

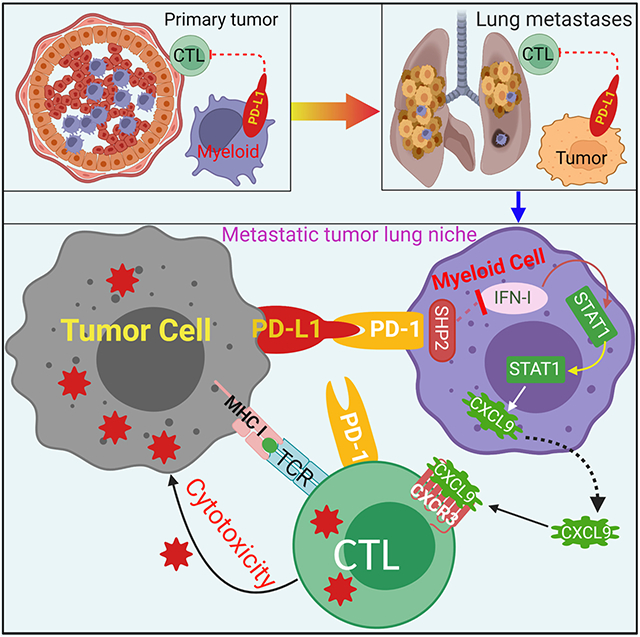
Summary
The cellular and molecular mechanisms underlying tumor cell PD-L1 (tPD-L1) function in tumor immune evasion are incompletely understood. We report here that tPD-L1 does not suppress cytotoxic T lymphocyte (CTL) activity in co-cultures of tumor cells and tumor-specific CTLs and exhibits no effect on primary tumor growth. However, deleting tPD-L1 decreases lung metastasis in a CTL-dependent manner in tumor-bearing mice. Depletion of myeloid cells or knocking out PD-1 in myeloid cells (mPD-1) impairs tPD-L1 promotion of tumor lung metastasis in mice. scRNA-seq reveals that tPD-L1 engages mPD-1 to activate SHP2 to antagonize the type I interferon (IFN-I) and STAT1 pathway to repress Cxcl9 and impair CTL recruitment to lung metastases. Human cancer patient response to PD-1 blockade immunotherapy correlates with IFN-I response in myeloid cells. Our findings determine that tPD-L1 engages mPD-1 to activate SHP2 to suppress the IFN-I-STAT1-CXCL9 pathway to impair CTL tumor recruitment in lung metastasis.
Keywords: PD-L1, PD-1, lung metastasis, immune checkpoint blockade, type I interferon, CXCL9
Graphical Abstract
Introduction
PD-L1 and PD-1 immune checkpoint inhibitor (ICI) immunotherapy is believed to function by blocking the engagement of PD-L1 to its co-inhibitory receptor PD-1 expressed by activated and exhausted cytotoxic T lymphocytes (CTLs).1 PD-1 engagement on CTLs has been shown to result in decreased activation and effector functions due to SHP2 mediated dephosphorylation of the T cell receptor and CD28 signaling cascades, driving CTLs toward an exhausted fate.2 PD-(L)1 blockade reactivates and expands tumor-reactive CTLs, leading to tumor suppression.3 Particular attention has been paid to the role of tumor cell PD-L1 (tPD-L1), which has been hypothesized to serve as a direct molecular shield against CTL lysis of tumor cells.4 However, although a study shows that tPD-L1 is sufficient to inhibit CTL cytotoxicity to promote tumor immune evasion in colon tumor-bearing mice,5 other studies determine that it is PD-L1 of the host immune cells, including dendritic cells (DCs) and macrophages, that plays the dominant role in limiting T cell anti-tumor response to promote tumor immune evasion in tumor-bearing mice.6-10 These findings thus indicate that the response of CTLs to PD-(L)1 blockade may depend on the host tumor microenvironment.11-14 However, these findings in preclinical mouse models do not correlate with human cancer patient response to ICI immunotherapy. Meta-analysis of 17 phase III randomized clinical trials, comprising of a total of 11,166 patients with advanced gastroesophageal cancer, reveals that tPD-L1 level is the strongest predictor of a better overall survival of squamous cell carcinoma after ICI immunotherapy,15 but the combined tumor cell and immune cell PD-L1 level is the strongest predictor of ICI benefit in patients with adenocarcinoma.15 Another meta-analysis of 17 clinical trials comprising of a total of 1,746 patients with metastatic breast cancer indicates that patients with PD-L1-positive tumors had an increased overall survival after ICI immunotherapy.16
A defining hallmark of PD-(L)1 blockade immunotherapy in human cancer patients is the sustained remission of disease experienced by responders.17 As the dominant cause of mortality is metastatic growth,18 the durable survival benefit conferred by PD-(L)1 blockade in human cancer patients thus arises from coordinated suppression of metastases.19 Recognition of this fact has led to increased interest in PD-(L)1 ICI immunotherapy in the neoadjuvant and the adjuvant setting.20,21 Neoadjuvant and adjuvant therapy is administered at an earlier point in disease course, either before or immediately following primary tumor resection, respectively. In doing so, these therapies extend survival by containing metastases.19,22 Robust tumor expression of PD-L1 has been shown to promote both micro- and macro-metastatic tumor growth, with enhancement at metastatic tumor sites as compared to primary tumor sites.23,24
The discrepancy in the function of tPD-L1 between human cancer patients and tumor-bearing mice thus might at least in part be due to the tumor anatomical sites. For human cancer patients, the major contributor to death is metastasis, whereas most of the mouse tumor models are subcutaneous tumors that more resemble the primary tumors.5-10 In this regards, tPD-L1 might play a more dominant role in the metastatic tumors than in the primary tumor. However, whether and how tPD-L1 might differentially drive metastasis from the primary tumor is unknown. PD-1 is a T cell co-repressive receptor.25 Emerging experimental data over the past decade has highlighted the importance of PD-1 expression in other cell populations, including myeloid, natural killer, and tumor cells in tumor immune evasion.26-30 It has recently been shown that myeloid cell PD-1 (mPD-1) also negatively regulates T cell function.28 mPD-1 suppresses T cell anti-tumor function through an extrinsic mechanism to promote the accumulation of immune suppressive myeloid cells in the tumor microenvironment.28 However, whether mPD-1 intrinsic signaling pathway directly regulates CTL activation and tumor infiltration is unknown. We therefore aimed at testing the hypothesis that PD-(L)1 function is tumor anatomical site-dependent by elucidating the cellular and molecular mechanisms underlying tPD-L1 and mPD-1 functions in tumor immune evasion.
Results
tPD-L1 exhibits no direct protection of tumor cells from CTL cytotoxicity
To determine whether tPD-L1 directly inhibits CTL anti-tumor cytotoxicity when only tumor cells and tumor-specific CTLs are present, we deleted Cd274, which encodes PD-L1, in 4T1 (Fig S1A-C), a murine triple negative breast cancer (TNBC) cell line, CT26, a murine microsatellite stable (MSS) colon tumor cell line,31 MC38-met, a metastatic derivative from the murine microsatellite instable (MSI) colon tumor cell line MC38,32 and B16-F10, a murine melanoma cell line. The choice was made to mimic TNBC and MSS colon cancer, which show minimal response to ICI immunotherapy in clinically advanced disease.33,34 The MC38 and B16-F10 tumors were used as C57BL/6 background tumors. All of these four tumor cell lines express PD-L1 and PD-L1 expression can be further up-regulated by IFNγ (Fig. S1C). In contrast, none of these four tumor cell lines express PD-L2, the only alternative ligand for PD-1, even after IFNγ treatment (Fig. S1D). Knocking out PD-L1 therefore renders tumor cells unable to engage CTL-expressed PD-1.
To directly investigate the functional consequences of PD-1:tPD-L1 interaction, we utilized a defined in vitro CTL-tumor interaction system. 2/20 is a tumor-specific CTL line sensitive to PD-1 mediated inhibition (Fig S1E) that recognizes the H2-Ld-restricted AH1 epitope (residues 6-14) of gp70 protein endogenously expressed by 4T1 and CT26 cells.35 As with effector CTLs, 2/20 cells exert antigen-specific cytotoxicity and do not need to undergo differentiation from a naïve state before acquiring effector function, allowing for recapitulation of the effector CTL:tumor interaction with high fidelity. This tumor model mimics the tPD-L1:CTL PD-1 interaction in the effector phase in the tumor microenvironment. Furthermore, 2/20 are directed against an endogenously expressed antigen – rather than artificially over-expressed at supraphysiological levels antigen – that undergoes silencing and thus effectively models a tumor-expressed antigen (Fig 1A).31
Figure 1. tPD-L1 does not confer direct protection from CTL killing.

A. Schematic of co-culture system. B. Representative images of tumor cell apoptosis following co-culture with 2/20 CTLs at indicated ratios for approximately 24 h. Plots are gated on CD8α− cells. C. Cell death quantification of target cells (CD8−AV+PI+) by flow cytometry following co-culture. N = 3/condition. D. Surface protein expression of PD-1 on 2/20 and PD-L1 on tumor cells following overnight co-culture at indicated Effector/Target (E/T) ratios. N=3/condition. E-F. Cell death after overnight co-culture following no pretreatment (E) or pretreatment with IFNγ (F). N=6/condition. G. Proportion of early apoptotic cells after four hours of co-culture at indicated ratios. H. Cell death of MC38-met derivatives stimulated with OVA peptide (SIINFEKL) after 28 h of co-culture with activated OT-1 cells. N=4/condition. I-K. Gating strategy (I) and cell death of CT26 (J) and 4T1 (K) following co-culture in 2/20 in the presence of indicated neutralizing antibodies. N=4-6/condition. All data shown as mean ± SD. See also Figures S1 and S2.
2/20 CTLs displayed dose-dependent cytotoxicity against target cell lines (Fig 1B-C). Co-culture enhanced PD-1 expression level in 2/20 CTLs and PD-L1 expression on tumor cells (Fig 1D). However, tPD-L1 expression conferred no protection against 2/20 CTL cytotoxicity in the 4T1 or CT26 cell models. This phenomenon was not rescuable by pretreatment with IFNγ or the CTL-conditioned medium to elevate surface PD-L1 expression (Fig. 1E-G, S1F-K). Knocking out β-2-microglobulin (B2M) in tumor cells abolished CTL function in killing the tumor cells (Fig. 1E. Fig. S1L). 2/20 CTLs failed to kill A20 cells, a murine tumor cell line without gp70 protein (Fig. S1M). These findings validate an antigen-specific killing of tumor cells by the 2/20 CTLs. Although knocking out tPD-L1 did not increase 2/20 CTL killing of target tumor cells, 2/20 CTLs did respond to tumor cells by upregulating PD-1 (Fig. 1D & S2) and increase the expression of activation markers CD25, CD44, GZMB, T-BET, and TIM-3 (Fig. S2). We next purified CD8+ T cells from OT-1 mouse spleens and stimulated the T cells with anti-CD3 and anti-CD28. The activated OT-1 T cells were then co-cultured with OVA peptide-loaded MC38-met cells. tPD-L1 did not protect MC38-met tumor cells from OT-1 T cell killing in vitro (Fig. 1H). PD-1 blockade also failed to alter 2/20 CTL killing of CT26 (Fig. 1I & J) and 4T1 tumor cells (Fig. 1K) in vitro. These observations indicate that tPD-L1 confers no survival advantage when an antigen-specific effector CTL encounters a tumor cell bearing a cognate peptide-MHC complex, free from any other tumor microenvironmental factors.
tPD-L1 enhances metastasis independent of primary tumor growth
We next sought to extend these in vitro findings to the in vivo setting. We first examined tumor growth in the site of tumor cell injection (primary tumor growth). Initial orthotopic injection of PD-L1 WT and PD-L1 KO 4T1 cells (Fig. S3A) into syngeneic mice led to tumor growth, but no difference in primary tumor growth were observed between the WT and PD-L1 KO tumors (Fig 2A, S3B). Similarly, no differences in primary tumor growth were observed in the WT and PD-L1 KO CT26, MC38-met and B16-F10 models (Fig. 2B & C Fig. S3C). To rule out that size of initial tumor bolus contributes to this phenomenon, we measured WT and PD-L1 KO CT26 tumor growth following subcutaneous injection of a decreased cell dose. No differences in tumor growth were observed in the WT and PD-L1 KO CT26 tumors (Fig. S3D).
Figure 2. tPD-L1 selectively enhances metastasis independently of primary tumor growth.

A. Tumor growth curves (left) and final mass (right) of 4T1.scramble (4T1.WT, 5x105 cells/mouse) and 4T1.PD-L1-KO (4T1.KO, 5x105 cells/mouse) following orthotopic injection. Representative of three independent experiments. N= 5-15/condition. 2-way ANOVA. B. Tumor growth curve of CT26.scramble (CT26.WT, 1x106 cells/mouse) and CT26.PD-L1.KO (CT26.KO, 1x106 cells/mouse) following subcutaneous injection. N=5/condition. 2-way ANOVA. C. Growth curves (left) and final mass (right) of MC38-met.scramble (MC38-met.WT, 1x106 cells/mouse) and MC38-met.PD-L1.KO (MC38-met.KO, 1x106 cells/mouse) following subcutaneous injection. Representative of two independent experiments. N=10/condition. 2-way ANOVA. D. 4T1.WT and 4T1.KO spontaneous metastasis to the lung following orthotopic injection. N= 5-10/condition, two independent experiments. 2-way ANOVA. E & F. Quantification of 4T1 experimental metastatic foci by visual (E, N=15/condition) and microscopic (F, N=4/condition) inspection. Scale: 1 mm. The arrows point to tumor nodules present on the pleural surface. G & H. CT26.WT and CT26.KO cells were injected intravenously to BALB/c mice. Quantification of CT26 experimental metastatic foci by visual (G, N=10/condition) and microscopic (H, N=5/condition) inspection. Scale: 1 mm. The arrows point to tumor nodules. I. MC38-WT and MC38-met.KO cells were injected intravenously to C57BL/6 mice. Quantification of MC38-met experimental metastatic foci by visual inspection (N=8-9/condition). All data are presented in mean ± SD. See also Figure S3.
4T1 tumor cells give rise to spontaneous lung metastasis when orthotopically transplanted to the mammary gland. Orthotopic injection of a minimal dose of cells allowed for dissemination and growth of metastatic tumor in the lung without resection of the primary tumor (Fig S3A). We then examined spontaneous lung metastasis. Although loss of tPD-L1 did not affect primary tumor growth, loss of tPD-L1 impaired spontaneous metastasis to the lung (Fig 2D). To extend this finding, we utilized an experimental lung metastasis model in which lung metastasis is quantified following tail vein injection (Fig S3E). Lung metastasis was observed in PD-L1-deficient 4T1 at both the gross and histological level (Fig 2E & F). To confirm these results were not 4T1- or TNBC-specific, we repeated these experiments in the MSS colon tumor model CT26, ICI responsive MSI colon tumor model MC38-met, and the B16-F10 melanoma model. Similarly, tPD-L1 promoted metastasis independent of primary tumor growth in CT26, MC38-met, and B16-F10 models (Fig 2G-I, Fig. S3F). Consistent with a hematologic process of metastatic dissemination, pathological analysis of lung specimens as shown in Figs 2F & H revealed tumor foci to be primarily localized to the pulmonary interstitium, with frequent demonstration of perivascular invasion. These findings held true for both spontaneous (4T1) and experimental (CT26) metastasis models. No evidence of a lepidic growth pattern, as would be seen in cases of alveolar invasion, was observed (Figs. 2F & H).
Myeloid cell-expressed PD-L1 compensates tPD-L1 function in primary tumor
The above in vivo finding is consistent with the in vitro finding that tPD-L1 does not shield T cells from CTL cytotoxicity in vitro. However, two essential questions arise: 1) what is the cellular mechanism underlying the different functions of tPD-L1 between the primary tumor and lung metastases; and 2) what are the relative functions of non-tumor cell-expressed PD-L1 in tumor growth promotion in the primary tumor site, since unlike the in vitro co-culture microenvironment in which only tumor cells and CTLs are present, primary tumors have other PD-L1+ cells such as PD-L1-expressing myeloid cells.9,10,28 To address these questions, we analyzed other PD-L1+ cells in the primary 4T1 tumor and the spontaneous lung metastases in the same mice. CD11b+ cells represent the largest non-tumor PD-L1+ cells (Fig. 3A & B). Quantification of CD11b+PD-L1+ cells determined that the percentage of CD11b+PD-L1+ cells is significantly higher in primary tumor cells than in lung metastases (Fig. 3C). This finding indicates that myeloid cell-expressed PD-L1 (mPD-L1) might compensate tPD-L1 function to promote tumor growth, which at least in part underlies tPD-L1 lack of function in primary tumor growth promotion. To test this hypothesis, we performed PD-1 blockade immunotherapy in the MC38-met colon tumor-bearing mice. PD-1 blockade should prevent engagement of all PD-L1 (e.g., both tPD-L1 and mPD-L1) and thus suppress tumor growth. Indeed, PD-1 blockade significantly suppresses established MC38-met tumor growth (Fig. 3D). To determine whether the above phenomenon depends on host PD-1, we transplanted MC38-met tumor cells to WT and PD-1 KO mice. MC38 tumor grew significantly slower in PD-1 KO mice than in WT mice (Fig. 3E).
Figure 3. The primary tumor harbors significant more PD-L1+ myeloid cells than lung metastases.

A-B. Gating strategy of primary tumor (A) and tumor-bearing lungs (B) to analyze myeloid PD-L1+ population. C. 4T1 derivatives were injected orthotopically into the mammary fat pad. Proportion of myeloid PD-L1+ cells in live (left) and immune (right) cell populations from primary tumor or metastasis-bearing lungs. Representative of two independent experiments. N = 5/condition. 2-way ANOVA. D. MC38-met (2.5x105 cells/mouse) was injected subcutaneously into C57BL/6 and treated with PD-1 blockade therapy or control IgG. Tumor volume was measured every 3 days following detection of palpable mass. N=5/condition. E. MC-38-met was injected into C57BL/6 (WT) or Pdcd1−/− mice. Tumor volume was measured every three days following detection of palpable mass. All data represent mean ± SD.
tPD-L1 promotes lung metastases through suppressing inflammatory and CTL-driven responses
Increased infiltration of CD3+ cells after loss of tPD-L1 in lung, but not in the primary tumor, were observed (Fig. 4A), indicating that tPD-L1 blocked T cell infiltration at the metastatic site. No difference in initial tumor cell colonization as measured by tumor cell gp70 level in the lungs was seen one day after tail vein injection (Fig. 4B), demonstrating that PD-L1 does not enhance disseminated tumor cell (DTC) colonization efficiency in the lung. However, WT and PD-L1 KOT1 tumor cells formed lung metastases equally in lymphocytes-deficient congenic RAGII-KO mice (Fig 4C), indicating that tPD-L1 suppresses lymphocytes, either directly or indirectly, to promote lung metastasis. Genetic ablation of MHC class I antigen presentation, as well as antibody-mediated depletion of CD8+ T cells, revealed that tPD-L1 promotes lung metastasis in a CTL-dependent manner (Fig 2F, 4D). Taken together, these results indicate that lung-resident DTCs and metastases utilize tPD-L1 to evade CTL-mediated clearance, rather than driving extravasation or seeding from the primary tumor site.
Figure 4. Loss of tPD-L1 limits metastasis by amplifying inflammatory and CTL-driven responses.

A. Representative images (left) and quantification (right) of IHC for CD3 protein in the indicated tumor tissues. Pooled from two independent experiments. N=8/condition. Scale: 1mm. B. 4T1.WT and 4T1.KO cells were injected intravenously to BALB/c mice. Lungs were harvested 24 h after tumor cell injection and analyzed by qPCR for gp70 mRNA level. Two independent experiments. N=5/condition. C. Representative image (left) and quantification (right) of experimental metastasis of 4T1 in BALB/c RAGIKO mice (N=4/condition), two independent experiments. D. Representative image (left) and quantification (right) of tumor nodules following CD8 depletion. (N=4/condition). E. Heatmap of top 125 most variably expressed genes detected in lungs colonized by 4T1.WT or 4T1.KO. F. Principal component analysis. G. Top 5 enriched gene signatures from MSigDB Hallmarks signature set in 4T1.WT (blue) or 4T1.KO (red) colonized lungs based on normalized enrichment score (NES). H. Enrichment plot of indicated signatures. All data represent mean ± SD. See also Figure S4.
Given our observation showing that tPD-L1 does not directly inhibit CTL killing of 4T1, CT26 and MC38-met cells in vitro (Fig. 1), we therefore hypothesized that a third factor, only present at lung metastatic niche, was necessary for tPD-L1 to exert its indirect protective role in vivo. To test this hypothesis and identify this third factor, we performed RNA sequencing of lungs colonized by WT and PD-L1 KO 4T1. Differentially expressed transcripts include those involved in antigen presentation (B2m, Cd74, Tap1), interferon response (Stat1, Bst2, Irf1) and CTL recruitment (Cxcl9, Cxcl10 and Cxcl11) (Fig 4E, S4A). Dimensionality reduction by principal component analysis separated on the first principal component (Fig 4F). Gene set enrichment analysis revealed this response to be driven by enhanced type I interferon (IFN-I) signaling (Fig 4G-H, S4B). This was validated at the protein level by elevated STAT1 phosphorylation (Fig S4C).
tPD-L1 loss drives transcriptional changes in myeloid cells, not CTLs, in the metastatic niche
We then sought to explore how tPD-L1 suppressed IFN-I signaling. We did so through single-cell RNA sequencing of CD45+ leukocytes from 4T1 WT and PD-L1 KO tumor colonized lungs (Fig 5A). Following shared nearest neighbor clustering and dimensional reduction of ~7500 cells by UMAP, cell populations were identified by comparison to publicly available datasets (Fig. 5B). Most of the PD-1 expression was in T cells. PD-1 expression was also observed in the macrophage-myeloid population (Fig. 5B). We examined the expression of genes that were found to be differentially expressed in our bulk RNA-experiment in each cell population at the single-cell level. Surprisingly, analysis of differentially expressed genes showed that expression changes resulted from increased transcription by macrophages (Fig 5C). Similarly, application of a signature composed of genes repressed by tPD-L1 in bulk RNA sequencing data (referred to as tPD-L1 signature, Table S1) showed the greatest magnitude change centered on macrophages (Fig 5D).
Figure 5. CTL activation and differentiation is independent of tPD-L1.

A. Schematic for scRNA-sequencing of 4T1-colonized lungs. B. UMAP projection (left) and barplot of identities of the subpopulation of the CD45+ cells (right) in 4T1 colonized lungs. C. Heatmap of indicated transcript expression in listed cell populations. For each cell population, left column represents WT expression, while right column represents KO expression. D. Single-cell scoring of tPD-L1 loss (top) and IFNα (bottom) signature. Left plot includes cells derived from WT-colonized lungs, while right column represents KO-colonized lungs. E. UMAP projection of k-means clusters of T Cells (left) and barplot of identities of the subpopulations of T cells (Right). F. Violin plots of indicated transcripts. NKT: NK T cells, T.4: CD4+ T cells, T.8.EFF: Effector CD8+ T cells, T.8.MEM: Memory CD8+ T cells, T.8.NVE: Naïve CD8+ T cells, T.TREG: Treg cells. G. Dot plots of the indicated transcripts in the indicated cell subpopulations. H. Expression of all transcripts in WT (x-axis) and KO (y-axis) T cells of indicated populations. Green lines represent 1.5-fold difference in transcription between populations. I. Expression of exhaustion-associated signatures in each cluster. J. CTL infiltration of primary tumor or lung metastases. N=10/condition K-L. Surface marker expression of CTLs isolated from lung metastases (K) or primary tumor (L). N=5/condition. **** p<0.0001. All data represent mean ± SD. See also Figure S5.
Contrastingly, tPD-L1 had minimal impact on the CTL transcriptome. Sub-clustering of the T cell compartment showed no changes in the relative proportions of activated versus naïve CTLs (Fig 5E). However, markedly enhanced expression of the integrin Itgal (encodes LFA-1), which mediates firm adhesion to vascular endothelium,36 provides a rationale for increased CTL infiltration as detected on IHC (Fig 5F). In addition to the increased homing-related gene, loss of tPD-L1 resulted in dramatic increases in myeloid expression of the chemokines Cxcl9 and Cxcl10, which are ligands for CXCR3 that promotes T cell tumor infiltration (Fig. 5G). To examine whether tPD-L1 drove T cell exhaustion in the experimental metastasis model, we scored each T cell subset against a variety of transcriptional signatures associated with exhaustion or PD-1-mediated dysfunction. No consistent difference was observed in signature scores or transcriptomes, suggesting that, in the metastatic setting, tPD-L1 does not drive CTL exhaustion (Fig 5H-I). Consistent with our histologic and transcriptomic data, flow cytometric analysis showed selectively enhanced infiltration of CTLs in lungs colonized with tPD-L1 deficient 4T1 (Fig 5J). We then sought to validate our transcriptome-level data in CTLs at the protein level. No difference was observed in the expression of exhaustion (PD-1, TIM-3) or effector (GZMB) associated markers by CTLs infiltrating the primary tumor or lung-resident metastases (Fig 5K-L, S5A & B). Together, these findings demonstrate that tPD-L1 restrains the recruitment, not the functionality, of CTLs.
As myeloid cells show the greatest transcriptional change after PD-L1 loss, we then investigated the myeloid compartment. A small fraction of myeloid cells expressed PD-1, while a majority of T cell expressed PD-1 (Fig S5C). To analyze the differentiation trajectory of myeloid cells, we performed PHATE analysis,37 followed by k-means clustering, which revealed a central progenitor population (cluster Ma) that diverged into two branches based on low (cluster Mb) and high (cluster Mc) IFN-I signaling (Fig 6A-B, S6A-B). tPD-L1 antagonized myeloid cell differentiation into cluster Mc, reducing the expression of antigen presentation machinery (B2m, H2-DMb1) and T cell-recruiting chemokines (Cxcl9, Cxcl10) (Fig 6C, S6C). The alternative cellular fate adopted by tPD-L1 resident macrophages correlated with enhanced myeloid SHP2 signaling, a downstream effector of PD-1, and decreased IFN-I, indicating that tPD-L1 may directly restrict myeloid differentiation by engaging mPD-1 (Fig 6D).
Figure 6. tPD-L1 restrains IFN-I-driven myeloid cell activation.

A. PHATE projection of non-alveolar macrophage population (left) and proportion of cells belonging to each cluster (right). B. Dimensional heatmap of top ten most variably expressed ISG genes in each cluster. C-D. Expression of indicated genes (C) and signatures (D) in cells. P-value calculated by Seurat “FindMarkers” function of cluster Mc vs Mb expression. E-F. Myeloid composition (E) and MHC class II expression (F) fourteen days post-colonization. Two independent experiments. N = 3,5/condition. Two-way ANOVA with Sidak’s multiple comparison. G. Differential expression of PD-L2 in lungs colonized by 4T1 WT or PD-L1-KO. H. Representative histograms of PD-L2 surface expression on indicated cell populations after colonization by 4T1 WT (blue) or 4T1 PDL1-KO (red). I. Quantification of % PD-L2+ cells in indicated cell populations. N=5/condition. J-K. Representative plots (J) and quantification (K) of % PD-L2+ antigen-presenting cells (CD45+CD11c+Ly6G−) at indicated time points. Results are representative of two independent experiments. Two-way ANOVA. N=3,5/condition. L. 4T1.WT and 4T1.KO cells were injected to mice intravenously. The 4T1.KO mice were then treated with IgG or anti-PD-L2 5 days later every 2 days for 3 times. Shown are quantification of lung tumor nodule number. M. C57BL6 and IFNAR1 KO mice were injected with 100 μg MCA subcutaneously. Tumors were collected and analyzed by qPCR for Cxcl9 and Cxcl10 expression level using β-actin as internal control. N. BALB/c mice were subjected to experimental metastasis model with indicated 4T1 derivatives (5x105 cells/mouse, i.v.). Mice were treated with anti-CXCL9 or control IgG (200ug, i.p) every 3 days. Lungs were harvested and metastatic foci visually quantified. O. & P. Quantification (O) and PD-1 expression (P) of gp70-tetramer+ cells in 4T1 WT or PDL1-KO colonized lungs at indicated timepoints. N=6/condition. Dotted line represents Fluorescence Minus One (FMO) baseline. Results are representative of two independent experiments. Fisher’s Exact Test. ** p<0.01; **** p<0.0001. All data represent mean ± SD. See also Figure S6 and S7.
To confirm these transcript-level observations, we performed flow cytometric analysis of 4T1-colonized lungs. Validating our transcriptomic data, no change in bulk lung myeloid cell composition was observed, though enhanced antigen presentation machinery was detected in macrophages (Fig 6E-F). Given that IFN-I signals in an autocrine or distance-dependent paracrine manner,38 we sought to confirm that tPD-L1 was directly acting on myeloid cells to suppress IFN-I production and signaling. Transcriptomic and proteomic analysis identified Pdcd1lg2, which encodes PD-L2, as a marker for both IFN-I-stimulated myeloid cells after tPD-L1 loss (Figure 6G-I, S6D-F). Differential expression of PD-L2 was observed in 4T1-colonized lungs as early as one day after injection (Figure 6J-K). However, PD-L2 blockade did not affect PD-L1 KO tumor cell lung metastasis (Fig. 6L), suggesting that PD-L2 plays no role in the type I interferon signaling after PD-L1 loss on tumor cells.
To determine the link between IFN-I and CXCL9 and CXCL10, the two essential CTL recruiting chemokines,39,40 we analyzed Cxcl9 and Cxcl10 expression in WT and IFNAR1 KO tumors. Indeed, deficiency of type I interferon signaling in the tumor tissues diminished Cxcl9 and Cxcl10 expression (Fig. 6M). To functionally determine CXCL9 role in tPD-L1 lung metastases promotion, 4T1 lung metastases-bearing mice with treated with a CXCL9 blocking mAb. Blocking CXCL9 significantly increased lung tumor nodule numbers (Fig. 6N). Consistent with this essential role of IFN-I in CXCL9 and CXCL10 expression, increased type I interferon signaling in PD-L1 KO tumor-bearing lung is accompanied by a robust infiltration and activation of 4T1 tumor-specific CTLs, further indicating remodeling of myeloid cell population occurred independently of CTL PD-1 (Fig. 6O & P, S6G-H).
tPD-L1 engagement with mPD-1, not tPD-1, creates an immunosuppressive niche
Given previous findings of myeloid cell-expressed PD-1 function in mediating PD-L1 immune suppressive function,26,28 as well as the enhanced SHP2 signaling observed in tPD-L1 colonized lungs, we hypothesized that this attenuation of IFN-I signaling was mediated by tPD-L1 engagement with PD-1 found on lung-resident myeloid cells. Myeloid cell surface PD-1 expression was enhanced following tPD-L1 loss (Fig 7A-B, S7A & B). To assess the consequences of the tPD-L1:mPD-1 interaction, 4T1 tumor cells were co-cultured in vitro with WT and PD-1 KO BM-macrophages (BMDM). PD-1 deficiency resulted in increased activation of STAT1 (Fig. 7C), and increased expression of the IFN-I-regulated H2Kb expression (Fig. 7D) in BMDM. We then used a complementary approach and inhibited the PD-1 signaling pathway in BMDM using the pharmacological SHP2 inhibitor RMC-4550. RMC-4550 treatment increased IFN-I-regulated MHC I and MHC II proteins (Fig. 7E), as well as the expression of Cxcl9 and Cxcl10 genes (Fig. 7F).
Figure 7. Myeloid PD-1 connects tumor PD-L1 to CTL suppression through suppressing IFN-I signaling following tPD-L1 engagement.

A. Myeloid cell composition in mouse lung fourteen days following injection of indicated 4T1 cell line derivatives. B. Representative histograms (left) and quantification (right) of PD-1 expression on the indicated myeloid cell subsets isolated from 4T1.WT or 4T1.KO colonized lungs fourteen days after injection. N=5/condition. Two-way ANOVA with multiple comparisons. Two independent experiments. C. Mouse bone-marrow-derived macrophages (BMDM) from WT or Pdcd1 KO mice were co-cultured with 4T1.WT or 4T1.KO for two days, and stained intracellularly for STAT1 phosphorylation. Left: Representative histogram. Right: Summary statistics. Data representative of two independent experiments. D. Mouse BMDM from WT B6 or Pdcd1 KO mice were co-cultured with 4T1.WT or 4T1.KO for two days. Surface marker H2Kb expression was analyzed on live CD11b+F4/80+ cells. N = 3-4. Data representative of two independent experiments. E. Mouse BMDM from WT BALB/c mice were co-cultured with 4T1.WT and treated with vehicle (DMSO) or RMC-4550 (1μM) for thirty-six hours, then assayed for cell surface marker expression by flow cytometry. Representative of two independent experiments. F. qPCR of indicated gene targets from RNA harvested from BMDM from WT BALB/c mice that were co-cultured with 4T1.WT and treated with vehicle (DMSO) or RMC-4550 (1μM) for eighteen hours. Representative of two independent experiments. G. Mice were injected with 4T1.WT (i.v., 5x105 cells/mouse) and treated with vehicle or RMC-4550. Lung tissue was harvested at fourteen-days post-injection, and RNA was harvested from cell lysate. qPCR of indicated gene targets. H. MC38-met.WT and MC38-met.KO cells were injected intravenously to Pdcd1 KO mice. Mice were sacrificed 15 days after tumor cell injection. Tumor nodules were quantified. Each dot represents lung tumor nodule number in a mouse. I. 4T1.WT and 4T1. tumor-bearing mice (n=5-8) were treated with control liposomes or clodronate liposomes, respectively, one day prior tumor cell injection. The tumor-bearing mice were treated every 3 days with control or clodronate liposomes for 3 times. Mouse lungs were quantified for tumor nodule numbers. Shown are one of two representative experiments. J. Left panel: MC38-met.WT cells were injected i.v. to WT mice (Pdcd1f/f, n=5) and mice with myeloid cell-specific Pdcd1 deletion (Pdcd1f/f lyzMCre, n=4), respectively. Mice were sacrificed 15 days after tumor cell injection and tumor nodules were quantified. Right panel: MC38-met.WT and MC38-met.KO cells were injected i.v. to mice with myeloid cell-specific Pdcd1 deletion (Pdcd1f/f lyzMCre, n=4 each group). Lung tumor nodules were quantified as in the left panel. All statistical tests performed by Student's T-test or two-way ANOVA with Holm-Sidaks test for multiple comparisons unless otherwise stated, ns: p>0.05; * p<0.05; ** p<0.01; *** p<0.001. All data represent mean ± SD. See also Figure S7.
We identified the myeloid cell intrinsic IFN-I signaling pathway as the tPD-L1 target in lung metastases (Fig. 6). To further validate this pathway, we analyzed IFN-I signaling pathway in the 4T1 lung metastasis model. SHP2 inhibition with RMC-4550 suppressed lung tumor growth as measured by the 4T1 tumor marker MuLV transcript level (Fig. 7G). As expected, RMC-4550 increased Stat1 level (Fig. 7G). The B16-F10 murine melanoma cell line was then used to validate the above findings in vivo. Myeloid cells in the tPD-L1 KO B16-F10 lung metastases exhibit higher MHC I and II levels as compared to B16-F10 WT tumor (Fig. S7C). Knocking out PD-L1 in B16-F10 cells also increased STAT1 activation in myeloid cells in the lung metastases (Fig. S7D). These finding further strengthen the finding that tPD-L1 engages mPD-1 to suppress IFN-I signaling in myeloid cells in lung metastases as observed in the 4T1 tumor model (Fig. 6A-F, 6M-P, & S7C &S7D).
To validate the function of PD-1 in vivo, we injected MC38-met.WT and MC38-met.KO cells to PD-1 KO mice. The rationale is that in the absence of host PD-1, tPD-L1 should have no effect on lung tumor growth. Analysis of lung metastasis revealed that in the absence of host PD-1, tPD-L1 lost its function in promoting lung metastasis (Fig. 7H). We then hypothesized that depletion of myeloid cells and macrophage should diminish tPD-L1 function in promotion of lung tumor growth. To test this hypothesis, we made use of the 4T1 lung metastasis model and clodronate. It has been shown that clodronate liposomes deplete macrophages and myeloid cells to increase lung tumor metastasis.41 Treatment of WT 4T1 lung tumor-bearing mice with clodronate liposomes indeed increased lung tumor nodule numbers. Clodronate liposomes treatment diminished PD-L1 function in promotion of 4T1 tumor lung metastasis since the treatment diminished the difference in tumor lung metastasis between 4T1.WT and 4T1.KO tumor-bearing mice.(Fig. 7I).
Myeloid cell-specific deletion of Pdcd1 in mice leads to rejection of the transplanted MC38 tumor and promotes differentiation of mature effector myeloid cells and antigen-presenting cells to enhance antigen presentation, resulting in increased T cell anti-tumor functionality to suppress tumor growth.28 Knocking out PD-1 in myeloid cells decreased MC38-met.WT tumor lung metastasis (Fig. 7J), validating the function of mPD-1 in promotion of tumor immune evasion.28 We then injected MC38-met.WT and MC38-met PD-L1 KO tumor cells intravenously to mPD-1 KO mice and analyzed lung tumor growth. No difference was observed in lung metastases between the WT and PD-L1 KO tumors in the mPD-1 KO mice (Fig. 7J), indicating of role of tPD-L1 in engagement of mPD-1 in promoting lung metastasis.
Autocrine IFN-I signaling in myeloid cells drives CTL recruitment in the metastatic tumor and response to ICI immunotherapy in human cancer patients.
PD-L1 is expressed in both tumor cells (Fig. S8A & B) and myeloid cells (Fig. S8C & D) in human cancer patients. 42 To determine the human relevance of our findings, we extended our above findings to a cohort of human patients with metastatic basal cell carcinoma who had single cell sequencing performed before and after PD-1 blockade (Table S1).43 Given the transcriptional heterogeneity between mouse and human myeloid cells, we transferred mouse myeloid cell cluster identities to a human lung myeloid cell scRNA-seq dataset to develop transcriptomic signatures of cluster-identifying genes (Fig. 8A, Table S2).44 The human myeloid cell cluster with high IFN-I signaling (Mc signature) was validated by correlation analysis across the TCGA database that demonstrated an inverse relationship between the Mc gene signature and myeloid SHP2 activity in all tumor types represented (Fig S8E). Myeloid cell PHATE analysis revealed differentiation restriction that was released following PD-1 blockade, leading to enhanced interferon signaling, antigen presentation, and CTL-recruiting chemokine expression (Fig. 8B-D). Consistent with what was observed in the metastatic mouse tumor models, the expression levels of CXCL9 and CXCL10 were higher post PD-1 blockade immunotherapy in the metastatic human tumor (Fig. 8D). These findings thus suggest that myeloid cells similarly drive CTL recruitment in human cancer patients as in the tumor-bearing mice. We extended these findings further by analyzing a recently published scRNA-seq dataset of locally invasive and metastatic triple negative breast cancer in human patients treated with chemotherapy and/or immunotherapy (Table S3).45 Analysis of locally-invasive and metastatic infiltrating myeloid cells revealed a macrophage population with PDCD1 transcripts detected (Figure 8E). This population did not overlap significantly with expression of transcripts encoding for PD-L1 or PD-L2 (CD274 and PDCD1LG2). The presence of these PD-1hi macrophages correlated favorably with patient response to therapy, suggesting that PD-1hi macrophages may mediate response to chemoimmunotherapy in the metastatic setting (Figure 8F). Similar to our results in mouse models implicating IFN-I as a major driver of PD-1 expression in myeloid cells, PD-1hi macrophages demonstrated evidence of prolonged exposure of IFN-I by elevated expression of both an IFNα gene signature and interferon-stimulated gene (ISGs)(Figures 8G-H). Furthermore, in patients responding to therapy, these macrophages show both elevated IFN signaling and decreased SHP2 activity, similar to what we observed in our murine models following loss of tPD-L1 (Figure 8I).
Figure 8. PD-1 blockade releases macrophage interferon signaling and production in human cancer patients.

A. Schematic for generation of tPD-L1 loss genomic signature. B. PHATE projection of metastatic basal cell cancer pre- and post-PD-1 blockade (left) and average cell displacement (see methods) ent (right). Right box plot: top line: 3rd quartile, low line: 1st quartile, middle line: medium. Student’s t test. C. Single cell scoring of indicated signatures (left) and heatmap of sample averages (right). D. Single cell expression of indicated genes, with expression imputed by Rmagic. E. UMAP plot of re-analysis of locally invasive or metastatic human breast cancer patients detailing expression of indicated transcripts. F. Proportion of PD-1+ macrophages in patients with indicated response to atezolizumab therapy. PD: Progressive Disease; SD: Stable Disease; PR: Partial Response. G. Expression of indicated transcripts in PD-1lo(no PDCD1transcript detected) macrophages compared to PD-1hi(PDCD1transcript detected) macrophages. H. Expression of Ifna signature in indicated cell populations. I. Expression of gene signatures in PD-1hi macrophages following treatment with atezolizumab in indicated patient populations. J. Correlation of change in indicated transcripts after nivolumab treatment in metastatic melanoma cohort (BMS038). K. Change in tPD-L1 loss and SHP2 activity signature (BMS038). Paired t-test. L. Correlation between change in indicated signatures pre- and post-nivolumab therapy (BMS038). M. Survival analysis of IFN-I responsive and nonresponsive tumors pre and post PD-1 blockade. Two-sided Log-rank survival test. (BMS038).
Analysis of a pre- and post-treatment RNA sequencing dataset of metastatic melanoma patients treated with nivolumab revealed positive associations between the Mc gene signature and markers of CTL infiltration (CD8B) and cytotoxic activity (Fig. 8J, Table S4). This correlation was validated across all tumor types represented in the TCGA database (Fig S8E-G). Resembling our findings in the 4T1 model, however, when samples were normalized to CD8B levels to correct for CTL infiltration levels, the correlation was no longer significant (Fig S8H). This indicates that tPD-L1 may repress CTL recruitment, rather than directly modulating CTL activation in human cancer patients with metastatic disease.
Enhanced expression of the tPD-L1 loss signature and repression of myeloid SHP2 activity was observed exclusively in responders (Fig 8K-L). We therefore hypothesized that tumors that possess infiltration by SHP2 inhibited myeloid cell progenitors (high Ma, PTPN11) and rich capacity for interferon signaling (high IFNAR1/2) would perform superiorly following checkpoint blockade. Indeed, these patients demonstrated enhanced overall survival following checkpoint blockade (Fig 8M). Survival benefit was observed at later, not initial, time points, indicating that these factors may impact the emergence of metastatic growths and may be required for a durable response. Taken together, these findings suggest that PD-L1 engages mPD-1 to activate SHP2 to antagonize the IFN-I-STAT1-CXCL9 pathway to suppress CTL tumor infiltration in human cancer patients with metastatic disease.
Discussion
Work in the past decade has increasingly highlighted the role of PD-1 in non-T cell populations in creating an immunosuppressive niche.27,46,47 Myeloid cells have long been noted to contribute to the formation of an immunosuppressive metastatic niche.48,49 These prometastatic functions have historically been attributed to alternatively activated or M2 suppressive macrophage populations.50 mPD-1 has been shown to promote tumor immune evasion through either macrophage phagocytosis or immune suppression of T cells.28,30 Therefore, mPD-1 may function via this extrinsic mechanism to suppress T cell anti-tumor function.28 In this study, we determined that the mPD-1-SHP2 axis suppresses the intrinsic IFN-I-STAT1-CXCL9 pathway in myeloid cells to promote tumor immune evasion.
The relative roles of tPD-L1 and host non-tumor cell-expressed PD-L1 in tumor immune evasion is still controversial. At the functional level, conflicting findings for the role of tPD-L1 and mPD-L1 in contribution to PD-1 blockade efficacy have been reported and remain an active research area.5,8,9 The function of mPD-L1 in CTL function was not investigated in this study. Our results indicate that tPD-L1 may control CTL recruitment versus functionality, which also depend on the tumor anatomical sites. Our finding is supported by recent observations of simultaneous infiltration of metastases by CTLs following PD-1 blockade.51 Recent literatures have also highlighted the importance of dendritic cell expressed PD-L1 in controlling T cell function, suggesting the effect of PD-1 blockade on CTLs may occur in secondary and tertiary lymphoid structures, rather than at the tumor site.43,52 We have thus demonstrated here that tPD-L1 has an independent role in metastasis.
IFN- plays a broad role in suppression of tumor development through both immunological and non-immunological mechanisms.53-58 IFNα was the first FDA-approved cancer immunotherapy for adjuvant treatment in human cancer and shown efficacy in regulating patient anti-tumor immune response.59 IFNα therapy induced long-term, metastasis-free remission in a significant fraction of patients with late-stage metastatic disease, similar to checkpoint blockade.60,61 Emerging experimental data indicate that IFN-I mediates crosstalk between immune cells in the tumor microenvironment.14,62-65 Our work and others have shown that IFN-I induces both PD-L1 and PD-1 in myeloid cells, though PD-L1 expression is up-regulated more rapidly.66 This supports the hypothesis that PD-(L)1 axis functions as a negative feedback circuit for IFN-I signaling and highlights the intriguing possibility that low response rates in IFNα therapies were due to compensatory PD-(L)1 upregulation. Supporting this, phase I/II clinical trials have shown early success for direct administration of interferon, or indirect upregulation through toll-like receptor (TLR) and stimulator of interferon genes (STING) agonists, in combination with anti-PD-(L)1.67,68 Furthermore, forced overexpression of Irf7, which mediates the second wave of interferon production in response to IFN-I exposure, abrogated triple negative breast cancer bone metastasis and was silenced at spontaneously occurring metastasis, including at the lung.69,70 Similarly, prostate cancer metastases display a silencing of IFN-I production.64 Interestingly, mutations in the type I and II interferon pathway (JAK1/JAK2, etc.) are associated with adaptive resistance to immunotherapy, suggesting that suppression of interferon signaling is a primary mechanism by which tumors achieve successful immune evasion and adaptive resistance.71,72 In this study, we determine that IFN-I signaling pathway is essential for myeloid cell function in CTL recruitment to the lung metastasis niche. Mechanistically, we determine that the tPD-L1 engages mPD-1 to activate SHP2 to suppress the IFN-I-STAT1-CXCL9 pathway in myeloid cells to suppress CTL tumor infiltration in lung metastases to promote lung metastasis, which underlies the efficacy of ICI immunotherapy in the neoadjuvant and the adjuvant setting.19-24
One limitation in this study is that we exclusively analyzed lung metastases, which due to its location in barrier tissue, may have a uniquely inflamed microenvironment.73. Metadata analysis indicates that breast cancer patients with lung metastasis have a more favorable response to ICI immunotherapy than breast cancer patients with liver metastasis.16 Similarly, ICI immunotherapy is much less effective in non-small cell lung cancer and melanoma patients with liver metastasis.74,75 Myeloid cells are the primary sources of CXCL9 and CXCL10 in the human tumor microenvironment.39,40 CXCL9 and CXCL10 are the predominant chemokines that recruit CTL tumor infiltration in response to ICI immunotherapy in human cancer patients.39,40 Further investigation is therefore needed to analyze whether tPD-L1-mPD-1-SHP2-IFN-I-STAT1-CXCL9 mechanism operates analogously at liver metastases and other non-lung metastatic sites in human cancer patients. Further studies are also needed to determine whether mPD-L1 regulates the mPD-1-SHP2-IFN-I-STAT1-CXCL9 pathway to suppress CTL recruitment in the metastatic tumor site. Metastasis accounts for over 90% mortality in human cancers. Such studies may elucidate the molecular mechanism underlying mPD-1-IFN-I pathway in immune suppression and tumor metastasis in other metastatic settings and are thus of high significance in human ICI immunotherapy.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and request should be directed to directed to: Kebin Liu (Kliu@augusta.edu).
Material and datasets availability
Bulk and single-cell RNA sequencing data generated in this studies are deposited in the National Institutes of Health Gene Expression Omnibus (GEO) database (GSE150743, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150743) and are publicly available. Genomic datasets were extracted from the public databases as described in the key resources table above in this study. The annotated data are presented in Figures as presented. Only annotated data are used in the figures and no large datasets are re-generated from these publicly available original raw data. All other materials, including the plasmid DNA, cell lines, generated during the current study are available from the lead contact upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC-Annexin V | Biolegend | 640941 |
| PE-B2M (A16041A) | Biolegend | 154503; RRID:AB_2721339 |
| FITC-BST2 (eBio927) | eBioscience | 12-3172-82; RRID:AB763417 |
| FITC-CD11b (M1/70) | Biolegend | 101206; RRID:AB_312789 |
| BV711-CD11b (M1/70) | Biolegend | 101242; RRID:AB_2563310 |
| APC/Fire-CD11c (N418) | Biolegend | 117352; RRID:AB_2572124 |
| APC/Cy7-CD25 (PC61) | Biolegend | 102012; RRID:AB_312861 |
| FITC-CD3e (145-2C11) | Biolegend | 100306; RRID:AB_312671 |
| PE-CD3e (145-2C11) | Biolegend | 100308; RRID:AB_312673 |
| PE/Cy7-CD3e (145-2C11) | Biolegend | 100320; RRID:AB_312685 |
| APC/Cy7-CD4 (RM4-5) | Biolegend | 100566; RRID:AB_2563684 |
| PE/Dazzle-CD4 (RM4-5) | Biolegend | 100526;RRID:AB_312727 |
| PE/Cy5-CD44 (IM7) | Biolegend | 103010; AB_312961 |
| PerCP-CD45.2 (104) | Biolegend | 109826; RRID:AB_893349 |
| PE/Cy7-CD45.2 (104) | Biolegend | 109830; RRID:AB_1186098 |
| PB-CD45.2 (104) | Biolegend | 109820; RRID:AB_492872 |
| FITC-CD64 (X54-5/7.1) | Biolegend | 139315; RRID:AB_2566555 |
| PE/Dazzle-CD64 (X54-5/7.1) | Biolegend | 139320; RRID:AB_2566559 |
| FITC-CD69 (H1.2F3) | Biolegend | 104506; RRID:AB_313109 |
| FITC-CD8a (53-6.7) | Biolegend | 100706; RRID:AB_312745 |
| AF700-CD8a (53-6.7) | Biolegend | 100730; RRID:AB_493703 |
| PE/Cy7-F4/80 (BM8) | Biolegend | 123114; RRID:AB_893478 |
| PB-GZMB (GB11) | Biolegend | 515408; RRID:AB_2562196 |
| FITC-H-2Kb (AF6-88.5) | Biolegend | 116505; RRID:AB_313732 |
| PE-H-2Kb (AF6-88.5) | Biolegend | 116505; RRID:AB_313732 |
| APC-H-2Ld MuLV gp70 Tetramer (N/A) | MBL International | TB-M521-2 |
| FITC-MHCII (I-A/I-E) (M5/114.15.2) | Biolegend | 107605; RRID:AB_313320 |
| FITC-Isotype Rat IgG1, lambda (G0114F7) | Biolegend | 401913 |
| FITC-Isotype Rat IgM, kappa (R4-22) | BD Pharmingen | 553942 |
| AF700-Ly6C | Biolegend | 128024; RRID:AB_10643270 |
| PerCP-Ly-6G (1A8) | Biolegend | 127654; RRID:AB_2616999 |
| PerCP-MHCII (I-A/I-E) (M5/114.15.2) | Biolegend | 107624; RRID:AB_893586 |
| PE-Mouse IgG2a, kappa (MOPC-21) | Biolegend | 400113 |
| PE/Cy7-Mouse IgG2a, kappa (MOPC-21) | Biolegend | 400125 |
| PE-PD-1 (29F.1A12) | Biolegend | 135206; RRID:AB_1877231 |
| FITC-PD-1 (29F.1A12) | Biolegend | 135214; RRID:AB_10680238 |
| APC-PD-1 (RMP1-30) | Biolegend | 109112; RRID:AB_10612938 |
| APC-PD-1 (29F.1A12) | Biolegend | 135210; RRID:AB_2159183 |
| APC/Cy7-PD-1 (29F.1A12) | Biolegend | 135224; RRID:AB_2563523 |
| APC-PD-L1 (10F.9G2) | Biolegend | 124312; RRID:AB_10612741 |
| PE-PD-L1 (10F.9G2) | Biolegend | 124308; RRID:AB_2073556 |
| APC-PD-L2 (TY25) | Biolegend | 107210; RRID:AB_2566345 |
| PE-PD-L2 (TY25) | Biolegend | 107206; RRID:AB_2162011 |
| APC-Rat IgG2a, kappa (RTK2758) | Biolegend | 400512 |
| PE/Cy7-Rat IgG2a, kappa (RTK2758) | Biolegend | 40522 |
| APC/Fire-Rat IgG2a, kappa (RTK2758) | Biolegend | 400567 |
| PE-Rat IgG2b, kappa (RTK4530) | Biolegend | 400636 |
| PerCP-Rat IgG2b, kappa (RTK4530) | Biolegend | 400640 |
| AF647-pSTAT1 (ICFC) | Biolegend | 666403; RRID:AB_2618938 |
| Mouse monoclonal anti-STAT1 (pY701) | Biolegend | 612133; RIDD: AB_399504 |
| PE-SHP2(pY542) (L99-921) | BD Pharmingen | 560389 |
| AF647-T-BET (4B10) | Biolegend | 644804; RRID:AB_1595466 |
| PE-TIM3 (B8.2C12) | Biolegend | 134003; RRID:AB_1626181 |
| APC-CD24 (M1/69) | Biolegend | 101813; RRID:AB_439715 |
| InVivoPlus anti-mCD3e (145-2C11) | BioXcell | BP0001-1 |
| InVivoMAb anti-mouse CD28 (37.51) | BioXcell | BP0015-1 |
| InVivoPlus mIgG (MOPC-21) | BioXcell | BP0083 |
| InVivoMAb anti-mouse CXCL9 (MIG) | BioXcell | BE0309 |
| InVivoPlus anti-mouse PD-L1 (10F.9G2) | BioXcell | BP0101 |
| InVivoMAb anti-mouse CD8 (53-6.7) | BioXcell | BE0004-1 |
| InVivoPlus anti-mouse PD-1 (RMP1-14) | BioXcell | BP0146 |
| InVivoPlus rat IgG2a (2A3) | BioXcell | BP0089 |
| InVivoMAb Armenian Hamster IgG | BioXcell | BE0091 |
| Bacterial and Virus Strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | ThermoFisher Scientific | C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TRIzol Reagent | Invitrogen | 15596018 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | 11668019 |
| Opti-MEM Reduced Serum Medium, GlutaMAX Supplement | Invitrogen | 51985034 |
| HyClone Fetal Bovine Serum | ThermoFisher | SH3039603 |
| Isopropanol, Molecular Biology Grade | ThermoFisher | BP2618500 |
| Chloroform | Sigma-Aldrich | C2432 |
| Blotting-Grade Blocker | Bio-Rad | 1706404 |
| S.O.C. Medium | Invitrogen | 15544034 |
| Paraformaldehyde, 96% | ACROS Organics | 416785000 |
| EDTA (0.5 M), pH 8.0 | Invitrogen | 15575038 |
| Corning_ Trypsin EDTA 1X | Corning | 25-052-CI |
| DNase I, RNase-free | ThermoFisher | EN0521 |
| Collagenase from Clostridium histolyticum | Sigma-Aldrich | C0130 |
| Hyaluronidase from bovine testes | Sigma-Aldrich | H3506 |
| Clodronate liposome and control liposoma | Liposoma | CP-005-005 |
| 3-Methylcholanthrene (MCA) | Sigma-Aldrich | 213942 |
| Propidium Iodide (PI) | Thermal Fisher Scientific | P1304MP |
| M-MLV Reverse Transcriptase | Promega | M170A |
| Corning 10% SDS | Corning | MT-46040-CI |
| Corning_ 500 mL RPMI 1640 | Corning | 10-040-CV |
| HEPES Buffer | Corning | MT-25060-CI |
| Corning_ 100 mL Penicillin-Streptomycin Solution, 100x | Corning | 30-002-CI |
| Sodium Pyruvate (100 mM) | GIBCO | 11360070 |
| MEM Non-Essential Amino Acids Solution (100X) | HyClone | SH3023801 |
| Hydrogen peroxide solution | Sigma-Aldrich | 216763 |
| VectaMount Permanent Mounting Medium | Vector Laboratories | H-5000 |
| Hematoxylin Solution, Harris Modified | Sigma-Aldrich | HHS16 |
| VECTOR Methyl Green | Vector Laboratories | H-3402 |
| issue-Tek_ O.C.T. Compound | Sakura | 4583 |
| 1-Butanol,anhydrous, 99.8% | Sigma-Aldrich | 281549 |
| Xylenes, histological grade | Sigma-Aldrich | 534056 |
| Antigen Unmasking Solution, Citrate Based | Vector Laboratories | H-3300 |
| ImmPACT DAB Peroxidase (HRP) Substrate | Vector Laboratories | SK-4105 |
| mGM-CSF | Biolegend | 576306 |
| mIFNα | R&D | 12100-1 |
| mIFNβ | Biolegend | 581302 |
| mIFNγ | Biolegend | 575306 |
| Formalin, 10% (Phosphate Buffer). | ThermoFisher | SF100-4 |
| Zombie UV | Biolegend | 423108 |
| Mouse FcBlock | Biolegend | 101320 |
| mPD-L1-FC | Biolegend | 758208 |
| Universal-Agarose, peqGOLD | Peqlab | 351020 |
| Critical Commercial Assays | ||
| Mini-PROTEAN_ TGX Gels | Bio-Rad | 4561096 |
| Power SYBR Green PCR Master Mix | ThermoFisher | 4367659 |
| ZymoPURE II Plasmid Maxiprep Kit | Zymo Research | D4203 |
| Fixation/Permeabilization Solution Kit with BD GolgiPlug | BD Biosciences | 555028 |
| Western Lightning Plus-ECL, Enhanced Chemiluminescence Substrate | PerkinElmer | NEL105001EA |
| Xray Film Processor Konica SRX-101A | Konica Minolta | N/A |
| StepOne Plus Real Time PCR System | Applied Biosystems | N/A |
| Chromium Single Cell 3’ Reagent Kit v3 | 10x Genomics | N/A |
| Direct-zol RNA microprep kit | Zymo Research | R2061 |
| FACSCalibur | BD Biosciences | SCR_000401 |
| LSRFortessa | BD Biosciences | N/A |
| Deposited Data | ||
| BMS038: Tumor and Microenvironment Evolution during Immune Checkpoint Blockade Therapy with Nivolumab | Riaz et al., 201782 | https://github.com/riazn/bms038_analysis |
| Biomarkers of response and resistance to checkpoint blockade immunotherapy in metastatic melanoma | Gide TN, et al., 2019 | PRJEB23709 |
| Single-cell RNA-seq of melanoma ecosystems reveals sources of T cell exclusion linked to immunotherapy clinical outcomes | Jerby-Anon, et al. 2018 | Study: Melanoma immunotherapy resistance, Broad Single-Cell Portal |
| Single cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species | Zilionis et al., 201944 | GSE127465 |
| Bulk and scRNA-seq data | this paper | GSE150743 |
| Experimental Models: Cell Lines | ||
| Cell Line: KEK293FT | ThermoFisher Scientific | R70007 |
| Cell line: 4T1 | American Type Culture Collection | CRL-2539 |
| Cell line: CT26 | American Type Culture Collection | CRL-2638 |
| Cell line: B16-F10 | American Type Culture Collection | CRL-6475 |
| Cell line: MC38-met | A derivative of MC38 cell line, provided by Dr. Scott Abrams at Roswell Park Comprehensive Cancer Center, Buffalo, NY. | Hodge and Schlom, 199932 |
| Cell line: 2/20 CTL | Provided by Dr. Scott Abrams at Roswell Park Comprehensive Cancer Center, Buffalo, NY. | Ryan et al., 200135 |
| Experimental Models: Organisms/Strains | ||
| Mouse: BALB/cJ | The Jackson Laboratory | 000651 |
| Mouse: C57BL/6J | The Jackson Laboratory | 000664 |
| Mouse: C.129S7(B6)-Rag1tm1Mom/J | The Jackson Laboratory | 003145 |
| Mouse: B6(Cg)-Ifnar1tm1.2Ees/J | The Jackson Laboratory | 028288 |
| Mouse: B6.Cg-Pdcd1tm1.1Shr/J | The Jackson Laboratory | 028276 |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J | The Jackson Laboratory | 003831 |
| Mouse: Pdcd1f/f lyzMCre | Straus et al., 202028 | |
| Oligonucleotides | ||
| Cd274-sgRNA 5’TCCAAAGGACTTGTACGTGG 3’ | Genscript | |
| Control-sgRNA 5’CTCGTATCTTTTCCCACGGC 3’ | Genscript | |
| B2m-sgRNA 5’AGTATACTCACGCCACCCAC 3’ | Genscript | |
| Pdcd1-sgRNA 5’CAGCTTGTCCAACTGGTCGG 3’ | Genscript | |
| Mulv-gp70-FW 5’TGACCTTGTCCGAAGTGACC 3’ | IDT | |
| Mulv-gp70-RV 5’TAGGACCCATCGCTTGTCTT 3’ | IDT | |
| Actb-FW 5’ATTGTTACCAACTGGGACGACATG 3’ | IDT | |
| Actb-RV 5’CTTCATGAGGTAGTCTGTCAGGTC 3’ | IDT | |
| Cxcl9-FW 5'TCATTGCTACACTGAAGAACGGAG-3' | IDT | |
| Cxcl9-RV 5'ACGACGACGACTTTGGGGTG-3' | IDT | |
| Cxcl10-FW 5'TCTCTCCATCACTCCCCTTTACC-3' | IDT | |
| Cxcl10-RV 5'CTTGCTTCGGCAGTTACTTTTGTC-3' | IDT | |
| Ifna-FW 5'CTGAAGGACAGGAAGGACTTTGG-3' | IDT | |
| Ifna-RV 5'CTGCTGGTGGAGGTCATTGC-3' | IDT | |
| Ifnb-FW 5'CTGCGTTCCTGCTGTGCTTC-3' | IDT | |
| Ifnb-RV 5'TCTTCTCCGTCATCTCCATAGGG-3' | IDT | |
| Stat1-FW 5'CCTGGAACTCAGAAATCCGCC-3' | IDT | |
| Stat1-RV 5'CAGAAGAACCAACCCACGACC-3' | IDT | |
| Recombinant DNA | ||
| pCMV-VSV-G | Addgene | RRID: Addgene_8454 |
| psPAX2 | Addgene | RRID: Addgene_12260 |
| lentiCRISPRv2 | Genscript | RRID: Addgene_52961 |
| PX459-sgRNA | Addgene | RRID: Addgene_62988 |
| Software and Algorithms | ||
| UCSC Xena Browser | University of California Santa Cruz | https://xenabrowser.net/heatmap/ |
| Rstudio v1.2.1335 | Rstudio | RRID:SCR_000432 |
| Seurat 3.1.1 | R | RRID:SCR_016341 |
| PHATE | Moon, et al., 201937 | https://github.com/KrishnaswamyLab/PHATE |
| quPATH v0.2 | https://qupath.github.io | |
| ImageJ | NIH | https://imagej.nih.gov |
| Harmony | https://github.com/immunogenomics/harmony | |
| Kallisto | https://github.com/pachterlab/kallisto | |
| SingleR | https://github.com/dviraran/SingleR | |
| DoubletFinder | https://github.com/chris-mcginnis-ucsf/DoubletFinder | |
| Cell Ranger 3.1.0 | 10xGenomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| STAR | https://github.com/alexdobin/STAR | |
| DESeq2 | Love et al., 201477 | https://github.com/mikelove/DESeq2 |
| GSEA4.0 | Broad | https://www.gsea-msigdb.org |
| HTSeqv0.6.1 | https://github.com/simon-anders/htseq | |
| clusterprofiler | Yu, et al., 201278 | https://github.com/YuLab-SMU/clusterProfiler |
| CellQuestPro | BD Biosciences | N/A |
| FlowJo v10.6.0 | BD Biosciences | N/A |
| Prism8 | Graphpad | N/A |
| BD Diva 8.01 | BD Biosciences | N/A |
| VISION | https://github.com/YosefLab/VISION | |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice.
BALB/c and C57BL/c were purchased from Jackson Laboratory (Bar Harbor, ME). PD-1 knock out (Pdcd1−/−, B6.Cg-Pdcd1tm1.1Shr/J), Rag1 knock out (B6.129S7-Rag1tm1Mom/J), and IFNAR1 knock out (B6(Cg)-Ifnar1tm1.2Ees/J) mice were purchased from Jackson Laboratory. Pdcd1flox/flox (Pdcd1f/f) were generated as previously described 28. Pdcd1f/f mice were mated with LysMcre mice [B6.129P2– Lyz2tm1(cre)Ifo/J] (The Jackson laboratory, Bar Harbor, MA) to generate Pcd1f/fLyzMCre mice in at Beth Israel Deaconess Medical Center (Boston, MA) as previously described 28. Use of animal studies were approved in advance by Augusta University Institutional Animal Care and Use Committee and the Institutional Animal Care and Use Committee at Beth Israel Deaconess Medical Center.
Cell lines.
CT26, 4T1 and B16-F10 cell lines were obtained from American Type Culture Collection (ATCC) (Manassas, VA). MC38-met is a derivative of MC3832 and is selected by collecting MC38 tumors from mouse lungs to establish the stable cell subline MC38-met (Provided by Dr. Scott Abrams at Roswell Park Comprehensive Cancer Center, Buffalo, NY). 293FT cells were obtained from (ThermoFisher Scientific). AH1 antigen-specific T cells (2/20 CTLs) were generated and maintained as previously described.35 (Provided by Dr. Scott Abrams). The CTLs (1.3x105 cells/ml) were cultured with AH1 peptide (SPSYVYHQF, 1 μg/ml), recombinant IL-2 protein (10 Units/ml), and lethally irradiated BALB/c mouse spleen cells (1/36 spleen/ml) weekly. Cell lines were tested bi-monthly for mycoplasma and were mycoplasma-free at time of experiments.
METHOD DETAILS
Gene knockout cell line generation
HEK293FT cells (ThermoFisher) were co-transfected with pCMV-VSV-G (Addgene #8454), psPAX2 (Addgene #12260) and lentiCRISPRv2 (Genscript, Piscataway, NJ) plasmids using Lipofectamine 2000 (Thermofisher). The lentiCRISPRv2 plasmid (4 μg), pCMV-VSV-G plasmid (1.4 μg), and psPAX2 plasmid (2.6 μg) were added to OPTI-MEM medium (ThermalFisher, Cat#51985034) with a final volume of 500 μl. Lipofectamine 2000 (20 μl) was diluted with 480 μl OPTI-MEM medium. The plasmid DNA mix and the diluted lipofectamine were then mixed and cultured at room temperature for 30 min, gently added to the HEK 293 FT cells in a 6-cm dish (2x106 cells/dish) using drop by drop method. Cells were incubated for 5-6 hours in tissue culture incubator. The medium was aspirated and 2.5 mL of prewarmed (37°C) 1:1 mix of DMEM medium (+10% FBS):OPTI-MEM medium was added and incubated at 37°C for 48-60 hours. Cell culture supernatant was collected and filtered through a 0.45 μm syringe filter (Fisher Scientific, Cat 09-719D). Tumor cells to be transduced were cultured with the viral particles/culture supernatant in 12-well plate (5x105 per well) until cells reach 30-50% confluence. Diluted medium (2.5 ml 50% OPTI-MEM+50% DMEM) was added and cultured at 37°C overnight. Medium was aspirated off from wells of the plate, A mix of 0.5 ml of viral solution and 0.5 ml of RPMI medium was added to each well with 0.6 μl of polybrene (10mg/ml stock, Santa Cruz Biotech, Cat# sc-134220), mixed well and incubated overnight at 37°C for 48-60 hours. Cells were harvested and cultured in a 10-cm dish. Puromycin (5 μg/mL) was added to the culture for three days to select for stable cell line.
Cells were then stimulated with 30 ng/mL mIFNγ (for PD-L1 andB2M KO cell lines) or 50 ng/mL mIFNβ (for PD-1 KO cell line) overnight, and bulk cells were sorted for marker-negative cells using a BD FACSARIA (BD Biosciences). This process was repeated 1-2 times to generate stable marker-negative cell lines. Alternatively, some cell lines were generated by transient transfection of PX459-sgRNA constructs (Addgene #62988) by Lipofectamine 2000 (ThermoFisher). Cells then underwent an identical selection and sorting process.
Cell phenotype was confirmed as follows. CT26.WT, CT26.PD-L1.KO, CT26.B2M.KO, 4T1.WT, 4T1.PD-L1.KO, MC38-met.WT, MC38-met.PD-L1.KO, B16-F10.WT, B16-F10.PD-L1.KO, RAW264.7.WT, RAW264.7.PD1-KO cell lines were cultured and either untreated or treated with 30 ng/mL mIFNγ or 50 ng/mL mIFNβ overnight to induce PD-L1/B2M or PD-1 expression, respectively. After 24 hours, the cells were harvested, stained with marker mAbs or isotype control mAb and analyzed by flow cytometry.
Tumor-CTL Co-culture system
Unless otherwise stated, CT26, 4T1, and MC38-met cells were seeded 1x105/well into a 96-well U-bottom plate. The gp70 antigen-specific T-cell line 2/20 was added at 0:1, 0.1:1, 0.25:1, 0.5:1, and 1:1 ratios (E:T) and cultured overnight. After approximately 22 hours, floating and adherent cells were collected, stained, and analyzed by flow cytometry. For early apoptosis experiments, cells were harvested after four hours of co-culture.
CT26.WT, CT26.PD-L1.KO, CT26.B2M.KO, 4T1.WT, 4T1.PD-L1.KO cells were co-cultured with 2/20 CTLs as described above. MC38-met.WT and MC38-met.PD-L1.KO cells were c-cultured with OT-1 T cells. The tumor cells were either 1) untreated, 2) treated with 30 ng/mL mIFNγ overnight one day prior to the start of the co-culture, or 3) treated with a 1:1 mixture of complete RPMI and supernatant from Day 4 2/20 CTLs harvested overnight one day prior to the start of the co-culture. Pretreated cells were initially seeded at a density of 5x104 cells/well. After 22 hours of co-culture, floating and adherent cells were collected, stained, and analyzed by flow cytometry.
To assay PD-L1 mediated inhibition of 2/20 CTLs, 96-well flat-bottom plates were coated with 1 μg/mL 145-2C11 (Bio X cell) and indicated concentrations of mIgG (MOPC-21, Bio X cell) or mPD-L1-FC (Biolegend, Cat#758208) and incubated at 4°C overnight. Wells were washed three times with PBS, then 1x105 2/20 cells were seeded and incubated overnight. Surface activation marker expression was assayed by flow cytometry.
In vivo models
WT and IFNAR1 KO tumors were established by injecting 100 μg methylcholanthrene (MCA, Sigma-Aldrich, Cat# 213942) in peanut oil subcutaneously to C57BL/6J and IFNAR1 KO mice. Tumors were resected from mice for analysis 3 months after MCA injection. For primary tumor and orthotopic models, 5x105 (4T1.WT and 4T1.PD-L1.KO), 1x106 (CT26.WT and CT26.PD-L1.KO), 2x105 (MC38-met), 1x106 (MC38-met.WT and MC38-met.PD-L1.KO) and 1x106 (B16-F10.WT and B16-F10.PD-L1.KO) cells were injected at the indicated site. 4T1 orthotopic tumors were established by injection at the right second mammary gland. CT26, MC38-met, and B16-F10 subcutaneous tumors were established by injection at the right flank. Tumor volume was measured every 2-3 days and calculated by the formula [(major axis x minor axis)*)/2]. At experimental endpoints, mice were sacrificed, tumors were harvested, a section was formaldehyde-fixed for microscopy, and the remaining portion was digested in a solution consisting of collagenase (1 mg/mL, Sigma), hyaluronidase (0.1 mg/mL, Sigma), and DNaseI (30 U/mL, ThermoFisher) and complete RPMI for 30 minutes, shaking at 37C. Tumors were then mechanically homogenized through a 100 μm cell strainer (Corning, Cat#431752) and underwent red cell lysis. For metastases models, lungs were harvested, inflated with an India ink solution, and quantified as previously described 79.
For experimental metastasis models, 2.5x105 (4T1.WT and 4T1.KO), 1x106 (CT26.WT and CT26.KO),1x106 (MC38-met.WT and MC38-met.KO), and 1x106 (B16-F10.WT and B16-F10.KO), 2x105 (MC38-met) cells were injected into the lateral tail vein. For quantification experiments, mice were sacrificed at the experimental endpoint and tumor nodules were quantified. For flow cytometric analysis of infiltrating immune cells, lungs were perfused with 20 mL of PBS immediately post-sacrifice and were digested and homogenized analogously to tumor samples.
For neutralization experiments, 200 μg of anti-CD8α (Clone 53-6.7, Bio X Cell Corp), anti-CXCL9 (Clone MIG-2F5.5), or isotype control (Clone 2A3, Bio X Cell) were injected i.v. every other day during the course of the experiment.
Bone marrow-derived macrophage generation and treatment.
To isolate bone marrow cells, mice were sacrificed and femur and tibia were harvested and sterilized in 70% ethanol for 10 min, rinsed in PBS. Bone marrow cells were collected by flushing bone marrow cavity RPMI medium. Bone marrow cell mixture was then processed to a single cell suspension through a 100μM filter (Cirning) and lysed of red cells. 5x106 bone marrow cells were plated in a 10-cm culture dish in complete RPMI containing 50ng/mL of mM-CSF (Cat: 576402, BioLegend). Medium was refreshed on day 3. On day 6-7, macrophages were harvested with PBS+1mM EDTA. Flow cytometry analysis revealed >90% purity of CD45+CD11b+F4/80+ cells. For co-culture studies, macrophages were plated at a 1:1 ratio with indicated tumor cell lines. For treatment with RMC-4550 (ChemieTek, CT-RMC4550), co-cultures were treated with 1 μM RMC-4550 or vehicle (DMSO).
Flow cytometry
General flow cytometry staining protocol is as follows. All samples (excepting CTL:Tumor cell co-cultures) were first blocked in FACS Buffer (PBS + 2%FBS) containing mouse FcBlock (Biolegend, Cat #101320, 1 μg/mL) for ten minutes at room temperature. Antibodies were added at indicated concentrations and samples were incubated at room temperature for 30 minutes. For samples stained with tetramer, incubation period was extended to one hour at room temperature. Samples were then washed with PBS and incubated with Zombie UV (1:1000, Biolegend, Cat#423108). Samples were then washed with FACS Buffer, then fixed in 2% paraformaldehyde.
For co-culture cytotoxicity experiments, supernatant and cells were harvested and resuspended in AnnexinV-binding buffer (10 mM Hepes, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Samples were stained with APC-AnnexinV (1:100, Biolegend, Cat #640941) and other indicated antibodies on ice for twenty minutes. Cells were then washed with AnnexinV-binding buffer and resuspended in AnnexinV-binding buffer containing propidium iodide.
For intracellular staining, samples were stained for surface antigens and viability, then cells were fixed and permeabilized according to manufacturer’s instructions (BD Biosciences, Cat#554714). Cells were resuspended in FACS buffer prior to acquisition. For nuclear antigen staining, cells were immediately ethanol fixed, washed in FACS Buffer, then stained at room temperature for one hour.
Samples were acquired on a FACSCalibur with CellQuestPro or LSRFortessa with BD Diva 8.01 (BD Biosciences). All flow cytometry data analysis was conducted with FlowJo v10.6.0 (BD Biosciences).
Bulk RNA Sequencing
Balb/c mice were injected i.v. through the lateral tail vein with 2.5x105 4T1 WT or PDL1-KO cells. After seven days, mice were sacrificed, lungs were perfused with 20 mL of PBS. Lungs were mechanically homogenized and RNA was extracted using Direct-zol RNA microprep kit according to manufacturer’s instructions (Zymo Research, Irvine CA). RNA was sequenced by Novogene. Reference genome (mm10) and gene model annotation files were downloaded from Ensembl directly. Indices were built and paired-end clean reads were aligned to the reference genome using STARv2.5. Reads were quantified using HTSeqv0.6.1. Rank-log transformed normalized counts from DESeq2 were used as inputs for GSEA, GO and PCA analysis.77 GSEA was performed using GSEA4.0 (Broad) with gene-set permutation. GO pathway enrichment was performed with clusterprofiler 78.
Single-Cell RNA Sequencing
Balb/c mice were injected i.v. with 2.5x105 4T1 WT or PDL1-KO cells. After fourteen days, mice were sacrificed, lungs were perfused with PBS and harvested. Following collagenase digestion, leukocytes were positively selected using anti-CD45 mAb-conjugated magnetic nanobeads (Cat# 480027, Biolegend). Cells were suspended in PBS with 0.04% BSA. Cell viability was assessed using trypan blue and assured of ≥ 80% viable cells. Cells were loaded at a concentration to capture approximately 2-3 x 103 targeted cells on a Chromium Chip B (10x Genomics). scRNA-seq libraries were generated using the Chromium Single Cell 3’ Reagent Kit v3 (10x Genomics) according to the manufacturer’s instructions. The libraries were sequenced on Illumina NextSeq 500 platform under the following sequencing protocol: 28 bp (Read 1), 8 bp (indexing Run), and 91 bp (RNA Read 2). Reads from the raw fastq files were mapped to mm10 Mouse Genome reference by STAR aligner in Cell Ranger 3.1.0 pipeline. Alignment generated 30-39K reads/cell with 75-102 million reads per sample at a target of 2.3-2.7 x 103 cells per sample identified at 88% in Q30 Bases in RNA read as well as greater than 91% genome-mapping rates along with approximately 1600 median genes per cell.
Filtered counts data was loaded into Seurat. High quality cells (200< feature count<4000, percent mitochondrial reads < 10) were extracted for downstream analysis. Doublet clusters were manually identified and removed, then cells were subjected to doublet removal by DoubletFinder. Cells were then reclustered and remaining doublet clusters were again manually identified and removed based on the expression of lineage-defining genes. Cells were clustered using the first fifty principal components, and clusters were annotated by comparison to pre-existing RNA-sequencing datasets using SingleR. Myeloid and T cells clusters were then subsetted and reclustered based off of top twenty principal components for further analysis. MSigDB and signature sets generated during the course of this study were loaded into VISION for single-cell scoring.
Signature Generation
tPD-L1 loss gene signature was created by isolating genes whose expression increased by at least two-fold with a padj of less than 0.05 calculated by DESeq2 following tPD-L1 loss from our bulk RNA-sequencing dataset. SHP2 activation signature was calculated by isolating genes whose expression respectively increased two-fold (padj <0.05) in myeloid cells following treatment with the SHP2 inhibitor SHP09980. Score generated was then inversed.
MA, MB and MC gene signatures were taken by using the Seurat “TransferData” function to transfer labels from our macrophage PHATE clusters to a previously published dataset of human lung tumor-resident macrophages.44 Cluster-identifying signatures were calculated by the Seurat “FindAllMarkers” function. All signatures generated in the course of this study can be found in Table S1.
Patient Dataset Analysis
TCGA RNA sequencing data was accessed through Xena Browser. Survival data was accessed from previously published analyses.81 Survival analyses were performed using the R survival and survminer packages.
Data from the BMS038 was accessed from authors’ processed files (https://github.com/riazn/bms038_analysis) and normalized counts were generated following author’s scripts, which were used for downstream analyses 82. Previously published dataset of response following PD-1 treatment (PRJEB23709) was accessed from EMBL and pseudo-aligned with kallisto using default settings.76 Counts were loaded into DESEQ2 for downstream analysis, which was conducted using rlog values.
To analyze tumor-infiltrating macrophages in a melanoma cohort, single cells annotated by the authors to be macrophages were subsetted and analyzed using Seurat pipeline 43. Batch effects were corrected by Harmony. To assess intrasample variation, mean displacement was calculated as follows:
Changes in melanoma cells resistant or naïve to ICB were accessed through the author’s dataset.83 Counts were extracted and analyzed analogously. Patient clinical data used for the above analysis is included in Table S2-4.
Colonization Assay
Balb/c mice were injected i.v. with 2.5x105 4T1 WT or 4T1 KO cells. After twenty four hours, mice were sacrificed, lungs were perfused with PBS and harveted. Lungs were minced with scissors, then homogenized in Trizol using an electric homogenizer. RNA was isolated according to the manufacturer’s instructions, and used for the first strand cDNA synthesis using the MMLV reverse transcriptase (Promega, Madison, WI). The cDNA was then used as template for real-time PCR analysis with Power SYBR Green PCR Master Mix (ThermoFisher, Waltham, MA) using the StepOne Plus Real Time PCR System (Applied Biosystems, Foster City, CA). Nucleic acid sequences can be found in STAR Method table.
Western Blotting
Lungs were collected and homogenized into single cell suspension in PBS using an electric homogenizer. Alternatively, cells were harvested by scraping, following two washes in PBS. Subsequently, cells were lysed in total cell lysis buffer (20 mM HEPES, pH 7.4, 20mM NaCl, 10% glycerol, 1% Triton X-100) for one hour on ice. Lysates were subjected to standard SDS-polyacrylamide gel electrophoresis and western blotting procedures using the following primary antibodies: anti-STAT1 (#610186, BDBiosciences, San Diego, CA, 1:1000) and anti-pSTAT1 (#612312, BDBiosciences, San Diego, CA, 1:1000), Anti-β-actin (Sigma-Aldrich, St Louis,MO, 1:2500) followed by anti-Mouse-HRP (1:5000) secondary antibodies (Cell Signaling, Danvers, MA). Signal was detected using the enhanced chemiluminescence system (ECL, Perkin Elmer, Waltham, MA) and the Xray Film Processor Konica SRX-101A (Konica Minolta, Tokyo, Japan). Band intensity was quantified using ImageJ (NIH).
Immunohistochemistry
Lung and tumor tissue was fixed in 10% formalin overnight. The fixed tissues were processed into paraffin blocks and cut into sections. Sections were dewaxed with Xylene and hydrated in 100%, 90%, 70% and 50% ethanol sequentially. Antigen retrieval was performed using Antigen Unmasking Solution (Vector Lab, Burlingame, CA). Slides were then incubated in 0.3% hydrogen peroxidide for 20 min at room temperature, washed in water, blocked with 2.5% Normal Horse Serum (Vector Lab, Burlingame, CA) for 1 hr, then probed with anti-CD3 antibody (Novus, Littleton, CO) overnight at 4C. Slides were washed three times with PBS-Tween and incubated with HRP Universal Anti-Mouse IgG/Anti-Rabbit IgG Antibody (Vector Lab, Burlingame, CA) for 1 hr. Sections were exposed to ImmPACT DAB Peroxidase Substrate (Vector Lab, Burlingame, CA) for 5 min, washed and counterstained with Harris Hematoxylin (Sigma Aldrich, St. Louis, MO), and incubated for 1 min with 0.1% Sodium Bicarbonate as a bluing reagent. Slides were mounted in VectaMount Permanent Mounting Medium (Vector Lab, Burlingame, CA). Tumor nodules in hematoxylin and eosion stained sections were visually identified and quantified. Images were aquired using LAS4.1 software (Leica). CD3+ cells were quantified by quPATH v0.2. Stained samples were ana;lyzed by a board-certified Pathologist (N.M.S).
Cytokine stimulation
For cytokine stimulation experiments, bone marrow-derived macrophage cells were harvested and cultured as previously described.84 Cells were cultured with the indicated factors overnight: IFNα (R&D, 50 ng/mL), IFNβ (Biolegend, 50 ng/mL), IFNγ (Biolegend, 50 ng/mL).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was conducted using Prism8 (Graphpad) and p-values were calculated by a two-tailed Student’s t-test. Significance between survival groups were computed by two-sided log-rank test (Survival package, R).
Supplementary Material
Highlights.
Tumor PD-L1 (tPD-L1) does not suppress CTL cytotoxicity in vitro.
TPD-L1 promotes metastatic, but not primary, tumor growth via a CTL-dependent mechanism.
TPD-L1 engages myeloid PD-1 (mPD-1) to repress IFN-I to impair CTL tumor recruitment.
Human patient response to nivolumab correlates with IFN-I response in myeloid cells.
In Brief.
Klement et al. use single-cell RNA-Seq, PD-L1/PD-1-deficient cell lines and mouse tumor models to elucidate the cellular and molecular mechanism underlying tumor cell PD-L1 function in tumor immune evasion. They find that tumor cell PD-L1 does not promote tumor immune evasion in the primary tumor site but engages myeloid cell PD-1 to activate SHP2 to antagonize the IFN-I-STAT1-CXCL9 intrinsic pathway in myeloid cells to suppress T cell recruitment in lung metastases.
ACKNOWLEDGEMENTS
We thank Drs. Aliazis Konstantinos, Sasitorn Yenyuwadee and Vassiliki Boussiotis in Beth Israel Deaconess Medical Center at Harvard Medical School for providing the Pdcd1f/f and Pdcd1f/fLyzMCre mice. We thank Ms. Penny Roon and Donna Kumiski at the Medical College of Georgia Electron Microscopy and Histology Core for technical assistance in tissue preparation and histology work. 10x Genomics single-cell sequencing was performed by Sam Chang-Sheng, Ph.D. at the Integrated Genomic and High-Performance Computing core facility at Georgia Cancer Center. This work was supported by National Institutes of Health grants NIH R01 CA133085 and NIH R01 CA227433 (to K.L.), NIH F30 CA236436 (to J.D.K.), NIH F31 CA257212 (to A.D.M.), and VA Merit Review Award I01CX001364 to K.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interest.
Conflict of interest: None
REFERENCES
- 1.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8, 793–800. 10.1038/nm730nm730 [pii]. [DOI] [PubMed] [Google Scholar]
- 2.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, and Vale RD (2017). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433. 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211.e110. 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 4.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, and Sharpe AH (2017). PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 214, 895–904. 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh SA, Wu DC, Cheung J, Navarro A, Xiong H, Cubas R, Totpal K, Chiu H, Wu Y, Comps-Agrar L, et al. (2020). PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat Cancer 1, 681–691. 10.1038/s43018-020-0075-x. [DOI] [PubMed] [Google Scholar]
- 7.Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, Guo J, Peng H, Chen M, Fu YX, and Tang H (2020). PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun 11, 4835. 10.1038/s41467-020-18570-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lau J, Cheung J, Navarro A, Lianoglou S, Haley B, Totpal K, Sanders L, Koeppen H, Caplazi P, McBride J, et al. (2017). Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat Commun 8, 14572. 10.1038/ncomms14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al. (2018). Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest 128, 805–815. 10.1172/JCI96113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang H, Liang Y, Anders RA, Taube JM, Qiu X, Mulgaonkar A, Liu X, Harrington SM, Guo J, Xin Y, et al. (2018). PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Invest 128, 580–588. 10.1172/JCI96061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, van Nimwegen M, Lau SP, Latupeirissa K, Schetters S, et al. (2020). The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell 38, 685–700 e688. 10.1016/j.ccell.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, Nordquist E, Cruz-Monserrate Z, Yu L, Young G, et al. (2018). IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 67, 320–332. 10.1136/gutjnl-2016-311585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perry CJ, Munoz-Rojas AR, Meeth KM, Kellman LN, Amezquita RA, Thakral D, Du VY, Wang JX, Damsky W, Kuhlmann AL, et al. (2018). Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J Exp Med 215, 877–893. 10.1084/jem.20171435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zanker DJ, Owen KL, Baschuk N, Spurling AJ, and Parker BS (2021). Loss of type I IFN responsiveness impairs natural killer cell antitumor activity in breast cancer. Cancer Immunol Immunother 70, 2125–2138. 10.1007/s00262-021-02857-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoon HH, Jin Z, Kour O, Kankeu Fonkoua LA, Shitara K, Gibson MK, Prokop LJ, Moehler M, Kang YK, Shi Q, and Ajani JA (2022). Association of PD-L1 Expression and Other Variables With Benefit From Immune Checkpoint Inhibition in Advanced Gastroesophageal Cancer: Systematic Review and Meta-analysis of 17 Phase 3 Randomized Clinical Trials. JAMA Oncol 8, 1456–1465. 10.1001/jamaoncol.2022.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou Y, Zou X, Zheng S, Tang H, Zhang L, Liu P, and Xie X (2020). Efficacy and predictive factors of immune checkpoint inhibitors in metastatic breast cancer: a systematic review and meta-analysis. Ther Adv Med Oncol 12, 1758835920940928. 10.1177/1758835920940928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, et al. (2019). Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 381, 1535–1546. 10.1056/NEJMoa1910836. [DOI] [PubMed] [Google Scholar]
- 18.Giancotti Filippo G. (2013). Mechanisms Governing Metastatic Dormancy and Reactivation. Cell 155, 750–764. 10.1016/j.cell.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Topalian SL, Taube JM, and Pardoll DM (2020). Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 367, eaax0182. 10.1126/science.aax0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, Zahurak M, Yang SC, Jones DR, Broderick S, et al. (2018). Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. New England Journal of Medicine 378, 1976–1986. 10.1056/NEJMoa1716078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, Denkert C, Park YH, Hui R, Harbeck N, et al. (2020). Pembrolizumab for Early Triple-Negative Breast Cancer. New England Journal of Medicine 382, 810–821. 10.1056/NEJMoa1910549. [DOI] [PubMed] [Google Scholar]
- 22.Rosner S, Reuss JE, and Forde PM (2019). PD-1 Blockade in Early-Stage Lung Cancer. Annual Review of Medicine 70, 425–435. 10.1146/annurev-med-050217-025205. [DOI] [PubMed] [Google Scholar]
- 23.Alves AM, Paredes J, and Schmitt F (2019). Expression of PD-L1 in primary breast carcinoma and lymph node metastases. Surgical and Experimental Pathology 2, 7. 10.1186/s42047-019-0033-z. [DOI] [Google Scholar]
- 24.Tarhini AA, Zahoor H, Yearley JH, Gibson C, Rahman Z, Dubner R, Rao UNM, Sander C, and Kirkwood JM (2015). Tumor associated PD-L1 expression pattern in microscopically tumor positive sentinel lymph nodes in patients with melanoma. Journal of Translational Medicine 13, 319. 10.1186/s12967-015-0678-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boussiotis VA (2016). Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med 375, 1767–1778. 10.1056/NEJMra1514296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, Xu H, Ruff W, Broadwater M, Choi IH, et al. (2009). PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood 113, 5811–5818. 10.1182/blood-2009-02-203141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al. (2015). Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 162, 1242–1256. 10.1016/j.cell.2015.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, Pal R, Yuan M, Asara J, Patsoukis N, and Boussiotis VA (2020). Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol 5. 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trefny MP, Kaiser M, Stanczak MA, Herzig P, Savic S, Wiese M, Lardinois D, Laubli H, Uhlenbrock F, and Zippelius A (2020). PD-1(+) natural killer cells in human non-small cell lung cancer can be activated by PD-1/PD-L1 blockade. Cancer Immunol Immun 69, 1505–1517. 10.1007/s00262-020-02558-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. (2017). PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499. 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, et al. (2014). Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics 15, 190. 10.1186/1471-2164-15-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodge JW, and Schlom J (1999). Comparative studies of a retrovirus versus a poxvirus vector in whole tumor-cell vaccines. Cancer Res 59, 5106–5111. [PubMed] [Google Scholar]
- 33.Adams S, Loi S, Toppmeyer D, Cescon DW, De Laurentiis M, Nanda R, Winer EP, Mukai H, Tamura K, Armstrong A, et al. (2019). Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase II KEYNOTE-086 study. Annals of Oncology 30, 405–411. 10.1093/annonc/mdy518. [DOI] [PubMed] [Google Scholar]
- 34.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. (2015). PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. New England Journal of Medicine 372, 2509–2520. 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan MH, Bristol JA, McDuffie E, and Abrams SI (2001). Regression of extensive pulmonary metastases in mice by adoptive transfer of antigen-specific CD8(+) CTL reactive against tumor cells expressing a naturally occurring rejection epitope. J Immunol 167, 4286–4292. [DOI] [PubMed] [Google Scholar]
- 36.Walling BL, and Kim M (2018). LFA-1 in T Cell Migration and Differentiation. Frontiers in Immunology 9. 10.3389/fimmu.2018.00952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, Elzen A.v.d., Hirn MJ, Coifman RR, et al. (2019). Visualizing structure and transitions in high-dimensional biological data. Nature Biotechnology 37, 1482–1492. 10.1038/s41587-019-0336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wimmers F, Subedi N, van Buuringen N, Heister D, Vivie J, Beeren-Reinieren I, Woestenenk R, Dolstra H, Piruska A, Jacobs JFM, et al. (2018). Single-cell analysis reveals that stochasticity and paracrine signaling control interferon-alpha production by plasmacytoid dendritic cells. Nat Commun 9, 3317. 10.1038/s41467-018-05784-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J, Fang Y, Chen K, Li S, Tang S, Ren Y, Cen Y, Fei W, Zhang B, Shen Y, and Lu W (2022). Single-cell RNA sequencing reveals the tissue architecture in human high-grade serous ovarian cancer. Clin Cancer Res. 10.1158/1078-0432.CCR-22-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, Todd KL, Henderson MA, Giuffrida L, Petley EV, et al. (2020). Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin Cancer Res 26, 487–504. 10.1158/1078-0432.CCR-19-1868. [DOI] [PubMed] [Google Scholar]
- 41.Tacconi C, Commerford CD, Dieterich LC, Schwager S, He Y, Ikenberg K, Friebel E, Becher B, Tugues S, and Detmar M (2021). CD169(+) lymph node macrophages have protective functions in mouse breast cancer metastasis. Cell Rep 35, 108993. 10.1016/j.celrep.2021.108993. [DOI] [PubMed] [Google Scholar]
- 42.Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, Thennavan A, Wang C, Torpy JR, Bartonicek N, et al. (2021). A single-cell and spatially resolved atlas of human breast cancers. Nat Genet 53, 1334–1347. 10.1038/s41588-021-00911-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259. 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, et al. (2019). Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334 e1310. 10.1016/j.immuni.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Chen H, Mo H, Hu X, Gao R, Zhao Y, Liu B, Niu L, Sun X, Yu X, et al. (2021). Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell. 10.1016/j.ccell.2021.09.010. [DOI] [PubMed] [Google Scholar]
- 46.Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, Ramnarain A, Gasmi B, Gururajan M, Redmond D, et al. (2020). ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 579, 130–135. 10.1038/s41586-020-2015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwartz C, Khan AR, Floudas A, Saunders SP, Hams E, Rodewald HR, McKenzie ANJ, and Fallon PG (2017). ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J Exp Med 214, 2507–2521. 10.1084/jem.20170051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, and Coussens LM (2009). CD4+ T Cells Regulate Pulmonary Metastasis of Mammary Carcinomas by Enhancing Protumor Properties of Macrophages. Cancer Cell 16, 91–102. 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Condeelis J, and Pollard JW (2006). Macrophages: Obligate Partners for Tumor Cell Migration, Invasion, and Metastasis. Cell 124, 263–266. 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 50.Sinha P, Clements VK, and Ostrand-Rosenberg S (2005). Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol 174, 636–645. 174/2/636 [pii]. [DOI] [PubMed] [Google Scholar]
- 51.Osorio JC, Arbour KC, Le DT, Durham JN, Plodkowski AJ, Halpenny DF, Ginsberg MS, Sawan P, Crompton JG, Yu HA, et al. (2019). Lesion-Level Response Dynamics to Programmed Cell Death Protein (PD-1) Blockade. Journal of Clinical Oncology 37, 3546–3555. 10.1200/jco.19.00709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayoux M, Roller A, Pulko V, Sammicheli S, Chen S, Sum E, Jost C, Fransen MF, Buser RB, Kowanetz M, et al. (2020). Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Science Translational Medicine 12, eaav7431. 10.1126/scitranslmed.aav7431. [DOI] [PubMed] [Google Scholar]
- 53.Gozgit JM, Vasbinder MM, Abo RP, Kunii K, Kuplast-Barr KG, Gui B, Lu AZ, Molina JR, Minissale E, Swinger KK, et al. (2021). PARP7 negatively regulates the type I interferon response in cancer cells and its inhibition triggers antitumor immunity. Cancer Cell 39, 1214–1226 e1210. 10.1016/j.ccell.2021.06.018. [DOI] [PubMed] [Google Scholar]
- 54.Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, and Fu YX (2014). Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 25, 37–48. 10.1016/j.ccr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Palma M, Mazzieri R, Politi LS, Pucci F, Zonari E, Sitia G, Mazzoleni S, Moi D, Venneri MA, Indraccolo S, et al. (2008). Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell 14, 299–311. 10.1016/j.ccr.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 56.Ortiz A, Gui J, Zahedi F, Yu P, Cho C, Bhattacharya S, Carbone CJ, Yu Q, Katlinski KV, Katlinskaya YV, et al. (2019). An Interferon-Driven Oxysterol-Based Defense against Tumor-Derived Extracellular Vesicles. Cancer Cell 35, 33–45 e36. 10.1016/j.ccell.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Carbone CJ, Beiting DP, Girondo MA, Peck AR, et al. (2017). Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 31, 194–207. 10.1016/j.ccell.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu C, Klement JD, Ibrahim ML, Xiao W, Redd PS, Nayak-Kapoor A, Zhou G, and Liu K (2019). Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J Immunother Cancer 7, 157. 10.1186/s40425-019-0635-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duggan MC, Jochems C, Donahue RN, Richards J, Karpa V, Foust E, Paul B, Brooks T, Tridandapani S, Olencki T, et al. (2016). A phase I study of recombinant (r) vaccinia-CEA(6D)-TRICOM and rFowlpox-CEA(6D)-TRICOM vaccines with GM-CSF and IFN-alpha-2b in patients with CEA-expressing carcinomas. Cancer Immunol Immunother 65, 1353–1364. 10.1007/s00262-016-1893-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hauschild A, Dummer R, Ugurel S, Kaehler KC, Egberts F, Fink W, Both-Skalsky J, Laetsch B, and Schadendorf D (2008). Combined treatment with pegylated interferon-alpha-2a and dacarbazine in patients with advanced metastatic melanoma: a phase 2 study. Cancer 113, 1404–1411. 10.1002/cncr.23722. [DOI] [PubMed] [Google Scholar]
- 61.Mocellin S, Pasquali S, Rossi CR, and Nitti D (2010). Interferon Alpha Adjuvant Therapy in Patients With High-Risk Melanoma: A Systematic Review and Meta-analysis. JNCI: Journal of the National Cancer Institute 102, 493–501. 10.1093/jnci/djq009. [DOI] [PubMed] [Google Scholar]
- 62.Brockwell NK, Owen KL, Zanker D, Spurling A, Rautela J, Duivenvoorden HM, Baschuk N, Caramia F, Loi S, Darcy PK, et al. (2017). Neoadjuvant Interferons: Critical for Effective PD-1-Based Immunotherapy in TNBC. Cancer Immunol Res 5, 871–884. 10.1158/2326-6066.CIR-17-0150. [DOI] [PubMed] [Google Scholar]
- 63.Saleiro D, and Platanias LC (2019). Interferon signaling in cancer. Non-canonical pathways and control of intracellular immune checkpoints. Semin Immunol 43, 101299. 10.1016/j.smim.2019.101299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Owen KL, Gearing LJ, Zanker DJ, Brockwell NK, Khoo WH, Roden DL, Cmero M, Mangiola S, Hong MK, Spurling AJ, et al. (2020). Prostate cancer cell-intrinsic interferon signaling regulates dormancy and metastatic outgrowth in bone. EMBO reports 21, e50162, e50162. doi: 10.15252/embr.202050162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boukhaled GM, Harding S, and Brooks DG (2021). Opposing Roles of Type I Interferons in Cancer Immunity. Annu Rev Pathol 16, 167–198. 10.1146/annurev-pathol-031920-093932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiao W, Klement JD, Lu C, Ibrahim ML, and Liu K (2018). IFNAR1 Controls Autocrine Type I IFN Regulation of PD-L1 Expression in Myeloid-Derived Suppressor Cells. J Immunol 201, 264–277. 10.4049/jimmunol.1800129 jimmunol.1800129 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, et al. (2015). STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Science Translational Medicine 7, 283ra252–283ra252. 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davar D, Wang H, Chauvin J-M, Pagliano O, Fourcade JJ, Ka M, Menna C, Rose A, Sander C, Borhani AA, et al. (2018). Phase Ib/II Study of Pembrolizumab and Pegylated-Interferon Alfa-2b in Advanced Melanoma. J Clin Oncol 36, JCO1800632–JCO1800632. 10.1200/JCO.18.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, Andrews D, Mikeska T, Mangan NE, Samarajiwa SA, et al. (2012). Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nature Medicine 18, 1224–1231. 10.1038/nm.2830. [DOI] [PubMed] [Google Scholar]
- 70.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, and Taniguchi T (2005). IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777. 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 71.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. (2017). Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov 7, 188–201. 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 375, 819–829. 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, McGraw T, and Mittal V (2019). The lung microenvironment: an important regulator of tumour growth and metastasis. Nature Reviews Cancer 19, 9–31. 10.1038/s41568-018-0081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tumeh PC, Hellmann MD, Hamid O, Tsai KK, Loo KL, Gubens MA, Rosenblum M, Harview CL, Taube JM, Handley N, et al. (2017). Liver Metastasis and Treatment Outcome with Anti-PD-1 Monoclonal Antibody in Patients with Melanoma and NSCLC. Cancer Immunol Res 5, 417–424. 10.1158/2326-6066.CIR-16-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pires da Silva I, Lo S, Quek C, Gonzalez M, Carlino MS, Long GV, and Menzies AM (2020). Site-specific response patterns, pseudoprogression, and acquired resistance in patients with melanoma treated with ipilimumab combined with anti-PD-1 therapy. Cancer 126, 86–97. 10.1002/cncr.32522. [DOI] [PubMed] [Google Scholar]
- 76.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 77.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS: A Journal of Integrative Biology 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zimmerman M, Hu X, and Liu K (2010). Experimental metastasis and CTL adoptive transfer immunotherapy mouse model. J Vis Exp, 2077. 10.3791/2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pandey R, Ramdas B, Wan C, Sandusky G, Mohseni M, Zhang C, and Kapur R (2019). SHP2 inhibition reduces leukemogenesis in models of combined genetic and epigenetic mutations. The Journal of Clinical Investigation 129, 5468–5473. 10.1172/JCI130520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416.e411. 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martín-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949.e916. 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, Leeson R, Kanodia A, Mei S, Lin J-R, et al. (2018). A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e24. ttps://doi.org/ 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Redd PS, Ibrahim ML, Klement JD, Sharman SK, Paschall AV, Yang D, Nayak-Kapoor A, and Liu K (2017). SETD1B Activates iNOS Expression in Myeloid-Derived Suppressor Cells. Cancer Research 77, 2834–2843. 10.1158/0008-5472.Can-16-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.