

Abstract
The COVID-19 pandemic highlights the need for effective vaccines against respiratory viruses. An ideal vaccine should induce robust and long-lasting responses with high manufacturing scalability. We used an adjuvant comprised of a Stimulator of Interferon Genes (STING) agonist incorporated in a microparticle (MP) platform that can be produced in a scalable manner to achieve durable protection against influenza virus challenge. This formulation overcomes the challenges presented by the cytosolic localization of STING and the hydrophilicity of its agonists. We evaluated a monoaxial formulation of polymeric acetalated dextran MPs to deliver the STING agonist cyclic GMP-AMP (cGAMP) in the context of innate immune activation and subunit vaccination against influenza hemagglutinin. In mice, acetalated dextran cGAMP MPs potently activated antigen presenting cells in vitro and lead to protective humoral and early T cell immune responses with >10X dose-sparing effects compared to other published work. Efficacy was also evaluated in ferrets, the larger animal model of choice for influenza vaccine testing. cGAMP MPs with recombinant hemagglutinin reduced viral shedding and improved vaccine outcomes compared to a seasonal influenza vaccine formulation. Importantly, sustained protection against influenza infection was detected a year after a single dose of the vaccine with cGAMP MPs adjuvant.
Introduction
The present SARS-CoV-2 (COVID-19) pandemic underscores the high probability and impact of a respiratory infection worldwide. It draws global attention to the importance of vaccines against these respiratory infections and emphasizes the need for continuous improvement to vaccine platforms. Influenza viruses remain a significant burden to society, with up to 650,000 deaths attributed to the virus each year worldwide [1], and an annual economic impact of $90 billion in the U.S. alone [2]. While vaccination is one of the greatest preventative measures against infection, the influenza vaccine has a poor estimated effectiveness ranging from 13–60% [3]. This low efficacy is often due to antigenic mismatch resulting from antigenic drift that occurs during the lengthy production process and flu season [3, 4], as well as strain adaptation due to growth in eggs [5, 6]. Additionally, protection provided by the current seasonal influenza vaccine approach is relatively short lived, on the order of three to four months, while the flu season spans six months or more [7]. This highlights the need to generate influenza vaccines with more durable responses.
The subunit influenza vaccine Flublok, the only FDA-approved influenza vaccine consisting of recombinant protein antigens, may address some of these concerns, but still has limitations. For instance, due to its lack of immunogenicity it requires hemagglutinin (HA) protein doses that are three-fold higher than what is used in conventional flu shots (45 versus 15 μg/strain). Moreover, Flublok has induced lower antibody titers in adults aged 18–49 (its recommended age group) compared to conventional inactivated viral influenza vaccines [8].
An effective way to improve the durability of a vaccine response is to include an adjuvant in the formulation. Squalene-based emulsions (e.g., microfluidization 59 [MF59], adjuvant system 3 [AS03]) are used clinically with inactivated influenza viruses as a vaccine, but they have a limited half-life in vivo [9] and alone do not drive strong type 1 helper T cell (Th1)-biased cellular responses [10]. Similarly, traditional aluminum salt (alum) adjuvants are unable to drive strong cellular immune responses required for protection against influenza [11]. Pathogen-associated molecular patterns (PAMPs), which stimulate pattern recognition receptors (PRRs) on antigen-presenting cells (APCs), are an exciting alternative because they drive a balanced humoral and cellular immune response [12, 13]. Existing FDA-approved vaccine adjuvants target PRRs on either plasma or endosomal membranes [14], and include monophosphoryl lipid A (MPL) with alum, MPL with liposomal QS-21, or cytosine phosphoguanine (CpG). In contrast, there is still a significant need to improve agonists for cytosolic PRRs such as Stimulator of Interferon Genes (STING), whose stimulation can contribute significantly to antiviral responses [15]. The difficulty in reaching cytosolic PRRs typically forces the need for high agonist doses that increase the potential for toxicity (e.g., 3,000 μg CpG in the hepatitis B vaccine Heplisav-B).
STING agonist delivery can be enhanced using formulations such as polymeric particles, micelles, or liposomes [16–25]. Polymeric particles have advantages over liposomes because of their extended shelf-life and enhanced stability [26, 27]. STING’s cytosolic location, however, requires use of polymers that can enhance intracellular release of its agonists. Poly(lactic-co-glycolic acid) (PLGA) microparticles (MPs) are not sensitive to degradation in the acidic endosomal environment, limiting the extent of cytosolic delivery. Additionally, the polymer hydrolyzes into acidic byproducts that can be detrimental to vaccine components. Alternatively, the biopolymer, acetalated dextran (Ace-DEX), also degrades through hydrolysis but into pH-neutral biocompatible byproducts dextran, acetone, and ethanol. Additionally, Ace-DEX degrades more rapidly in the acidic endosomal environment (~pH 5.0) of APCs than at neutral pH [28–31]. This leads to triggered degradation of Ace-DEX MPs once endocytosed by APCs, resulting in release of the MP cargo. The targeted intracellular release leads to enhanced adjuvant innate signaling as demonstrated by the higher bioactivity of the STING agonist cyclic GMP-AMP (cGAMP) when delivered in Ace-DEX MPs compared to PLGA MPs or liposomes [22, 32, 33]. Another added advantage of Ace-DEX compared to PLGA MPs is their stability outside the cold-chain [34].
We have previously reported the use of coaxial electrospray for the encapsulation of cGAMP into Ace-DEX MPs as a vaccine adjuvant [22]. We have recently improved this technology by using monoaxial electrospray [35]. Monoaxial electrospray is a simpler method that addresses the issue of ease of scalability but has not been validated for vaccine use. In the current work, the monoaxial electrospray method for encapsulation of cGAMP into Ace-DEX MPs was validated for biological efficacy as compared to the previously published coaxial method. Furthermore, a subunit recombinant HA (HA) influenza vaccine adjuvanted with monoaxial electrospray Ace-DEX cGAMP MPs (cGAMP MPs) was evaluated against current influenza vaccine formulations in multiple ferret studies. Ferrets were used because they are the preferred animal model for assessing influenza vaccine efficacy and durability [36–38], and they exhibit similar disease pathogenesis to humans. This report demonstrates that the monoaxial cGAMP Ace-DEX MPs adjuvant can induce a durable and robust response over the span of a year and achieved a >10X dose sparing of cGAMP compared to a recent paper using a different particle formulation [39]. Together these studies show that in the context of recombinant HA subunit vaccination, monoaxial cGAMP MPs are a potent and well-tolerated adjuvant that elicit robust protective antiviral immunity against influenza respiratory challenge.
Results
Coaxial and monoaxial electrospray cGAMP MPs have comparable physical characteristics and in vitro bioactivity
Previously, we have demonstrated that an influenza subunit vaccination adjuvanted with cGAMP MPs formulated through coaxial electrospray provided complete and long lasting protection against lethal PR8 influenza challenge in mice [22]. Although effective, the coaxial electrospray setup is complex and therefore less ideal for scale up than a monoaxial electrospray system, which is an important consideration for increasing production and translation to the clinic. To test the efficacy of coaxial versus monoaxial electrospray systems, Ace-DEX cGAMP MPs were made by either the coaxial electrospray or single-fluid monoaxial electrospray system. As depicted in the scanning electron micrographs (SEMs; Fig 1A–B), each set of MPs had a size on the order of sub-micron to approximately 1 μm and remain stable for up to a week in solution under physiologic conditions with 10% v/v FBS (Fig S1A). The hydrodynamic mean particle diameters by volume, per dynamic light scattering, were very similar, with values of 902 and 1,060 nm for the coaxial and monoaxial MPs, respectively. Although the scanning electron micrographs would suggest the monoaxial MPs had a more monodisperse population in a dehydrated state, the monoaxial MPs were only slightly less hydrodynamically polydisperse, with a polydispersity index of 0.23 ± 0.03, versus the coaxial MPs’ PDI of 0.27 ± 0.03. The coaxial particles displayed a collapsed appearance, while the monoaxial MPs had a more sphere-like, porous morphology. Under physiological conditions, the monoaxial MPs exhibited moderately less burst release of cGAMP (47.6 ± 0.6%) than the coaxial MPs (60.1 ± 0.4), and a relatively linear release out to 1 week even in the presence of 10% v/v FBS (Fig 1C; Fig S1). While the reason for decreased burst release of the monoaxial cGAMP MPs has not been fully elucidated, likely there is better and perhaps more homogenous incorporation of the cGAMP in the MP with monoaxial spraying over coaxial spraying. This improved distribution maybe the result of decreased volumes of water-based solutions (preferred by hydrophilic cGAMP) in the monoaxial spray over the coaxial one. Regardless, a decreased burst release is highly desirable as a greater proportion of agonist remain within the intact particle allowing improved intracellular delivery.
Figure 1. Structural and functional comparison of Ace-DEX cGAMP MPs formulated via monoaxial or coaxial electrospray to activate BMDCs in vitro.
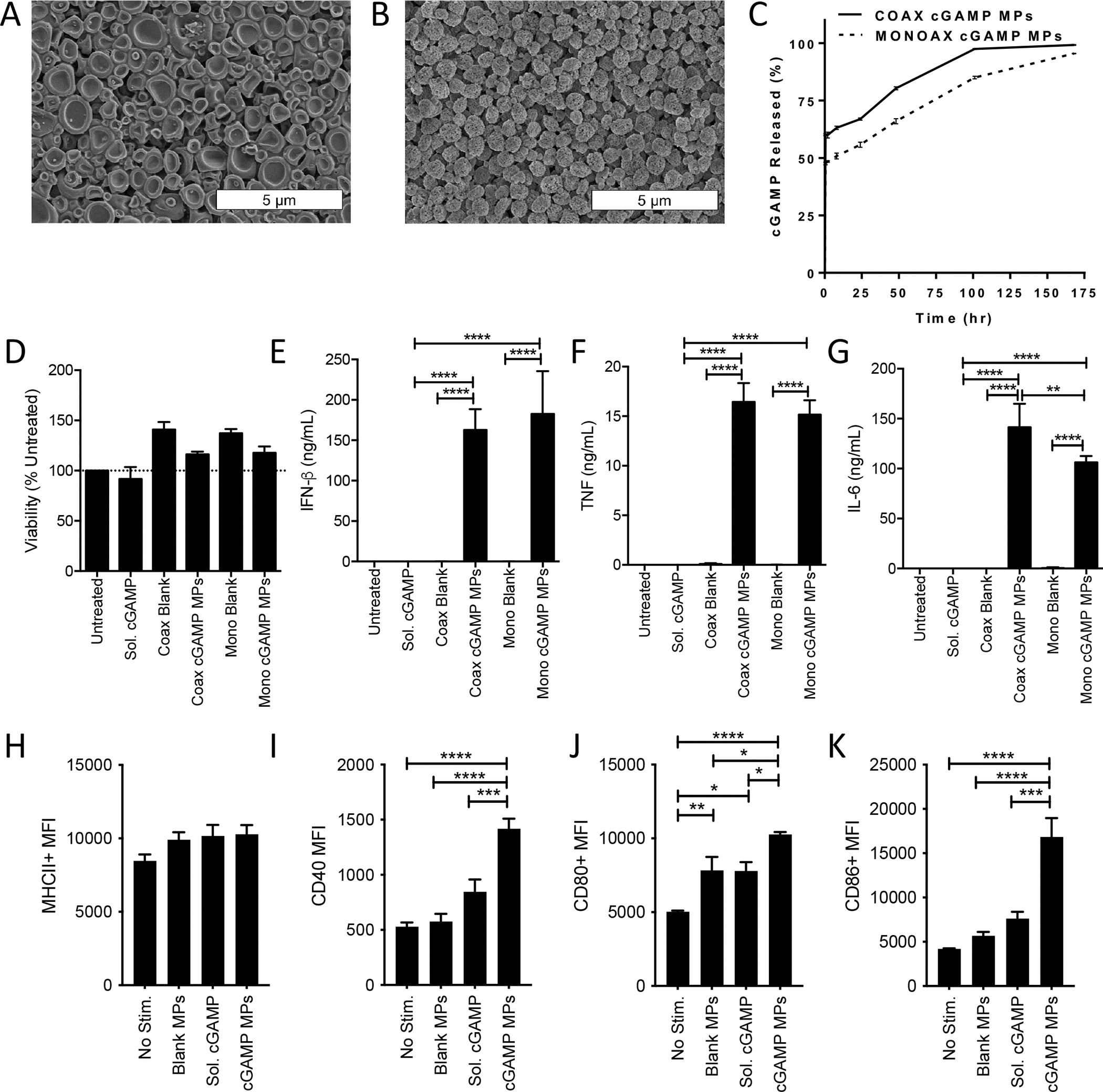
(A-B) Scanning electron microscopy (SEM) images were taken of cGAMP MPs made by (A) coaxial (coax) or (B) monoaxial (mono) electrospray. (C) Percent of free cGAMP released into solution from the initial calculated cGAMP encapsulated within coaxial (solid line) and monoaxial (segmented line) MPs while incubated at 37 °C and pH 7.4 (n=3 ± SD). (D-K) Bone marrow derived dendritic cells (BMDCs) from C57BL/6 mice were treated with 1 μg/mL cGAMP delivered as soluble CDN or encapsulated within Ace-DEX cGAMP MPs at a loading of 10 μg/mg MP through coaxial or monoaxial electrospray. Empty particle controls were also tested. (D-G) Twenty-four hours later cell supernatants were collected and assayed for (D) viability (E) IFN-β, (F) TNF, or (G) IL-6. (n=3 ± SEM). (H-K) Forty-eight hours later, flow cytometry was used to analyze upregulation of surface activation markers of antigen presenting cells including (H) MHCII, (I) CD40, (J) CD80, and (K) CD86. (n=6 ± SEM. *p<0.05, **p<0.01, ***p<0.001, ****p < 0.0001).
Biological activity of coaxial and monoaxial cGAMP MPs was first compared in vitro using cultured murine bone marrow derived dendritic cells (BMDCs). Importantly, in vitro stimulation of BMDCs with either formulation of blank MPs or cGAMP MPs did not lead to decreased viability of the treated cells (Fig. 1D). Both coaxial and monoaxial cGAMP MPs induced a strong IFN-β response (Fig. 1E) as well as the inflammatory cytokines TNF (Fig 1F) and IL-6 (Fig 1G) significantly enhanced over soluble cGAMP. No differences in the magnitude of response were observed between monoaxial and coaxial cGAMP MPs for IFN-β or TNF, while a small, but significant decrease in IL-6 production was observed with the monoaxial formulation.
Monoaxial MPs were also tested for the ability to activate BMDCs through upregulation of proteins involved with antigen presentation and co-stimulation of T cells including major histocompatibility complex II (MHCII) (Fig 1H), CD40 (Fig 1I), CD80 (Fig 1J), and CD86 (Fig 1K). The relative expression levels of these surface markers were observed by flow cytometry at 4, 24, and 48 hours, with 48 hours being sufficient to observe significant upregulation in expression (Fig S2 A–E). Upregulation of MHCII after 48-hour stimulation with cGAMP MPs was not statistically significant compared to unstimulated, blank MP, or soluble cGAMP stimulated BMDCs (Fig 1H). Comparatively, BMDCs treated with cGAMP MPs had significant upregulation of expression of the costimulatory molecules CD40 (Fig 1I), CD86 (Fig 1J), and CD86 (Fig1K). Of these proteins, only CD80 expression was significantly elevated with treatment with blank MPs or soluble cGAMP as compared to untreated, but not to the same extent as cGAMP MP stimulation. Together these data indicate that although there are minor morphologic differences between monoaxial and coaxial electrospray loaded cGAMP MPs, monoaxial cGAMP MPs maintain strong stimulatory activation of antigen presenting cells in vitro.
Coaxial or monoaxial electrospray Ace-DEX cGAMP MPs achieve similar protection against lethal infection in mice
Having observed comparable biological activity between coaxial and monoaxial formulations in vitro, we next assessed whether monoaxial cGAMP MPs maintained their adjuvant activity in a mouse influenza immunogenicity and challenge model. Eight-week-old female C57BL/6 mice were immunized with PBS alone, or recombinant HA protein from the mouse-adapted influenza strain PR8 adjuvanted with equivalent doses of soluble cGAMP, or cGAMP MPs formulated through coaxial or monoaxial electrospray (Table 1). Mice received a boost with the same formulation 21 days later. Serum was collected on day 28 post-prime to assess PR8 neutralizing titers (Fig 2A–E). Both coaxial and monoaxial MP formulations generated robust virus neutralizing titers that were significantly enhanced over PBS treated animals (Fig 2A). HA-specific total IgG endpoint titer levels were comparable between coaxial and monoaxial formulation and achieved levels almost 100-fold higher than observed with soluble cGAMP (Fig 2B). Furthermore, both coaxial and monoaxial formulations generated significantly more of the antiviral Th1-skewed IgG2c isotype compared to PBS and soluble cGAMP controls (Fig 2C). Both cGAMP MP formulations also led to increased levels of Th2-skewed IgG1 isotype as compared to PBS, but IgG1 levels were not significantly different between the two formulations and soluble cGAMP (Fig 2D). Using the ratio of IgG2c:IgG1 as an indicator of Th1:Th2 response skewing, vaccination with coaxial or monoaxial cGAMP MPs as an adjuvant drives a balanced immune response with an increased Th1 bias compared to soluble cGAMP (Fig 2E).
Table 1.
Antigen and adjuvant doses for vaccination studies.
| Figure | Species | Group | Antigen Dose (μg) |
Adjuvant Dose (μg) |
MP Dose (mg) |
|---|---|---|---|---|---|
| 2 | Mouse | PBS | - | - | - |
| HA-PR8 + Sol cGAMP | 1 | 0.2 | - | ||
| HA-PR8 + Coaxial cGAMP MPs | 1 | 0.2 | 0.020 | ||
| HA-PR8 + Monoaxial cGAMP MPs | 1 | 0.2 | 0.020 | ||
| 3 | Ferret | PBS | - | - | - |
| Fluarix Quadrivalent | 15 | - | - | ||
| HA-Cali + Sol cGAMP | 15 | 15 | - | ||
| HA-Cali + Monoaxial cGAMP MPs | 15 | 15 | 1.5 | ||
| 4–5 | Ferret | PBS | - | - | - |
| HA-Cali + AddaVax (1X) | 15 | 9.75 mg | - | ||
| HA-Cali + Monoaxial cGAMP MPs (Prime) | 15 | 15 μg | 1.5 | ||
| HA-Cali + Monoaxial cGAMP MPs (Boost) | 15 | 15 μg | 1.5 |
Figure 2. Monoaxial and Coaxial Ace-DEX MP adjuvants protect equally against lethal influenza challenge in mice:
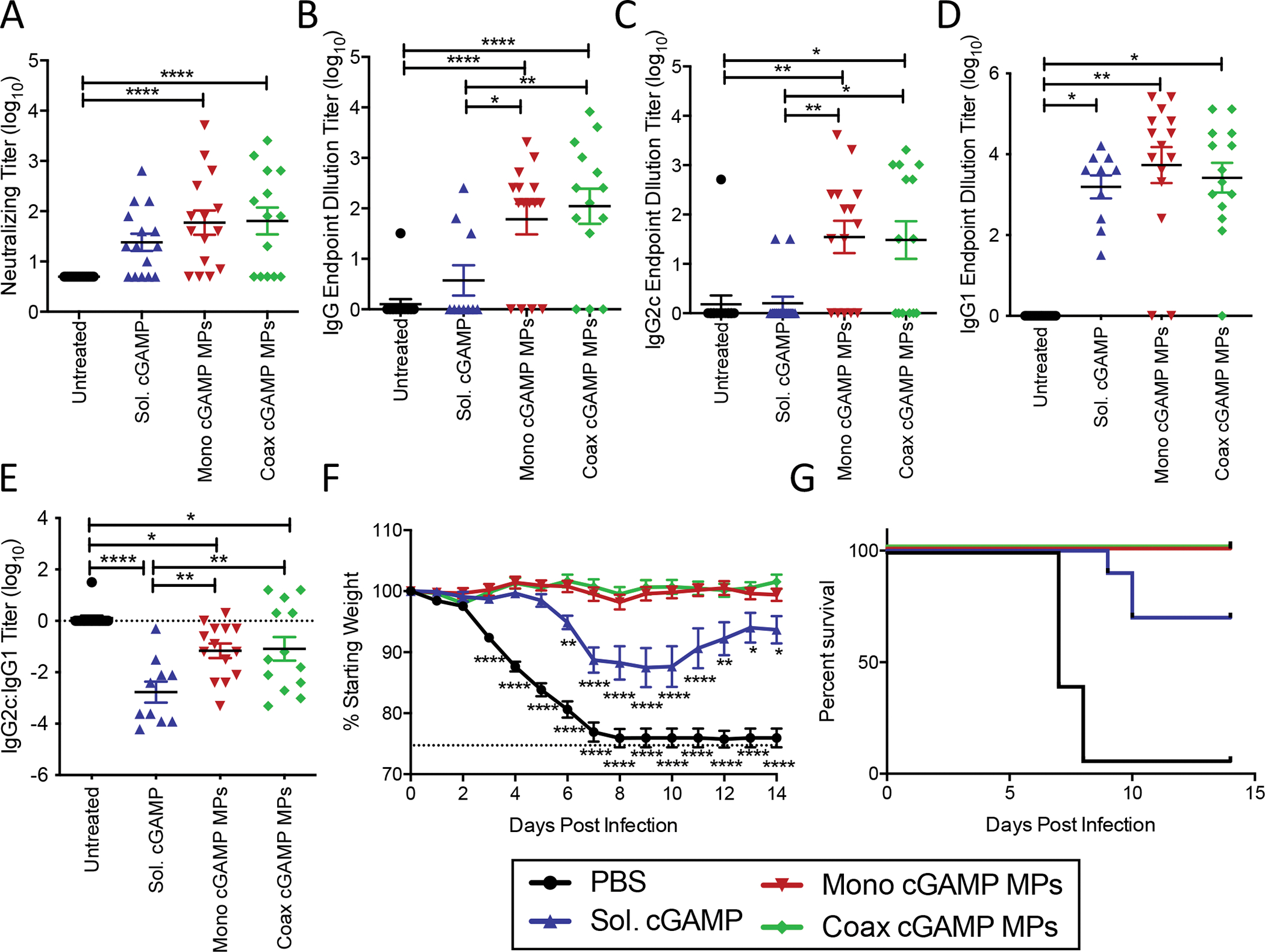
Eight-week-old female C57BL/6 mice were immunized i.m. with PBS, or recombinant HA from influenza strain A/Puerto Rico/8/34 (1 μg) adjuvanted with soluble cGAMP (0.2 μg), or Ace-DEX cGAMP MPs (0.2 μg cGAMP in 20 μg MPs) formulated through coaxial (coax) or monoaxial (mono) electrospray. Animals received a boost with the same formulation 21 days post-prime. (A) Serum was collected 28 days post-prime and virus neutralizing titers against A/Puerto Rico/8/34 were assessed. (C-D) The serum antibody endpoint titers at day 28 of HA specific (B) total IgG, (C) anti-viral Th1-skewed IgG2c, and (D) Th2-skewed IgG1 antibody isotypes were also assessed. (E) The ratio of IgG2c:IgG1 titers. (F-G) Following challenge with 5,000 ffu of influenza strain A/Puerto Rico/8/34, (F) weight loss, (G) and survival were monitored for 14 days. (n=10=15 ± SEM, *p < 0.05, **p < 0.01, ****p < 0.0001).
To test for in vivo biological activity of both types of cGAMP MPs, mice were challenged intranasally (i.n.) 56 days post-prime with a lethal dose of 5,000 FFU of PR8. Weight loss (Fig 2F) and survival (Fig 2G) were monitored for 14 days post challenge. Un-immunized mice succumbed to the infection within 7 to 8 days following infection. Mice immunized with soluble cGAMP showed partial protection against both mortality and weight loss, while mice immunized with either monoaxial or coaxial cGAMP MPs were completely protected from both weight loss and mortality. These results indicated that coaxial and monoaxial cGAMP MPs have similar bioactivity in this model. Changes in early cellular T cell responses were also observed in the draining popliteal lymph node (PLN) five days after boost with blank monoaxial electrospray MPs with HA, or monoaxial cGAMP MPs with or without HA antigen (Fig S3A–D). As compared to the other groups, mice immunized with HA + cGAMP MPs had elevated numbers of total CD8 T cells (Fig S3A), CD8 memory precursor effector cells (MPEC) (Fig S3B), total CD4 T cells (Fig S3C), and follicular helper (Tfh) CD4 T cells (Fig S3D). Increased numbers of MPECs and Tfh cells has been shown to correlate with improved T and B cell memory responses respectively [40, 41]. Due to the ease of manufacturing and potential enhanced scalability for monoaxial relative to coaxial electrospray, this former approach was used in all subsequent studies, and all cGAMP MPs hereafter refer to this form of the particles.
Subunit influenza vaccine adjuvanted with cGAMP MPs provides improved protection from influenza challenge compared to inactivated seasonal influenza vaccine in ferrets
We next compared a recombinant protein vaccine adjuvanted with cGAMP MPs to an inactivated seasonal influenza vaccine in ferrets. Ferrets are an important larger animal model for influenza infection. They were used in subsequent studies because, unlike mice, they can be infected with clinically relevant influenza A virus strains, allowing direct comparison to seasonal influenza vaccines. Furthermore, ferrets represent the most widely accepted model for assessing influenza vaccines as they display similar disease symptoms as humans, including fever, nasal discharge, and sneezing [36]. Ferrets were immunized intramuscularly with PBS, HA from strain A/California/07/09 adjuvanted with either soluble cGAMP or cGAMP MPs, or the 2016–17 formulation of the quadrivalent seasonal influenza vaccine Fluarix®, which contained A/California/07/09-like vaccine strain A/Christchurch/16/2010 (Table 1). Antigen doses were increased to account for the larger size of ferrets compared to mice and matched to the commercial vaccine (15 μg HA). The recommended pediatric 4-week prime/boost regimen was used for all immunizations. Serum was collected 21 and 49 days post-immunization for assessment of neutralizing titers (Fig. 3A). A time-dependent increase in neutralizing antibody titers was observed for all three vaccine formulations, while no virus-neutralizing activity was observed in PBS controls. The greatest neutralizing titers were generated by the HA + cGAMP MP vaccine formulation, which were 6.5-fold higher than the soluble cGAMP formulation, and 182-fold higher than the commercial Fluarix vaccine.
Figure 3. Ace-DEX cGAMP MP adjuvanted vaccine provides superior protection against influenza challenge in ferrets than a standard influenza vaccine:
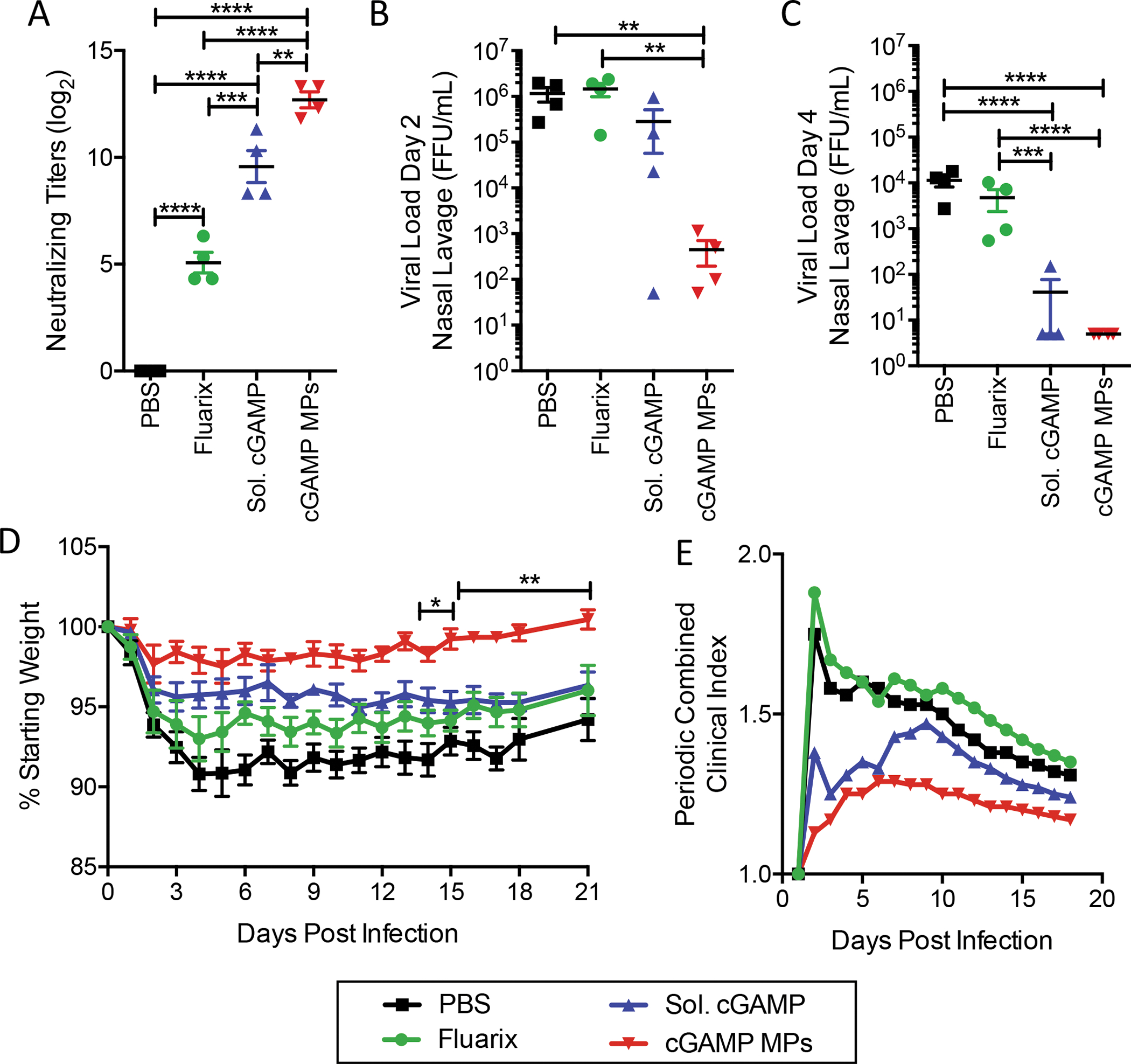
Four- to six-month-old male ferrets were injected i.m. with PBS, Fluarix (2016–2017 formulation), or HA from A/California/07/09 adjuvanted with soluble cGAMP (15 μg) or monoaxial Ace-DEX cGAMP MPs (15 μg cGAMP in 1.5 mg MPs). Animals received a boost with the same formulation 4 weeks later. (A) Serum was collected post boost (Day 49) and virus neutralizing titers against A/California/07/2009 were assessed. Ferrets were challenged intranasally with 1.25 × 107 FFU of Cali/07/09. Nasal lavage was collected (B) day 2 and (C) day 4 post challenge and viral load was assessed by foci forming assay. (D) Animal weights and (E) the periodic combined clinical index, a composite measure of animal activity and respiratory index (sneezing and nasal discharge), were assessed daily for 18 days following infection following infection. (n=4 ± SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
In order to assess protection, animals were infected i.n. with A/California/07/09, which was confirmed through assessment of serum neutralizing titers three weeks post infection (Figure S4A). Viral shedding was monitored in nasal lavage collected from ferrets on days two (Fig 3B) and four (Fig 3C) post-infection. PBS control animals showed significant viral shedding, with upwards of 106 FFU/mL lavage fluid recovered on day two post-infection, which decreased to approximately 104 FFU/mL by day four. The commercial quadrivalent Fluarix vaccine, which includes an A/California/07/09-like strain, did not impact viral shedding at either time point. In contrast, on day two the cGAMP MP formulation had significantly reduced viral shedding by greater than 2,500- and 3,200-fold compared to PBS and Fluarix, respectively. By day four, no viral shedding was detected in the cGAMP MP group. The soluble cGAMP vaccine formulation resulted in an intermediate reduction of viral shedding four days post-infection.
Weight loss was monitored for 21 days post-infection (Fig 3D). Control animals displayed rapid weight loss over the first five days, losing almost 10% of their starting weights. Both the commercial Fluarix vaccine and the HA + soluble cGAMP formulation partially mitigated weight loss. However, the HA + cGAMP MP formulation provided the greatest protection and nearly completely prevented infection-induced weight loss, which was significantly lower compared to all other groups. The combined clinical index, a cumulative measure of respiratory symptoms and activity level scores, was also tracked (Fig 3E). Control animals immunized with PBS showed a rapid onset of symptoms on day one post-infection, which gradually resolved over the course of the study. The commercial Fluarix vaccine did not impact the combined clinical index. However, both the soluble cGAMP and cGAMP MP vaccine formulations greatly decreased the early onset of clinical symptoms observed in the PBS and Fluarix groups.
Evaluation of cGAMP MP adjuvant dosing compared to a squalene adjuvant used in a seasonal flu vaccine
Although the HA adjuvanted with monoaxial cGAMP MP formulation was superior to the quadrivalent conventional seasonal vaccine in the context of a prime/boost vaccination regimen, there are many confounding factors that may have reduced the efficacy of Fluarix. Furthermore, the seasonal flu vaccine is normally given as a single dose, thus it is important to assess whether cGAMP MPs could achieve protection against influenza in this context. To expand our understanding of the efficacy of cGAMP MPs, we compared them head-to-head with AddaVax, an MF59-like squalene oil-in-water adjuvant that is considered one of the best FDA-approved adjuvants. This class of adjuvant is currently approved for use in FLUAD™, a seasonal inactivated influenza vaccine indicated for individuals over 65 years of age. Ferrets were immunized intramuscularly with PBS or a single (1x) dose of HA from the A/California/07/09 strain adjuvanted with either cGAMP MPs or a dose of AddaVax chosen to match the FLUAD squalene dose (9.75 mg). Alternatively, animals received both a prime and boost (2x) with HA + cGAMP MPs (Table 1). Serum was collected eight days prior to immunization, as well as 21 and 49 days post-prime immunization for assessment of neutralizing titers (Fig 4A). A 1x dose of HA adjuvanted with cGAMP MPs or AddaVax provided similar neutralizing titers while animals that received both a prime and boost with the cGAMP MP formulation displayed the most robust neutralizing titers. Serum collected pre-immunization, post-prime, post-boost, and post-infection showed no elevation in liver ALT enzymes, indicating that all formulations were well-tolerated (Fig S5).
Figure 4. Ace-DEX cGAMP MP adjuvanted vaccine provides decreases viral load following influenza challenge in ferrets:
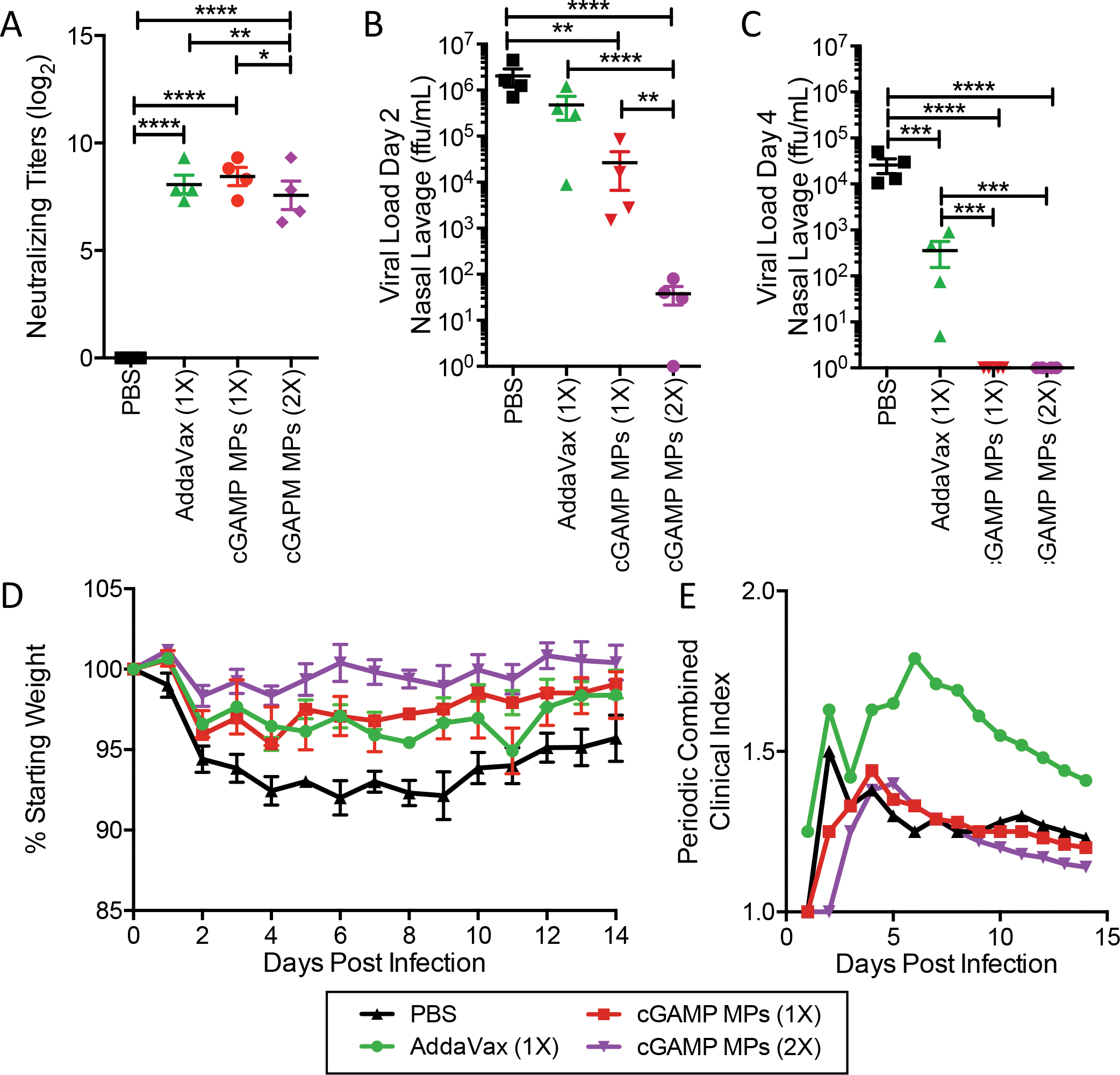
Four- to six-month-old male ferrets were injected i.m. with PBS, or immunized with HA from A/California/07/09 adjuvanted with AddaVax (9.75 mg squalene), or monoaxial Ace-DEX cGAMP MPs (15 μg cGAMP in 1.5 mg MPs). Animals in the cGAMP MPs (2X) group received a boost with the same formulation 4 weeks later. (A) Serum was collected post boost (Day 49) and virus neutralizing titers against A/California/07/09 were assessed. Ferrets were challenged intranasally with 1.25 × 107 FFU of influenza strain A/California/07/09. Nasal lavage was collected (B) day 2 and (C) day 4 post challenge and viral load was assessed by foci forming assay. (D) Animal weights and (E) the periodic combined clinical index, a composite measure of animal activity and respiratory index (sneezing and nasal discharge), were assessed daily following infection. (n=4 ± SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
A single dose of cGAMP MP adjuvant provides more rapid protection than a single dose of squalene adjuvant
To assess protection, animals were infected i.n. with A/California/07/09 and infection was confirmed through assessment of serum neutralizing titers three weeks post infection (Figure S4B). Viral shedding was assessed by nasal lavage on days two (Fig 4B) and four (Fig 4C) post-infection. Animals which received both a 2x dose of HA + cGAMP MPs displayed a drastic reduction in viral shedding, which was significantly lower than all other groups two days post-infection (Fig 4C). Animals receiving a 1x dose of the cGAMP MPs had significantly less viral shedding compared to untreated mice, while animals receiving the AddaVax formulation showed no reduction in viral shedding on day two. By day four, all animals receiving either a 1x or 2x dose with HA + cGAMP MPs did not shed any detectable virus. The animals immunized with HA + AddaVax showed a reduction in viral shedding compared to the unimmunized PBS controls but showed levels significantly higher than either the prime only or prime/boost HA + cGAMP MP groups.
Weight loss was monitored for 14 days post-infection (Fig 4D). Consistent with the first study, PBS control animals displayed significant weight loss following infection. Animals that received a 1x of HA adjuvanted with cGAMP MPs or AddaVax showed similar moderate levels of weight loss while animals which received a 2x with the HA + cGAMP MPs maintained their starting body weight. Animals immunized with the HA + AddaVax displayed more severe clinical symptoms, quantified using a combined clinical index, driven primarily by a worsened respiratory index (Fig 4E). Conversely, animals receiving either one or two doses of HA + cGAMP MPs displayed fewer clinical symptoms. These data show that although a prime and boost with HA + cGAMP MPs leads to greater protection short-term after vaccination, a single dose of cGAMP MP-adjuvanted HA is superior to a single dose of AddaVax-adjuvanted HA in clinical symptoms and the rate of viral load reduction.
cGAMP MPs promote a durable virus neutralizing vaccine response that lasted over a year
A common issue with the seasonal influenza vaccine is the relatively short-lived (3–4 months) protection afforded by the vaccine [7]. To assess the durability of the virus neutralizing response induced by vaccination with HA plus cGAMP MPs or a conventional squalene adjuvant, ferrets were immunized intramuscularly with HA (15 μg) adjuvanted with either a 1x dose or 2x dose of cGAMP MPs (15 μg cGAMP), or a 1x dose of AddaVax (9.75 mg squalene). Serum was collected monthly for a year to assess serum virus neutralizing titers (Fig. 5A). Similar to the findings in Figure 4, 2x of cGAMP MP formulations had increased neutralizing antibody titers short-term, but interestingly the 1x of HA + cGAMP MPs was sufficient to produce similar levels of neutralizing titers one year after vaccination. Importantly, both the 1x dose and 2x with HA + cGAMP MPs generated significantly higher neutralizing antibody titers than vaccination using 1x HA formulated with AddaVax (Fig 5A).
Figure 5. Prime-Boost of cGAMP MPs and AddaVax provide comparable protection:
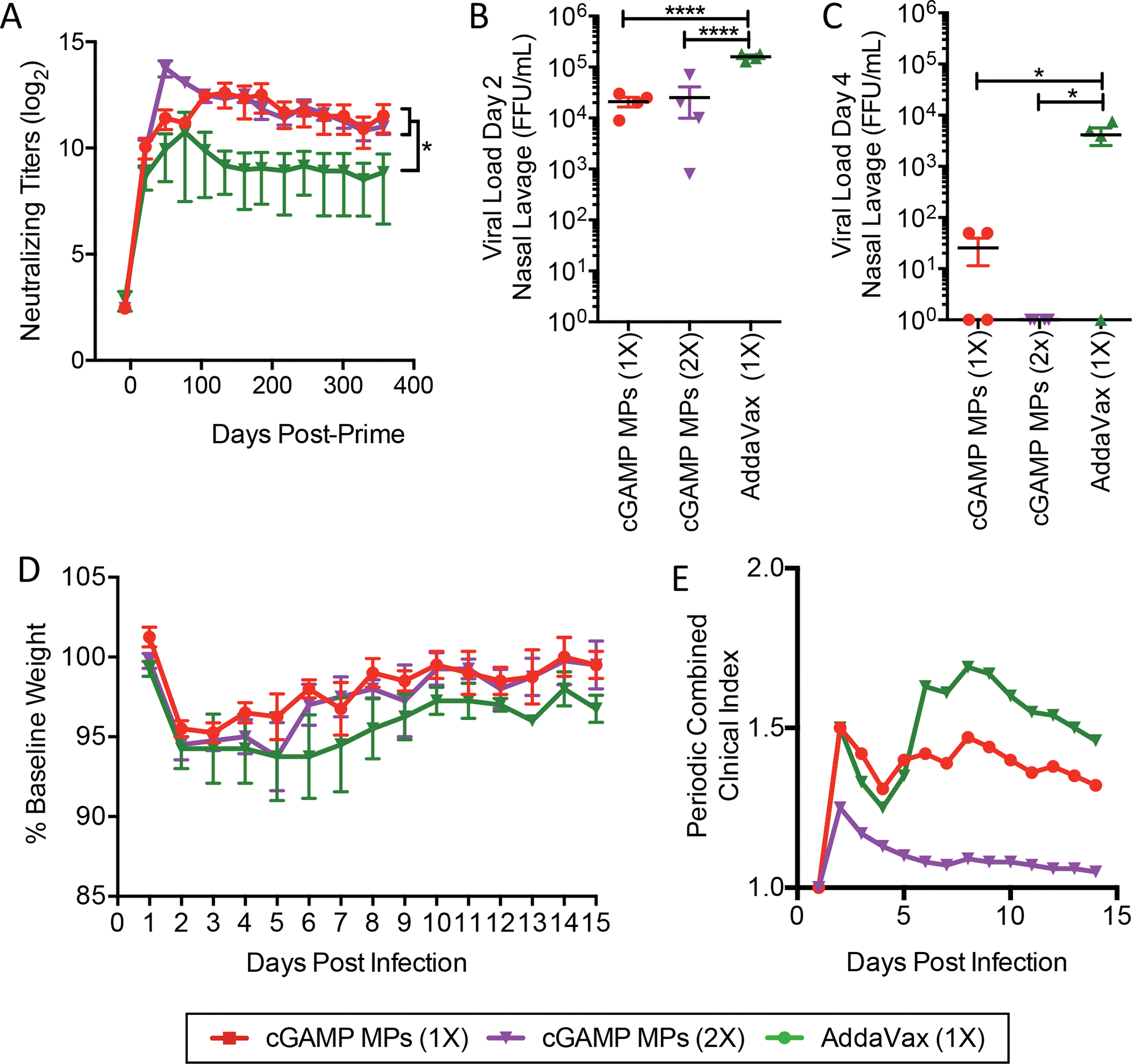
Four- to six-month-old male ferrets were immunized i.m. with a single dose of HA from A/California/07/09 (15 μg) adjuvanted with either monoaxial Ace-DEX cGAMP MPs (15 μg cGAMP in 1.5 mg MPs) or AddaVax (9.75 mg squalene). Animals in the cGAMP MPs (2X) group received a boost with the same formulation 4 weeks later. (A) Serum was collected monthly and virus neutralizing titers against A/California/07/09 were assessed. (n=4 ± SD, *p < 0.05). One year after immunization, ferrets were challenged intranasally with 1.25 × 107 FFU of influenza strain A/California/07/09. Nasal lavage was collected (B) day 2 and (C) day 4 post challenge and viral load was assessed by foci forming assay. (D) Animal weights and (E) the periodic combined clinical index, a composite measure of animal activity and respiratory index (sneezing and nasal discharge), were assessed daily following infection. (n=4 ± SD, *p < 0.05, ****p < 0.0001).
To assess the ability of HA + cGAMP MPs to generate long-term protection against influenza, ferrets were infected i.n. with A/California/07/09 one year after vaccination and infections were confirmed through assessment of serum neutralizing titers three weeks post infection (Fig S4C). No significant difference in viral shedding was observed between the 1x or 2x dose vaccination with HA + cGAMP MPs, two (Fig 5B) and four days (Fig 5C) post-infection. However, the cGAMP MPs led to significantly lower viral shedding two days post-infection compared to AddaVax. Furthermore, while viral shedding persisted in ferrets vaccinated with HA + AddaVax four days post-infection, viral shedding was nearly eliminated in ferrets that received HA vaccination with cGAMP MPs (Fig 5C). Of note, although 2x with the AddaVax formulation did increase neutralizing antibody titers similar to cGAMP MPs after one year, most of these ferrets still had low level viral shedding four days post infection (Fig S6A–C). There was no significant difference in the loss of body weight between the 1x HA + cGAMP MP vaccinated group compared to the 2x HA with AddaVax treated ferrets (Fig 5D). However, the 1x and 2x HA + cGAMP MP vaccinated ferrets appeared to recover quicker as shown by a lower periodic combined clinical index compared to 1x HA + AddaVax vaccinated ferrets (Fig 5E). Vaccination with 2x HA + AddaVax leads to a similar weigh loss profile (Fig S6D) and periodic clinical score (Fig S6E) as an 2x HA + cGAMP MP. These data show that HA vaccination with cGAMP MPs as an adjuvant is better than a state-of-the-art flu vaccine adjuvant class as measured by significantly enhanced neutralizing antibody titers and reduced viral load.
Discussion
The ultimate goal for an influenza vaccine is to generate robust and long-lived protective immunological memory, similar to the response after infection, using a platform that is easily scaled for clinical manufacturing. An advantage of subunit vaccines is that they avoid the use of live or attenuated virus that has the risk of becoming reactivated. Another preferred characteristic of a subunit vaccine is their ability to generate a robust immune response that similarly mimics host response after natural viral infection by including innate immune pathway-targeting adjuvants. This study was conducted to address these crucial issues. The monoaxial platform is used here for the first time in a vaccine and it provides a simpler formulation than the coaxial formulation used previously and allows for potentially more rapid and scalable production of the highly effective cGAMP MP adjuvant. Furthermore, it exhibits moderately less undesirable burst release of cGAMP than the MP produced using the coaxial platform. The monoaxial cGAMP MPs produce biologic effects that are indistinguishable from the coaxial cGAMP MPs both in vitro and in vivo. It also serves as a strong adjuvant that generates a robust and durable neutralizing immune response against HA, a non-infectious protein subunit of influenza virus. It achieves dose-sparing of cGAMP over particle formulations used in other reports, which is an important consideration because cGAMP is relatively costly. A final strength of the work is that the vaccine-induced immunity was measured in both mice and ferrets. The latter represents a gold-standard model of influenza infection [36] and this is the only report that has followed vaccine potency a year after immunization. These features significantly increase the translatability of the findings of this report.
In a side-by-side comparison, the monoaxial cGAMP MP formulation provided superior virus neutralizing titers, decreased disease symptoms, and reduced viral shedding compared to an inactivated influenza vaccine used in the clinic, and almost completely abrogated weight loss associated with influenza challenge (Fig 3–Fig 5). These results suggest that the monoaxial cGAMP MP formulation may address three critical shortcomings that afflict current influenza vaccines. First, the results clearly demonstrate improved efficacy over the clinical influenza vaccine, which is currently estimated to protect only 13–60% of immunized individuals [3]. Second, using a recombinant protein in lieu of inactivated virus allows for more rapid vaccine production compared to traditional egg-based methods, and eliminated the risk of mutations due to egg-adaptation [5, 6]. Finally, the lack of an inactivated virus may allow for improved plug-and-play of a variety of viral protein subunits for broader application of the formulation [42]. Importantly, immune responses generated by influenza vaccination with cGAMP MPs was illustrated to be long-lasting, leading to decreased viral shedding and clinical score during influenza challenge one year after vaccination, in the gold-standard ferret model of influenza.
Previously, Major et al. reported a vaccine adjuvanted with 50 μg of the soluble CDN cyclic di-GMP protected ferrets but did not reduce weight loss or viral load following influenza infection [43]. In contrast, our current findings show soluble cGAMP enhanced survival in mice and reduced both weight loss and viral shedding in ferrets. This may have been due to differences in the CDNs used, as Major et al. used cyclic di-GMP while the current study employed 3’3’ cGAMP. These two CDNs likely differ significantly in their ability to activate ferret STING, with reported EC50 values of 40 nM for 3’3’ cGAMP and 537 nM for cyclic di-GMP in digitonin-permeabilized mouse fibroblasts [44]. While their affinity for ferret STING has not been characterized, differences in STING binding and potency may exist. More recently, Wang et al. showed that subunit or inactivated virus vaccination adjuvanted with 2’3’ cGAMP-loaded nanoparticles that mimic a pulmonary surfactant for delivery to alveolar epithelial cells can elicit protection against homosubtypic and heterosubtypic influenza challenge [39]. While highly encouraging, these studies demonstrated only shorter-term protection in ferrets tested up to only 30 days after vaccination using a relatively high dose of cGAMP (200 μg of 2’3’ cGAMP) compared to our studies (15 μg of 3’3’ cGAMP). Also, both previous studies vaccinated the ferrets intranasally, while the current study used an intramuscular route, the most common administration route used clinically. Previous reports have indicated that mucosal immunization with CDNs drives a TH17 skewed response [45], generates IgA in the mucosa [46], and that the route of CDN administration greatly impacts the immune response [47]. Furthermore, no adjuvanted subunit intranasal influenza vaccines are currently approved for clinical use, making the intramuscular approach reported here more feasible for translation than the other formulations.
The in vitro results in the current study are very similar to what we previously observed with both type I IFN and pro-inflammatory cytokines IL-6 and TNF (Fig 1E–G) [22], and further strengthens the Ace-DEX MP platform’s ability to deliver STING agonists intracellularly. Others have reported similarly enhanced in vitro results of formulated compared to naked CDNs, yielding increased uptake [17], cytokine expression [17, 18, 48], and interferon regulatory factor 3 (IRF3) activation [20]. This study further demonstrates that stimulation with cGAMP MPs in vitro activates BMDCs to upregulate the molecular machinery needed for antigen presentation and co-stimulation with T cells (Fig 1H–K; Fig S2). Potent activation of antigen presenting cells may also drive the early increase in key T cell numbers seen in the draining PLN after immunization (Fig S3), but more work is needed to understand the cellular responses elicited by cGAMP MP adjuvanted vaccines. In the current study, the small but significant decrease in IL-6 following monoaxial cGAMP MP treatment compared to the coaxial MPs may be due to minor differences in the observed particle characteristics including differences in release kinetics [49], as well as particle size [50], shape [51], or surface topography [52], all of which can impact cellular uptake and/or inflammatory responses.
The in vivo mouse studies demonstrated that the monoaxial Ace-DEX cGAMP MP formulation showed comparable adjuvant activity to the previously described coaxial formulation (Fig 2) [22]. Both MP formulations drove a strong neutralizing and Th1 driven IgG2c humoral response and completely protected mice against a lethal influenza challenge. These results are consistent with various other studies that have used CDN STING agonists as influenza vaccine adjuvants to increase serum IgG titers [43, 47, 53–60], enhance Th1-biased IgG subtype titers [47, 53, 56–59], generate potent virus neutralizing antibody responses [53, 59], and protect against homosubtypic challenges [43, 47, 53, 57, 58]. Based on our previous studies, the cGAMP MP adjuvant effect is also largely cGAMP dependent as blank MPs had minimal vaccine adjuvant activity alone and Tmem173−/− mice that lack functional STING, the immune agonist effect was lost [22, 35]. As we have discussed previously [22], in contrast to these other mouse studies, the results we have observed with Ace-DEX cGAMP MPs are using 10 to 100-fold lower CDN doses via the clinically relevant intramuscular immunization route. This speaks to the strength of the Ace-DEX MP delivery system and demonstrates that the biological results can be recapitulated using the more recently designed monoaxial electrospray formulation method.
Previous studies using the MF59-like adjuvant AddaVax in ferret influenza vaccines, which employed similar squalene doses (10 mg), have shown only modest increases in (hemagglutination inhibition) HAI titers [61, 62] and decreases in viral shedding [61]. While it is difficult to directly compare HAI and virus neutralizing titers, these findings are consistent with our observation that the AddaVax-adjuvanted HA vaccine formulation did not reduce early viral shedding at day two, and only modestly reduced viral shedding at day four (Fig 4B and C). This is in contrast to the monoaxial cGAMP MP formulation that, after just a single dose, dramatically reduce early virus shedding and completely eliminate viral shedding by day four post challenge. Despite improved viral clearance, no differences in virus neutralizing antibody titers were observed between the AddaVax and the single dose of monoaxial cGAMP MPs. These findings are consistent with our previous observations that influenza virus neutralizing titers were poorly predictive of protection from lethal influenza challenge in Ace-DEX cGAMP MP-vaccinated mice[22]. This suggests that Ace-DEX cGAMP MPs promote protective immunity against influenza infection through mechanisms extending beyond direct virus neutralization, possibly through T cell memory responses or alternative antibody-dependent functions such as antibody induced cellular cytotoxicity, opsonization, or complement fixation. Future studies will be needed to better understand these mechanisms. Adding a secondary boost to the vaccination further decreased viral shedding and disease score, and a single dose of the vaccine with the cGAMP MPs was sufficient to drive robust immunity and outperform an MF59-like squalene vaccine adjuvant.
Together, these findings demonstrate that monoaxial Ace-DEX cGAMP MPs are an efficacious adjuvant for subunit influenza vaccines and offer improvements over both conventional influenza vaccines, as well as an influenza vaccine adjuvant class currently used clinically. The monoaxial method is simpler and more scalable than the coaxial method and has a dose-sparing effect compared to other particle-delivery methods in the literature. This study is also the first to address vaccine efficacy and durability for over a year in a large animal model. The formulation advances reported here set the groundwork for scale-up of monoaxial Ace-DEX cGAMP MP manufacturing, paving the way for further development of this promising vaccine adjuvant.
Methods
Materials
Unless otherwise noted, all materials were purchased from Sigma Aldrich (St. Louis, MO).
Synthesis of acetalated dextran polymer
Ace-DEX was synthesized from dextran (average molecular weight of 70 kDa) and characterized by 1H-nuclear magnetic resonance (NMR) spectroscopy according to Kauffman et al. [30]. Briefly, dextran was dissolved in anhydrous dimethyl sulfoxide (DMSO), and using a pyridinium p-toluenesulfonate catalyst, was reacted with 2-ethoxypropene (Matrix Scientific, Columbia, SC). To prepare Ace-DEX for the monoaxial electrospray MP formulation, an additional purification step was used. Polymer was dissolved in a 90/10 % v/v solution of ethanol/water, insolubilities were removed by centrifugation (20 minutes, 20,000 × g, 4 °C), the supernatant was evaporated off using a R-210 rotary evaporator (BUCHI Corporation, New Castle, DE), and the remaining solution was freeze-dried using a VirTis BenchTop K unit (SP Industries, Warminster, PA). The Ace-DEX used in all particle formulations had a relative cyclic acetal coverage of 40 ± 3% as assessed by NMR.
Formulation of coaxial cGAMP microparticles
The coaxial cGAMP particles were generated as previously reported[22]. Briefly, Ace-DEX was dissolved in an ethyl acetate/butanol/ethanol solution and 3’3’ cGAMP (InvivoGen, San Diego, CA) was dissolved in molecular grade water. The two solutions were driven by separate syringe pumps (Scientific Lab Supply, Acton, MA) through the sheath and core of a custom coaxial needle (Ramé-Hart Instrument Co., Succasunna, NJ). Using high voltage power sources (Gamma High Voltage Research, Inc., Ormond Beach, FL), the needle was charged with a negative polarity, while the stainless steel collection plate (McMaster Carr, Elmhurst, IL) was charged with a positive polarity. The plate was UV-treated and baked at 260 °C prior to use. A thin film of egg phosphatidylcholine (Avanti Polar Lipids, Alabaster, AL) was cast on the plate. Corresponding blank coaxial MPs were made by the same process without any cGAMP present.
Formulation of monoaxial cGAMP microparticles
The monoaxial cGAMP particles were generated as previously reported32. Briefly, Ace-DEX and 3’3’ cGAMP were dissolved together in an ethanol/molecular grade water solution. A single 20G blunt stainless-steel needle (Hamilton Company, Reno, NV) was used in place of the coaxial needle, and the particles were generated via a similar setup as the one used to make the coaxial particles. However, in this case no egg phosphatidylcholine was pre-cast on the collection plate. Corresponding blank monoaxial MPs were made by the same process without any cGAMP present.
Characterization of cGAMP microparticles
All particle characterization methods were completed as previously reported [22, 27, 35]. Briefly, all particles were deemed to have undetectable endotoxin (<0.125 EU/mg) per the LAL Chromogenic Endotoxin Quantitation Kit (Waltham, MA). For imaging by scanning electron microscopy (SEM), particles were cast on an aluminum stub (Ted Pella, Redding, CA), and after being sputter coated with palladium, were imaged on a S-4700 Cold Cathode Field Emission SEM (Hitachi, Tokyo, Japan). The particle hydrodynamic size by volume was acquired using a Nanobrook 90Plus Zeta (Brookhaven Instruments Corporation, Holtsville, NY). cGAMP loading was determined as previously described[22] using a high-performance liquid chromatography (HPLC) method by detecting cGAMP at 254 nm. Briefly, HPLC was run using an isocratic mobile phase (80% water:20% methanol v/v) at a rate of 0.6 mL/min through a Aquasil C18 (Thermo Fisher Scientific, Waltham, MA). To determine the cGAMP release profile, the particles were incubated in PBS in the presence or absence of 10% v/v FBS pH 7.4 at 37 °C and at predetermined time points the same HPLC method was used to measure percent cGAMP released.
In vitro assessment of particle bioactivity and cytocompatibility
Murine bone marrow-derived dendritic cells (BMDCs) were prepared according to Lutz et al. [63]. Cells were seeded in a 96-well plate at a density of 1 × 105 cells/well in 100 μL of RPMI containing 10% FBS and 50 U/mL of each penicillin and streptomycin and allowed to adhere overnight. Following a media change, cells were treated with 1 μg/mL soluble cGAMP, or a total dose of 1 μg/mL cGAMP delivered in monoaxial or coaxial cGAMP MPs for 24 hours for cytokine production and up to 48 hours for upregulation of MHCII and costimulatory molecules. Untreated cells, and cells treated with an equivalent dose of unloaded blank monoaxial and coaxial MPs, were included as controls. For cytokine analysis supernatants were collected, and cytokines assessed by ELISA. IL-6 and TNF were assessed using kits from BD Biosciences (San Jose, CA). As previously described by Junkins et al. [22], the IFN-β murine ELISA was completed using mouse-specific IFN-β antibodies from Santa-Cruz Biotechnology (Dallas, TX) and R&D Systems (Minneapolis, MN), anti-Rabbit IgG HRPO from Cell Signaling Technology (Danvers, MA), and recombinant IFN-β standard from R&D Systems. To examine cytocompatibility, once the supernatants were removed for analysis by ELISA, cell metabolic activity was assessed by the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay per Gallovic et al. [64]. Cells were incubated with 0.6 mg/mL of the MTT reagent, and absorbance was read at 560/670 nm after resulting formazan crystals were dissolved in isopropanol. To analyze upregulation of BMDC activation markers, cells were resuspended in PBS with 10% FBS and 1 mM EDTA, treated with an Fc Block (anti-CD16/32 clone 2.4.G2 antibody from BioXcell in PBS with 10% rat serum and 2% FBS), and then fluorescently labeled with: anti-CD11c-APC (Clone HL3, BD Bioscience), anti-IA/IE-FITC (Clone G29, BD Biosciences), anti-CD40-PE (Clone 1C10, eBiosciences), anti-CD80-PerCP-Cy5.5 (Clone 16–10A1, BioLegend), anti-CD86-APC-Cy7 (Clone GL1, BioLegend). Sample data were collected on an LSRFortessa (BD Biosciences) and analyzed by FlowJo V10.8.1.
Institutional Animal Care and Use Committee statement
All studies were conducted in accordance with the National Institutes of Health’s guidelines for the care and use of laboratory animals. Mouse procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina (UNC) and ferret procedures were approved by the Duke University IACUC.
Recombinant hemagglutinin protein production and purification
Hemagglutinin proteins including amino acids 1–519 from Influenza A/Puerto Rico/8/1934 (H1N1) or A/California/04/2009 (H1N1) fused to a C-terminal TEV protease cleavage site, a T4 bacteriophage fibritin foldon trimerization domain, and a 6x His tag were expressed from CHO DG44 stable cell lines. Cells were scaled to final expression volumes in OptiCHO media supplemented with 8 mM glutamine in shaker flasks, grown to 1–2 million cells per ml, followed by addition of 30% (v/v) CHO CD EfficientFeed A Nutriant Supplement. Cultures were then transferred to 32°C, and incubated for 10–14 days. Supernatants were harvested by centrifugation at 12,000×g for 45 minutes, and filtered through 0.45 μm membranes into sterile bottles.
Supernatants were concentrated and buffer-exchanged via tangential-flow filtration into nickel-binding buffer (50 mM sodium phosphate pH 7.4, 500 mM NaCl, 20 mM imidazole, 0.02% sodium azide), and subjected to nickel-affinity chromatography on HisTrap HP columns (GE Life Sciences). After washing with nickel binding buffer, proteins were eluted with binding buffer supplemented with 500 mM imidazole. Fractions containing hemagglutinin protein were pooled and concentrated, followed by removal of C-terminal tags by incubation with TEV protease (1 mg protease/100 mg substrate) in a dialysis cassette dialyzing against nickel binding buffer. Cleaved proteins were rerun through the nickel-affinity column, with tagless hemagglutinin protein collected in the flowthrough fractions. Proteins were subsequently subjected to size-exclusion chromatography on a Superdex S200 16/600 column equilibrated with PBS with 10% glycerol to remove residual protein contaminants and confirm trimerization. Hemagglutinin-containing fractions were pooled, concentrated to 1–5 mg/ml, flash frozen in liquid nitrogen, and stored at −80°C. Final protein purity was assessed as >95% pure by SDS-PAGE.
In vivo immunization
Mice were injected intramuscularly (50 μL) with each of the groups indicated in Table 1 on day 0 and 21 using hemagglutinin (HA-PR8) from H1N1 strain A/Puerto Rico/8/1934. Antigen was given in soluble form, and when delivered with either coaxaxial or monoaxial cGAMP MPs, premixed with the MPs immediately prior to injection. The monoaxial MPs were suspended in 1 mg/mL egg phosphatidylcholine in PBS to achieve homogenous dispersion. The mice were bled submandibularly on day 14 and 28 to collect serum for assessing IgG antibody endpoint titers. Barrier-raised, influenza-free, four-to-six-month-old, de-scented, and reproductively intact male ferrets (Mustela putorius furo) were purchased from Marshall BioResources. As required by experimental procedure, ferrets were anesthetized using inhalant isoflurane (Patterson Veterinary Supply) or an injectable mixture of ketamine HCL (30 mg/kg, Henry Schein) and xylazine (3 mg/kg, AKORN) or ketamine HCL (6 mg/kg) and dexmedetomidine HCL (75 mg/kg, Putney). Prior to immunization all ferrets were evaluated by a Duke Division of Laboratory Animal Research veterinarian and subcutaneously implanted with an identification/temperature transponder (Bio Medic Data Systems).
Ferrets were injected intramuscularly (≤ 500 μL) with each of the groups indicated in Table 1 on day 0 only, or days 0 and 28, as indicated. For soluble cGAMP, monoaxial cGAMP MP, and AddaVax-adjuvanted groups, recombinant H1N1 strain A/California/07/2009 hemagglutinin was used as the antigen. The inactivated FDA-approved quadrivalent Fluarix from the 2016–17 flu season included inactivated A/California/07/2009-like virus A/Christchurch/16/2010 (H1N1) NIB-74XP (15 μg HA), A/Hong Kong/4801/2014 (H3N2) NYMC X-263B (15 μg HA), B/Phuket/3073/2013 (15 μg HA), and B/Brisbane/60/2008 (15 μg HA). The AddaVax dose was matched with the clinical dose of squalene found in the FDA-approved FLUAD™ vaccine. For immunological assessments, ferrets were bled via jugular venipuncture, vena cava bleed, or terminal cardiac puncture.
Preparation of influenza viruses
Purified influenza virus A/Puerto Rico/8/1934 H1N1 (PR8; Charles River) was diluted in ultra-pure phosphate-buffered saline to generate mouse infection stocks. PR8 was also propagated in embryonated specific-pathogen-free chicken hen eggs (Sunrise Farms Inc.) to generate stocks for use in the influenza microneutralization assay. Stocks of A/California/07/2009 (H1N1)pdm09 (Cali/07/09; IRR, FR-458) were generated via propagation in MDCK CCL-34 cells. All influenza virus stocks were aliquoted and stored at −80 °C until use.
In vivo challenge and monitoring
Mice were anesthetized with 400 mg/kg of Avertin. Upon induction, mice were challenged i.n. in a single nare with 5,000 FFU of the PR8 flu strain (Charles River) in 20 μL of 0.9% w/v saline, followed by a 10 μL saline wash. Mice were monitored for body weight loss and overall condition up to 14 days post-challenge. Morbidity or a 25% loss in body weight was used as a survival endpoint per the protocol approved by the IACUC at UNC.
Ferrets were anesthetized with isoflurane and upon induction challenged with A/California/07/09, 3.4×106 - 1.25×107 FFU (i.n.) diluted in 0.9% USP saline in 1.0 mL distributed equally between both nares. Ferrets were monitored daily for body weight loss, changes in activity, and respiratory symptoms (coughing, sneezing, nasal discharge, difficulty breathing, etc.) for at least 14 days post-challenge. Respiratory and activity scores were assigned, and indices calculated similar as to previously described[65, 66]. Virus shedding in respiratory secretions was assessed at days two and four post-infection via nasal lavage with 0.9% USP saline. At 21 days post-challenge ferrets were anesthetized and administered Euthasol (Virbac) intracardiac, euthanasia was confirmed by bilateral thoracotomy.
Influenza foci forming assay
Influenza stock titers and viral load in ferret nasal lavage samples were determined by a standard focus forming assay on MDCK CCL-34 cells [67, 68]. Briefly, serial dilutions of test sample were inoculated onto Mandin-Darby Canine Kidney (MDCK; ATCC® CCL-34™) cells and incubated in the presence of an Avicel (FMC BioPolymer) based overlay. Plates were then fixed and viral foci detected immunocytochemically via sequential incubation with anti-influenza A nucleoprotein clones A1 and A3, a HPR conjugated goat anti-mouse IgG (SeraCare), and TrueBlue™ peroxidase substrate (SeraCare). Viral titers are expressed as focus forming units per mL (FFU/mL).
Influenza microneutralization assay
Influenza endpoint neutralization titers were determined using an adaptation of the CDC/WHO influenza MN assay[69, 70] similar to as previously described [22, 71]. Briefly, serial dilutions of heat-inactivated sera were incubated with influenza virus followed by infection of MDCK London cells. Plates were then incubated, acetone fixed, and influenza virus infected cells were detected using either biotin-conjugated (mouse sera) or un-labeled (ferret sera) anti-influenza A NP antibodies (clones A1 and A3) and a streptavidin-HRP or goat anti-mouse IgG-HRP, respectively. Plates were developed using an o-Phenylenediamine dihydrochloride based substrate, absorbance (490 nm) measured and neutralization cutoffs established using assay controls.
Antigen-specific endpoint binding titers
Antigen-specific endpoint immunoglobulin (Ig) isotyping were determined by standard ELISA similar to previous studies [22, 33, 72]. Briefly serial dilutions of sera (two-fold) were performed in HA-PR8 coated plates (384-well format), plates were then blocked, washed, incubated with a horseradish peroxidase-conjugated species-specific detection antibody (Southern Biotech), developed using a TMB peroxidase substrate (SeraCare), and absorbance (450 nm) measured using a Synergy H1 (BioTek) plate reader.
Statistics
All statistical analyses were performed with GraphPad Prism Version 6, 8, or 9. Analysis of groups was performed using one-way, or two-way ANOVA followed by post hoc Tukey’s multiple comparison’s test. Comparison of vaccine durability over time was assessed using area under the curve analysis followed by an unpaired t-test.
Supplementary Material
Figure S1. Ace-DEX cGAMP MPs formulated via monoaxial or coxial electrospray remain stable when suspended in PBS with 10% FBS. (A) Scanning electron microscopy (SEM) images were taken of the coaxial and monoaxial MPs at 0, 48, and 168 hr timepoints. (B) The release of cGAMP from coaxial or monoaxial electrospray MPs when incubated at 37°C in PBS with 10% (v/v) FBS (n=3 ±SD) was measured.
Figure S2. Ace-DEX cGAMP MPs formulated via monoxial upregulation of antigen presentation and costimulatory molecules: Bone marrow derived dendritic cells (BMDCs) from C57BL/6 mice were treated with 1 μg/mL cGAMP delivered as soluble cGAMP or encapsulated within Ace-DEX cGAMP MPs at a loading of 10 μg/mg MP monoaxial electrospray. After culture for 4, 24, and 48 hours, expression of antigen presentation and co-stimulation molecules were analyzed by flow cytometry. A) Representative histograms of MHCII, CD40, CD80, CD86 antibody staining on treated BMDCs gated on singlets, debris negative, and CD11c+ cells at 4 (black), 24 (blue) and 48 (red) hours. (B-C) Quantification of mean fluorescent intensity (MFI) of (B) MHCII, (C) CD40, (D) CD80, (E) CD86 over time after treatment. (n=6 ± SEM).
Figure S3. Cellular T cell responses early after prime-boost immunization with Ace-DEX cGAMP MPs generated by monoaxial electrospray. Eight-week-old female C57BL/6 mice were immunized i.m. with PBS, or recombinant HA from influenza strain A/Puerto Rico/8/34 (1 μg) adjuvanted with soluble cGAMP (0.2 μg), or Ace-DEX cGAMP MPs (0.2 μg cGAMP in 20 μg MPs) formulated through monoaxial (mono) electrospray. Animals received a boost with the same formulation 21 days post-prime and T cell responses in the draining PLN were analyzed by flow cytometry on day 26. (A-D) Quantification of total number of (A) CD8 (defined as CD3+CD8+), (B) MPEC CD8 (defined as CD3+CD8+CD44hiCD127+KLRG1−), (C) total CD4 (defined as CD3+CD4+), and (D) Tfh CD4 T cells (defined as CD3+CD4+CXCR5+PD-1+) per draining popliteal lymph node (PLN). (n=10 ± SEM. Samples also pre-gated for live singlet lymphocytes.)
Figure S4. Infection was confirmed with post-infection neutralizing titers. Ferrets were immunized according to the experimental design described in (A) Figure 3, (B) Figure 4 or (C) Figure 5. Serum was collected 21-28 days post influenza challenge and infection was confirmed by virus neutralizing titer. (n=4 ± SD. All data are n.s.).
Figure S5. Immunization does not increase serum ALT activity. Ferrets were immunized according to experimental outline in Figure 4. Serum was collected on day −7 (Pre-Bleed), Day 21 (Post-Prime) and day 49 (Post-Boost) and on day 84 (Post-Infection) ALT activity was assessed. (n=4 ± SD).
Figure S6. Prime-Boost of cGAMP MPs and AddaVax provide comparable protection: Four to six month old male ferrets were immunized IM with a single dose of HA from A/California/07/09 (15 μg) adjuvanted with either monoaxial Ace-DEX cGAMP MPs (15 μg cGAMP @ 1% weight loading) or AddaVax (9.75 mg squalene). (A) Serum was collected monthly and virus neutralizing titers against A/California/07/09 were assessed. (n=4 ± SD, *p < 0.05). One year after immunization, ferrets were challenged intranasally with 1.25 × 107 FFU of influenza strain A/California/07/09. Nasal lavage was collected (B) day 2 and (C) day 4 post challenge and viral load was assessed by foci forming assay. (D) Animal weights and (E) the periodic combined clinical index, a composite measure of animal activity and respiratory index (sneezing and nasal discharge), were assessed daily following infection. (n=4 ± SD, *p < 0.05, ****p < 0.0001).
Acknowledgements
We thank the UNC Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) for SEM usage. This work was supported by U19 AI109784 (to JPYT, GDS and EMB), UNC internal funds (to KMA), Lineberger Comprehensive Cancer Center internal funding (JPYT and KMA), T32-AI007151 (to RDJ), T32-CA196589 (to AS), and a Biotechnology Innovation Grant from the North Carolina Biotechnology Center # 2018-BIG-6504 (to JPYT and KMA). Antigen production for this work was performed at the UNC Protein Expression and Purification (PEP) core by John Forsberg, Michael Miley and Nathan Nicely, and partially supported by the National Cancer Institute of the National Institutes of Health under award number P30CA016086.
Influenza virus propagation, micro-neutralization assays, and ferret studies were done by the Virology Unit of the Duke Regional Biocontainment Laboratory (RBL) under the management of Dr. Charles E. McGee. The Duke RBL received partial support for construction from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (UC6-AI058607).
This work was performed in part at the Chapel Hill Analytical and Nanofabrication Laboratory, CHANL, a member of the North Carolina Research Triangle Nanotechnology Network, RTNN, which is supported by the National Science Foundation, Grant ECCS-1542015, as part of the National Nanotechnology Coordinated Infrastructure, NNCI.
The following reagents were obtained through BEI Resources, NIAID, NIH: Monoclonal Anti-Influenza A Virus Nucleoprotein (NP), Clones A1 and A3 (ascites blend, Mouse), NR-4282 and Ferret Hyperimmune Sera to Influenza A/California/07/2009 (H1N1)pdm09, NR-19261. Influenza A Virus, A/California/07/2009 (H1N1)pdm09 Antiviral Resistance (AVR) - Reference Virus M2: S31N NA: wild type (wt), FR-458 and Madin-Darby Canine Kidney (MDCK) Cells, London Line, FR-58 were obtained through the International Reagent Resource (formerly the Influenza Reagent Resource), Influenza Division, WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza, Centers for Disease Control and Prevention, Atlanta, GA, USA.
References
- [1].World Health Organization, Up to 650 000 people die of respiratory diseases linked to seasonal flu each year, in, 2017.
- [2].Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB, The annual impact of seasonal influenza in the US: measuring disease burden and costs, Vaccine, 25 (2007) 5086–5096. [DOI] [PubMed] [Google Scholar]
- [3].Centers for Disease Control and Prevention, Influenza (flu), in, 2018.
- [4].Paules CI, Sullivan SG, Subbarao K, Fauci AS, Chasing Seasonal Influenza - The Need for a Universal Influenza Vaccine, N Engl J Med, (2017). [DOI] [PubMed] [Google Scholar]
- [5].Wu NC, Zost SJ, Thompson AJ, Oyen D, Nycholat CM, McBride R, Paulson JC, Hensley SE, Wilson IA, A structural explanation for the low effectiveness of the seasonal influenza H3N2 vaccine, PLoS Pathog, 13 (2017) e1006682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zost SJ, Parkhouse K, Gumina ME, Kim K, Diaz Perez S, Wilson PC, Treanor JJ, Sant AJ, Cobey S, Hensley SE, Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains, Proc Natl Acad Sci U S A, 114 (2017) 12578–12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kissling E, Valenciano M, Larrauri A, Oroszi B, Cohen JM, Nunes B, Pitigoi D, Rizzo C, Rebolledo J, Paradowska-Stankiewicz I, Jimenez-Jorge S, Horvath JK, Daviaud I, Guiomar R, Necula G, Bella A, O’Donnell J, Gluchowska M, Ciancio BC, Nicoll A, Moren A, Low and decreasing vaccine effectiveness against influenza A(H3) in 2011/12 among vaccination target groups in Europe: results from the I-MOVE multicentre case-control study, Euro surveillance : bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin, 18 (2013). [DOI] [PubMed] [Google Scholar]
- [8].Dunkle LM, Izikson R, Patriarca PA, Goldenthal KL, Muse D, Cox MMJ, Randomized Comparison of Immunogenicity and Safety of Quadrivalent Recombinant Versus Inactivated Influenza Vaccine in Healthy Adults 18–49 Years of Age, J Infect Dis, 216 (2017) 1219–1226. [DOI] [PubMed] [Google Scholar]
- [9].O’Hagan D, Ott G, De Gregorio E, Seubert A, The mechanism of action of MF59–an innately attractive adjuvant formulation, Vaccine, 30 (2012) 4341–4348. [DOI] [PubMed] [Google Scholar]
- [10].O’Hagan DT, MF59 is a safe and potent vaccine adjuvant that enhances protection against influenza virus infection, Expert review of vaccines, 6 (2007) 699–710. [DOI] [PubMed] [Google Scholar]
- [11].Del Giudice G, Rappuoli R, Didierlaurent AM, Correlates of adjuvanticity: A review on adjuvants in licensed vaccines, in: Seminars in immunology, Elsevier, 2018. [DOI] [PubMed] [Google Scholar]
- [12].Maisonneuve C, Bertholet S, Philpott DJ, De Gregorio E, Unleashing the potential of NOD-and Toll-like agonists as vaccine adjuvants, Proceedings of the National Academy of Sciences, 111 (2014) 12294–12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hogner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, Walmrath HD, Bodner J, Gattenlohner S, Lewe-Schlosser P, Matrosovich M, Seeger W, Lohmeyer J, Herold S, Macrophage-expressed IFN-beta contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia, PLoS Pathog, 9 (2013) e1003188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].O’Neill LA, Golenbock D, Bowie AG, The history of Toll-like receptors—redefining innate immunity, Nature Reviews Immunology, 13 (2013) 453. [DOI] [PubMed] [Google Scholar]
- [15].Zevini A, Olagnier D, Hiscott J, Crosstalk between cytoplasmic RIG-I and STING sensing pathways, Trends in immunology, 38 (2017) 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S, Irvine DJ, Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants, The Journal of clinical investigation, 125 (2015) 2532–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ, Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy, Advanced Biosystems, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee E, Jang HE, Kang YY, Kim J, Ahn JH, Mok H, Submicron-sized hydrogels incorporating cyclic dinucleotides for selective delivery and elevated cytokine release in macrophages, Acta biomaterialia, 29 (2016) 271–281. [DOI] [PubMed] [Google Scholar]
- [19].Nakamura T, Miyabe H, Hyodo M, Sato Y, Hayakawa Y, Harashima H, Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma, Journal of controlled release : official journal of the Controlled Release Society, 216 (2015) 149–157. [DOI] [PubMed] [Google Scholar]
- [20].Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, Kim YJ, Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy, Nanomedicine: Nanotechnology, Biology and Medicine, 14 (2018) 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Goodwin TJ, Huang L, Investigation of phosphorylated adjuvants co-encapsulated with a model cancer peptide antigen for the treatment of colorectal cancer and liver metastasis, Vaccine, 35 (2017) 2550–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Junkins RD, Gallovic MD, Johnson BM, Collier MA, Watkins-Schulz R, Cheng N, David CN, McGee CE, Sempowski GD, Shterev I, A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination, Journal of Controlled Release, 270 (2018) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Van Herck S, Feng B, Tang L, Delivery of STING agonists for adjuvanting subunit vaccines, Adv Drug Deliv Rev, 179 (2021) 114020. [DOI] [PubMed] [Google Scholar]
- [24].Garland KM, Rosch JC, Carson CS, Wang-Bishop L, Hanna A, Sevimli S, Van Kaer C, Balko JM, Ascano M, Wilson JT, Pharmacological Activation of cGAS for Cancer Immunotherapy, Front Immunol, 12 (2021) 753472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF, STING pathway agonism as a cancer therapeutic, Immunol Rev, 290 (2019) 24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Irvine DJ, Hanson MC, Rakhra K, Tokatlian T, Synthetic Nanoparticles for Vaccines and Immunotherapy, Chemical reviews, 115 (2015) 11109–11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kohane DS, Microparticles and nanoparticles for drug delivery, Biotechnology and bioengineering, 96 (2007) 203–209. [DOI] [PubMed] [Google Scholar]
- [28].Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Frechet JM, Acetal-derivatized dextran: an acid-responsive biodegradable material for therapeutic applications, Journal of the American Chemical Society, 130 (2008) 10494–10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Broaders KE, Cohen JA, Beaudette TT, Bachelder EM, Frechet JM, Acetalated dextran is a chemically and biologically tunable material for particulate immunotherapy, Proceedings of the National Academy of Sciences of the United States of America, 106 (2009) 5497–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kauffman KJ, Do C, Sharma S, Gallovic MD, Bachelder EM, Ainslie KM, Synthesis and characterization of acetalated dextran polymer and microparticles with ethanol as a degradation product, ACS applied materials & interfaces, 4 (2012) 4149–4155. [DOI] [PubMed] [Google Scholar]
- [31].Bachelder EM, Pino EN, Ainslie KM, Acetalated dextran: a tunable and acid-labile biopolymer with facile synthesis and a range of applications, Chemical reviews, 117 (2016) 1915–1926. [DOI] [PubMed] [Google Scholar]
- [32].Peine KJ, Bachelder EM, Vangundy Z, Papenfuss T, Brackman DJ, Gallovic MD, Schully K, Pesce J, Keane-Myers A, Ainslie KM, Efficient delivery of the toll-like receptor agonists polyinosinic:polycytidylic acid and CpG to macrophages by acetalated dextran microparticles, Mol Pharm, 10 (2013) 2849–2857. [DOI] [PubMed] [Google Scholar]
- [33].Collier MA, Junkins RD, Gallovic MD, Johnson BM, Johnson MM, Macintyre AN, Sempowski GD, Bachelder EM, Ting JP, Ainslie KM, Acetalated Dextran microparticles for co-delivery of STING and TLR7/8 agonists, Mol Pharm, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kanthamneni N, Sharma S, Meenach SA, Billet B, Zhao J-C, Bachelder EM, Ainslie KM, Enhanced stability of horseradish peroxidase encapsulated in acetalated dextran microparticles stored outside cold chain conditions, International journal of pharmaceutics, 431 (2012) 101–110. [DOI] [PubMed] [Google Scholar]
- [35].Watkins-Schulz R, Tiet P, Gallovic MD, Junkins RD, Batty C, Bachelder EM, Ainslie KM, Ting JPY, A microparticle platform for STING-targeted immunotherapy enhances natural killer cell- and CD8(+) T cell-mediated anti-tumor immunity, Biomaterials, 205 (2019) 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Belser JA, Katz JM, Tumpey TM, The ferret as a model organism to study influenza A virus infection, Dis Model Mech, 4 (2011) 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Matsuoka Y, Lamirande EW, Subbarao K, The ferret model for influenza, Curr Protoc Microbiol, Chapter 15 (2009) Unit 15G 12. [DOI] [PubMed] [Google Scholar]
- [38].Maher JA, DeStefano J, The ferret: an animal model to study influenza virus, Lab Anim (NY), 33 (2004) 50–53. [DOI] [PubMed] [Google Scholar]
- [39].Wang J, Li P, Yu Y, Fu Y, Jiang H, Lu M, Sun Z, Jiang S, Lu L, Wu MX, Pulmonary surfactant-biomimetic nanoparticles potentiate heterosubtypic influenza immunity, Science, 367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM, Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor, Immunity, 27 (2007) 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mastelic Gavillet B, Eberhardt CS, Auderset F, Castellino F, Seubert A, Tregoning JS, Lambert PH, de Gregorio E, Del Giudice G, Siegrist CA, MF59 Mediates Its B Cell Adjuvanticity by Promoting T Follicular Helper Cells and Thus Germinal Center Responses in Adult and Early Life, J Immunol, 194 (2015) 4836–4845. [DOI] [PubMed] [Google Scholar]
- [42].Yeung MP, Lam FL, Coker R , Factors associated with the uptake of seasonal influenza vaccination in adults: a systematic review, J Public Health (Oxf), 38 (2016) 746–753. [DOI] [PubMed] [Google Scholar]
- [43].Major D, Chichester JA, Pathirana RD, Guilfoyle K, Shoji Y, Guzman CA, Yusibov V, Cox RJ, Intranasal vaccination with a plant-derived H5 HA vaccine protects mice and ferrets against highly pathogenic avian influenza virus challenge, Human vaccines & immunotherapeutics, 11 (2015) 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhang X, Shi H, Wu J, Sun L, Chen C, Chen ZJ, Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING, Mol Cell, 51 (2013) 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Skrnjug I, Guzman CA, Rueckert C, Cyclic GMP-AMP displays mucosal adjuvant activity in mice, PLoS One, 9 (2014) e110150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Varma DM, Batty CJ, Stiepel RT, Graham-Gurysh EG, Roque JA 3rd, Pena ES, Hasan Zahid MS, Qiu K, Anselmo A, Hill DB, Ross TM, Bachelder EM, Ainslie KM, Development of an Intranasal Gel for the Delivery of a Broadly Acting Subunit Influenza Vaccine, ACS Biomater Sci Eng, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wang J, Li P, Wu MX, Natural STING agonist as an “ideal” adjuvant for cutaneous vaccination, The Journal of investigative dermatology, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Miyabe H, Hyodo M, Nakamura T, Sato Y, Hayakawa Y, Harashima H, A new adjuvant delivery system ‘cyclic di-GMP/YSK05 liposome’ for cancer immunotherapy, Journal of controlled release : official journal of the Controlled Release Society, 184 (2014) 20–27. [DOI] [PubMed] [Google Scholar]
- [49].Chen N, Gallovic MD, Tiet P, Ting JP, Ainslie KM, Bachelder EM, Investigation of tunable acetalated dextran microparticle platform to optimize M2e-based influenza vaccine efficacy, J Control Release, 289 (2018) 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Champion JA, Walker A, Mitragotri S, Role of particle size in phagocytosis of polymeric microspheres, Pharm Res, 25 (2008) 1815–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Champion JA, Mitragotri S, Role of target geometry in phagocytosis, Proc Natl Acad Sci U S A, 103 (2006) 4930–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang J, Hui-Jiuan C, Hang T, Yu Y, Guishi L, He G, Xiao S, Yang B.-r., Yang C, Lui F, Tao J, Wu MX, Xie X, Physical Activation of Innate Immunity by Spiky Particles, in, Nature Nanotechnology, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ebensen T, Debarry J, Pedersen GK, Blazejewska P, Weissmann S, Schulze K, McCullough KC, Cox RJ, Guzman CA, Mucosal Administration of Cycle-Di-Nucleotide-Adjuvanted Virosomes Efficiently Induces Protection against Influenza H5N1 in Mice, Front Immunol, 8 (2017) 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Madhun AS, Haaheim LR, Nøstbakken JK, Ebensen T, Chichester J, Yusibov V, Guzman CA, Cox RJ, Intranasal c-di-GMP-adjuvanted plant-derived H5 influenza vaccine induces multifunctional Th1 CD4+ cells and strong mucosal and systemic antibody responses in mice, Vaccine, 29 (2011) 4973–4982. [DOI] [PubMed] [Google Scholar]
- [55].Neuhaus V, Chichester JA, Ebensen T, Schwarz K, Hartman CE, Shoji Y, Guzmán CA, Yusibov V, Sewald K, Braun A, A new adjuvanted nanoparticle-based H1N1 influenza vaccine induced antigen-specific local mucosal and systemic immune responses after administration into the lung, Vaccine, 32 (2014) 3216–3222. [DOI] [PubMed] [Google Scholar]
- [56].Pedersen GK, Ebensen T, Gjeraker IH, Svindland S, Bredholt G, Guzmán CA, Cox RJ, Evaluation of the sublingual route for administration of influenza H5N1 virosomes in combination with the bacterial second messenger c-di-GMP, PLoS One, 6 (2011) e26973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sanchez MV, Ebensen T, Schulze K, Cargnelutti D, Blazejewska P, Scodeller EA, Guzmán CA, Intranasal delivery of influenza rNP adjuvanted with c-di-AMP induces strong humoral and cellular immune responses and provides protection against virus challenge, PLoS One, 9 (2014) e104824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schulze K, Ebensen T, Babiuk LA, Gerdts V, Guzman CA, Intranasal vaccination with an adjuvanted polyphosphazenes nanoparticle-based vaccine formulation stimulates protective immune responses in mice, Nanomedicine, 13 (2017) 2169–2178. [DOI] [PubMed] [Google Scholar]
- [59].Svindland SC, Pedersen GK, Pathirana RD, Bredholt G, Nøstbakken JK, Jul-Larsen Å, Guzmán CA, Montomoli E, Lapini G, Piccirella S, Jabbal-Gill I, Hinchcliffe M, Cox RJ, A study of Chitosan and c-di-GMP as mucosal adjuvants for intranasal influenza H5N1 vaccine, Influenza Other Respir Viruses, 7 (2013) 1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Takaki H, Takashima K, Oshiumi H, Ainai A, Suzuki T, Hasegawa H, Matsumoto M, Seya T, cGAMP Promotes Germinal Center Formation and Production of IgA in Nasal-Associated Lymphoid Tissue, Med Sci (Basel), 5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Shim SM, Song EJ, Song D, Lee TY, Kim DJ, Nam JH, Gwin Jeong D, Lee CK, Kim SH, Kim JK, Nontoxic outer membrane vesicles efficiently increase the efficacy of an influenza vaccine in mice and ferrets, Vaccine, 35 (2017) 3741–3748. [DOI] [PubMed] [Google Scholar]
- [62].Hilgers LAT, Platenburg PPLI, Bajramovic J, Veth J, Sauerwein R, Roeffen W, Pohl M, van Amerongen G, Stittelaar KJ, van den Bosch JF, Carbohydrate fatty acid monosulphate esters are safe and effective adjuvants for humoral responses, Vaccine, 35 (2017) 3249–3255. [DOI] [PubMed] [Google Scholar]
- [63].Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G, An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow, Journal of immunological methods, 223 (1999) 77–92. [DOI] [PubMed] [Google Scholar]
- [64].Gallovic MD, Montjoy DG, Collier MA, Do C, Wyslouzil BE, Bachelder EM, Ainslie KM, Chemically modified inulin microparticles serving dual function as a protein antigen delivery vehicle and immunostimulatory adjuvant, Biomaterials science, 4 (2016) 483–493. [DOI] [PubMed] [Google Scholar]
- [65].Reuman PD, Keely S, Schiff GM, Assessment of signs of influenza illness in the ferret model, J Virol Methods, 24 (1989) 27–34. [DOI] [PubMed] [Google Scholar]
- [66].Huang SS, Banner D, Fang Y, Ng DC, Kanagasabai T, Kelvin DJ, Kelvin AA, Comparative analyses of pandemic H1N1 and seasonal H1N1, H3N2, and influenza B infections depict distinct clinical pictures in ferrets, PLoS One, 6 (2011) e27512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gambarian AS, Marinina VP, Solodar TA, Bovin NV, Tuzikov AB, Pazynina GV, Iamnikova SS, L’Vov D K, Klimov AI, Matrosovich MN, [Different receptor specificity of influence viruses from ducks and chickens and its reflection in the composition of sialosides on host cells and mucins], Vopr Virusol, 51 (2006) 24–32. [PubMed] [Google Scholar]
- [68].McGee CE, Sample CJ, Kilburg-Basnyat B, Gabor KA, Fessler MB, Gowdy KM, Influenza-Mediated Lung Infection Models, Methods Mol Biol, 1960 (2019) 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Immunology and Pathogenesis Branch/Influenza Division/CDC. Influenza Virus Microneutralization Assay. Updated September 2007.
- [70].World Health Organization. Serological Diagnosis of Influenza by Microneutralization Assay. Updated December 2010.
- [71].Koutsonanos DG, Vassilieva EV, Stavropoulou A, Zarnitsyn VG, Esser ES, Taherbhai MT, Prausnitz MR, Compans RW, Skountzou I, Delivery of subunit influenza vaccine to skin with microneedles improves immunogenicity and long-lived protection, Scientific reports, 2 (2012) 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Samo M, Choudhary NR, Riebe KJ, Shterev I, Staats HF, Sempowski GD, Leduc I, Immunization with the Haemophilus ducreyi trimeric autotransporter adhesin DsrA with alum, CpG or imiquimod generates a persistent humoral immune response that recognizes the bacterial surface, Vaccine, 34 (2016) 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ace-DEX cGAMP MPs formulated via monoaxial or coxial electrospray remain stable when suspended in PBS with 10% FBS. (A) Scanning electron microscopy (SEM) images were taken of the coaxial and monoaxial MPs at 0, 48, and 168 hr timepoints. (B) The release of cGAMP from coaxial or monoaxial electrospray MPs when incubated at 37°C in PBS with 10% (v/v) FBS (n=3 ±SD) was measured.
Figure S2. Ace-DEX cGAMP MPs formulated via monoxial upregulation of antigen presentation and costimulatory molecules: Bone marrow derived dendritic cells (BMDCs) from C57BL/6 mice were treated with 1 μg/mL cGAMP delivered as soluble cGAMP or encapsulated within Ace-DEX cGAMP MPs at a loading of 10 μg/mg MP monoaxial electrospray. After culture for 4, 24, and 48 hours, expression of antigen presentation and co-stimulation molecules were analyzed by flow cytometry. A) Representative histograms of MHCII, CD40, CD80, CD86 antibody staining on treated BMDCs gated on singlets, debris negative, and CD11c+ cells at 4 (black), 24 (blue) and 48 (red) hours. (B-C) Quantification of mean fluorescent intensity (MFI) of (B) MHCII, (C) CD40, (D) CD80, (E) CD86 over time after treatment. (n=6 ± SEM).
Figure S3. Cellular T cell responses early after prime-boost immunization with Ace-DEX cGAMP MPs generated by monoaxial electrospray. Eight-week-old female C57BL/6 mice were immunized i.m. with PBS, or recombinant HA from influenza strain A/Puerto Rico/8/34 (1 μg) adjuvanted with soluble cGAMP (0.2 μg), or Ace-DEX cGAMP MPs (0.2 μg cGAMP in 20 μg MPs) formulated through monoaxial (mono) electrospray. Animals received a boost with the same formulation 21 days post-prime and T cell responses in the draining PLN were analyzed by flow cytometry on day 26. (A-D) Quantification of total number of (A) CD8 (defined as CD3+CD8+), (B) MPEC CD8 (defined as CD3+CD8+CD44hiCD127+KLRG1−), (C) total CD4 (defined as CD3+CD4+), and (D) Tfh CD4 T cells (defined as CD3+CD4+CXCR5+PD-1+) per draining popliteal lymph node (PLN). (n=10 ± SEM. Samples also pre-gated for live singlet lymphocytes.)
Figure S4. Infection was confirmed with post-infection neutralizing titers. Ferrets were immunized according to the experimental design described in (A) Figure 3, (B) Figure 4 or (C) Figure 5. Serum was collected 21-28 days post influenza challenge and infection was confirmed by virus neutralizing titer. (n=4 ± SD. All data are n.s.).
Figure S5. Immunization does not increase serum ALT activity. Ferrets were immunized according to experimental outline in Figure 4. Serum was collected on day −7 (Pre-Bleed), Day 21 (Post-Prime) and day 49 (Post-Boost) and on day 84 (Post-Infection) ALT activity was assessed. (n=4 ± SD).
Figure S6. Prime-Boost of cGAMP MPs and AddaVax provide comparable protection: Four to six month old male ferrets were immunized IM with a single dose of HA from A/California/07/09 (15 μg) adjuvanted with either monoaxial Ace-DEX cGAMP MPs (15 μg cGAMP @ 1% weight loading) or AddaVax (9.75 mg squalene). (A) Serum was collected monthly and virus neutralizing titers against A/California/07/09 were assessed. (n=4 ± SD, *p < 0.05). One year after immunization, ferrets were challenged intranasally with 1.25 × 107 FFU of influenza strain A/California/07/09. Nasal lavage was collected (B) day 2 and (C) day 4 post challenge and viral load was assessed by foci forming assay. (D) Animal weights and (E) the periodic combined clinical index, a composite measure of animal activity and respiratory index (sneezing and nasal discharge), were assessed daily following infection. (n=4 ± SD, *p < 0.05, ****p < 0.0001).