

Summary
Somatic cell reprogramming using the microRNAs miR-200c, miR-302s, and miR-369s leads to increased expression of cyclin-dependent kinase inhibitors in human colorectal cancer (CRC) cells and suppressed tumor growth. Here, we investigated whether these microRNAs inhibit colorectal tumorigenesis in CPC;Apc mice, which are prone to colon and rectal polyps. Repeated administration of microRNAs inhibited polyp formation. Microarray analysis indicated that c-MAF, which reportedly shows oncogene-like behavior in multiple myeloma and T cell lymphoma, decreased in tumor samples but increased in microRNA-treated normal mucosa. Immunohistochemistry identified downregulation of c-MAF as an early tumorigenesis event in CRC, with low c-MAF expression associated with poor prognosis. Of note, c-MAF expression and p53 protein levels were inversely correlated in CRC samples. c-MAF knockout led to enhanced tumor formation in azoxymethane/dextran sodium sulfate–treated mice, with activation of cancer-promoting genes. c-MAF may play a tumor-suppressive role in CRC development.
Subject areas: Molecular biology, Molecular mechanism of gene regulation, Cancer
Graphical abstract
Highlights
-
•
miR-200c, miR-302s, and miR-369s inhibited polyp formation in CPC;Apc mice
-
•
c-MAF induces p53 and p21 which lead to inhibition of cell proliferation
-
•
In CRC specimens, reciprocal expression between p53 and c-MAF was observed
-
•
c-MAF KO mice exhibited extensive colorectal tumor formation with chemicals
Molecular biology; Molecular mechanism of gene regulation; Cancer
Introduction
Colorectal cancer (CRC) is among the most prevalent cancers worldwide, ranking as the third most common cancer and the fourth leading cause of cancer death.1,2,3 Despite considerable progress in surgery and chemotherapy in the past decade and the development of molecular targeted therapies, 5-year survival remains at 50%–65%.2,4,5,6
Cancer is a genetic disease, but epigenetic alterations also are involved in its initiation and progression. By introducing Yamanaka reprogramming factors, i.e., Oct3/4, Sox2, Klf4, and c-Myc, for generation of induced pluripotent stem cells,7 we previously showed that reprogramming of CRC cells reduces their malignant potential.8 In this way, reprogramming could prove useful as a cancer therapy. Other work has highlighted potential risks with virus vectors and the oncogenic c-myc gene.7,9,10,11,12 To sidestep these risks, we have used the microRNAs (miRNAs) miR-200c, miR-302s, and miR-369s to reprogram differentiated human and mouse somatic cells.13 Our results showed that these miRNAs trigger increased expression of cyclin-dependent kinase inhibitors such as p16Ink4a and p21Waf1/Cip1 and histone methylation of H3K4 in human CRC cells and suppress tumor growth in vitro and in vivo.14,15
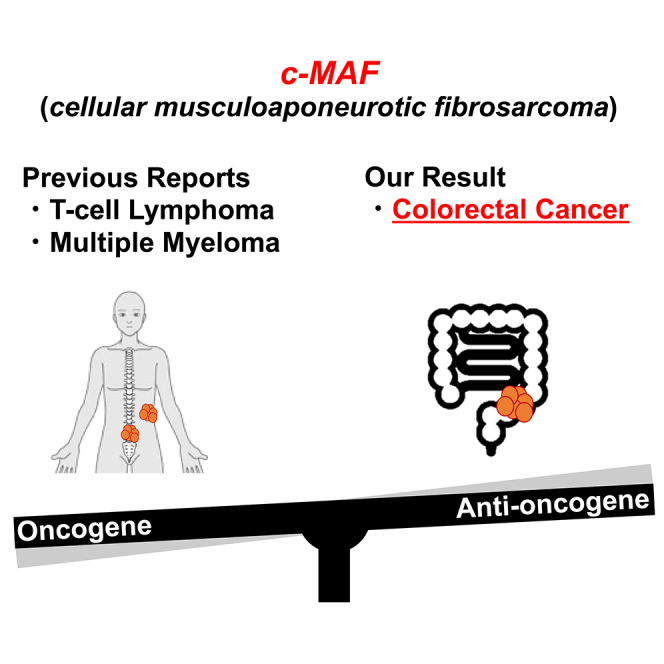
In this study, we sought to clarify whether miR-200c, miR-302s, and miR-369s would inhibit tumorigenesis in the colorectum of CPC;Apc mice, in which colon and rectal polyps are preferentially produced.16 We then performed microarray analysis to assess differential gene expression between normal colon mucosa and tumors from CPC;Apc mice. Based on the results, we focused on c-MAF (musculoaponeurotic fibrosarcoma) gene.17
The MAF family protein functions as a transcription factor of AP-1 family. c-MAF constitutes a large MAF family, together with MAFA and MAFB. It is reported that MAF may have tumor-suppressive roles through regulating p53-dependent cell death, inhibition of MYB, and induction of the cell cycle inhibitor p27Kip1.18 c-MAF is a human analog of v-MAF which was identified as a retroviral oncoprotein in 1989, from avian retrovirus AS42 derived from a chicken musculoaponeurotic fibrosarcoma.17
To elucidate the role of c-MAF in CRC, we evaluated its expression in normal mucosa, early cancer, and advanced cancer samples from patients with CRC and performed in vitro mechanistic studies in intestinal IEC-18 cells and CRC cell lines. Finally, we generated c-MAF knockout mice and investigated how tumor formation would be affected during chemical carcinogenesis with azoxymethane (AOM)/dextran sodium sulfate (DSS) treatment. Overall, our findings highlight c-MAF as a potential tumor suppressor in CRC.
Results
miRNA treatment suppresses colorectal tumor formation in CPC;Apc mice
We intravenously injected mouse miR-200c-3p, miR-302–3p (-a,-b,-c,-d), and miR-369 (-3p, -5p) simultaneously,13,14 or negative control (NC) miRNA (Table S1) into the tail vein of CPC;Apc mice,16 three times weekly for 8 weeks from 8 weeks of age, using the super carbonate apatite delivery system19 (Figure 1A). Colorectal tumor burden was surveyed by rectosigmoid endoscopy at 9, 11, and 13 weeks (Figure 1B) and directly confirmed postmortem at 15 weeks (Figure 1C). The incidence of polyps in mice treated with the trio of miRNAs was significantly lower than in the NC animals (0.5 ± 0.2 vs. 3.3 ± 1.5 polyps/mouse, respectively; p = 0.026; Figure 1D).
Figure 1.

MicroRNA treatment suppressed colorectal tumor formation in CPC;Apc mice
(A) Experimental design. miR-200c-3p, miR-302–3p (-a,-b,-c,-d), and miR-369 (-3p, -5p) or negative control (NC) miRNA was intravenously injected into the tail vein of CPC;Apc mice, three times per week for 8 weeks using the super carbonate apatite delivery system.
(B) Images of distal colon observed by a rectosigmoid endoscopy at 13 weeks. Red arrowheads indicate polyp formation.
(C) Mice were sacrificed at 15 weeks, and the colorectum was opened. Red arrowheads indicate polyp formation.
(D) The incidence of polyps in mice treated with miRNAs was significantly lower than in negative control (NC)-treated mice (0.5 ± 0.2 vs. 3.3 ± 1.5 polyps/mouse, respectively; p = 0.026).
(E) Microarray analysis revealed that 53 genes in NC-treated normal mucosa were downregulated compared with the miRNA-treated normal mucosa and upregulated compared with NC-treated polyps.
(F) qRT-PCR results indicated that c-MAF mRNA expression in the miRNA-treated normal mucosa was significantly higher than in control normal mucosa, and c-MAF mRNA was significantly decreased in polyps compared with control normal mucosa (∗∗p < 0.001). Data are expressed as the mean ± standard deviation. Statistical differences were analyzed by the Student’s t test. miRs, microRNA; NC, negative control miR; N, normal; P; polyp. ∗p < 0.05, ∗∗p < 0.01.
In a subset of samples, we next performed microarray analysis for differential mRNA expression between normal mucosa and colorectal polyps. Heatmap analysis showed that 15 genes were highly expressed in polyps, and 53 genes showed stepwise downregulation from miRNA-treated normal mucosa to NC normal mucosa to NC polyps (Figure S1, Table S2, and Figure 1E). Among these genes, we confirmed by qRT-PCR that c-MAF mRNA expression in the miRNA-treated normal mucosa was significantly higher than in NC normal mucosa, and that c-MAF mRNA was significantly decreased in polyps as compared with NC normal mucosa (p < 0.01 for both; Figure 1F).
A database survey indicated that c-MAF mRNA expression was decreased in adenocarcinoma of the colon and rectum compared with normal mucosa (Figure S2A; ONCOMINE [https://www.oncomine.org].20 Moreover, several human malignancies, including colon and rectal cancer, have been found to express less c-MAF mRNA than normal tissues (Figure S2B; FIREBROWSE21 [http://firebrowse.org/] and C).
c-MAF expression in normal epithelial and CRC tissues
We found by qRT-PCR that c-MAF mRNA expression in CRC tissues was significantly lower than in paired normal mucosa samples (p = 0.045; Figure 2A). Immunohistochemistry for c-MAF with duodenum samples as a positive control (Figure 2B) showed intense nuclear staining of the c-MAF protein in normal mucosa from the bottom to the top of the glands. In contrast, we detected heterogeneous c-MAF expression in advanced CRC tissue samples (Figure 2C). High nuclear expression of the c-MAF protein in advanced CRC tissues was significantly decreased compared with expression in normal epithelium (p < 0.05; Figure 2C), and this downregulation occurred at the early cancer stage (p = 0.038; Figure 2D).
Figure 2.

c-MAF expression in clinical samples of CRC
(A) c-MAF mRNA level in human CRC tissues was significantly lower than in their pair-matched adjacent normal mucosal tissues (p = 0.045).
(B) Immunostaining of c-MAF protein in the duodenum as a positive control. Nuclear staining of c-MAF was noted in the duodenal epithelium. Scale bar: left panel, 500 μm; right panel, 50 μm.
(C) Immunostaining of c-MAF protein in normal mucosa and advanced CRC tissue; depth of invasion defined as T2 (deep or deeper than the muscularis propria). c-MAF expression was noted in the nucleus of normal epithelium and tumor cells. When the c-MAF positive staining cutoff was set at > 50%, the positive proportion in CRC tissues was significantly lower compared with normal mucosa (p < 0.05). Scale bar: 100 μm.
(D) c-MAF expression in early cancer and adjacent normal mucosa; early cancer defined as T0 and T1 (invasion into lamina propria or submucosa). Cancer cells showed low c-MAF expression, whereas normal epithelial cells had strong c-MAF staining. Scale bars: left panel, 500 μm; right panels, 100 μm. Investigation of 20 early cancer samples revealed downregulation of c-MAF at an early cancer stage (p = 0.038).
(E) Setting c-MAF expression in normal mucosa; as a basis, we divided the CRC cases into two groups: strong (tumor > normal, n = 35) and weak (tumor < normal, n = 63). The Kaplan–Meier survival curve shows better prognosis for overall survival in the strong expression group (p = 0.039; median follow-up 66.4 [range 41.2–791.8] months). Relapse-free survival was examined after exclusion of eight patients with stage IV disease, yielding a similar result (p = 0.040; median follow-up 63.5 [range 32.2–89.8] years). Statistical differences were analyzed using Student’s t test for continuous variables and the Chi-squared test for non-continuous data. Survival curves were developed with the Kaplan–Meier method and compared using the log rank test. ∗p < 0.05.
Survival analysis in patients with advanced CRC revealed significantly prolonged overall survival (OS) in the group with strong c-MAF expression compared with those showing weak c-MAF expression (p = 0.039, median follow-up 66.4 months; Figure 2E). Similarly, Kaplan–Meier survival curves showed significantly better relapse-free survival (RFS) in the group with strong c-MAF expression compared with the group showing low c-MAF expression (p = 0.040, median follow-up 63.5 months; Figure 2E). A clinicopathological survey indicated that lymphatic duct invasion was significantly associated with weak c-MAF expression (p = 0.002; Table S3). Multivariate analysis using a Cox proportional hazard model showed that c-MAF expression tends to be an independent prognostic factor for OS (hazard ratio [HR] 3.084, 95% confidence interval [CI] 0.921–10.320, p = 0.068) and that c-MAF was a significant independent indicator of better prognosis with RFS (HR 3.935, 95% CI 1.038–14.919, p = 0.044) (Table 1).
Table 1.
Univariate and multivariate analysis of clinicopathological characteristics associated with overall survival and relapse-free survival
| Univariate and multivariate analysis of clinicopathological characteristics associated with OSa | ||||||
|---|---|---|---|---|---|---|
| Univariate |
Multivariate |
|||||
| HRb | 95% CIc | p value | HR | 95% CI | p value | |
| Age (≥65/< 65) | 3.331 | 1.319–8.411 | 0.011 | 4.37 | 1.461–13.072 | 0.008 |
| Gender (female/male) | 0.642 | 0.275–1.501 | 0.307 | |||
| Location (rectum/colon) | 0.405 | 0.161–1.020 | 0.055 | 0.250 | 0.082–0.764 | 0.015 |
| Depth (T4/Tis, T1, T2, T3) | 2.34 | 0.799–6.851 | 0.121 | 1.875 | 0.493–7.122 | 0.356 |
| Lymph node metastasis (positive/negative) | 2.649 | 1.186–5.917 | 0.018 | 1.544 | 0.608–3.923 | 0.361 |
| Histological type (por, sig, muc/tub1, tub2) | 3.204 | 1.093–9.396 | 0.034 | 7.830 | 2.000–30.651 | 0.003 |
| Lymphatic duct invasion (positive/negative) | 3.173 | 0.946–10.644 | 0.062 | 1.398 | 0.334–5.857 | 0.646 |
| Venous invasion (positive/negative) | 2.666 | 1.195–5.944 | 0.017 | 1.971 | 0.763–5.094 | 0.161 |
| MAF expression (weak/strong) | 2.934 | 1.002–8.585 | 0.049 | 3.084 | 0.921–10.320 | 0.068 |
| Univariate and multivariate analysis of clinicopathological characteristics associated with RFSd | ||||||
|---|---|---|---|---|---|---|
| Univariate |
Multivariate |
|||||
| HR | 95% CI | p value | HR | 95% CI | p value | |
| Age (≥65/< 65) | 6.355 | 1.878–21.502 | 0.003 | 10.464 | 2.693–40.659 | <0.001 |
| Gender (female/male) | 0.795 | 0.333–1.895 | 0.604 | |||
| Location (rectum/colon) | 0.510 | 0.208–1.251 | 0.141 | 0.432 | 0.165–1.133 | 0.088 |
| Depth (T4/Tis, T1, T2, T3)e | 4.081 | 1.502–11.092 | 0.006 | 6.388 | 1.570–25.989 | 0.010 |
| Lymph node metastasis (positive/negative) | 2.787 | 1.207–6.436 | 0.016 | 1.853 | 0.648–5.299 | 0.250 |
| Histological type (por, sig, muc/tub1, tub2)f | 1.441 | 0.336–6.177 | 0.623 | |||
| Lymphatic duct invasion (positive/negative) | 2.544 | 0.861–7.520 | 0.091 | 1.124 | 0.286–4.416 | 0.867 |
| Venous invasion (positive/negative) | 2.697 | 1.163–6.253 | 0.021 | 1.341 | 0.444–4.051 | 0.602 |
| MAF expression (weak/strong) | 2.945 | 0.996–8.706 | 0.051 | 3.935 | 1.038–14.919 | 0.044 |
A Cox proportional hazard regression model was used to estimate HRs and 95% CIs. p values <0.05 were considered to indicate statistical significance.
OS, overall survival.
HR, hazard ratio.
CI, confidence interval.
RFS, relapse-free survival.
Tis, carcinoma in situ; T1, involvement of submucosa; T2, involvement of muscularis propria; T3, involvement of subserosa; T4, involvement of serosal surface or direct invasion to other organs.
tub1, well differentiated adenocarcinoma; tub2, moderately differentiated adenocarcinoma; muc, mucinous carcinoma; por, poorly differentiated adenocarcinoma.
Anti-tumor function of c-MAF in rat intestinal epithelial cells and CRC cells
To investigate the fundamental function of c-MAF, we performed knockdown experiments using siRNA. c-MAF mRNA expression significantly decreased after transduction of c-MAF siRNA into the IEC-18 rat intestinal cell line22,23,24 (Figure 3A). Knockdown of c-MAF mRNA led to a significant increase in cell proliferation and colony-forming ability (p < 0.05 for both; Figures 3B and 3C).
Figure 3.

Small interfering (si)RNA knockdown of c-MAF in IEC-18 cells
(A) c-MAF siRNA or negative control siRNA was transfected into rat intestinal IEC-18 cells at 30 nM c-MAF mRNA expression was measured by qRT-PCR. (n = 3).
(B) Knockdown of c-MAF expression led to increased cell proliferation. (n = 3).
(C) Colony-forming ability was significantly increased in c-MAF knockdown cells compared with negative control cultures. (n = 3) Left panel: representative pictures of Giemsa staining of colonies in the 6-well plate. Data are expressed as the mean ± standard deviation. Statistical differences were analyzed by the Student’s t test. ∗p < 0.05. Si-MAF, c-MAF-siRNA; NC, negative control siRNA.
We then transduced c-MAF cDNA into the HCT116 and LS174T CRC cell lines, both of which harbor wild-type p53. c-MAF-overexpressing CRC cells showed considerably higher c-MAF mRNA expression than empty vector (EV)-transduced control cells. In both cell lines, cell proliferation was significantly inhibited in the c-MAF-overexpressed cells compared with EV control cells (p < 0.05; Figure 4A). In contrast, exogenous c-MAF transduction did not affect cell proliferation in p53-null HCT116 cells (Figure 4B). In the HCT116 cells retaining wild-type p53, c-MAF overexpression induced cyclin-dependent kinase inhibitor p21Waf1/Cip1 expression as well as p53 expression at the RNA and protein levels, but did not do so in p53-null HCT116 cells (Figures 4C and 4D). We also found that c-MAF overexpression enhanced the sensitivity to fluorouracil (5-FU) in HCT116 cells (Figure 4E). Results of an Annexin V assay indicated that transduction of c-MAF significantly enhanced apoptosis with 24-h treatment with 100 μM 5-FU (p < 0.05; Figure 4F).
Figure 4.

Effects of c-MAF overexpression on proliferative ability and 5-FU-induced apoptosis in CRC lines
(A) Overexpression of c-MAF in HCT116 and LS174T cells was validated by qRT-PCR. Cell proliferation of c-MAF-overexpressed cells (MAF) was decreased compared with empty vector (EV)-transduced control cells (n = 3 for each).
(B) Overexpression of c-MAF in HCT116 p53-null cells was validated by qRT-PCR. Cell proliferation was not changed between c-MAF-overexpressed cells and control cells (n = 3).
(C and D) Expression of p53 and p21Waf1/Cip1 in c-MAF-overexpressed HCT116 and HCT116 p53-null cells was measured by (C) qRT-PCR (n = 3) and (D) immunoblotting. Cells were collected 48 h after transfection. Uncropped Western Blot data are shown in Figure S10.
(E) Cells were harvested 24 h after transfection with either c-MAF expression vector (MAF) or empty vector control (EV) and seeded to 96-well plates for cytotoxicity assay under 5-FU treatment for 24 h. Sensitivity to 5-FU was significantly enhanced in c-MAF-overexpressed HCT116 cells (n = 6 for each concentration).
(F) Annexin V apoptosis assay was performed with Annexin V and propidium iodide staining and flow cytometry analysis. c-MAF-overexpressed HCT116 cells showed increased 5-FU-induced apoptosis compared with vector control cells (n = 3 for each). Data are presented as mean ± standard deviation. Statistical differences were analyzed by the Student’s t test. ∗p < 0.05.
Effect of c-MAF knockout on colorectal tumor formation
To investigate the effect of c-MAF on carcinogenesis of the colorectum, we generated c-MAF KO mice in which one or eight nucleotides were deleted in 3′ flanking region of ATG transcription start codon (Figures S3A and S3B). Among 187 c-MAF KO mice (1 nucleotide deletion homo mice; male/female: ⅔, 1 nucleotide deletion hetero mice; 63/70, 8 nucleotide deletion homo mice; 0/1, 8 nucleotide deletion hetero mice; 27/21) and 37 wild-type mice (male/female; 37/0), only 3 KO mice eventually developed tumors by 2 years after birth (Figure 5A). A rectal adenocarcinoma arose in one c-MAF KO mouse bearing the eight-nucleotide depletion (hetero status) (Figure S4A). Tumor formation also was observed in the thorax and abdominal cavity in the other two mice. Based on immunohistochemistry results showing S100-positive and SOX10-negative cells, the tumor in the thorax was diagnosed as a malignant schwannoma (Figure S4B). The tumor in the abdominal cavity showed weakly positive staining for BCL6 and was positive for CD45R and was diagnosed as Burkitt’s lymphoma (Figure S4C).
Figure 5.

Spontaneous or colitis-associated tumor formation
(A) Tumors that spontaneously occurred are summarized. Only three tumors were generated in c-MAF KO mice around 2 years after birth.
(B) Details of colitis-associated tumor formation; n = 32 mice (19 c-MAF KO and 13 wild type). The majority of c-MAF KO mice (18 of 19, 94.7%) developed colorectal tumors whereas 9 of 13 (69.2%) of wild-type mice did. The incidence of tumor formation was significantly higher in c-MAF KO mice. The average tumor number in each group is also shown.
(C) Number of tumors per mouse. c-MAF KO mice produced significantly more tumors than did wild-type mice.
(D) Tumor size was significantly larger in c-MAF KO mice compared with wild-type mice. When stratified by tumor diameter at 1, 3, and 5 mm, c-MAF KO mice had significantly more tumor formation >3 mm.
(E) Immunohistochemical staining for Ki-67 in tumors. c-MAF KO tumors had a significantly higher Ki-67 index as compared with wild-type tumors. Scale bars: 100 μm. Data are expressed as mean ± standard deviation, or the median and interquartile range (IQR). Statistical differences were analyzed using Student’s t test. The incidence of tumor formation and the size of tumor in c-MAF KO mice and wild-type mice were analyzed using the Wilcoxon signed-rank test. ∗p < 0.05, ∗∗p < 0.01. KO, knock out; WT, wild type.
c-MAF deficiency increases tumor formation under chemical carcinogenesis
c-MAF KO mice (n = 19) and wild-type mice (n = 13) were treated with initial i.p. administration of 10 mg/kg AOM and two cycles of drinking water containing 2.0% DSS according to the protocol shown in Figure S5A. 1 nucleotide deletion hetero mice included two males and four females. 8 nucleotide deletion homo mice included three males and three females. 8 nucleotide deletion mice included three males and four females. All wild-type mice were males. As a whole, the tumors were positioned at the rectum and distal colon in either c-MAF KO or wild-type mice (Figure S5B). In one c-MAF KO mouse, a rectal tumor prolapsed from the anus approximately 100 days after i.p. injection of AOM (Figure S5C, I–III (a)). H&E staining revealed a moderately differentiated adenocarcinoma (Figure S5C, IV and V).
Figure 5B summarizes tumor formation, which was significantly higher in c-MAF KO than in wild-type mice (p < 0.05). c-MAF KO mice produced significantly more tumors than did wild-type mice (1.8 ± 0.8 vs. 4.0 ± 0.7, p < 0.05; Figure 5C). Regarding tumor size, c-MAF KO mice had significantly larger tumors than their wild-type counterparts (p < 0.01; Figure 5D), as was especially evident for tumors larger than 3 mm (p < 0.01; Figure 5D). The Ki-67 index, a proliferation marker, was significantly higher in c-MAF KO than wild-type tumors (p < 0.01; Figure 5E).
Comparative gene expression analysis of AOM/DSS-treated c-MAF KO and wild-type mice
To elucidate the underlying mechanism by which AOM/DSS treatment resulted in a significant increase in tumor formation in c-MAF KO mice, we performed RNA sequencing (wild-type normal, n = 4; wild-type tumor, n = 9; c-MAF KO normal, n = 6; c-MAF KO tumor, n = 5). The heatmap showed numbers of downregulated or upregulated gene expression between c-MAF KO and wild-type mice in normal mucosa or tumors (Figures S6A and S6B). Ingenuity Pathway Analysis (Qiagen Redwood City, CA, USA; www.qiagen.com/ingenuity) of normal mucosa indicated that many cancer-promoting growth factors, transcription factors, kinases, and cytokines were activated as the upstream regulators in c-MAF KO mice (Table 2), as also was the case with tumor samples from c-MAF KO mice (Table S4). Downstream analysis for disease and function showed that many cancer-related factors were activated in c-MAF KO tumors (Figure S7).
Table 2.
Lists of activated molecules in normal mucosa of c-MAF KO mice compared with wild-type mice after treatment with AOM/DSS
| Upstream Regulator | Molecule Type | Predicted Activation State | Activation Z score | p value |
|---|---|---|---|---|
| ESR1 | Ligand-dependent nuclear receptor | Activated | 4.146 | 0.068 |
| ERBB2 | Kinase | Activated | 3.762 | 0.000 |
| EGF | Growth factor | Activated | 3.253 | 0.029 |
| E2F1 | Transcription regulator | Activated | 3.179 | 0.002 |
| IL4 | Cytokine | Activated | 3.160 | 0.016 |
| Insulin | Group | Activated | 3.098 | 0.239 |
| MITF | Transcription regulator | Activated | 3.073 | 0.003 |
| CD3 | Complex | Activated | 3.054 | 0.012 |
| CEBPB | Transcription regulator | Activated | 3.035 | <0.001 |
| RABL6 | Other | Activated | 2.828 | < 0.001 |
| ESR2 | Ligand-dependent nuclear receptor | Activated | 2.724 | 0.358 |
| NTRK2 | Kinase | Activated | 2.599 | < 0.001 |
| IL13 | Cytokine | Activated | 2.576 | 0.024 |
| CKAP2L | Other | Activated | 2.449 | < 0.001 |
| MYB | Transcription regulator | Activated | 2.449 | 0.010 |
| TAL1 | Transcription regulator | Activated | 2.449 | 0.081 |
| FOXM1 | Transcription regulator | Activated | 2.441 | 0.024 |
| INSR | Kinase | Activated | 2.433 | 0.019 |
| CD38 | Enzyme | Activated | 2.425 | 0.025 |
| CCR2 | G-protein coupled receptor | Activated | 2.412 | 0.004 |
| AIRE | Transcription regulator | Activated | 2.401 | 0.001 |
| RORA | Ligand-dependent nuclear receptor | Activated | 2.236 | 0.027 |
| CYP1B1 | Enzyme | Activated | 2.219 | 0.018 |
| IL33 | Cytokine | Activated | 2.170 | 0.005 |
| CCND1 | Transcription regulator | Activated | 2.101 | < 0.001 |
| E2f | Group | Activated | 2.034 | < 0.001 |
| STAT5a/b | Group | Activated | 2.000 | 0.090 |
| Eldr | Other | Activated | 2.000 | 0.007 |
| TFDP1 | Transcription regulator | Activated | 2.000 | < 0.001 |
| NKX2-3 | Transcription regulator | Activated | 2.000 | 0.339 |
| TPH1 | Enzyme | Activated | 2.000 | 0.025 |
| BRCA1 | Transcription regulator | Activated | 2.000 | 0.100 |
| CDK2 | Kinase | Activated | 2.000 | < 0.001 |
Cancer-promoting growth factors, transcription factors, kinases, and cytokines are shown in bold.
Ingenuity Pathway Analysis of normal mucosa showed that many cancer-promoting growth factors, transcription factors, kinases, and cytokines were activated as upstream regulators in c-MAF KO mice.
Inverse relationship between c-MAF protein and p53 protein expression
We then performed comparative immunohistochemical analysis for c-MAF and p53 protein because the c-MAF transcription factor can activate p53 transcription.25 We found an inverse staining pattern between c-MAF and p53 expression (Figure 6A, Case A and Case B). Of note, this reciprocal staining pattern was found even within identical CRC tissue samples (Figure 6B), so that cells that accumulated p53 protein lost c-MAF expression, and vice versa. As a whole, we found an inverse relationship between c-MAF and p53 expression in CRC tissue samples when the cutoff point was set at 10% for p53 positivity (p = 0.024; Figure 6C). When we transduced two types of mutant p53 (R175H and R248W) into p53-null HCT116 cells, the clones displayed decreased c-MAF expression and increased miR-155 (Figures S8A–S8C). Previous studies had shown that miR-155 directly bound to the 3′-untranslated region of c-MAF,26,27,28 and we confirmed that transfection of mature miR-155 suppressed c-MAF expression (Figure S8D). The schemes of the relationship between c-MAF and p53 in normal mucosa and in p53-mutated tumors are shown in Figures S8E and S8F.
Figure 6.

Inverse relationship between c-MAF and p53 protein expression
(A) Representative pictures of p53 staining in two human CRC tissue samples. Left: c-MAF; right, p53. c-MAF and p53 showed complementarily positive findings.
(B) A set of pictures showing an inverse staining pattern between c-MAF and p53 expression in serial sections.
(C) A significantly inverse correlation was observed between the expression of c-MAF and p53 (p = 0.024). Statistical differences were analyzed using Student’s t test for continuous variables and the Chi-squared test for non-continuous data. scale bar; 100 μm.
We could not find direct binding sites between miR-200c, miR-302s, and miR-369s and the 3′ untranslated region of c-MAF mRNA through searches of public databases (TargetScan human version 7.229 [http://www.targetscan.org/vert_72/], miRwalk30 [http://mirwalk.umm.uni-heidelberg.de/], miRabel [http://bioinfo.univ-rouen.fr/mirabel/view/result.php?page=mir], miRmap31 [https://mirmap.ezlab.org/app https://mirmap.ezlab.org/app/]). To explore a possibility of indirect upregulation of c-MAF, we performed a survey by interaction network in Ingenuity Pathway Analysis and identified GATA3 as an upstream activator of c-MAF (Figure S9A) and the GATA3-cMAF axis was previously reported.32 To confirm this route, we examined differential gene expression in human MRC5 lung fibroblasts transfected with human miR-200c, miR-302s, and miR-369s and RNA sequencing indicated upregulation of GATA3 mRNA by 3.07-fold in transfected compared with control cells (p = 0.035; data deposited at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE210980). When we introduced human miR-200c, miR-302s, and miR-369s into MRC5 lung fibroblasts, HBEC3-KT bronchial epithelial cells, and two colon cancer cell lines HCT116 and RKO, qRT-PCR showed increased expression of GATA3 and c-MAF mRNA (Figure S9B).
To further reveal underlying mechanism, we performed ChIP-PCR to prove direct binding of GATA3 to c-MAF promoter region as a transcription factor. This is because public database ChIP-atlas33 [https://chip-atlas.org/] showed a peak of GATA3 in the promoter region of c-MAF (Figure S9C). Moreover, the transcription site prediction software JASPAR34 [https://jaspar.genereg.net/] suggested a possibility of direct binding of GATA3 to the promoter region of c-MAF (Figure S9C). With setting the primers in the promoter region of c-MAF (Figure S9C), ChIP-PCR assay indicated that GATA3 significantly bound to the promoter region of c-MAF compared to control IgG in normal cell lines and a colon cancer cell line (Figure S9D).
Discussion
MiRNAs are involved not only in progression of human cancers but also in carcinogenesis. It is reported that miR-26a can overcome potential oncogenic activity in intestinal tumorigenesis of Apcmin/+ mice and that tumor formation is abundant in miR-10a-deficient mice.35,36 We previously reported an inhibitory effect of administering mixture of miR-200c, miR-302s, and miR-369s on in vivo tumor growth of CRC cells.14,15 In this study, we employed CPC;Apc mice because polyp formation can be readily monitored using periodical rectosigmoid endoscopy, and found that systemic administration of the miRNAs simultaneously suppressed tumor formation in the colorectum of these animals. We previously administered miR-302s and miR-369s or these miRNAs plus miR-200c to colon cancer cell lines according to their expression levels. In an instance, RKO colon cancer cells did not express three miRNAs and we administered miR-200c, miR-302s, and miR-369s.14 It is reported that miR-200c expression is significantly lower in normal colorectum than in tumor.37 In this study, we attempted to deliver 3 miRNAs to normal colonic mucosa in order to suppress tumorigenesis, as we previously introduced 3 miRNAs into human normal cells.13 Of note, our approach using intravenous injection of the miRNAs steps around virus-derived genome integration, so that it is safe and suitable for clinical application, and we and others have confirmed its efficacy in animal studies.19,38,39,40,41,42,43,44,45,46,47,48
To explore the underlying mechanism of how the miRNAs suppress tumor formation, we analyzed expression of genes that were upregulated in miRNAs-treated normal mucosa and downregulated in control tumors compared with NC normal mucosa. MiRNAs-treated polyps showed similar gene expression profile to NC-treated polyps rather than those of normal mucosa. We postulate that tumor inhibitory effect endowed by the miRNAs is no longer sustained in the polyps that were generated despite administration of miRNAs. Among 53 genes that showed stepwise downregulation in normal to tumor process, we were interested in c-MAF because c-MAF is supposed to be an oncogene. Its bird homolog v-MAF is considered an oncogene17 and its increased mRNA expression and an association with malignant properties have been reported in multiple myeloma49 and T cell lymphoma,50 which is contradictory to our initial results of stepwise downregulation. In addition, MAF has been reported to be a mediator of breast cancer bone metastasis through regulating parathyroid hormone-related protein.51 According to a public database, however, human c-MAF mRNA expression is downregulated in carcinomas of the cervix and uterus, colorectum, stomach, bile duct, breast, bladder, and organs (FIREBROWSE21 [http://firebrowse.org/]). Based on these findings, we hypothesized that c-MAF could function as either an oncogene or a tumor suppressor, depending on cancer type, and made it the focus of our further investigation.
As one of the mechanisms how c-MAF is induced by human miR-200c, miR-302s, and miR-369s, we explored a possibility of involvement of GATA3 according to Ingenuity Pathway Analysis and a literature.32 We found that the three miRs increased GATA3 and c-MAF RNA expression and ChIP-PCR assay proved that the GATA3 protein directly bound to the MAF promoter region. Collectively, these findings suggest that the transcription factor GATA3 binds to the promoter region of c-MAF and regulates c-MAF expression.
Although c-MAF has been investigated in some cancers,49,50,51,52 the expression profile and function of c-MAF in human cancers are still largely unknown. Moreover, our database survey showed that c-MAF mRNA expression was downregulated more often than not in a series of human malignancies including CRC, as shown in Figures S2A–S2C. At the protein level, we confirmed by immunohistochemistry an intense expression of c-MAF in colonic epithelial cells, with a decrease in samples from early stage CRC and a poor prognosis associated with low expression. Overall, our findings in CPC;Apc mice and clinical CRC samples suggest that c-MAF as a transcription factor may have a tumor-suppressive role in the generation and progression of CRC.
Our in vitro mechanistic studies showed that silencing c-MAF expression resulted in increased cell proliferation and enhanced colony-forming ability in the IEC-18 intestinal cell line, further highlighting a tumor-suppressive role of c-MAF in intestinal cells. c-MAF binds the MAF recognition element in the mouse p53 promoter and causes cell death through induction of the p53 protein. This region is conserved between mouse and human.25 p53 is a predominant gene in controlling apoptosis in response to abnormal cell proliferation and stress53,54 and may cooperate with c-MAF to exert a tumor-inhibitory effect. Using CRC cell lines, we showed that overexpression of c-MAF led to reduced cell proliferation and enhancement of 5-FU-dependent apoptosis in CRC cell lines harboring wild-type p53. Overexpression of c-MAF indeed increased the expression of p53 and its downstream target p21waf1/cip1, a negative cell cycle regulator55,56,57 in the HCT116 cells with wild-type p53. In contrast, forced expression of c-MAF did not induce p53 or p21waf1/cip1 and had no effect on cell proliferation in p53-null HCT116 cells. These findings suggest that c-MAF may play an anti-oncogenic role through p53 upregulation in intestinal and CRC cells.
In our in vivo experiment using c-MAF KO mice, we rarely observed spontaneous tumor formation, with an incidence of 3/187 (1.6%), including one rectal adenocarcinoma. Therefore, c-MAF alone is unlikely to be a definitive tumor-suppressor gene. However, our chemical carcinogenesis experiment made it clear that c-MAF supports suppression of tumor formation: c-MAF KO mice had a significantly higher incidence of colorectal tumors and larger tumor size, with a higher Ki-67 index. Ingenuity Pathway Analysis further demonstrated that c-MAF KO normal mucosa bears higher potential for tumor formation via activation of genes that reportedly promote carcinogenesis (Table 2). For example, ERBB2 is an oncogene amplified in breast cancer58,59; E2F1 facilitates carcinogenesis in liver, brain, skin, and testis60,61,62,63; and TFDP1, a heterodimer partner of E2F, is reported to facilitate carcinogenesis in skin tissue.64 Other molecules are reported to have a role in carcinogenesis, such as MITF in kidney angiomyolipoma, Myb and FOXM1 in colon cancer, TAL-1 in T cell acute lymphoblastic leukemia, and NKX2-3 in B cell lymphoma.65,66,67,68,69 A comparison of c-MAF KO tumors and wild-type tumors showed that c-MAF KO tumors exhibited activation of many tumor-promoting growth factors, such as VEGF, IGF1, HGF, EGF, TGFβ1, and FGF2, as well as the transcription factors Jun and STAT3, which activate signal transduction for tumor growth and survival.70,71 Taken together, our data suggest that c-MAF behaves like a tumor suppressor in tumorigenesis of CRC.
c-MAF also may coordinate cells in differentiating into retina, sensory nerve, and immune T cells and maintain cell quiescence in the lens. This transcription factor thus also is considered to induce gene expression during the tissue-specific differentiation process.52,72,73,74 Brundage et al. showed that c-MAF expression was downregulated in a malignant peripheral nerve sheath tumor cell line and that c-MAF suppressed cell proliferation and anchorage-independent growth, and induced cell differentiation and apoptosis.18 They also observed that c-MAF promoted in vivo tumor growth of NF1 (neurofibromatosis type 1) patient-derived malignant peripheral nerve sheath tumor cells.18 These findings suggest that as a transcription factor, c-MAF may cooperate with optimal downstream targets according to the cell context, surrounding microenvironment, and/or cell type, so that it can act either as an oncogene or anti-oncogene.
The mechanism of how c-MAF is downregulated in CRC remains to be addressed. A few studies have described mechanisms of MAF regulation through deletion, loss-of-function mutations, and promoter methylation,75,76,77 but such modifications are not reported in CRC. Here, we observed impressive staining results showing a reciprocal expression pattern between c-MAF and p53 in CRC tissue samples (Figure 6B). This pattern implied that the role of c-MAF could extend beyond mouse or cell culture systems and be relevant in clinical CRC tissues. By immunohistochemistry, we found that the wild-type p53 protein was basically undetectable; therefore, a c-MAF-positive/p53-negative CRC pattern seems to make sense considering that c-MAF is a transcription factor that positively regulates p53 (Figure 6A, Case A). On the other hand, a mutated p53 product is known to be detectable in the nucleus because of its prolonged half-life.78 Of considerable interest is that c-MAF somehow lost its expression when mutated p53 protein was accumulated in the CRC cells (Figure 6A, Case B; Figure 6B). One possible underlying mechanism could be related to the mutant p53–miR-155-c–MAF axis. Neilsen et al. reported that transduction of mutant p53 upregulated miR-155 expression through p63 in breast cancer.79 miR-155 is one of the representative oncomirs and targets c-MAF by direct binding.26,27,28 We confirmed this scenario by transduction of mutated p53 into p53-null HCT116 CRC cells (Figure S8). p53 is an important tumor suppressor acting at a critical transition point from adenoma to cancer.80 It is assumed that p53 mutation could be one reason for c-MAF inhibition and that mutated p53-mediated abolition of c-MAF may further accelerate carcinogenesis and progression of CRC. However, we should emphasize that c-MAF could have a tumor-suppressive effect via mechanisms other than p53; some studies have shown that p53 gene mutation is not detectable in AOM/DSS-induced CRC,81,82 whereas c-MAF KO revealed many other candidate factors facilitating carcinogenesis, as we show in Tables 2 and S4.
Taken together, the present findings imply a tumor-suppressive role of c-MAF in tumorigenesis and progression of CRC. Our data would provide c-MAF as a marker for prognosis of patients with CRC and may lead to development of a therapeutic option against CRC.
Limitations of the study
We have demonstrated that thec-MAF plays a tumor-suppressive role in the colon. However, its role in other organs is not elucidated yet. In addition, although clinical CRC samples suggest that mutated p53 gene may affect c-MAF, mouse chemical carcinogenesis model does not include mutations in p53, and further information on p53 pathway was not obtained.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-MAF | Abcam | Catalog # ab72584| RRID:AB_1268172 |
| Anti-p53 | Dako | Catalog # M7001 | RRID:AB_2206626 |
| Anti-p21 Waf/Cip1 | Cell Signaling Technology | Catalog # 2947 | RRID:AB_823586 |
| Anti-Actin | Sigma-Aldrich | Catalog # A2066 | RRID:AB_476693 |
| Anti-Ki67 | Cell Signaling Technology | Catalog # 12202 | RRID:AB_2620142 |
| anti-BCL6 | Invitrogen | Catalog # PA5-27390 | RRID:AB_2544866 |
| anti-SOX10 | Abcam | Catalog # ab227680 |
| anti-S100 | NICHIREI BIOSCIENCE INC. | Catalog # 422091 |
| anti-CD45R | Thermo Fisher Scientific | Catalog # 14045182 | RRID: AB_467251 |
| Normal Rabbit IgG | Cell Signaling Technology | Catalog # 2729 | RRID: AB_1031062 |
| anti-GATA3 | Abcam | Catalog # ab199428 | RRID: AB_2819013 |
| Biological samples | ||
| Colorectal cancer Patient Samples | Osaka University | IRB Permission No. #15144 |
| Chemicals, peptides, and recombinant proteins | ||
| Sodium Dextran Sulfate | MP BIOMEDICALS | Catalog # 191-08365 |
| Azoxymethane | Sigma-Aldrich | Catalog # A5486-25MG |
| 5-fluorouracil | Nacalai Tesque Inc. | Catalog # 16220-01 |
| Lipofectamine™ RNAiMAX Transfection Reagent | Thermo Fisher Scientific | Catalog # 13778150 |
| Lipofectamine 3000 Regent | Thermo Fisher Scientific | Catalog # L3000075 |
| Cas9 protein, Alt-R® S.p. Cas9 Nuclease 3NLS | Integrated DNA Technologies | Catalog # 1074182 |
| Precision gRNA Synthesis Kit | Thermo Fisher Scientific | Catalog # A29377 |
| KSOM medium | ARK Resource | Catalog # I0BAIK500 |
| KAPA Express Extract DNA Extraction Kit | Kapa Biosystems | Catalog # KK7103 |
| Critical commercial assays | ||
| Cell Counting Kit-8 | DOJINDO | Catalog # CK04 |
| Annexin V-FITC Apoptosis Kit | BioVision | Catalog # K101 |
| miRNeasy Mini Kit | Qiagen | Catalog # 217004 |
| High Capacity RNA-to-cDNA Kit | Applied Biosystems | Catalog # 4387406 |
| TaqMan Universal PCR Master Mix | Applied Biosystems | Catalog # 4304437 |
| LightCycler-DNA Master SYBR Green I | Roche | Catalog # 3003230 |
| RNAlater | Thermo Fisher Scientific | Catalog # AM7020 |
| Vectastain Elite ABC Kit | Vector | Catalog # PK-6101, # PK-6102, # PK-6104 |
| Animal Tissue Direct PCR Amplification Kit (with TL) | FineGene | Catalog # DT01 |
| Mini-Gel extraction kit (One-step) | FineGene | Catalog # FG209P |
| Bradford protein assay | Bio-Rad | Catalog # 5000006 |
| Deposited data | ||
| CPC; APC mice Microarray Data | This paper | GEO: GSE92944 |
| MAF KO mice RNA Sequencing Data | This paper | GEO: GSE210970 |
| MRC5 RNA Sequencing Data | This paper | GEO: GSE210980 |
| Experimental models: Cell lines | ||
| LS174T | American Type Culture Collection | RRID:CVCL_1384 |
| IEC-18 | American Type Culture Collection | RRID:CVCL_0342 |
| HCT116 (p53 wild type) | Gifted from Dr. Bert Vogelstein (Johns Hopkins University School of Medicine) | N/A |
| HCT116 (p53 null) | Gift from Dr. Bert Vogelstein (Johns Hopkins University School of Medicine) | N/A |
| MRC5 | American Type Culture Collection | RRID:CVCL_0440 |
| HBEC-3-KT | American Type Culture Collection | RRID:CVCL_X491 |
| HT29 | American Type Culture Collection | RRID:CVCL_A8EZ |
| Experimental models: Organisms/strains | ||
| C57BL/6JJCL | CLEA Japan, Inc. | RRID:IMSR_JCL:JCL:mIN-0003 |
| C57BL/6JJCL cryopreserved zygotes | CLEA Japan, Inc. | N/A |
| Jcl:ICR pseudopregnant | CLEA Japan, Inc. | N/A |
| CPC;APC mice | Hinoi et al., 2007 | N/A |
| MAF KO mice | This paper | N/A |
| Oligonucleotides | ||
| siRNA c-MAF | Thermo Fisher Scientific | Catalog # 4390771 |
| siRNA Negative Control | Thermo Fisher Scientific | Catalog # 4390843 |
| pCMV6-MAF plasmid DNA | OriGene | Catalog # SC116772 |
| pCMV6-XL4 Mammalian Expression Vector | OriGene | Catalog # PCMV6XL4 |
| pCMV-Neo-Bam p53 R175H | Addgene | Catalog # 16436 | RRID:Addgene_16436 |
| pCMV-Neo-Bam p53 R248W | Addgene | Catalog # 16437 | RRID:Addgene_16437 |
| pCMV-Neo-Bam | Addgene | Catalog # 16440 | RRID:Addgene_16440 |
| microRNAs | see Table S1 | N/A |
| PCR or sanger sequence primer | see Table S5 | N/A |
| Software and algorithms | ||
| ImageJ | National Institutes of Health | RRID:SCR_003070 |
| Subio | Subio inc | https://www.subioplatform.com/ |
| Ingenuity Pathway Analysis (IPA) | Qiagen | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/?gclid = Cj0KCQjwpcOTBhCZARIsAEAYLuU2uBoPYEwAjfKm5S0IeMMKdT2e6A9WQDKXBNipkzh0QNE9wNgH-mQaAspZEALw_wcB |
| CRISPR DESIGN | Massachusetts Institute of Technology ZHANG LAB | http://crispr.mit.edu/ |
| JMP ver. 14.0 | SAS Institute, Inc. | RRID:SCR_014242 |
| TargetScan human version 7.2 | Agarwal et al., 201577 | http://www.targetscan.org/vert_72/ |
| miRwalk | Sticht et al., 201878 | http://mirwalk.umm.uni-heidelberg.de/ |
| miRabel | University of Rouen LITIS Lab | http://bioinfo.univ-rouen.fr/mirabel/ |
| miRmap | Vejnar et al., 201379 | https://mirmap.ezlab.org/app/ |
| JASPAR | Castro-Mondragon et al.80 | http://jaspar.genereg.net/ |
| ChIP-Atlas | Oki et al.81 | https://chip-atlas.org/ |
| FIREBROWSE | Deng et al.82 | http://firebrowse.org/ |
| ONCOMINE | Rhodes et al.20 | https://www.oncomine.org |
Resource availability
Lead contact
Further information and requests for resources, reagents and samples should be directed to and will be fulfilled by the lead contact, Hirofumi Yamamoto (hyamamoto@sahs.med.osaka-u.ac.jp).
Materials availability
Materials and reagents used in this study are listed in the key resources table. Reagents generated in our laboratory in this study or previous studies are available upon request.
Experimental model and subject details
Cell lines
Human lung fibroblast (MRC5), human bronchial epithelial (HBEC3-KT), and human CRC (LS174T, RKO) cell lines and a rat intestinal cell line, IEC-18, were obtained from the American Type Culture Collection (Rockville, MD, USA). HCT116 p53+/+ cells retained the wild-type p53 gene, whereas both alleles of the p53 gene were deleted through homologous recombination in HCT116 p53−/− cells. This genetically impaired HCT116 cell line and the parental line with wild-type genes were generous gifts from Dr. Bert Vogelstein (Johns Hopkins University School of Medicine, Baltimore, MD, USA). HBEC3-KT cells were maintained in Airway Epithelial Cell Basal Medium (ATCC PCS-300-030) supplemented with Bronchial Epithelial Cell Growth Lit (ATCC PCS-300-040). Other cell lines were maintained in Dulbecco’s modified Eagle medium (Sigma-Aldrich, St. Louis, MO, USA) containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cultures were maintained at 37°C in a humid incubator with 5% CO2.
Animals
All studies were conducted using male or female mice over 8 weeks of age. All animals were housed and maintained under conditions of 50% humidity and a 12:12-h light:dark cycle. They were fed a standard pellet diet (MF, Oriental Yeast Co., Tokyo, Japan) and tap water ad libitum. The Osaka University Animal Experiment Committee approved all animal experiments.
Clinical tissue samples
CRC samples were collected from 98 patients (Stage 0/I/II/III/IV: 5/28/29/28/8), of which 20 were early cancers. These patients underwent surgery between 2003 and 2010 at Osaka University Hospital. The Union for International Cancer Control classification was used for patient staging.83 For transcriptome analysis, tissue samples were immediately frozen in RNAlaterTM (Ambion, Austin, TX, USA) and stored at −80°C until RNA extraction. For immunohistochemistry, tissue samples were fixed in 10% buffered formalin at 4°C overnight, processed through graded ethanol solutions, and embedded in paraffin.
Ethics Approval
This study was approved by the institutional review board of our institution (Permission No. #15144). Written informed consent was obtained from all patients. The study protocol was in accordance with the Declaration of Helsinki, the Japanese Ethical Guidelines for Human Genome/Gene Analysis Research, and the Ethical Guidelines for Medical and Health Research Involving Human Subjects in Osaka University.
Method details
Chemicals
5-FU was purchased from Nacalai Tesque Inc. (Kyoto, Japan). AOM saline was purchased from Sigma-Aldrich. DSS was purchased from MP Biomedicals (Santa Ana, CA, USA).
siRNA and miRNA
siRNA for rat-c-MAF (4390771) and its negative control siRNA (4390843) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). miR-155 mimic and its negative control sequence, and mouse and human miR-200c-3p, 302a-3p, 302b-3p, 302c-3p, 302d-3p, 369–3p, and 369–5p mimics and their negative control sequences were purchased from Gene Design, Inc. (Osaka, Japan). The sequence information is shown in Table S1. Lipofectamine RNAiMax (Thermo Fisher Scientific) was used for transfection of siRNA or miRNA according to the manufacturer’s instructions.
Plasmid DNA
pCMV6-c-MAF plasmid DNA was purchased from OriGene (Rockville, MD, USA). pCMV6-empty vector was used as a control. pCMV-Neo-Bam p53 R175H (R175H), pCMV-Neo-Bam p53 R248W (R248W), and pCMV-Neo-Bam (Empty) were purchased from Addgene ( a gift from Bert Vogelstein, addgene, #16436; http://n2t.net/addgene:16436; RRID:Addgene_16436, #16437; http://n2t.net/addgene:16437; RRID:Addgene_16437, #16440; http://n2t.net/addgene:16440; RRID:Addgene_16440, respectively).84 Transfection was performed with Lipofectamine 3000 Reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol.
Western blot analysis
Western blotting was performed according to our protocol.85 Approximately each sample were homogenized in 1 ml of lysis buffer [50 mM Tris (pH 8.0), 150 mM NaCl, and 0.5% NP40] with protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, and 10 μg/ml leupeptin). The homogenate was centrifuged at 14,000 rpm for 20 min at 4°C. The resulting supernatant was collected, and total protein concentration was determined using the Bradford protein assay (Bio-Rad, Hercules, CA). The protein lysates (20 μg) from each sample were separated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane. The membrane was blocked with 5% skim milk and incubated with the primary antibody at a concentration of 1–2 μg/mL, as follows: anti-human c-MAF polyclonal antibody (ab72584, Abcam, Cambridge, UK), anti-human p53 monoclonal antibody (M7001, Dako, Santa Barbara, CA, USA), anti-human p21Waf1/Cip1 monoclonal antibody (ab80633, Abcam), and anti-human ACTB polyclonal antibody (A2066, Sigma-Aldrich). The membrane was incubated with secondary antibodies and visualized with the ECL Detection System (GE Healthcare, Little Chalfont, UK).
Proliferation assay
Cells were seeded at a density of 3–5 × 103 cells per well in 96-well plates. After culture for 24, 48, or 72 h, cell viability was determined using the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) by measuring the absorbance at 450 nm using an iMarkTM microplate absorbance reader (Bio-Rad, Hercules, CA, USA).
Colony-formation assay
Cells were seeded at a density of 500 cells per well in a 6-well plate. After incubation at 37°C for 10 days, cells were washed with phosphate-buffered saline, fixed with 10% formalin, and stained with Giemsa solution. The number of colonies was counted with ImageJ software (National Institutes of Health).
Annexin V assay
Apoptosis was evaluated by flow cytometry with the Annexin V-FITC Apoptosis Kit (BioVision, Milpitas, CA, USA) according to the instructions of the manufacturer. Briefly, cells were harvested and stained with Annexin V-FITC and propidium iodide. Each sample was analyzed using the BD FACS Aria IIu (BD Biosciences, San Jose, CA, USA).
RNA isolation
Total mRNA was isolated with TRIzol Reagent (Invitrogen) for mouse samples and cell lines or with miRNeasy Mini Kit (Qiagen, Hilden, Germany) for human tissue samples, according to the respective manufacturer’s protocol. RNA quality was assessed with a NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific).
Real-time quantitative RT-PCR (qRT-PCR) analysis
For quantitative analysis of mRNA, total RNA was reverse transcribed using the High Capacity RNA-to-cDNA Kit (Applied Biosystems). qRT-PCR was performed with a LightCycler 480 Real-Time PCR system (Roche Diagnostics, Mannheim, Germany) using the specific primers and LightCycler-DNA Master SYBR Green I (Roche Diagnostics) or with ABI Prism 7900HT Sequence Detection System (Applied Biosystems) using TaqMan Gene Expression Assays (Applied Biosystems) and TaqMan Universal PCR Master Mix (Applied Biosystems). The specifically designed primers and the product numbers of the TaqMan Gene Expression Assay were listed in Table S5. Each gene expression value was normalized to the mRNA expression level of β-actin. Relative expression was quantified with the ΔΔCT method.86
Immunohistochemistry
Tissue sections of 4-μm thickness were prepared from paraffin-embedded blocks. H&E staining was performed for histological examination. Immunostaining was carried out with the Vectastain ABC Peroxidase Kit (Vector Laboratories, Burlingame, CA, USA), after antigen retrieval treatment in 10 mM citrate buffer (pH 6.0) at 95°C for 40 min. The slides were incubated overnight at 4°C with anti-human polyclonal antibody against MAF (#ab72584, Abcam), anti-mouse monoclonal antibody against p53 (#M7001, Dako, Glostrup, Denmark), anti-rabbit monoclonal antibody against SOX10 (#ab227680, Abcam), anti-rabbit polyclonal antibody against BCL6 (#PA5-27390, Invitrogen), anti-rabbit polyclonal antibody against S-100 (#422091, NICHIREI BIOSCIENCE Inc., Tokyo, Japan), anti-rabbit polyclonal antibody against CD45R (#14-0451-82, Invitrogen), and anti-rabbit monoclonal antibody against Ki-67 (#12202, Cell Signaling Technology, Danvers, MA, USA) at the following dilutions: anti-MAF antibody, 1:200; anti-p53, 1:50, anti-S100, 1:1; anti-SOX10, 1:100; anti-BCL6, 1:100; and anti-CD45R, 1:100.
Tissue sections of 8-μm thickness were prepared from OCT compound–embedded blocks. They were incubated overnight at 4°C with anti-mouse monoclonal rabbit antibody against Ki-67 (#12202, Cell Signaling Technology) at dilutions of 1:200. Counter nuclear staining was performed with a hematoxylin solution.
Systemic administration of miRNAs to CPC;Apc mice
Apcmin mice produce polyps mainly in the small intestine, whereas CPC;Apc mice produce colorectal tumors in which conditional knockout of the Apc gene was accomplished under the CDX2 promoter (∼9.5-kb 5′-flanking region from the human CDX2 gene), specifically acting at the mouse colorectum.16 Male mice were treated with formulated miRNAs (miR-200c-3p, miR-302a/b/c/d-3p, and miR-369–3p/-5p) or negative control miRNA, three times a week for 8 weeks from 8 weeks of age. miRNAs were injected via tail vein using sCA as a drug delivery system.19 The preparation of a sCA transfection mixture for in vivo use was previously described.19 Briefly, to prepare a CA transfection mixture, 25 μg of each miRNA (a total amount of nucleic acid is 175 μg) or NC miRNA (175 μg) was mixed with 350 μL of 1 M CaCl2 in 87.5 mL of serum-free bicarbonate–buffered inorganic solution (NaHCO3 44 mM, NaH2PO4 0.9 mM, CaCl2 1.8 mM, pH 7.5), and incubated at 37°C for 30 min. The solution was centrifuged at 12,000 rpm for 3 min and the pellet dissolved with saline containing 0.5% albumin. The products were sonicated (38 kHz, 80 W) in a water bath for 10 min to generate sCA plus 0.5% albumin, which was intravenously injected (approximately 70 μg per mouse) within 10 min. All miRNAs used in this study were purchased from Gene Design Inc. (Osaka, Japan; Table S1).
At 15 weeks after birth, mice were sacrificed, and normal mucosa and polyps were collected, immediately frozen in RNAlaterTM (Ambion), and stored at −80°C until RNA extraction. All experiments were performed in strict accordance with the prescribed guidelines and protocols approved by the Committee on the Ethics of Animal Experiments of Osaka University (No. 30011026). Generation of colorectal polyps was monitored by a small-diameter rectosigmoid scope (Natsume Seisakusho, Tokyo, Japan).
Microarray analysis
Total RNA from mouse tissue samples was reverse transcribed with oligo-dT primers containing the T7 RNA polymerase promoter sequence. The resulting cDNA was subjected to in vitro transcription with T7 RNA polymerase for Cy3 labeling (CyDye; Amersham Pharmacia Biotech). Cy3-labeled cRNAs (600 ng) were hybridized onto Agilent Sure Print G3 Mouse GE 8 × 60K (G4852A). The signal intensity of Cy3 was calculated for every probe, and the results were analyzed with the Subio Basic Plug-in (v1.6; Subio Inc.), which allows for visualization of microarray data in the form of a heat map. The microarray raw data are available in the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) database with accession number GSE92944.
RNA sequencing
We conducted RNA sequencing as previously described.38 The library was prepared using a TruSeq Stranded mRNA Sample Prep Kit (Illumina, San Diego, CA, USA). Sequencing was performed using the Illumina HiSeq 2500 platform in 75-base single-end mode. Illumina Casava 1.8.2 software was used for base calling, and the sequenced reads were mapped to human reference genome sequences (hg19) using TopHat version 2.0.13 combined with Bowtie2 version 2.2.3 and SAMtools version 0.1.19. We calculated the fragments per kilobase of exon per million mapped fragments using Cuffnorm version 2.2.1. We identified a series of genes that were enhanced or reduced (tumor: 1.5 fold; normal mucosa: 1.3 fold) for further gene expression analysis. The raw data were deposited in the NCBI Gene Expression Omnibus database under GEO accession number GSE210970. We identified enhanced or suppressed pathways using Qiagen’s Ingenuity Pathway Analysis (IPA; Qiagen Redwood City, CA, USA; www.qiagen.com/ingenuity) with the default settings.
Generation of c-MAF KO mice
Mouse species
Jcl:ICR pseudopregnant female mice and C57BL/6JJcl cryopreserved zygotes were purchased from CLEA Japan Inc. (Tokyo, Japan).
Preparation of Cas9 and gRNA
The following reagents were purchased: Cas9 protein, Alt-R® S.p. Cas9 Nuclease 3NLS (Integrated DNA Technologies, Inc. USA); guide RNA (gRNA), and GeneArt Precision gRNA Synthesis Kit (Thermo Fisher Scientific). To design the gRNA sequence (5′-CAGGAGGATGGCTTCAGAAC-3′), we used a software tool (http://crispr.mit.edu/) to predict unique target sites throughout the mouse genome.
Electroporation into mouse embryos
Pronuclear-stage mouse embryos were prepared by thawing frozen embryos in KSOM medium (ARK Resource, Kumamoto, Japan). For electroporation, 150 embryos at 1 h after thawing were placed into a chamber with 40 μL of serum-free medium (Opti-MEM, Thermo Fisher Scientific) containing 100 ng/μL Cas9 protein and 200 ng/μL gRNA. They were electroporated with a 5-mm gap electrode (CUY505P5 or CUY520P5 Nepa Gene, Chiba, Japan) in a NEPA21 Super Electroporator (Nepa Gene, Chiba, Japan). The poring pulses for the electroporation were voltage 225 V, pulse width 1 ms, pulse interval 50 ms, and number of pulses 4. The first and second transfer pulses were voltage 20 V, pulse width 50 ms, pulse interval 50 ms, and number of pulses 5. Mouse embryos that developed to the two-cell stage after the introduction of Cas9 and gRNA were transferred into the oviducts of female surrogates anesthetized with sevoflurane (Mylan Pfizer Japan Inc.). Male and female mice with c-MAF heterogeneous KO (MAF+/-) were mated so that homogeneous c-MAF KO (MAF-/-) c-MAF and heterogeneous KO (MAF+/-) mice were produced.
Genotyping analysis
Genomic DNA was extracted from the tail tip using the KAPA Express Extract DNA Extraction Kit (Kapa Biosystems, London, UK) and Animal Tissue Direct PCR Amplification Kit (with TL) (FineGene, Shanghai, CN). For PCR and sequence analysis, we used primers that amplified the targeted region. PCR was performed under the following conditions: 1 cycle of 94°C for 1 min; 30 cycles of 98°C for 10 s, 60°C for 15 s, and 68°C for 30 s; and 1 cycle of 72°C for 3 min. The PCR products were sequenced immediately or after purification using the Mini-Gel extraction kit (One-Step) (FineGene) with BigDye Terminator v3.1 cycle sequencing mix and the standard protocol for an Applied Biosystems 3130 DNA Sequencer (Life Technologies).
Examination of tumorigenesis in mice
Spontaneous carcinogenesis
c-MAF KO mice (n = 187) and wild-type mice (n = 37) were observed for 24 months for tumor generation. 1 nucleotide deletion homo mice included two males and three females, 1 nucleotide deletion hetero mice included sixty-three males and seventy females, 8 nucleotide deletion homo mice included a female, 8 nucleotide deletion hetero mice included twenty-seven males and twenty-one females and 37 wild-type mice are all males. Tumors were histologically examined with H&E staining and the specific immunostaining.
Chemical carcinogenesis
AOM and DSS treatment was used to produce CRCs, as previously reported.87 c-MAF KO mice (n = 19) and wild-type mice (n = 13) were treated. 1 nucleotide deletion hetero mice included two males and four females. 8 nucleotide deletion homo mice included three males and three females. 8 nucleotide deletion mice included three males and four females. All wild type mice were males. On day 100 after administration of AOM, all mice were examined by an animal endoscope for tumor formation in the colorectum (Natsume Seisakusho). On 136 ± 8 day, mice were sacrificed, and the colorectum was removed. After gross observation, normal mucosa and tumors of diameter >1 mm were collected and subjected to histological examination and RNA sequencing.
ChIP-qPCR
ChIP experiment was performed as previously reported.88 Cells were fixed and sheared by using Covaris M200 (Covaris, Woburn, MA, USA). The fragmented chromatin was incubated with the following primary Abs; GATA3 (ab199428, Abcam, Cambridge, UK). The Ab against normal rabbit IgG (#2729, Cell Signaling Technology, Danvers, MA, USA) was used as a negative control. The purified DNA was subjected to qPCR. qPCR was performed as described in the qPCR section. The sequences of the primers are listed in Table S5.
Quantification and statistical analysis
All data are expressed as mean ± standard deviation, or the median and interquartile range (IQR). Statistical differences were analyzed using Student’s t test for continuous variables and the Chi-squared test for non-continuous data. The incidence of tumor formation and the size of tumor in c-MAF KO mice and wild type mice were analyzed using the Wilcoxon signed-rank test. Survival curves were developed with the Kaplan–Meier method and compared using the log-rank test. A Cox proportional hazard regression model was used to estimate HRs and 95% CIs. All statistical analyzes were conducted with JMP ver. 14.0 (SAS Institute, Inc., Cary, NC, USA) and Subio platform ver. 1.24.5853 (Subio Inc., Aichi, Japan) for analyzing microarray and RNA-seq data. All p values <0.05 were considered to indicate statistical significance.
Acknowledgments
HCT116 and p53-null derivatives were kind gifts from Prof. Bert Vogelstein (Johns Hopkins University, Baltimore, MD, USA). We are grateful to other collaborators Y. Kotani for generating c-MAF knockout mice, and H. Ogawa, M. Konno, M. Nomura, A Eto, K Kitagawa, A. Toyama, and K. Asai for their support in animal experiments, and genotyping. We also thank prof. M. Futakuchi (Department of Pathological Diagnosis, Yamagata University Faculty of Medicine) for the useful guidance of autopsy of mice.
This work was supported by JSPS Grant-in-Aid for Exploratory Research Grant Number 26670605 to H.Y., JSPS KAKENHI Grant Number 20K08356 to D.O. and Research Grants of Princess Takamatsu Cancer Research Funds 2017 to H.Y.
Author contributions
H.I. and H.Y. designed the study. T.Hata, Y.D., H.E., and M.M. supervised the study. H.Y., Y.Y., T.A., and T.Hinoi are responsible for methodology. H.I., T.Hata, Y.Y., and H.Y. analyzed and interpreted the data and confirm the authenticity of all the raw data. H.I., T.Hata, D.O., K.T., and K.I. performed the experiments. H.I., Y.Y., and H.Y. are responsible for the statistical analysis. T.O., N.M., H.T., M.U., and T.M. collected and provided the normal and colorectal cancer tissue samples and their clinical data. H.I. wrote the original draft. H.Y., H.T., and Y.Y. reviewed and edited the manuscript. H.I., K.T., Y.M., Q.Y., T.Hata, and H.H. performed animal experiments. All authors have read and approved the final manuscript.
Declaration of interests
The authors have no conflict of interest to disclose.
Published: March 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106478.
Supplemental information
Data and code availability
-
•
Data are available on Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) database with accession numbers: GSE92944, GSE210970 and GSE210980.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Abbasi O., Mashayekhi F., Mirzajani E., Fakhriyeh Asl S., Mahmoudi T., Saeedi Saedi H. Soluble VEGFR1 concentration in the serum of patients with colorectal cancer. Surg. Today. 2015;45:215–220. doi: 10.1007/s00595-014-0886-4. [DOI] [PubMed] [Google Scholar]
- 2.Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet-Tieulent J., Jemal A. Global cancer statistics, 2012. CA. Cancer J. Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Brenner H., Kloor M., Pox C.P. Colorectal cancer. Lancet. 2014;383:1490–1502. doi: 10.1016/s0140-6736(13)61649-9. [DOI] [PubMed] [Google Scholar]
- 4.Colvin H.S., Nishida N., Koseki J., Konno M., Kawamoto K., Tsunekuni K., Doki Y., Mori M., Ishii H. Cancer stem cells of the digestive system. Jpn. J. Clin. Oncol. 2014;44:1141–1149. doi: 10.1093/jjco/hyu146. [DOI] [PubMed] [Google Scholar]
- 5.Dienstmann R., Vermeulen L., Guinney J., Kopetz S., Tejpar S., Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer. 2017;17:79–92. doi: 10.1038/nrc.2016.126. [DOI] [PubMed] [Google Scholar]
- 6.Biller L.H., Schrag D. Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA. 2021;325:669–685. doi: 10.1001/jama.2021.0106. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 8.Miyoshi N., Ishii H., Nagai K.i., Hoshino H., Mimori K., Tanaka F., Nagano H., Sekimoto M., Doki Y., Mori M. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc. Natl. Acad. Sci. USA. 2010;107:40–45. doi: 10.1073/pnas.0912407107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okita K., Nakagawa M., Hyenjong H., Ichisaka T., Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- 10.Kaji K., Norrby K., Paca A., Mileikovsky M., Mohseni P., Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woltjen K., Michael I.P., Mohseni P., Desai R., Mileikovsky M., Hämäläinen R., Cowling R., Wang W., Liu P., Gertsenstein M., et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia F., Wilson K.D., Sun N., Gupta D.M., Huang M., Li Z., Panetta N.J., Chen Z.Y., Robbins R.C., Kay M.A., et al. A nonviral minicircle vector for deriving human iPS cells. Nat. Methods. 2010;7:197–199. doi: 10.1038/nmeth.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyoshi N., Ishii H., Nagano H., Haraguchi N., Dewi D.L., Kano Y., Nishikawa S., Tanemura M., Mimori K., Tanaka F., et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell. 2011;8:633–638. doi: 10.1016/j.stem.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Miyazaki S., Yamamoto H., Miyoshi N., Wu X., Ogawa H., Uemura M., Nishimura J., Hata T., Takemasa I., Mizushima T., et al. A cancer reprogramming method using MicroRNAs as a novel therapeutic approach against colon cancer: Research for reprogramming of cancer cells by MicroRNAs. Ann. Surg Oncol. 2015;22:S1394–S1401. doi: 10.1245/s10434-014-4217-1. [DOI] [PubMed] [Google Scholar]
- 15.Ogawa H., Wu X., Kawamoto K., Nishida N., Konno M., Koseki J., Matsui H., Noguchi K., Gotoh N., Yamamoto T., et al. MicroRNAs induce epigenetic reprogramming and suppress malignant phenotypes of human colon cancer cells. PLoS One. 2015;10 doi: 10.1371/journal.pone.0127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinoi T., Akyol A., Theisen B.K., Ferguson D.O., Greenson J.K., Williams B.O., Cho K.R., Fearon E.R. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67:9721–9730. doi: 10.1158/0008-5472.Can-07-2735. [DOI] [PubMed] [Google Scholar]
- 17.Nishizawa M., Kataoka K., Goto N., Fujiwara K.T., Kawai S. v-maf, a viral oncogene that encodes a "leucine zipper" motif. Proc. Natl. Acad. Sci. USA. 1989;86:7711–7715. doi: 10.1073/pnas.86.20.7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brundage M.E., Tandon P., Eaves D.W., Williams J.P., Miller S.J., Hennigan R.H., Jegga A., Cripe T.P., Ratner N. MAF mediates crosstalk between Ras-MAPK and mTOR signaling in NF1. Oncogene. 2014;33:5626–5636. doi: 10.1038/onc.2013.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu X., Yamamoto H., Nakanishi H., Yamamoto Y., Inoue A., Tei M., Hirose H., Uemura M., Nishimura J., Hata T., et al. Innovative delivery of siRNA to solid tumors by super carbonate apatite. PLoS One. 2015;10 doi: 10.1371/journal.pone.0116022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rhodes D.R., Yu J., Shanker K., Deshpande N., Varambally R., Ghosh D., Barrette T., Pandey A., Chinnaiyan A.M. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng M., Brägelmann J., Kryukov I., Saraiva-Agostinho N., Perner S. FirebrowseR: an R client to the broad institute's firehose pipeline. Database. 2017;2017 doi: 10.1093/database/baw160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arber N., Han E.K., Sgambato A., Piazza G.A., Delohery T.M., Begemann M., Weghorst C.M., Kim N.H., Pamukcu R., Ahnen D.J., et al. A K-ras oncogene increases resistance to sulindac-induced apoptosis in rat enterocytes. Gastroenterology. 1997;113:1892–1900. doi: 10.1016/s0016-5085(97)70008-8. [DOI] [PubMed] [Google Scholar]
- 23.Arber N., Sutter T., Miyake M., Kahn S.M., Venkatraj V.S., Sobrino A., Warburton D., Holt P.R., Weinstein I.B. Increased expression of cyclin D1 and the Rb tumor suppressor gene in c-K-ras transformed rat enterocytes. Oncogene. 1996;12:1903–1908. [PubMed] [Google Scholar]
- 24.Takayama O., Yamamoto H., Damdinsuren B., Sugita Y., Ngan C.Y., Xu X., Tsujino T., Takemasa I., Ikeda M., Sekimoto M., et al. Expression of PPARdelta in multistage carcinogenesis of the colorectum: implications of malignant cancer morphology. Br. J. Cancer. 2006;95:889–895. doi: 10.1038/sj.bjc.6603343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hale T.K., Myers C., Maitra R., Kolzau T., Nishizawa M., Braithwaite A.W. Maf transcriptionally activates the mouse p53 promoter and causes a p53-dependent cell death. J. Biol. Chem. 2000;275:17991–17999. doi: 10.1074/jbc.M000921200. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez A., Vigorito E., Clare S., Warren M.V., Couttet P., Soond D.R., van Dongen S., Grocock R.J., Das P.P., Miska E.A., et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su W., Hopkins S., Nesser N.K., Sopher B., Silvestroni A., Ammanuel S., Jayadev S., Möller T., Weinstein J., Garden G.A. The p53 transcription factor modulates microglia behavior through microRNA-dependent regulation of c-Maf. J. Immunol. 2014;192:358–366. doi: 10.4049/jimmunol.1301397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolf L., Gao C.S., Gueta K., Xie Q., Chevallier T., Podduturi N.R., Sun J., Conte I., Zelenka P.S., Ashery-Padan R., et al. Identification and characterization of FGF2-dependent mRNA: microRNA networks during lens fiber cell differentiation. G3 (Bethesda) 2013;3:2239–2255. doi: 10.1534/g3.113.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agarwal V., Bell G.W., Nam J.W., Bartel D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4 doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sticht C., De La Torre C., Parveen A., Gretz N. miRWalk: an online resource for prediction of microRNA binding sites. PLoS One. 2018;13 doi: 10.1371/journal.pone.0206239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vejnar C.E., Blum M., Zdobnov E.M. miRmap web: comprehensive microRNA target prediction online. Nucleic Acids Res. 2013;41:W165–W168. doi: 10.1093/nar/gkt430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naito T., Tanaka H., Naoe Y., Taniuchi I. Transcriptional control of T-cell development. Int. Immunol. 2011;23:661–668. doi: 10.1093/intimm/dxr078. [DOI] [PubMed] [Google Scholar]
- 33.Oki S., Ohta T., Shioi G., Hatanaka H., Ogasawara O., Okuda Y., Kawaji H., Nakaki R., Sese J., Meno C. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018;19 doi: 10.15252/embr.201846255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castro-Mondragon J.A., Riudavets-Puig R., Rauluseviciute I., Lemma R.B., Turchi L., Blanc-Mathieu R., Lucas J., Boddie P., Khan A., Manosalva Pérez N., et al. Jaspar 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022;50 doi: 10.1093/nar/gkab1113. D165–d173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stadthagen G., Tehler D., Høyland-Kroghsbo N.M., Wen J., Krogh A., Jensen K.T., Santoni-Rugiu E., Engelholm L.H., Lund A.H. Loss of miR-10a activates lpo and collaborates with activated Wnt signaling in inducing intestinal neoplasia in female mice. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeitels L.R., Acharya A., Shi G., Chivukula D., Chivukula R.R., Anandam J.L., Abdelnaby A.A., Balch G.C., Mansour J.C., Yopp A.C., et al. Tumor suppression by miR-26 overrides potential oncogenic activity in intestinal tumorigenesis. Genes Dev. 2014;28:2585–2590. doi: 10.1101/gad.250951.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roh M.S., Lee H.W., Jung S.B., Kim K., Lee E.H., Park M.I., Lee J.S., Kim M.S. Expression of miR-200c and its clinicopathological significance in patients with colorectal cancer. Pathol. Res. Pract. 2018;214:350–355. doi: 10.1016/j.prp.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Fukata T., Mizushima T., Nishimura J., Okuzaki D., Wu X., Hirose H., Yokoyama Y., Kubota Y., Nagata K., Tsujimura N., et al. The supercarbonate apatite-MicroRNA complex inhibits dextran sodium sulfate-induced colitis. Mol. Ther. Nucleic Acids. 2018;12:658–671. doi: 10.1016/j.omtn.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takeyama H., Yamamoto H., Yamashita S., Wu X., Takahashi H., Nishimura J., Haraguchi N., Miyake Y., Suzuki R., Murata K., et al. Decreased miR-340 expression in bone marrow is associated with liver metastasis of colorectal cancer. Mol. Cancer Ther. 2014;13:976–985. doi: 10.1158/1535-7163.Mct-13-0571. [DOI] [PubMed] [Google Scholar]
- 40.Morimoto Y., Mizushima T., Wu X., Okuzaki D., Yokoyama Y., Inoue A., Hata T., Hirose H., Qian Y., Wang J., et al. miR-4711-5p regulates cancer stemness and cell cycle progression via KLF5, MDM2 and TFDP1 in colon cancer cells. Br. J. Cancer. 2020;122:1037–1049. doi: 10.1038/s41416-020-0758-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inoue A., Mizushima T., Wu X., Okuzaki D., Kambara N., Ishikawa S., Wang J., Qian Y., Hirose H., Yokoyama Y., et al. A miR-29b byproduct sequence exhibits potent tumor-suppressive activities via inhibition of NF-κB signaling in KRAS-mutant colon cancer cells. Mol. Cancer Ther. 2018;17:977–987. doi: 10.1158/1535-7163.Mct-17-0850. [DOI] [PubMed] [Google Scholar]
- 42.Hiraki M., Nishimura J., Takahashi H., Wu X., Takahashi Y., Miyo M., Nishida N., Uemura M., Hata T., Takemasa I., et al. Concurrent targeting of KRAS and AKT by MiR-4689 is a novel treatment against mutant KRAS colorectal cancer. Mol. Ther. Nucleic Acids. 2015;4:e231. doi: 10.1038/mtna.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J., Yokoyama Y., Hirose H., Shimomura Y., Bonkobara S., Itakura H., Kouda S., Morimoto Y., Minami K., Takahashi H., et al. Functional assessment of miR-1291 in colon cancer cells. Int. J. Oncol. 2022;60 doi: 10.3892/ijo.2022.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamai K., Mizushima T., Wu X., Inoue A., Ota M., Yokoyama Y., Miyoshi N., Haraguchi N., Takahashi H., Nishimura J., et al. Photodynamic therapy using indocyanine green loaded on super carbonate apatite as minimally invasive cancer treatment. Mol. Cancer Ther. 2018;17:1613–1622. doi: 10.1158/1535-7163.Mct-17-0788. [DOI] [PubMed] [Google Scholar]
- 45.Abd-Aziz N., Kamaruzman N.I., Poh C.L. Development of MicroRNAs as potential therapeutics against cancer. J. Oncol. 2020;2020 doi: 10.1155/2020/8029721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merhautova J., Demlova R., Slaby O. MicroRNA-Based therapy in animal models of selected gastrointestinal cancers. Front. Pharmacol. 2016;7:329. doi: 10.3389/fphar.2016.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi R.U., Prieto-Vila M., Kohama I., Ochiya T. Development of miRNA-based therapeutic approaches for cancer patients. Cancer Sci. 2019;110:1140–1147. doi: 10.1111/cas.13965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forterre A., Komuro H., Aminova S., Harada M. A comprehensive review of cancer MicroRNA therapeutic delivery strategies. Cancers. 2020;12 doi: 10.3390/cancers12071852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hurt E.M., Wiestner A., Rosenwald A., Shaffer A.L., Campo E., Grogan T., Bergsagel P.L., Kuehl W.M., Staudt L.M. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004;5:191–199. doi: 10.1016/s1535-6108(04)00019-4. [DOI] [PubMed] [Google Scholar]
- 50.Morito N., Yoh K., Fujioka Y., Nakano T., Shimohata H., Hashimoto Y., Yamada A., Maeda A., Matsuno F., Hata H., et al. Overexpression of c-Maf contributes to T-cell lymphoma in both mice and human. Cancer Res. 2006;66:812–819. doi: 10.1158/0008-5472.Can-05-2154. [DOI] [PubMed] [Google Scholar]
- 51.Pavlovic M., Arnal-Estapé A., Rojo F., Bellmunt A., Tarragona M., Guiu M., Planet E., Garcia-Albéniz X., Morales M., Urosevic J., et al. Enhanced MAF oncogene expression and breast cancer bone metastasis. J. Natl. Cancer Inst. 2015;107:djv256. doi: 10.1093/jnci/djv256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eychène A., Rocques N., Pouponnot C. A new MAFia in cancer. Nat. Rev. Cancer. 2008;8:683–693. doi: 10.1038/nrc2460. [DOI] [PubMed] [Google Scholar]
- 53.Benchimol S. p53-dependent pathways of apoptosis. Cell Death Differ. 2001;8:1049–1051. doi: 10.1038/sj.cdd.4400918. [DOI] [PubMed] [Google Scholar]
- 54.Levine A.J. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 55.Abbas T., Dutta A. p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.el-Deiry W.S., Tokino T., Velculescu V.E., Levy D.B., Parsons R., Trent J.M., Lin D., Mercer W.E., Kinzler K.W., Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 57.el-Deiry W.S., Harper J.W., O'Connor P.M., Velculescu V.E., Canman C.E., Jackman J., Pietenpol J.A., Burrell M., Hill D.E., Wang Y., et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–1174. [PubMed] [Google Scholar]
- 58.Calogero R.A., Cordero F., Forni G., Cavallo F. Inflammation and breast cancer. Inflammatory component of mammary carcinogenesis in ErbB2 transgenic mice. Breast Cancer Res. 2007;9:211. doi: 10.1186/bcr1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ursini-Siegel J., Schade B., Cardiff R.D., Muller W.J. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat. Rev. Cancer. 2007;7:389–397. doi: 10.1038/nrc2127. [DOI] [PubMed] [Google Scholar]
- 60.Conner E.A., Lemmer E.R., Omori M., Wirth P.J., Factor V.M., Thorgeirsson S.S. Dual functions of E2F-1 in a transgenic mouse model of liver carcinogenesis. Oncogene. 2000;19:5054–5062. doi: 10.1038/sj.onc.1203885. [DOI] [PubMed] [Google Scholar]
- 61.Olson M.V., Johnson D.G., Jiang H., Xu J., Alonso M.M., Aldape K.D., Fuller G.N., Bekele B.N., Yung W.K.A., Gomez-Manzano C., Fueyo J. Transgenic E2F1 expression in the mouse brain induces a human-like bimodal pattern of tumors. Cancer Res. 2007;67:4005–4009. doi: 10.1158/0008-5472.Can-06-2973. [DOI] [PubMed] [Google Scholar]
- 62.Pierce A.M., Gimenez-Conti I.B., Schneider-Broussard R., Martinez L.A., Conti C.J., Johnson D.G. Increased E2F1 activity induces skin tumors in mice heterozygous and nullizygous for p53. Proc. Natl. Acad. Sci. USA. 1998;95:8858–8863. doi: 10.1073/pnas.95.15.8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Agger K., Santoni-Rugiu E., Holmberg C., Karlström O., Helin K. Conditional E2F1 activation in transgenic mice causes testicular atrophy and dysplasia mimicking human CIS. Oncogene. 2005;24:780–789. doi: 10.1038/sj.onc.1208248. [DOI] [PubMed] [Google Scholar]
- 64.Wang D., Russell J., Xu H., Johnson D.G. Deregulated expression of DP1 induces epidermal proliferation and enhances skin carcinogenesis. Mol. Carcinog. 2001;31:90–100. doi: 10.1002/mc.1044. [DOI] [PubMed] [Google Scholar]
- 65.Zarei M., Giannikou K., Du H., Liu H.J., Duarte M., Johnson S., Nassar A.H., Widlund H.R., Henske E.P., Long H.W., Kwiatkowski D.J. MITF is a driver oncogene and potential therapeutic target in kidney angiomyolipoma tumors through transcriptional regulation of CYR61. Oncogene. 2021;40:112–126. doi: 10.1038/s41388-020-01504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malaterre J., Pereira L., Putoczki T., Millen R., Paquet-Fifield S., Germann M., Liu J., Cheasley D., Sampurno S., Stacker S.A., et al. Intestinal-specific activatable Myb initiates colon tumorigenesis in mice. Oncogene. 2016;35:2475–2484. doi: 10.1038/onc.2015.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshida Y., Wang I.C., Yoder H.M., Davidson N.O., Costa R.H. The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology. 2007;132:1420–1431. doi: 10.1053/j.gastro.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 68.Condorelli G.L., Facchiano F., Valtieri M., Proietti E., Vitelli L., Lulli V., Huebner K., Peschle C., Croce C.M. T-cell-directed TAL-1 expression induces T-cell malignancies in transgenic mice. Cancer Res. 1996;56:5113–5119. [PubMed] [Google Scholar]
- 69.Robles E.F., Mena-Varas M., Barrio L., Merino-Cortes S.V., Balogh P., Du M.Q., Akasaka T., Parker A., Roa S., Panizo C., et al. Homeobox NKX2-3 promotes marginal-zone lymphomagenesis by activating B-cell receptor signalling and shaping lymphocyte dynamics. Nat. Commun. 2016;7 doi: 10.1038/ncomms11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fang J.Y., Richardson B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6:322–327. doi: 10.1016/s1470-2045(05)70168-6. [DOI] [PubMed] [Google Scholar]
- 71.Yu H., Lee H., Herrmann A., Buettner R., Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer. 2014;14:736–746. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 72.Kataoka K., Noda M., Nishizawa M. Maf nuclear oncoprotein recognizes sequences related to an AP-1 site and forms heterodimers with both Fos and Jun. Mol. Cell Biol. 1994;14:700–712. doi: 10.1128/mcb.14.1.700-712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Andris F., Denanglaire S., Anciaux M., Hercor M., Hussein H., Leo O. The transcription factor c-maf promotes the differentiation of follicular helper T cells. Front. Immunol. 2017;8:480. doi: 10.3389/fimmu.2017.00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wende H., Lechner S.G., Cheret C., Bourane S., Kolanczyk M.E., Pattyn A., Reuter K., Munier F.L., Carroll P., Lewin G.R., Birchmeier C. The transcription factor c-Maf controls touch receptor development and function. Science. 2012;335:1373–1376. doi: 10.1126/science.1214314. [DOI] [PubMed] [Google Scholar]
- 75.Ivascu C., Wasserkort R., Lesche R., Dong J., Stein H., Thiel A., Eckhardt F. DNA methylation profiling of transcription factor genes in normal lymphocyte development and lymphomas. Int. J. Biochem. Cell Biol. 2007;39:1523–1538. doi: 10.1016/j.biocel.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 76.Johnson R.L., Fleet J.C. Animal models of colorectal cancer. Cancer Metastasis Rev. 2013;32:39–61. doi: 10.1007/s10555-012-9404-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perveen R., Favor J., Jamieson R.V., Ray D.W., Black G.C.M. A heterozygous c-Maf transactivation domain mutation causes congenital cataract and enhances target gene activation. Hum. Mol. Genet. 2007;16:1030–1038. doi: 10.1093/hmg/ddm048. [DOI] [PubMed] [Google Scholar]
- 78.Finlay C.A., Hinds P.W., Tan T.H., Eliyahu D., Oren M., Levine A.J. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol. Cell Biol. 1988;8:531–539. doi: 10.1128/mcb.8.2.531-539.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neilsen P.M., Noll J.E., Mattiske S., Bracken C.P., Gregory P.A., Schulz R.B., Lim S.P., Kumar R., Suetani R.J., Goodall G.J., Callen D.F. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene. 2013;32:2992–3000. doi: 10.1038/onc.2012.305. [DOI] [PubMed] [Google Scholar]
- 80.Ohue M., Tomita N., Monden T., Fujita M., Fukunaga M., Takami K., Yana I., Ohnishi T., Enomoto T., Inoue M., et al. A frequent alteration of p53 gene in carcinoma in adenoma of colon. Cancer Res. 1994;54:4798–4804. [PubMed] [Google Scholar]
- 81.De Robertis M., Massi E., Poeta M.L., Carotti S., Morini S., Cecchetelli L., Signori E., Fazio V.M. The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J. Carcinog. 2011;10:9. doi: 10.4103/1477-3163.78279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tanaka T., Kohno H., Suzuki R., Yamada Y., Sugie S., Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94:965–973. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amin M., Edge S., Greene F., Byrd D., Brookland R., Washington M., Gershenwald J., Compton C., Hess K., Sullivan D., et al. AJCC Cancer Staging Manual. Springer International Publishing AG; 2017. [Google Scholar]
- 84.Baker S.J., Markowitz S., Fearon E.R., Willson J.K., Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249:912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- 85.Hamabe A., Konno M., Tanuma N., Shima H., Tsunekuni K., Kawamoto K., Nishida N., Koseki J., Mimori K., Gotoh N., et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA. 2014;111:15526–15531. doi: 10.1073/pnas.1407717111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 87.Parang B., Barrett C.W., Williams C.S. AOM/DSS model of colitis-associated cancer. Methods Mol. Biol. 2016;1422:297–307. doi: 10.1007/978-1-4939-3603-8_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee T.I., Johnstone S.E., Young R.A. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 2006;1:729–748. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data are available on Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) database with accession numbers: GSE92944, GSE210970 and GSE210980.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.