

Abstract
Angelman syndrome (AS) is a neurogenetic disorder caused by the loss of ubiquitin ligase E3A (UBE3A) gene expression in the brain. The UBE3A gene is paternally imprinted in brain neurons. Clinical features of AS are primarily due to the loss of maternally expressed UBE3A in the brain. A healthy copy of paternal UBE3A is present in the brain but is silenced by a long non-coding antisense transcript (UBE3A-ATS). Here, we demonstrate that an artificial transcription factor (ATF-S1K) can silence Ube3a-ATS in an adult mouse model of Angelman syndrome (AS) and restore endogenous physiological expression of paternal Ube3a. A single injection of adeno-associated virus (AAV) expressing ATF-S1K (AAV-S1K) into the tail vein enabled whole-brain transduction and restored UBE3A protein in neurons to ∼25% of wild-type protein. The ATF-S1K treatment was highly specific to the target site with no detectable inflammatory response 5 weeks after AAV-S1K administration. AAV-S1K treatment of AS mice showed behavioral rescue in exploratory locomotion, a task involving gross and fine motor abilities, similar to low ambulation and velocity in AS patients. The specificity and tolerability of a single injection of AAV-S1K therapy for AS demonstrate the use of ATFs as a promising translational approach for AS.
Keywords: Angelman syndrome, AS, UBE3A, imprinting, artificial transcription factor, ATF, zinc finger, epigenome editing, AAV therapy, brain-wide delivery
Graphical abstract
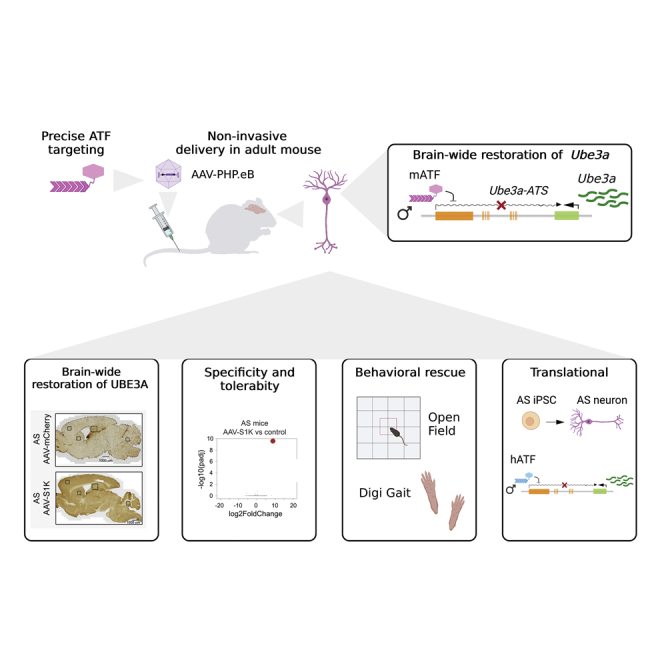
Non-invasive adeno-associated virus (AAV) treatment with a zinc finger artificial transcription factor (ATF) restores UBE3A brain-wide and improves behavior phenotypes in an adult Angelman syndrome mouse. AAV-ATF treatment is specific and well tolerated, supporting the use of ATFs for the treatment of Angelman syndrome and other genetic disorders.
Introduction
Angelman syndrome (AS) is a rare (1 in 15,000 births) neurogenetic disorder caused by the loss of ubiquitin ligase E3A (UBE3A) gene expression in the brain.1,2 Loss of UBE3A expression leads to severe developmental delays, deficiencies in expressive communication, deficits in movement and coordination, recurrent uncontrollable seizures, sleep disturbances, and other characteristic behaviors. The UBE3A gene is paternally imprinted during neuronal maturation, and the clinical features of AS are primarily due to the loss of maternally expressed UBE3A in the brain. A healthy copy of paternal UBE3A is present in the brain but is silenced by an antisense transcript (UBE3A-ATS). UBE3A-ATS and other transcripts are processed from SNHG14, a long non-coding RNA (lncRNA).
Several different approaches have been taken to reinstate Ube3a expression in AS. For example, gene therapy has been used to deliver Ube3a isoforms to the brain of AS mice and rats by intrathecal or intrahippocampal injection of Ube3a-AAV,3,4,5 but behavioral rescue was limited to an improvement in associative learning tested with contextual and cued tone-shock fear conditioning, Morris water maze, and nest building. More recently, mouse Ube3a isoform 3 was delivered via hematopoietic stem cells to an immunocompromised mouse model of AS, lacking the interleukin-2γ (IL-2γ) chain.6 This approach rescued several behavioral and neurophysiological phenotypes in both neonatal and adult animals, including motor coordination, gait coordination, and novel object recognition. The treatment also prevented elevated delta power on electroencephalogram, an established known AS biomarker.7,8
The caveat of gene therapy approaches is a potential overexpression of UBE3A, which in turn can lead to another devastating neurologic disorder, Dup15q syndrome, caused by duplication of the UBE3A locus.9 In fact, overexpression of Ube3a isoform 2 in transgenic mice caused neurodevelopmental dysfunction and a variety of behavioral and neurophysiological deficits.10 Alternative approaches focus on the paternal allele and activation of its fully functional but silenced copy of UBE3A. Antisense oligonucleotides (ASOs) have demonstrated efficacy in the downregulation of the Ube3a antisense transcript (Ube3a-ATS) that is responsible for Ube3a silencing.11 In 2020, ASOs entered clinical trials for the treatment of AS (ClinicalTrials.gov identifiers: NCT04259281, NCT04428281, NCT05127226). This demonstrates that targeting UBE3A-ATS holds potential for therapeutic intervention in AS. A drawback to ASO therapy is the comparatively short half-life of this molecule; therefore, repeated intrathecal injections over the lifetime of the individual are required.
More recently, two independent studies used adeno-associated virus (AAV) delivery of CRISPR-Cas9 to unsilence the paternal Ube3a allele by altering genomic DNA.12,13 In the first study, CRISPR-Cas9 targeting of the genome between Snord115 and Ube3a 3′ UTR created indels in the genome that interfered with the extension of the Ube3a-ATS and resulted in lasting Ube3a paternal allele expression.13 In a second study, CRISPR-Cas9 was used to target the repetitive Snord115 cluster located upstream of the Ube3a-ATS, which also unsilenced paternal Ube3a expression.12 In addition to upregulating the expression of the paternal Ube3a allele, additional undesired changes to the genome were observed at the target and off-target sites. A substantial caveat in this approach is that these changes are permanent, carrying the risk of unexpected outcomes. Undesired consequences include indels, large deletions,14 translocations,15,16,17,18 chromothripsis,19 integration of vector sequences into the genome,20 and other chromosomal abnormalities.21
We have taken a different approach for downregulating Ube3a-ATS, thereby unsilencing the paternal allele and restoring endogenous Ube3a expression. Zinc finger proteins (ZFPs) are prevalent transcription factors in eukaryotic cells and often contain a Kruppel-associated box (KRAB) domain that facilitates gene-specific repression. ZFPs can be programmed to target specific sequences in the genome. Appending a KRAB domain derived from zinc finger families turns the engineered ZFPs into gene-specific transcriptional repressors.22,23,24,25 These artificial transcription factors (ATFs) are engineered versions of human proteins, are well tolerated by human cells, and have entered clinical trials for HIV-1 and β-thalassemia (e.g., ClinicalTrials.gov identifiers: NCT02388594, NCT03432364). We have previously shown that subcutaneous injection of the artificial transcription factor ATF-S1K could upregulate Ube3a expression in a mouse model of AS.26 Limitations to this S1K approach include extensive purification, protein degradation, and the requirement of repeated injections/treatments. Thus, we desired to evaluate the therapeutic potential of a delivery approach that would allow a more robust and brain-wide expression of the ATF.
AAV9 capsids have been modified to increase neuronal tropism, and the evolved capsid AAV.PHP.eB provides a more efficient transfer through the blood-brain barrier in the brain of mice.27 Gene therapy using similar ATFs (ZFP-KRAB fusions) delivered with a single injection of AAV.PHP reduced tau expression in an Alzheimer’s disease animal model.25 The authors demonstrated long-term and highly specific downregulation of tau with a single injection of AAV carrying the ATF.
In this study, we used AAV-PHP.eB to deliver more efficiently ATF-S1K to the brain of an adult AS mouse model and downregulate the Ube3a antisense transcript. A single AAV-S1K injection into the tail vein was sufficient for brain-wide delivery and restoration of endogenous UBE3A in the brain. ATF-S1K was highly specific to its endogenous target and was not accompanied by global transcriptomic changes. In addition, AAV-S1K was well tolerated with no detectable immune response. Overall, only a few genes were differentially expressed between wild-type (WT) and AS mice, suggesting that the underlying cause for the disease phenotype was not based on the transcriptome but might be better reflected in differences in the proteome, protein modifications, or metabolome.4,28 To this end, we demonstrate that restoration of UBE3A to 26% of WT level is sufficient to rescue several motor deficits in young adult mice in a mouse model of AS.
Results
Engineering of artificial transcription factors that specifically target and downregulate Snurf/Snrpn expression in mouse and human cells
In the brain, the paternal UBE3A allele is silenced by the lncRNA SNHG14, which originates from the SNURF/SNRPN locus (Figure 1A) and is subject to extensive alternative splicing and promoter usage. LncRNA SNHG14 is processed into several other poly(A) RNAs including two small nucleolar RNAs, C/D box clusters (SNORD116 and SNORD115), and UBE3A-ATS, which is responsible for the silencing of the paternal UBE3A allele (Figure 1A). The human and mouse SNURF/SNRPN locus have similar architectures. The SNURF (SNRPN upstream reading frame) gene shares a promoter and exons with SNRPN (small nuclear ribonucleoprotein polypeptide N). In addition, SNRPN transcripts with upstream exons have been identified (Figures 1B and S1A). In this study, we used the AS mouse model29 that carries a transgene insertion in coding exon 2 of the maternal Ube3a gene (Ube3amat−/pat+). To reach a better transcript-level understanding of this complex locus, we performed strand-specific RNA sequencing (RNA-seq) analysis of whole-brain tissue from young adult (∼11 weeks old) mice. Data from two WT C57BL/6J mice (one male and one female) were analyzed. Isoform-specific analysis shows that the lncRNA Snhg14 originates from the Snurf/Snrpn promoter (99.6%) and not from Snrpn upstream exons. We have previously engineered an artificial transcription factor ATF-S1K26 that specifically binds to the mouse Snurf/Snrpn transcription start site (TSS) to downregulate transcription in a target-specific manner (Figures 1B and S1B).26 The mouse ATF-S1K is based on a DNA-binding ZFP that carries an SV40 nuclear localization sequence at the N-terminus and the KRAB repressor domain at the C- terminus (Figures 1A and 1B).
Figure 1.

Transcriptional reprogramming of Angelman syndrome locus in human and mouse
(A) Diagram depicting neuron-specific imprinting of Angelman syndrome (AS) locus. Genomic architecture is comparable for humans and mice. Ube3a expression is lacking from the maternal allele, while the healthy paternal allele is silenced by a Ube3a antisense transcript (Ube3a-ATS), which is processed from the long non-coding RNA (lncRNA) Snhg14. Snhg14 originates from the Snurf/Snrpn promoter and is processed into several transcripts including Snord116 and Snord115 gene clusters. (B) Artificial transcription factor (ATF) S1K binds to the mouse Snurf/Snrpn promoter. The ATF consists of a nuclear localization signal (NLS), a zinc finger protein (ZFP) programmed to bind its genomic target site, and the KRAB repressor domain. Diagram of mouse Snurf/Snrpn gene isoforms and Snrpn isoform including upstream exons (Uexons) (adapted from UCSC genome browser mm10 mouse genome assembly). Normalized expression levels (transcripts per million [TPM]) of individual Snurf/Snrpn transcripts were determined by strand-specific mRNA-seq of brain tissue from WT mice. (C) Downregulation of Snurf/Snrpn expression was evaluated by RT-qPCR after transient transfection with ATF expressing plasmids (n = 3, mean ± SEM, one-way ANOVA with Dunnett’s multiple comparisons test, ∗∗p < 0.01, ∗∗∗p < 0.001). Relative expression has been normalized to GAPDH. Neuro-2a cells were transfected with a plasmid expressing the mouse targeting ATF (S1K), while LNCaP cells were transfected with the human targeting hATF-555. A plasmid expressing a non-targeting SR6K and a GFP-expressing plasmid were used as controls. (D) AS iPSC-derived neurons were transduced with lentivirus hATF-555. RT-qPCR of treated neurons was performed after 10 weeks of neuronal differentiation from NSCs to assess downregulation of SNORD116, SNORD115, and UBE3A-ATS (n = 10, mean ± SEM, one-way ANOVA with Tukey’s multiple comparisons test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). As a result, UBE3A expression was upregulated from the paternal allele (one-way ANOVA with Tukey’s multiple comparisons test, ∗∗p < 0.01). Relative expression (mRNA) has been normalized to GAPDH. hATF-555-treated neurons were compared with the control treated only with protamine sulfate (PS).
To evaluate that this approach is translatable to the human locus, we designed 63 ZFP sequences to a ∼1-kb region around the SNURF/SNRPN TSS in the human genome. We selected and screened 11 of the 63 designed human ZFPs that were predicted to bind closest to the SNURF/SNRPN TSS (Figure S1C). All ZFPs were fused to the nuclear localization signal (NLS) and the KRAB repressor domain to shape human ATFs (hATFs). hATFs were screened by transient transfection of LNCaP cells with plasmids expressing individual hATFs. As a control, cells were transfected with a GFP-expressing plasmid or with a plasmid encoding the scrambled ATF-SR6K that has no predicted target sites in the mouse or human genomes. Three days after transfection, SNURF/SNRPN expression levels were measured by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and normalized to GAPDH expression. The top candidate hATF-555 reduced SNURF expression by 87% in LNCaP cells, which is comparable with the 81% reduction achieved by transient transfection with S1K-expressing plasmid in Neuro-2a mouse neuroblastoma cells (Figure 1C). hATFs 567, 604, and 667 reduced SNURF/SNRPN transcription by 46%–55% (Figure S1). All other hATFs had no detectable effect on SNURF/SNRPN expression. The non-targeting control SR6K did not reduce SNURF/SNRPN transcription and was indistinguishable from GFP-transfected control cells.
In recent years, programmable CRISPR-Cas9 has gained significant interest as a therapeutic intervention.30,31,32 We, therefore, wanted to determine how the lead mouse and human ATFs (ATF-S1K and hATF-555, respectively) compared with CRISPR-mediated repression. We used KRAB-dCas9, a fusion of the catalytically dead Cas9 (dCas9) DNA targeting module with the KRAB repressor domain, in combination with guide RNAs (gRNAs) targeting the mouse Snurf/Snrpn TSS and promoter. Plasmids expressing KRAB-dCas9 and gRNAs were cotransfected into mouse or human cell lines (Neuro-2a and LNCaP, respectively), and reduction of Snurf/Snrpn expression was determined by RT-qPCR (Figure S1). As expected, gRNA S1 that localizes to the TSS (the same as ATF-S1K) showed the highest repressive capacity with a 90% reduction of mouse Snurf/Snrpn expression. dCas9 without effector domain was also able to repress Snurf/Snrpn transcription. Similarly, KRAB-dCas9 repressed human SNURF expression by 88% with a pool of gRNAs targeting the TSS. Taken together, ATFs demonstrated efficient downregulation of SNURF/SNRPN expression that is comparable with targeting with CRISPR-KRAB-dCas9 (Figures S1B and S1C). However, ZFP-based ATFs offer advantages for therapeutic delivery because of their small size (S1K ∼0.8 kb) compared with KRAB-dCas9 (∼4.7 kb), which creates limitations for packaging and delivery via AAV.33,34,35
Human artificial transcription factor upregulates UBE3A expression from the paternally silenced allele in neurons
We then sought to evaluate the functionality of the top candidate hATF-555 in neurons and in cell models of AS. We therefore used the patient-derived AS induced pluripotent stem cell (iPSC) line (ASdel1-0, AGI-0) that carries a large deletion (303 Mb) of chromosome 15q11-q13 on the maternal allele.36 UBE3A expression occurs only from the paternal allele in AGI-0 iPSCs. Imprinting of UBE3A is established during neuronal maturation, leading to expression of the lncRNA SNHG14 and downregulation of UBE3A expression in mature neurons. We confirmed differentiation of the AGI-0 iPSCs into neurons by the presence of NeuN, synaptobrevin, and MAP2 markers and a low abundance of glial fibrillary acidic protein positive (GFAP+) cells (Figure S2A). RT-qPCR assays were used to measure the increase of UBE3A-ATS and the reduction of UBE3A in iPSC-derived neurons 10 weeks following neuronal maturation from neuronal stem cells (Figure S2B). After neuronal differentiation, we observed an 11-fold increase in UBE3A-ATS (two-tailed unpaired t test, p < 0.001) accompanied by a 1.3-fold decrease in UBE3A expression (two-tailed unpaired t test, p = 0.02). Lentiviral transduction with hATF-555 successfully downregulated transcripts processed from the lncRNA SNHG14 (Figure 1D). UBE3A-ATS expression was downregulated 1.25-fold compared with control cells treated with protamine sulfate (transduction control; two-tailed unpaired t test, p = 0.03) while the expression of SNORD116 and SNORD115 was reduced by 1.7- and 1.8-fold, respectively (two-tailed unpaired t test, p < 0.001). Most importantly, UBE3A expression was partially restored from the paternal allele with a 1.33-fold increase (two-tailed unpaired t test, p < 0.001, Figure 1D).
A boundary element located between the SNORD116 and SNORD115 clusters is required to maintain neuron-specific expression of lncRNA SNHG14 transcripts beyond the boundary. As a result, SNORD115 and UBE3A-ATS are only expressed in mature neurons. A 4-kb deletion encompassing this boundary element allows transcription beyond the boundary and leads to constitutive expression of SNORD115 and UBE3A-ATS even in non-neurons, but has no effect on UBE3A imprinting.37 However, a 303-kb deletion spanning from SNRPN intron 1 to SNORD115-47 leads to complete repression of paternal UBE3A in addition to constitutive UBE3A-ATS expression in iPSCs with this deletion. The iPSC line AS ΔS-115 was created by superimposing the 303-kb deletion onto the AS genotype.37 AS ΔS-115 iPSCs were differentiated into neural stem cells (NSCs) over a 31-day period and displayed distinct morphology of NSCs (Figure S3A). Transduction of AS ΔS-115 NSCs with hATF-555 partially rescued UBE3A expression relative to protamine sulfate controls (Figure S3B, two-tailed unpaired t test, p < 0.01).
In summary, we have demonstrated that the human targeting ATF (hATF-555) targets and downregulates lncRNA SNHG14 in neurons from AS-patient-derived cells, supporting the view that the ATF-S1K approach in mice is directly translatable to human cells.
Brain-wide delivery of ATF-S1K restores UBE3A protein in the brain of an AS mouse model after a single systemic administration of AAV-S1K
In our previous study, we delivered an injectable version of S1K protein to the mouse brain.26,38 Although we were able to detect upregulation of the UBE3A protein in the brain, this approach had several limitations. Injectable S1K protein treatment was readministered every 2–3 days and after subcutaneous injection, intact S1K protein was barely detectable in the brain.26 Although modest upregulation of Ube3a was observed, injectable S1K protein did not result in detectable downregulation of Snurf/Snrpn expression in the mouse brain (Figure S4). In this study, we sought to establish a more robust presence of S1K in the brain to investigate its potential to rescue molecular and phenotypic deficits in an AS mouse model. ATF-S1K was packaged with the AAV-PHP.eB capsid, which has improved its ability to pass through the blood-brain barrier and transduce neurons in the brain of mice after systemic delivery.27
For durable expression of ATF-S1K, we transferred the S1K coding gene into an AAV expression vector (Figure 2A) under the control of the constitutive CBA promoter and the WPRE3 element that has been shown to increase transgene expression from viral vectors.39 AAV packaging has limited capacity, but the small size of the ATF-S1K allowed us to add an mCherry cassette to monitor AAV delivery. AAV-S1K was delivered systemically by injecting 1 × 1012 vector genomes (vg) into the tail vein of young adult AS mice (6 weeks old). AAV-mCherry-injected AS and WT mice served as controls. We did not observe an inflammatory response or other adverse effects at the site of injection. Mouse brains were extracted 5 weeks after transduction, and expression of S1K was confirmed by RT-qPCR (Figures 2B and 2C). RT-qPCR assays determined that several transcripts originating from lncRNA Snhg14 were downregulated approximately 2-fold (Figures 2B and 2C). The Snurf transcript was reduced 2.4-fold (one-way ANOVA with Dunnett’s multiple comparison test, p < 0.001), and Ube3a-ATS was reduced 2.2-fold (one-way ANOVA with Dunnett’s multiple comparison test, p = 0.01). Inadvertently, Snord115 and Snord116 genes were also downregulated 3-fold (one-way ANOVA with Dunnett’s multiple comparison test, p = 0.02) and 1.9-fold (one-way ANOVA with Dunnett’s multiple comparison test, p = 0.04), respectively.
Figure 2.

Single AAV-S1K treatment restores UBE3A in the brain of AS mice
(A) Diagram of AAV-S1K and control AAV-mCherry flanked by AAV-ITRs. ATF-S1K and mCherry expression cassettes are under the control of their own promoters (CBA and pGK, respectively). Nuclear localization signal (NLS) and termination elements (WPRE3) are indicated. Timeline of AAV-S1K treatment with PhP.eB capsid is shown. (B) Diagram depicting the mode of action for ATF-S1K. (C) Expression of ATF-S1K and downregulation of Ube3a-ATS and other transcripts (Snurf, Snord116, Snord115) processed from the lncRNA Snhg14 were evaluated by RT-qPCR 5 weeks after a single AAV-S1K administration (n ≥ 4, mean ± SEM, one-way ANOVA with Dunnett’s multiple comparisons test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). AS mice treated with AAV-S1K were compared with control AS mice and wild-type (WT) mice, both treated with AAV-mCherry. (D) UBE3A protein in treated AS mice (AAV-S1K), AS mice, and WT mice was analyzed by western blot. Cytosolic and nuclear extracts from whole-brain tissue were analyzed. UBE3A protein was normalized to ACTIN signal and then quantified relative to WT UBE3A levels (n = 4, mean ± SEM, two-tailed unpaired t test). #Loading in this lane was at one-fifth the volume of that in other lanes.
Downregulation of Ube3a-ATS is expected to result in Ube3a expression from the silenced, but intact, paternal allele. We performed a Western blot analysis to determine UBE3A protein levels in whole-brain lysates (Figure 2D). After a single administration of AAV-S1K, UBE3A protein in AS animals was restored to 26% of WT animals. Brain-wide restoration of UBE3A was visualized in sagittal brain sections using diaminobenzidine (DAB) immunohistochemical analysis (Figure 3A). The number of UBE3A-expressing cells per selected region was counted in the brains of at least four mice and averaged per genotype and treatment. The average number of UBE3A-positive cells in brains of AAV-S1K-treated mice was comparable with that in WT controls (AAV-mCherry): 76% in the prefrontal cortex, 83% in the hippocampus, 80% in the cerebellum, and 45% in the striatum (Figure 3B). There was no further increase in UBE3A-positive cells when WT mice were treated with AAV-S1K (Figure 3B).
Figure 3.

Immunohistochemical analysis demonstrates brain-wide restoration of UBE3A in an AS mouse model
(A) UBE3A labeling of sagittal brain sections using diaminobenzidine (DAB). Representative immunohistochemical images are shown for WT (top), AS (middle), and AS mice 5 weeks after a single AAV-S1K treatment (bottom). Zoom-in regions for prefrontal cortex, striatum, hippocampus, and cerebellum are indicated by squares in the sagittal section. Scale bars represent 1,000 μm for sagittal sections, 50 μm for zoom-in on cortex, striatum, and cerebellum, and 100 μm for zoom-in on hippocampus. (B) The average number of UBE3A-expressing cells per selected region was counted per genotype and treatment. A significant increase in the average number of UBE3A-positive cells, similar to the WT level, was observed in AAV-S1K-treated AS mice (n = 4, mean ± SEM, one-way ANOVA with Tukey’s multiple comparisons test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
It is imperative for the treatment of AS that UBE3A upregulation occurs in mature neurons. Using immunofluorescence microscopy (Figures 4A and S5), we labeled sagittal brain slices for the neuronal marker NeuN and counted the average number of NeuN-positive (NeuN+) cells per region. WT and AS mice had comparable numbers of NeuN+ cells for treatment with AAV-S1K and control treatment with AAV-mCherry (one-way ANOVA with Dunnett’s multiple comparisons test, Figure 4B). Uptake of AAV in mature neurons was visualized by mCherry expression for treatment with AAV-S1K or control AAV-mCherry (Figure 2A). As expected, AAV delivery was observed brain-wide. We next measured UBE3A fluorescence intensities in NeuN+/mCherry+ cells. AAV-S1K treatment increased UBE3A fluorescence in the brains of AS animals compared with the control AS group (one-way ANOVA with Dunnett’s multiple comparisons test, p < 0.01) to an average of 58.5% relative to WT. We did notice a range of UBE3A intensities in the brains of different AAV-S1K-treated animals. We hypothesized that lower levels of AAV-S1K may result in low levels of UBE3A restoration. Correlation analysis of AAV delivery (presence of mCherry) and UBE3A signal demonstrates a positive correlation between mCherry and UBE3A signal (Pearson’s r = 0.86, p = 0.027, Figure 4C). This illustrates that UBE3A restoration is dependent on AAV-S1K dosage and suggests that a minimal threshold of S1K expression is critical for treatment efficiency.
Figure 4.

Brain-wide and dose-dependent restoration of UBE3A in mature neurons
(A) Immunostaining of sagittal brain sections (scale bar, 1,000 μm) including cortex isolated regions (scale bar, 100 μm) was performed 5 weeks after AAV-S1K treatment. Mice were evaluated at 11 weeks of age. Representatives are shown for WT (top row), AS mice (second row), and AAV-S1K-treated AS mice (bottom two rows). AS mice with low mCherry intensity are labeled AAV-S1K (lo), while AS mice with mid to high mCherry intensity are labeled AAV-S1K (hi). Brain tissue was labeled for mature neurons (NeuN+, red), ATF-S1K (mCherry+, violet) and UBE3A (green). Merged images are shown for the sagittal section and the cortex region (last column). Zoom-in images are shown in Figure S5. (B) Number of NeuN-positive cells is comparable across different genotypes (AS, WT) and treatments (AAV-mCherry, AAV-S1K). Restoration of UBE3A was quantified by UBE3A intensity analysis in NeuN+/mCherry+ neurons in the prefrontal cortex after AAV-S1K treatment in AS mice and control groups (n ≥ 4, mean ± SEM, one-way ANOVA with Dunnett’s multiple comparisons test, ∗∗∗p < 0.001 ∗∗p < 0.01). (C) Dose-dependent UBE3A increase was identified by correlation analysis of UBE3A and mCherry intensities in NeuN+/mCherry+ neurons (Pearson correlation, ∗p < 0.05).
Specificity and tolerability of AAV-S1K treatment in vivo
We have demonstrated on-target activity in vivo after a single administration of AAV-S1K that results in upregulation of UBE3A. To evaluate the specificity of ATF-S1K, we compared gene expression by strand-specific RNA-seq in brain tissue from AS mice either treated with AAV-S1K or with AAV-mCherry as the control. After differential gene expression analysis, we identified significant upregulation of S1K transcription (adjusted p < 0.05) (Figures 5A and S6A). We observed upregulation of the pseudogene Gm2436 (adjusted p < 0.05), but it was only detected in two out of four mice (Figure S6B). We did not detect any additional off-target impact on global gene expression, demonstrating that even prolonged AAV-S1K expression is highly specific to the intended target site. In tissues outside the central nervous system (CNS), Ube3a is biallelically expressed, and the paternal Ube3a-ATS transcript is not present. Although not directly examined in this study, we do not expect our treatment to affect UBE3A levels in peripheral tissues because we are specifically targeting the Ube3a-ATS, which is not present outside the brain.
Figure 5.

Specificity and tolerability of AAV-S1K treatment in vivo
(A) AAV-S1K treatment is highly specific as determined by strand-specific mRNA-seq. Volcano plot shows differential gene expression of AS mice 5 weeks after AAV-S1K treatment (n = 4) with AAV-mCherry-treated AS mice as controls (n = 3). p values were adjusted by Benjamini-Hochberg correction (padj). AAV-S1K-treated mice show no global transcriptomic changes. Red dots indicate genes that were significantly differentially expressed (padj < 0.05). Gray dots represent genes without significant difference. (B) Volcano plot shows genes differentially expressed between AS (n = 3) and WT (n = 3) mice. Red dots indicate genes that were significantly different (fold change >1.5, padj < 0.05). (C) Normalized expression levels (TPM) for immune-response genes were obtained from RNA-seq analysis. AS mice treated with AAV-S1K or control (AAV-mCherry) and WT mice treated with control (AAV-mCherry) were compared with untreated WT mice (n ≥ 2, mean is indicated, one-way ANOVA with Dunnett’s multiple comparisons test). (D) Normalized expression of Gfap and Iba-1 (TPM) evaluates neuroinflammatory response by the presence of astrocyte or microglia markers (GFAP or IBA1) (n ≥ 2, mean is indicated, Dunnett’s multiple comparisons test, ∗p = 0.02).
We then determined differential gene expression between AS and WT mice, whereby both groups were administered the control AAV-mCherry. We only observed significant differences for 11 genes (adjusted p < 0.05) (Figure 5B). A few of these differentially expressed genes (DEGs) showed expression levels closer to WT after AAV-S1K treatment of AS mice (Figure S6B). Although only a small set of DEGs was observed between WT and AS animals, the functional annotation tool DAVID found significant enrichment for brain development (p = 0.05; Nes, Nefh) and positive regulation of neural precursor cell proliferation (p = 0.007; Nes, Cdon). However, the adjusted p values did not show significance after Benjamini-Hochberg correction. The small transcriptomic changes suggest that the AS disease phenotype is not primarily due to gene expression differences but might be better reflected in changes in the proteome.4,28
A single AAV-S1K treatment of young adult AS mice resulted in a 2-fold downregulation of Ube3a-ATS sufficient to reinstate expression of Ube3a. Importantly, our treatment did not lead to a complete knockout of Ube3a-ATS and its spliced transcripts (e.g., Snord116, Figure 2C). The full prenatal loss of Snord116 RNAs that are normally spliced from the Ube3a-ATS transcript is associated with Prader-Willi syndrome (PWS).40 This raises the question of whether the amount of Snord116 knockdown achieved by AAV-S1K would lead to a molecular PWS phenotype. We therefore performed a more targeted analysis of PWS-related transcripts (Figure S6C), which have been identified from RNA-seq data comparing whole neonate brains of WT mice with a PWS mouse model.41 The PWS mouse model carries a paternal deletion of the Prader-Willi (PW) imprinting center encompassing Snrpn, Necdin, Mkrn3, and Magel2. Notable DEGs outside the PW region are the circadian clock regulator Per1, Plp1 and insulin growth factor Igf1. We then evaluated changes in gene expression of these PWS-related transcripts in AS mice treated with AAV-S1K. Snrpn and Snhg14 transcripts are only modestly downregulated in our treatment (1.4- and 1.8-fold change, respectively) when compared with the PWS phenotype (41- and 8-fold change, respectively) (Figure S6C). As a result, we did not observe major changes in any of the PWS-related transcripts. In summary, the level of downregulation of Ube3a-ATS transcripts was sufficient to restore Ube3a expression but did not lead to the dramatic expression changes associated with the PWS phenotype.
In addition, differential gene expression analysis did not detect the upregulation of genes related to the immune response after treatment with AAV-S1K. Upregulation of immune-related genes has been found in response to treatment with SadCas9 in mice and SpCas9 in large animals.34,42 This response was specific to Cas9 and was not observed with non-Cas9 AAV. All animals in our study were treated with AAV, either AAV-S1K or control AAV-mCherry. Using our RNA-seq data, we took a closer look at immune-response genes by comparing 11-week-old mice from our study with untreated WT mice of the same age. We compared the normalized expression of gene transcripts (transcripts per million) associated with cytokine response, and we did not detect expression changes for IL2, IL15, IL18, Ifn-γ, or Tnf-α between animals treated with AAV (AAV-S1K or AAV-mCherry) and untreated WT animals (one-way ANOVA with Dunnett’s multiple comparison test, Figure 5C). We then evaluated neuroinflammatory response by the presence of astrocyte or microglia markers (GFAP or IBA1, Figure 5D). There was no change in Iba1 expression, but we detected a modest increase in Gfap expression 5 weeks after AAV-S1K administration (one-way ANOVA with Dunnett’s multiple comparison test, p = 0.02).
In summary, we did not detect a cytokine response 5 weeks after a single administration of AAV-S1K, suggesting that the ATF-S1K treatment is not only specific but also well tolerated in vivo.
AAV-S1K improves reduced exploratory motor abilities in the AS mouse
Motor function is highly translational, utilizes species conserved circuitry, and consists of many nuanced components. The most robust and clear examples are exploration and velocity in a novel open field arena. As previously published by our group and others,6,43,44,45,46 AS mice had reduced activity across all three parameters during the open field assay—horizontal (Figures 6A and 6B), vertical (Figures 6C and 6D), and total activity (Figures 6E and 6F)—compared with WT mice treated with AAV-mCherry. AS mice exhibited a large main effect of genotype for horizontal, vertical, and total activity, exhibiting substantially fewer movements in the arena compared with WT (two-way repeated-measures ANOVA, p < 0.05). AS mice treated with AAV-mCherry (AS) differed significantly from WT during five of the six 5-min time bins across the 30-min assay for horizontal activity (Figure 6A; Dunnett’s multiple comparisons test: 6–10 min, p < 0.0048; 11–15 min, p < 0.0183; 16–20 min, p < 0.0062; 21–25 min, p < 0.028; and 26–30 min, p < 0.009) and vertical activity (Figure 6C; Dunnett’s multiple comparisons test: 0–5 min, p < 0.019; 6–10 min, p < 0.0055; 11–15 min, p < 0.0396; 16–20 min, p < 0.0012; and 21–25 min, p < 0.0414). In corroboration, the total distance also illustrated robust differences between AS and WT in three of the six 5-min time bins across the 30-min activity assay (Figure 6E; Dunnett’s multiple comparisons test: 6–10 min, p < 0.047; 16–20 min, p < 0.0159; 26–30 min, p < 0.0259).
Figure 6.

AAV-S1K improves reduced exploratory motor abilities in the AS mouse
Assessment of motor translational phenotypes in AAV-S1K-treated AS mice (n = 12, red, Ube3amat−/pat+) compared with control groups that received control AAV (AAV-mCherry), AS (n = 14, blue, Ube3amat−/pat+), and WT (n = 13, gray/black, Ube3amat+/pat+). Two and three weeks after a single AAV-S1K treatment, treated and control mice were subjected to (A–G) open field locomotion and (H) treadmill walking, respectively. Deficits in open field activity were improved in AAV-S1K-treated AS mice for (A and B) horizontal activity, (C and D) vertical activity, and (E and F) total distance. Data are presented as mean ± SEM. Bar graphs represent the summed activity for the 15- to 30-min time window. p values in (B), (D), and (F) were determined using one-way ANOVA with Dunnett’s multiple comparisons tests (∗p < 0.05). (G) In the novel open field arena, AS mice showed reduced velocity compared with WT when exploring for the 30-min time window (Dunnett’s multiple comparisons tests, ∗∗p < 0.01). The AAV-S1K-treated AS group showed a trend of increased velocity compared with the AS control group. (H) Latency to fall from the rotarod was not significantly improved in AAV-S1K-treated AS mice compared with the AS control group. (I) Box plot evaluates differences in DigiGait analysis. The AS control group exhibited lower stride frequency compared with WT. AAV-S1K treatment improved motor deficits of the AS group, most notably for hindlimb (one-way ANOVA with Dunnett’s multiple comparisons tests, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Most importantly, activity summed over the 15- to 30-min session demonstrated improvement of motor deficits in AS mice treated with AAV-S1K compared with the AS control group. AS mice also exhibited reduced horizontal locomotion when compared with WT (Figure 6B; p = 0.016) while AS mice treated with AAV-S1K exhibited improvement on this metric when compared with the control AS group (Figure 6B; Dunnett’s multiple comparisons test, p = 0.16). AS mice exhibited less vertical rearing in the arena compared with WT (Dunnett’s multiple comparisons test, p = 0.031), whereas the AS group treated with AAV-S1K demonstrated typical vertical rearing (Figure 6D; Dunnett’s multiple comparisons test, p = 0.051). Summed total activity illustrated robust rescue of motor deficit in AS mice treated with AAV-S1K (Dunnett’s multiple comparisons test, Figure 6F; p = 0.037).
In addition to the reduced activity in a novel open field exploration task, AS mice showed reduced velocity while traversing the open field during the 30-min session compared with WT (Figure 6G; F(2,36) = 4.588, p = 0.0168), a consistent finding in our laboratory and in children with AS.45,47,48 We observed a trend of increased velocity in the AS group treated with AAV-S1K when compared with control AS mice (Figure 6G; Dunnett’s multiple comparisons test, p = 0.129). Taken together, these data highlight improvements in a variety of components of motor function in the AAV-S1K-treated AS group.
Nuanced motoric behaviors, such as gait, balance, posture, stride, and motor coordination, can be measured using the standard rotarod and the innovative automated contrast tracking software alongside treadmill systems, such as DigiGait. On the rotarod coordination and motor learning assay, as expected, genotype affected performance as measured by latency to fall from the accelerating rod (Figure 6H, two-way repeated-measures ANOVA; F(2, 35) = 3.321, p < 0.0478). AS mice had substantially worse performance than WT (Dunnett’s multiple comparisons test; day 1, p = 0.043), whereas on average the AAV-S1K-treated AS group showed slight improvement (Dunnett’s multiple comparisons test; day 1, p = 0.521). Motor learning deficits were not apparent in either the AS control group (Dunnett’s multiple comparisons test; day 3, p = 0.131) or the AS group treated with AAV-S1K (Dunnett’s multiple comparisons test; day 3, p = 0.510).
We also investigated the more nuanced spatial and temporal gait indices. Spatial metrics, including stride length and stride frequency, showed less frequent but longer strides in AS mice, similar to deficits previously observed preclinically and clinically.45 The AS group exhibited fewer strides in frequency (Figure 6I) and longer strides in length (Figure S7A). For stride frequency, AS and WT groups exhibited significant differences in hindlimb and forelimb (Figure 6I, Dunnett’s multiple comparisons test; hindlimb p = 0.0012 and forelimb p = 0.0002). AAV-S1K treatment improved motor deficits in the AS group, most notably for hindlimb (Figure 6I, Dunnett’s multiple comparisons test; hindlimb p = 0.046 and forelimb p = 0.083). For stride length, we observed significant differences in hindlimb and forelimb (Figure S7A, Dunnett’s multiple comparisons test; hindlimb p = 0.014 and forelimb p = 0.033) between AS and WT mice. We did not observe dramatic improvement by AAV-S1K (Figure S7A). Temporal metrics, including stride duration and swing duration, showed differences between AS and WT mice (Figures S7B and S7C), similar to the deficits previously observed in AS mice.45 AAV-S1K treatment did not rescue longer stride and swing duration observed in AS mice (Figures S7B and S7C).
In summary, rescue of AS phenotypes in AAV-S1K-treated adult AS mice was observed in multiple assays of motor deficits 2–4 weeks after a single treatment with AAV-S1K. This was in contrast to the AS control group, which illustrated typical AS deficits of reduced motor activity and impaired motor coordination.
Discussion
While lack of UBE3A in the brain causes AS, too much UBE3A causes 15q11-q13 duplication syndrome, another devastating neurologic disorder highly associated with autism spectrum disorders.9 In fact, several mouse models of overexpression of Ube3a in transgenic mice result in neurodevelopmental and behavioral deficits.10,49,50 Therefore, the level of UBE3A restoration is critical, and increasing the expression from the healthy but silenced paternal allele has become an attractive target for the treatment of AS. In our study, a single tail vein injection with AAV-S1K specifically targeted and repressed transcription of the long non-coding Ube3a-ATS, responsible for paternal UBE3A silencing. Consequently, UBE3A protein was restored to 26% of normal levels in WT brains. In our study, 26% of the whole UBE3A protein level in the brain was sufficient to rescue the robust exploratory motor deficit in the open field assay. Another study reported that gene editing of the Ube3a-ATS encoding locus with CRISPR-SaCas9 in neonates was able to restore UBE3A to levels similar to those in our study (27% of normal), which led to the behavioral rescue on the rotarod at 2 and 28 weeks,12 a mild increase in the number of marbles buried (from ∼4 to ∼6), and a 1-point increase in the nest rank score.13 Yet, in contrast to our report, the standardized exploratory motor task of open field locomotion was unimproved. This is unusual given the substantial motor component involved in both the marble-burying and nest-building assays.51 In our study, AAV-S1K not only improved exploratory locomotion in the open field but the treatment also improved stride frequency, a spatial component of the nuanced motor index of gait, which has been shown to have high clinical relevance as an outcome measure for AS. Children with AS have exhibited gait deficiencies measured by various equipment devices, including pressure-sensitive mats48 to wearable bracelets with activity-monitoring technology, such as Fitbits and Actimyos, as in the KIK-AS clinical trial (ClinicalTrials.gov identifier: NCT04259281). This report is only the second study to show improvements in gait metrics. Given its (gait’s) translatability, conserved neural circuitry, and reasonable sensitivity for observable deficits and clear improvements, we are optimistic that this improved outcome measure will inform apropos outcomes for IND-enabling studies and trial design going forward.
One of the main questions that remain is the optimal age for AS interventions. Using a conditional AS mouse model, reinstatement of UBE3A at different ages suggested a critical window for intervention.46 Reinstatement of UBE3A in adolescent mice (6 weeks) rescued motor deficits, while reinstatement in early development was able to rescue additional phenotypes such as anxiety-like, repetitive behavior, and seizures. In contrast, hippocampal synaptic plasticity measured by long-term potentiation was restored at any age. Therefore, many recent studies of therapeutic treatment of AS have focused on the treatment of neonates while still observing partial behavioral rescue.3,4,6,11,12,13 Although earlier intervention may have some bona fide advantages, this dogma has been challenged by a recent study6 that treated adult immunodeficient AS mice (Ube3amat−/pat+ IL2rg−/y) with a stem cell gene therapy that is based on Ube3a expressing hematopoietic stem and progenitor cells (HSCs). HSC engraftment at 4–5 weeks of age led to improvement in motor and cognitive-behavioral assays as well as normalized delta power measured by electroencephalogram. AAV and ASO behavioral and weight rescues in AS mice were also observed in adult AS mice. AS is most commonly diagnosed in early childhood between 1 and 4 years of age, underlining the importance of postnatal therapeutic intervention. Our study aimed to treat adolescent mice (6 weeks) and evaluated behavioral rescue in young adult mice (8–11 weeks). AAV-S1K treatment rescued the most profound motor deficits. However, we were unable to rescue finer motor skill phenotypes such as coordination, balance, and gait nor evaluated the rescue of cognitive phenotypes. It has been reported that the genetic background affects the severity of AS phenotypes,8 and it would therefore be of interest how AAV-S1K treatment performs in cognitive assays when using the immunodeficient AS model (Ube3amat−/pat+ IL2rg−/y).
In recent years CRISPR-Cas9 has revolutionized the field of gene editing, with AAV-CRISPR therapy holding great promise to therapeutically correct human genetic diseases. However, Cas9-specific immune responses have been a concern and present a significant hurdle for CRISPR-Cas9 therapy. Pre-existing immunity has been reported in human donors with antibodies to SaCas9 and SpCas9 in 78% and 58% of tested individuals, respectively.52,53 In addition, CRISPR-Cas9 gene-editing components themselves elicit immune, stress, and apoptotic responses in human CD34+ HSCs, with the delivery of Cas9 mRNA causing the largest response.54 Interestingly, AAV6 alone did not cause a detectable transcriptional response in these cells. More recently, AAV-CRISPR treatment of a canine disease model for Duchenne muscular dystrophy triggered humoral and cytotoxic T lymphocyte responses that were specific to the bacterial-derived Cas9.42 Cas9-specific immune response was not prevented by transient immunosuppression with prednisolone. Importantly, this response was not observed with other AAV-delivered therapeutics.
AAV variants such as AAV-PHP.B have great transduction efficiency throughout the CNS after intravenous delivery in adult mice.27 However, intravenously delivered AAV-PHP.B does not demonstrate the same capabilities in non-human primates (NHPs).55,56 In this study, we used an enhanced AAV-PHP.B variant (AAV-PHP.eB) that shows further improvements in CNS transduction efficiency in mice27 but has limited penetrance in NHPs.57 Although translating AAV variants from mice to NHPs has been challenging, there has been considerable development in this area. New capsids have been engineered for more effective and non-invasive gene delivery to the CNS of NHPs.58,59,60 The emergence of new AAV variants with the ability to cross the blood-brain barrier and with neuronal specificity in NHPs enables unprecedented translatability of AAV-based therapeutic approaches.
Gene editing introduces double-strand breaks and bears the risk of undesired permanent events at the target site such as rearrangements and AAV vector integration.12,20 Our approach has the advantage of downregulating Ube3a-ATS expression without altering the genetic content and avoids issues associated with traditional gene editing. Engineered ATFs are based on ZFPs that are prevalent in human cells. We did not detect upregulation of gene expression consistent with inflammatory response, such as interleukins (IL2, IL15, IL18), tumor necrosis factor α, or interferon-γ 5 weeks after administration of AAV-ATF (S1K). Another study that targets the tau gene in an Alzheimer’s disease mouse model reported a transient increase of microglia and astrocyte marker transcripts Gfap and Iba-1 (AIF1) at the highest AAV doses 5 weeks after hippocampal injection with a zinc finger transcription factor (ZFP-TF).25 Although glial marker levels increased in the first 4 weeks after injection, they slowly decreased between 4 and 12 weeks and subsided after prolonged expression of ZFP for 11 months. Intrastriatal treatment with AAV to deliver a ZFP-TF targeting the mutant Huntingtin allele also did not increase GFAP and IBA1 markers,24 further supporting the view that ATF treatments are well tolerated. In our study, we did not detect activation of Iba-1 mRNA and only observed a modest increase in Gfap expression 5 weeks after a single tail vein injection of AAV-S1K or control AAV (AAV-mCherry) when compared with untreated mice. This probably represents a transient increase previously reported by other laboratories.25 Taken together, our data suggest good tolerability of AAV-S1K treatment.
A single AAV-S1K injection was able to downregulate the lncRNA Snhg14 and its processed transcripts. Ube3a-ATS was downregulated 2-fold, which led to upregulation of the functional paternal copy of Ube3a. However, silencing of the entire Ube3a-ATS also reduced Snord116 and Snord115 clusters of small RNAs, which are spliced from the large UBE3A-ATS transcript. In another UBE3A restoration approach, CRISPR-Cas9 was targeted to the repetitive Snord115 gene cluster, and transcription of the lncRNA was disrupted.12 The authors did not detect changes in expression or splicing patterns and suggested that downregulation or mutation of certain Snord115 genes does not compromise Snord115 function(s). In our study, AAV-S1K administration also did not result in global changes in gene expression. Meanwhile, clinically, the loss of SNORD116 expression causes PWS, which presents as a failure to thrive at birth but in adolescence changes to a hyperphagic disorder in which food satiation does not occur and affected individuals become highly obese with severe cardiovascular disease risk.61 Lack of SNORD116 in the germline results in some PWS phenotypes in a mouse model.62 However, in another study, the onset of Snord116 loss in young adult mice (8 weeks) led to reduced food intake and increased fat mass, which is the opposite of the PWS phenotype.62,63 It is, therefore, unclear which (if any) phenotype might be associated with partial downregulation of SNORD116 at the age of diagnosis and intervention. Our treatment resulted in a knockdown of the Ube3a-ATS sufficient to reinstate expression of Ube3a. This was not a complete knockdown as observed in PWS. RNA-seq analysis after AAV-S1K treatment of AS mice did not lead to dramatic expression changes in PWS-related transcripts. Unfortunately, there are no good animal models to determine whether there will be phenotypic consequences for this amount of Ube3a-ATS knockdown. We did not observe any adverse phenotypic consequences to the treatment in our study, but such adverse phenotypes, especially phenotypes of PWS associated with full prenatal loss of Snord116 RNAs, will need to be carefully monitored in subsequent experiments.
In summary, we demonstrate that a single injection of an AAV delivering ATF-S1K (AAV-S1K) into the tail vein rescues molecular and behavioral phenotypes in young adult AS mice. The AAV-S1K treatment is highly specific to the target site, with no detectable inflammatory response 5 weeks after AAV-S1K administration. Partial reduction of Snord116 expression at the time of treatment did not result in transcriptomic changes associated with PWS. In conclusion, the specificity and tolerability of a single AAV-S1K treatment in mice support the use of ATFs for the treatment of AS and other genetic disorders.
Materials and methods
Plasmid construction
The plasmid-expressing mouse ATF-S1K for cell assays (pGK-S1K) was previously described.26 ZF proteins targeting human DNA were designed for a 1.2-kb region that included the 5′ UTR of SNRPN and flanking sequences (hg19; chr15: 25,199,515–25,200,747). 44 designs were provided by MilliporeSigma and 19 designs by the Segal lab using extended modular assembly with a B-score cutoff of 7.23 Eleven designs were selected for testing based on proximity to the SNRPN transcriptional start site and minimal off-target sites differing by 1–3 mismatches. Control ATF-SR6K was composed of a 6-zinc-finger protein in which all DNA-contacting residues of helix positions −1, 3, and 6 were mutated to serine and therefore are not expected to bind DNA (LEASEKPYMCAECGKSFSSRASLESHQRTHTGEKPYKCPECGKSFSSKNSLTSHQRTHTGEKPYKCPECGKSFSSKKSLTSHQRTHTGEKPYKCPECGKSFSSCRSLASHQRTHTGEKPYKCPECGKSFSSSGSLVSHQRTHTGEKPYKCPECGKSFSSRASLRSHQRTHTG).
ZF hATFs were synthesized as gBlocks (IDT) with human codon optimization and cloned into an HIV-1-derived lentiviral backbone containing the HA-NLS-ZF-KRAB-WPRE (pCCLc-MNDU3-X2-WPRE) via NheI and AgeI restriction sites. Clones were subsequently confirmed by Sanger sequencing (Quintara Biosciences, South San Francisco, CA). Lentivirus was generated by complexing 25 μg of transgene vector, 25 μg of delta8.9, and 5 μg of vesicular stomatitis virus G with 145 μg of polyethylinimine in 3 mL of serum-free high-glucose DMEM (Thermo Fisher, Waltham, MA) for 20 min prior to application to 2.5 × 106 Lenti-X 293 cells (Takara Bio, Kusatsu, Japan). Twenty-four hours after transfection, cells were switched to serum-free Ultraculture medium (Lonza, Basel, Switzerland) for 48 h. Viral harvest was performed by transferring the supernatant of transfected Lenti-X 293 cells into 50-mL conicals, which were spun at 500 rpm for 5 min to pellet cellular debris, and the virus was isolated in a 100-kDa Centricon (MilliporeSigma, Burlington, MA). Concentrated lentivirus was titrated with the ABM Titer Kit according to the manufacturer’s protocols. AGI-0 and AS(Del) cells were transduced with 2.5 μL/10,000 cells (1 × 109 vg/mL) and 1 μL/mL protamine sulfate in respective complete cell media. All cells were harvested 4 days following transduction for molecular analysis.
Plasmids expressing KRAB-dCas9 and the dCas9 cloning vector without any effector domain have previously been described33 and are available through Addgene (KRAB-dCas9 catalog #112195, dCas9 #100091). gRNA Cloning Vector was a gift from George Church (Addgene plasmid #41824).64 Nineteen-basepair gRNA target sequences were designed using the online tool CHOPCHOP v2.65 Each gRNA sequence was cloned as G-N19 into the AflII-linearized plasmid using Gibson cloning. The target site sequences used to create target-specific gRNA vectors are listed in Table S1.
AAV plasmid construction and AAV packaging
Cloning of AAV-S1K was performed by the Vector Core at the Gene Therapy Center at UC Davis using the vector backbone from pTR12.1 MCS,3 a generous gift from Dr. Kevin Nash, USF. AAV-S1K was created in a two-step process. First, a gene fragment (synthesized by IDT) containing the Kozak consensus sequence, the cell-penetrating peptide TatK, HA tag, and an NLS was inserted into BamHI/NarI-digested vector pTR12.1 MCS. Second, a larger gene fragment (synthesized by GenScript) was inserted to introduce a modified and truncated post-translational regulatory signal CW3SL that was derived from WPRE and poly(A),39 and the mCherry cassette driven by its own pGK promoter and a shortened WPRE element (WPRE3) with a poly(A) signal. The final construct AAV-S1K consists of the following elements: AAV-CBA:TatK-HA-NLS-S1ZF-KRAB-CW3SL; PGK-mCherry-WPRE3-poly(A).
The AAV-mCherry control was created at the Molecular Construct and Packaging Core at UC Davis by exchanging GFP from pAAV.CMV.PI.EGFP.WPRE.bGH (Addgene #105530) with mCherry. AAV-S1K and AAV-mCherry were then packaged by the Molecular Construct and Packaging Core at UC Davis with envelope plasmid AAV-PHP.eB (Addgene, #103005).27 AAV titers were subsequently determined by measuring the number of DNaseI-resistant vector genomes using qPCR and comparing it with the used genome plasmid.66
Cell culture and transfection
Neuro-2a (ATCC #XXCCL-131) cells were maintained in high-glucose DMEM (Life Technologies) supplemented with 10% FBS and 1× penicillin/streptomycin at 37°C under 5% CO2. LNCaP cells (ATCC #CRL-1740) were grown in RPMI (Life Technologies) supplemented with 10% FBS and 1× penicillin/streptomycin at 37°C under 5% CO2. Transfections were carried out in 12-well plates using Lipofectamine 3000 (Qiagen, Hilden, Germany). A total of 1.25 μg of plasmid DNA per well was used for transfection, and a tenth of the amount (125 ng) was pBabe-Puro to allow for enrichment of transfected cells with puromycin-containing medium. Twenty-four hours after transfection, medium was switched to puromycin (3 μg/mL)-containing medium. After 48 h of selection in puromycin-containing medium, RNA was extracted directly from a 12-well plate using the DirectZol RNA Isolation kit (Zymo Research, Irvine, CA).
Neuronal differentiation of iPSC cells
Differentiation of AGI-0 iPSC cells was performed according to published protocols.67 Colonies were grown on mouse embryonic fibroblast plates in medium containing 30 ng/mL basic fibroblast growth factor iPSC medium. When clones had grown to large colonies, the medium was removed and replaced with 1 mL of PBS. Marked colonies were manually dissected out with a P10 pipette tip. Colonies were pooled in a 15-mL conical tube (Corning) and centrifuged for 3 min at 850 rpm. The pellet was incubated for 3 min at 37°C with Accutase, and cells were triturated afterward with a P1000 pipette tip. Subsequently, 2 mL of mTeSR1 medium (STEMCELL Technologies, Vancouver, Canada) was added to the cells. Cells were centrifuged again at 850 rpm and resuspended in 1 mL of mTeSR1 medium. Cells were plated at a concentration of 25,000 cells per well on poly-L-ornithine (100 μg/mL; Sigma) and laminin (3 μg/mL; Sigma) coated 48-well plates (Corning) in mTeSR1 and 10 μM ROCKi (Calbiochem, San Diego, CA). Cells were incubated for 2 days at 37°C at 5% CO2. On day 3, cells were incubated in STEMdiff APEL medium (STEMCELL Technologies) with 10 μM SB421542 (Stemgent, Cambridge, MA) and 250 nM LDN193189 (Stemgent). LDN193189 and SB421542 were used for dual SMAD inhibition to rapidly differentiate iPSCs into early neuroectoderm. 68 Medium was changed every other day. On day 11, half of the medium was replaced with neural differentiation medium (neurobasal medium [Thermo Fisher Scientific, Waltham, MA], B27 without retinoic acid [1×; Thermo Fisher], 2 mM GlutaMAX, and penicillin/streptomycin [100 U/mL and 0.1 mg/mL]). On day 13, another half of the medium was replaced with a neuronal differentiation medium. After day 15, the medium was completely exchanged for neuronal differentiation medium every other day. On day 22, cells were fixed with 4% paraformaldehyde for 12 min. Neuronal markers were labeled according to previous protocols.38 Images were taken immediately with a Ti-U microscope (Nikon) using a CoolSnap camera (Photometrics).
Reverse transcription-quantitative PCR from cell assays
RNA was extracted directly from plates using the standard protocol with a DirectZol RNA Isolation kit (Zymo Research). cDNA synthesis was performed using the RevertAid Random Hexamer Kit (ThermoFisher). qPCR was performed with primers for hSNURF (hSNURF-F 5′ CTG TCT GAG GAG CGG TCA GT 3′ and hSNURF-R 5′ CAG GTA CTT GCT GCT GCT GA 3′), UBE3A-ATS (hATS-F 5′ GCA CTG AAA ATG TGG CAT CCA GTC 3′ and hATS-R 5′ GGT GTG TCA GCT GTG CTG GTG TCA 3′), UBE3A (UBE3A-F 5′ ATG ACG GTG GCT ATA CCA GG 3′ and UBE3A-R 5′ CCT TTC TGT GTC TGG GCA TTT TTG G 3′), and GAPDH (GAPDH-F 5′ AAT CCC ATC ACC ATC TTC CA 3′ and GAPDH-R 5′ CTC CAT GGT GGT GAA GAC G 3′) in the Applied Biosystems StepOne Plus Real-Time PCR System (Thermo Fisher). Data are presented using the ΔΔCt method, normalized to GAPDH and the indicated groups in figure legends.
Mice
Ube3a deletion mice were generated by crossing Ube3a deletion females (JAX stock #016590) with inbred C57BL/6J males. Breeders were replenished every 4–5 generations. To identify mice, neonates were labeled by paw tattoo on postnatal day (PND) 2–3 using non-toxic animal tattoo ink (Ketchum Manufacturing, Brockville, ON, Canada). At PND 5–7, tails of pups were clipped (0.5 cm) for genotyping, following the UC Davis Institutional Animal Care and Use Committee (IACUC) policy regarding tissue collection. Genotyping was performed with RED Extract-N-Amp (Sigma-Aldrich, St. Louis, MO) using primers R1965 5′ GCT CAA GGT TGT ATG CCT TGG TGC T 3′, WTF1966 5′ AGT TCT CAA GGT AAG CTG AGC TTG C 3′, and ASF1967 5′ TGC ATC GCA TTG TCT GAG TAG GTG TC 3′ for Ube3a. After weaning on PND21, mice were socially housed in groups of 2–4 by sex.
All procedures were approved by the UC Davis IACUC, protocol #21644.
Tissue preparation
Intravenous administration of an average of 1 × 1012 vg of AAV-PHP.eB per animal was performed by injection into the tail vein of adult mice (5–6 weeks of age). The viral dose was determined by previous observations as the highest well-tolerated dose for the best ATF-S1K brain-wide distribution (data not shown). The volume range (100–120 μL) of viral preparation was adjusted based on the animal’s weight. After allowing time for expression and behavioral testing at age 9–11 weeks, mice were anesthetized with isoflurane and transcardially perfused with ice-cold 0.1 M phosphate buffer (pH 7.4) at room temperature. For RNA and protein extractions, one hemisphere from each brain was flash-frozen in liquid nitrogen until further processing.
RNA extraction from mouse brains and RT-qPCR
Whole-brain halves were ground in liquid nitrogen before homogenization in TRIzol. RNA was extracted using the DirectZol RNA Miniprep Kit (Zymo Research). Total RNA (500–1,000 μg) was reverse transcribed using the VILO kit (Invitrogen) according to the manufacturer’s instructions. Primers for qPCR were designed using Primer3.33 RT-qPCR was performed in triplicate using PowerUp SYBR Green Master Mix (Thermo Fisher) with the CFX384 Real-Time System C1000 Touch system (Bio-Rad). Gene expression analysis was performed with gene-specific primers (Table S1) using three biological replicates. Relative target gene expression was calculated as the difference between the target gene and the Gapdh reference gene (dCq = Cq[target] − Cq[Gapdh]). Statistical significance was evaluated with a one-way ANOVA and Dunnett’s multiple comparisons test.
RNA-seq analysis
Poly(A) mRNA was isolated from 800 ng of total RNA from whole mouse brain tissue using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs, Ipswich, MA). Samples were processed from the following female mice 5 weeks after AAV treatment: AS control mice treated with AAV-mCherry (n = 3), AS mice treated with AAV-S1K (n = 4), and WT mice treated with AAV-mCherry (n = 3). Mice were 11 weeks old. Additionally, RNA-seq libraries were prepared from two untreated WT mice as a no-treatment control. Strand-specific RNA-seq libraries were prepared using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England BioLabs) according to the manufacturer’s protocol. Unique 6-nt barcodes were incorporated during the library amplification step, and equal library amounts were pooled for sequencing on the HiSeq 4000. AAV-treated samples were sequenced PE150 while the no-treatment sample was sequenced SR50. Sequencing and demultiplexing of Illumina reads were performed by the DNA Technologies and Expression Analysis Core at the UC Davis Genome Center. Fastq reads were aligned to the mm10 mouse genome assembly using GENCODE vM23 annotation files with the STAR aligner.69 BAM output files were used for gene and transcript counts using RSEM.69,70 Differential gene expression was determined using DESeq271 (Table S2).
Western blot analysis
Flash-frozen brain hemispheres were ground in liquid nitrogen. Cytosolic and nuclear protein was extracted with the CelLytic NuCLEAR Extraction Kit (MilliporeSigma, Burlington, MA) using the hypotonic lysis buffer protocol. Protein lysates were quantified using the BCA assay (Life Technologies). Protein (25 μg) was loaded onto SDS-PAGE gels (Bolt 4%–12% Bis-Tris Plus, Invitrogen) and separated using MOPS buffer (Invitrogen). Proteins were transferred to the nitrocellulose membrane overnight at 4°C using the Wet/Tank Blotting Systems (Bio-Rad). Total protein was stained with Ponceau S stain before blocking the membrane in 5% dry milk in TBST (20 mmol/L Tris [pH 7.5], 150 mmol/L NaCl, 0.1% Tween). Anti-UBE3A antibody (1:1,000; E8655, MilliporeSigma) and anti-β-actin antibody (1:3,000; A5441, MilliporeSigma) were used overnight at 4°C and subsequently incubated with rabbit anti-mouse antibody conjugated to horseradish peroxidase (1:2,000; #7076S, Cell Signaling Technology, Beverly, MA). Western blot was incubated with the ECL Plus Reagent (Cytiva, MilliporeSigma), and the chemiluminescent signal was visualized using the Molecular Imager Gel Doc XR system (Bio-Rad). Intensities of Ube3a protein bands were quantified using the volume box tool and normalized to intensities of β-actin.
Immunohistochemistry
Mice used for immunohistochemistry (IHC) were euthanized using CO2 and transcardially perfused with 0.1 M PBS and fresh, ice-cold 4% paraformaldehyde (Avantor [J.T. Baker], Radnor, PA; #S898-07) at pH 7.4. All brains were extracted and post-fixed in 4% paraformaldehyde for 36–48 h at 4°C and transferred to 30% sucrose in 0.1 M PBS for 48 h at 4°C. The brains were then cryopreserved in isopropanol on dry ice (−65°C) for 5 min and subsequently stored at −80°C or lower. Sagittal oriented serial sections at 30 μm thickness were obtained using a cryostat microtome (CF-6100; Precisionary, Natick, MA), starting from the midline and continuing laterally to a depth of ∼1,800 μm (10 sections in 6 wells), and were stored prior to labeling in a 0.02% sodium azide and 0.1 M PBS solution at 4°C.
Sagittal sections were washed in 0.1% Triton X-100 in PBS for 30 min, followed by incubation in a 10% SEABLOCK + 0.1% PBS-Triton blocking solution for 1 h. Primary antibody labeling utilized a multiplex of 1:1,000 monoclonal mouse (ms)-UBE3A (Sigma-Aldrich, #SAB1404508), a 1:1,000 polyclonal rabbit (rb)-mCherry (Abcam, Boston, MA, #Ab167543), a 1:1,000 guinea pig (gp)-NeuN (Sigma-Aldrich, #ABN90), incubated at 4°C overnight on gentle agitation. Tissue was then washed with PBS + Tween 20 (PBST) three times for 5 min before a 1-h incubation with 1:1,000 goat anti-ms-488, 1:1,000 goat anti-rb-594, and 1:1,000 goat anti-gp-647 AlexaFluor secondary antibodies (Thermo Fisher Scientific, #A106830, #A111012, and A21450), followed immediately with 1:1,000 Hoechst (Cell Signaling Technology, #4082) for nuclear visualization for 5 min. The tissue was then washed three times with PBST. Tissue was counterstained with 0.025% Sudan black in 70% ETOH for 1–2 min (to aid in automated scanning) before mounting, subsequently adding coverslips with Fluoromount (Sigma-Aldrich).
Fluorescently labeled tissues were imaged using a Zeiss Axioscan at 20× magnification for both stitched whole-segment images and isolated regions of interest. Isocortex regions of interest (ROIs) were isolated and analyzed using Zen Blue image analysis software (v. 3.4) to determine the relative expression of UBE3A and S1K across treatment groups in NeuN-positive cells. In brief, neurons were spatially isolated via NeuN labeling (Alexa Fluor 647) using intensity thresholds, and the intensity of 488/Ube3a and 594/mCherry S1K was captured for those same cells in isolation. The intensity values for all NeuN-positive cells for each channel within an ROI were averaged and compared across treatment groups. Statistical significance was evaluated with a one-way ANOVA and Dunnett’s multiple comparisons test.
DAB immunohistochemistry
Sagittal sections were labeled with ImmPACT DAB Peroxidase Substrate (Vector Labs, Burlingame, CA, #SK-4150), utilizing the Vectastain ABC Kit (Vector Labs, #HP1-26), following the manufacturer’s protocol. Tissues underwent endogenous peroxidase quenching using a 0.3% hydrogen peroxide solution in water for 30 min, followed by immersion in a 10% blocking solution in 0.1% PBS-Triton + SEABLOCK blocking buffer (Thermo Fisher Scientific, #37527), for 1 h, followed by incubation in primary antibody monoclonal anti-UBE3A clone E3 in mouse at a concentration of 1:1,000 (Sigma-Aldrich, #SAB1404508-100UG), and incubated overnight at 4°C. On the second day, tissues were washed in PBST three times for 15 min, followed by immersion in a biotinylated goat anti-mouse immunoglobulin G secondary antibody (Vector Labs, #BA-9200) solution at a concentration of 1:200 with incubation for 1 h, followed by Vectastain ABC reagent (Vector Labs, #HP1-26) incubation for 30 min, and concluded with immersion into ImmPACT DAB Peroxidase Substrate (#SK-4105) at the manufacturer’s recommended concentration for 8 min. Serial sections were mounted onto uncharged slides and coverslipped using Permount Mounting Medium (Thermo Fisher Scientific, #SP15-100).
Whole-brain imaging of UBE3A-DAB-labeled tissues was captured using a Zeiss Axioscan.Z1 slide scanner at 20× magnification. ROIs within the prefrontal cortex, striatum, hippocampus, and cerebellum were isolated from stitched whole-segment images using Zen Blue image analysis software (v. 3.4). Neural cell counts to determine the relative number of UBE3A-expressing cells across treatment groups were obtained using ImageJ (NIH, Bethesda, MD). Statistical significance was evaluated with one-way ANOVA and Dunnett’s multiple comparisons test.
Behavioral assays
Subjects for behavior
All mice were housed in Techniplast cages (Techniplast, West Chester, PA). Cages were housed in ventilated racks in a temperature (20°C–22.2°C) and humidity (∼25%) controlled colony room on a 12:12-h light/dark cycle. Standard rodent chow and tap water were available ad libitum. In addition to standard bedding, a Nestlet square, shredded brown paper, and a cardboard tube (Jonesville Corp, Jonesville, MI) were provided in each cage. All experimental procedures were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the IACUC of UC Davis protocol #21644 (PI: Silverman).
Order of testing
Two cohorts of mice were tested as follows. Cohort 1 of Ube3a deletion mice (AS) was sampled from five litters. The order and age of testing were as follows: (1) open field at 8 weeks of age, (2) DigiGait at 9 weeks of age, and (3) rotarod at 10 weeks of age. Cohort 2 of Ube3a deletion mice was sampled from 11 litters. Results of statistical analysis for all behavioral assays are summarized in Table S3.
Open field
General exploratory locomotion in a novel open field arena was evaluated as previously described.72,73 In brief, each subject was tested in a VersaMax Animal Activity Monitoring System (Accuscan, Columbus, OH) for 30 min in a ∼30-lux testing room. The total distance traversed, horizontal activity, vertical activity, and time spent in the center were automatically measured to assess gross motor abilities in mice. Open field parameters (total distance traveled, horizontal activity, vertical activity, and center time) were analyzed using two-way repeated-measures ANOVA, with genotype as the between-group factor and time as the within-group factor. Dunnett’s multiple comparisons tests for time bins were conducted.
Gait analysis
Treadmill gait analysis was performed using the DigiGait system (Mouse Specifics, Framingham, MA).6,45 Mouse paws were painted with non-toxic red food coloring to augment dark-green paw tattoos that generated conflict in DigiGait analysis 1 min prior to introduction to the walking chamber to reliably capture the entire paw surface area. Before data collection, each subject was acclimated to the Perspex walking chamber for 1 min, and the treadmill was slowly accelerated to the final speed of 20 cm/s to allow mice to adjust to walking on the belt. Digital images of paw placement were recorded through a clear treadmill from the ventral plane of the animal. Mice were tested in a single session at a 20 cm/s treadmill speed maintaining a normal pace walk for WT mice. Non-performers were defined as mice unable to sustain walking at 20 cm/s without colliding with the posterior bumper for at least 3 s. There is no practice effect or repeated exposure and, therefore, mice were allowed retrial and retest if they were unable to adjust to walking on the belt easily. The treadmill belt and the encasing Perspex chamber were cleaned with 70% (v/v) ethanol between tests. For each mouse, videos of ∼5 s duration of all sessions were analyzed using the DigiGait Imaging and Analysis software v12.2 (Mouse Specifics). Contrast filters were determined on a mouse-by-mouse basis to facilitate consistent recognition of all four paws. All analyses were conducted in a single session by an experimenter blind to genotype. Stride length (distance a paw makes during a single stride) and frequency (number of strides per second to maintain pace) were automatically calculated. Data were averaged between left and right paws for fore and hind paws.
Rotarod
Motor coordination, balance, and motor learning were tested with an accelerating rotarod (Ugo Basile, Gemonio, Italy), as previously described,6,45,72,74 for balance motor coordination and motor learning. Mice were placed on a rotating cylinder that slowly accelerated from 5 to 40 rpm over 5 min. Mice were given three trials per day with a 60-min intertrial rest interval and tested for three consecutive days for a total of nine trials. Performance was scored as latency to fall off the cylinder. Latency to fall was analyzed with two-way repeated-measures ANOVA, with genotype as the between-group factor and day as the within-group factor. Multiple comparisons for the day were conducted.
Statistics
All statistical analysis was carried out using Prism 9 software (GraphPad Software, San Diego, CA). All significance levels were set at p < 0.05. Outliers of behavioral assays were identified and excluded using Grubb’s test, and D'Agostino and Pearson tests were used to check assumptions of normality. Post hoc comparisons were performed following a significant main effect or interaction and were conducted using Dunnett’s multiple comparisons test. Data are presented as mean ± standard error of the mean (SEM) unless otherwise noted.
In addition, one-way ANOVA followed by Dunnett’s multiple comparisons test was carried out for RT-qPCR and immunohistochemistry analyses.
Study approval
Animal studies followed NIH guidelines and were performed in strict compliance with animal protocols approved by the IACUC #21644 of UC Davis.
Data availability
All data associated with this study are available in the main text or the supplemental material. All high-throughput sequencing datasets have been submitted to GEO (BioProject ID GEO: PRJNA867496).
Acknowledgments
This work was supported by the Foundation for Angelman Syndrome Therapeutics (FAST; D.J.S. and J.L.S.), NINDS (grant R01NS097808), Eunice Kennedy Shriver National Institute of Child Health and Human Development (P50HD103526; PI: Abbedutto) and CIRM (DISC2-09032). P.D. was supported by the Diversity Supplement 3R24OD021606-02ZF, NIGMS Pharmacology T32 GM099608, and NIH NCATS UL1 TR001860 and linked award TL1 TR001861. High-throughput sequencing was carried out by the DNA Technologies and Expression Analysis Core at the UC Davis Genome Center, supported by NIH Shared Instrumentation grant 1S10OD010786-01. Some images were created with BioRender.com.
Author contributions
H.O. and U.B. designed, performed, and analyzed experiments. H.O. and U.B. contributed equally to experiments. H.O. conceptualized and wrote the manuscript. M.S.G. maintained mouse colonies, performed genotyping, and processed tissues. D.L.C. and I.J.V. performed IHC experiments and analyses. H.O., P.D., and J.H. performed and analyzed human ATF experiments. S.L. maintained and differentiated AS iPSC lines. A.A., T.A.F., and N.A.C. performed and analyzed behavioral studies. J.L., D.W., and D.J.S. designed human targeting zinc fingers. K.D.F., J.L.S., and D.J.S. guided experiments and contributed to data analysis. All authors edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.01.013.
Supplemental information
References
- 1.Magenis R.E., Brown M.G., Lacy D.A., Budden S., LaFranchi S., LaFranchi S., Opitz J.M., Reynolds J.F., Ledbetter D.H. Is Angelman Syndrome an alternate result of del(15)(qllql3)? Am. J. Med. Genet. 1987;28:829–838. doi: 10.1002/ajmg.1320280407. [DOI] [PubMed] [Google Scholar]
- 2.Buckley R.H., Dinno N., Weber P. Angelman syndrome: are the estimates too low? Am. J. Med. Genet. 1998;80:385–390. doi: 10.1002/(sici)1096-8628(19981204)80:4<385::aid-ajmg15>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 3.Daily J.L., Nash K., Jinwal U., Golde T., Rogers J., Peters M.M., Burdine R.D., Dickey C., Banko J.L., Weeber E.J. Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS One. 2011;6:e27221. doi: 10.1371/journal.pone.0027221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Judson M.C., Shyng C., Simon J.M., Davis C.R., Punt A.M., Salmon M.T., Miller N.W., Ritola K.D., Elgersma Y., Amaral D.G., et al. Dual-isoform hUBE3A gene transfer improves behavioral and seizure outcomes in Angelman syndrome model mice. JCI Insight. 2021;6:e144712. doi: 10.1172/jci.insight.144712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dodge A., Morrill N.K., Weeber E.J., Nash K.R. Recovery of Angelman syndrome rat deficits with UBE3A protein supplementation. Mol. Cell. Neurosci. 2022;120:103724. doi: 10.1016/j.mcn.2022.103724. [DOI] [PubMed] [Google Scholar]
- 6.Adhikari A., Copping N.A., Beegle J., Cameron D.L., Deng P., O’Geen H., Segal D.J., Fink K.D., Silverman J.L., Anderson J.S. Functional rescue in an Angelman syndrome model following treatment with lentivector transduced hematopoietic stem cells. Hum. Mol. Genet. 2021;30:1067–1083. doi: 10.1093/hmg/ddab104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sidorov M.S., Deck G.M., Dolatshahi M., Thibert R.L., Bird L.M., Chu C.J., Philpot B.D. Delta rhythmicity is a reliable EEG biomarker in Angelman syndrome: a parallel mouse and human analysis. J. Neurodev. Disord. 2017;9:17. doi: 10.1186/s11689-017-9195-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Born H.A., Dao A.T., Levine A.T., Lee W.L., Mehta N.M., Mehra S., Weeber E.J., Anderson A.E. Strain-dependence of the Angelman Syndrome phenotypes in Ube3a maternal deficiency mice. Sci. Rep. 2017;7:8451. doi: 10.1038/s41598-017-08825-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker E.K., Butler M.G., Hartin S.N., Ling L., Bui M., Francis D., Rogers C., Field M.J., Slee J., Gamage D., et al. Relationships between UBE3A and SNORD116 expression and features of autism in chromosome 15 imprinting disorders. Transl. Psychiatry. 2020;10:362. doi: 10.1038/s41398-020-01034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Copping N.A., Christian S.G.B., Ritter D.J., Islam M.S., Buscher N., Zolkowska D., Pride M.C., Berg E.L., LaSalle J.M., Ellegood J., et al. Neuronal overexpression of Ube3a isoform 2 causes behavioral impairments and neuroanatomical pathology relevant to 15q11.2-q13.3 duplication syndrome. Hum. Mol. Genet. 2017;26:3995–4010. doi: 10.1093/hmg/ddx289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meng L., Ward A.J., Chun S., Bennett C.F., Beaudet A.L., Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–412. doi: 10.1038/nature13975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolter J.M., Mao H., Fragola G., Simon J.M., Krantz J.L., Bazick H.O., Oztemiz B., Stein J.L., Zylka M.J. Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature. 2020;587:281–284. doi: 10.1038/s41586-020-2835-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmid R.S., Deng X., Panikker P., Msackyi M., Breton C., Wilson J.M. CRISPR/Cas9 directed to the Ube3a antisense transcript improves Angelman syndrome phenotype in mice. J. Clin. Invest. 2021;131:e142574. doi: 10.1172/jci142574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kosicki M., Tomberg K., Bradley A. Erratum: repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:899. doi: 10.1038/nbt0918-899c. [DOI] [PubMed] [Google Scholar]
- 15.Giannoukos G., Ciulla D.M., Marco E., Abdulkerim H.S., Barrera L.A., Bothmer A., Dhanapal V., Gloskowski S.W., Jayaram H., Maeder M.L., et al. UDiTaS™, a genome editing detection method for indels and genome rearrangements. BMC Genomics. 2018;19:212. doi: 10.1186/s12864-018-4561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stadtmauer E.A., Fraietta J.A., Davis M.M., Cohen A.D., Weber K.L., Lancaster E., Mangan P.A., Kulikovskaya I., Gupta M., Chen F., et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367:eaba7365. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turchiano G., Andrieux G., Klermund J., Blattner G., Pennucci V., El Gaz M., Monaco G., Poddar S., Mussolino C., Cornu T.I., et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell. 2021;28:1136–1147.e5. doi: 10.1016/j.stem.2021.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Webber B.R., Lonetree C.-L., Kluesner M.G., Johnson M.J., Pomeroy E.J., Diers M.D., Lahr W.S., Draper G.M., Slipek N.J., Smeester B.A., et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat. Commun. 2019;10:5222. doi: 10.1038/s41467-019-13007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leibowitz M.L., Papathanasiou S., Doerfler P.A., Blaine L.J., Sun L., Yao Y., Zhang C.-Z., Weiss M.J., Pellman D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021;53:895–905. doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H., et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alanis-Lobato G., Zohren J., McCarthy A., Fogarty N.M.E., Kubikova N., Hardman E., Greco M., Wells D., Turner J.M.A., Niakan K.K. Frequent loss of heterozygosity in CRISPR-Cas9-edited early human embryos. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2004832117. e2004832117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beerli R.R., Segal D.J., Dreier B., Barbas C.F., 3rd Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl. Acad. Sci. USA. 1998;95:14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhakta M.S., Henry I.M., Ousterout D.G., Das K.T., Lockwood S.H., Meckler J.F., Wallen M.C., Zykovich A., Yu Y., Leo H., et al. Highly active zinc-finger nucleases by extended modular assembly. Genome Res. 2013;23:530–538. doi: 10.1101/gr.143693.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeitler B., Froelich S., Marlen K., Shivak D.A., Yu Q., Li D., Pearl J.R., Miller J.C., Zhang L., Paschon D.E., et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 2019;25:1131–1142. doi: 10.1038/s41591-019-0478-3. [DOI] [PubMed] [Google Scholar]
- 25.Wegmann S., DeVos S.L., Zeitler B., Marlen K., Bennett R.E., Perez-Rando M., MacKenzie D., Yu Q., Commins C., Bannon R.N., et al. Persistent repression of tau in the brain using engineered zinc finger protein transcription factors. Sci. Adv. 2021;7:eabe1611. doi: 10.1126/sciadv.abe1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bailus B.J., Pyles B., McAlister M.M., O’Geen H., Lockwood S.H., Adams A.N., Nguyen J.T.T., Yu A., Berman R.F., Segal D.J. Protein delivery of an artificial transcription factor restores widespread Ube3a expression in an angelman syndrome mouse brain. Mol. Ther. 2016;24:548–555. doi: 10.1038/mt.2015.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.-L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pandya N.J., Meier S., Tyanova S., Terrigno M., Wang C., Punt A.M., Mientjes E.J., Vautheny A., Distel B., Kremer T., et al. A cross-species spatiotemporal proteomic analysis identifies UBE3A-dependent signaling pathways and targets. Mol. Psychiatry. 2022;27:2590–2601. doi: 10.1038/s41380-022-01484-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Y.H., Armstrong D., Albrecht U., Atkins C.M., Noebels J.L., Eichele G., Sweatt J.D., Beaudet A.L. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 30.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 31.Lu Y., Xue J., Deng T., Zhou X., Yu K., Deng L., Huang M., Yi X., Liang M., Wang Y., et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020;26:732–740. doi: 10.1038/s41591-020-0840-5. [DOI] [PubMed] [Google Scholar]
- 32.Ruan G.-X., Barry E., Yu D., Lukason M., Cheng S.H., Scaria A. CRISPR/Cas9-Mediated genome editing as a therapeutic approach for leber congenital amaurosis 10. Mol. Ther. 2017;25:331–341. doi: 10.1016/j.ymthe.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Geen H., Ren C., Nicolet C.M., Perez A.A., Halmai J., Le V.M., Mackay J.P., Farnham P.J., Segal D.J. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Res. 2017;45:9901–9916. doi: 10.1093/nar/gkx578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thakore P.I., Kwon J.B., Nelson C.E., Rouse D.C., Gemberling M.P., Oliver M.L., Gersbach C.A. RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors. Nat. Commun. 2018;9:1674. doi: 10.1038/s41467-018-04048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moreno A.M., Fu X., Zhu J., Katrekar D., Shih Y.-R.V., Marlett J., Cabotaje J., Tat J., Naughton J., Lisowski L., et al. In situ gene therapy via AAV-CRISPR-cas9-mediated targeted gene regulation. Mol. Ther. 2018;26:1818–1827. doi: 10.1016/j.ymthe.2018.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chamberlain S.J., Chen P.-F., Ng K.Y., Bourgois-Rocha F., Lemtiri-Chlieh F., Levine E.S., Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. USA. 2010;107:17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsiao J.S., Germain N.D., Wilderman A., Stoddard C., Wojenski L.A., Villafano G.J., Core L., Cotney J., Chamberlain S.J. A bipartite boundary element restricts imprinting to mature neurons. Proc. Natl. Acad. Sci. USA. 2019;116:2181–2186. doi: 10.1073/pnas.1815279116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng P., Halmai J.A.N.M., Beitnere U., Cameron D., Martinez M.L., Lee C.C., Waldo J.J., Thongphanh K., Adhikari A., Copping N., et al. An in vivo cell-based delivery platform for zinc finger artificial transcription factors in pre-clinical animal models. Front. Mol. Neurosci. 2021;14:789913. doi: 10.3389/fnmol.2021.789913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi J.-H., Yu N.-K., Baek G.-C., Bakes J., Seo D., Nam H.J., Baek S.H., Lim C.-S., Lee Y.-S., Kaang B.-K. Optimization of AAV expression cassettes to improve packaging capacity and transgene expression in neurons. Mol. Brain. 2014;7:17. doi: 10.1186/1756-6606-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mendiola A.J.P., LaSalle J.M. Epigenetics in prader-willi syndrome. Front. Genet. 2021;12:624581. doi: 10.3389/fgene.2021.624581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zahova S.K., Humby T., Davies J.R., Morgan J.E., Isles A.R. Comparison of mouse models reveals a molecular distinction between psychotic illness in PWS and schizophrenia. Transl. Psychiatry. 2021;11:433. doi: 10.1038/s41398-021-01561-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hakim C.H., Kumar S.R.P., Pérez-López D.O., Wasala N.B., Zhang D., Yue Y., Teixeira J., Pan X., Zhang K., Million E.D., et al. Cas9-specific immune responses compromise local and systemic AAV CRISPR therapy in multiple dystrophic canine models. Nat. Commun. 2021;12:6769. doi: 10.1038/s41467-021-26830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang H.-S., Burns A.J., Nonneman R.J., Baker L.K., Riddick N.V., Nikolova V.D., Riday T.T., Yashiro K., Philpot B.D., Moy S.S. Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav. Brain Res. 2013;243:79–90. doi: 10.1016/j.bbr.2012.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berg E.L., Pride M.C., Petkova S.P., Lee R.D., Copping N.A., Shen Y., Adhikari A., Fenton T.A., Pedersen L.R., Noakes L.S., et al. Translational outcomes in a full gene deletion of ubiquitin protein ligase E3A rat model of Angelman syndrome. Transl. Psychiatry. 2020;10:39. doi: 10.1038/s41398-020-0720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petkova S.P., Adhikari A., Berg E.L., Fenton T.A., Duis J., Silverman J.L. Gait as a quantitative translational outcome measure in Angelman syndrome. Autism Res. 2022;15:821–833. doi: 10.1002/aur.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silva-Santos S., van Woerden G.M., Bruinsma C.F., Mientjes E., Jolfaei M.A., Distel B., Kushner S.A., Elgersma Y. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Invest. 2015;125:2069–2076. doi: 10.1172/jci80554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carson R.P., Herber D.L., Pan Z., Phibbs F., Key A.P., Gouelle A., Ergish P., Armour E.A., Patel S., Duis J. Nutritional formulation for patients with angelman syndrome: a randomized, double-blind, placebo-controlled study of exogenous ketones. J. Nutr. 2021;151:3628–3636. doi: 10.1093/jn/nxab284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grieco J.C., Ciarlone S.L., Gieron-Korthals M., Schoenberg M.R., Smith A.G., Philpot R.M., Heussler H.S., Banko J.L., Weeber E.J. An open-label pilot trial of minocycline in children as a treatment for Angelman syndrome. BMC Neurol. 2014;14:232. doi: 10.1186/s12883-014-0232-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith S.E.P., Zhou Y.-D., Zhang G., Jin Z., Stoppel D.C., Anderson M.P. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 2011;3:103ra97. doi: 10.1126/scitranslmed.3002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakatani J., Tamada K., Hatanaka F., Ise S., Ohta H., Inoue K., Tomonaga S., Watanabe Y., Chung Y.J., Banerjee R., et al. Abnormal behavior in a chromosome- engineered mouse model for human 15q11-13 duplication seen in autism. Cell. 2009;137:1235–1246. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silverman J.L., Thurm A., Ethridge S.B., Soller M.M., Petkova S.P., Abel T., Bauman M.D., Brodkin E.S., Harony-Nicolas H., Wöhr M., et al. Genes Brain Behav.; 2022. Reconsidering Animal Models Used to Study Autism Spectrum Disorder: Current State and Optimizing Future; p. e12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M., et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toral M.A., Charlesworth C.T., Ng B., Chemudupati T., Homma S., Nakauchi H., Bassuk A.G., Porteus M.H., Mahajan V.B. Investigation of Cas9 antibodies in the human eye. Nat. Commun. 2022;13:1053. doi: 10.1038/s41467-022-28674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cromer M.K., Vaidyanathan S., Ryan D.E., Curry B., Lucas A.B., Camarena J., Kaushik M., Hay S.R., Martin R.M., Steinfeld I., et al. Global transcriptional response to CRISPR/Cas9-AAV6-Based genome editing in CD34 hematopoietic stem and progenitor cells. Mol. Ther. 2018;26:2431–2442. doi: 10.1016/j.ymthe.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hordeaux J., Wang Q., Katz N., Buza E.L., Bell P., Wilson J.M. The neurotropic properties of AAV-PHP.B are limited to C57BL/6J mice. Mol. Ther. 2018;26:664–668. doi: 10.1016/j.ymthe.2018.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsuzaki Y., Konno A., Mochizuki R., Shinohara Y., Nitta K., Okada Y., Hirai H. Intravenous administration of the adeno-associated virus-PHP.B capsid fails to upregulate transduction efficiency in the marmoset brain. Neurosci. Lett. 2018;665:182–188. doi: 10.1016/j.neulet.2017.11.049. [DOI] [PubMed] [Google Scholar]
- 57.Arotcarena M.-L., Dovero S., Biendon N., Dutheil N., Planche V., Bezard E., Dehay B. Pilot study assessing the impact of intrathecal administration of variants AAV-PHP.B and AAV-PHP.eB on brain transduction in adult rhesus macaques. Front. Bioeng. Biotechnol. 2021;9:762209. doi: 10.3389/fbioe.2021.762209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goertsen D., Flytzanis N.C., Goeden N., Chuapoco M.R., Cummins A., Chen Y., Fan Y., Zhang Q., Sharma J., Duan Y., et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 2022;25:106–115. doi: 10.1038/s41593-021-00969-4. [DOI] [PubMed] [Google Scholar]
- 59.Stanton A.C., Lagerborg K.A., Tellez L., Krunnfusz A., King E.M., Ye S., Solomon I.H., Tabebordbar M., Sabeti P.C. Systemic administration of novel engineered AAV capsids facilitates enhanced transgene expression in the macaque CNS. Med. 2022 doi: 10.1016/j.medj.2022.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen X., Ravindra Kumar S., Adams C.D., Yang D., Wang T., Wolfe D.A., Arokiaraj C.M., Ngo V., Campos L.J., Griffiths J.A., et al. Engineered AAVs for non-invasive gene delivery to rodent and non-human primate nervous systems. Neuron. 2022;110:2242–2257.e6. doi: 10.1016/j.neuron.2022.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Angulo M.A., Butler M.G., Cataletto M.E. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J. Endocrinol. Invest. 2015;38:1249–1263. doi: 10.1007/s40618-015-0312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qi Y., Purtell L., Fu M., Lee N.J., Aepler J., Zhang L., Loh K., Enriquez R.F., Baldock P.A., Zolotukhin S., et al. Snord116 is critical in the regulation of food intake and body weight. Sci. Rep. 2016;6:18614. doi: 10.1038/srep18614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Purtell L., Qi Y., Campbell L., Sainsbury A., Herzog H. Adult-onset deletion of the Prader-Willi syndrome susceptibility gene in mice results in reduced feeding and increased fat mass. Transl. Pediatr. 2017;6:88–97. doi: 10.21037/tp.2017.03.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Labun K., Montague T.G., Gagnon J.A., Thyme S.B., Valen E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016;44:W272–W276. doi: 10.1093/nar/gkw398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Challis R.C., Ravindra Kumar S., Chan K.Y., Challis C., Beadle K., Jang M.J., Kim H.M., Rajendran P.S., Tompkins J.D., Shivkumar K., et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat. Protoc. 2019;14:379–414. doi: 10.1038/s41596-018-0097-3. [DOI] [PubMed] [Google Scholar]
- 67.Germain N.D., Chen P.-F., Plocik A.M., Glatt-Deeley H., Brown J., Fink J.J., Bolduc K.A., Robinson T.M., Levine E.S., Reiter L.T., et al. Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Mol. Autism. 2014;5:44. doi: 10.1186/2040-2392-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cary W.A., Hori C.N., Pham M.T., Nacey C.A., McGee J.L., Hamou M., Berman R.F., Bauer G., Nolta J.A., Waldau B. Efficient generation of induced pluripotent stem and neural progenitor cells from acutely harvested dura mater obtained during ventriculoperitoneal shunt surgery. World Neurosurg. 2015;84:1256–1266.e1. doi: 10.1016/j.wneu.2015.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gompers A.L., Su-Feher L., Ellegood J., Copping N.A., Riyadh M.A., Stradleigh T.W., Pride M.C., Schaffler M.D., Wade A.A., Catta-Preta R., et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci. 2017;20:1062–1073. doi: 10.1038/nn.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Copping N.A., Berg E.L., Foley G.M., Schaffler M.D., Onaga B.L., Buscher N., Silverman J.L., Yang M. Touchscreen learning deficits and normal social approach behavior in the Shank3B model of Phelan–McDermid Syndrome and autism. Neuroscience. 2017;345:155–165. doi: 10.1016/j.neuroscience.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Flannery B.M., Silverman J.L., Bruun D.A., Puhger K.R., McCoy M.R., Hammock B.D., Crawley J.N., Lein P.J. Behavioral assessment of NIH Swiss mice acutely intoxicated with tetramethylenedisulfotetramine. Neurotoxicol. Teratol. 2015;47:36–45. doi: 10.1016/j.ntt.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are available in the main text or the supplemental material. All high-throughput sequencing datasets have been submitted to GEO (BioProject ID GEO: PRJNA867496).