adhesion and proliferation of endometrial epithelial cells. Biol. Reprod 92, 99. [DOI] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcés J-J, Simicek M, Vicari M, Brozova L, Burgos L, Bezdekova R, Alignani D, Calasanz M-J, Growkova K, Goicoechea I, et al. (2020). Transcriptional profiling of circulating tumor cells in multiple myeloma: a new model to understand disease dissemination. Leukemia 34, 589–603. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Syed SM, and Tanwar PS (2017). In vivo genetic cell lineage tracing reveals that oviductal secretory cells self-renew and give rise to ciliated cells. Development 144, 3031–3041. [DOI] [PubMed] [Google Scholar]
- Givan AL, White HD, Stern JE, Colby E, Gosselin EJ, Guyre PM, and Wira CR (1997). Flow cytometric analysis of leukocytes in the human female reproductive tract: comparison of fallopian tube, uterus, cervix, and vagina. Am. J. Reprod. Immunol 38, 350–359. [DOI] [PubMed] [Google Scholar]
- Haas AR, Tanyi JL, O’Hara MH, Gladney WL, Lacey SF, Torigian DA, Soulen MC, Tian L, McGarvey M, Nelson AM, et al. (2019). Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol. Ther 27, 1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healey A (2012). Embryology of the female reproductive tract. In Imaging of Gynecological Disorders in Infants and Children, Mann GS, Blair JC, and Garden AS, eds. (Springer Berlin; Heidelberg: ), pp. 21–30. [Google Scholar]
- Hu Z, Artibani M, Alsaadi A, Wietek N, Morotti M, Shi T, Zhong Z, Santana Gonzalez L, El-Sahhar S, KaramiNejadRanjbar M, et al. (2020). The repertoire of serous ovarian cancer non-genetic heterogeneity revealed by single-cell sequencing of normal fallopian tube epithelial cells. Cancer Cell 37, 226–242.e7. [DOI] [PubMed] [Google Scholar]
- Karst AM, and Drapkin R (2012). Primary culture and immortalization of human fallopian tube secretory epithelial cells. Nat. Protoc 7, 1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst AM, Levanon K, and Drapkin R (2011). Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc. Natl. Acad. Sci. USA 108, 7547–7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst AM, Jones PM, Vena N, Ligon AH, Liu JF, Hirsch MS, Etemadmoghadam D, Bowtell DDL, and Drapkin R (2014). Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube-derived high-grade serous ovarian cancers. Cancer Res. 74, 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köbel M, Kalloger SE, Boyd N, McKinney S, Mehl E, Palmer C, Leung S, Bowen NJ, Ionescu DN, Rajput A, et al. (2008). Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 5, e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak EL, Palit S, Miranda-Rodríguez JR, Janjic A, Böttcher A, Lickert H, Enard W, Theis FJ, and López-Schier H (2020). Epithelial planar bipolarity emerges from Notch-mediated asymmetric inhibition of Emx2. Curr. Biol 30, 1142–1151.e6. [DOI] [PubMed] [Google Scholar]
- Lawrenson K, Fonseca MAS, Liu AY, Segato Dezem F, Lee JM, Lin X, Corona RI, Abbasi F, Vavra KC, Dinh HQ, et al. (2019). A study of high-grade serous ovarian cancer origins implicates the SOX18 transcription factor in tumor development. Cell Rep. 29, 3726–3735.e4. [DOI] [PubMed] [Google Scholar]
- Lee Y, Miron A, Drapkin R, Nucci MR, Medeiros F, Saleemuddin A, Garber J, Birch C, Mou H, Gordon RW, et al. (2007). A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol 211, 26–35. [DOI] [PubMed] [Google Scholar]
- Lee CWL, Chuang LSH, Kimura S, Lai SK, Ong CW, Yan B, SaltoTellez M, Choolani M, and Ito Y (2011). RUNX3 functions as an oncogene in ovarian cancer. Gynecol. Oncol 122, 410–417. [DOI] [PubMed] [Google Scholar]
- Levanon K, Ng V, Piao HY, Zhang Y, Chang MC, Roh MH, Kindelberger DW, Hirsch MS, Crum CP, Marto JA, and Drapkin R (2010). Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene 29, 1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallette FA, Calabrese V, Ilangumaran S, and Ferbeyre G (2010). SOCS1, a novel interaction partner of p53 controlling oncogene-induced senescence. Aging (Albany NY) 2, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros F, Muto MG, Lee Y, Elvin JA, Callahan MJ, Feltmate C, Garber JE, Cramer DW, and Crum CP (2006). The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am. J. Surg. Pathol 30, 230–236. [DOI] [PubMed] [Google Scholar]
- Meserve EEK, Brouwer J, and Crum CP (2017). Serous tubal intraepithelial neoplasia: the concept and its application. Mod. Pathol 30, 710–721. [DOI] [PubMed] [Google Scholar]
- Nevadunsky NS, Barbieri JS, Kwong J, Merritt MA, Welch WR, Berkowitz RS, and Mok SC (2009). RUNX3 protein is overexpressed in human epithelial ovarian cancer. Gynecol. Oncol 112, 325–330. [DOI] [PubMed] [Google Scholar]
- Ozcan A, Shen SS, Hamilton C, Anjana K, Coffey D, Krishnan B, and Truong LD (2011). PAX 8 expression in non-neoplastic tissues, primary tumors, and metastatic tumors: a comprehensive immunohistochemical study. Mod. Pathol 24, 751–764. [DOI] [PubMed] [Google Scholar]
- Paik DY, Janzen DM, Schafenacker AM, Velasco VS, Shung MS, Cheng D, Huang J, Witte ON, and Memarzadeh S (2012). Stem-like epithelial cells are concentrated in the distal end of the fallopian tube: a site for injury and serous cancer initiation. Stem Cells 30, 2487–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, Chen JYH, Ohman AW, Stepule CD, Kwak S, et al. (2013). Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 24, 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reade CJ, McVey RM, Tone AA, Finlayson SJ, McAlpine JN, Fung-Kee-Fung M, and Ferguson SE (2014). The fallopian tube as the origin of high grade serous ovarian cancer: review of a paradigm shift. J. Obstet. Gynaecol. Can 36, 133–140. [DOI] [PubMed] [Google Scholar]
- Reddy J, Fonseca MAS, Corona RI, Nameki R, Segato Dezem F, Klein IA, Chang H, Chaves-Moreira D, Afeyan L, Malta TM, et al. (2019). Predicting master transcription factors from pan-cancer expression data. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K, Yin X, Cai M-C, Yan Y, Jia C, Ma P, Zhang S, Zhang Z, Gu Z, Zhang M, et al. (2019). PAX8 regulon in human ovarian cancer links lineage dependency with epigenetic vulnerability to HDAC inhibitors. eLife 8, e44306. 10.7554/eLife.44306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M, Toki T, Takagi Y, Konishi I, and Fujii S (2000). Immunohistochemical detection of the Wilms’ tumor gene (WT1) in epithelial ovarian tumors. Int. J. Gynecol. Pathol 19, 158–163. [DOI] [PubMed] [Google Scholar]
- Smith JM, Wira CR, Fanger MW, and Shen L (2006). Human fallopian tube neutrophils–a distinct phenotype from blood neutrophils. Am. J. Reprod. Immunol 56, 218–229. [DOI] [PubMed] [Google Scholar]
- Snegovskikh V, Mutlu L, Massasa E, and Taylor HS (2014). Identification of putative fallopian tube stem cells. Reprod. Sci 21, 1460–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, and Robinson MD (2018). Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 15, 255–261. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B, et al. ; Australian Ovarian Cancer Study Group (2008). Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res 14, 5198–5208. [DOI] [PubMed] [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, and Fuchs E (2004). Defining the epithelial stem cell niche in skin. Science 303, 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilas-Boas V, Cooreman A, Gijbels E, Van Campenhout R, Gustafson E, Ballet S, Annaert P, Cogliati B, and Vinken M (2019). Primary hepatocytes and their cultures for the testing of drug-induced liver injury. Adv. Pharmacol 85, 1–30. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhao Y, Wu X, Yin S, Chuai Y, and Wang A (2015a). The utility of human fallopian tube mucosa as a novel source of multipotent stem cells for the treatment of autologous reproductive tract injury. Stem Cell Res. Ther 6, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li L, Wang Y, Tang SN, and Zheng W (2015b). Fallopian tube secretory cell expansion: a sensitive biomarker for ovarian serous carcinogenesis. Am. J. Transl. Res 7, 2082–2090. [PMC free article] [PubMed] [Google Scholar]
- Wang X, Park J, Susztak K, Zhang NR, and Li M (2019). Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun 10, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zheng Y, Li J, Yu Y, Zhang W, Song M, Liu Z, Min Z, Hu H, Jing Y, et al. (2020). Single-cell transcriptomic atlas of primate ovarian aging. Cell 180, 585–600.e19. [DOI] [PubMed] [Google Scholar]
- White HD, Crassi KM, Givan AL, Stern JE, Gonzalez JL, Memoli VA, Green WR, and Wira CR (1997). CD3+ CD8+ CTL activity within the human female reproductive tract: influence of stage of the menstrual cycle and menopause. J. Immunol 158, 3017–3027. [PubMed] [Google Scholar]
- You J-L, Wang W, Tang M-Y, Ye Y-H, Liu A-X, and Zhu Y-M (2018). A potential role of galectin-1 in promoting mouse trophoblast stem cell differentiation. Mol. Cell. Endocrinol 470, 228–239. [DOI] [PubMed] [Google Scholar]
- Yu G, and He Q-Y (2016). ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst 12, 477–479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-Seq data generated during this study are available at NCBI GEO under the following accession numbers: GSE151214, GSE150443 and GSE151316.