
Fig. 6 |. Predicted GT-A fold of RGGAT1.
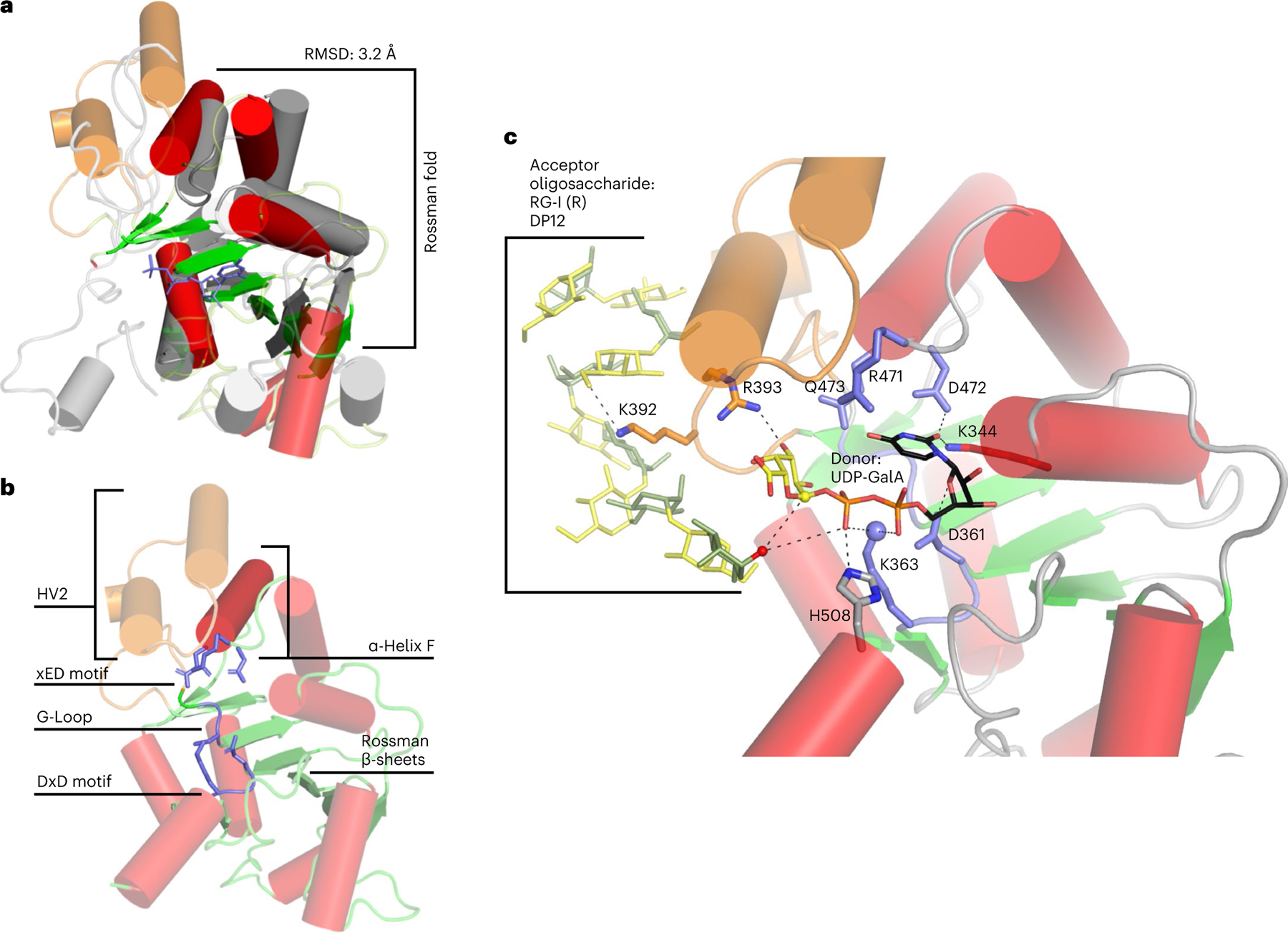
a, Structural alignment of RGGAT1 AlphaFold2 structure (red and green) to a GT31 structure (grey) (pdb: 6wmo) at an RMSD of 3.2 Å across 155 residues, validating that the 3D structure matches a GT-A fold topology. The secondary structure matching algorithm64 in the molecular graphics programme Coot 0.9.7 was used to produce an alignment that was restricted to the core Rossman α-helices (red) and β-sheets (green) shown as opaque structures. b, The AlphaFold2 structure of RGGAT1 has elements of the canonical GT-A fold structure that includes β-sheets of the Rossman fold (green), α-Helix F (dark red) and three conserved motifs of the GT-A fold core (xED, G-Loop and DxD motifs, blue). Hypervariable region 2 (HV2, orange) has helices that are poorly aligned to the template. c, Docked structure of RGGAT1 with the donor UDP-GalA and acceptor RG-I (R) DP12 oligosaccharide. Selected amino acids predicted to interact with the donor and acceptor substrates are shown in stick representation. Dashed lines represent putative hydrogen bonding interactions within 2.7–3.4 Å distance. Residues in blue indicate putative GT-A motifs. Residues in orange are residues present on the HV2 region in contact with the acceptor. Based on a retaining mechanism, the acceptor nucleophile (red sphere) is deprotonated by the β-phosphate oxygen of the UDP donor, allowing a nucleophilic attack on the anomeric carbon of the GalA (yellow sphere)33. The side-chain amine of K363 (blue sphere) may function in place of a divalent cation to interact with the nucleotide phosphate diester of UDP-GalA.