

Abstract
Bladder cancer is a prevalent but currently understudied cancer type and patient outcomes are poor when it progresses to the muscle-invasive stage. Current research in bladder cancer focuses on the genetic and epigenetic alterations occurring within the urothelial cell compartment; however, the stromal compartment receives less attention. Dynamic changes and intercellular communications occur in the tumour microenvironment (TME) of the bladder — a new concept and niche that we designate as the bladder TME (bTME) — during tumour evolution, metastatic progression and in the context of therapeutic response. Collagens and their cognate receptors, the discoidin domain receptors, have a role in various steps of the metastatic cascade and in immune checkpoint resistance. Furthermore, the presence of another TME niche, the metastatic TME (met-TME), is a novel concept that could support divergent progression of metastatic colonization in different organs, resulting in distant metastases with distinct characteristics and genetics from the primary tumour. The stroma has divergent roles in mediating therapeutic response to BCG immunotherapy and immune checkpoint inhibitors, as well as conventional chemotherapy or trimodality therapy (that is, maximal transurethral resection of bladder tumour, chemotherapy and radiotherapy). The local bTME and distant met-TME are currently conceptually and therapeutically unexploited niches that should be actively investigated. New biological insights from these TMEs will enable rational design of strategies that co-target the tumour and stroma, which are expected to improve the outcomes of patients with advanced bladder cancer.
Bladder cancer is the second most common urological malignancy worldwide after prostate cancer1. Despite its prevalence, bladder cancer is considerably understudied and, therefore, the fundamental understanding of this cancer, especially research studies investigating the tumour microenvironment (TME) in the bladder, has considerably lagged behind investigations into the TME of other cancer types2. Advanced bladder cancers that progress to metastatic disease and colonize distant organs are difficult to cure, and less than one-third of patients with metastatic bladder cancer exhibit response to chemotherapy or durable responses to immune checkpoint inhibitor (ICI) therapies3,4, posing a major clinical challenge. Previous studies on human bladder cancer have primarily focused on characterizing the molecular alterations that accumulate within the urothelial cell compartment, which often defines the intrinsic properties of tumour cells5. In other epithelial cancer types, such as pancreatic, prostate and breast, the active contribution of the stromal and immune microenvironment is becoming widely accepted6–8, but the roles of the bladder tumour microenvironment (bTME) in influencing the tumorigenic process are not as well characterized.
In the nonmalignant bladder, the multilayered urothelium (the mucosa)9 lies on top of a vascularized, thin basement membrane that separates it from the underlying interstitial cell compartment (also called the lamina propria)5,9 (FIG.1a). In the area proximal to the urothelial basement membrane, the lamina propria contains an extensive network of nerve fibres and a capillary network that is surrounded by pericytes, fibroblasts with myoid features, myofibroblasts and a complex extracellular matrix (ECM)5,10–12 (FIG.1a). Under the lamina propria are inner longitudinal, middle and outer circular smooth muscle layers (the muscularis propria, also known as the detrusor muscle)13, a layer of perivesical adipose tissue and the peritoneum (FIG.1a). The interstitial cells of Cajal are located within the lamina propria and muscularis propria layers; these unique cells interact with nerve cells and smooth muscle cells to act as a conduit for suburothelial sensory processing and modulation of detrusor activities11,14 (FIG.1a). Cellular communication between nonmalignant bladder urothelial cells and the underlying stroma that maintains healthy bladder physiology is mediated by a variety of growth factors and pathways, including epidermal growth factor (EGF), transforming growth factor β1 (TGFβ1), sonic hedgehog (SHH) and WNT signalling15,16 (FIG.1a). In premalignant bladder lesions or carcinoma in situ, interstitial fibroblasts initially exert inhibitory signals (such as TGFβ1) and urothelial differentiation signals (such as bone morphogenetic protein 4 (BMP4) and BMP5) to impede uncontrolled urothelial proliferation and aberrant differentiation, respectively17 (FIG.1b). As tumour progression proceeds, these tissue-resident fibroblasts and other cell types (such as bone marrow-derived fibrocytes) are converted into cancer-associated fibroblasts (CAFs), such as myofibroblasts, via a signalling cascade that is very similar to scarring in response to tissue injury18 (FIG.1b,c). The similarity between the fibrotic reactions that occur during tissue injury and the tumorigenic process highlight the relevance of the stroma in promoting a wound that never heals18. During the early stage of the metastatic cascade, cancer cells are able to invade the muscle layers, facilitated by the loss and modification of their ability to adhere to the ECM components of the basement membrane and interstitium, as well as their evolution to evade responses to stromal-derived inhibitory factors19. After surviving anoikis in the circulation — a form of programmed cell death induced by cell detachment from ECM — as well as evading immune surveillance, these metastatic cancer cells (the ‘seed’) will then interact with the receptive organ microenvironment (the ‘soil’)20 to facilitate metastatic colonization. Evidence is emerging that reveals that the metastatic TMEs differ from the primary bTME21. This difference is important as these distinct TMEs could contribute to the divergent progression of primary and metastatic tumour clones. This theory adds further complexity to the mechanistic development and therapeutic targeting of distant metastases, which probably means molecular targets for metastasis are different from the primary tumour.
Fig. 1 |. Bladder TME components and their interactions with urothelial cells during bladder tumour progression.
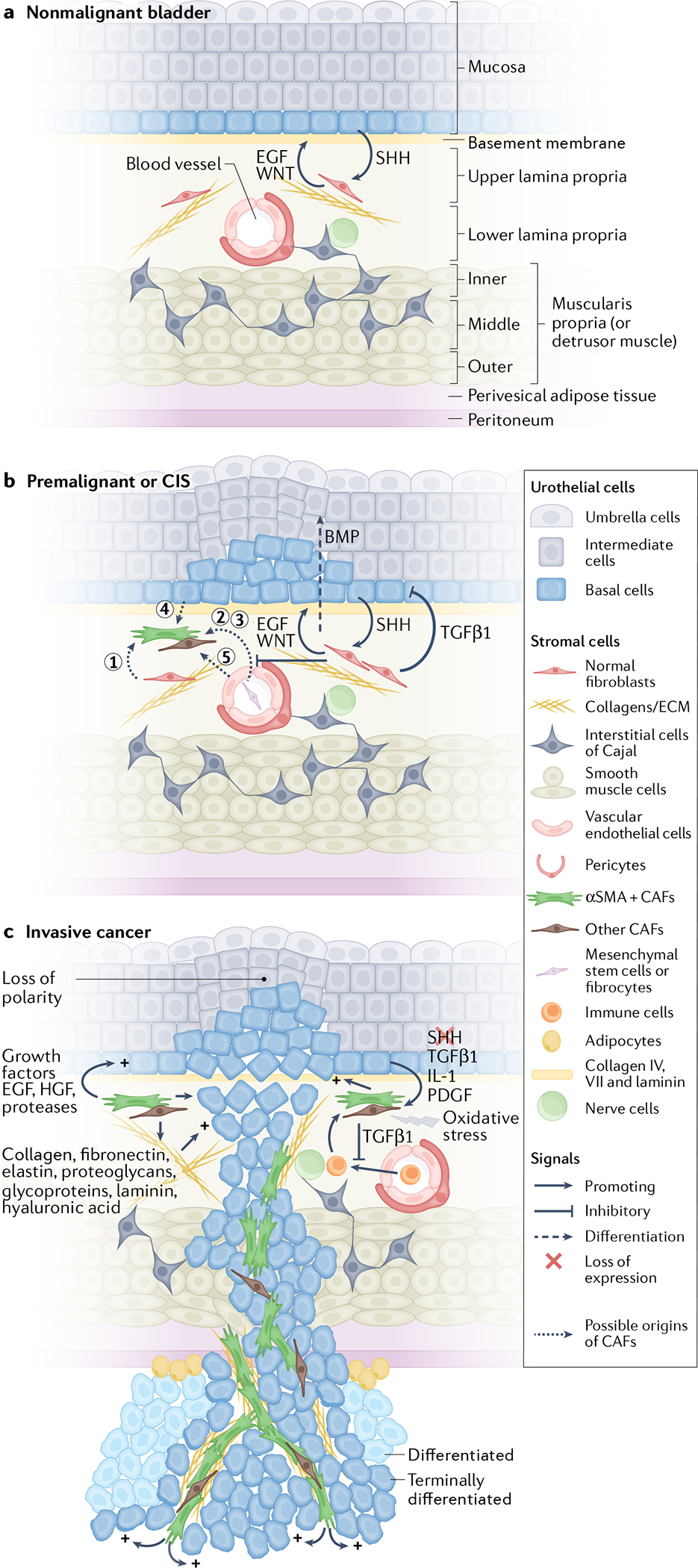
a | In a nonmalignant bladder, the multilayered urothelium is supported by a stroma (the lamina propria) that contains nerve fibres, vasculature, interstitial extracellular matrix (ECM), intercalated by a few fibroblasts and interstitial cells of Cajal within the stroma. The lamina propria is further surrounded by inner longitudinal, middle circular and outer longitudinal smooth muscle layers (the muscularis propria or detrusor muscle), as well as perivesical adipose tissue and the peritoneum. b | In premalignant lesions or carcinoma in situ (CIS), stromal fibroblasts initially secrete inhibitory signals (such as transforming growth factor β1 (TGFβ1)) and differentiation signals (such as bone morphogenetic proteins (BMPs)) to impede uncontrolled cellular proliferation and aberrant differentiation, respectively. As the tumour progresses, these stromal fibroblasts and/or other cell types (types 1–5, that is type 1, normal fibroblasts; types 2 and 3, fibrocytes or monocytes; type 4, epithelial cancer cells; and type 5, endothelial cells) are converted into myofibroblasts or other cancer-associated fibroblast (CAF) types in a cascade highly reminiscent of the wound-healing response. c | At an advanced stage (that is, invasive cancer), stromal cells and ECM in the tumour microenvironment (TME) co-evolve and communicate with cancer cells to promote cancer progression and cause drug resistance. CAFs promote bladder cancer progression through secreting chemoattractants, growth factors (such as basic fibroblast growth factor 2 (FGF2), epidermal growth factor receptor (EGFR) ligands, colony-stimulating factors, TGFβ1 and hepatocyte growth factor (HGF)57–59, angiogenic factors (vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF)), and ECM-degrading proteases (such as matrix metalloproteinase (MMP)) and ECM. αSMA, α-smooth muscle actin; SHH, sonic hedgehog.
In this Review, we summarize current advances in our understanding of the roles of stroma and associated ECM during tissue injury and cancer development, with an emphasis on bladder cancer. We discuss the key cellular and non-cellular components comprising the bTME, and their dynamic interactions with bladder cancer cells. We also introduce the concept of a metastatic TME (met-TME) in supporting divergent progression of metastatic colonization in different organs and discuss their relationships to the bladder TME (bTME). Finally, we highlight the clinical significance and therapeutic vulnerability of the bladder TME for novel treatment approaches.
The bladder TME
The bTME is composed of both cellular (that is, stromal cells) and non-cellular components (such as ECM). The cellular components include CAFs, vascular endothelial cells, pericytes, immune cells and adipocytes22 (FIG.1c), whereas the ECM includes fibrous proteins (such as collagen and fibronectin), glycoproteins (including fibulins, fibrillins and thrombospondins), elastin, proteoglycans and hyaluronic acid (which is a non-protein glycosaminoglycan polymer23) (FIG.1). Historically, the stromal cells and ECM of the bTME had been thought of as a passive scaffold enclosing neighbouring cancer cells, supporting tissue architecture, and acting as a barrier to impede tumour spread. However, evidence gathered over the past decade has revealed that stromal cells and ECM in the TME co-evolve during tumorigenesis as non-autonomous drivers of cancer progression and drug resistance via crosstalk with cancer cells24,25. Dynamic communication between tumour cells and the TME are increasingly accepted as important drivers of almost every stage of tumour progression, from local invasion of the primary tumour to distant metastatic colonization21.
The concept of a metastatic TME
Metastatic tumour cells continually remodel the microenvironment of their recipient organ to facilitate their outgrowth. This observation is important because it provides insights into the mechanisms of metastatic outgrowth and their divergent progression from the primary tumour, affecting current conceptual thinking in co-targeting the primary tumour and metastases26. Study results demonstrate that tumour-secreted factors and extracellular vesicles from primary tumours prime a pre-metastatic niche at distant organs to create a permissive environment for metastatic colonization. For example, integrin β-like protein 1 (ITGBL1)-rich extracellular vesicles activate resident fibroblasts and pre-metastatic niche formation in distant organs, which promotes metastatic colonization by secreting pro-inflammatory cytokines27,28. Intriguingly, results from a study in lung cancer indicated that tumour cells could bring along their own fibroblasts (shown using fluorescent labelling) from the primary TME as their own ‘soil’ to facilitate metastatic seeding at distant organs29. The above mechanisms could also be used by bladder cancer cells during their metastatic cascade and colonization.
Despite the widely acknowledged clinical benefits to improve survival by resecting the primary tumour, emerging evidence suggests that surgical removal of the primary tumour triggers a systemic inflammatory response30, and chemotherapy induces a local wound-healing response at the primary tumour31,32; both could lead to metastatic spread30,33. However, how bladder tumour cells interact with the cellular and non-cellular TME to mediate these biological phenomena remains elusive and much work is still needed.
Cellular components of the bladder TME
The major cellular components of the TME include, but are not limited to, CAFs, endothelial cells and pericytes of the vasculature, and immune cells. Fibroblasts are the most frequently studied TME component, but other cell types within the TME are also important. Crosstalk and interactions (including cancer cell–fibroblast, cancer cell–vascular cell and cancer cell–immune cell) between cells in the TME and the urothelial cancer cell compartment co-contribute to the tumorigenic process.
Cancer-associated fibroblasts.
CAFs are generally referred to as activated or reactive fibroblasts34 surrounding cancer cells and are a major cellular component of the TME (FIG.1b,c). Fibroblasts are activated by biochemical signals from urothelial cells (such as TGFβ1 and platelet-derived growth factor (PDGF)) and from the TME (such as oxidative stress)35, as well as inflammatory cytokines (such as IL-1) secreted by immune or urothelial cells36,37 (FIG.1c). Activated fibroblasts secrete growth factors, including hepatocyte growth factor (HGF), EGF and pro-inflammatory cytokines (such as IL-1β, IL-6 and IL-8), or deposit ECM proteins, such as collagens, whose components can promote tumour progression, drug resistance and immune evasion38 (FIG.1c). Several precursors have been proposed as the origins of CAFs, including tissue-resident fibroblasts37,39,40, bone marrow-derived mesenchymal stem cells41,42, haematopoietic stem cells43, monocyte-derived fibrocytes44–46, epithelial cells (derived from epithelial–mesenchymal transition (EMT)47,48 and endothelial cells (from endothelial– mesenchymal transition)49 (FIG.1b). However, in various cancer models, lineage-tracing studies using haematopoietic cell promoter-driven Cre mice have indicated that only a small fraction of CAFs come from bone marrow or haematopoietic-derived cells, and largely concluded that the bulk of CAFs arise from tissue-resident fibroblasts or pericytes6. These findings indicate that CAFs primarily arise from fibrotic events occurring at the local TME, although a small fraction can come from the bone marrow or a haematopoietic cell lineage.
The most commonly used biomarkers to detect CAFs include α-smooth muscle actin (αSMA), fibroblast activation protein-α (FAP), collagens type I and type III, tenascin C50, platelet-derived growth factor receptor-α (PDGFRα) and PDGFRβ, fibroblast-specific protein 1 (FSP1)38,51,52 and podoplanin53 (TABLE 1). However, some of these markers can also be expressed by other cell types and are not necessarily CAF specific when used as single markers6. Unlike normal fibroblasts, CAFs undergo continuous activation without initiating apoptosis or returning to their quiescent or resting state38. This observation is important, as the activated state of CAFs results in the alteration in important biological properties, such as matrix-secreting and matrix-remodelling functions, leading to tissue stiffness as a mechanism to exert pro-tumorigenic roles38. Early studies focused on αSMA+ myofibroblasts as the predominant population of CAFs in cancers6, including bladder cancer53, but emerging evidence has shown that CAFs are more heterogeneous than was originally thought6. Different subtypes of CAFs express different cellular markers, for example, myofibroblastic CAFs (myCAFs) are αSMA+/high and inflammatory CAFs (iCAFs) are IL-6+, PDGFRαhigh54. These distinct CAF subpopulations have been shown to have different roles in supporting tumour development in other epithelial cancer types, such as pancreatic cancer54,55. In bladder cancers, distinct subtypes of CAFs, such as myCAF and iCAF, have only just begun to be immunohistochemically identified53 (TABLE 1). State-of-the-art single-cell RNA sequencing (scRNAseq) has also been used to profile human bladder tumours. Using this technique, to date, seven subpopulations of fibroblasts have been discovered based on COL1A1-positive expression (that is COL1A1+ CAFs), which could be broadly subdivided into two major subtypes: RGS5+ myCAFs and PDGFRA+ iCAFs56, revealing a similarity to the CAF subtypes identified from pancreatic cancer55. CAFs have been reported to promote bladder cancer progression through secreting chemoattractants (such as CC-chemokine ligand 5 (CCL5; also known as RANTES)) and connective tissue growth factor (CTGF), growth factors (such as basic fibroblast growth factor 2 (FGF2) and EGF receptor (EGFR) ligands), colony-stimulating factors (CSFs), TGFβ157,58, HGF59, angiogenic factors (such as vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF)), and ECM-degrading proteinases (such as matrix metalloproteinases (MMPs))60 (FIG. 1c). By contrast, stromal signals such as BMPs have been reported to hinder bladder cancer progression by inducing urothelial differentiation17, which is lost upon tumour progression (FIG. 1b,c). Apart from directly acting on bladder cancer cells, CAFs manipulate the immune environment by producing TGFβ1, which is immunosuppressive and is associated with a T cell exclusion phenotype in human bladder cancer61. This finding suggests that bladder tumour CAFs probably contribute to the confining of T cells within the stromal region via additional mechanisms, excluding them from infiltrating into the epithelial tumour regions to exert their cytotoxic function.
Table 1 |.
Markers of cellular components in the bladder tumour microenvironment
| Cell types | Markers | Refs | |
|---|---|---|---|
| Positive | Negative | ||
| Nonmalignant bladder | |||
| In the lamina propria: fibroblasts with myoid features and sparse myofibroblasts, and interstitial cells of Cajal | Vimentin αSMA Caveolin 1, caveolin 2 PDGFRα CD34 Connexin 43 |
CD34 KIT αSMA Caveolin 1, caveolin 2 PDGFRα |
11 |
| Myofibroblasts and smooth muscle cells | Cadherin 11 | NA | 13 |
| Pericytes | PDGFRβ NG2 |
CD31 | 66 |
| Bladder cancer | |||
| Cancer-associated fibroblasts | αSMA | CD31 | 53,117,118 |
| FAP | NA | 53,117,118 | |
| Tenascin C | NA | 50 | |
| PDGFRα, PDGFRβ | NA | 53 | |
| CD90 | NA | 53,117,118 | |
| FSP1 | NA | 117 | |
| Vimentin | NA | 117 | |
| Tumour vasculature: endothelial cells | CD34 DLL4 |
NA | 73 |
| Tumour vasculature: pericytes | αSMA (mouse), αSMA NG2 (rat) |
NA | 66,67,73,163 |
| Monocyte-derived fibrocytes in the lamina propria | αSMA | CD34 | 44 |
| Monocyte-derived fibrocytes in the lamina propria and muscularis propria | CD34 | NA | 47 |
DLL4, delta-like protein 4; FAP, fibroblast activation protein-α; FSP1, ferroptosis suppressor protein 1; NA, not applicable; PDGFR, platelet-derived growth factor receptor.
In addition to the cellular composition of the bTME, CAFs are also key drivers of the deposition of non-cellular TME components (that is, the ECM) during cancer progression. Bladder tumour CAFs produce fibrillar collagens such as types I, III and VI, which are principal components of the ECM in the TME21. Collagens, laminin, fibronectin, tenascin C and hyaluronic acid create a dense ECM that promotes tumour progression and acts as a physical barrier to limit immune cell infiltration6 (FIG. 1c). Certain CAFs also express FAP, a serine protease that has been proposed to cleave type I collagen, triggering ECM remodelling, and is positively associated with poor disease-specific survival in patients with bladder cancer62,63. In other tumour models, particularly breast tumours, the degradation of type I collagen in the TME has been shown to be mediated by MMP14 or MT1-MMP on the surface of epithelial tumour cells64,65, implying that active ECM remodelling within the TME could be mediated by both CAFs and tumour cells.
Endothelial cells and pericytes.
In a nonmalignant bladder, the urothelium and its underlying lamina propria and muscularis propria receive nourishment from an organized microvasculature that consists of an endothelial cell layer supported by a basement membrane, that is further surrounded by perivascular cells (pericytes)66,67. Suburothelial capillaries are a network of dense, planar meshwork, whereas in the trigone region and the urethral orifice the capillaries are organized in a loose network with elongated meshes68, which are essential for maintaining nutrient supplies to the bladder tissues.
The tumour vasculature is dynamically modified through angiogenesis (the formation of new vessels from pre-existing vessels)69, de novo vasculogenesis (the emergence of a new vascular network via the recruitment of endothelial progenitors from bone marrow and differentiating into endothelial cells)70, and vessel co-option via hijacking pre-existing blood vessels within surrounding tissue71.
Similar to nonmalignant tissues, vessels in tumours comprise endothelial cells surrounded by pericytes. However, endothelial cells in tumour vasculature are immature, characterized by a lower pericyte coverage than those from nonmalignant tissues, and loose and leaky interendothelial cell junctions that often collapse upon interstitial pressure, whereas mature vessels from non-malignant tissues have higher pericyte coverage72. Leaky tumour vasculature affects the bTME by limiting nutrient supply, ultimately leading to hypoxia within tumour regions73. Hypoxia-induced nutrient deprivation could then lead to metabolic rewiring of bladder tumour cells and enhance their invasive properties in this context. Pericyte coverage was found to be considerably lower in vessels from non-muscle invasive bladder cancer (NMIBC) than in the nonmalignant mucosa, and >15% pericyte coverage is predictive of significantly shorter progression-free survival in patients with NMIBC than <15% pericyte coverage (n = 47, P = 0.0036) Bladder cancer cells also secret VEGFA to stimulate endothelial cell release of von Willebrand Factor (VWF), which induces platelet aggregation; and VWF-mediated blood vessel occlusions are also associated with poor patient outcome, linking vascular secreted factors from endothelial cells and pericytes in the bTME with clinical outcomes74.
Differential drug penetration from the existing tumour vasculature can effect drug-induced killing of cancer cells. During chemotherapy or targeted drug treatment, a gradient of drug concentration from the vasculature caused by differential drug penetration from the vasculature is created, meaning that tumours cells further from the vasculature are exposed to lower drug concentrations than those that are closer and, consequently, drug-induced killing is heterogeneous75. Studies using patient-derived bladder cancer xenografts showed that gemcitabine–cisplatin chemotherapy was initially effective in debulking tumour volume during early treatment cycles31,32; however, residual tumours were enriched with chemoresistant cancer stem cells, which were found to localize within hypoxic regions that are far away from existing vasculature and have poor nutrient supply, or these chemoresistant cancer stem cells localize at urothelial–stromal junctions adjacent to collagen-rich CAFs (K.S.C., unpublished work). These findings illustrate that the therapeutic efficacy of chemotherapy is not only determined by their direct cytotoxic effects on urothelial tumour cells but can also be affected by other components within the bTME, such as vasculature maturation and CAF-mediated paracrine effects. Leaky tumour vasculature caused by low pericyte coverage and loose interendothelial cell junctions affects the bTME by causing inadequate nutrient supply and hypoxia, which promotes enrichment of cancer stem cells, and limiting chemotherapeutic response.
Immune cells.
The bTME can be broadly characterized into T cell-inflamed, immune-excluded, or immune-dessert phenotypes, primarily based on the frequency of tumour-infiltrating T cells. In additional to T cells, the bTME is also heavily infiltrated with various innate or myeloid cells, which can have protumoural (for example, immunosuppressive) or antitumoural (such as antigen-presenting) roles dependent on their phenotypes and activation status.
In a healthy bladder, the lumen is open to the environment; thus, the urothelium is constantly challenged by uropathogens, such as Gram-positive or Gram-negative bacteria and fungi76. Thus, unsurprisingly, a healthy bladder in a steady state is normally colonized by tissue-resident immune cells and protected by both innate and adaptive immune cells77. The types of innate immune cells in a healthy bladder include tissue-resident macrophages, dendritic cells, mast cells, neutrophils and natural killer cells78. For instance, dendritic cells with a phenotype similar to that of skin-resident Langerhans cells have been reported to reside within the lamina propria in human79 and mouse bladders80. In addition, tissue-resident macrophages in mouse bladders were broadly categorized into CD11c+ and F480+ myeloid cells81,82 and two functionally distinct resident macrophage subsets, MacM and MacL, with distinct transcriptomes and response to UTI, were identified, which is important, as a history of recurrent UTI elevates the risk of bladder cancer.83. Furthermore, resident αβ and γδ T cells have also been observed within the urothelium and submucosa of naive human bladders, but not in the detrusor muscle84. These γδ T cells are unconventional T cells capable of recognizing and lysing cancer cells in a MHC-unrestricted manner.
Immune cells have been extensively studied in bladder cancer, which is highly responsive to conventional immunotherapy, that is, BCG85. BCG, an antituberculosis vaccine, has been used for the clinical management of high-risk NMIBCs since the 1970s86, long before the success and accelerated FDA approval of ICIs in 201787. Muscle-invasive bladder cancer (MIBC) is molecularly heterogeneous between patients and is characterized by the existence of different molecular subtypes and/or differentiation status88, which can be further subdivided into those with high T cell infiltration (inflamed) and those with a low T cell infiltration (termed ‘immune desert’) or exclusion of immune cells (termed ‘immune-excluded’)87,88. A consensus report published in 2020 on the molecular classification of MIBC described a transcriptionally stroma-rich tumour subtype with overexpression of smooth muscle, endothelial, fibroblast and myofibroblast gene signatures that is also enriched with T cells and B cells, using MCPcounter analysis89. In The Cancer Genome Atlas (TCGA) data set, a high immune gene signature indicative of immune infiltration in the bTME stroma is associated with a remarkably improved 5-year DSS of 80% versus <25% for patients with an uninflamed subtype90. These findings are supported by IHC results from another study that showed that a high level of CD8a T cell infiltration within a specific CD90+ stroma is associated with an exceptionally good prognosis in treatment-naive MIBCs, compared with tumours containing other types of stroma53. These findings indicate that certain stromal components within MIBC probably influence immune infiltration and, therefore, patient prognosis53. This theory is supported by another study in which TCGA data were used to stratify patients with bladder cancer into those with high or low stromal or immune scores, or a combination of both91; immune score did not correlate significantly with tumour stage whereas the stromal score significantly positively correlated (P = 1.5 × 10−8). Additionally, in patients with combined low stromal and immune scores, the common downregulated genes in patients with low immune score as well as those with low stromal score were analysed and found to be enriched for gene ontology categories such as ‘ECM’ and ‘collagen-containing ECM’91. These transcriptomic data provide initial prediction of immune cell components within each bladder cancer subtype and their association with clinical outcome. However, the precise tumour-infiltrating immune landscape illustrated by cell-surface markers at the protein level and how they interact with the bTME remains an active area of research. This observation is important, as the spatial and subcellular colocalization of T cells and myeloid cells with the stroma will reveal important insights into their mechanistic interactions during cancer development and therapeutic response of current and future research.
Non-cellular components
The non-cellular components of the bTME include a basement membrane that separates urothelial cells from the stroma in the steady state, and remodelled ECM components that are modified from a normal core matrisome.
The basement membrane (or basal lamina).
In a healthy bladder, the multilayered urothelial cells are separated from the underlying lamina propria and the muscularis propria by a basement membrane, which is composed of a meshwork of ECM components, including collagen type IV, laminin, nidogen, entactin, perlecan, heparan-sulfate proteoglycan and the anchoring fibril collagen VII (FIG. 1a). In a healthy bladder, urothelial basal cells attach to basement membrane ECM protein via integrins binding to type VII collagen, anchoring fibrils and laminins, which provide the crucial biological signals for maintaining epithelial apical-basal polarity18.
In cancer, degradation of the basement membrane ECM proteins by matrix metalloproteinases (MMPs) and subsequent loss of urothelial polarity are hallmark characteristics of invasive tumours18 (FIG. 1b,c). In the context of bladder cancer, several historical studies were conducted to establish a link between the loss of basement membrane ECM proteins (specifically collagen IV and VII) and bladder tumour cell invasion into the underlying stroma and muscle layers19,92. In MIBCs, collagen IV staining is widely fragmented or absent in >5% of tumour areas (which correlates significantly with a worse 3-year survival, n = 29, P < 0.001) unlike in NMIBC and non-cancerous urothelium, in which the staining pattern of collagen IV is continuous, indicating an intact basement membrane that is not being breached by invasive cancer cells. This observation is interesting, as NMIBCs are usually exophytic and papillary in nature, and are not accompanied by a breach in basement membrane until later tumour stages92. However, analysis of MIBCs revealed that the loss of the anchoring fibril collagen VII is associated with derangement and depolarized localization of the α6β4 integrin subunits, which are normally expressed at the basolateral surface of urothelial basal cells19. Promoter methylation of genes encoding laminin 5 (LAMA3, LAMB3 and LAMC2) silences these genes, which occurs in bladder tumours and exfoliated cells in the urine. The methylation frequency of genes encoding laminin 5 is between 21% and 45% and is associated with poor prognosis (n = 128, both NMIBC and MIBC). Nonmalignant urothelium lacked promoter methylation, LAMA3 and the LAMB3 methyl ation index were significantly higher in MIBC than in NMIBC samples (P < 0.0001), and high LAMC2 methylat ion index was significantly associated with reduced patient survival (n = 91, P = 0.002)93. Collectively, these studies support the idea that alterations in basement membrane components (such as collagen IV, VII, laminin 5 and the anchoring integrins), either by enzymatic degradation (by MMPs, for example) or via gene promoter methylation lead to basement membrane modification and loss of tumour cell polarity, which precedes muscle invasion (FIG. 1c). Importantly, these alterations are considered poor prognostic indicators for patients with bladder cancer.
The core matrisome.
Beyond the basement membrane, the bTME is composed of ECM components that are derived from a ‘core matrisome’ of ECM proteins from the non-malignant interstitium. This list of core matrisome proteins was defined by analysing protein extracts enriched for ECM using liquid chromatography combined with mass spectrometry followed by in silico definition via bioinformatics prediction and gene ontology94. Using these approaches, genes encoding all components constituting the ECM were defined as the ‘core matrisome’ and those components associated with it were defined as ‘matrix-associated’ proteins, which constitute 1.0–1.5% of the mammalian proteome. The ECM core matrisome comprises ~300 proteins that can be categorized into collagen subunits (>40 subunits), glycoproteins (>200 proteins, including laminins, fibronectin, tenascins, secreted protein acidic and rich in cysteine (SPARC), thrombospondin 1 and many others), proteoglycans (>35 proteins with large glycosaminoglycan chains), ECM-bound growth factors and cytokines, and ECM-modifying enzymes95 The bladder tumour ECM components must be modified from this core matrisome; however, studies comprehensively characterizing the non-malignant and tumour bladder matrisome have not be performed to date.
The ECM had been thought to be a passive barrier and physical scaffold; however, the ECM is now known to provide a range of important biochemical and biomechanical signals that influence many cellular processes and functions, such as cell spreading, growth, proliferation, migration, differentiation and organoid formation96. Importantly, degradation of ECM by MMPs during tumour progression is one mechanism by which ECM-bound growth factors, such as FGFs, are released and function as biochemical signals that drive cancer cell invasion. On the other hand, SPARC — an ECM glycoprotein — impedes bladder carcinogenesis, partly owing to its role in inhibiting the acquisition of an inflammatory phenotype in macrophages and CAFs, through inhibiting NF-κB activation97. Loss of SPARC in a mouse model significantly enhanced urothelial neoplasia and metastasis in response to a chemical carcinogenesis regimen (P < 0.05), which is concordant with the progressive loss of SPARC expression when NMIBC progresses to MIBC97. Collectively, these findings illustrate key examples of the core matrisome components, their dynamic alterations and functions in regulating cell signalling and stromal cells within the bTME during tumorigenesis.
Collagen as the major ECM within bladder TME
Collagens are a major ECM component within the TME, which comprises a family of 43 collagen or collagen-associated proteins out of a total of 274 (mouse) or 278 (human) core matrisome proteins94.
The structure of collagen fibres.
Fibrillar collagens, which are the key structural components of the ECM, are helical heterotrimers or homotrimers98. Triple-helical collagen monomers are assembled and covalently crosslinked into tightly packed fibrils with a diameter of 10–300 nm (FIG. 2A), and the fibrils are bundled into large fibres. Both homotrimeric (three identical α subunits, for example, collagen type III) and heterotrimeric (2 α subunits and 1 β subunit, such as collagen type I) triple helices are found in ECM98 (FIG. 2A). Collagen subunits have three domains: the N-terminal non-triple helical (N-telopeptide) domain; the triple helical domain; and the C-terminal non-triple helical (C-telopeptide) domain98 (FIG. 2B). The central triple helical domain of most collagens contains more than 300 Gly-X-Y repeats, accounting for more than 95% of the polypeptide; X is frequently proline and Y is frequently 4-hydroxyproline. The short segments of N-telopeptides and C-telopeptides do not assume the triple-helical conformation (FIG. 2B).
Fig. 2 |. Collagen as the major extracellular matrix in bladder tumour microenvironment.
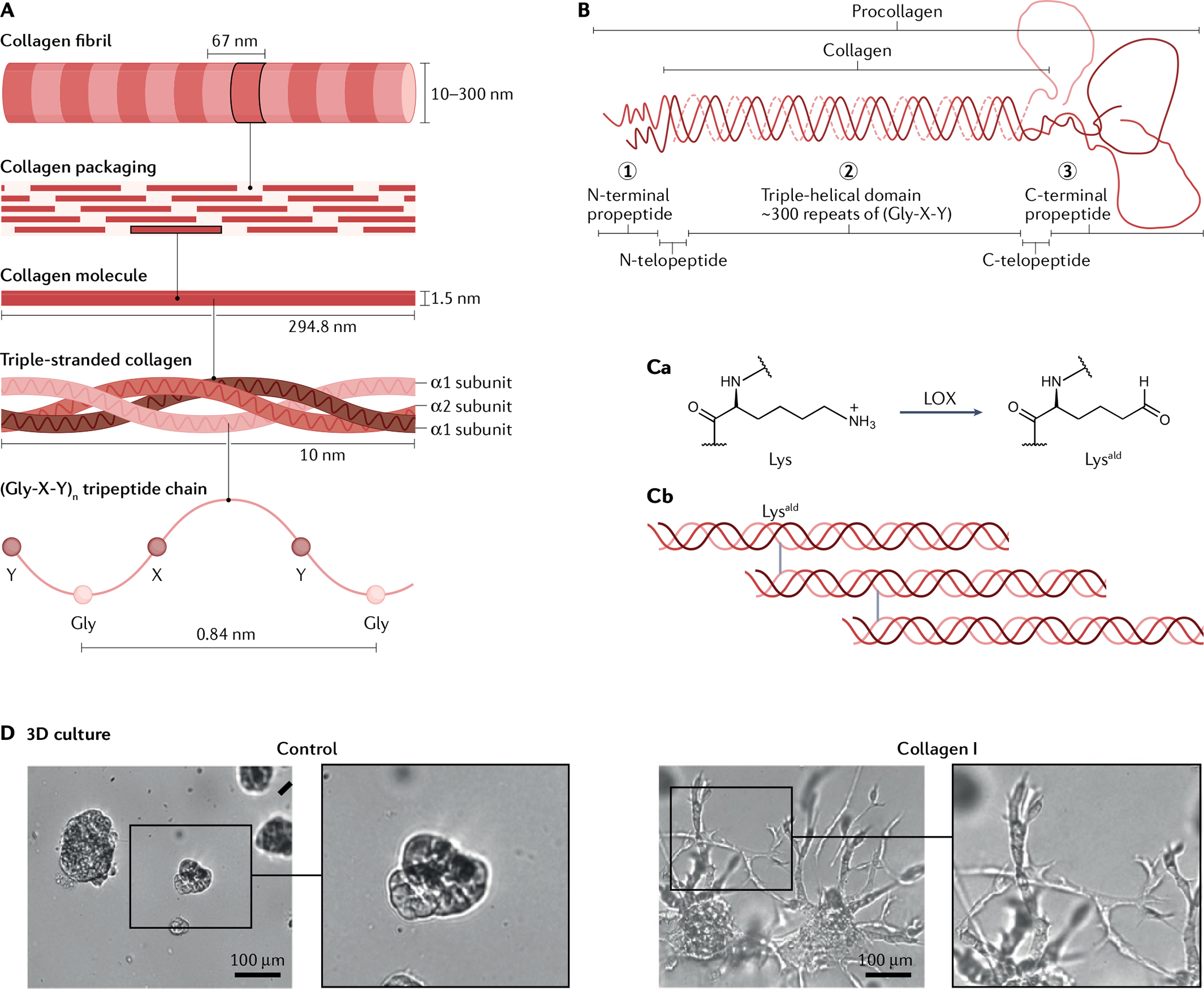
A | Packaging of triple-helical collagen monomers by covalent crosslinking into tightly packed fibrils. B | Collagens consist of three domains: the N-terminal non-triple helical (N-telopeptide) domain (1), the triple helical domain (2), and the C-terminal non-triple helical (C-telopeptide) domain (3), which are surrounded by terminal pro peptide domains. C | Lysyl oxidase (LOX)-mediated collagen crosslinking. Ca | Lys residues on the N-telopeptides and C-telopeptides can be oxidatively deaminated by the extracellular LOXs, a family of extracellular enzymes to form reactive aldehydes Lysald. Cb | Lysald forms covalent crosslinks, which greatly increase the tensile strength of collagen and, therefore, tissue stiffness. Stiffened collagen promotes integrin clustering and focal adhesions that facilitate tumour cell invasion. D | Addition of collagen I into a 3D microenvironment created by growth factor-reduced Matrigel induces invasive sprouting of bladder cancer cells in vitro, compared with control (Matrigel without collagen I). Part D reprinted from REF.21, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Collagen crosslinking to increase ECM stiffness.
Among the interesting characteristics of collagen are its extensive post-translational modifications. For instance, the telopeptidyl Lys residues can be hydroxylated to form hydroxylysine (Hyl) — a unique modification only found in collagens98 (FIG. 2C). In many cancer types, Lys and Hyl residues at the N-telopeptides and C-telopeptides can be oxidatively deaminated by the extracellular lysyl oxidases (LOX), a family of extracellular enzymes, to form reactive aldehydes Lysald and Hylald, respectively98,99 (FIG. 2Ca). These modifications initiate a series of condensation reactions to form various covalent intermolecular crosslinks involving juxtaposed Lys, Hyl and His residues on the neighbouring collagen trimers, resulting in the formation of Hylald-derived collagen covalent crosslinks99,100 (FIG. 2Cb). These covalent crosslinks greatly increase the tensile strength of collagen and, therefore, tissue stiffness. Stiffened collagen promotes integrin clustering and focal adhesions that facilitate tumour cell invasion101. A subset of patients with Ta or T1 (that is, NMIBC) bladder cancer exhibited straighter collagen fibres (that is a low curvature ratio) than other NMIBCs, shown using second harmonic imaging microscopy102, indicating increased tensile strength or collagen stiffness owing to collagen crosslinking, and these patients with NMIBC experienced an increased rate of invasive progression compared with those who did not have straighter collagen fibres102. Currently, the mechanism leading to the increased collagen stiffness in these patients remains unclear, although LOX-mediated enzymatic crosslinking was implicated in other cancer types, such as breast cancer101. Alternatively, collagen can be modified by a non-enzymatic crosslinking mechanism103. During aging, non-enzymatic glycation of Lys and Hyl residues on collagen leads to advanced glycation end products — a type of non-enzymatic collagen crosslink that also results in stiffness103. In vitro, glycation-mediated non-enzymatic collagen crosslinking enhanced breast tumour cell invasion. This type of aging-associated collagen crosslinking (that is non-enzymatic glycation) could contribute to ageing-associated collagen stiffness and bladder cancer progression. This speculation is important as bladder cancer predominantly occurs in older people (9 out of 10 patients with bladder cancer are >55 years old104), and the underlying mechanisms or contribution of the ageing bTME remains unclear. Similar to enzymatic collagen crosslinking, this less-studied and age-associated form of non-enzymatic collagen crosslinking will probably become an interesting future research topic to connect ageing and bladder tumorigenesis.
Collagens as signalling molecules.
Collagens can trigger cell signalling by activating collagen receptors and their downstream signalling cascades in adjacent cells and releasing biologically active collagen fragments that mediate signalling at distant sites.7 This signalling is accomplished by direct or indirect binding to collagen receptors. For example, integrins (including α1β1, α2β1, α10β1 and α11β1) recognize the triple-helical GFOGER amino acid sequences on major collagen types via their α1 domain105,106, whereas discoidin domain receptors (DDR) recognize the GVMGFO amino acid motif on fibrillar collagens through their discoidin domain107,108.
Few mechanistic studies have been performed to evaluate the functional roles of collagens in bladder cancer cells. Results from one study revealed that collagen type I acts as a functional ligand to induce migration of bladder cancer cells (the T24 cancer cell line) and patient-derived bladder cancer cells in a monolayer wound closure assay21. Soluble collagen I was added to the supernatant on top of these monolayer cultured bladder cancer cells, which effectively induced a dose-dependent enhancement of cell migration, measurable by wound closure. Although exogenous collagen I treatment consistently and significantly induces wound closure in this 2D environment (T24, P < 0.01; patient-derived xenograft (PDX), P < 0.005), its effect is less pronounced than other specialized migration-inducing growth factors, such as HGF. This observation is important as the biological effects of collagen I in a 3D microenvironment is more pronounced than in a 2D environment. In a 3D microenvironment created using growth factor-reduced Matrigel, collagen type I induces significantly more extensive sprouting and invasive phenotype in the same bladder cancer cell models (T24 and PDX, P < 0.005) than its effects in a 2D monolayer microenvironment21 (FIG. 2D). The high potency of collagen I to induce invasion in a 3D environment compared with a 2D environment is highly relevant biologically, as collagen type I typically exists and functions in a 3D ECM microenvironment in vivo. These findings suggest that collagen type I has a role as a signalling molecule in promoting bladder cancer cell migration and invasion. At the lung metastatic site, collagen production in a newly identi fied metastatic niche (the airway smooth muscle cells) (FIG. 3) creates a permissive microenvironment for the adhesion and outgrowth of metastatic bladder cancer cell foci, by activating signalling downstream of collagen receptors within the cancer cells21. Collagen type III secreted from these airway smooth muscle cells activates the collagen receptor DDR1 (or CD167a) in bladder T24 cancer cells and PDXs and its signalling via a molecular chaperone containing HSP90 and CDC37 through its client protein STAT3 (REF.21), which maintains the survival of these metastatic foci.
Fig. 3 |. The bTME and the met-TMEs in various organs.
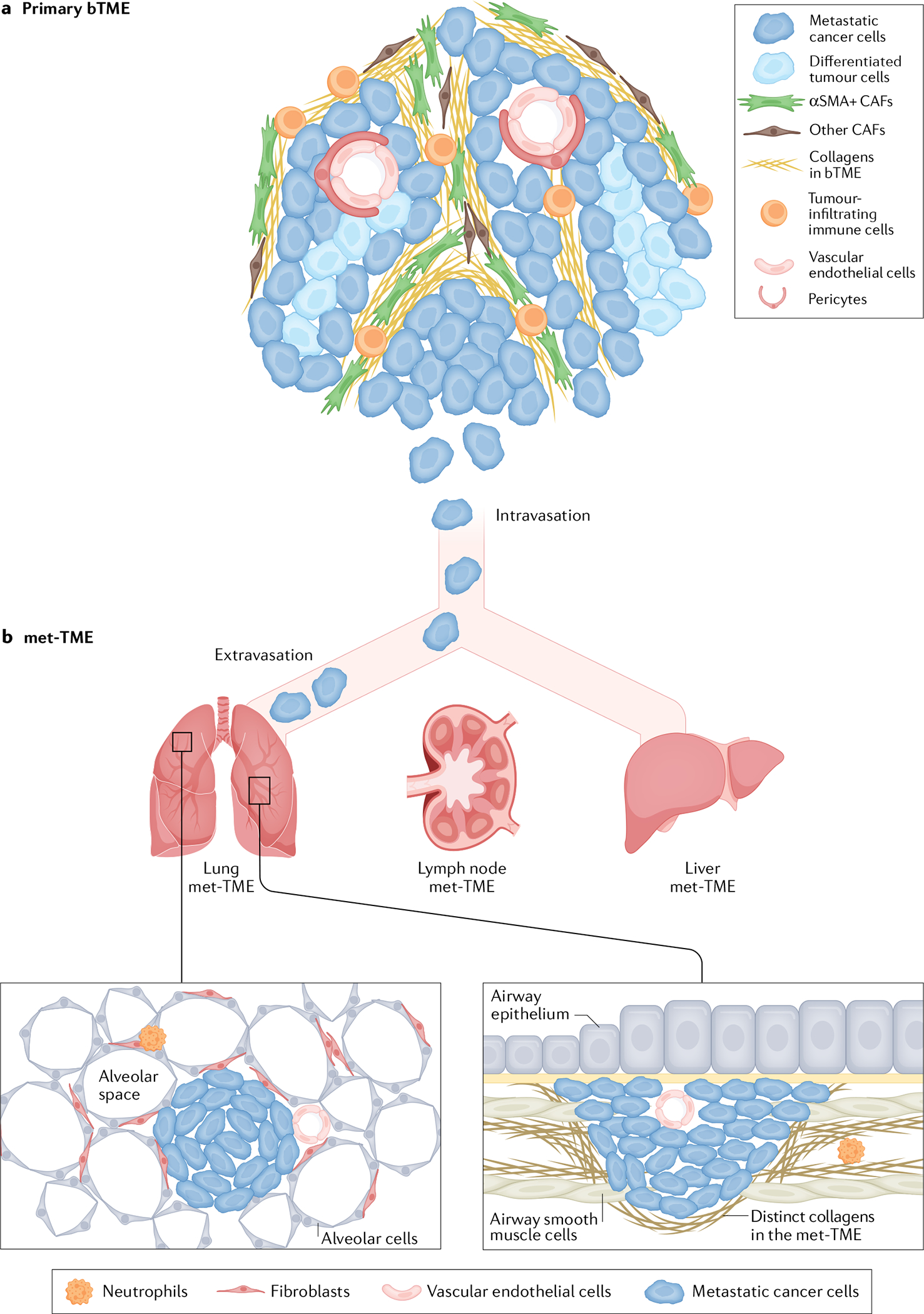
a | The primary bladder tumour microenvironment (bTME), its cellular and non-cellular components supporting metastatic cancer cells to intravasate into the circulation. b | Extravasation of metastatic cancer cells and their colonization into distinct metastatic tumour microenvironments (met-TMEs) at different organs, such as lung, non-regional lymph nodes and liver. Airway smooth muscle cells are a newly identified lung met-TME that secrete collagens (distinct from the bTME) to support the preferential colonization of cancer cells that expresses the collagen receptor discoidin domain receptor 1 (DDR1). The metastatic colonies within airway smooth muscle cells have a different morphological phenotype from the typical lung metastatic foci within the alveoli space. The lymph node and liver met-TME remain largely uncharacterized; however, they could contain different TME components that support metastatic tumour outgrowth and are probably distinct from the primary bTME.
Other ECM components in the bTME
Other major non-collagenous ECM proteins within the bTME include laminin and fibronectin. Laminins are heterotrimeric glycoproteins composed of 15 possible α, β and γ trimeric combinations109; they are the most abundant structural non-collagenous glycoproteins, which are a major component of the basement membrane and also surround smooth muscle bundles in the detrusor muscles. Laminins are composed of a long arm for integrin binding, and a short arm for mediating cell signalling through laminin–nidogen complex formation and Ca2+-dependent interactions109. In bladder cancer, genes encoding laminin 5 — a basement membrane laminin — such as LAMC2 were found to be methylated (or silenced), and increased methylation correlated with reduced patient survival (n = 91, P = 0.002)93. Fibronectin is another major ECM glycoprotein within the urinary bladder, which has a central role in collagen fibrillogenesis in vivo109. In the nonmalignant bladder, fibronectin is primarily expressed in the lamina propria underneath the basement membrane and surrounding muscle fibres110. In bladder cancer, fibronectin was found to be expressed in the tumour stroma in 89% of bladder tumours (n = 103) and correlated positively with tumour stage and proliferative activity111. These findings reveal that other non-collagenous ECM proteins also have important roles during bladder tumorigenesis.
Communication between the TME and tumour cells
Cell–cell communication between the cellular components of the bTME and tumour cells demonstrates various biological functions during bladder tumorigenesis. Wound healing and cancer — a wound that never heals — have been shown to have many similarities. Key epithelial–stromal interactions occur during wound healing and bladder tumorigenesis, with the collagen ECM and its effects being central in this process.
Epithelial–stromal interactions: wound healing
Collagen has a central role throughout multiple phases of the wound repair process in most tissues112. During the initial phase of tissue injury, vascular endothelial lesion or rupture leads to the exposure of collagen and tissue factors from the subendothelial matrix. Platelets adhere to the subendothelial surface within minutes of blood vessel rupture, and become activated when their cell-surface integrins and glycoprotein VI receptors are exposed to extravascular type I collagen, releasing growth factors, cyclic AMP and adhesive glycoproteins (such as fibrinogens, fibronectin, thrombospondin and VWF), resulting in the deposition of a fibrin clot or thrombus formation (that is, platelet aggregation)112. Damaged cells and aggregated platelets then release growth factors such as PDGF and TGFβ, as well as pro-inflammatory cytokines, such as IL-1β, IL-6 and IL-8, causing vasodilation and the recruitment of inflammatory immune cells112. Neutrophils are the first immune cells recruited by chemotactic factors into the wound site to kill and clear pathogens, followed by an influx of macrophages, T cells and other immune cells, and eventually inflammation resolves112. Epithelial cells at the wound edge dedifferentiate and stretch out over the exposed wound bed, followed by epithelial cell proliferation that drives re-epithelialization and wound closure113. Within the wound bed, activated fibroblasts synthesize collagen and other ECM proteins to replace the fibronectin and fibrin-rich provisional ECM with a new ECM (known as the granulation tissue) and mediate wound contraction, facilitating re-epithelialization113. The granulation tissue becomes a collagen-rich scar that provides temporary strength to the damage site. Scar remodelling (which typically occurs long after re-epithelialization is complete and the immune response is largely resolved) is characterized by a decrease in fibroblast density and MMP-mediated proteolysis by resident macrophages114,115. During bladder wound healing, urothelial cell-derived SHH, which is a ligand in the hedgehog signalling pathway, induces bidirectional crosstalk with the underlying stromal fibroblast cells to induce the secretion of paracrine factors such as WNT and BMPs that signal back to the urothelium. Urothelial SHH-induced stromal factors include WNT2 and WNT4 (which are short-range mitogenic factors that mediate wound-induced proliferation of urothelial basal cells in close proximity to the stromal fibroblasts16) and BMP4 and BMP5 (which are long-range pro-differentiation factors that mediate umbrella cell differentiation)17. WNT and BMP signalling seem to have contrasting functions; however, their effects manifest at different distances from the origin of their secretion during wound healing, which enables the proper regeneration of a full-thickness bladder urothelium comprising basal, intermediate and differentiated cells16,17.
Some of these urothelial–stromal interactions during wound healing, for example, the roles of collagen during various stages of wound healing, and the reciprocal signalling of urothelial SHH and stromal WNT or BMP, either remain conserved or become aberrantly expressed during bladder tumorigenesis, and serve as key drivers during bladder cancer development.
Epithelial–stromal interactions: tumorigenesis
In bladder tumorigenesis, urothelial SHH expression is gradually lost during progression from carcinoma in situ to MIBC (FIG. 1b,c). Basal cell-derived SHH was shown to induce the reciprocal secretion of growth-restraining and differentiation signalling molecules from stromal fibroblasts, such as BMPs, to inhibit tumour formation17. Pharmaceutical activation of BMP via the small molecule FK506 inhibited bladder tumour formation in a classical nitrosamine-induced carcinogenesis mouse model17. These observations support the notion that during early bladder tumour development, transformed urothelial cells need to overcome stromal-derived inhibitory signals (FIG. 1b).
Collagen type I secreted by CAFs induces bladder cancer cell migration and invasion21, and this effect is significantly more pronounced in a 3D matrix (which better recapitulates tumour cell movement within the bTME in vivo than a 2D in vitro environment), promoting invasive sprouting of metastatic bladder cancer cells21 (FIG. 2D). Collagen type I activates DDRs, DDR1 (or CD167a) and DDR2 (or CD167b) on bladder cancer cells, which are collagen receptors and important mediators of cell migration and invasion through 3D matrices21. Interestingly, overexpression of DDR1 in bladder cancer cells independent of collagen stimulation also enhances local invasion and metastasis to lung21. This observation is interesting, as only a fraction of patients with DDR1+ bladder cancer also have high collagen expression. These functional studies are, therefore, clinically relevant, supporting the notion that overexpression of DDR1 itself could result in DDR1 signalling pathway activation independent of its binding to collagen21. High collagen deposition in human bladder cancer correlates with high expression of DDR2. Amplification of DDR2 is observed in ~20% of human bladder cancers116; however, its functional role during tumour progression and metastasis is currently unknown. Cancer cells also secrete certain collagen types or express membrane-associated collagens, whereas CAFs are the major source of most fibrillar collagens (K.S.C., unpublished work), as well as other growth factors such as TGFβ1 that increase bladder cancer cell invasion via inducing EMT within the bTME117. In addition to these conventional ligand–receptor interactions between urothelial and stromal cells within bladder cancer, exosome-based cell–cell communication has been reported118,119. Bladder cancer cell-derived exosomes, which are internalized by fibroblasts, contain TGFβ1, which induces their proliferation and differentiation into CAFs118. Conversely, CAF-derived exosomes transfer long non-coding RNA from fibroblasts to bladder cancer cells, promoting bladder cancer cell proliferation and invasion119. Collectively, these studies are key examples of epithelial–stromal cell communication during early and advanced stages of bladder tumorigenesis.
Cancer–stromal–immune cell interaction
Sequential recruitment and interactions between urothelial, stromal and immune cells have key roles during the wound healing process. Similarly, cytokines and chemokines secreted by bladder cancer cells promote the recruitment of immune cells to the bTME120. High tissue expression of the chemokine CXCL1 is associated with increased tumour stage in bladder cancer, and its detection in urine is a biomarker for early detection121. Interestingly, highly invasive bladder cancer cell lines such as T24 and UMUC3 also express and secrete CXCL1 (REF.121), which recruits tumour-associated macro phages (TAMs) and CAFs into the bTME in coculture assays, initiating a feedforward loop to induce CXCL1 secretion from TAMs and CAFs that enhances the invasive properties of bladder tumour cells in coculture assays122,123. scRNAseq of human bladder tumours in conjunction with bioinformatics analyses is beginning to help elucidate such complicated cellular crosstalk networks56. For example, bladder tumour cells have been shown to downregulate MHC-II compared with nonmalignant urothelial cells, suggesting one mechanism employed by tumour cells to evade immune surveillance. Network analysis further revealed that iCAFs have increased expression of CXCL12, which can interact with its receptors (such as CXCR4 and CXCR3) on a wide variety of immune cells (such as CD8+ T cells, CD4+ T cells, regulatory T cells, NKT cells, dendritic cells, TAMs and B cells) and endothelial cells. Particularly, CXCL12 correlated positively with a TAM signature and high CXCL12 expression correlated with poor prognosis in TCGA MIBC cohort, implying a connection between CXCL12-expressing iCAFs to TAMs56. scRNAseq platforms provide a new technology for predicting cell–cell communications within the bTME globally, which will create unique opportunities for studying the complex biological crosstalk between these distinct cell types within bladder cancer.
Cancer–endothelial cell interaction
Endothelial cells within the bTME could also communicate with neighbouring bladder cancer cells. Specifically, when bladder cancer cell lines such as RT4, T24 and TCCSUP were co-cultured with human umbilical vein endothelial cells (HUVECs) as a commonly used model of vascular endothelial cells, bladder cancer cells secreted soluble ephrin A1, a regulator of angiogenesis, causing the downregulation and internalization of its receptor EPHA2 on endothelial cells, resulting in endothelial cell activation and promoting angiogenesis124. In another study using T24 and 253 J human bladder cancer cells and HUVEC coculture assays, bladder cancer cells secreted VEGFA and VEGFC that activated VEGFR2 on endothelial cells, which then released EGFR ligands such as EGF, amphiregulin, and TGFα125. These ligands reciprocally activate EGFR–AKT pro-survival signalling in bladder cancer cells and simultaneously triggered CXCL signalling in bladder cancer cells, which positively feedback to cause recruitment of more endothelial cells to promote tumour migration and invasion in vitro, indicative of such reciprocal signalling during tumour progression125. Similarly, another study revealed VEGFA secreted by immortalized bladder cancer cells could induce VWF secretion from endothelial cells (using HUVECs as a model), causing platelet aggregation74. Further data obtained from patient tissue sections revealed that VWF-mediated vessel occlusion was associated with poor clinical outcome, indicative of a role for VWF-mediated hypercoagulation during the metastatic process in bladder cancer patients74. Collectively, these studies illustrate that intricate crosstalk occurs between various cellular components of the bTME that contributes to bladder cancer development and metastatic progression.
The metastatic tumour microenvironment
A genomic profiling study in which primary human bladder tumours, matched lymph nodes and metastatic tumour foci at distant sites were compared revealed intriguing observations that could be extrapolated to the bTME126. Despite common mutations being shared between the primary tumours, matched lymph nodes and metastatic tumours, distinct mutations were observed, indicating a divergent or parallel progression of tumour evolution at the primary and metastatic sites126,127. One plausible explanation for this observation is that the TME at the primary bladder tumour site (FIG. 3a) is distinct — both qualitatively and functionally — from the TME associated with the metastatic foci (FIG. 3b). Distinct ECM and the associated growth factor components in the bTME and met-TME could drive divergent evolutionary pathways in the primary tumour and the metastatic foci (M.L. and K.S.C., unpublished work). Early studies revealed that the ECM of the primary bladder tumour is predominated by collagen type I, whereas in a metastatic niche in the lung airway smooth muscle cells secreted different collagens, including collagen types III21, VI and XII (K.S.C., unpublished work) (FIG. 3b) that supported the colonization of DDR1+ metastatic tumour cells21. Such initial characterization of collagen subtypes within the bTME and lung met-TME imply that fundamental differences exist between the two niches, which harbour clonal selection of distinct tumour characteristics. Obviously, the ECM components within the bTME and the lung met-TME are more complex than just the collagens it contains; moreover, the concept of distinct met-TMEs could be extrapolated to other metastatic sites including non-regional lymph nodes, liver and bone (FIG. 3b). This observation of divergent TMEs between primary and metastatic sites provides new opportunities to study unique met-TMEs and their roles in supporting metastatic colonization. Historically, the met-TME has been extremely difficult to study owing to the limited clinical specimens and models available. With the expanding acquisition of patient tissue from metastatic bladder cancers, such as establishment of rapid autopsy programmes, and development of PDX models that recapitulate spontaneous metastasis21, defining clinically relevant met-TMEs for potential therapeutic intervention in metastatic bladder cancer will become possible.
Clinical significance of the bladder TME
The clinical relevance of bTME components has diagnostic and prognostic value, as well as considerable therapeutic implications.
Diagnostics
Early stromal alterations in the TME probably precede urothelial changes, which can be used as urine biomarkers for early detection. A validated urine-based bladder cancer diagnostic signature for NMIBC and MIBC with 85% sensitivity and 81% specificity for both cancer types has been reported128. This signature is composed of ten biomarkers; APOE, ANG, A1AT, CA9, IL8, MMP9, MMP10, PAI1, SDC1 and VEGFA129, which have a varied range of biological functions, including factors secreted by or affecting stromal cells (IL-8 and VEGFA)130,131, enzymes directly degrading or affecting the ability to degrade ECM proteins (MMP9, MMP10 and PAI1)132,133, and pro-angiogenic cytokines (IL8, VEGFA and ANG)134,135, all of which are associated with the bTME and could promote tumour growth. The origin of these biomarkers is an area of speculation. These ten biomarkers were found to be present in both the urothelial136 and stromal137 components of bladder tumours using immunohistochemical staining. Furthermore, overexpression of MMP10, PAI1 and ANG were associated with increased tumour grade or tumour stage137, indicating that these markers are not only diagnostic markers but could also exhibit prognostic value and probably have certain biological roles during bladder tumorigenesis. For example, one of the diagnostic biomarkers, SDC1 (a cell-adhesion molecule implicated in epithelial cell migration)138, demonstrated a marked transition from its usual expression in the cell membrane to the cytoplasm as bladder tumour grade and stage increased139. The loss of this crucial membrane adhesion protein in high-grade and/or high-stage bladder tumours might facilitate tumour growth and metastasis.
Furthermore, urine-derived lymphocytes are an easily accessible source of T cells from the bTME. Effector CD8+ and CD4+ cells and regulatory T cells detected within the urine accurately recapitulate the immune cell landscape in bladder tumour and were used to identify the immune checkpoint and T cell receptor repertoire within the tumours140. Chemokines, cytokines and other secreted proteins in the urine could also provide information concerning the bladder tumour, giving details about the urothelial and stromal components of the tumour. Thus, non-invasive evaluation of urine samples might provide a glimpse highly representative of the bTME; conversely, stromal alterations within the bTME and associated extracellular release of TME components could be detected in the urine and used as diagnostic markers on prospective validation.
Prognostics
The discovery of distinct CAF subtypes, their relative abundance and associated secreted products (such as collagen deposition) have considerable prognostic value in bladder cancer.
Secreted collagens and clinical outcome.
One key clinical issue for NMIBC is the unidentified mechanism causing its invasive progression into MIBC and collagens are the most extensively studied ECM in bladder cancer prognosis. Patients with NMIBC containing collagen fibres that have implied increased tensile strength caused by collagen crosslinking (that is, a low curvature ratio, based on second harmonic imaging) experienced an increased frequency of invasive progression102. These observations are consistent with functional studies indicating that collagen crosslinking increases stiffness and invasive properties of other epithelial cancer cell types101. In additional to COL1A1 and COL1A2 (REF.102), COL4A1 and COL18A1 are amongst a 12-gene signature that is predictive of invasive progression of NMIBC (P < 0.001)141,142. High mRNA expression of other collagens, such as COL5A2, COL6A1, COL6A2 and COL6A3, has been reported to correlate with poor overall and recurrence-free survival143. In the context of MIBC, increased expression of COL1A1 and COL1A2 correlates with increasing pathological tumour T stages, indicative of a relationship with local invasion to the muscularis propria and into perivascular tissues21. At the protein level, immunohistochemical analysis of collagen type I staining in NMIBC samples revealed complex staining patterns, which include papillary tumour stroma staining; vascular tumour stromal staining; reticular stromal staining; and dense lamina propria staining near the tumour–ECM boundary, showing that the pattern of collagen staining is also an important factor in prognosis102. Dense collagen I staining in lamina propria near the tumour–ECM boundary correlated positively with poor progression-free survival (P = 0.0145) suggesting its biological role as a ligand to induce cell invasion102.
Cancer-associated fibroblasts and clinical outcome.
CAFs are the principal cell types that produce collagens in the bTME. The association of several commonly used CAF or fibroblast markers (such as αSMA, CD90, FAP, and PDGFRα and PDGFRβ) with clinical outcome in NMIBC and MIBC has been evaluated. αSMA+ fibroblasts are considered to be myofibroblasts, with reported tumour-inhibiting or tumour-promoting properties and have been reported to be associated with poor prognosis in several epithelial tumour types53. FAP positivity in the bladder stroma is an independent poor prognostic indicator, predicting reduced 5-year-survival (HR (95% CI) 2.25 (1.08–4.67), P = 0.030) in patients with either NMIBC or MIBC53. This observation is consistent with the results of another study showing that co-expression of FAP, CK5 or CK6 and CD44, which label basal tumour cells, is a strong prognostic indicator for disease-specific survival (HR = 2.3; P = 0.001), muscle invasion (HR = 2.47; P = 0.02) and nodal involvement (HR = 3.47; P < 0.0001) in patients with bladder cancer62, implying the presence of functional crosstalk between FAP+ stromal cells and cancer stem cells with a basal phenotype9,141,144, CD90 positivity in the bladder stroma was associated with high CD8a+ T cell infiltration and an improved, but not statistically significant, 5-year overall survival (0.58 (0.27–1.25), P = 0.165) in patients with either NMIBC or MIBC53, suggesting a role for certain CAFs in recruiting CD8+ antitumour T cells to the bTME. The focal adhesion protein kindlin 2 that controls bidirectional signalling of integrins is expressed at a higher level in bladder CAFs than in normal stromal fibroblasts, and stromal kindlin 2 expression correlated positively with advanced stage and grade and recurrence of bladder cancer142. Loss of syndecan, a proteoglycan that acts as a co-receptor enabling interaction with a large variety of ligands, including growth factors, in epithelial cells, but gain in stromal cells, is an independent risk factor for poor survival and muscle invasion in patients with bladder cancer145. Collectively, these findings show that collagens and CAF markers have prognostic value in bladder cancer (TABLE 2)
Table 2 |.
Cancer-associated fibroblast and extracellular matrix markers and clinical outcome
| Stromal or extracellular matrix components | Marker | Expression level or pattern (mRNA or protein) | NMIBC or MIBC (cohort size) | Clinical outcomea | Ref. |
|---|---|---|---|---|---|
| Basement membrane components | Collagen IV | Fragmented or absent in >5% of tumours | NMIBCs (n = 27) and MIBCs (n = 48) | Reduced OS | 92 |
| Collagen VII | Loss of expression | MIBCs (n = 30) | NA | 19 | |
| Laminin 5 encoding genes: LAMA3, LAMB3, LAMC2 | Promoter methylation | NMIBCs and MIBCs (n = 128 total) | Reduced OS | 93 | |
| Extracellular matrix | COL4A1, COL18A1 (part of a 12-gene signature) | High (mRNA) | NMIBC (115) | Reduced PFS | 164 |
| High (mRNA) | NMIBC (750) | Reduced PFS | 165 | ||
| Collagen I | High (protein); dense staining in lamina propria near tumour-ECM boundary | NMIBC (80) | Reduced PFS | 102 | |
| Second harmonic generation imaging | Median fibre curative ratio (collagen tensile strength) | NMIBC (80) | Progression to MIBC | 102 | |
| COL1A1, COL1A2 | High (mRNA) | NMIBC (189) | Reduced PFS and OS | 102 | |
| COL1A1, COL1A2, COL5A2, COL6A1, COL6A2, COL6A3 | High (protein) | Integrated analysis on GSE13507: NMIBC (103), MIBC (61) or GSE32548: NMIBC (92), MIBC (38) or GSE89: NMIBC (30), MIBC (10) | Reduced RFS and OS | 143 | |
| Cancer-associated fibroblasts | CD90, PDGFRα, PDGFRβ, FAP | High (protein) | NMIBC and MIBC (384) | Reduced OS | 53 |
| CD90 or CD8a | High or low (protein) | NMIBC and MIBC (384) | Increased OS | 53 | |
| FAP | Positive (protein) | MIBC (121) | Reduced DSS | 62 | |
| FAP and CK5 or CK6 (tumour cells) or CD44 (tumour cells) | Positive & positive or positive (protein) | MIBC (110) | Reduced DSS | ||
| Kindlin 2 | High (protein) | NMIBC and MIBC (203) | Reduced DFS, DSS, and OS | 142 | |
| SDC1 (also known as CD138) | Positive (protein) | NMIBC and MIBC (119) | Reduced DSS | 145 |
CK, cytokeratin; DFS, disease-free survival; DSS, disease-specific survival; FAP, fibroblast activation protein-α; MIBC, muscle-invasive bladder cancer; NA, not available; NMIBC, non-muscle-invasive bladder cancer; OS, overall survival; PFS, progression-free survival; RFS, recurrence-free survival; SDC1, syndecan 1.
All studies listed here have a P value < 0.05.
Therapeutics
The contribution of the TME to the therapeutic response has been implied in other cancer types and also reported in bladder cancer. bTME components can be involved in regulating therapeutic response to the major treatment modalities in bladder cancer in the context of NMIBC (BCG) and MIBC (ICI therapy, chemotherapy and trimodality therapy).
BCG.
BCG immunotherapy is the standard-of-care therapy for preventing the recurrence of high-risk NMIBC85. Mechanistically, BCG has been demonstrated to induce fibroblast proliferation and their differentiation into αSMA+ myofibroblasts, either directly or indirectly through macrophage-secreted FGF2 (REF.146) A pro-fibrotic stromal bTME phenotype was associated with an improved response to BCG immunotherapy in patients with NMIBC146, whereas a high stroma core signature or stromal bTME was associated with poor response to ICIs in patients with MIBC147. Differential contributions of the stromal TME to distinct immunotherapies, such as BCG and ICIs, probably indicate that different TME components within the stroma could function as protumoural or antitumoural factors. Thus, future dissection of the stromal TME will be important to elucidate which stromal components drive protumoural or antitumoural activity. However, a major technical limitation of the NMIBC study is the use of NIH3T3 fibroblasts rather than patient-derived CAFs, confounding the evaluation of clinically relevant stromal response in the bTME146.
The importance of various immune cell types in the response to BGC has been extensively studied, contributions of both the innate and adaptive immunity have been reported85,148. In brief, BCG has been shown to induce trained immunity — a non-specific memory of innate immune cells, such as monocytes and macrophages, which is mediated through their epigenetic and/or metabolic reprogramming. These processes cause BCG-trained innate immune cells to increase production of pro-inflammatory cytokines and, therefore, antitumoural effects, when challenged by a second stimuli either related to or unrelated to BCG. Furthermore, BCG can be internalized by urothelial cancer cells or antigen-presenting cells, which cross-prime T cells to induce a TH1 response. In fact, both CD4+ and CD8+ T cells are important mediators of this adaptive response, as functional depletion studies using CD4-neutralizing or CD8-neutralizing antibodies both abrogated BCG-induced antitumoural activity149. However, whether the stroma TME connects to these immune cells and how their crosstalk influences BCG-induced response remain to be explored.
Immune checkpoint inhibitor therapies.
ICIs, such as anti-PDL1 and anti-PD1 drugs, are emerging as a highly tolerable treatment modality for patients with advanced bladder cancer87, leading to the expedited FDA approval of the anti-PDL1 drug atezolizumab in 2016 and the anti-PD1 drug pembrolizumab in 2017 (REFS.150–152). In the KEYNOTE-045 phase III trial including patients with cisplatin-refractory advanced urothelial carcinoma (n = 542), participants were randomized 1:1 to receive pembrolizumab or chemotherapy (paclitaxel, docetaxel or vinflunine). Pembrolizumab treatment resulted in a substantially improved objective response (OR) compared with chemotherapy (21.1% for pembrolizumab versus 11.4% for chemotherapy)153. However, in the IMvigor211 phase III trial (n = 931)154, IMvigor130 (n = 1,213)3 and the KEYNOTE-361 (n = 1,010)4, neither the anti-PDL1 atezolizumab nor the anti-PD1 pembrolizumab improved overall response or survival compared with chemotherapy. In IMvigor 211, the OR in the atezolizumab group was 23% compared with an OR of 21.6% in the chemotherapy group, but a more durable response was observed in the ICI group (15.9 months for atezolizumab versus 8.3 months for chemotherapy)154. In IMvigor 130, an OR of 23% was observed in the atezolizumab group compared with a 44% OR in the chemotherapy group3. In KEYNOTE-361, an OR of 30.3% was observed in the pembrolizumab group compared with a 44.9% OR in the chemotherapy group4. These clinical trials revealed an overall response rate of 21.1–30.3% for patients with advanced bladder cancer to both anti-PD1 or anti-PDL1 ICIs, which is initially encouraging. However, 70–80% of the patients are considered non-responders to ICIs and the underlying mechanisms conferring resistance are still under intense investigation.
A potential role for the stromal microenvironment and the cognate receptors for ECM components in the modulation of immune checkpoint resistance in bladder cancer has been observed. In the cohort of patients in CheckMate 275, a phase II, single-arm clinical trial in which patients with metastatic bladder cancer were treated with the anti-PD1 drug nivolumab, an eight-gene EMT/stroma signature (including FLNA, EMP3, CALD1, FN1, FOXC2, LOX, FBN1 and TNC) was derived bioinformatically147. The investigators demonstrated that a high CD8+ T cell infiltration together with low EMT/stromal_core signature was associated with the highest response rates, longest progression-free and overall survival to the anti-PD1 drug nivolumab147 (TABLE 3). Conversely, patients with a high CD8+ T cell infiltration concurrently with a high EMT/stromal_core signature showed considerably worse progression-free and overall survival147. These findings suggest that the stromal compartment of the bTME could have a role in impeding T cell function and, therefore, driving ICI resistance. Indeed, in another study in which patients with advanced bladder cancer were treated with atezolizumab, high TGFβ1 pathway gene expression was associated with lack of response to atezolizumab (that is, stable disease and progressive disease), and an increased pan-fibroblast TGFβ response signature was significantly associated with poor response within immune-excluded tumours (P = 0.0066), with no association with response within tumours with an inflamed or immune-desert phenotype61. Further preclinical experiments using anti-TGFβ antibody blockade significantly reduced expression of fibroblast genes associated with matrix remodelling (P < 0.01) and synergized with anti-PDL1 to produce a 70% complete response versus a 10% complete response in anti-PDL1-treatment alone arm. These findings implicate stromal TGFβ signalling in restricting T cell movement in the TME, producing an immune-exclusion phenotype in advanced MIBCs that is associated with poor response to ICIs61.
Table 3 |.
Association between stromal markers and therapeutic response
| Marker | Disease (n analysed) | Therapeutic modality | Prognosisa | Ref. |
|---|---|---|---|---|
| High CD8 T cell infiltration plus low eight-gene EMT/ stroma signature (FLNA, EMP3, CALD1, FN1, FOXC2, LOX, FBN1 and TNC) | Metastatic or unresectable, platinum-resistant MIBC (214) | Nivolumab (anti-PD1) | High response rate, increased PFS and OS | 147 |
| COL1A2, FN1 and THBS1 | MIBC (103) | Neoadjuvant chemotherapy | Chemoresistance | 158 |
| Signatures of T cell activation (HLA-DMA, DMB, HLA-DOA DOB, GZMK, ICOS, CCL2, CCL3, CCL4, CXCL9, CXCL10 and CD8A) and interferon-γ signalling (STAT1, STAT2, CXCL9, CXCL10, CXCL11, GZMA, IDO1, CCL2, CCL5, ICAM1 and IL-6) | MIBC (136) | Bladder-sparing trimodality therapy | Increased DSS | 159 |
| Stromal signature (MYH11, CNN1, DES, PCP4, ACTC1, C7, PGM5, MFAP4 and SGCD) | MIBC (223) | Neoadjuvant chemotherapy and radical cystectomy | Reduced DSS | 159 |
DSS, disease-specific survival; MIBC, muscle-invasive bladder cancer; NMIBC, non-muscle-invasive bladder cancer; OS, overall survival; PFS, progression-free survival.
All studies listed here have a P value < 0.05.
These results clearly indicate that certain stromal components of the bTME have an immunosuppressive role; however, the results of another study were contradictory, suggesting that other ECM components could instead create an immune stimulatory environment. A decellularized ECM-based bioscaffold generated from a porcine urinary bladder unexpectedly created an immune stimulatory environment that inhibited tumour formation in multiple mouse tumour models155. Thus, other ECM components or ECM-associated growth factors within the scaffold could be responsible for eliciting immune stimulatory functions. Intriguingly, conventionally, an increased TH1:TH2 intratumoural T cell ratio is thought to be important for driving antitumoural activities, but in this study, a T cell ratio skewed towards TH2 T cells together with non-classical CD206+ macrophages and eosinophils were found to be responsible for the antitumoural effects impeding tumour growth, which is an alternative and important observation that needs to be validated by future studies155. Nonetheless, as the bladder bioscaffold comprises hundreds of ECM-related proteins within the core matrisome, precisely which ECM components are responsible for triggering the immune stimulatory role remains unclear. In another study that employed an unbiased in vivo short-hairpin (sh)RNA functional screen to identify ICI resistance mechanisms, relevant information was provided to explain this intriguing phenomenon. Using a shRNA-pooled library, DDR2, a fibrillar collagen receptor, was found to be a key modulator of anti-PD1 resistance in bladder cancer116. Genetic knockdown of DDR2 in bladder tumour cells (NA13), as well as treatment with dasatinib, a receptor tyrosine kinase inhibitor with cross-reactivity towards DDR2, sensitizes bladder cancer tumours to anti-PD1 treatment via increasing splenic and tumour-infiltrating CD8+ T cells, demonstrated using CyTOF and multicolour flow cytometry116. DDR2 is a collagen receptor; thus, the observation that a collagen-crosslinking enzyme lysyl oxidase (LOX) was amongst the EMT/stroma_core signature that was associated with poor ICI response is interesting, indicating a potential connection between collagen modifications (such as crosslinking) and the activation of its downstream collagen receptor signalling in mediating ICI resistance. Increased expression of another collagen receptor, DDR1, could be associated with immunologically cold or immune-excluded bladder tumours, which also correlates with poor immune checkpoint response in MIBCs. Collectively, these results reveal a central role for stromal components of the bTME, such as collagen ECM and its receptors, in modulating immune checkpoint response in advanced MIBCs. Future investigations are needed to elucidate the distinct stromal or ECM components within the bTME that drive divergent BCG and ICI responses in NMIBC and MIBC.
Chemotherapy and trimodality therapy.
Few functional studies have been published in which the mechanistic connection between the bTME components and conventional therapies were investigated. Neoadjuvant chemotherapy remains the standard-of-care treatment for locally advanced bladder cancer before proceeding to radical cystectomy156. However, chemotherapy provides a minimal survival advantage for patients (demonstrating an overall survival benefit of 5% at 5 years), except for those whose disease has exhibited pathological downstaging156. Results from one study demonstrated a role for CAFs in mediating chemoresistance: when bladder CAFs isolated from human MIBC tissue were co-cultured with the human bladder cancer cell lines T24 and 5637, they enhanced the capacity of bladder cancer cells to survive cisplatin chemotherapy, demonstrated using the MMT assay, colony formation assay, flow cytometry measuring propidium iodide and annexin V, as well as western blot evaluating the apoptotic effector cleaved caspase-3. Mechanistically, CAFs induced upregulation of the antiapoptotic protein BCL-2 in the same human bladder cancer cell lines through IGF1 and ERβ signalling157. Further observations support the role of CAFs in promoting the survival and repopulation of residual bladder cancer cells (that is, cancer stem cells)31,32 through enhancing collagen depositions to exclude drug penetration into epithelial regions, as well as activating collagen receptor signalling in bladder cancer cells (K.S.C. unpublished work). Currently, how these cell-extrinsic mechanisms connect to other anti-apoptotic proteins, such as BCL-2, or other cell death mechanisms, remains an active area of investigation.
These laboratory findings together with clinical studies involving patients with bladder cancer, demonstrate that chemoresistance is also associated with increased deposition of collagens and other ECM components within the bTME31 (TABLE 3). In patients with bladder cancer whose disease is chemoresistant, analysis of matched pre-chemotherapy and post-chemotherapy tissues showed that non-responders exhibited significant upregulation of several ECM-associated genes, including COL1A2, FN1 and THBS1 (P < 0.001), which was associated with sustained MTOR signalling in the peritumoural and surrounding stroma158 (TABLE 3). These findings were independently supported by results of another study demonstrating that a high stromal gene signature is associated with resistance to neoadjuvant chemotherapy in patients with MIBC158. Conversely, a similar stromal signature had no association with the response of patients treated with bladder-sparing trimodality therapy (TMT), which involves maximal transurethral resection of bladder tumour followed by chemotherapy and radiotherapy. Instead, a high immune signature indicative of T cell activation and interferon γ signalling was associated with improved survival in patients with MIBC patients treated using TMT159 (TABLE 3), but has no significant association in patients with MIBC treated with neoadjuvant chemotherapy160.
Collectively, these findings show that the bTME components are dynamically modified in response to different treatment modalities. Different stromal bTME components probably act as divergent determining factors that contribute to the therapeutic response to chemotherapy or TMT, respectively; immune infiltration seems to be a favourable factor for TMT but not chemotherapy. These findings are consistent with preclinical studies in mice, the results of which support the observation that the standard-of-care gemcitabine and cisplatin chemotherapies are insufficient to induce immunogenic cell death — a mode of cell death that depends on the extracellular release of damage-associated molecular patterns (DAMPs) and inhibitory DAMP (iDAMP) that act as immunological adjuvants to activate professional antigen-presenting cells (such as dendritic cells) for priming an adaptive CD8+ T cell response161,162. The results of these studies help to explain why immune infiltration is not associated with chemotherapeutic response in MIBC. Furthermore, these results suggest that therapeutic targeting of the iDAMP axis reinvigorates dendritic cell activation and vasculature maturation, converting the bTME from an immune-excluded TME into one that enables CD8+ T cell access (that is, T cell inflamed) to target bladder tumour cells161,162.
Perspectives and future directions
Targeted therapies such as erdafitinib or cabozantinib and conventional cancer therapies such as chemotherapy are thought to exert their cytotoxic effects by directly targeting malignant tumour cells. However, the bTME has been shown to demonstrate active supporting and/or driving roles in tumour development and progression, as well as determining clinical response to different types of therapies, highlighting the bTME as an understudied area for biomarker development and therapeutic targeting. Stromal cells are thought to be genetically stable; therefore, the bTME components are attractive targets that are less likely than cancer cells to acquire mutations that confer resistance when subjected to selective pressure of therapies.
Current studies investigating the TME, including the bTME, largely focus on the primary tumour. The lack of patient specimens from distant metastatic sites and preclinical models of metastasis has hindered investigations into met-TMEs, although studies have been conducted that initially define the characteristics of the lung met-TME. The TME milieu and its alterations in response to treatment-induced wounding, TME-driven treatment resistance or susceptibility and the tumour-TME crosstalk at metastatic sites (such as lung, liver and bone) are yet to be defined; thus, defining the characteristics of these TMEs and met-TMEs will reveal new vulnerabilities for novel drug discovery to control this cancer type at the advanced or metastatic stage. Therapeutic targeting of the tumour alone possibly limits therapeutic efficacy owing to the fast acquisition of resistance; conversely, targeting the TME alone is ineffective owing to the aggressive nature of the tumour. Thus, the concept of co-targeting the tumour and its TME or met-TME is an intriguing idea and warrants further evaluation. Targeting the TME can disrupt tumour growth in several ways: by directly abrogating growth-promoting signalling to tumour cells; by disrupting the tumour-supporting ECM in the bTME; and by unleashing effector immune cells to act against the tumour. Co-targeting TME and the tumour could maximally affect the tumour at an ecosystem level, thereby reducing tumour growth and delaying the development of treatment resistance.
Conclusions
Tumour cell-directed therapy remains the standard of care for treating advanced bladder cancer. The introduction of ICI therapy shows early promise by unleashing the bladder tumour immune microenvironment to treat bladder cancer. Despite increasing evidence showing the complex interplay between the cellular and non-cellular components of the bTME, its biological roles remain poorly understood. Furthermore, the met-TME is currently largely unexplored. Thus, further research into the local (bTME) and met-TMEs is required to aid discovery of new therapeutic targets for this disease. Future mechanistic investigation of the dynamic interaction between the tumour and its TMEs at each stage of bladder cancer progression will enable rational design of next-generation therapies that co-target both the tumour and the TME.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Urology thanks Lars Dyrskjøt, François Radvanyi and Edmund Chiong for their contribution to the peer review of this work.
References
- 1.Antoni S et al. Bladder cancer incidence and mortality: a global overview and recent trends. Eur. Urol. 71, 96–108 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Boormans JL & Zwarthoff EC Limited funds for bladder cancer research and what can we do about it. Bladder Cancer 2, 49–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galsky MD et al. Atezolizumab with or without chemotherapy in metastatic urothelial cancer (IMvigor130): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 395, 1547–1557 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Powles T et al. Pembrolizumab alone or combined with chemotherapy versus chemotherapy as first-line therapy for advanced urothelial carcinoma (KEYNOTE-361): a randomised, open-label, phase 3 trial. Lancet Oncol. 22, 931–945 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Knowles MA & Hurst CD Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 15, 25–41 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Sahai E et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winkler J, Abisoye-Ogunniyan A, Metcalf KJ & Werb Z Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 11, 5120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turley SJ, Cremasco V & Astarita JL Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 15, 669–682 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Ho PL, Kurtova A & Chan KS Normal and neoplastic urothelial stem cells: getting to the root of the problem. Nat. Rev. Urol. 9, 583–594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabella G. Lamina propria: the connective tissue of rat urinary bladder mucosa. Neurourol. Urodyn. 38, 2093–2103 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Gevaert T et al. Identification of different phenotypes of interstitial cells in the upper and deep lamina propria of the human bladder dome. J. Urol. 192, 1555–1563 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Neuhaus J et al. 3D-electron microscopic characterization of interstitial cells in the human bladder upper lamina propria. Neurourol. Urodyn. 37, 89–98 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Kuijpers KAJ, Heesakkers JPFA, Jansen CFJ & Schalken JA Cadherin-11 is expressed in detrusor smooth muscle cells and myofibroblasts of normal human bladder. Eur. Urol. 52, 1213–1221 (2007). [DOI] [PubMed] [Google Scholar]
- 14.McCloskey KD Interstitial cells in the urinary bladder — localization and function. Neurourol. Urodyn. 9, 82–87 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Baskin LS et al. Cellular signaling in the bladder. Front. Biosci. 2, d592–d595 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Shin K et al. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature 472, 110–114 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin K et al. Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer Cell 26, 521–533 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flier JS, Underhill LH & Dvorak HF Tumors: wounds that do not heal. N. Engl. J. Med. 315, 1650–1659 (1986). [DOI] [PubMed] [Google Scholar]
- 19.Liebert M, Washington R, Wedemeyer G, Carey TE & Grossman HB Loss of co-localization of alpha 6 beta 4 integrin and collagen VII in bladder cancer. Am. J. Pathol. 144, 787–795 (1994). [PMC free article] [PubMed] [Google Scholar]
- 20.Paget S The distribution of secondary growths in cancer of the breast. Lancet 133, 571–573 (1889). [PubMed] [Google Scholar]
- 21.Lee Y-C et al. Collagen-rich airway smooth muscle cells are a metastatic niche for tumor colonization in the lung. Nat. Commun. 10, 2131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanahan D & Coussens LM Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Egeblad M, Nakasone ES & Werb Z Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell 18, 884–901 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Junttila MR & de Sauvage FJ Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501, 346–354 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Bhowmick NA, Neilson EG & Moses HL Stromal fibroblasts in cancer initiation and progression. Nature 432, 332–337 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sleeman JP The metastatic niche and stromal progression. Cancer Metastasis Rev. 31, 429–440 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji Q et al. Primary tumors release ITGBL1-rich extracellular vesicles to promote distal metastatic tumor growth through fibroblast-niche formation. Nat. Commun. 11, 1211 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peinado H et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Duda DG et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl Acad. Sci. USA 107, 21677–21682 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krall JA et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl Med. 10, eaan3464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurtova AV et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 517, 209–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan KS Molecular pathways: targeting cancer stem cells awakened by chemotherapy to abrogate tumor repopulation. Clin. Cancer Res. 22, 802–806 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panigrahy D et al. Preoperative stimulation of resolution and inflammation blockade eradicates micrometastases. J. Clin. Invest. 129, 2964–2979 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuxhorn JA et al. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 8, 2912–2923 (2002). [PubMed] [Google Scholar]
- 35.Toullec A et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med. 2, 211–230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nielsen SR et al. Corrigendum: macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 18, 822 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Albrengues J et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 6, 10204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalluri R The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Kojima Y et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl Acad. Sci. USA 107, 20009–20014 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arina A et al. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl Acad. Sci. USA 113, 7551–7556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jung Y et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 4, 1795 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quante M et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19, 257–272 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonald LT et al. Hematopoietic stem cell-derived cancer-associated fibroblasts are novel contributors to the pro-tumorigenic microenvironment. Neoplasia 17, 434–448 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nimphius W, Moll R, Olbert P, Ramaswamy A & Barth PJ CD34+ fibrocytes in chronic cystitis and noninvasive and invasive urothelial carcinomas of the urinary bladder. Virchows Arch. 450, 179–185 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Bellini A & Mattoli S The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab. Invest. 87, 858–870 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Reilkoff RA, Bucala R & Herzog EL Fibrocytes: emerging effector cells in chronic inflammation. Nat. Rev. Immunol. 11, 427–435 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwano M et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 110, 341–350 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeisberg M et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 282, 23337–23347 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Zeisberg EM, Potenta S, Xie L, Zeisberg M & Kalluri R Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 67, 10123–10128 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Brunner A et al. Prognostic significance of tenascin-C expression in superficial and invasive bladder cancer. J. Clin. Pathol. 57, 927–931 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kalluri R & Zeisberg M Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Shiga K et al. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers 7, 2443–2458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mezheyeuski A et al. Fibroblasts in urothelial bladder cancer define stroma phenotypes that are associated with clinical outcome. Sci. Rep. 10, 281 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elyada E et al. Cross-species single-cell analy sis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Helms E, Onate MK & Sherman MH Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov. 10, 648–656 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Z et al. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat. Commun. 11, 5077 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hung T-T, Wang H, Kingsley EA, Risbridger GP & Russell PJ Molecular profiling of bladder cancer: involvement of the TGF-beta pathway in bladder cancer progression. Cancer Lett. 265, 27–38 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Liang Y et al. Conditional ablation of TGF-β signaling inhibits tumor progression and invasion in an induced mouse bladder cancer model. Sci. Rep. 6, 29479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang P et al. Bladder cancer cell invasion is enhanced by cross-talk with fibroblasts through hepatocyte growth factor. Urology 69, 780–784 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Szarvas T, Vom Dorp F, Ergün S & Rübben H Matrix metalloproteinases and their clinical relevance in urinary bladder cancer. Nat. Rev. Urol. 8, 241–254 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Mariathasan S et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calvete J et al. The coexpression of fibroblast activation protein (FAP) and basal-type markers (CK 5/6 and CD44) predicts prognosis in high-grade invasive urothelial carcinoma of the bladder. Hum. Pathol. 91, 61–68 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Park JE et al. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem. 274, 36505–36512 (1999). [DOI] [PubMed] [Google Scholar]
- 64.Hotary KB et al. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 114, 33–45 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Feinberg TY et al. Divergent matrix-remodeling strategies distinguish developmental from neoplastic mammary epithelial cell invasion programs. Dev. Cell 47, 145–160.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hashitani H, Mitsui R, Shimizu Y, Higashi R & Nakamura K Functional and morphological properties of pericytes in suburothelial venules of the mouse bladder. Br. J. Pharmacol. 167, 1723–1736 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hashitani H, Mitsui R, Miwa-Nishimura K & Lam M Role of capillary pericytes in the integration of spontaneous Ca2+ transients in the suburothelial microvasculature in situ of the mouse bladder. J. Physiol. 596, 3531–3552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miodoński AJ & Litwin JA Microvascular architecture of the human urinary bladder wall: a corrosion casting study. Anat. Rec. 254, 375–381 (1999). [DOI] [PubMed] [Google Scholar]
- 69.Sherwood LM, Parris EE & Folkman J Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182–1186 (1971). [DOI] [PubMed] [Google Scholar]
- 70.Risau W Mechanisms of angiogenesis. Nature 386, 671–674 (1997). [DOI] [PubMed] [Google Scholar]
- 71.Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS & Reynolds AR Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 16, 469–493 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Patel NS et al. Up-regulation of endothelial delta-like 4 expression correlates with vessel maturation in bladder cancer. Clin. Cancer Res. 12, 4836–4844 (2006). [DOI] [PubMed] [Google Scholar]
- 73.McDonald DM & Baluk P Significance of blood vessel leakiness in cancer. Cancer Res. 62, 5381–5385 (2002). [PubMed] [Google Scholar]
- 74.John A et al. Urothelial carcinoma of the bladder induces endothelial cell activation and hypercoagulation. Mol. Cancer Res. 18, 1099–1109 (2020). [DOI] [PubMed] [Google Scholar]
- 75.Trédan O, Galmarini CM, Patel K & Tannock IF Drug resistance and the solid tumor microenvironment. J. Natl Cancer Inst. 99, 1441–1454 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Flores-Mireles AL, Walker JN, Caparon M & Hultgren SJ Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 13, 269–284 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ingersoll MA & Albert ML From infection to immunotherapy: host immune responses to bacteria at the bladder mucosa. Mucosal Immunol. 6, 1041–1053 (2013). volume 19 [DOI] [PubMed] [Google Scholar]
- 78.Abraham SN & Miao Y The nature of immune responses to urinary tract infections. Nat. Rev. Immunol. 15, 655–663 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gardiner RA et al. Immunohistochemical analysis of the human bladder. Br. J. Urol. 58, 19–25 (1986). [DOI] [PubMed] [Google Scholar]
- 80.Hart DNJ & Fabre JW Demonstration and characterization of ia-positive dendritic cells in the interstitial connective tissues of rat heart and other tissues, but not brain. J. Exp. Med. 154, 347–361 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schilling JD, Martin SM, Hung CS, Lorenz RG & Hultgren SJ Toll-like receptor 4 on stromal and hematopoietic cells mediates innate resistance to uropathogenic Escherichia coli. Proc. Natl Acad. Sci. USA 100, 4203–4208 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Engel DR et al. CCR2 mediates homeostatic and inflammatory release of Gr1 high monocytes from the bone marrow, but is dispensable for bladder infiltration in bacterial urinary tract infection. J. Immunol. 181, 5579–5586 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Mariano LL et al. Functionally distinct resident macrophage subsets differentially shape responses to infection in the bladder. Sci. Adv. 6, eabc5739 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Christmas TJ Lymphocyte sub-populations in the bladder wall in normal bladder, bacterial cystitis and interstitial cystitis. Br. J. Urol. 73, 508–515 (1994). [DOI] [PubMed] [Google Scholar]
- 85.Pettenati C & Ingersoll MA Mechanisms of BCG immunotherapy and its outlook for bladder cancer. Nat. Rev. Urol. 15, 615–625 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Lobo N et al. 100 years of Bacillus Calmette–Guérin immunotherapy: from cattle to COVID-19. Nat. Rev. Urol. 18, 611–622 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schneider AK, Chevalier MF & Derré L The multifaceted immune regulation of bladder cancer. Nat. Rev. Urol. 16, 613–630 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Mo Q et al. Prognostic power of a tumor differentiation gene signature for bladder urothelial carcinomas. J. Natl Cancer Inst. 110, 448–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kamoun A et al. A consensus molecular classification of muscle-invasive bladder cancer. Eur. Urol. 77, 420–433 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pfannstiel C et al. The tumor immune microenvironment drives a prognostic relevance that correlates with bladder cancer subtypes. Cancer Immunol. Res. 7, 923–938 (2019). [DOI] [PubMed] [Google Scholar]
- 91.Luo Y, Zeng G & Wu S Identification of microenvironment-related prognostic genes in bladder cancer based on gene expression profile. Front. Genet. 10, 1187 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Daher N, Abourachid H, Bove N, Petit J & Burtin P Collagen IV staining pattern in bladder carcinomas: relationship to prognosis. Br. J. Cancer 55, 665–671 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sathyanarayana UG et al. Molecular detection of noninvasive and invasive bladder tumor tissues and exfoliated cells by aberrant promoter methylation of laminin-5 encoding genes. Cancer Res. 64, 1425–1430 (2004). [DOI] [PubMed] [Google Scholar]
- 94.Naba A et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteom. 11, M111.014647 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hynes RO & Naba A Overview of the matrisome-an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 4, a004903 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chaudhuri O, Cooper-White J, Janmey PA, Mooney DJ & Shenoy VB Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 584, 535–546 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Said N, Frierson HF, Sanchez-Carbayo M, Brekken RA & Theodorescu D Loss of SPARC in bladder cancer enhances carcinogenesis and progression. J. Clin. Invest. 123, 751–766 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamauchi M & Sricholpech M Lysine post-translational modifications of collagen. Essays Biochem. 52, 113–133 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen Y et al. Lysyl hydroxylase 2 induces a collagen cross-link switch in tumor stroma. J. Clin. Invest. 125, 1147–1162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yamauchi M, Barker TH, Gibbons DL & Kurie JM The fibrotic tumor stroma. J. Clin. Invest. 128, 16–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Levental KR et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brooks M et al. Positive association of collagen type I with non-muscle invasive bladder cancer progression. Oncotarget 7, 82609–82619 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reiser KM, Amigable M & Last JA Nonenzymatic glycation of type I collagen. J. Biol. Chem. 267, 24207–24216 (1992). [PubMed] [Google Scholar]
- 104.American Cancer Society. Key statistics for bladder cancer. cancer.org; https://www.cancer.org/cancer/bladder-cancer/about/key-statistics.html (2022). [Google Scholar]
- 105.Knight CG et al. The collagen-binding a-domains of integrins α1/β1 and α2/β1 recognize the same specific amino acid sequence, GFOGER, in native (triple-helical) collagens. J. Biol. Chem. 275, 35–40 (2000). [DOI] [PubMed] [Google Scholar]
- 106.Emsley J, Knight CG, Farndale RW, Barnes MJ & Liddington RC Structural basis of collagen recognition by integrin α2β1. Cell 101, 47–56 (2000). [DOI] [PubMed] [Google Scholar]
- 107.Ichikawa O et al. Structural basis of the collagen-binding mode of discoidin domain receptor 2. EMBO J. 26, 4168–4176 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu H et al. Collagen binding specificity of the discoidin domain receptors: binding sites on collagens II and III and molecular determinants for collagen IV recognition by DDR1. Matrix Biol. 30, 16–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aitken KJ & Bägli DJ The bladder extracellular matrix. Part I: architecture, development and disease. Nat. Rev. Urol. 6, 596–611 (2009). [DOI] [PubMed] [Google Scholar]
- 110.Wilson CB, Leopard J, Cheresh DA & Nakamura RM Extracellular matrix and integrin composition of the normal bladder wall. World J. Urol. 14 (Suppl. 1), S30–S37 (1996). [DOI] [PubMed] [Google Scholar]
- 111.Ioachim E et al. A clinicopathological study of the expression of extracellular matrix components in urothelial carcinoma. BJU Int. 95, 655–659 (2005). [DOI] [PubMed] [Google Scholar]
- 112.Eisinger F, Patzelt J & Langer HF The platelet response to tissue injury. Front. Med. 5, 317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rousselle P, Montmasson M & Garnier C Extracellular matrix contribution to skin wound re-epithelialization. Matrix Biol. 75–76, 12–26 (2019). [DOI] [PubMed] [Google Scholar]
- 114.Rohani MG et al. MMP-10 regulates collagenolytic activity of alternatively activated resident macrophages. J. Invest. Dermatol. 135, 2377–2384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rohani MG & Parks WC Matrix remodeling by MMPs during wound repair. Matrix Biol. 44–46, 113–121 (2015). [DOI] [PubMed] [Google Scholar]
- 116.Tu MM et al. Targeting DDR2 enhances tumor response to anti-PD-1 immunotherapy. Sci. Adv. 5, eaav2437 (2019).Sci. Rep. 5, 11924 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhuang J et al. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 5, 11924 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Goulet CR et al. Exosomes induce fibroblast differentiation into cancer-associated fibroblasts through TGFβ signaling. Mol. Cancer Res. 16, 1196–1204 (2018). [DOI] [PubMed] [Google Scholar]
- 119.Yan L, Wang P, Fang W & Liang C Cancer-associated fibroblasts-derived exosomes-mediated transfer of LINC00355 regulates bladder cancer cell proliferation and invasion. Cell Biochem. Funct. 38, 257–265 (2020). [DOI] [PubMed] [Google Scholar]
- 120.Joseph M & Enting D Immune responses in bladder cancer — role of immune cell populations prognostic factors and therapeutic implications. Front. Oncol. 9, 1270 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kawanishi H et al. Secreted CXCL1 is a potential mediator and marker of the tumor invasion of bladder cancer. Clin. Cancer Res. 14, 2579–2587 (2008). [DOI] [PubMed] [Google Scholar]
- 122.Miyake M et al. CXCL1-mediated interaction of cancer cells with tumor-associated macrophages and cancer-associated fibroblasts promotes tumor progression in human bladder cancer. Neoplasia 18, 636–646 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Said N, Sanchez-Carbayo M, Smith SC & Theodorescu D RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. J. Clin. Invest. 122, 1503–1518 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chu M & Zhang C Inhibition of angiogenesis by leflunomide via targeting the soluble ephrin-A1/EphA2 system in bladder cancer. Sci. Rep. 8, 1539 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Huang Z et al. Bladder cancer cells interact with vascular endothelial cells triggering EGFR signals to promote tumor progression. Int. J. Oncol. 54, 1555–1566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Faltas BM et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat. Genet. 48, 1490–1499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Winters BR Genomic distinctions between metastatic lower and upper tract urothelial carcinoma revealed through rapid autopsy. JCI Insight 4, e128728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Goodison S et al. A multiplex urinary immunoassay for bladder cancer detection: analysis of a Japanese cohort. J. Transl Med. 14, 287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shimizu Y et al. A multiplex immunoassay for the non-invasive detection of bladder cancer. J. Transl Med. 14, 31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Arici A et al. Interleukin-8 induces proliferation of endometrial stromal cells: a potential autocrine growth factor. J. Clin. Endocrinol. Metab. 83, 1201–1205 (1998). [DOI] [PubMed] [Google Scholar]
- 131.O’Connell JT et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl Acad. Sci. USA 108, 16002–16007 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lu P, Takai K, Weaver VM & Werb Z Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 3, a005058 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tashiro Y et al. Inhibition of PAI-1 induces neutrophil-driven neoangiogenesis and promotes tissue regeneration via production of angiocrine factors in mice. Blood 119, 6382–6393 (2012). [DOI] [PubMed] [Google Scholar]
- 134.Bancroft CC et al. Coexpression of proangiogenic factors IL-8 and VEGF by human head and neck squamous cell carcinoma involves coactivation by MEK-MAPK and IKK-NF-κB signal pathways. Clin. Cancer Res. 7, 435–442 (2001). [PubMed] [Google Scholar]
- 135.Metheny-Barlow LJ & Li LY The enigmatic role of angiopoietin-1 in tumor angiogenesis. Cell Res. 13, 309–317 (2003). [DOI] [PubMed] [Google Scholar]
- 136.Zhang G et al. Validation and clinicopathologic associations of a urine-based bladder cancer biomarker signature. Diagn. Pathol. 9, 200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Furuya H et al. Prognostic significance of lymphocyte infiltration and a stromal immunostaining of a bladder cancer associated diagnostic panel in urothelial carcinoma. Diagnostics 10, 14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen P et al. MMP7 shedding of syndecan-1 facilitates re-epithelialization by affecting α2β1 integrin activation. PLoS ONE 4, e6565 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Miyake M et al. Clinical implications in the shift of syndecan-1 expression from the cell membrane to the cytoplasm in bladder cancer. BMC Cancer 14, 86 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wong YNS et al. Urine-derived lymphocytes as a non-invasive measure of the bladder tumor immune microenvironment. J. Exp. Med. 215, 2748–2759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Volkmer JP et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proc. Natl Acad. Sci. USA 109, 2078–2083 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu J et al. Effects of increased Kindlin-2 expression in bladder cancer stromal fibroblasts. Oncotarget 8, 50692–50703 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhu H, Chen H, Wang J, Zhou L & Liu S Collagen stiffness promoted non-muscle-invasive bladder cancer progression to muscle-invasive bladder cancer. Onco Targets Ther. 12, 3441–3457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chan KS et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Natl Acad. Sci. USA 106, 14016–1421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Szarvas T et al. Enhanced stromal syndecan-1 expression is an independent risk factor for poor survival in bladder cancer. Hum. Pathol. 45, 674–682 (2014). [DOI] [PubMed] [Google Scholar]
- 146.Lodillinsky C et al. Bacillus Calmette Guerin induces fibroblast activation both directly and through macrophages in a mouse bladder cancer model.PLoS ONE 5, e13571 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang L et al. EMT-and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat. Commun. 9, 3503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.van Puffelen JH et al. Trained immunity as a molecular mechanism for BCG immunotherapy in bladder cancer. Nat. Rev. Urol. 17, 513–525 (2020). [DOI] [PubMed] [Google Scholar]
- 149.Ratliff TL, Ritchey JK, Yuan JJ, Andriole GL & Catalona WJ T-cell subsets required for intravesical BCG immunotherapy for bladder cancer. J. Urol. 150, 1018–1023 (1993). [DOI] [PubMed] [Google Scholar]
- 150.Balar AV et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet 389, 67–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.[No authors listed.] Nod for atezolizumab in advanced bladder cancer. Cancer Discov. 7, OF4 (2017). [DOI] [PubMed] [Google Scholar]
- 152.Inman BA, Longo TA, Ramalingam S & Harrison MR Atezolizumab: a PD-L1-blocking antibody for bladder cancer. Clin. Cancer Res. 23, 1886–1890 (2017). [DOI] [PubMed] [Google Scholar]
- 153.Bellmunt J et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N. Engl. J. Med. 376, 1015–1026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Powles T et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet 391, 748–757 (2018). [DOI] [PubMed] [Google Scholar]
- 155.Wolf MT et al. A biologic scaffold-associated type 2 immune microenvironment inhibits tumor formation and synergizes with checkpoint immunotherapy. Sci. Transl Med. 11, eaat7973 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nguyen DP & Thalmann GN Contemporary update on neoadjuvant therapy for bladder cancer. Nat. Rev. Urol. 14, 348–358 (2017). [DOI] [PubMed] [Google Scholar]
- 157.Long X et al. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERβ/Bcl-2 signalling. Cell Death Dis. 10, 375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Winters BR et al. Mechanistic target of rapamycin (MTOR) protein expression in the tumor and its microenvironment correlates with more aggressive pathology at cystectomy. Urol. Oncol. 36, 342.e7–342.e14 (2018). [DOI] [PubMed] [Google Scholar]
- 159.Efstathiou JA et al. Impact of immune and stromal infiltration on outcomes following bladder-sparing trimodality therapy for muscle-invasive bladder cancer. Eur. Urol. 76, 59–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Taber A et al. Molecular correlates of cisplatin-based chemotherapy response in muscle invasive bladder cancer by integrated multi-omics analysis. Nat. Commun. 11, 4858 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hayashi K et al. Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. Nat. Commun. 11, 6299 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nikolos F et al. Cell death-induced immunogenicity enhances chemoimmunotherapeutic response by converting immune-excluded into T-cell inflamed tumors. Nat. Commun. 13, 1487 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.O’Keeffe MB et al. Investigation of pericytes, hypoxia, and vascularity in bladder tumors: association with clinical outcomes. Oncol. Res. 17, 93–101 (2008). [DOI] [PubMed] [Google Scholar]
- 164.Dyrskjot L et al. Analysis of molecular intra-patient variation and delineation of a prognostic 12-gene signature in non-muscle invasive bladder cancer; technology transfer from microarrays to PCR. Br. J. Cancer 107, 1392–1398 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dyrskjøt L et al. Prognostic impact of a 12-gene progression score in non-muscle-invasive bladder cancer: a prospective multicentre validation study. Eur. Urol. 72, 461–469 (2017). [DOI] [PubMed] [Google Scholar]