

Abstract
The identification and proper naming of microfungi, in particular plant, animal and human pathogens, remains challenging. Molecular identification is becoming the default approach for many fungal groups, and environmental metabarcoding is contributing an increasing amount of sequence data documenting fungal diversity on a global scale. This includes lineages represented only by sequence data. At present, these taxa cannot be formally described under the current nomenclature rules. By considering approaches used in bacterial taxonomy, we propose solutions for the nomenclature of taxa known only from sequences to facilitate consistent reporting and communication in the literature and public sequence repositories.
The kingdom Fungi forms a highly diverse lineage of eukaryotes that shares a common ancestor with animals. Both comprise heterotrophic organisms, but Fungi form (chitinous) cell walls and are exclusively osmotrophic; that is, nutrient uptake is extracellular1. Although nearly 150,000 species of Fungi have been described, between 2.2 and 3.8 million are estimated to exist2. Fungi are ubiquitous and perform essential ecosystem processes and are of economic importance as agents of diseases or sources of biocontrol agents, biofuel, food and food additives, industrial enzymes and pharmaceuticals.
Fungi follow a simple body plan that underwent convergent evolution3, which makes it challenging to identify them with accuracy and precision4. Methods in fungal taxonomy depend on whether a species is in culture, available as a dried fungarium sample or assessed in situ, and diagnostic tools encompass phenotype-based identification, physiological profiling and sequence-based DNA barcoding or phylogenetic reconstruction, including phylogenomics. In addition, the recent past has seen a shift towards laboratory-based approaches for many fungal groups, which has resulted in an increasing methodological overlap with prokaryote taxonomy4.
Some unrelated microbial groups have evolved life forms similar to Fungi and are studied by mycologists5. The most important are the Oomycota (‘egg fungi’), which include the causal agents of late blight on potatoes (Phytophthora infestans) and white rust on mustards (Albugo candida) (Fig. 1a,b). Oomycota belong to the Straminipila, forming a heterotrophic lineage closely related to brown algae and diatoms5,6. The non-taxonomic term ‘fungi’, without italics and capitalization, is used to encompass Fungi, Oomycota and several other unrelated but fungus-like organisms. This is similar to the usage of ‘algae’ for unrelated organisms with an algal-like habit, for example, in the Archaeplastida, Straminipila and Rhizaria.
Fig. 1 |. Fungal diversity.
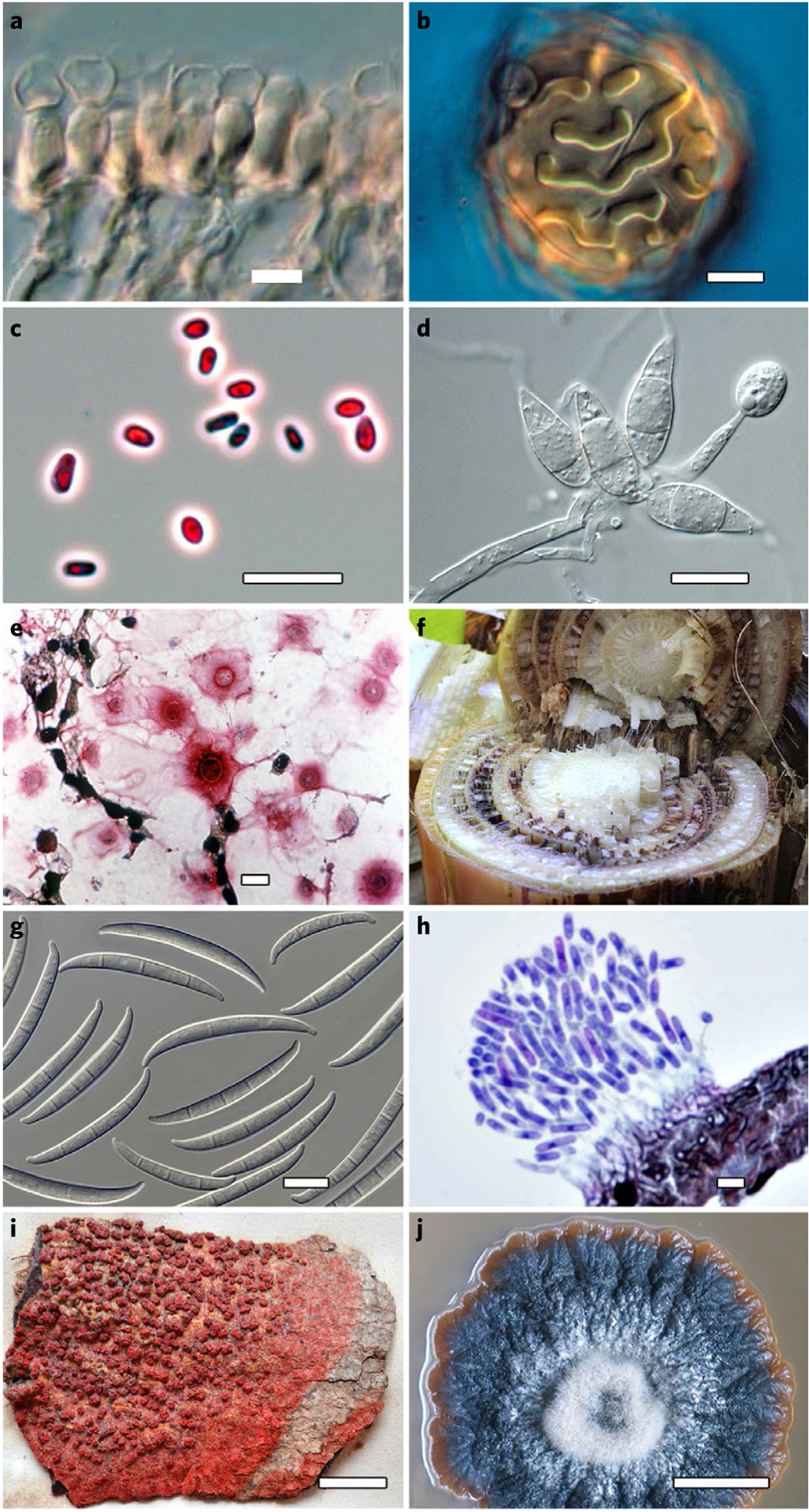
Although Fungi and fungus-like organisms exhibit striking phenotypic diversity, accurate and precise identification often requires molecular approaches or specific tools such as metabolic profiling owing to widespread cryptic diversification and a lack of diagnostic features in microscopic vegetative structures. a, Albugo laibachii (Oomycota) sporogenous hyphae. b, Albugo candida oospore. c, Candida auris cells. d, Pyricularia oryzae conidiophore with conidia. e, Cryptococcus neoformans cells in tissue. f, Banana plant infected with fusarium wilt. g, Fusarium odoratissimum macroconidia. h, Colletotrichum siamense section of acervulus. i, Trypethelium purpurinum (also known as Marcelaria purpurina) physical type specimen. j, Chaetocapnodium tanzanicum culture. Credit: photographs courtesy of Young-Joon Choi (a,b), Nani Maryani (f,g), Min Fu (h) and Jafar Abdollahzadeh (j). Scale bars, 10 μm (a–e, g and h) or 10 mm (i and j).
Fungi and fungus-like organisms are treated under the same nomenclatural rules set forth in the International Code of Nomenclature for algae, fungi, and plants7 (ICNafp; Box 1). The ICNafp reflects advancements made along a historical timeline spanning three centuries (Fig. 2 and Supplementary Information). With fungal taxonomy beginning before the official start of scientific nomenclature in the middle of the eighteenth century, the nineteenth century brought substantial advancements in cataloguing fungal diversity and witnessed the discovery of lichen symbiosis. During the twentieth century, the mycological community became increasingly networked, and fungal systematics organized itself in platforms such as Index of Fungi and Systema Ascomycetum. The increasing importance and refinement of molecular approaches after the turn of the millennium led to substantial changes in fungal classifications and nomenclature (Fig. 2 and Supplementary Information).
Box 1 |. Regulating the naming of fungi.
The nomenclature of Fungi and fungus-like organisms is regulated by the ICNafp (https://www.iapt-taxon.org/nomen/main.php), which historically goes back to the first Lois de la Nomenclature Botanique (Laws of Botanical Nomenclature) published in 1867. A new edition of the ICNafp is prepared every 6 years following the deliberations of the Nomenclature Section of an International Botanical Congress. Since 2018, provisions that specifically relate to Fungi and organisms treated as fungi have been included in a special Chapter F of the ICNafp, which can be modified only during an International Mycological Congress (IMC), which occurs every 4 years. The IMC now appoints the Nomenclature Committee for Fungi (NCF) with around 20 members, which operates between IMCs and considers and votes on formal nomenclatural matters submitted to it, including those related to the conservation or rejection of names (http://www.ima-mycology.org/nomenclature/nomenclature-committee-fungi).
The International Commission on the Taxonomy of Fungi (ICTF), established in 1982, complements the NCF and has the primary remit of promoting sound taxonomic practice through disseminating guidelines and recommendations as to best practice and developing proposals for consideration by the NCF or IMCs. Membership is also decided at IMCs, and the ICTF now has 23 members supplemented by five subcommissions and a series of start-and-finish task-related working groups (https://www.fungaltaxonomy.org).
The ICNafp contains provisions that allow retention of names of fungi in current use even if they do not have priority, either through proposals to conserve or reject names or through lists of names proposed for protection, all of which are reviewed by the NCF. Other fungal-specific provisions include the sanctioning of names; that is, giving priority to names of fungi (except slime moulds) adopted in Persoon’s Synopsis Methodica Fungorum (1801) and Fries’ Systema Mycologicum (1821–1832), and the obligate registration of new fungal names (since 2012) and new typifications (since 2019).
Fig. 2 |. Timeline of important events in fungal taxonomy and nomenclature.

See Supplementary Information for detailed references regarding each event.
Many fungi form phenotypically and biologically divergent life stages that reproduce sexually or asexually. Sexual and asexual morphs were traditionally named and classified separately under the concept of ‘dual nomenclature’8. Molecular systematics has documented numerous instances whereby differently named fungi represent different morphs of the same species. Therefore, mycologists adopted the principle of ‘one fungus = one name’8. Consequently, well-known fungal names, for example, in clinical mycology or plant pathology, may end up as synonyms of older, unfamiliar names. Such changes can, however, be held to a minimum due to new provisions in the ICNafp that allow greater flexibility in selecting the preferred name (Box 1).
Providers and users of fungal taxonomy
Owing to the diversity of fungal biology, ranging from simple to complex life cycles and including multiple morphs within one species, a universal approach to unambiguously identify all fungi is currently not feasible. Identification is also often confused with the underlying processes of species delimitation and recognition4. Species delimited through molecular phylogenies may exhibit diagnostic phenotypic characteristics, which makes identifications feasible without molecular data. Likewise, resolving species complexes may require phylogenomic approaches, while the individual taxa may be identifiable with DNA barcoding markers. Consequently, species identification by a broad user community should require fewer, more readily available resources than species delimitation performed by a small number of taxonomic experts.
The level of accuracy and precision in fungal taxonomy depends on the group under study, the sample size, the available tools and the underlying objectives, and is particularly critical in plant pathology, food safety and clinical mycology4. While species name labels imply accuracy and precision, identifications may be incorrect or imprecise, which has consequences for understanding the biology, the distribution ranges and the conservation status of the underlying taxa. Therefore, verification of identifications is of crucial importance4. While physiological profiling is routinely used in food microbiology and clinical settings, molecular identification has become a major tool in fungal taxonomy. In addition to accuracy and precision, it offers universal coverage and broad applicability9. Regrettably, the increasing availability of molecular tools generates the misleading impression that DNA barcoding can replace taxonomic expertise, thereby overlooking the fact that such expertise remains indispensable to establish comprehensive and accurate reference databases for molecular identifications, a task that is still in its infancy.
A particular challenge for DNA-based identifications of fungi is the choice of genetic markers. While mycologists routinely use the nuclear internal transcribed spacer (ITS) region of the ribosomal RNA operon as a standard DNA barcode9, the Fusarium and Trichoderma communities employ translation elongation factor 1 alpha (TEF-1α) as secondary barcode. In arbuscular mycorrhizal (AM) and in rust fungi, the small subunit (nuSSU) and the large subunit (nuLSU) of the nuclear ribosomal RNA operon are preferred, whereas the Oomycota community focuses on the mitochondrial cytochrome oxidase c subunits (COX1 and COX2)4. Users must be aware of such community-specific approaches to prevent inaccurate or imprecise identifications, which is a problem arising particularly in environmental metabarcoding approaches4.
Species identification in applied mycology
Problems of correct identification and nomenclature are notorious in economically and medically important species, whereby different methods may result in conflicting identifications. The use of physiological profilers, such as API 20C, API ID32C and VITEK 2, has led to misidentification of the emerging, multidrug-resistant yeast Candida auris (Fig. 1c). It can accurately be identified using matrix-assisted laser desorption/ionization–time of flight (MALDI–TOF) mass spectrometry or the ITS barcoding marker10. This example also shows how outdated taxonomy may result in misinformation. Classification of yeasts in vast, polyphyletic genera, such as Candida, gives the misleading impression that these species are closely related11. Multimarker and genome data indicate that C. auris is a member of the Candida haemulonis species complex, which is more closely related to Clavispora than to Candida in the strict sense, which is typified by Candida albicans12. Although the introduction of new genus names to accurately reflect such relationships may cause short-term confusion for users, it provides long-term, community-wide benefits. For instance, antifungal-resistance profiles of yeasts currently classified under the genus name Candida correlate with their phylogenetic position in different families, including C. albicans (Debaryomycetaceae), C. auris (Metschnikowiaceae) and Candida glabrata (Saccharomycetaceae)10. Separating distantly related species into different genera or families therefore provides a clear signal that these fungi can be expected to have different properties and treatment options.
The causal agent of the invasive Asian soybean rust, Phakopsora pachyrhizi, posed a similar problem in plant pathology, since species classified in Phakopsora represent a polyphyletic assemblage13. Revised tools provided by the ICNafp (Box 1) allow these issues to be resolved, but name changes are sometimes unavoidable or deemed acceptable by the user community. In the case of P. pachyrhizi, the genus and species name were proposed for conservation so that this fungus will retain its name. A contrasting case is the rice blast fungus, which has been placed in the genus Magnaporthe based on its sexual stage. Molecular studies showed that Magnaporthe is polyphyletic and that the fungus, previously known under the name Magnaporthe grisea, encompasses distinct, host-specific lineages. The rice blast fungus is now correctly referred to by its original name Pyricularia oryzae14 (Fig. 1d).
Taxonomic interpretations may also be challenging in a broader context. The human pathogen Cryptococcus neoformans (Fig. 1e), best identified with MALDI–TOF mass spectrometry and molecular tools15,16, is classified in a genus that traditionally included diverse members of Tremellomycetes; however, that genus is now restricted to C. neoformans and its relatives17. As a result, fungi in plant and soil samples that are identified with the name Cryptococcus may not actually represent that genus, and may give the erroneous impression that these substrata bear human-pathogenic yeasts. Therefore, ecological tools, such as the FUNGuild pipeline18, require continuous updates to reflect revised phylogenetic classifications.
Resolving species complexes remains a major challenge when it comes to obtaining accurate identifications. Panama disease (fusarium wilt) of banana (Fig. 1f) is caused by a species of Fusarium, in particular tropical race 4 (TR4) of Fusarium oxysporum f. sp. cubense (Foc), which affects the popular Cavendish cultivar of banana. Using secondary barcoding markers, this Fusarium lineage was recognized as a separate species, Fusarium odoratissimum (Fig. 1g), which is phenotypically characterized by conidial septation and peculiar volatile secondary compounds19. However, it may be unclear where to draw the limit between species and infraspecific lineages. In Colletotrichum siamense (Fig. 1h), a plant pathogen that causes postharvest rot of tropical crops, an integrative taxonomic approach encompassing phenotyping, multiple molecular markers and mating tests found evidence of genetic recombination and cross fertility, which therefore did not support the separation of lineages as separate species20.
Oomycota exhibit similar challenges as Fungi when it comes to accurate identifications. Basil downy mildew has become a global pandemic that was at first attributed to a common species, Peronospora lamii, with an assumed broad host range. Through morphological and phylogenetic analyses, it was shown that the pandemic was caused by a previously unrecognized species, Peronospora belbahrii21, which has led to more effective phytosanitary precautions. These findings suggest that fine-scaled diversification in fungi is triggered by dynamic pathogen–host interactions that generate conditions for abundant diversification but also require sophisticated approaches to properly delimit, recognize and identify these species.
Classification of fungal ‘dark taxa’
Environmental metabarcoding via high-throughput sequencing (HTS) has added a new dimension to assessing the diversity of fungi22. Many ecologically cryptic groups, such as anaerobic gut fungi and chytrids, have seen improved sampling, and environmental sequencing has uncovered new lineages of evolutionary importance: while early diverging clades have expanded the range of organisms included in Fungi23,24, others have emerged within the better studied Ascomycota and Basidiomycota25. For new lineages discovered through environmental metabarcoding, taxonomy is operating metaphorically in the dark because sequence-based identifications cannot be verified with voucher specimens, and culturing is necessary to bring these fungi ‘to life’.
The term ‘dark taxa’ was originally coined for unnamed sequences26. However, some of these may represent known species but have not been linked to a published name. Therefore, dark taxa in a fungal context denote new lineages known from sequence data only, but for which no individual voucher specimens or cultures exist. Consequently, a lineage is no longer considered a dark taxon when a matching specimen or culture becomes available.
Environmental sequencing has generated more than 99.9% of the available molecular sequence data for fungi. The Sequence Read Archive (SRA), which collects data from metabarcoding studies separately from the nucleotide archive in GenBank27, has amassed more than 16,000,000,000 HTS-derived fungal ITS reads (https://ftp.ncbi.nih.gov/sra/fungi). Compared with about 1,400,000 mostly Sanger-derived fungal ITS sequences in GenBank, this results in a ratio of over 11,000:1. These numbers are not comparable, as HTS reads represent multiple instances from single individuals, but even so, only a fraction of fungal sequence data are derived from voucher specimens or cultures.
While the staggering amount of HTS data encompass substantial fungal diversity not captured by specimen-based or culture-based sequencing (Fig. 3), environmental metabarcoding faces important challenges4. Because this approach targets a broad range of higher taxa across fungal phyla, accuracy and precision cannot be achieved for all fungal groups with a single DNA barcode, and different markers sequenced from the same environmental samples cannot be linked back to the same individual sources. Analytical HTS pipelines are optimized to rapidly process hundreds of thousands of sequences28; however, they are not able to satisfactorily recognize species-level lineages or derive formal classifications, thereby resulting in a trade-off between speed and accuracy4. A substantial proportion of sequences in public repositories is incorrectly or incompletely named and should be treated with caution4. Matching but misidentified or unidentified sequences will lead to inaccurate names or imprecise identifications, and new lineages may not be properly recognized.
Fig. 3 |. Visualization of new lineages.

Environmental samples of ITS amplicons generated from the fungal microbiome of switchgrass (Panicum virgatum) at a site in North Carolina, USA (SRA accession number SRS7144269; see ref.50 for the corresponding paper). The approximately 60,000 original fungal ITS reads from the switchgrass sample (black dots) were analysed together with 84,000 ITS sequences (blue dots) from the UNITE General FASTA Release 8.2. (https://unite.ut.ee/repository.php). Visualization was done in the rgl package in R (https://r-forge.r-project.org/projects/rgl) after employing a sparse similarity matrix approach, with multidimensional ordination using LargeVis51 based on fast multilevel clustering (fMLC; thresholds of 0.95 and 0.98)52. The two ellipses indicate the approximate location of Ascomycota (separate smaller cluster represents Eurotiomycetes) and Basidiomycota sequences; the large centre cluster also contains representatives of the other phyla present in Fungi. The reads from the Panicum sample form two additional clusters (A and B) in the periphery of the UNITE reference sequences, which indicate phyla or classes only sparsely captured in the UNITE General FASTA Release.
Curated sequence databases, such as UNITE, MaarjAM and NCBI RefSeq Targeted Loci, attempt to overcome this problem. UNITE has emerged as the most widely used resource for curated fungal ITS sequences, including data from vouchered specimens (indexed with a UDB prefix) and reference sequences for so-called ‘species hypotheses’, that is, phylogroups of sequences that are interpreted as species. These data are curated by taxonomy experts during taxonomic workshops29. Its General FASTA Release (https://unite.ut.ee/repository.php) provides vetted reference sequences. Using the nuSSU marker, MaarjAM provides curated reference sequences for phylogroups of arbuscular mycorrhizal fungi, so-called ‘virtual taxa’30. The NCBI RefSeq Targeted Loci project (https://www.ncbi.nlm.nih.gov/bioproject/41209) provides curated reference sequences from type material annotated as part of the NCBI Taxonomy31 and keeps track of biocollections and collection identifiers in the sequence records to facilitate verification.
A major issue regarding the documentation of fungal diversity from environmental metabarcoding is their formal naming. The ICNafp requires fungal names to have a type: either a physical voucher specimen (Fig. 1i) or alternatively a culture (Fig. 1j) cryopreserved in a metabolically inactive state or, in certain cases, an illustration. The requirement for a type enforces the permanent linking of a name to an individual, thereby providing context for the stability of nomenclature and for taxonomic comparisons. Similarly, the International Code of Nomenclature of Prokaryotes (ICNP)32 requires a preserved pure culture to represent a new species. Since dark taxa do not immediately provide such types, there is no possibility to formally name species known from sequence data alone under the current ICNafp rules33,34. New lineages first detected through metabarcoding can only be validly described using laboratory approaches. One option is subsequent culturing from the environment (Table 1, Option 1), as was done for Archaeorhizomyces25 and Bifiguratus35. Another is visualizing fungal structures corresponding to sequence data, thereby resulting in an illustration as the type (Table 1, Option 2), for example, with fluorescent in situ hybridization (FISH), which was employed in the case of the new phylum Cryptomycota24. Additionally, fluorescence-activated cell sorting (FACS) can be utilized to obtain an actual sample from the environment.
Table 1 |.
Proposed alternatives to name fungi discovered through sequence data (dark taxa) and their assessment based on selected parameters
| Parameter | Option 1 | Option 2 | Option 3 | Option 4 | Option 5 |
|---|---|---|---|---|---|
| Approach to obtain or visualize fungus | Culturing | FISH | None (options 3–5) | ||
| Nature of type | Metabolically inactive culture | FISH image | DNA sequence (options 3–5) | ||
| Nature of name | Formal binomial (with priority) | Formal binomial (with priority) | Formal binomial (with priority) | Provisional binomial (nom. seq. without priority) | Informal (non-binomial with identifier) |
| Compliant with ICNafp | Yes | Yes | No (pending further action) | No (pending further action) | Not required |
| Enforcing rules | ICNafp Divisions I-III, Chapter F | ICNafp Divisions I-III, Chapter F | ICNafp Divisions I-III, Chapter F | ICNafp Appendix to Chapter F | Community agreement (for example, through UNITE) |
| Integration of names into fungal classifications | Immediate | Immediate | Immediate | Adoption as formal binomial through matching specimen or culture or vetted lists | Provisional, does not replace formal binomial |
| Competition with specimen-based names | Yes (desired) | Yes (not necessarily desired) | Yes (not necessarily desired) | No (desired) | No (desired) |
| Resource requirements | Very high (laboratory facilities, space, labour) | High (laboratory facilities) | Low (computational) | ||
| Speed of cataloguing dark taxa | Very low | Low | High | ||
| Taxonomic bias | Low to medium | Low | Absent | ||
| Risk of establishing synonyms | Low | Low to medium | Low to medium if restricted to specific marker (or whole-genome approach) (medium to high if more than one marker is used in parallel) | ||
| Risk of establishing artefactual species | Absent | Very low | Low to medium (depending on quality control) | ||
| Quality control | Intrinsic in approach | Intrinsic in approach | Methodological guidelines (can be enforced through registration) | ||
| Phenotype assessment | Limited (culture characteristics) | Strongly limited (cell or hyphal outlines) | Not possible (metadata only) | ||
| Multigene or genome-wide assessment | Unlimited | Strongly limited (matching FISH patterns) | Currently not feasible (will probably become possible with technological advancements) | ||
Options 1 and 2 are implemented in the ICNafp and available for formal descriptions of dark taxa, whereas options 3–5, which share similar methodological approaches and challenges, require amendments to the ICNafp (options 3 and 4) or a community-wide agreement outside the ICNafp (option 5).
Fungal taxonomists may argue that techniques such as FISH or FACS are difficult to access, but since they are widely used in microbiology, the formal naming of dark taxa may offer new opportunities for interdisciplinary collaboration. While culturing has a long history of use in microbiology and mycology, it faces various challenges. Not all fungi grow readily in culture, especially in early diverging fungal clades such as Aphelidiomycota and Cryptomycota, or in arbuscular mycorrhizal Glomeromycotina or parasitic rust fungi (Pucciniomycotina). Since the physiological requirements of dark taxa are not known, culturing requires resource-intensive approaches, a situation that is comparable to prokaryote microbiology. For example, the cultivation of an Asgard archaeon took more than 5 years36, and cultivation of a bacterial strain may cost 10,000 euros37. Even for fungi that can be readily cultured, the quantity of predicted undiscovered species poses a logistical challenge. According to the World Directory of Culture Collections (http://www.wfcc.info/ccinfo/statistics), culture collections currently hold about 870,000 fungal strains, representing around 30,000 known species. This number represents just 3% compared with the 1 million unrecognized fungi estimated to be hidden in environmental samples2. During the past two decades, about 10,000 new fungi have been described based on cultures (Index Fungorum; http://www.indexfungorum.org). While this is an impressive number, a comparable effort would need another 2,000 years to formally catalogue dark fungal taxa through culturing alone, or else the underlying resources would have to be increased by two orders of magnitude. Prokaryote taxonomy faces the same problem: some 16,000 Bacteria and Archaea have been formally described38, whereas around 1 million are estimated to exist39, thereby challenging their formal classification based on pure cultures alone37,40.
While these issues limit the options for formally naming dark taxa under the ICNafp, unambiguous naming of all fungal species is desirable for precise communication in science, conservation and applications41. It also complies with the obligation to make associated data findable, accessible, interoperable and reusable (FAIR data principles42). Therefore, alternative options for sequence-based nomenclature are being explored, for which proof-of-concept studies are needed to gain broad acceptance among the mycological community. Some earlier proposed options either circumvented the ICNafp, creating invalid names, or attempted a flexible interpretation of existing provisions. de Beer et al.43 deliberately described a new species with a non-ICNafp-compliant, invalid name, Hawksworthiomyces sequentia sp. nov. ENAS, adding the acronym ENAS (environmental nucleic acid sequences). Khan et al.44 established two invalid names in the genus Archaeorhizomyces using the denomination ‘nom. seq.’. Arguing that a sequence illustration would constitute a valid type under the ICNafp, Lücking and Moncada45 introduced seven new species in the newly proposed genus Lawreymyces. This approach has been rejected because a DNA sequence is an abstract letter code, and so its depiction does not represent an actual fungal feature7. A further strategy consisted of the deposition of the environmental sample, a so-called ‘bag type’. The species thus published, Piromyces cryptodigmaticus46, was ruled invalid by the Nomenclature Committee for Fungi (Box 1) because of an insufficient diagnosis, but it was based on a technically permissible type as the ICNafp allows admixtures with other organisms. However, the ‘bag type’ is not a desired strategy as the individual fungus corresponding to the sequence data is not discernible within the sample or might have even been used up for DNA extraction.
The straightforward solution would be to amend the ICNafp to allow formal names based on sequence types (Table 1, option 3). Two such proposals have been rejected and referred to internationally appointed committees, with reports expected at the International Mycological Congress (IMC) in Amsterdam 2022 or the next International Botanical Congress in Rio de Janeiro 2023 (ref.47). This is comparable to the situation in prokaryotes, for which proposals for the formal naming of uncultivated taxa based on sequence data have likewise been rejected, thereby prompting an alternative proposal for a separate nomenclatural code for uncultivated species40. The main arguments against formal sequence-based nomenclature in fungi are the potential establishment of artefactual taxa based on compromised sequence data, the inability to link sequences from different markers back to the same lineage and the notion that sequence-based nomenclature cannot be subjected to verification based on mycological expertise34.
The Candidatus status in prokaryote nomenclature is one model that aims to overcome this conflict. It allows provisional names under the guidance of an appendix to a nomenclatural code, and such names do not have priority until a pure culture becomes available32. Analogously, the ICNafp could, either in a commentary in the introductory matter to Chapter F or in an appendix, refer to provisional fungal names that have no priority over names based on currently permissible types (Table 1, option 4). Recommendations could then be put forward through a resolution at an IMC plenary session, under governance by the International Commission on the Taxonomy of Fungi (ICTF; Box 1). As outlined above, such names could be indicated by a specific denomination such as nom. seq. A corresponding amendment to the ICNafp was proposed, but has been rejected and referred for further discussion to a special purpose committee47.
A fifth option would be to establish provisional names independently of the ICNafp (Table 1, option 5). UNITE offers the tools to implement such an informal classification29, and the strings applied to its species hypotheses could be used as a naming convention. For instance, the unnamed species hypothesis SH1566369.08FU (https://unite.ut.ee/bl_forw_sh.php?sh_name=SH1566369.08FU), represented by sequences in the genus Archaeorhizomyces, could be given the informal dark taxon name Archaeorhizomyces SH1566369.08FU, with a corresponding digital object identifier (doi:10.15156/BIO/SH1566369.08FU). Such alphanumeric identifiers are not meant to do away with formal Latinized names, but they speed up the recognition of taxa, thereby allowing precise communication while awaiting formal solutions and avoiding invalid Latinized binomials.
Databases such as GlobalFungi48 have adopted the UNITE species hypotheses as searchable name strings, so there is potential for broad acceptance of this option. This approach is analogous to species clusters in the prokaryotic Genome Taxonomy Database (https://gtdb.ecogenomic.org), which provides species hypotheses based on genome sequences in the form of alphanumeric designations49.
Formal requirement for registration of fungal names of dark taxa under options 4 and 5 would allow quality control measures at the registration step that are difficult to enforce through peer review. The mycological community would have to agree on the requirements, with rules put forward by the ICTF (Box 1). These could include a minimum length for underlying sequence data, a minimum number of independent recoveries and an underlying phylogenetic analysis based on multiple sequence alignments, together with metadata to assess distribution and ecology. A subcommittee of the ICTF could then check published names and add them to a vetted list. There are therefore several instruments that the mycological community can use to advance the naming of dark taxa while maintaining high quality standards and opening new pathways to cataloguing fungal biodiversity.
Conclusions
Fungal taxonomy has entered a new phase with methodological and conceptual advances. A single, universal approach to the identification of fungi is not feasible because of different needs by communities focusing on different groups, species limits remaining in flux and the majority not having been catalogued yet. Although the increased availability of whole-genome data will shift approaches towards genome-based identification, phenotype-based identifications, physiological profiling and DNA barcoding will remain important, which emphasizes the importance of broadly employed standards for these tools. The ICNafp has become increasingly flexible in dealing with nomenclatural challenges, mediating between revised phylogenetic classifications of fungi and the need for stable nomenclature.
Environmental metabarcoding allows for the rapid documentation of unknown fungal diversity. The mycological community is divided as to how this diversity should be named. Obtaining physical types, cultures or visualization of these fungi to meet formal requirements for valid publication of names may be impracticable, and discovery rates of dark taxa greatly exceed the current culturing capacities. Proposals to allow DNA sequence data as types have met with limited support, but sequence-based nomenclature appears to be the only alternative to name the vast diversity of dark taxa within a reasonable time frame. Therefore, alternative solutions that allow provisional names under a standardized approach need to be sought, either as an appendix to or outside the ICNafp, mirroring challenges in prokaryotic taxonomy. Before the adoption of a unified approach for fungi, case studies should assess potential problems. The mycological community would then put forward a strategy leading up to a decision at the 12th or the 13th IMCs planned for 2022 and 2026, respectively. Simultaneously, further technical advancements are expected that will allow substantially longer sequence reads of higher quality in metabarcoding, therefore improving the basis for sequence-based nomenclature.
Supplementary Material
Acknowledgements
Work by C.L.S. and B.R. was supported by the Intramural Research Program of the National Library of Medicine at the National Institutes of Health in Bethesda, Maryland, USA. D.M.G. received support through the National Science Foundation (NSF) grant DEB-1655980 and Project 4655 of the Pennsylvania State Agricultural Experiment Station. E.M. acknowledges CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil) and FACEPE (Fundação de Amparo à Ciência e Tecnologia de Pernambuco, Brazil). K.D.H. thanks the Thailand Research Fund, grant RDG6130001, entitled “Impact of Climate Change on Fungal Diversity and Biogeography in the Greater Mekong Subregion”. The USDA Hatch project 1010662 is acknowledged for support to M.C.A. M.Ö. was supported by the European Regional Development Fund (Centre of Excellence EcolChange). M.T. acknowledges LOEWE for funding in the framework of the Centre for Translational Biodiversity Genomics (TBG) and the German Science Foundation. N.Z. acknowledges the NSF of the United States (DEB-1452971). P.R.J. was supported through the Manaaki Whenua Biota Portfolio with funding from the Science and Innovation Group of the New Zealand Ministry of Business, Innovation and Employment. R.J. thanks the University of Mauritius for research support. We thank S. Redhead for nomenclatural advice. R. Sanders provided the update for the fungal ITS data in the SRA.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41564-021-00888-x.
References
- 1.Richards TA, Leonard G & Wideman JG What defines the “kingdom” Fungi? Microbiol. Spectr 5, 57–77 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Hawksworth DL & Lücking R in The Fungal Kingdom (eds Heitman J et al. ) 79–95 (ASM Press, 2017). [Google Scholar]
- 3.Berbee ML, James TY & Strullu-Derrien C Early diverging fungi: diversity and impact at the dawn of terrestrial life. Annu. Rev. Microbiol 71, 41–60 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Lücking R et al. Unambiguous identification of fungi: where do we stand and how accurate and precise is fungal barcoding? IMA Fungus 11, 14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wijayawardene NN et al. Outline of Fungi and fungus-like taxa. Mycosphere 11, 1060–1456 (2020). [Google Scholar]
- 6.Beakes GW & Thines M in Handbook of the Protists 2nd edn (eds Archibald JM et al. ) 435–505 (Springer, 2017). [Google Scholar]
- 7.Turland NJ et al. International Code of Nomenclature for Algae, Fungi, and Plants (Shenzhen Code) 2018 (Koeltz Botanical Books, 2018). [Google Scholar]
- 8.Hawksworth DL et al. The Amsterdam declaration on fungal nomenclature. IMA Fungus 2, 105–112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schoch CL et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl Acad. Sci. USA 109, 6241–6246 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forsberg K et al. Candida auris: the recent emergence of a multidrug-resistant fungal pathogen. Med. Mycol 57, 1–12 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Daniel H-M, Lachance M-A & Kurtzman CP On the reclassification of species assigned to Candida and other anamorphic ascomycetous yeast genera based on phylogenetic circumscription. Antonie van Leeuwenhoek 106, 67–84 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Shen XX et al. Tempo and mode of genome evolution in the budding yeast subphylum. Cell 175, 1533–1545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rush TA et al. Variation in the internal transcribed spacer region of Phakopsora pachyrhizi and implications for molecular diagnostic assays. Plant Dis. 103, 2237–2245 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Zhang N et al. Generic names in Magnaporthales. IMA Fungus 7, 155–159 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cogliati M et al. Genotypes and population genetics of Cryptococcus neoformans and Cryptococcus gattii species complexes in Europe and the Mediterranean area. Fungal Genet. Biol 129, 16–29 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Firacative C, Trilles L & Meyer W MALDI–TOF MS enables the rapid identification of the major molecular types within the Cryptococcus neoformans/C. gattii species complex. PLoS ONE 7, e37566 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu XZ et al. Towards an integrated phylogenetic classification of the Tremellomycetes. Stud. Mycol. 81, 85–147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen NH et al. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248 (2016). [Google Scholar]
- 19.Maryani N et al. Phylogeny and genetic diversity of the banana Fusarium wilt pathogen Fusarium oxysporum f. sp. cubense in the Indonesian centre of origin. Stud. Mycol 92, 155–194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu F, Wang M, Damm U, Crous PW & Cai L Species boundaries in plant pathogenic fungi: a Colletotrichum case study. BMC Evol. Biol 16, 81–81 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thines M, Telle S, Ploch S & Runge F Identity of the downy mildew pathogens of basil, coleus, and sage with implications for quarantine measures. Mycol. Res 113, 532–540 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Ruppert KM, Kline RJ & Rahman MS Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: a systematic review in methods, monitoring, and applications of global eDNA. Glob. Ecol. Conserv 17, e00547 (2019). [Google Scholar]
- 23.James TY, Stajich JE, Hittinger CT & Rokas A Toward a fully resolved fungal tree of life. Annu. Rev. Microbiol 74, 291–313 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Jones MDM et al. Discovery of novel intermediate forms redefines the fungal tree of life. Nature 474, 200–203 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Rosling A et al. Archaeorhizomycetes: unearthing an ancient class of ubiquitous soil Fungi. Science 333, 876–879 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Page RD DNA barcoding and taxonomy: dark taxa and dark texts. Philos. Trans. R. Soc. B 371, 20150334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sayers EW et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. D1, D9–D16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anslan S et al. Great differences in performance and outcome of high-throughput sequencing data analysis platforms for fungal metabarcoding. MycoKeys 39, 29–40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilsson RH et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Öpik M et al. The online database MaarjAM reveals global and ecosystemic distribution patterns in arbuscular mycorrhizal fungi (Glomeromycota). New Phytol. 188, 223–241 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Schoch CL et al. NCBI taxonomy: a comprehensive update on curation, resources and tools. Database 2020, baaa062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parker CT, Tindall BJ & Garrity GM International Code of Nomenclature of Prokaryotes. Prokaryotic Code (2008 revision). Int. J. Syst. Evol. Microbiol 69, S7–S111 (2019). [Google Scholar]
- 33.Lücking R & Hawksworth DL Formal description of sequence-based voucherless Fungi: promises and pitfalls, and how to resolve them. IMA Fungus 9, 143–166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thines M et al. Ten reasons why a sequence-based nomenclature is not useful for fungi anytime soon. IMA Fungus 9, 177–183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torres-Cruz TJ et al. Bifiguratus adelaidae, gen. et sp nov., a new member of Mucoromycotina in endophytic and soil-dwelling habitats. Mycologia 109, 363–378 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Imachi H et al. Isolation of an archaeon at the prokaryote–eukaryote interface. Nature 577, 519–525 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Overmann J Significance and future role of microbial resource centers. Syst. Appl. Microbiol 38, 258–265 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Parte AC, Sarda Carbasse J, Meier-Kolthoff JP, Reimer LC & Goker M List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol 70, 5607–5612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Louca S, Mazel F, Doebeli M & Parfrey LW A census-based estimate of Earth’s bacterial and archaeal diversity. PLoS Biol. 17, e3000106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray AE et al. Roadmap for naming uncultivated Archaea and Bacteria. Nat. Microbiol 5, 987–994 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryberg M & Nilsson RH New light on names and naming of dark taxa. MycoKeys 30, 31–39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilkinson MD et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Beer ZW et al. Hawksworthiomyces gen. nov. (Ophiostomatales), illustrates the urgency for a decision on how to name novel taxa known only from environmental nucleic acid sequences (ENAS). Fungal Biol. 120, 1323–1340 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Khan F et al. Naming the untouchable—environmental sequences and niche partitioning as taxonomical evidence in fungi. IMA Fungus 11, 23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lücking R & Moncada B Dismantling Marchandiomphalina into Agonimia (Verrucariaceae) and Lawreymyces gen. nov. (Corticiaceae): setting a precedent to the formal recognition of thousands of voucherless fungi based on type sequences. Fungal Divers. 84, 119–138 (2017). [Google Scholar]
- 46.Kirk PM Nomenclatural novelties. Index Fungi 1, 1 (2012). [Google Scholar]
- 47.May TW, Redhead SA, Lombard L & Rossman AY XI International Mycological Congress: report of Congress action on nomenclature proposals relating to fungi. IMA Fungus 9, xxii–xxvii (2018). [Google Scholar]
- 48.Větrovský T et al. GlobalFungi: global database of fungal records from high-throughput-sequencing metabarcoding studies. Sci. Data 7, 228 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parks DH et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol 38, 1098–1098 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Lee MR, & Hawkes CV Plant and soil drivers of whole-plant microbiomes: variation in switchgrass fungi from coastal to mountain sites. Phytobiomes J. 10.1094/PBIOMES-07-20-0056-FI (2020). [DOI] [Google Scholar]
- 51.Tang J, Liu J, Zhang MY & Mei Q Visualizing large-scale and high-dimensional data. In WWW ‘16: Proc. 25th International Conference on World Wide Web 287–297 (International World Wide Web Conferences Steering Committee, 2016); 10.1145/2872427.2883041 [DOI] [Google Scholar]
- 52.Vu D, Groenewald M & Verkley G Convolutional neural networks improve fungal classification. Sci. Rep 10, 12628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.