
FIGURE 6.
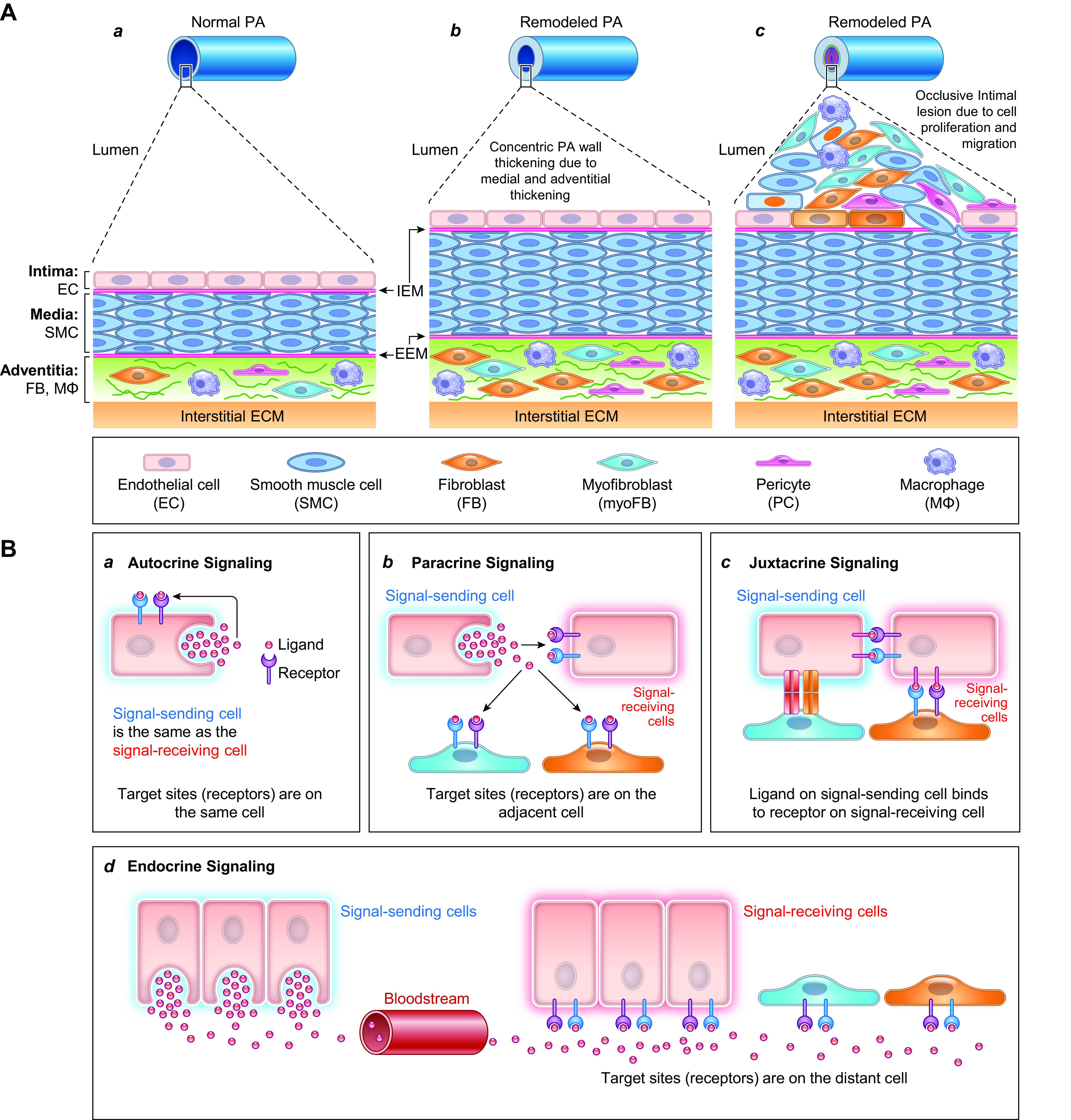
Schematic diagram depicting the progression of pulmonary vascular remodeling caused by changes in adventitial fibroblasts (FBs) and macrophages (MΦs), medial smooth muscle cells (SMCs), and intimal endothelial cells (ECs) (A) and the pathogenic interaction among different cells through the autocrine, paracrine, and juxtacrine mechanisms (B). A: normal pulmonary artery (PA) is thin and composed of the intima or the endothelium (with a monolayer of ECs), the media (mainly contains SMCs and may contain SMC-like pericytes), and the adventitia [mainly contains FBs, MΦs, progenitor cells, and extracellular matrix (ECM)] (a). The intima and media are separated by the internal elastic membrane (IEM), whereas the media is separated by the external elastic membrane (EEM) from the adventitia. Because of the stimulation of pathogenic triggers and self-defects (e.g., somatic mutation, genetic manifestation), increased SMC/FB proliferation results in concentric PA wall thickening (b), whereas EC injury, phenotypical change [e.g., endothelium-to-mesenchymal transition (EndMT)], and SMC migration contribute to the development occlusive intimal lesions (c). All cell types in the PA contribute to the development and progression of the pathological changes that narrow the lumen and increase pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP). B: diagram showing cell interactions through different mechanisms including autocrine (a), paracrine (b), juxtacrine (c), and endocrine (d) signaling. All cells can use the autocrine signaling mechanism for self-regulation or -stimulation. Adjacent cells can regulate each other through paracrine and juxtacrine signaling mechanism in the same layers (e.g., EC↔EC, SMC↔SMC, FB↔FB), whereas EC and SMC (or SMC and FB) can also interact with each other through the myoendothelial junction as well as the internal (and external) elastic lamina.