

Abstract
Briefly considered extinct in the wild, the future of the Wyoming toad (Anaxyrus baxteri) continues to rely on captive breeding to supplement the wild population. Given its small natural geographic range and history of rapid population decline at least partly due to fungal disease, investigation of the diversity of key receptor families involved in the host immune response represents an important conservation need. Population decline may have reduced immunogenetic diversity sufficiently to increase the vulnerability of the species to infectious diseases. Here we use comparative transcriptomics to examine the diversity of toll-like receptors and major histocompatibility complex (MHC) sequences across three individual Wyoming toads. We find reduced diversity at MHC genes compared to bufonid species with a similar history of bottleneck events. Our data provide a foundation for future studies that seek to evaluate the genetic diversity of Wyoming toads, identify biomarkers for infectious disease outcomes, and guide breeding strategies to increase genomic variability and wild release successes.
Keywords: Amphibian, Bufonidae, Immunogenetics, MHC, TLR, Transcriptomics
Introduction
Over the next century, shifts in environmental conditions are predicted to alter the global distribution of wildlife and their pathogens (Hof et al. 2011; Rohr et al. 2013; Palmer 2018; St Leger 2021; Haver et al. 2022). When coupled with already dramatic declines in biodiversity worldwide (Berger et al. 1998; Daszak et al. 2003; Lips et al. 2006; Pounds et al. 2006; Allendorf et al. 2010; Conde et al. 2019), this forecast makes understanding the diversity of core genetic loci involved in host-pathogen interactions an urgent research frontier vital to the conservation of critically endangered species. Assessing the immunogenetic diversity in species that are recovering from recent severe genetic bottlenecks represents an opportunity to assess the degree to which diversity in these highly variable genomic regions is maintained. For many taxonomic classes experiencing historically unprecedented population declines such as Amphibia, immunogenetic assessments remain a relatively unexplored area of research. This lack of immunogenetic studies represents a knowledge gap that is critical for determining how a species is equipped to handle future pathogens and should be included in conservation genetic assessments, especially for species at high risk for future disease.
The Wyoming toad (Anaxyrus baxteri, formerly Bufo baxteri), a species endemic only to the Laramie Basin in Albany County, Wyoming, United States, represents an exemplary model for such immunogenetic surveys. This species maintained a stable population up until the 1970s when it experienced a rapid decline, was declared endangered in 1984, and thought to be extinct by 1985 (Vincent et al. 2015). While the cause for the rapid decline of the species is not definitively known, dramatic changes in land use practices and the amphibian chytrid fungus, Batrachochytrium dendrobatidis (Bd) likely played a role (Dickerson et al. 2003; Dreitz 2006; Burton et al. 2009; Hornyak 2012; Polasik et al. 2016). Two years following the presumed extinction of the species, a small population was discovered at Mortenson Lake in Albany County. In 1989 some surviving wild toads were brought into captivity to found breeding colonies and considerable advances have been made in both breeding management and approaches to reintroduction into the wild (Jennings et al. 2001). Rearing toads for release, rather than tadpoles, has reduced release mortality and shortened the time to reach breeding size (Jennings and Anderson 1997) resulting in an ex situ population of around 500 individuals (Vincent et al. 2015). Expansion to multiple, privately owned release sites under Safe Harbor Agreements offers further hope for increasing the wild population (Hornyak 2012).
Diversity at immune loci is recognized as a critical axis of defense against infections from pathogens (Sommer 2005), including limiting the immunosuppressive effects of Bd (Ellison et al. 2014; Lips 2016). However, assessments of the genetic diversity of the immune system have yet to be widely adopted in most breeding programs (Woodhams et al. 2011). Several studies have indicated a link between the deeply conserved toll-like receptors (TLRs), a family of innate immune receptors that bind well conserved pathogen associated molecular patterns (PAMPs) from a wide array of pathogens, and immunity against important amphibian pathogens (Richmond et al. 2009; Chen and Robert 2011; Varga et al. 2018). TLRs are one of the first defenses in pathogen recognition and immune response that, upon recognition of a PAMP, trigger signal cascades that initiate an immune response (Khan et al. 2019). TLRs bind PAMPs via an extracellular or endosomal ectodomain, comprised of leucine-rich repeat domains (LRRs), and transduce an activation signal via a cytoplasmic toll-IL-1 receptor (TIR) domain. Most research on anuran TLRs has focused on Xenopus, with nearly 20 TLRs identified in both X. laevis and X. tropicalis. As very little is known about anuran TLRs outside of Xenopus, quantifying the TLRs present in Wyoming toads and identifying any genetic variation in them will not only be of utility for immediate conservation and breeding efforts, but also improve the understanding of Anuran innate immunity.
An additional set of genes essential for immunity are those of the major histocompatibility complex (MHC). The primary role of cell surface-bound MHC class I and class II proteins is to present cytosolic (class I) and extracellular (class II) pathogen associated antigens to immune cells which can then initiate an immune response. Although genetic differences in MHC have been associated with chytrid susceptibility and resistance across anurans (Richmond et al. 2009; Savage and Zamudio 2011; Bataille et al. 2015) the diversity of the MHC within Wyoming toads is not known. These molecules are present on nucleated cells throughout the vertebrate body (class I) or on professional antigen presenting cells (class II). MHC class I complexes include a class I protein that heterodimerizes with β−2-microglobulin (B2M). The role of B2M is primarily structural. Class I proteins possess three extracellular domains (α1, α2 and α3). The α1 and α2 domains bind peptide antigens for displaying to immune cells and these domains exhibit the most sequence diversity. The α3 domain promotes interactions with B2M and tends to be more conserved. MHC class II protein complexes consist of heterodimers of an α chain and a β chain, each of which possess two extracellular domains (α1 and α2 for the class IIα chain and β1 and β2 for the class IIβ chain). Similar to class I, the α1 and β1 domains of class II complexes bind and display peptide antigens and are typically the most diverse in sequence. Correspondingly, the α2 and β2 domains promote dimerization and tend to be more conserved. Given the critical role of MHC in the immune response and its identified importance in vertebrate captive breeding programs (Fulton et al. 2017; Grogan et al. 2017; Schenekar and Weiss 2017; Russell et al. 2018; Smallbone et al. 2021), defining the diversity of these receptors in Wyoming toads would provide an invaluable resource for the optimization of strategies to mitigate the impact of disease in recovery plans.
Here we deploy RNA-seq based analyses to describe the diversity of TLR and MHC transcripts in Wyoming toads. We first assembled de novo transcriptomes from three retired breeders and curated transcripts encoding TLR and MHC proteins. We then assessed the immunogenetic variability between the three individual toads. Finally, we compared our Wyoming toad results to two Xenopus species and two closely related bufonid species; the cane toad (Rhinella marina) that has undergone severe recent bottleneck events as a result of introductions to Australia and the common toad (Bufo bufo) that has not (Lillie et al. 2014; Thörn et al. 2021). We also compare the Wyoming toad genome at a chromosomal level to other anurans through karyotyping to identify potential chromosomal or genome duplications. Our analyses of these components of the immune system provide the first insights into how variability of immune receptors that are typically characterized as “conserved” or “highly polymorphic” is reflected within species that have experienced population declines in the last century.
Materials and Methods
Sampling protocol
All experiments involving live animals were performed in accordance with relevant institutional and national guidelines and regulations and were approved by the North Carolina State University Institutional Animal Care and Use Committee (protocol 12–127-O). Individual Wyoming toads 6691, 7039 and 7092 were retired male breeders from US Fish and Wildlife Service managed breeding facilities at the Saratoga National Fish Hatchery and Red Buttes Biological Laboratory in Wyoming. Historically, toads were kept in separate subpopulations (A, B & M) in an attempt to increase genetic diversity (Vincent et al. 2015). Group A toads were descended directly from the wild individuals brought into captivity in the late 1980s while group B toads are offspring collected from Mortenson Lake known to be derived from group A. Group M toads are derived from A and B parents. The toads in our study are derived from two of the groups from two breeding facilities. Toads 6691 and 7039 were group A toads from the Saratoga National Fish Hatchery and Red Buttes Biological Laboratory, respectively. Toad 7092 was an M toad from the Saratoga National Fish Hatchery.
Toads were evaluated for chytridiomycosis by quantitative PCR (qPCR). Samples were collected by rolling a dry polyester-tipped swab across the ventral surface of both hindlimbs ten times each and then rolled along the plantar surface of each hindfoot and between the metatarsals five times. A second sample was collected by scraping a scalpel blade 3–4 times across the ventral surface of both hindlimbs and collecting the surface epithelium onto a dry swab. Tips of swabs were placed into cryovials, air dried and stored at −20 °C. Samples were subjected to qPCR as described (Boyle et al. 2004). Toads were euthanized with buffered tricaine methane sulfonate (10 g/L) and a suite of tissues including lungs, liver, kidney, and small intestine were dissected and stored in TRIzol Reagent (Life Technologies) at −80 °C for RNA extraction. Kidney and liver samples were also harvested for primary cell culture.
Chromosomal spreads
Kidney and/or liver tissues were finely chopped and added to a 15 mL centrifuge tube containing 2 mL of Hanks Balanced Salt Solution supplemented with 2 mg/mL Collagenase-B, 3 mM CaCl2, and 0.1 mg/mL Primocin (Invivogen). The tube was rocked at 32 °C for 14 h and the resulting cell suspension used to seed a T25 tissue culture flask (with 2 μm filtered cap) containing 10 mL of RPMI-1640 supplemented with 10% fetal bovine serum, 2 mM Glutamax (Thermo Fisher), and 0.1 mg/mL Primocin. The primary culture was incubated at 32 °C, 5% CO2 and observed daily until 80% confluent, at which time dividing cells were arrested at metaphase by exposure to Karyomax (50 ng/mL) for 4 h and then harvested using routine hypotonic exposure and methanol/glacial acetic fixation. Fixed cells were dropped onto clean glass slides, air-dried, dehydrated, and stained with 80 ng/mL DAPI. Chromosome images were acquired using an Olympus BX61 microscope equipped with Smart Capture 3, (Digital Scientific, Cambridge, UK) as described previously (Breen et al. 2004).
Transcriptome Sequencing, Assembly, & Annotation
RNA was extracted from Wyoming toad lungs, liver, kidney, and small intestine using Qiagen RNeasy Kit. RNA quantity and integrity was assessed with an Agilent Bioanalyzer. For each toad, 0.25 μg of high quality RNA (RNA Integrity numbers >7) from each tissue were pooled for sequencing. RNA was prepared for multiplex sequencing with the TruSeq RNA kit (Illumina) and sequenced (2 × 100 bp PE reads) on two lanes of HiSeq2000 (Illumina). Raw data was deposited into NCBI’s Sequence Read Archive (SRA) under study number SRP156163 and accession numbers SRX4501368 (toad 6691), SRX4501367 (toad 7039), and SRX4501369 (toad 7092). Raw reads were trimmed to remove adaptors and poor quality sequences using Trimmomatic (Bolger et al. 2014) and assembled into transcripts using Trinity de novo assembler version 02–25-2013 (Grabherr et al. 2011). Transcripts containing bacterial, viral, plasmid or adaptor sequences identified by NCBI’s vector trim tool were removed. The resulting assembled transcriptomes were deposited in NCBI’s Transcriptome Shotgun Assembly (TSA) database under accession numbers GGUS00000000 (toad 6691), GGUR00000000 (toad 7039), and GGUQ00000000 (toad 7092), and BUSCO v3.0.2 was used to identify ultraconserved, single copy genes within the transcriptomes (Simão et al. 2015) employing the tetrapoda ortholog database, odb9 (Zdobnov et al. 2017). Orthologs identified here are expected to be found in all species belonging to a selected order. To assess transcriptome quality, Bowtie2 v2.3.4.1 (Langmead and Salzberg 2012) was used to estimate the percent of raw reads able to be mapped back onto each respective de novo transcriptome.
The identity of individual transcripts from de novo assembled transcriptomes were predicted using the Trinnotate pipeline (Bryant et al. 2017). Transcriptomes were assigned annotations with Kegg, Eggnog, Uniprot, and Uniref90. BlastX and BlastP were performed for the Uniprot and Uniref90 portions of the pipeline to assign gene ontology (GO) terms. An e-value threshold of 10 e-6 was used for each of the annotation stages. To assess the breakdown of transcripts into their biological processes, GO terms and their relative proportions were charted using Bioconductor package GSEABase (Morgan et al. 2018) and custom scripts. Visualizations of GO terms and variants between each individual toad were generated using the ggplot2 package in R studio (Wilkinson 2011). The annotated transcriptome files are available via Dryad (https://doi.org/10.5061/dryad.n2z34tmz9).
Manual Identification of Immune Genes
TLR and MHC sequences were identified from the three Wyoming toad de novo assembled transcriptomes using an internal custom BLAST server (Priyam et al. 2019). For both TLR and MHC, X. tropicalis protein sequences from Xenbase (Fortriede et al. 2020) and NCBI were used as queries (see figures for accession numbers). For TLR genes, well-conserved TIR domains were used as blast queries while full sequences were used as queries for MHC genes. Putative TLR and MHC transcripts were imported into Geneious Prime 2021.2 (https://www.geneious.com) where they were translated into amino acid sequences. Protein sequences were assigned as TLR (1–21) or MHC (class I or II) using a combination of BLAST (Altschul et al. 1990), SMART (Letunic et al. 2021), Pfam (Punta et al. 2012), and phylogenetic analysis. X. laevis, X. tropicalis, common toad and cane toad sequences were acquired from NCBI and Xenbase to use as comparison groups in both TLR and MHC analyses (see figures for accession numbers). For common and cane toads, both predicted genes, as well as sequences present in the reference genome that were identified via BLAST searches, were used in our analyses.
Alignments were generated using the Clustal Omega (Sievers and Higgins 2014) plugin in Geneious Prime with no adjustments. We generated alignments for both full length sequences (MHC) and domain only sequences (TLR TIR domains; MHC α or β domains). Domains were determined through a combination of SMART and Pfam analysis as well as manual annotation based on previously published domain assignments (Lillie et al. 2014, 2016; Dirscherl et al. 2014). The evolutionary relationships among genes were estimated using maximum likelihood based phylogenetic analyses in IQ-TREE version 1.6.10 (Minh et al. 2020). Support for inferred relationships was evaluated using 1000 bootstrap replicates. Xenbase nomenclature for TLRs was used to assign names to the putative Wyoming toad TLR sequences. Predicted peptide binding regions for MHC alignments are based on those used by Lillie et al. (2014, 2016).
Annotation of Major Histocompatibility Complex (MHC) sequences
As MHC class I and class II have been described in the closely related cane toads (Lillie et al. 2014, 2016), we used these sequences for identifying and cataloging Wyoming toad MHC sequences. We employed the MHC nomenclature guidelines (Klein et al. 1990) for the initial classification of Wyoming toad sequences. For example, class I sequences were named Anabax-UX where the six letter abbreviation, Anabax, refers to the first three letters of the genus and species, Anaxyrus baxteri. The letter “U” (named for “Uno”) corresponds to the classical MHC class I lineage, and the variable second letter “X” designates the locus (named alphabetically). Similarly, class II sequences were named Anabax-DXA or Anabax-DXB, with the first letter D (named for “duo”) corresponding to the classical class II lineage, the variable second letter “X” designates the locus (named alphabetically), and the third letter reflecting an α chain (A) or β chain (B). Predicted alleles of the same sequence are indicated with a numeric suffix (e.g. *01, *02).
Results
Chromosomal Spreads
We find that Wyoming toads possess eleven pairs of chromosomes (Fig. 1), as determined by karyotype analyses using DAPI stained metaphase preparations. This is consistent with at least 20 other diploid bufonid species with which Wyoming toads share recent common ancestry including the American toad (Bufo americanus), common toad (Bufo bufo), and green toad (Bufo viridis) (Gregory 2021). These results suggest Wyoming toads have a similar genome size to most bufonids, demonstrating no evidence for cryptic polyploidy.
Fig. 1. Wyoming toad chromosomes.

a Chromosomal spread and b karyotype of a metaphase preparation of a Wyoming toad (Anaxyrus baxteri) 7039. Wyoming toads possess 11 pairs of chromosomes with no evidence of cryptic polyploidy.
Transcriptome assembly and annotation
The final de novo transcriptomes consisted of 331,616, 199,011 and 209,390 transcripts, for toads 6691, 7039, and 7092, respectively. More than 90% of cleaned reads were accurately mapped onto the respective transcriptome assembly, suggesting high quality transcriptome assemblies. (Supplementary Table 1). More than 75% of tetrapod orthologs could be assigned to Wyoming toad transcripts by BUSCO analysis (Supplementary Fig. S1) (Manni et al. 2021), suggesting these transcriptomes from selected tissues represent a sufficient survey of the full Wyoming toad transcriptome. Additionally, 15 genes were found to be duplicated in all three toads (Supplementary Table 2), though further Gene Ontology enrichment analyses are not possible on so few genes. Analysis of automated transcriptome annotation revealed little variation between the three individuals in terms of sequences annotated. (Supplementary Fig. S2).
Toll Like Receptor Sequences
Vertebrate TLRs can be classified into six major families: TLR1, TLR3, TLR4, TLR5, TLR7 and TLR11 with some families including multiple TLRs (e.g. TLR7, TLR8 and TLR9 are all within the TLR7 family) (Roach et al. 2005; Ishii et al. 2007; Nie et al. 2018). Although TLR sequence information is publicly available for Xenopus and other amphibian species, automated gene annotations have not been verified for Anuran taxa outside of X. tropicalis. We find all bufonids surveyed to express members of all six TLR families (Fig. 2 and Supplementary Table S3). In the TLR1 family, Wyoming toad, common toad, and cane toad were found to have TLR1, TLR2, and TLR14 sequences. Wyoming toad and cane toad expressed two TLR1 sequences: TLR1a and TLR1b. This parallels the multiple TLR1 sequences found in X. tropicalis (Roach et al. 2005; Ishii et al. 2007; Nie et al. 2018). In our Wyoming toad transcriptomes, full-length TLR1a and TLR1b were identified in all three individuals while complete TLR2 and TLR14 sequences were only detected in individuals 6691 and 7092. Although truncated transcripts of TLR2 and TLR14 were identified from toad 7039 with identical ectodomains to toads 6691 and 7092, these transcripts were missing a majority of their TIR domain and thus were not included in the phylogeny (Fig. 2).
Fig. 2. Wyoming toad transcriptomes reveal 13 toll-like receptor genes.

Phylogenetic tree comparing TLR TIR domains of three Wyoming toads to those in five other ranid species, including two additional bufonids. Sequences are from Wyoming toad (Anabax), common toad (Bufbuf), cane toad (Rhimar), Xenopus laevis (Xenlae) and Xenopus tropicalis (Xentro). Sequence identifiers for Wyoming toad TLRs are provided in Supplemental Table S3. If available, GenBank or Xenbase identifiers are included in the figure with sequence names. As TLR LRR ectodomains are typically variable between species, the more conserved TIR domains were employed for phylogenetic analyses (Quiniou et al. 2013; Boudinot et al. 2014; Wcisel et al. 2017). Wyoming toads express TLRs in all six of the major TLR families with identical or nearly identical sequences across the three individuals. Circles at nodes indicate bootstrap support values (BSS) with filled black circles indicating BSS=100, gray circles indicating BSS values equal to or greater than 90 but less than 100, and white circles indicating BSS values greater than 70 but less than 90. Scale bar indicates substitutions over time.
The single gene TLR3, TLR4 and TLR5 families are known from Xenopus species, and were also found in all bufonid species surveyed, with no variation between the three individual Wyoming toad sequences. Within the TLR7 family, Xenopus encodes TLR7, TLR8, and TL9 (Roach et al. 2005; Ishii et al. 2007; Nie et al. 2018). TLR7 was not identified in any bufonid examined. In contrast, two TLR8 sequences (TLR8a and TLR8b) were found across all bufonids examined. However, each of the Wyoming toads expressed only one of the TLR8 sequences. TLR8a was found in toad 7039 and TLR8b was found in toad 6691 and toad 7092. It is currently unclear if TLR8a and TLR8b represent different genes or, perhaps, reflect different haplotypes of the same gene. The common toad also encodes TLR9, but orthologs were not identified in the Wyoming toad or cane toad.
Within the TLR11 family, Xenopus encodes TLR12, TLR13, TLR21 and TLR22 (Roach et al. 2005; Ishii et al. 2007; Nie et al. 2018). TLR12, TLR13 and TLR21 were identified in all bufonids examined. All Wyoming toad sequences were identical at TIR domains across all individual genes within the TLR11 family, with the exception of TLR12 where individual 6691 and 7039 were identical, but differed from 7092 at one amino acid residue. Although Xenopus are known to encode a TLR22 sequence, this was not found in any bufonid species surveyed. For all identified sequences in the TLR11 family, there is evidence of only one gene copy per species (Fig. 2).
MHC class I sequences
Wyoming toads were found to express identical B2M sequences across all individual toads (Supplemental Table S4) and four different groups of MHC class I sequences (Anabax-UA, Anabax-UB, Anabax-UC, Anabax-UD) that likely indicate the presence of at least two and up to four MHC class I gene loci in this species. Two to three transcripts were identified from each group that may reflect haplotypic variation and/or alternative mRNA splicing. Phylogenetic analyses of the Wyoming toad MHC class I α1 - α3 domains with those from cane toad, common toad, X. laevis and X. tropicalis demonstrate that the Wyoming toad Anabax-UA sequences are the most similar to the cane toad MHC class I UA sequence, Rhimar-UA (Lillie et al. 2014), whereas Anabax-UB, Anabax-UC, and Anabax-UD sequences are more similar to common toad “F10-like” sequences (Fig. 3). Transcripts of Anabax-UA are the only ones encoding a prototypical MHC class I structure with a transmembrane domain and we identified two full-length Anabax-UA sequences: Anabax-UA*01 from toad 7039, and Anabax-UA*02 from toads 6691 and 7092. These have identical signal peptide, α1, α3, and transmembrane domains and cytoplasmic tails, and differ only by four residues in the α2 domain (Fig 4, Supplemental Table S4, and Supplemental Fig S3).
Fig. 3. Phylogenetic comparison of Wyoming toad MHC class I sequences to other anurans.

Phylogenetic trees comparing Wyoming toad (Anabax) MHC class I protein sequences to those of common toad (Bufbuf), cane toad (Rhimar), Xenopus laevis (Xenlae) and Xenopus tropicalis (Xentro). Trees were generated using alignments of (a) contiguous α1-α2-α3 domains or by using individual protein domains; (b) α1, (c) α2, and (d) α3. Note that Bufbuf “F10-like(b)” is likely a “Q9-like” sequence and inaccurately annotated. Circles at nodes indicate bootstrap support values (BSS) with filled black circles indicating BSS=100, gray circles indicating BSS values equal to or greater than 90 but less than 100, and white circles indicating BSS values greater than 70 but less than 90. Scale bar indicates substitutions over time.
Fig. 4. Wyoming toad transcriptomes reveal four distinct groups of MHC class I sequences.
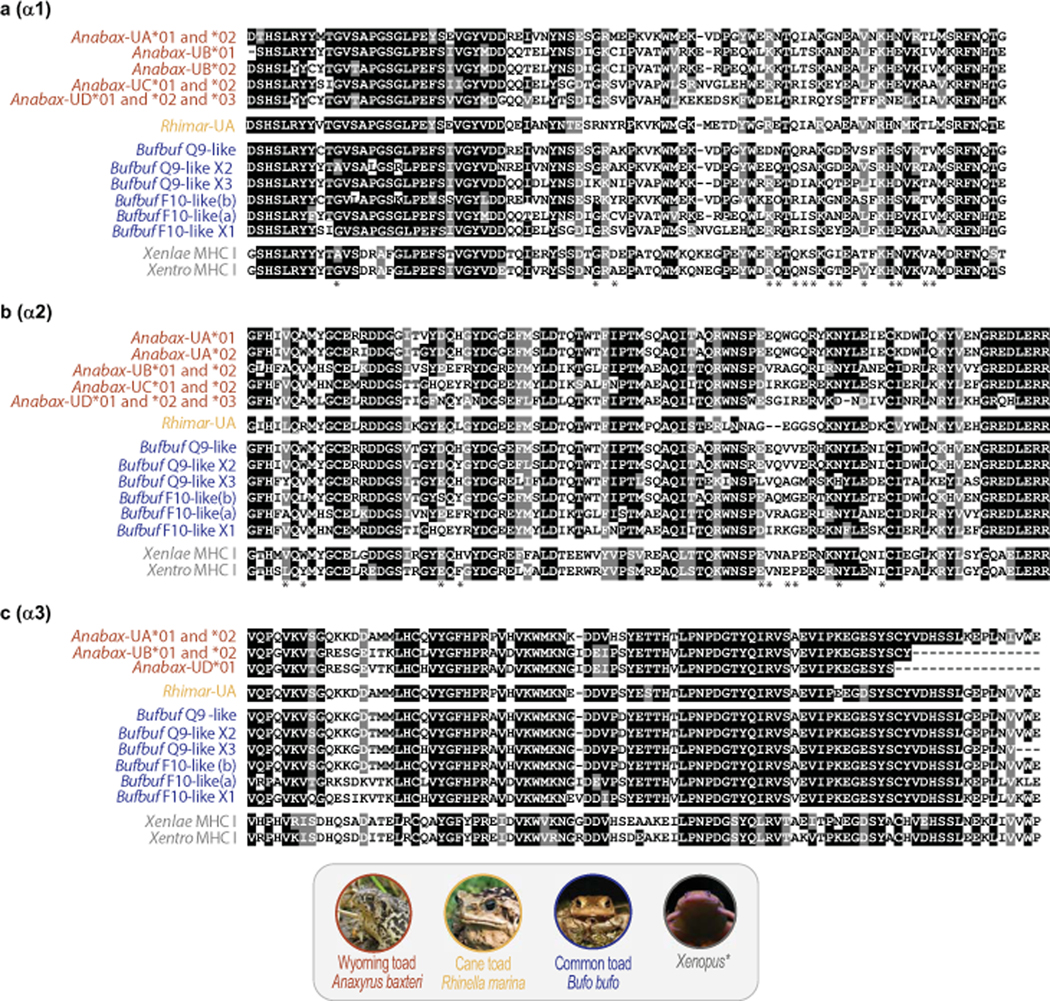
Annotated alignments of individual MHC class I (a) α1, (b) α2, and (c) α3 domains from Wyoming toad (Anabax) with common toad (Bufbuf), cane toad (Rhimar), Xenopus laevis (Xenlae) and Xenopus tropicalis (Xentro). Asterisks indicate putative peptide binding sites as described by Lillie et al (2014).
In contrast to Anabax-UA, all other class I sequences either encode a stop codon after the α2 (Anabax-UC) or are 3’ truncated (Anabax-UB and Anabax-UD). We identified two partial Anabax-UB sequences: Anabax-UB*01 from toads 7039 and 7092, and Anabax-UB*02 from toad 6691. Both sequences encode identical α2 and α3 domains but differ by three residues in the α1 domain. Anabax-UB*01 encodes a signal peptide sequence, but is truncated on the 3’ lacking both a TM domain and a stop codon. Anabax-UB*02 is truncated at both the 5’ and 3’ ends and encodes only α1, α2 and α3 domains (Fig 4, Supplemental Table S4, and Supplemental Fig S4). Regarding Anabax-UC, we identified two sequences: Anabax-UC*01 from toads 6691 and 7092, and Anabax-UC*02 from toads 7039 and 7092. These sequences encode identical signal peptides, as well as α1 and α2 domains, but lack significant carboxyl-terminal sequences. Anabax-UC*01 encodes a stop codon 16 residues after the α2 domain, whereas Anabax-UC*02 lacks a stop codon and may be 3’ truncated. (Fig 4, Supplemental Table S4, and Supplemental Fig S5). Finally, we identified three partial Anabax-UD sequences: Anabax-UD*01 from toads 6691 and 7039, Anabax-UD*02 from toad 7092 and Anabax-UD*03 from toad 6691. These sequences encode identical signal peptides, as well as α1 and α2 domains, but differ in the carboxyl-terminus. Anabax-UD*01 encodes a complete α3 domain, but no stop codon. The protein encoded by Anabax-UD*02 encodes nearly the identical sequence to Anabax-UD*01 but only includes approximately half of the α3 domain. In contrast, Anabax-UD*03 encodes what may reflect a partial sequence of an unusual α3 domain that shares little resemblance to the α3 domain of Anabax-UD*01 (Fig 4, Supplemental Table S4, and Supplemental Fig S6).
MHC class II sequences
Compared to the MHC class I transcripts, Wyoming toad MHC class II sequences revealed much less diversity displaying only two distinct MHC class IIα chains (Anabax-DAA and Anabax-DBA) (Fig. 5) and two putative haplotypes of a single MHC IIβ chain (Anabax-DAB*01 and Anabax-DAB*02) (Fig. 6 and Supplemental Table S5). Two additional, but partial, class IIβ sequences were also identified, but not included in this analysis (Supplemental Table S5).
Fig. 5. Wyoming toad transcriptomes reveal two distinct groups of MHC class IIα sequences.

Phylogenetic trees comparing Wyoming toad (Anabax) MHC class IIα chain protein sequences to common toad (Bufbuf), cane toad (Rhimar), Xenopus laevis (Xenlae) and Xenopus tropicalis (Xentro). Trees were generated using alignments of (a) contiguous α1-α2 domains or by using individual protein domains; (b) α1, and (c) α2. Annotated alignments of individual (d) α1, and (e) α2 domains from Wyoming toad with other anuran species. Asterisks indicate putative binding sites as described by Lillie et al (2016). Circles at nodes indicate bootstrap support values (BSS) with filled black circles indicating BSS=100, gray circles indicating BSS values equal to or greater than 90 but less than 100, and white circles indicating BSS values greater than 70 but less than 90. Scale bar indicates substitutions over time.
Fig. 6. Wyoming toad transcriptomes reveal a single group of MHC class IIβ sequences.
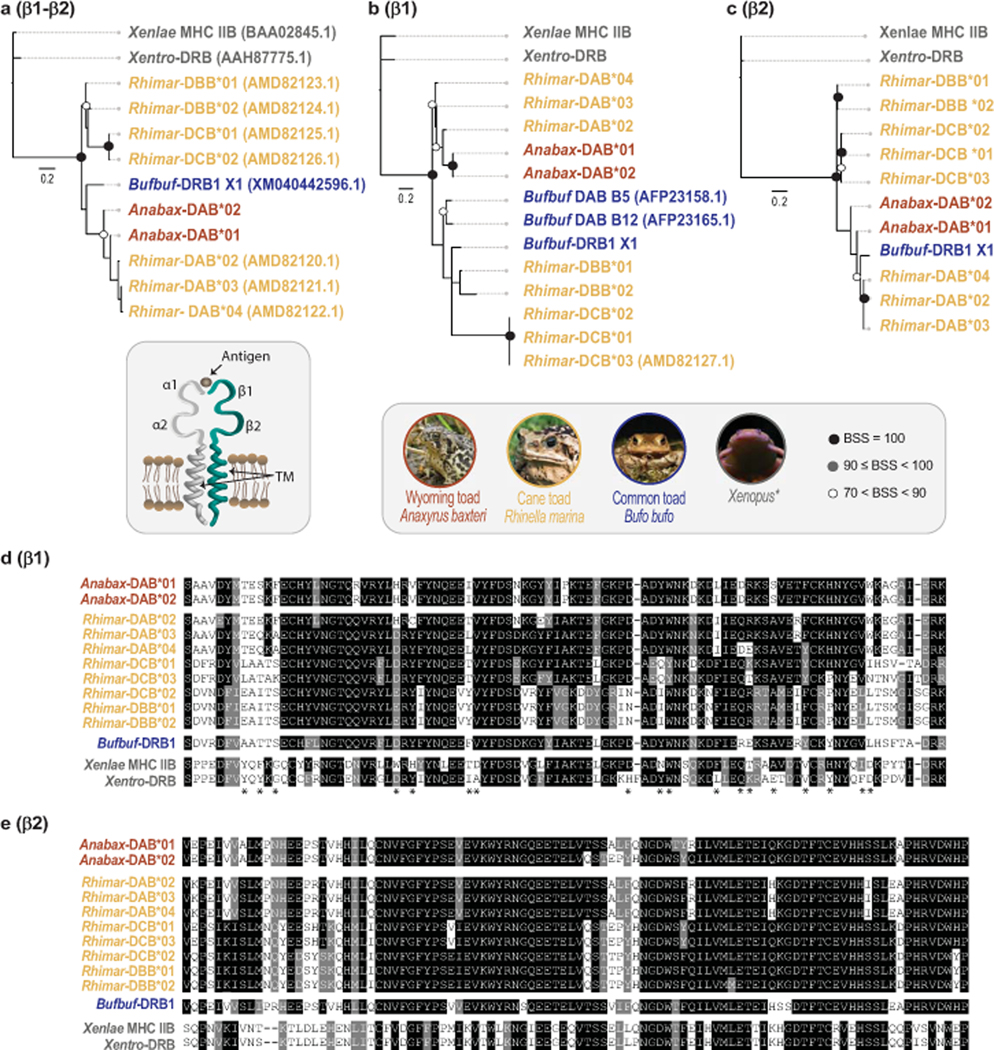
Phylogenetic trees comparing Wyoming toad (Anabax) MHC class IIβ chain protein sequences to common toad (Bufbuf), cane toad (Rhimar), Xenopus laevis (Xenlae) and Xenopus tropicalis (Xentro). Trees were generated using alignments of (a) contiguous β1-β2 domains or by using individual protein domains; (b) β1, and (c) β2. Annotated alignments of individual (d) β1, and (e) β2 domains from Wyoming toad with other anuran species. Asterisks indicate putative binding sites as described by Lillie et al (2016). Circles at nodes indicate bootstrap support values (BSS) with filled black circles indicating BSS=100, gray circles indicating BSS values equal to or greater than 90 but less than 100, and white circles indicating BSS values greater than 70 but less than 90. Scale bar indicates substitutions over time.
We found all Anabax-DAA transcripts to encode identical full-length proteins between individuals, a finding that was repeated across Anabax-DBA transcripts (Supplemental Table S5). The Anabax-DAA and Anabax-DBA proteins possess signal peptide sequence, α1, α2 and transmembrane domains (Supplementary Figure S7). A one-to-one comparison revealed that the α1 domains from Anabax-DAA and Anabax-DBA are 52% identical and that the α2 domains are more similar at 68% identity. In our analysis, Anabax-DAA is most similar to cane toad Rhimar-DAA, whereas Anabax-DBA is most similar to Rhimar-DCA (Fig. 5). In contrast to the homogeneity within Anabax-DAA or Anabax-DBA transcripts, Anabax-DAB transcripts encode full-length proteins that represent two distinct groups: Anabax-DAB*01 and Anabax-DAB*02 that are collectively most similar to cane toad Rhimar-DAB (Fig. 6). Both Anabax-DAB*01 and Anabax-DAB*02 encode identical signal peptide and β1 domains. However, the β2 domains between groups differ at 7 residues (92% identity) and each group possesses distinct transmembrane domains (Supplementary Fig. S8). We identified identical Anabax-DAB*01 coding sequences in all three individuals, while Anabax-DAB*02 sequences were only recovered from toads 7039 and 7092 (Supplementary Table S5).
Discussion
Genetic management tools for the Wyoming toad are severely lacking, necessitating breeding facilities to utilize studbooks and mean kinship analyses in hopes of maintaining diversity within the captive populations (Vincent et al. 2015). While recent studies have shown reduced diversity at microsatellite loci (Martin et al. 2019), no studies to date have assessed variation at adaptive loci or genes responsible for immune function that may confer an adaptive advantage to Bd. Our results indicate that Wyoming toads do not exhibit a high level of variability between individuals at TLR or MHC loci. In particular, sequence diversity of TLR and MHC class II transcripts is nearly homogenous between individuals. This sequence similarity highlights the need for future investigations that assess whether Wyoming toad, or even bufonid species in general, have reduced sequence diversity in these core loci of the innate immune system relative to other anurans. In contrast, MHC class I transcripts display enough sequence variability to predict that at least two MHC class I genes are present in the Wyoming toad genome and that each gene likely has two, or possibly three, haplotypes that may influence immune function.
The conserved nature of toll-like receptor genes and their role in the innate immune response within species necessitates higher levels of sequence conservation than those exhibited by MHC loci. From our transcriptome analyses, we identified transcripts representing twelve different TLRs (TLR1a, TLR1b, TLR2, TLR3, TLR4, TLR5, TLR8a, TLR8b, TLR12, TLR13, TLR14 and TLR21) (Fig. 2). These TLR receptors are also present in Xenopus species. However, relative to Xenopus, the Wyoming toad is missing TLR7, TLR9 and TLR22 and does not appear to encode any additional TLRs. TLR7 is known to bind single stranded RNA and TLR9 recognizes unmethylated CpG DNA sequences (Akira and Takeda 2004; Vidya et al. 2018). TLR22 has been described from amphibians and teleost fishes and studies with teleost TLR22 suggest it recognizes microbial RNA (Samanta et al. 2014; Ji et al. 2019; Du et al. 2019). Further studies of anuran TLR function are needed to better characterize the implications of these gene losses for the Wyoming Toads.
Within Wyoming toads, we identified full-length transcripts encoding identical proteins across all individuals for nearly all TLRs. The exceptions to sequence homogeneity comprised TLR2 (7039 encodes a truncated sequence), TLR8a (7039 encodes a different TIR domain), TLR12 (the 7092 TIR domain differs by a single residue, 6691 encodes a truncated ectodomain, and all three individuals vary within the ectodomain at numerous residues) and TLR14 (7039 encodes a truncated sequence). Further, TLR8a and TLR8b transcripts identified from Wyoming toad encode identical ectodomains (Supplemental Table S6). This result stands in contrast to expectations from common toad and cane toad where these genes are predicted to encode distinctly different ectodomains that presumably bind different PAMPs. We classify Wyoming toad TLR8a and TLR8b as different sequences as their TIR domains are only 89% identical (Fig. 2 and Supplemental Table S7). Although this observation might suggest that the TLR8 paralogs in Wyoming toad have not diversified as observed in common toad and cane toad, we cannot exclude the possibility that what we identify as TLR8a and TLR8b from the Wyoming toad are different haplotypes of the same gene, and that Wyoming toad encodes a second TLR gene that was not detected in our transcriptome analyses. Collectively, the loss of three TLRs and lack of diversity between TLR8a and TLR8b ectodomains may impact the ability of the Wyoming toad to mount a successful immune defense against viral pathogens. It is our hope that this classification of Wyoming toad TLRs, combined with sequences from the common toad and cane toad may facilitate future studies on their role in early immune responses to infection.
Genes within the MHC are hotspots for polymorphisms and are some of the most diverse regions within the vertebrate genome (Eizaguirre et al. 2012; Radwan et al. 2020). Because of their high level of variability and their importance in adaptive immunity, peptide binding regions of MHC molecules are often targets for selection with heterozygosity typically conveying an advantage, as they are able to present a wider array of peptides (McClelland Erin E. et al. 2003; Sommer 2005; Savage and Zamudio 2011). However, Wyoming toad transcripts identified as MHC class I revealed four distinct groups that were nearly identical between individuals. In general, these groups have different residues across the peptide binding regions for each domain. In instances where their transcripts had both identical and non identical domains (UA α2 domain and UB α1 domain), putative binding regions shared identical amino acid residues except for the α2 domain in UA where the two transcripts had different amino acids at the second peptide binding region (Fig. 4 and Supplementary Figures S3 and S4). Compared to cane toads, Wyoming toads have a greater number of expressed MHC class I transcripts, however we found only one full length sequence, Anabax-UA, that we feel confidently acts as a true MHC class I molecule. Other MHC class I sequences were truncated, which could indicate that these sequences are from genes encoding non-classical or secreted MHCI molecules or indicate sequencing or assembly error. Increased sampling beyond the scope of this project, including genomic analyses and inclusion of additional individuals, would be needed to distinguish between these two alternatives.
The three Wyoming toads examined in this study also revealed a reduced MHC class II repertoire relative to their MHC class I sequences, as well as in comparison to other bottlenecked bufonids, such as cane toads (Lillie et al. 2014; Selechnik et al. 2019). We found two class IIα transcripts, Anabax-DAA and Anabax-DBA, that were present across all individuals with 100% sequence identity. Out of 18 putative peptide binding residues, eight are different between the two transcript classes. Similarly, two class IIβ transcripts, both Anabax-DAB, were identified which are likely haplotypes of the same gene. These sequences encode identical β1 domains and thus possess identical peptide binding regions. In several amphibian species, specific changes within the peptide binding regions of MHC IIβ (DAB) have been associated with chytridiomycosis susceptibility or tolerance through either immune response regulation or direct interaction with Bd peptides (Richmond et al. 2009; Savage and Zamudio 2011; Bataille et al. 2015; Savage et al. 2020). This linkage between MHC class II and Bd suggests the possibility for strong selective pressures towards specific MHC class II conformations in anurans. Our finding of near homogeneity at MHC class II highlights a significant genotypic challenge facing this species in regard to overall disease pressures. Further studies across larger numbers of individuals from the breeding program are critically needed to investigate whether selective breeding for more diverse MHC class II genotypes is possible within the captive population.
A limitation of our study is the small number of individuals that could be used due to the lethal nature of taking samples from immune tissues. In hopes of increasing diversity, toads are now managed as one population rather than three smaller subpopulations (Vincent et al. 2015). While no B strain toads were available for this study, having M (7092) and A (6691 & 7039) toads (see Methods) should provide insights into the overall genetic diversity that formed the basis of the breeding program. Future genomics research evaluating genomic markers that confer an advantage for wild persistence in reintroduced individuals would be warranted.
Bottleneck events are not uncommon in amphibian species, and are forecast to increase due to climate change, habitat fragmentation and loss, and the introduction of disease (Allentoft and O’Brien 2010; Pabijan et al. 2020). While the Wyoming toads examined in this study display a low level of variation at immune loci, the ability to recover from a bottleneck relies on more than immunogenetic diversity. A number of highly invasive Anuran species have repeatedly undergone multiple bottleneck events across the world (Beard and Pitt 2005; Ficetola et al. 2008; Forti et al. 2017). For example, the Cane toad is one of the world’s most successful invasive species of amphibians (Phillips et al. 2007; Brown et al. 2015) yet has a reduced classical MHC Class I repertoire. Additionally, MHC diversity can vary naturally across populations. In amphibian lineages that span Hochstetter’s frogs (Leiopelma hochstetteri) to crested newts (Triturus cristatus) MHC class II diversity varies between populations from high to very little. In these cases, the immunological consequences of such differences remain unknown (Babik et al. 2009; Lillie et al. 2015). In other species, reduced diversity at MHC loci, due to either natural variation or bottlenecking events, can correlate with increased infection and differential survivorship (Belasen et al. 2019; Kosch et al. 2019). This is exemplified by common toads in Sweden, where overall MHC class II diversity increases from north to south, with northern populations showing a higher susceptibility to, and mortality from, disease relative to more diverse southern populations (Thörn et al. 2021). These examples highlight the complexity in correlating diversity of immune loci to the role of disease in the recovery of populations from bottlenecking events. Future studies of the selective pressures placed on the immunome of Wyoming toads are critically needed.
As they continue to recover from near extinction, the outlook for Wyoming toads no longer looks as grim as it once did. However, there are still numerous challenges facing the species including the threat of Bd and other pathogens, as well as reduced genetic diversity. We recommend the inclusion of immune associated genetic markers to evaluate genetic variation within captive and wild Wyoming toad populations to improve management decisions. With the devastating effects of infectious diseases like Bd, it has become vital for conservation management practices to incorporate immunogenetics and broader “omics” techniques to better understand the interplay between variation at immune loci and disease outcomes. The results of this study highlight the need for more in depth research on the immunome of not only this species, but also others that are threatened with extinction.
Conclusion
Our analyses indicate that overall immunogenetic diversity within the Wyoming toad species is minimal at best, reflecting overall expectations of genomic diversity in studies using microsatellite markers (Martin et al. 2019). While overall genetic diversity may be low, our results provide potential immune gene markers that could be combined with other markers to create a genetic panel to better monitor species-wide diversity. The wide application of immunogenetics in conservation (including studies on diversity, mate selection, and disease tolerance), coupled with greater accessibility for next generation sequencing, underscores the potential for incorporating such assays into the management of species of concern. With more than 40% of amphibians experiencing decline due to disease or anthropogenic pressures (Pabijan et al. 2020), understanding the relationship between core functional genomic regions and species persistence is critical for the future of a diverse Amphibia. As population loss trends continue across the planet, captive breeding programs have become increasingly common (Farquharson et al. 2021) and “omics” techniques may be key tools for understanding host pathogen interactions and guiding management decisions in species of concern.
Supplementary Material
Acknowledgements
We are indebted to the Wyoming Toad Recovery Team for assistance and providing the toads used in this study. We thank Debra Tokarz (North Carolina State University), Iván Rodríguez-Nunez (North Carolina State University), Amanda Kortum (North Carolina State University) and Jacques Robert (University of Rochester Medical Center) for initial sequence analyses and helpful discussions and Karen Gore (North Carolina State University) for chytridiomycosis qPCR. Photograph sources: https://commons.wikimedia.org/wiki/File:African_clawed_frogs;_Xenopus_laevis.jpg, https://commons.wikimedia.org/wiki/File:Bufo-bufo-erdkroete-maennlich-front.jpg, https://commons.wikimedia.org/wiki/File:Cane-toad.jpg, https://commons.wikimedia.org/wiki/File:Anaxyrus_baxteri-3.jpg
Funding.
This report was supported by grants from the National Science Foundation (IOS-1755242 to AD and IOS-1755330 to JAY) and by funds from the North Carolina State University Research and Innovation Seed Funding (RISF) Program and from the North Carolina State University Center for Comparative Medicine and Translational Research (CCMTR). H.D.A. and J.R. were supported by a National Institutes of Health Biotechnology Traineeship (T32 GM008776). H.D.A. also was supported by a Joseph E. Pogue Fellowship through the UNC Royster Society of Fellows. D.J.W. was supported by a Graduate Student Fellowship from the Triangle Center for Evolutionary Medicine (TriCEM)/National Evolutionary Synthesis Center (NESCent).
Footnotes
Competing Interests. The authors have no competing interests to declare that are relevant to the content of this article.
Data Availability.
Transcriptome sequence data has been archived on NCBI as BioProject PRJNA484136; SRA accession numbers SRX4501368 (toad 6691), SRX4501367 (toad 7039); and SRX4501369 (toad 7092); and TSA accession numbers accession numbers GGUS00000000 (toad 6691), GGUR00000000 (toad 7039), GGUQ00000000 (toad 7092). Annotated transcriptome data are archived on Dryad (https://doi.org/10.5061/dryad.n2z34tmz9).
References
- Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4:499–511. 10.1038/nri1391 [DOI] [PubMed] [Google Scholar]
- Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709. 10.1038/nrg2844 [DOI] [PubMed] [Google Scholar]
- Allentoft ME, O’Brien J (2010) Global Amphibian Declines, Loss of Genetic Diversity and Fitness: A Review. Diversity 2:47–71. 10.3390/d2010047 [DOI] [Google Scholar]
- Altschul SF, Gish W, Miller W, et al. (1990) Basic local alignment search tool. J Mol Biol 215:403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Babik W, Pabijan M, Arntzen JW, et al. (2009) Long-term survival of a urodele amphibian despite depleted major histocompatibility complex variation. Mol Ecol 18:769–781. 10.1111/j.1365-294X.2008.04057.x [DOI] [PubMed] [Google Scholar]
- Bataille A, Cashins SD, Grogan L, et al. (2015) Susceptibility of amphibians to chytridiomycosis is associated with MHC class II conformation. Proc Biol Sci. 282(1805): 20143127. 10.1098/rspb.2014.3127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard KH, Pitt WC (2005) Potential consequences of the coqui frog invasion in Hawaii. Divers Distrib 11:427–433. 10.1111/j.1366-9516.2005.00178.x [DOI] [Google Scholar]
- Belasen AM, Bletz MC, Leite D da S, et al. (2019) Long-Term Habitat Fragmentation Is Associated With Reduced MHC IIB Diversity and Increased Infections in Amphibian Hosts. Frontiers in Ecology and Evolution 6:236. 10.3389/fevo.2018.00236 [DOI] [Google Scholar]
- Berger L, Speare R, Daszak P, et al. (1998) Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc Natl Acad Sci U S A 95:9031–9036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudinot P, Zou J, Ota T, et al. (2014) A tetrapod-like repertoire of innate immune receptors and effectors for coelacanths. J Exp Zool B Mol Dev Evol 322:415–437. 10.1002/jez.b.22559 [DOI] [PubMed] [Google Scholar]
- Boyle DG, Boyle DB, Olsen V, et al. (2004) Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis Aquat Organ 60:141–148. 10.3354/dao060141 [DOI] [PubMed] [Google Scholar]
- Breen M, Hitte C, Lorentzen TD, et al. (2004) An integrated 4249 marker FISH/RH map of the canine genome. BMC Genomics 5:65. 10.1186/1471-2164-5-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GP, Kelehear C, Shilton CM, et al. (2015) Stress and immunity at the invasion front: a comparison across cane toad (Rhinella marina) populations. Biol J Linn Soc Lond 116:748–760. 10.1111/bij.12623 [DOI] [Google Scholar]
- Bryant DM, Johnson K, DiTommaso T, et al. (2017) A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep 18:762–776. 10.1016/j.celrep.2016.12.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton EC, Gray MJ, Schmutzer AC, Miller DL (2009) Differential responses of postmetamorphic amphibians to cattle grazing in wetlands. J Wildl Manage 73:269–277. 10.2193/2007-562 [DOI] [Google Scholar]
- Chen G, Robert J (2011) Antiviral immunity in amphibians. Viruses 3:2065–2086. 10.3390/v3112065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde DA, Staerk J, Colchero F, et al. (2019) Data gaps and opportunities for comparative and conservation biology. Proc Natl Acad Sci U S A 116:9658–9664. 10.1073/pnas.1816367116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daszak P, Cunningham AA, Hyatt AD (2003) Infectious disease and amphibian population declines. Divers Distrib 9:141–150. 10.1046/j.1472-4642.2003.00016.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson KK, Hooper MJ, Huang T, Allen M (2003) Determination of Malathion Aerial Drift and Associated Effects to the Endangered Wyoming Toad (Bufo baxteri) Using Surrogate Woodhouse’s Toads (Bufo woodhousii) at Mortenson and Hutton Lake National Wildlife Refuges and Potential Reintroduction Sites. In: Multiple Stressor Effects in Relation to Declining Amphibian Populations. ASTM STP 1443. Linder G, Krest S, Sparling D, and Little E, Eds., American Society for Testing and Materials, West Conshohocken, PA [Google Scholar]
- Dirscherl H, McConnell SC, Yoder JA, de Jong JLO (2014) The MHC class I genes of zebrafish. Dev Comp Immunol 46:11–23. 10.1016/j.dci.2014.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreitz VJ (2006) Issues in species recovery: an example based on the Wyoming toad. BioScience 56:765–771. 10.1641/0006-3568(2006)56[765:IISRAE]2.0.CO;2 [DOI] [Google Scholar]
- Du X, Wu J, Li Y, et al. (2019) Multiple subtypes of TLR22 molecule from Schizothorax prenanti present the functional diversity in ligand recognition and signal activation. Fish Shellfish Immunol 93:986–996. 10.1016/j.fsi.2019.08.042 [DOI] [PubMed] [Google Scholar]
- Eizaguirre C, Lenz TL, Kalbe M, Milinski M (2012) Rapid and adaptive evolution of MHC genes under parasite selection in experimental vertebrate populations. Nat Commun 3:621. 10.1038/ncomms1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison AR, Tunstall T, DiRenzo GV, et al. (2014) More than skin deep: functional genomic basis for resistance to amphibian chytridiomycosis. Genome Biol Evol 7:286–298. 10.1093/gbe/evu285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquharson KA, Hogg CJ, Grueber CE (2021) Offspring survival changes over generations of captive breeding. Nat Commun 12:3045. 10.1038/s41467-021-22631-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficetola GF, Bonin A, Miaud C (2008) Population genetics reveals origin and number of founders in a biological invasion. Mol Ecol 17:773–782. 10.1111/j.1365-294X.2007.03622.x [DOI] [PubMed] [Google Scholar]
- Forti LR, Becker CG, Tacioli L, et al. (2017) Perspectives on invasive amphibians in Brazil. PLoS One 12:e0184703. 10.1371/journal.pone.0184703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortriede JD, Pells TJ, Chu S, et al. (2020) Xenbase: deep integration of GEO & SRA RNA-seq and ChIP-seq data in a model organism database. Nucleic Acids Res 48:D776–D782. 10.1093/nar/gkz933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton JE, Berres ME, Kantanen J, Honkatukia M (2017) MHC-B variability within the Finnish Landrace chicken conservation program. Poult Sci 96:3026–3030. 10.3382/ps/pex102 [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory TR (2021) Animal Genome Size Database. http://www.genomesize.com. Accessed 3 Nov 2021
- Grogan KE, Sauther ML, Cuozzo FP, Drea CM (2017) Genetic wealth, population health: Major histocompatibility complex variation in captive and wild ring-tailed lemurs (Lemur catta). Ecol Evol 7:7638–7649. 10.1002/ece3.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haver M, Le Roux G, Friesen J, et al. (2022) The role of abiotic variables in an emerging global amphibian fungal disease in mountains. Sci Total Environ. 815:152735. 10.1016/j.scitotenv.2021.152735 [DOI] [PubMed] [Google Scholar]
- Hof C, Araújo MB, Jetz W, Rahbek C (2011) Additive threats from pathogens, climate and land-use change for global amphibian diversity. Nature 480:516–519. 10.1038/nature10650 [DOI] [PubMed] [Google Scholar]
- Hornyak V (2012) The Wyoming Toad: Partnerships for Recovery. World Association of Zoos and Aquariums Magazine 366:33–35 [Google Scholar]
- Ishii A, Kawasaki M, Matsumoto M, et al. (2007) Phylogenetic and expression analysis of amphibian Xenopus Toll-like receptors. Immunogenetics 59:281–293. 10.1007/s00251-007-0193-y [DOI] [PubMed] [Google Scholar]
- Jennings M, Anderson A (1997) The Wyoming Toad. Endangered Species Bulletin 22:16–17 [Google Scholar]
- Jennings M, Beiswenger R, Corn PS, et al. (2001) Population and habitat viability assessment for the Wyoming toad (Bufo baxteri): Final workshop report. Conservation Breeding Specialist Group, Apple Valley, MN [Google Scholar]
- Ji J, Liao Z, Rao Y, et al. (2019) Thoroughly Remold the Localization and Signaling Pathway of TLR22. Front Immunol 10:3003. 10.3389/fimmu.2019.03003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I, Maldonado E, Silva L, et al. (2019) The vertebrate TLR supergene family evolved dynamically by gene gain/loss and positive selection revealing a host-pathogen arms race in birds. Diversity 11.: 10.3390/d11080131 [DOI] [Google Scholar]
- Klein J, Bontrop RE, Dawkins RL, et al. (1990) Nomenclature for the major histocompatibility complexes of different species: a proposal. Immunogenetics 31:217–219. 10.1007/BF00204890 [DOI] [PubMed] [Google Scholar]
- Kosch TA, Silva CNS, Brannelly LA, et al. (2019) Genetic potential for disease resistance in critically endangered amphibians decimated by chytridiomycosis. Anim Conserv 22:238–250. 10.1111/acv.12459 [DOI] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Khedkar S, Bork P (2021) SMART: recent updates, new developments and status in 2020. Nucleic Acids Research 49:D458–D460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie M, Cui J, Shine R, Belov K (2016) Molecular characterization of MHC class II in the Australian invasive cane toad reveals multiple splice variants. Immunogenetics 68:449–460. 10.1007/s00251-016-0919-9 [DOI] [PubMed] [Google Scholar]
- Lillie M, Grueber CE, Sutton JT, et al. (2015) Selection on MHC class II supertypes in the New Zealand endemic Hochstetter’s frog. BMC Evol Biol 15:63. 10.1186/s12862-015-0342-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie M, Shine R, Belov K (2014) Characterisation of major histocompatibility complex class I in the Australian cane toad, Rhinella marina. PLoS One 9.: 10.1371/journal.pone.0102824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips KR (2016) Overview of chytrid emergence and impacts on amphibians. Philos Trans R Soc Lond B Biol Sci 371.: 10.1098/rstb.2015.0465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips KR, Brem F, Brenes R, et al. (2006) From The Cover: Emerging infectious disease and the loss of biodiversity in a Neotropical amphibian community. Proceedings of the National Academy of Sciences 103:3165–3170. 10.1073/pnas.0506889103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manni M, Berkeley MR, Seppey M, et al. (2021) BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38:4647–4654. 10.1093/molbev/msab199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RM, Meador H, Bender L, Hopper L (2019) Isolation and characterization of 27 novel microsatellite loci in critically endangered Wyoming toad. Journal of Fish and Wildlife Management 10:563–566. 10.3996/042019-JFWM-029 [DOI] [Google Scholar]
- McClelland Erin E, Penn Dustin J, Potts Wayne K. (2003) Major Histocompatibility Complex Heterozygote Superiority during Coinfection. Infect Immun 71:2079–2086. 10.1128/IAI.71.4.2079-2086.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Schmidt HA, Chernomor O, et al. (2020) IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 37:1530–1534. 10.1093/molbev/msaa015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan M, Falcon S, Gentleman R (2018) GSEABase: Gene set enrichment data structures and methods. R package version 1: [Google Scholar]
- Nie L, Cai S-Y, Shao J-Z, Chen J (2018) Toll-Like Receptors, Associated Biological Roles, and Signaling Networks in Non-Mammals. Front Immunol 9:1523. 10.3389/fimmu.2018.01523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabijan M, Palomar G, Antunes B, et al. (2020) Evolutionary principles guiding amphibian conservation. Evol Appl 13:857–878. 10.1111/eva.12940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CV (2018) Immunity and the coral crisis. Commun Biol 1:91. 10.1038/s42003-018-0097-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips BL, Brown GP, Greenlees M, et al. (2007) Rapid expansion of the cane toad (Bufo marinus) invasion front in tropical Australia. Austral Ecol 32:169–176. 10.1111/j.1442-9993.2007.01664.x [DOI] [Google Scholar]
- Polasik JS, Murphy MA, Abbott T, Vincent K (2016) Factors limiting early life stage survival and growth during endangered Wyoming toad reintroductions: Wyoming Toad Early Life Stage Limits. Jour Wild Mgmt 80:540–552. 10.1002/jwmg.1031 [DOI] [Google Scholar]
- Pounds JA, Bustamante MR, Coloma LA, et al. (2006) Widespread amphibian extinctions from epidemic disease driven by global warming. Nature 439:161–167. 10.1038/nature04246 [DOI] [PubMed] [Google Scholar]
- Priyam A, Woodcroft BJ, Rai V, et al. (2019) Sequenceserver: A Modern Graphical User Interface for Custom BLAST Databases. Mol Biol Evol 36:2922–2924. 10.1093/molbev/msz185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, et al. (2012) The Pfam protein families database. Nucleic Acids Res 40:D290–301. 10.1093/nar/gkr1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiniou SMA, Boudinot P, Bengtén E (2013) Comprehensive survey and genomic characterization of Toll-like receptors (TLRs) in channel catfish, Ictalurus punctatus: identification of novel fish TLRs. Immunogenetics 65:511–530. 10.1007/s00251-013-0694-9 [DOI] [PubMed] [Google Scholar]
- Radwan J, Babik W, Kaufman J, et al. (2020) Advances in the Evolutionary Understanding of MHC Polymorphism. Trends Genet 36:298–311. 10.1016/j.tig.2020.01.008 [DOI] [PubMed] [Google Scholar]
- Richmond JQ, Savage AE, Zamudio KR, Rosenblum EB (2009) Toward immunogenetic studies of amphibian chytridiomycosis: Linking innate and acquired immunity. Bioscience 59:311–320. 10.1525/bio.2009.59.4.9 [DOI] [Google Scholar]
- Roach JC, Glusman G, Rowen L, et al. (2005) The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci U S A 102:9577–9582. 10.1073/pnas.0502272102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr JR, Raffel TR, Blaustein AR, et al. (2013) Using physiology to understand climate-driven changes in disease and their implications for conservation. Conserv Physiol 1:cot022. 10.1093/conphys/cot022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell T, Lisovski S, Olsson M, et al. (2018) MHC diversity and female age underpin reproductive success in an Australian icon; the Tasmanian Devil. Sci Rep 8:4175. 10.1038/s41598-018-20934-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta M, Swain B, Basu M, et al. (2014) Toll-like receptor 22 in Labeo rohita: molecular cloning, characterization, 3D modeling, and expression analysis following ligands stimulation and bacterial infection. Appl Biochem Biotechnol 174:309–327. 10.1007/s12010-014-1058-0 [DOI] [PubMed] [Google Scholar]
- Savage AE, Gratwicke B, Hope K, et al. (2020) Sustained immune activation is associated with susceptibility to the amphibian chytrid fungus. Mol Ecol 29:2889–2903. 10.1111/mec.15533 [DOI] [PubMed] [Google Scholar]
- Savage AE, Zamudio KR (2011) MHC genotypes associate with resistance to a frog-killing fungus. Proc Natl Acad Sci U S A 108:16705–16710. 10.1073/pnas.1106893108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenekar T, Weiss S (2017) Selection and genetic drift in captive versus wild populations: an assessment of neutral and adaptive (MHC-linked) genetic variation in wild and hatchery brown trout (Salmo trutta) populations. Conserv Genet 18:1011–1022. 10.1007/s10592-017-0949-3 [DOI] [Google Scholar]
- Selechnik D, Richardson MF, Shine R, et al. (2019) Increased Adaptive Variation Despite Reduced Overall Genetic Diversity in a Rapidly Adapting Invader. Front Genet 10:1221. 10.3389/fgene.2019.01221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Higgins DG (2014) Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol 1079:105–116. 10.1007/978-1-62703-646-7_6 [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, et al. (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212 [DOI] [PubMed] [Google Scholar]
- Smallbone W, Ellison A, Poulton S, et al. (2021) Depletion of MHC supertype during domestication can compromise immunocompetence. Mol Ecol 30:736–746. 10.1111/mec.15763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2:16. 10.1186/1742-9994-2-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Leger RJ (2021) Insects and their pathogens in a changing climate. J Invertebr Pathol 184:107644. 10.1016/j.jip.2021.107644 [DOI] [PubMed] [Google Scholar]
- Thörn F, Rödin-Mörch P, Cortazar-Chinarro M, et al. (2021) The effects of drift and selection on latitudinal genetic variation in Scandinavian common toads (Bufo bufo) following postglacial recolonisation. Heredity 126:656–667. 10.1038/s41437-020-00400-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga JFA, Bui-Marinos MP, Katzenback BA (2018) Frog Skin Innate Immune Defences: Sensing and Surviving Pathogens. Front Immunol 9:3128. 10.3389/fimmu.2018.03128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidya MK, Kumar VG, Sejian V, et al. (2018) Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol 37:20–36. 10.1080/08830185.2017.1380200 [DOI] [PubMed] [Google Scholar]
- Vincent K, Abbott T, Wyoming Toad Recovery Team (2015) Wyoming toad (Bufo hemiophrys baxteri now known as Anaxyrus baxteri) revised recovery plan. US Fish and Wildlife Service, Cheyenne, WY [Google Scholar]
- Wcisel DJ, Ota T, Litman GW, Yoder JA (2017) Spotted Gar and the Evolution of Innate Immune Receptors. J Exp Zool B Mol Dev Evol 328:666–684. 10.1002/jez.b.22738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson L (2011) Ggplot2: Elegant graphics for data analysis by WICKHAM, H. Biometrics 67:678–679. 10.1111/j.1541-0420.2011.01616.x [DOI] [Google Scholar]
- Woodhams DC, Bosch J, Briggs CJ, et al. (2011) Mitigating amphibian disease: strategies to maintain wild populations and control chytridiomycosis. Front Zool 8:8. 10.1186/1742-9994-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Tegenfeldt F, Kuznetsov D, et al. (2017) OrthoDB v9.1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res 45:D744–D749. 10.1093/nar/gkw1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptome sequence data has been archived on NCBI as BioProject PRJNA484136; SRA accession numbers SRX4501368 (toad 6691), SRX4501367 (toad 7039); and SRX4501369 (toad 7092); and TSA accession numbers accession numbers GGUS00000000 (toad 6691), GGUR00000000 (toad 7039), GGUQ00000000 (toad 7092). Annotated transcriptome data are archived on Dryad (https://doi.org/10.5061/dryad.n2z34tmz9).