

Abstract
Immunotherapy has demonstrated great success in clinical treatment, especially for cancer care. Here we review preclinical models, including cell lines, three dimensional (3D) cultures, and mouse models to support the need for tools enabling the development of novel immune–oncology (I–O) therapies. While in vitro studies have the advantage of being relatively simpler, faster, and higher throughput than in vivo models, they must be designed carefully to recapitulate the biological conditions that influence drug efficacy. The growing prevalence of 3D in vitro and ex vivo models has enabled screening and mechanistic studies in more complex, tissue-like environments containing multiple interacting cell types. On the other hand, syngeneic mouse models have been instrumental in the historical development of immunotherapies and remain an important tool in drug development, despite lacking fidelity to certain aspects of human physiology and pathology. Xenograft and humanized mouse models address some of these challenges, yet present limitations of their own. Successful development and translation of new I–O therapies will likely require thoughtful combination of several of these preclinical models, and we aim to help research and development scientists utilize the appropriate tools and technologies to facilitate rapid transition from preclinical evaluation to clinical trials.
INTRODUCTION
The field of immune–oncology (I–O) has transformed the care for cancer patients. In the late 19th century, William B. Coley, the father of immunotherapy, first attempted to harness the power of the immune system using ‘Coley’s toxin’ for treating cancer patients. This cocktail of live and inactivated bacteria achieved some durable complete remissions in a series of malignancies, including sarcoma, lymphoma, and testicular carcinoma [1]. In the 1980s, Rosenberg et al. demonstrated that administration of high dose cytokine IL-2 could lead to durable, complete, and apparently curative regressions in some patients with metastatic melanoma and renal cancer [2,3]. Inspired by Paul Ehrlich’s ‘magic bullets’ concept, in 1997 rituximab became the first approved monoclonal antibody (mAb) for the treatment of lymphoma [4,5]. The discovery of cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) and the antibody drugs targeting them, immune checkpoint inhibitors (ICIs), propelled the I–O field into the current era [6–10]. On the other hand, chimeric antigen receptor T (CAR-T) cell therapy rewires patient immune cells to target tumor antigens independent of major histocompatibility complex (MHC) and there are six CAR-T products that have been approved by the US food and drug administration (FDA) [11–17]. The first pediatric patient in the world to receive CAR-T cell therapy has been tumor free for 10 years.
Through years of breakthroughs, as well as challenges and struggles, I–O therapies have been embraced by the oncology community due to their great clinical success. In this review article, we highlight emerging preclinical models for I–O therapy development [Table 1, Figure 1] and describe their ability to recapitulate the tumor microenvironment (TME), inclusion of extracellular matrix (ECM), discuss specific applications in drug development, and compare the advantages and limitations of current models.
TABLE 1.
Preclinical immuno-oncology models.
| Applications | Advantages | Limitations | |
|---|---|---|---|
| 2D cultures | ► Drug screening ► In vitro evaluation |
► Easy access ► Fast readout ► Low cost |
► Can be poorly predictive |
| 3D cultures | ► Drug screening ► Ex vivo evaluation ► Study of TME and ECM |
► Relatively easy to prepare ► Can recapitulate TME and ECM ► Histological fidelity to original tumor |
► Lack of inter-organ communication |
| Syngeneic mouse models | ► In vivo efficacy and safety assessment ► Study of disease development |
► Can engineer specific genes | ► Failure to accurately mimic human disease phenotypes |
| Xenograft mouse models | ► In vivo efficacy and safety assessment | ► Includes cell-derived xenograft (CDX) and patient-derived xenograft (PDX) ► PDX can recapitulate patient tumor signatures |
► Costly |
| Humanized mouse models | ► In vivo efficacy and safety assessment ► Study of TME |
► Can recapitulate human TME | ► Costly |
FIGURE 1.
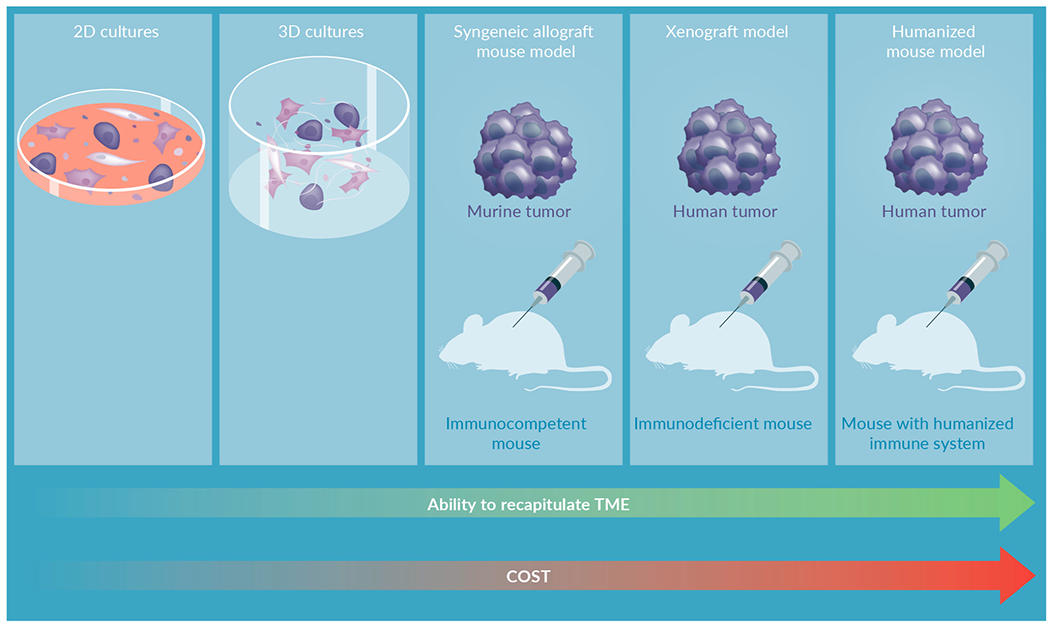
Preclinical immuno-oncology models.
2D cultures
Choosing a suitable cell line is critical for producing models reflective of tumor biology with appropriate antigenicity and driver mutations. Mutational statuses of cell lines used in I–O research should reflect tumor biology For example, the von Hippel–Lindau (VHL) gene is mutated in 90% of sporadic clear cell renal cell carcinoma (ccRCC) cases [18]. Other common mutations found in ccRCC include tumor suppressor genes, such as PBRM1, BAP1, SETD2 [19]. More than 20 cell lines are frequently used in renal cell carcinoma (RCC) research, including ACHN (uncertain RCC histotype), A-498 (used as a model of ccRCC and widely in cancer research), 786-O (used as a model of ccRCC), and SK-RC cell lines (obtained from ccRCC metastases) [20]. ACHN mRNA lacks mutations in VHL and hypoxia-inducible factor (HIF)-1α, 786-O bears mutated VHL, and SK-RC cell lines express either HIF-2α only or both HIF-1α and HIF-2α [20]. In order to use models most reflective of natural tumor biology, immunohistochemistry (IHC), gene sequencing, and histology analysis of tumors can provide insight into RCC subtypes to enhance the translational potential of experiments using 2D cultures [20].
Cell line choice is not only determined based on gene mutation status, but also on different antigen expression. For example, hormone receptor status plays an important role in determining a suitable model for breast cancer research. Estrogen receptor (ER), progesterone receptor (PR), and amplification of human epidermal growth factor receptor 2 (HER2) status provide information on tumor biology and therapeutic response, necessitating choosing cell lines for 2D culture that reflect tumor subtypes [21,22]. In addition, many of the long-established cell lines frequently used in research are derived from metastases, rather than primary tumors, which is not representative of varying stages of tumor progression [23]. Dai and colleagues categorized breast cancer cell lines into subtypes luminal A, luminal B, HER2+, triple negative breast cancer (TNBC) A (TNA), and TNBC B (TNB), from least to most aggressive, to better reflect differences in receptor statuses [21]. Luminal A cell lines, including MCF-7, BT-483, CAMA-1, HCC-1428, HCC-712, and IBEP-2, are ER+, HER2−, and have varying PR statuses [21]. Luminal B cell lines, including BSMZ, BT474, IBEP1, and IBEP3, are ER+, HER2+, and have varying PR statuses [21]. HER2+ cell lines, including 21MT2, HCC1008, HH315, and SKBR3 are ER−, HER2+, and typically PR− [21]. TNA cell lines, including DU4475, EMG3, HCC1937, MDAMB436, and MDAMB468, and TNB cell lines, including Hs578T, MDAMB157, MDAMB231, and SUM149PT are ER−, PR−, and HER2− [21].
One limitation of 2D culture is that standard incubators mimic atmospheric oxygen concentrations. These conditions are not reflective of the lower oxygen tension, termed hypoxia, or the insufficient delivery of oxygen to cells that is commonly found in solid tumors [24]. This important aspect of cell physiology can be achieved in 2D culture by using hypoxia mimetic agents to increase HIF-1α availability [24]. For example, CoCl2 is a commonly used hypoxia mimetic that competes with Fe2+ ions, inhibiting HIF-prolyl hydroxylases (PHDs) activity, which prevents the degradation of HIF-1α and thereby mimics hypoxia [24]. Adherent cell lines, even under ‘normoxic’ atmospheric concentrations, can experience hypoxia or anoxia, as oxygen exchange may only occur via diffusion from cell culture media, with oxygen availability and consumption rates periodically changing in response to one another [24]. Short-term, cyclic, ‘intermittent hypoxia’ (IH) experienced by cells in vivo may be mimicked in vitro through the use of flow-through systems supplying precise concentrations of oxygen through solenoid valves, growing adherent cells in a perfusion-based system of tube-like channels through which media is supplied, or using bioreactors with peristaltic pumps to periodically flow media with desired oxygen conditions [24]. Furthermore, cell culture media formulations also do not replicate the concentration of nutrients, amino acids, and electrolytes found in human plasma. The two most commonly used media formulations, Dulbecco’s modified Eagle’s medium and Roswell Park Memorial Institute-1640 (RPMI-1640), contain significantly higher glucose concentrations than physiologic and varying amounts of electrolytes [25]. Typically, cell culture media is supplemented with serum, often from fetal calves, to supply growth factors and other essential components lacking in the basal medium. However, serum is known to vary between batches, and there are now many efforts designed to reduce or eliminate the need for serum in cell culture. Among these reduced-serum or serum-free approaches, there are recently developed media designed to mimic human serum or plasma, with adjusted amino acid formulations. The balance of nutrients, metabolites, amino acids, electrolytes, vitamins, and trace elements inevitably impact cell metabolism and gene expression, and adoption of more physiologic media may improve the likeness of cell culture to in vivo conditions [26,27].
Another challenge for 2D studies of cancer research is the heterogeneity of the TME, which in addition to cancer cells, include endothelial cells, epithelial cells, immune cells, and cancer-associated fibroblasts (CAFs), which are not replicated using cancer cell lines [28]. CAFs found in the stroma of human cancers provide signaling and remodeling functions, and typically exhibit upregulated ECM production and remodeling (e.g. collagen) and secretion of soluble pro-tumor cytokines and growth factors [28]. Recent scRNAseq studies have revealed that the heterogeneity of these CAFs in the TME may derive from the variety of spatial subgroups found in normal fibroblasts [28], and further work will be required to characterize the cross-talk between CAFs and other cell types in the TME [28]. These goals cannot be accomplished through the use of 2D culture alone but will require the use of co-cultured cells (in trans wells, for example) or 3D organoid culture to mimic the TME.
In addition to the variety of cell types that make up the TME, CAFs function to produce and assemble the complex composition of the tumor ECM through the production of fibrous proteins, proteoglycans, glycosaminoglycans, and glycoproteins, which contribute signaling and support for tumor growth and migration [28] and can also impede immune cell movement and activation. The composition of the ECM and resulting crosslinking of the tumor stroma impacts drug penetration, with CAFs playing an important role of remodeling the ECM through the production of lysyloxidase (LOX) family and MMP enzymes [28]. LOX oxidases catalyze the crosslinking of collagen and ELN in the ECM, increasing tumor stroma stiffness [28]. LOX oxidases are overexpressed in CAFs, with LOXL2 expression in gastric CAFs having been associated with invasive potential [28,29]. Inhibition of LOXL2 and LOX in breast cancer has resulted in reduction of tumors, angiogenesis, and metastasis [30].
ECM proteins can also function as ligands, binding integrin receptors on cell membranes [28]. Interaction with the rigid ECM can lead to integrin molecule dimerization, activating the focal adhesion cascade [28]. Further, ECM rigidity can trigger SRC-YAP-MYL9/MYL2, leading to maintenance of the CAF phenotype with CAF function reinforcing ECM stiffness, promoting an environment that facilitates improved tumor cell invasion [28,30].
Targeting ECM proteins, therefore, is an attractive method for generating an environment that is more permissive to the delivery of anti-cancer therapies. Generating models that are reflective of the crosstalk between cell types and CAF-ECM protein interactions cannot be accomplished through 2D culture alone. However, the use of cell lines is beneficial as they are able to provide a relatively high number of cells for experiments, compared to primary cultures and animal models increasing the speed at which research can be conducted [20]. Further, 2D cultures are an unlimited self-replicating source [23]. An important drawback of the use of cell lines, however, is the inability of these simplified models to exhibit crosstalk between cells and interactions with the tumor microenvironment [20]. This limitation can be overcome through the use of 3D cultures or co-cultures [20].
Another key benefit of using established cancer cell lines is experimental consistency and repeatability between labs. Short tandem repeat (STR) DNA profiling can be used to identify human cell lines to ensure the absence of cross-contamination or misidentification thereby improving the accuracy of assays [31]. STR DNA profiling uses DNA hypervariable regions, consisting of variable number tandem repeat (VNTR) units, for identification of a unique DNA ‘fingerprint,’ through the analysis of 1–6 bp core sequences of STR microsatellite regions [31]. The eight core STR loci used for identification include D5S818, D13S317, D7S820, D16S539, vWA, Th01, TPOX, and CSF1PO [31]. Cell lines are authenticated if the STR profile is a greater than 80% match with the tissue from which it originates. A match of 56% or more is considered unrelated, and values between 56 and 80% require further analysis [31]. In this way, independent research groups are capable of repeating and validating published studies using the same cell lines.
3D organoids
The ultimate promise of organoid technology is to improve the accuracy and predictive value of I–O research. Organoids represent a compromise between the simplicity and straightforwardness of traditional cell culture and the more complex and physiological conditions provided by in vivo experiments. While both of these methodological approaches will remain components of any research and discovery efforts, organoids have begun to take a larger role in basic and translational research programs. Whether these organoids are generated from differentiated stem cells to resemble specific tissue types, assembled from cell aggregates to form tumor spheroids, or are collected from patient samples for ex vivo organoid studies, they possess several advantages for I–O studies.
First, the most obvious feature of organoids and spheroids that distinguish them from traditional cell culture is their 3D structure. While this difference might seem subtle or arbitrary, cellular organization and culture substrates can have significant impacts on cell phenotypes in ways that influence tumor growth and immunity Through the years, many research groups have reported how conversion from 2D to 3D culture format changed cell phenotypes, with distinct gene signatures that are required to support 3D tumor growth identified by a recent CRISPR screen study [32].
When a cancer cell line is aggregated into tumor spheroids, they adhere to each other and form connections more similar to in vivo architecture, including the generation of ECM. Cancer cells produce more ECM in 3D than in 2D, and the 3D format may also alter the ratios between different ECM proteins, including collagens (I, III, IV, V), fibronectin, and laminin [33–36]. ECM organization can also evolve in 3D, in ways that cannot be modeled by simple 2D monolayers of cells [37], with these changes in the ECM likely to alter the density and stiffness of the tumor spheroid. Tumor mechanical properties such as stiffness may dramatically impact response to immunotherapies because lymphocytes are supremely mechanosensitive cells and respond to the mechanical conditions of both the microenvironment and of their target cells.
Natural killer (NK) and T lymphocytes are mechanosensitive as a consequence of their mechanisms of cytotoxicity. The immunological synapse (IS) of a T cell consists of the joining of the T cell receptor (TCR) on the effector cell and the peptide-MHC on the target cell or antigen presenting cell [38]. T cell cytotoxicity is correlated to the force generated at the IS, largely through the efficiency of perforin delivery. Increasing the membrane tension of the target cell enhances the speed and efficiency of pore formation and perforin-mediated killing [39]. Stiff environments, such as tissue culture plastic surfaces, enhance T cell cytotoxicity due to increased membrane tension in monolayers of cancer cells [39–41]. Similarly, NK cells employ perforin-mediated cytotoxicity, and they have also demonstrated more rapid killing in higher density collagen gels [42]. Furthermore, T cells rely on stiffness cues to regulate their proliferation, migration, and activation, and T cell expansion methodology has been improved by optimizing the stiffness imposed by microparticles carrying activating antibodies [43,44].
Beyond the influence on cytotoxic efficiency of tumor infiltrating lymphocytes, stiffness can modulate immune checkpoint molecule expression in the spheroids, with higher stiffness upregulating the expression of programmed death ligand 1 (PD-L1) in breast cancer spheroids [45]. Simply culturing tumor spheroids in 3D has been shown to alter PD-L1 expression heterogeneously by tissue type. PD-L1 increased as a result of spheroid culture in colorectal cancer, renal cell carcinoma, and breast cancer cell lines, but was unchanged in gastric adenocarcinoma [45–48]. Therefore, models that faithfully recapitulate the mechanical properties of the native tissue are important to ensure realistic levels of lymphocyte cytotoxicity occur as would be seen in vivo.
In addition to PD-L1, additional phenotypic shifts occur in cancer cell lines cultured in 3D vs 2D monolayers, including changes in several cell surface molecules important to drug delivery and I–O studies. Studies examining NK or T cell killing in cancer spheroids have noted reduced activation and killing in 3D compared to 2D controls. Reduced T cell cytotoxicity was attributed, in part, to reduced expression of MHC-class I molecules by 3D spheroids [49], and these spheroids were less susceptible to cytokine-induced upregulation of MHC-class I [50]. On the other hand, HLA-E, an inhibitory ligand towards NK cells, was upregulated in cancer cells cultured in 3D [42,51]. Spheroids may also lose expression of death receptors required for apoptosis mediated by TNF-α -related apoptosis inducing ligand (TRAIL), through the upregulation of cyclooxygenase-2 and prostaglandin E2 (COX-2/PGE2) pathways [52]. 3D spheroids are also likely to increase expression of an efflux pump known as P-glycoprotein (P-gp), a recognized cause of multidrug resistance [53]. P-gp upregulation has been attributed to metabolic changes that occur in spheroids such as reactive oxygen species and activation of the HIF-1α pathway [54,55].
Hypoxia can be achieved in traditional cell culture using specialized equipment, but spatial gradients in oxygen tension occur naturally in spheroids due to the balance between diffusion and consumption. One study that measured the oxygen pressure in tumor spheroids found an average oxygen diffusion distance of 232 ± 22 μm [56]. Therefore, spheroids large enough to exhaust oxygen diffusion limits will develop concentric regions of oxygenation: from the well-oxygenated and proliferative outer shell, through a hypoxic transitional zone, and to a central anoxic, necrotic core [57,58]. Tumor spheroids have been observed to activate the HIF-1α pathway in cell lines that do not express it in 2D (e.g. HeLa, MCF-7) [59,60]. Hypoxia subsequently reduced the migration, infiltration and cytotoxicity of T cells in microfluidic models [50,61]. Like oxygen, nutrients must also diffuse sufficient distances to reach distal cells in 3D organoids. A study of NK cell activation established a nutrient gradient in a microfluidic device and found that in the distal, nutrient-deprived region, NK cells became less proliferative and less responsive to cytokines, while at the same time, more pro-inflammatory [62].
Solid tumors have long been known to shift their metabolism to favor aerobic glycolysis, a phenomenon known as the Warburg effect [63]. Cancer spheroids exhibit increased expression of the glucose transporter 1 (GLUT-1) and lactate dehydrogenase, the enzyme responsible for lactate production [64,65]. As expected, levels of lactic acid and lactate have been found to be higher in 3D spheroids than in 2D, impairing T cell function [49,66]. Acidification of the TME reduces lymphocyte efficacy in a number of ways including impaired cytotoxicity, reduced cytokine production, increased immunoinhibitory activity of the VISTA pathway [67], diminished expression of T cell receptors and CD25/IL-2Rα, and decreased activation of signal transducer and activator of transcription 5 and extracellular signal-regulated kinase [68–70].
Therefore, establishing 3D tumor geometries that allow realistic gradients of oxygen, nutrients, and pH will influence the results of I–O studies based on the altered response of lymphocytes to these conditions.
Patient-derived organoids
Tumor organoids derived from fresh patient tissue (patient-derived organotypic tumor spheroids or PDOTS), yield even more similarities to in vivo human tumors than organoids generated from cancer cell lines. PDOTS maintain the molecular characteristics of the native tumor sample, preserve intra-tumoral heterogeneity that does not exist in cell line models, and can retain the original stroma and immune cell populations, depending on the method of generation [71–73]. Sources for PDOTS include surgical resections, biopsies (both core-needle and fine-needle aspiration), or pleural effusion, and the PDOTS generated can be expanded, passaged, and cryopreserved [74–77]. Typically, mechanical and/or enzymatic digestion are used to break down tissue before straining to isolate small spheroids or single cells. Methods that fully dissociate samples into single cells then re-form spheroids by culturing in ultra-low attachment multi-well plates [78]. Several groups isolate small spheroids (<100 μm) using incomplete digestion of patient-derived tissue, which ensures that PDOTS generated in this way retain intact stroma from the native tumor, as well as a representative population of immune cells, including a matching repertoire of T cell receptors as the original tumor [73,79].
PDOTS may be immediately used in experiments or expanded using air-liquid interface or submerged hydrogel techniques [80,81]. With growing adoption of patient-derived organoid models, more groups have begun to use them for drug screening and validating that the response in PDOTS correlates to the response of the patient from which the tumor fragments were isolated [71,82]. Studies that obtain PDOTS from patients in clinical trials can compare the response rate observed in organoids to the patient response (generally using RECIST criteria or progression-free survival as metrics) using quantifications of spheroid size changes or viability [71]. While some such studies have only small numbers of samples, they often report clear concordance between organoid and patient responses to targeted therapies [83]. Larger studies have compared the molecular features of the native tumor to the PDOTS and found no significant differences between the genotype and phenotype of the tumor and PDOTS [84,85]. Furthermore, for immunotherapy testing, matched T cells can be obtained from peripheral blood mononuclear cells (PBMC) or from tumor infiltrating lymphocytes (TILs), and these can be added to PDOTS culture to assess spheroid infiltration and cytotoxicity by lymphocytes [73,86–88]. Further studies are required to determine the information gained from adding PBMC-derived immune cells vs. retaining the native immune population for immunotherapy efficacy. For example, PDOTS with intact stroma and immune cells were found to have a highly immunosuppressive environment [84]. PDOTS from colorectal cancer had high levels of myeloid-derived suppressor cells and low levels of effector lymphocytes such as NK cells and CD8+ T cells.
The exploration and development of patient-derived organoids presents the opportunity to use them for ‘personalized medicine’ or ‘precision clinical trials’ [72]. Obtaining tissue for PDOTS isolation at the start of a new trial will allow researchers to correlate the ex vivo response of PDOTS to the clinical response of each patient, which could increase the speed of determining drug response in the future, since organoid drug screening studies typically last for days to weeks rather than the weeks to months necessary to determine clinical responses. Such trials have reported good correlation between organoids and the clinic, with one study reporting 100% sensitivity and 93% specificity when testing immune checkpoint blockade in melanoma [89]. Beyond I–O therapy, many groups have used patient-derived organoid models for drug screening. This means that testing PDOTS should be able to identify ineffective therapies and point clinicians toward drugs more likely to be effective in an individual patient, such as a recent study involving breast cancer in which an organoid drug screen was used to identify the most effective drug for a patient experiencing early metastatic relapse [90]. Treating the patient with the drug identified resulted in disease-free progression 3-times longer than any other drug. Other studies have screened large drug libraries against PDOTS and validated the results with xenograft mouse models [91] or with correlation to clinical outcomes for chemotherapies currently in clinical use [92]. However, limitations still exist, and not all studies report high specificity, such as a trial in colorectal cancer that found that interferon γ (IFN-γ) production by T cells in PDOTS did not correlate well with patient response to immunotherapy [93]. This discordance between ex vivo and in vivo response may not be due to inherent differences in tumor phenotype, but rather due to the aspects of the microenvironment missing from PDOTS studies. For example, immune cell trafficking (adhesion to vasculature, extravasation, and migration to tumors) remains a significant barrier to mounting a productive immune response to tumors, even with the administration of immune checkpoint blockade therapies. Therefore, studies that combine microvascular models, patient-derived organoids, and circulating immune cells will be required to recapitulate the full TME and additional barriers to response produced by the stroma [94–96].
Limitations of tumor organoid methods
While tumor organoid models offer several advantages that will ensure their continued use for I–O studies, there are a number of limitations as well. First, organoid models are more complex than traditional 2D cultures, and thus will require additional training and resources, and potentially have reduced throughput. Cell line organoids generated in ultra-low attachment (ULA) plates do not require significantly more expertise that monolayer culture, but many other methods described here require more complex plating such as the air-liquid interface or submerged hydrogel methods for expansion of PDOTS, or microfluidic devices with compartments for tumor spheroids, stroma (vasculature, CAFs, etc.), immune cells, cell culture medium, etc. Many biological labs do not have equipment or expertise needed for soft-lithography fabrication of microfluidic devices. This limitation can be overcome by purchasing commercial microfluidic devices on the market, but at greater cost than tissue culture plastics and without the ability to customize device designs to suit specific needs.
Additionally, 3D organoid culture introduces additional variables that are not present in traditional cell culture, especially the choice of hydrogel for organoid embedding. Care must be taken to standardize and characterize these hydrogels. There is a growing desire to develop synthetic gels and culture conditions to eliminate these sources of uncertainty and variability [71]. Since lymphocytes are highly sensitive to mechanical cues, subtle changes in matrix density, stiffness, or composition could produce differences in therapeutic response that will be difficult to attribute to a single cause without thorough understanding of the role the microenvironment plays in lymphocyte behavior. However, this is also a key benefit of using micro physiological systems for basic science studies of interactions between tumor, stroma, and immune cells.
While many tumor spheroid models exist and have been described here, there are also increasingly sophisticated tissue-specific organoid models of normal tissue being developed. However, few groups have combined normal and tumor organoids [97]. Future cancer organoid models could integrate tumor spheroids with healthy organoids from the same tissue, which would enable us to model additional aspects of tumor growth and development such as invasion and metastasis. Similarly, micro physiological models of the immune system, such as lymph node on-a-chip, have been developed but not combined with tumor organoids, so there are opportunities to model features of lymphocyte maturation and proliferation that these platforms enable [98,99].
Finally, since the behavior of CD8+ effector T cells is critical to response to ICIs, multicellular organoid models must address mismatched human leukocyte antigen (HLA) types and the graft vs host response that can result from combining cells from multiple donors. Though syngeneic mouse cells circumvent this limitation and can be used in organoid platforms, the need for human models remains [100–103]. An alternative is to use HLA-matched cells, such as the combination of HLA-A*0201 melanoma and MART-1 specific, HLA-A*0201 restricted T cells [49], or engineered MHC-non-restricted CAR-T or TCR T cells [50,61]. For patient-derived models, T cells can be isolated from the same patient and re-introduced into the organoid model [86,87]. However, these approaches may not work for all pre-clinical immune-oncology studies and new approaches such as knockout of MHC molecules on cell types required to generate the microenvironmental architecture could be employed [104,105].
Mouse models
In the early-stage development of immunotherapies, researchers heavily depend on the in vitro models which lack of systemic immunity to provide response from endogenous immune cells. Using mouse models to assess immunotherapy efficacy provides researchers a means to inquiry the relationship between tumor cells and immune cells, as well as assess efficacy and safety of immunotherapies in presence of systemic immunity. Here, we summarize multiple mouse models for preclinical research, including syngeneic mouse model, tumor bearing immunodeficient mouse model, and humanized mouse model.
Syngeneic mouse models
The syngeneic mouse model is able to mimic the pathological transformation process of oncogenesis from normal cells into malignant cells [106], and can be categorized into three classes, subcutaneous tumor cell line, orthotopic tumor cell line and genetically engineered orthotopic tumor development. Kirsten rat sarcoma virus gene mutations are presented in approximately 25% of lung adenocarcinoma and are associated with a worse prognosis [107,108]. Tumor cells derived from Kraslox-stop-lox(lsl)-G12D/+; p53flox/flox (KP) inversion induced Joined neoantigen (NINJA) mice expressed neoantigens, were immunogenic and able to response to ICIs, including anti-PD1 and anti-CTLA4 mAbs [109,110]. In addition to NINJA, Cre-Lox system enables mammalian genome modification in vivo, carrying out deletions, insertions, translocations and inversions at specific tissues via tamoxifen induced Cre recombinase activation [111,112]. For example, ccRCC is characterized by inactivation of the VHL gene. The dysfunction of VHL leads to HIF hyperactivation, resulting in overexpression of many downstream genes involved in angiogenesis, metabolism, and cell-cycle regulation including which represent important therapy targets for patients with ccRCC [113,114]. A tamoxifen inducible ccRCC mouse model generated by renal epithelial cells with specific deletion from Vhl, Trp53, and Rb1 is able to mimic the cancer pathological process from proximal tubule epithelial cells and share similar transcriptional signatures with human ccRCC [115,116]. Overall, the cell lines have natural number of neoantigens and the spontaneous developed tumor has fewer neoantigens.
Immunodeficient mouse models
Immunodeficient mice were designed to overcome the rejection of human cancer cells as well as human immune cells mediated by the mouse adaptive and innate immune responses, and serve as powerful tools to assess I–O therapies [117]. For example, the fork head box N1 (Foxn1null) mutation, commonly known as nude, lacks a thymus and therefore is deficient in T cells but has functional B cells and NK cells [118,119]. Knocking out the recombination activating gene 1 (Rag1) [120], recombination activating gene 2 (Rag2) [121], protein kinase DNA-activated catalytic polypeptide (Prkdc) genes [122] that are essential for variable (V), diversity (D), and joining (J) rearrangements, results in murine T and/or B cell deficiency Depletion of interleukin 2 receptor subunit gamma (IL2rg) [123] or β2-microglobulin (B2m) [124] genes that are required in interleukin signaling and NK development, leads to the absence or functional impairment of murine NK cells in non-obese diabetic (NOD) mouse model [125]. Combinations of these genetic strategies have been applied to develop the popular immunodeficient mouse strains, such as NOD/Prkdcscid (NOD/SCID) [124], NOD/SCID IL2rg−/− (NSG or NOG) [126,127], and Balb/c Rag1−/− IL2rg−/− (BRG) that have all been used in human oncology studies [117].
To choose an appropriate immunodeficient mouse model for a specific project, a number of factors should be taken into consideration, including gene background, endogenous immune cell components, leakiness (B and T cell development), lifespan, and husbandry [128]. The table 2 summarizes the immune cell components (T cells, B cells, NK cells) in several commonly used immunodeficient mouse models. Leakiness refers to the tendency of some mouse strains to develop functional B and T cells as the mice age. In general, leakiness is higher in mice with the C57BL/6J and BALB/cByJ backgrounds, lower in the ones with C3H/HeSn-JSmn background [129]. Due to the severe immunodeficiency, Rag1null and Pkrdcscid mice have specific husbandry requirements including that they should be housed in specific pathogen-free (SPF) environments. In addition, due to lack of efficient DNA repair, the Prkdcscid mice are radiation sensitive [130] and therefore cannot be as intensively irradiated as other immunodeficient models before being engrafted.
TABLE 2.
Immunodeficient mouse strains for human cancer study.
| Name | Strain | T cells | B cells | NK cells |
|---|---|---|---|---|
| Nude [118] | Foxn1null | No | Yes | Yes |
| Scid [131] | B6.CB17-Prkdcscid/SzJ | No | No | Yes |
| BRG [117] | BALB/c.Rag2−/− IL-2Rg−/−c | No | No | No |
| NOD-scid [132] | NOD.CB17-Prkdcscid/J | No | No | Function impaired |
| NOD/SCID [124] | B2mnull NOD.Cg-B2mtm1UncPrkdcscid/SzJ | No | No | Function loss |
| NSG [126] | NOD.Cg-PrkdcscidIL2rgtm1Wjl/SzJ | No | No | No |
| NOG [127] | NOD.Cg-PrkdcscidIL2rgtm1Sug/JicTac | No | No | No |
| BRGS [133] | BALB/c.Rag2−/−IL-2Rg−/− c NOD.sirpa | No | No | No |
| hSIRPa-BRG [134] | BALB/c.Rag2−/−IL-2Rg−/− c human.sirpa | No | No | No |
| MISTRG [135] | C;129S4-Rag2tm1.1FlvCsf1tm1(CSF1)FlvCsf2/Il3tm1.1(CSF2,IL3)Flv Thpotm1.1(TPO)FlvIl2rgtm1.1FlvTg(SIRPA)1Flv/J | No | No | No |
Cell-derived xenograft (CDX) models and patient-derived xenograft (PDX) models
CDX [118] and PDX [136] models developed in immunodeficient mice are widely used in cancer studies. A cell-derived COLO205 colorectal cancer cell xenograft mouse model is able to assess the synergistic effect of combination therapy of anti-death receptor 5 antibody TRA-8 and SN-38, an active metabolite of antitumor agent irinotecan (CPT-11) [137]. Orthotopic, tumor-bearing, mouse models provide more relevant development environments compared to an ectopic model in evaluation of I–O therapies, such as antibody therapies [138] and CAR-T cell therapies [139–141], and could have a better predictive value of disease [142–145].
PDX established directly from patient tumor tissue, conserves patient tumor signatures as well as the complex interplay between cancer cells and TME and has a better prediction for response and prognosis [146]. It has been reported that PDX share remarkable similarity in response rates compared to respective clinical trials [147], and serve as a critical tool in personalized medicine [148,149]. The patient-derived colorectal cancer models can retain intratumoral clonal heterogeneity and chromosomal instability and can be used for prediction of the response to an anti-epidermal growth factor receptor (EGFR) antibody, cetuximab, in patients [150,151]. The RCC models maintain the ability to evaluate tumor angiogenesis, retain genetic and histological characteristics [152], and accurately represent their respective original patient tumors [153]. In 2016, US National Cancer Institute (NCI) decided to retire the NCI-60 (a panel of 60 human cancer cell lines), and preferentially use PDX models derived from patient clinical samples and tagged with their clinical information for drug screening because the TME in PDX mimics human tumor better [154].
Humanized mouse models
The application of CDX and PDX models remarkably facilitates human cancer research and antitumor drug development. However, recent studies have demonstrated that the absence of human immunity in these models severely compromise their value in translational research and the development of novel I–O therapies [106,155]. The construction of humanized animal models through transplanting human tissues (such as bone marrow-liver-thymus, aka BLT), PBMCs (such as Hu-PBL-SCID) or hematopoietic stem cells (HSCs) (such as SRC-Hu) into immunodeficient mice has allowed for the development a rudimentary level of innate and adaptive human immunity in small animals [156].
In hu-PBL-SCID mice, the human T cells are highly engrafted and expanded and the mice developed severe graft-versus-host disease (GVHD) [157]. Using PBMC-engrafted NSG and SGM3 mice, Ye et al. were able to capture alloreactivity in the form of cytokine release syndrome (CRS) from individual human PBMC donors [158]. Thus, hu-PBL-SCID mouse models serve as a rapid, sensitive, and reproducible platform to screen novel therapeutics for CRS, and provides a potential translational bridge for the study and prediction of CRS in vivo [159]. HSC-derived humanized mouse models derived from CD34+ progenitor cells are used to evaluate I–O therapies, such as anti-PD-1 mAb [160] and study antitumor effect in a physiologically relevant immune environment [161]. The humanization efficiency is determined by the mouse species, the CD45 cell resource, as well as the age of the mouse recipient [162]. The NSG-SGM3 strain is a particularly good mouse model for humanization to assess immunotherapies and to study the TME [117,163], as it expresses human stem cell factor, GM-CSF, and IL-3 transgenes, supporting HSCs engraftment and the development of myeloid cells in vivo [164–166, 167]. It has been reported that transferring cord blood or fetal liver derived HSCs results in a higher engraftment of human CD45 cells compared to engrafting the bone marrow or mobilized peripheral blood derived HSCs [168,169]. In general, newborn recipients exhibited a better reconstitution of human CD45 cells compared to adult recipients [167,170,171].
Due to the lack of human thymus in HSCs derived humanized mice, the T cell are educated in mouse thymus, leading to poor human thymopoiesis [160] and deficient HLA dependent antigen specific immune responses [172]. The Thy/HSC [173] and BLT [174] models can overcome this limitation, providing robust human thymopoiesis and generating HLA-restricted antigen specific human T cell reactions. However, this model is limited by the accessibility of fetal tissues and local policy regulation [106]. On the other hand, Chang et al. matured DCs to present tumor antigens to prime T cells in vitro, to assess cytotoxicity of CCR4 targeted mAb in vivo. Those tumor primed T (TP-T) cells had an increased IFN-γ expression reacting to the same tumor cells compared to unprimed T cells from the same donor in vitro and exhibited superior tumor control in combination with anti-CCR4 mAb in an ovarian cancer bearing mouse model [175].
CONCLUSION
Here, we summarize the applications of 2D culture, 3D cultures, and mouse models in I–O in order provide insights for research scientists trying to choose appropriate models in different phases of therapy development and to speed up the process of translating preclinical research to clinical trials. Selecting appropriate models will be critical to achieve robust results that enable accurate identification of effective and ineffective drugs and the successful clinical translation of new technologies. Therefore, researchers must carefully consider which features the TME are of key importance for testing a new therapeutic. Convincing I–O researchers to consider this additional layer of methodological scrutiny and fostering greater understanding of the relative strengths and weaknesses of each of these preclinical drug screening methods will benefit the field as a whole by improving the predictive power of preclinical studies.
Funding declaration:
Wang Y received financial support for the research, authorship and/or publication of this article by the Wong Family Award. Wang Y is also a NIH-funded author, grant number NIH P50 CA101942. Shelton S recieved financial support for the research, authorship and/or publication of this article by National Institute of Health (NIH) fellowship made to MIT (K00CA212227). Freeman G received financial support for the research, authorship and/or publication of this article by NIH P50 CA101942. Marasco WA received financial support for the research, authorship and/or publication of this article by the Assistant Secretary of Defense for Health Affairs endorsed by the Department of Defense, through the FY21 Translational Research Partnership Award (W81X-WH-21-1-0442) and FY21 Idea Development Award (W81XWH-21-1-0482) to W.A.M. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
Disclosure and potential conflicts of interest:
Barbie D discloses they are the co-founder of Xsphera Biosciences Freeman G discloses he has patents on PD-L1/PD-1 pathway at Roche, Merck MSD, Bristol Myers Squibb, Merck KGA, Boehringer-Ingelheim, AstraZeneca, Dako, Leica, Mayo Clinic, Eli Lilly, Novartis. He also has a patent at patents on TIM-3 pathway at Novartis. Also has consulting fees at Roche, Bristol-Myers-Squibb, Triursus, iTeos, NextPoint, IgM, Jubilant, Trillium, GV20, IOME and Geode. At Invaria he is on the board of directors. He also has stock options at Nextpoint, Triursus, Xios, iTeos, IgM, Trillium, Invaria, GV20, Geode.
Contributor Information
Yufei Wang, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA and Harvard Medical School, Boston, MA 02115, USA.
Sarah E Shelton, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA and Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Gabriella Kastrunes, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA.
David A Barbie, Harvard Medical School, Boston, MA 02115, USA and Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA.
Gordon J Freeman, Harvard Medical School, Boston, MA 02115, USA and Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA.
Wayne A Marasco, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA and Harvard Medical School, Boston, MA 02115, USA.
REFERENCES
- 1.McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft –tissue sarcomas. Iowa Orthop. J 2006;26, 154–158. [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA. IL –2: the first effective immunotherapy for human cancer. J. Immunol 2014. Jun 15; 192, 5451–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Packard BS, Aebersold PM et al. Use of Tumor –Infiltrating Lymphocytes and Interleukin –2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med 1988;319, 1676–1680. [DOI] [PubMed] [Google Scholar]
- 4.Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008; 8: 473–480. [DOI] [PubMed] [Google Scholar]
- 5.Grillo–López AJ, White CA, Varns C et al. Overview of the clinical development of rituximab: first monoclonal antibody approved for the treatment of lymphoma. Semin. Oncol 1999; 26, 66–73 [PubMed] [Google Scholar]
- 6.Brunet JF, Denizot F, Luciani MF et al. A new member of the immunoglobulin superfamily --CTLA −4.Nature 1987; 328, 267–270. [DOI] [PubMed] [Google Scholar]
- 7.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo. J 1992; 11, 3887–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA –4 blockade. Science. 1996; 271, 1734–1736. [DOI] [PubMed] [Google Scholar]
- 9.Freeman GJ, Long AJ, Iwai Y et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med 2000; 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med 1999; 5, 1365–1369. [DOI] [PubMed] [Google Scholar]
- 11.Maude SL, Laetsch TW, Buechner J et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med 2018; 378, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuster SJ, Bishop MR, Tam CS et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B Cell Lymphoma. N. Engl. J. Med 2019; 380, 45–56. [DOI] [PubMed] [Google Scholar]
- 13.Neelapu SS, Locke FL, Bartlett NL et al. Axicabtagene Ciloleucel CAR T Cell Therapy in Refractory Large B Cell Lymphoma. N. Engl. J. Med 2017; 377, 2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Locke FL, Ghobadi A, Jacobson CA, Miklos DB et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019; 20, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang M, Munoz J, Goy A et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med 2020; 382: 1331–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abramson JS, Palomba ML, Gordon LI et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020; 396, 839–852. [DOI] [PubMed] [Google Scholar]
- 17.Food and Drug Administeration (FDA). Approves First Cell-Based Gene Therapy for Adult Patients With Multiple Myeloma. FDA. 2021. (Mar 27). (Accessed Mar 27). [Google Scholar]
- 18.Nickerson ML, Jaeger E, Shi Y et al. Improved identification of von Hippel–Lindau gene alterations in clear cell renal tumors. Clin. Cancer Res 2008; 14, 4726–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brugarolas J. PBRM1 and BAP1 as novel targets for renal cell carcinoma. Cancer J. 2013; 19, 324 –332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brodaczewska KK, Szczylik C, Fiedorowicz M, Porta C, Czarnecka AM. Choosing the right cell line for renal cell cancer research. Mol. Cancer 2016; 15, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai X, Cheng H, Bai Z, Li J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017; 8(6), 3131 –3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chavez KJ, Garimella SV, Lipkowitz S. Triple negative breast cancer cell lines: one tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010;32, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burdall SE, Hanby AM, Lansdown MR, Speirs V. Breast cancer cell lines: friend or foe? Breast Cancer Res. 2003; 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavlacky J, Polak J. Technical Feasibility and Physiological Relevance of Hypoxic Cell Culture Models. Front Endocrinol. (Lausanne) 2020; 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKee TJ, Komarova SV. Is it time to reinvent basic cell culture medium? Am J Physiol Cell Physiol. 2017; 312, C624–C626. [DOI] [PubMed] [Google Scholar]
- 26.Ackermann T & Tardito S Cell Culture Medium Formulation and Its Implications in Cancer Metabolism. Trends Cancer. 2019; 5, 329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vande Voorde J, Ackermann T, Pfetzer N et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv 2019; 5, eaau7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu T, Zhou L, Li D, Andl T, Zhang Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front Cell Dev. Biol 2019; 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kasashima H, Yashiro M, Kinoshita H et al. Lysyl oxidase –like 2 (LOXL2) from stromal fibroblasts stimulates the progression of gastric cancer. Cancer Lett. 2014; 354, 438 –446. [DOI] [PubMed] [Google Scholar]
- 30.Chang J, Lucas MC, Leonte LE et al. Pre –clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 2017; 8, 26066–26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reid Y, Storts D, Riss T, Minor L. in Assay Guidance Manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences (Editors: Markossian S et al. ) 2004. [PubMed] [Google Scholar]
- 32.Han K, Pierce SE, Li A et al. CRISPR screens in cancer spheroids identify 3D growth –specific vulnerabilities. Nature. 2020; 580, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nederman T, Norling B, Glimelius B, Carlsson J, Brunk U. Demonstration of an extracellular matrix in multicellular tumor spheroids. Cancer Res. 1984; 44(7), 3090–3097 [PubMed] [Google Scholar]
- 34.Bai C, Yang M, Fan Z, Li S, Gao T, Fang Z. Associations of chemo-and radio-resistant phenotypes with the gap junction, adhesion and extracellular matrix in a three-dimensional culture model of soft sarcoma. J. Exper. Clin. Cancer Res 2015;34, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bjerkvig R, Laerum OD, Rucklidge GJ. Immunocytochemical characterization of extracellular matrix proteins expressed by cultured glioma cells. Cancer Res. 1989;49, 5424–5428 [PubMed] [Google Scholar]
- 36.Glimelius B, Norling B, Nederman T, Carlsson J. Extracellular matrices in multicellular spheroids of human glioma origin: Increased incorporation of proteoglycans and flbronectin as compared to monolayer cultures. Apmis. 1988;96, 433–444. [DOI] [PubMed] [Google Scholar]
- 37.Ackland ML, Ward J, Ackland CM, Greaves M, Walker M. Extracelluar matrix induces formation of organoids and changes in cell surface morphology in cultured human breast carcinoma cells PMC42-LA.In vitro Cell Dev Biol Anim. 2003;39, 428–433. [DOI] [PubMed] [Google Scholar]
- 38.Dustin ML. The immunological synapse. Cancer Immunol. Res 2014;2, 1023–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basu R, Whitlock BM, Husson J et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 2016;165, 100–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rossy J, Laufer JM, Legler DF. Role of mechanotransduction and tension in T cell function. Front. Immunol 2018; 2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Zhang T, Zhang H et al. Cell Softness Prevents Cytolytic T-cell Killing of Tumor-Repopulating Cells. Cancer Res. 2021; 81, 476–488. [DOI] [PubMed] [Google Scholar]
- 42.Park D, Son K, Hwang Y et al. High-throughput microfluidic 3D cytotoxicity assay for cancer immunotherapy (CACI-IMPACT platform). Front. Immunol 2019;10, 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dang AP, De Leo S, Bogdanowicz DR, et al. Enhanced activation and expansion of T cells using mechanically soft elastomer fibers. Adv. Biosyst 2018; 2, 1700167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lambert LH, Goebrecht GK, De Leo SE et al. Improving T cell expansion with a soft touch. Nano. letters 2017;17, 821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Azadi S, Es HA, Bazaz SR et al. Upregulation of PD-L1 expression in breast cancer cells through the formation of 3D multicellular cancer aggregates under different chemical and mechanical conditions. Biochim Biophys Acta Mol Cell Res. 2019;1866, 118526. [DOI] [PubMed] [Google Scholar]
- 46.Lanuza PM, Vigueras A, Olivan S et al. Activated human primary NK cells efficiently kill colorectal cancer cells in 3D spheroid cultures irrespectively of the level of PD-L1 expression. Oncoimmunol. 2018; 7, e1395123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rausch M, Blanc L, De Souza Silva O, Dormond O, Griffioen AW, Nowak-Sliwinska P. Characterization of renal cell carcinoma heterotypic 3D co –cultures with immune cell subsets.Cancers. 2021; 13, 2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou S, Zhu M, Meng F et al. Evaluation of PD-1 blockade using a multicellular tumor spheroid model. Am. J. Transl. Res 2019;11, 7471. [PMC free article] [PubMed] [Google Scholar]
- 49.Feder –Mengus C, Ghosh S, Weber W, Wyler S, Zajac P, Terracciano L et al. Multiple mechanisms underlie defective recognition of melanoma cells cultured in three –dimensional architectures by antigen –specific cytotoxic T lymphocytes.British journal of cancer. 2007. 96: 1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pavesi A, Tan AT, Koh S et al. A 3D microfluidic model for preclinical evaluation of TCR –engineered T cells against solid tumors. JCI Insight. 2017; 2, e89762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He W, Kuang Y, Xing X et al. Proteomic comparison of 3D and 2D glioma models reveals increased HLA-E expression in 3D models is associated with resistance to NK cell-mediated cytotoxicity. J. Proteome Res 2014; 13, 2272–2281. [DOI] [PubMed] [Google Scholar]
- 52.Chandrasekaran S, Marshall JR, Messing JA, Hsu JW, King MR. TRAIL-mediated apoptosis in breast cancer cells cultured as 3D spheroids. PLoS One. 2014; 9, e111487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bergman AM, Pinedo HM, Talianidis I et al. Increased sensitivity to gemcitabine of P-glycoprotein and multidrug resistance-associated protein-overexpressing human cancer cell lines. Br J Cancer. 2003; 88, 1963–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wartenberg M, Ling FC, Muschen M et al. Regulation of the multidrug resistance transporter P-glycoprotein in multicellular tumor spheroids by hypoxia-inducible factor (HIF-1) and reactive oxygen species. FASEB J. 2003; 17, 503–505. [DOI] [PubMed] [Google Scholar]
- 55.Wartenberg M, Richter M, Datchev A et al. Glycolytic pyruvate regulates P-Glycoprotein expression in multicellular tumor spheroids via modulation of the intracellular redox state. J. Cell. Biochem 2010; 109, 434–446. [DOI] [PubMed] [Google Scholar]
- 56.Grimes DR, Kelly C, Bloch K, Partridge MA. Method for estimating the oxygen consumption rate in multicellular tumour spheroids. J. R. Soc. Interface 2014;11. 20131124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sciumè G, Shelton S, Gray W et al. Tumor growth modeling from the perspective of multiphase porous media mechanics. Mol. Cell. Biomech 2012; 9, 193. [PMC free article] [PubMed] [Google Scholar]
- 58.Sciume G, Shelton S, Gray WG et al. A multiphase model for three-dimensional tumor growth. New J. Phys 2013;15, 015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian X, Wang W, Zhang Q et al. Hypoxia –inducible factor –1alpha enhances the malignant phenotype of multicellular spheroid HeLa cells in vitro. Oncol. Lett 2010; 1, 893–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doublier S, Belisario DC, Polimeni M et al. HIF-1 activation induces doxorubicin resistance in MCF7 3-D spheroids via P-glycoprotein expression: a potential model of the chemo-resistance of invasive micropapillary carcinoma of the breast. BMC Cancer 2012; 12, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ando Y, Siegler EL, Ta HP et al. Evaluating CAR-T Cell Therapy in a Hypoxic 3D Tumor Model. Adv. Healthc. Mater 2019; 8, 1900001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ayuso JM, Rehman S, Virumbrales-Munoz M et al. Microfluidic tumor-on-a-chip model to evaluate the role of tumor environmental stress on NK cell exhaustion. Sci. Adv 2021; 7, eabc2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Potter M, Newport E, Morten KJ. The Warburg effect: 80 years on. Biochem. Soc. Trans 2016; 44, 1499–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Longati P, Jia X, Eimer J et al. 3D pancreatic carcinoma spheroids induce a matrix –rich, chemoresistant phenotype offering a better model for drug testing. BMC cancer. 2013;13, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pereira PMR, Berisha N, Bhupathiraju N, Fernandes R, Tome JPC, Drain CM. Cancer cell spheroids are a better screen for the photodynamic efficiency of glycosylated photosensitizers. PLoS One. 2017; 12, e0177737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khaitan D, Chandna S, Arya MB, Dwarakanath BS. Establishment and characterization of multicellular spheroids from a human glioma cell line; Implications for tumor therapy. J. Transl. Med 2006; 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yuan L, Tatineni J, Mahoney KM, Freeman GJ. VISTA: A Mediator of Quiescence and a Promising Target in Cancer Immunotherapy. Trends Immunol. 2021; 42, 209–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakagawa Y, Negishi Y, Shimizu M, Takahashi M, Ichikawa M, Takahashi H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol. Lett 2015; 167, 72–86. [DOI] [PubMed] [Google Scholar]
- 69.Lardner A. The effects of extracellular pH on immune function. J. Leukoc. Biol 2001; 69, 522–530. [PubMed] [Google Scholar]
- 70.Calcinotto A, Filipazzi P, Grioni M, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012; 72, 2746–2756. [DOI] [PubMed] [Google Scholar]
- 71.Wensink GE, Elias SG, Mullenders J et al. Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. NPJ Precis. Oncol 2021; 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Veninga V, Voest EE. Tumor organoids: opportunities and challenges to guide precision medicine. Cancer Cell. 2021; 39, 1190–1201. [DOI] [PubMed] [Google Scholar]
- 73.Jenkins RW, Aref AR, Lizotte PH et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 2018; 8, 196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vilgelm AE, Bergdorf K, Wolf M et al. Fine-needle aspiration-based patient-derived cancer organoids. Iscience. 2020; 23, 101408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Phifer CJ, Bergdorf KN, Bechard ME et al. Obtaining patient-derived cancer organoid cultures via fine-needle aspiration. STAR protocols. 2021; 2, 100220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim S-Y, Kim S-M, Lim S et al. Modeling clinical responses to targeted therapies by patient –derived organoids of advanced lung adenocarcinoma. Clin. Cancer Res 2021; 27, 4397–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mazzocchi A, Devarasetty M, Herberg S et al. Pleural effusion aspirate for use in 3D lung cancer modeling and chemotherapy screening. ACS Biomat Sci Eng. 2019; 5, 1937–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Appleton KM, Elrod AK, Lassahn KA, Shuford S, Holmes LM, DesRochers TM. PD-1/PD-L1 checkpoint inhibitors in combination with olaparib display antitumor activity in ovarian cancer patient- derived three-dimensional spheroid cultures. Cancer Immunol., Immunother 2021; 70, 843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neal JT, Li X, Zhu J, Giangarra V et al. Organoid modeling of the tumor immune microenvironment. Cell 2018; 175, 1972–1988 e1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nagle PW, Plukker JTM, Muijs CT, van Luijk P, Coppes RP. Patient-derived tumor organoids for prediction of cancer treatment response. Semin. Cancer Biol 2018; 53, 258–264.81. [DOI] [PubMed] [Google Scholar]
- 81.Shelton SE, Nguyen HT, Barbie DA, Kamm RD. Engineering approaches for studying immune-tumor cell interactions and immunotherapy. Iscience 2021;24, 101985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Driehuis E, Kretzschmar K, Clevers H. Establishment of patient-derived cancer organoids for drug-screening applications. Nature Protocols. 2020; 15, 3380–3409. [DOI] [PubMed] [Google Scholar]
- 83.Puca L, Bareja R, Prandi D et al. Patient derived organoids to model rare prostate cancer phenotypes. Nature Commun. 2018; 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Finnberg NK, Gokare P, Lev A et al. Application of 3D tumoroid systems to define immune and cytotoxic therapeutic responses based on tumoroid and tissue slice culture molecular signatures. Oncotarget 2017. ; 8, 66747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vlachogiannis G, Hedayat S, Vatsiou A et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018; 359, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dijkstra KK, Cattaneo CM, Weeber F et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 2018; 174, 1586–1598 e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsai S, McOlash L, Palen K, Johnson B, Duris C, Yang Q. et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018; 18, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vellinga T, Den Uil S, Rinkes I et al. Collagen –rich stroma in aggressive colon tumors induces mesenchymal gene expression and tumor cell invasion. Oncogene. 2016; 35, 5263–5271. [DOI] [PubMed] [Google Scholar]
- 89.Votanopoulos KI, Forsythe S, Sivakumar H et al. Model of patient-specific immune-enhanced organoids for immunotherapy screening: feasibility study. Ann. Surg. Oncol 2020; 27, 1956–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guillen KP, Fujita M, Butterfield AJ et al. A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat. Cancer 2022; 3, 232–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.9Toshimitsu K, Takano A, Fujii M et al. Organoid screening reveals epigenetic vulnerabilities in human colorectal cancer. Nat. Chem. Biol 2022; 18, 605–614. [DOI] [PubMed] [Google Scholar]
- 92.Shi X, Li Y, Yuan Q et al. Integrated profiling of human pancreatic cancer organoids reveals chromatin accessibility features associated with drug sensitivity. Nat. Commun 2022; 13, 2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chalabi M, Fanchi LF, Dijkstra KK et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med 2020; 26, 566–576. [DOI] [PubMed] [Google Scholar]
- 94.Ayuso JM, Truttschel R, Gong MM et al. Evaluating natural killer cell cytotoxicity against solid tumors using a microfluidic model. Oncoimmunol. 2019; 8, 1553477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Campisi M, Sundararaman SK, Shelton SE et al. Tumor-derived cGAMP regulates activation of the vasculature. Front. Immunol 2020; 11. 2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shirure VS, Bi Y, Curtis MB et al. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab on a Chip. 2018; 18, 3687–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bian S, Repic M, Guo Z, A et al. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018; 15, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rosa PM, Gopalakrishnan N, Ibrahim H, Haug M, Halaas Ø. The intercell dynamics of T cells and dendritic cells in a lymph node-on-a-chip flow device. Lab on a Chip. 2016; 16, 3728–3740. [DOI] [PubMed] [Google Scholar]
- 99.Shim S, Belanger MC, Harris AR, Munson JM, Pompano RR. Two-way communication between ex vivo tissues on a microfluidic chip: Application to tumor-lymph node interaction. Lab on a Chip. 2019; 19, 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Businaro L, De Ninno A, Schiavoni G et al. Cross talk between cancer and immune cells: exploring complex dynamics in a microfluidic environment. Lab on a Chip. 2013; 13, 229–239. [DOI] [PubMed] [Google Scholar]
- 101.Agliari E, Biselli E, De Ninno A et al. Cancer-driven dynamics of immune cells in a microfluidic environment. Sci. Rep 2014; 4, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Phan-Lai V, Florczyk SJ, Kievit FM et al. Three-dimensional scaffolds to evaluate tumor associated fibroblast-mediated suppression of breast tumor specific T cells. Biomacromolecules 2013; 14, 1330–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mattei F, Schiavoni G, De Ninno A et al. A multidisciplinary study using in vivo tumor models and microfluidic cell-on-chip approach to explore the cross-talk between cancer and immune cells. J. Immunotoxicol 2014; 11, 337–346. [DOI] [PubMed] [Google Scholar]
- 104.Mattapally S, Pawlik KM, Fast VG et al. Human leukocyte antigen class I and II knockout human induced pluripotent stem cell-derived cells: universal donor for cell therapy. J. Am. Heart Assoc 2018; 7, e010239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thongsin N, Wattanapanitch M. ). CRISPR/Cas9 Ribonucleoprotein Complex-Mediated Efficient B2M Knockout in Human Induced Pluripotent Stem Cells (iPSCs). Induced Pluripotent Stem (iPS) Cells. Methods in Molecular Biology (Editors: Nagy A, Turksen K). 2021; 2454. [DOI] [PubMed] [Google Scholar]
- 106.Tian H, Lyu Y, Yang YG, Hu Z. Humanized Rodent Models for Cancer Research. Front Oncol. 2020; 10, 1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Riely GJ, Kris MG, Rosenbaum D et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res 2008; 14, 5731–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kellar A, Egan C, Morris D. Preclinical Murine Models for Lung Cancer: Clinical Trial Applications. BioMed. Res. Int 2015; 2015, 621324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Damo M, Fitzgerald B, Lu Y et al. Inducible de novo expression of neoantigens in tumor cells and mice. Nat. Biotechnol 2021. ; 39, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fitzgerald B, Connolly KA, Cui C et al. A mouse model for the study of anti-tumor T cell responses in Kras -driven lung adenocarcinoma. Cell Rep. Methods 2021; 27, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Orban PC, Chui D, Marth JD. Tissue-and site-specific DNA recombination in transgenic mice. Proc. Natl. Acad. Sci. USA 1992; 89, 6861–6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim H, Kim M, Im SK, Fang S. Mouse Cre-LoxP system: general principles to determine tissue-specific roles of target genes. Lab Anim. Res 2018; 34, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shen C, Kaelin WG Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin. Cancer Biol 2013; 23, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ruf M, Mittmann C, Nowicka AM et al. pVHL/HIF-regulated CD70 expression is associated with infiltration of CD27+ lymphocytes and increased serum levels of soluble CD27 in clear cell renal cell carcinoma. Clin. Cancer Res 2015; 21, 889–898. [DOI] [PubMed] [Google Scholar]
- 115.Harlander S, Schönenberger D, Toussaint NC et al. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice. Nat. Med 2017; 23, 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schmidt LS, Linehan WM. A mouse model of renal cell carcinoma. Nat. Med 2017; 23, 802–803. [DOI] [PubMed] [Google Scholar]
- 117.Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat. Rev. Immunol 2012; 12, 786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fogh J, Fogh JM, Orfeo T. One Hundred and Twenty-Seven Cultured Human Tumor Cell Lines Producing Tumors in Nude Mice23. J. Nat. Cancer Inst 1977; 59, 221–226. [DOI] [PubMed] [Google Scholar]
- 119.Fogh J, Fogh JM, Orfeo T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J. Natl. Cancer Inst 1977; 59, 221–226. [DOI] [PubMed] [Google Scholar]
- 120.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992; 68, 869–877. [DOI] [PubMed] [Google Scholar]
- 121.Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 1992; 68, 855–867. [DOI] [PubMed] [Google Scholar]
- 122.Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983; 301, 527–530. [DOI] [PubMed] [Google Scholar]
- 123.Ito M, Hiramatsu H, Kobayashi K et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 2002; 100, 3175–3182. [DOI] [PubMed] [Google Scholar]
- 124.Christianson SW, Greiner DL, Hesselton RA et al. Enhanced human CD4+ T cell engraftment in beta2-microglobulin-deficient NOD-scid mice. J. Immunol 1997; 158, 3578–3586. [PubMed] [Google Scholar]
- 125.Takenaka K, Prasolava TK, Wang JC, et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol 2007; 8, 1313–1323. [DOI] [PubMed] [Google Scholar]
- 126.Shultz LD, Lyons BL, Burzenski LM et al. Human lymphoid and myeloid cell development in NOD/LtSz –scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol 2005; 174, 6477–6489. [DOI] [PubMed] [Google Scholar]
- 127.Nakamura M, Suemizu H. Novel metastasis models of human cancer in NOG mice. Curr. Top Microbiol. Immunol 2008; 324, 167–177. [DOI] [PubMed] [Google Scholar]
- 128.The Jackson Laboratory. Choosing an immunodeficient mouse model. JAX Notes 2006. (March 20). (Accessed Sep 2022).
- 129.Nonoyama S, Smith FO, Bernstein ID, Ochs HD. Strain-dependent leakiness of mice with severe combined immune deficiency. J. Immunol 1993; 150, 3817–3824 [PubMed] [Google Scholar]
- 130.Mathieu AL, Verronese E, Rice GI et al. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator-dependent autoimmunity. J. Allergy Clin. Immunol 2015; 135, 1578–1588 e1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bendle GM, Linnemann C, Hooijkaas AI et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med 2010; 16, 565–570. [DOI] [PubMed] [Google Scholar]
- 132.Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood 2006; 108, 487–492. [DOI] [PubMed] [Google Scholar]
- 133.Yamauchi T, Takenaka K, Urata S et al. Polymorphic Sirpa is the genetic determinant for NOD –based mouse lines to achieve efficient human cell engraftment. Blood. 2013; 121, 1316–1325. [DOI] [PubMed] [Google Scholar]
- 134.Herndler-Brandstetter D, Shan L, Yao Y et al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc. Natl. Acad. Sci. USA 2017; 114, e9626–e9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rongvaux A, Willinger T, Martinek J et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol 2014; 32, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hidalgo M, Amant F, Biankin AV et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014; 4, 998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Oliver PG, LoBuglio AF, Zinn KR et al. Treatment of human colon cancer xenografts with TRA-8 anti-death receptor 5 antibody alone or in combination with CPT-11. Clin. Cancer Res 2008; 14, 2180–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chang DK, Moniz RJ, Xu Z et al. Human anti-CAIX antibodies mediate immune cell inhibition of renal cell carcinoma in vitro and in a humanized mouse model in vivo. Mol. Cancer 2015; 14, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang Y, Buck A, Grimaud M et al. Anti-CAIX BBζ CAR4/8 T cells exhibit superior efficacy in a ccRCC mouse model. Mol. Ther. Oncolytics 2022; 24, 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Suarez ER, Chang de K, Sun J et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016; 7, 34341–34355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.de Campos NSP, de Oliveira Beserra A, Pereira PHB et al. Immune Checkpoint Blockade via PD-L1 Potentiates More CD28-Based than 4-1BB-Based Anti-Carbonic Anhydrase IX Chimeric Antigen Receptor T Cells. Int. J. Mol. Sci 2022; 23, 5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kerbel RS. Human tumor xenografts as predictive preclinical models for anticancer drug activity in humans: better than commonly perceived-but they can be improved. Cancer Biol. Ther 2003; 2 (4 Suppl 2), S134–S139. [PubMed] [Google Scholar]
- 143.Killion JJ, Radinsky R, Fidler IJ. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev. 1998; 17, 279–284. [DOI] [PubMed] [Google Scholar]
- 144.Johnson JI, Decker S, Zaharevitz D et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 2001; 8, 1424–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.1Priolo C, Agostini M, Vena N et al. Establishment and genomic characterization of mouse xenografts of human primary prostate tumors. Am. J. Pathol 2010; 176, 1901–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Izumchenko E, Paz K, Ciznadija D et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol 2017. ; 28, 2595–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Garrido-Laguna I, Uson M, Rajeshkumar NV et al. Tumor engraftment in nude mice and enrichment in stroma-related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin. Cancer Res 2011; 17, 5793–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hidalgo M, Bruckheimer E, Rajeshkumar NV et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol. Cancer Ther 2011; 10, 1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stebbing J, Paz K, Schwartz GK et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer 2014; 120, 2006–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Guenot D, Guérin E, Aguillon-Romain S, et al. Primary tumour genetic alterations and intra –tumoral heterogeneity are maintained in xenografts of human colon cancers showing chromosome instability. J. Pathol 2006; 208, 643–652. [DOI] [PubMed] [Google Scholar]
- 151.Fichtner I, Slisow W, Gill J et al. Anticancer drug response and expression of molecular markers in early-passage xenotransplanted colon carcinomas. Eur. J. Cancer 2004; 40, 298–307. [DOI] [PubMed] [Google Scholar]
- 152.Grisanzio C, Seeley A, Chang M et al. Orthotopic xenografts of RCC retain histological, immunophenotypic and genetic features of tumours in patients. J. Pathol 2011; 225, 212–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Karam JA, Zhang XY, Tamboli P et al. Development and characterization of clinically relevant tumor models from patients with renal cell carcinoma. Eur. Urol 2011; 59, 619–628. [DOI] [PubMed] [Google Scholar]
- 154.Ledford H. US cancer institute to overhaul tumour cell lines. Nature. 2016; 530, 391–391. [DOI] [PubMed] [Google Scholar]
- 155.Aparicio S, Hidalgo M, Kung AL. Examining the utility of patient-derived xenograft mouse models. Nat. Rev. Cancer 2015; 15, 311–316. [DOI] [PubMed] [Google Scholar]
- 156.Brehm MA, Cuthbert A, Yang C et al. Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rgamma(null) mutation. Clin. Immunol 2010; 135, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ito R, Katano I, Kawai K et al. Highly sensitive model for xenogenic GVHD using severe immunodeficient NOG mice. Transplantation 2009; 87, 1654–1658. [DOI] [PubMed] [Google Scholar]
- 158.Ye C, Yang H, Cheng M et al. A rapid, sensitive, and reproducible in vivo PBMC humanized murine model for determining therapeutic-related cytokine release syndrome. FASEB. J 2020; 34, 12963–12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shultz LD, Keck J, Burzenski L et al. Humanized mouse models of immunological diseases and precision medicine. Mamm. Genome 2019; 30, 123–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang M, Yao LC, Cheng M et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB, J. 2018; 32, 1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhao Y, Shuen TWH, Toh TB et al. Development of a new patient –derived xenograft humanised mouse model to study human –specific tumour microenvironment and immunotherapy. Gut 2018; 67, 1845–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ito R, Takahashi T, Ito M. Humanized mouse models: Application to human diseases. J. Cell Physiol 2018; 233, 3723–3728. [DOI] [PubMed] [Google Scholar]
- 163.Morton JJ, Bird G, Refaeli Y, Jimeno A. Humanized Mouse Xenograft Models: Narrowing the Tumor-Microenvironment Gap. Cancer Res. 2016; 76, 6153–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nicolini FE, Cashman JD, Hogge DE, Humphries RK, Eaves CJ. NOD/SCID mice engineered to express human IL-3, GM-CSF and Steel factor constitutively mobilize engrafted human progenitors and compromise human stem cell regeneration. Leukemia 2004; 18, 341–347. [DOI] [PubMed] [Google Scholar]
- 165.Wunderlich M, Chou FS, Link KA et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3.Leukemia. 2010; 24, 1785–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jangalwe S, Shultz LD, Mathew A, Brehm MA. Improved B cell development in humanized NOD-scid IL2Rγnull mice transgenically expressing human stem cell factor, granulocyte-macrophage colony-stimulating factor and interleukin-3. Immun. Inflamm. Dis 2016; 4, 427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ishikawa F, Yasukawa M, Lyons B et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 2005; 106, 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lepus CM, Gibson TF, Gerber SA et al. Comparison of human fetal liver, umbilical cord blood, and adult blood hematopoietic stem cell engraftment in NOD-scid/gammac−/−, Balb/c-Rag1−/− gammac−/−, and C.B 17-scid/bg immunodeficient mice. Hum Immunol. 2009; 70, 790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Matsumura T, Kametani Y, Ando K et al. Functional CD5+ B cells develop predominantly in the spleen of NOD/SCID/gammac(null) (NOG) mice transplanted either with human umbilical cord blood, bone marrow, or mobilized peripheral blood CD34+ cells. Exp. Hematol 2003; 31, 789–797. [DOI] [PubMed] [Google Scholar]
- 170.Katano I, Ito R, Kamisako T et al. NOD-Rag2null IL-2Rγnull mice: an alternative to NOG mice for generation of humanized mice. Exp. Anim 2014; 63: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Traggiai E, Chicha L, Mazzucchelli L et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. 2004; 304, 104–107. [DOI] [PubMed] [Google Scholar]
- 172.Shultz LD, Saito Y, Najima Y et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc. Natl Acad. Sci. USA 2010; 107, 13022–13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lan P, Wang L, Diouf B et al. Induction of human T –cell tolerance to porcine xenoantigens through mixed hematopoietic chimerism. Blood 2004; 103, 3964–3969. [DOI] [PubMed] [Google Scholar]
- 174.Melkus MW, Estes JD, Padgett-Thomas A et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat. Med 2006; 12, 1316–1322. [DOI] [PubMed] [Google Scholar]
- 175.Chang DK, Peterson E, Sun J et al. Anti –CCR4 monoclonal antibody enhances antitumor immunity by modulating tumor-infiltrating Tregs in an ovarian cancer xenograft humanized mouse model. Oncoimmunol. 2016; 5, e1090075. [DOI] [PMC free article] [PubMed] [Google Scholar]