

Abstract
Objective:
Universal screening of endometrial carcinoma (EC) for mismatch repair deficiency (MMRd) and Lynch syndrome uses presence of MLH1 methylation to omit common sporadic cases from follow-up germline testing. However, this overlooks rare cases with high-risk constitutional MLH1 methylation (epimutation), a poorly-recognized mechanism that predisposes to Lynch-type cancers with MLH1 methylation. We aimed to determine the role and frequency of constitutional MLH1 methylation among EC cases with MMRd, MLH1-methylated tumors.
Methods:
We screened blood for constitutional MLH1 methylation using pyrosequencing and real-time methylation-specific PCR in patients with MMRd, MLH1-methylated EC ascertained from (i) cancer clinics (n=4, <60 years), and (ii) two population-based cohorts; “Columbus-area” (n=68, all ages) and “Ohio Colorectal Cancer Prevention Initiative (OCCPI)” (n=24, <60 years).
Results:
Constitutional MLH1 methylation was identified in three out of four patients diagnosed between 36 and 59 years from cancer clinics. Two had mono-/hemi-allelic epimutation (~50% alleles methylated). One with multiple primaries had low-level mosaicism in normal tissues and somatic “second-hits” affecting the unmethylated allele in all tumors, demonstrating causation. In the population-based cohorts, all 68 cases from the Columbus-area cohort were negative and low-level mosaic constitutional MLH1 methylation was identified in one patient aged 36 years out of 24 from the OCCPI cohort, representing one of six (~17%) patients <50 years and one of 45 patients (~2%) <60 years in the combined cohorts. EC was the first/dual-first cancer in three patients with underlying constitutional MLH1 methylation.
Conclusions:
A correct diagnosis at first presentation of cancer is important as it will significantly alter clinical management. Screening for constitutional MLH1 methylation is warranted in patients with early-onset EC or synchronous/metachronous tumors (any age) displaying MLH1 methylation.
Keywords: Endometrial cancer, MLH1 methylation, constitutional MLH1 epimutation, Lynch syndrome
Introduction
About 30% of endometrial cancers (EC) are mismatch repair (MMR) deficient (MMRd), detected by absence of immunoexpression of one or more MMR proteins,[1] microsatellite instability (MSI),[2] or high mutational burden.[3] About 70% of MMRd EC are associated with aberrant methylation of the MLH1 CpG island promoter, causing transcriptional silencing.[4] These cases are considered “sporadic” since tumor MLH1 methylation is typically somatic-in-origin.[5, 6] About 20% of MMRd EC are associated with Lynch syndrome (LS), caused by a germline pathogenic variant (PV) affecting a MMR gene (MSH2, MLH1, MSH6, PMS2).[7] LS-associated cancer risks vary by gene mutated and sex, although EC in females and colorectal cancer (CRC) in both sexes are most prevalent. Lifelong surveillance, including regular colonoscopies, is recommended for early cancer detection and prevention.[8]
Constitutional MLH1 epimutation is an alternative mechanism that predisposes to MMRd cancers. This defect manifests as methylation of a single allele of the MLH1 promoter throughout normal tissues, accompanied by transcriptional silencing of the methylated allele.[9] This serves as the “first-hit” to produce tumors displaying MSI, dual absence of MLH1/PMS2 immunoexpression, and MLH1 methylation.[9, 10] Carriers of constitutional MLH1 epimutation have presented with early-onset and/or multiple cancers akin to the MLH1-LS phenotype and about one-third of female carriers developed EC <60 years of age.[11] Familial cases of constitutional MLH1 epimutation have been reported to show either autosomal dominant inheritance of MLH1 methylation linked to a genetic variant within or nearby MLH1, or non-Mendelian inheritance in the absence of any apparent genetic variant.[12–23] However, most cases have no significant family history due to de novo occurrence of the constitutional MLH1 epimutation, and negative germline genetic tests.[9, 22, 24, 25] Therefore, cases with constitutional MLH1 epimutation present a clinical and molecular diagnostic challenge. These cases need to be distinguished from common “sporadic” cases with somatic-in-origin tumor MLH1 methylation, and diagnosed at first presentation of cancer, in order to receive appropriate genetic counseling and surveillance to prevent metachronous cancers that they are at high risk for. Additional testing for the presence of MLH1 methylation in peripheral blood leukocytes (PBL), or other source of germline DNA, is required for a molecular diagnosis. This in turn, requires recognition of patients warranting this additional testing.
“Universal screening” of all EC for MMRd is recommended as the standard-of-care to guide precision therapy and identify cases warranting germline testing for LS. Current algorithms involve stepwise testing of all incident EC for MMRd by immunohistochemistry (IHC) of the MMR proteins (or MSI testing if results are uninterpretable) in the first tier. Reflex testing of those exhibiting dual loss of MLH1/PMS2 (or MSI) for MLH1 methylation is performed to identify “sporadic” cases for exclusion from genetic testing for LS.[4, 26–28] According to current National Comprehensive Cancer Network (NCCN) guidelines for uterine neoplasms, MMRd EC cases that are unmethylated at MLH1 are eligible for LS testing, whilst no further testing is recommended for those whose tumor is MLH1-methylated.[29] NCCN guidelines for genetic/familial high risk assessment for CRC state that presence of tumor MLH1 methylation can be used to exclude genetically high-risk cases and that constitutional MLH1 methylation etiology is rare, but should be considered in cases with MLH1-methylated CRC or EC if early-onset (<50 years) or there is a family history.[30] In practice, universal screening may overlook cases whose MLH1 methylation was constitutional (under the presumption it was somatic-in-origin). This may be compounded by the frequent lack of a family history in carriers of constitutional MLH1 methylation, who may therefore, be misdiagnosed as a common sporadic case.
While constitutional MLH1 epimutation is rare, its frequency among incident MMRd cancers remains unclear. Prior screens have focused on CRC cases, which have found the prevalence among patients meeting the revised Bethesda Guidelines for MSI testing with an MLH1-deficient tumor, and negative germline genetic test result, to be 3–9%.[9, 17, 22, 31–35] Slightly higher frequencies have been reported with the inclusion of tumor MLH1 methylation as a selection feature, at 3.5–15.6%.[35–38] However, only one screen for constitutional MLH1 methylation in EC has been reported, conducted in a hospital-based unselected, consecutive series of EC cases in Japan, which identified 1/206 (0.49%).[39] The patient had a prior colon cancer and a family history. MMR activity was not assessed systematically in this case series, so the overall rate of constitutional MLH1 epimutation among MMRd cases could not be determined. To our knowledge, the frequency of constitutional MLH1 epimutation among incident EC cases with MLH1-methylated tumors identified through universal screening has not previously been evaluated. Evidence-based guidelines are needed to select EC patients warranting screening for constitutional MLH1 methylation.
We assessed the role of constitutional MLH1 epimutation in patients ascertained via cancer clinics who had presented with MLH1-methylated EC <60 years. To estimate the frequency and age distribution of constitutional MLH1 methylation among incident MMRd EC cases whose tumor was MLH1-methylated, we leveraged the clinical resources and prior molecular pathology findings from population-based EC cohorts in the “Columbus-area HNPCC Study” (Columbus) and the “Ohio Colorectal Cancer Prevention Initiative” (OCCPI), from Ohio, USA.
Materials and Methods
The overall study design is shown in Figure 1.
Figure 1. Schematic of overall study design and assays for constitutional MLH1 methylation analyses.
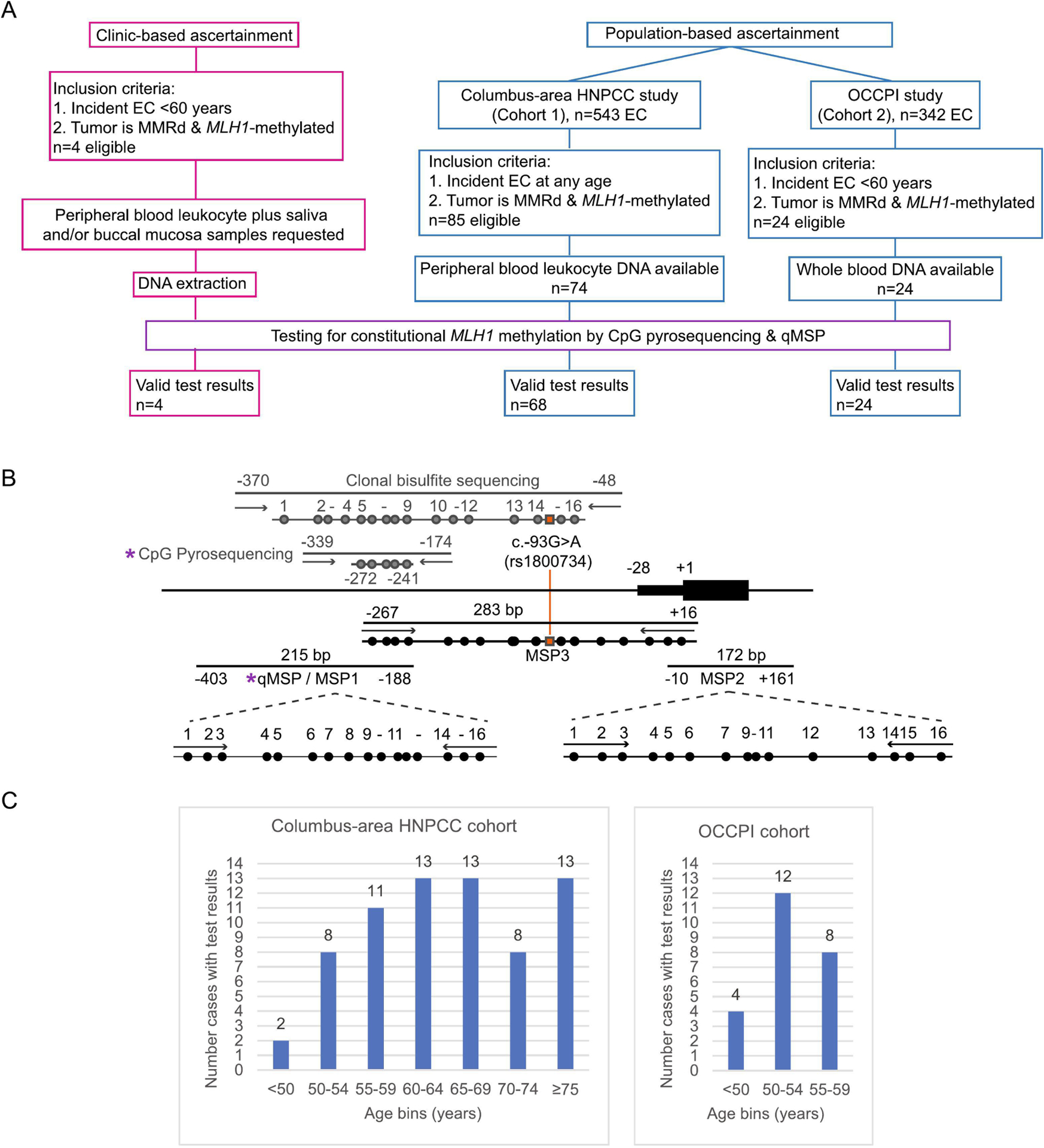
A: Flow diagram showing the ascertainment, selection, and numbers of patients eligible for testing for constitutional MLH1 epimutation. B: Map of the MLH1 CpG island proximal promoter region that corresponds with transcriptional activity,[6, 43] and assays designed to interrogate methylation status and levels in bisulfite-converted genomic DNA (not to scale). Purple asterix (*) indicates the quantitative CpG pyrosequencing and high-sensitivity real-time methylation-specific PCR (qMSP) assays used for first-pass screening to detect constitutional MLH1 methylation. Assays shown in gray are unbiased with respect to methylation status, such that PCR amplification will occur from both unmethylated and methylated templates. Assays in black are methylation-specific PCR-based (MSP) with primers overlapping designated CpG sites, hence amplification will occur only from methylated templates with high sensitivity. The MSP1 assay used for bisulfite-sequencing uses the same primers as the qMSP assay used for screening. MSP2 and MSP3 were used to confirm the presence of low-level methylation. Additional assays shown were used for confirmation of methylation. Horizontal bars show PCR products, circles show CpG sites interrogated within each amplicon. Orange line and squares indicate the location of the promoter c.−93G>A SNP (rs1800734) used to trace allele specificity for methylation in heterozygous cases positive for constitutional MLH1 methylation. The unbiased clonal bisulfite-sequencing assay was used to confirm and identify allelic methylation in germline DNA in patients with high levels of methylation measured by CpG pyrosequencing, and in tumor DNA. MSP3 was used to determine allelic methylation in heterozygous cases with low-level methylation. Locations of each assay are numbered with respect to the translation start site at +1 of MLH1 transcript, GenBank accession number NM_000249.3.
Cancer clinic-based ascertainment and sampling
Patients with MMRd, MLH1-methylated EC <60 years were referred by their treating physician or genetic counselor from cancer clinics in the USA (Figure 1A). First-degree relatives of probands with constitutional MLH1 epimutation were eligible to assess carrier status and potential inheritance. This study was approved by the Cedars-Sinai Medical Center institutional review board (Pro00049624) and subjects provided informed consent.
If PBL DNA was not already available, fresh samples were collected during a clinical appointment. Blood was collected into STRECK tubes and at least one additional non-circulating sample was requested, including saliva using Oragene-500 DNA Saliva Kit (DNA Genotek), buccal swab using DNA/RNA Shield Collection Kit (Zymo Research), or hair follicles. Buffy coat was separated by standard centrifugation and PBL nuclei isolated before DNA extraction using the Blood and Cell Culture DNA Mini kit (Qiagen). DNA was extracted from saliva and buccal mucosa post nuclease deactivation using the PT-L2P kit (DNA Genotek), and from hair follicles using the QuickExtract DNA Solution (Lucigen), according to the manufacturers’ instructions. Formalin-fixed paraffin-embedded (FFPE) tissue samples were macrodissected from 10 μM sections with reference to a hematoxylin and eosin-stained slide, following review and demarcation of areas of high tumor cellularity and normal cells by a pathologist. FFPE tissues were deparaffinized then DNA extracted using the QIAmp DNA FFPE Tissue Kit (Qiagen).
Population-based ascertainment
The first series of incident EC cases were derived from the Columbus cohort, which recruited unselected EC patients (n=543) who underwent surgery in a metropolitan hospital in the Columbus-area between 1999–2004. [40, 41] The second series was derived from the OCCPI cohort, which recruited unselected EC (n=342) patients who underwent surgery at Ohio State University between 2013–2016.[38] As previously described, MMR status was determined by IHC of the MMR proteins and/or MSI assessment using the NCI-designated pentaplex panel, whereby instability at 2/5 markers was classified MSI-high, 1/5 was MSI-low, and 0/5 was microsatellite stable (MSS).[40] MLH1 methylation status in tumors showing loss of MLH1 and/or MSI was previously assessed using established assays; methylation-specific PCR (MSP) (Columbus),[42, 43] or CpG pyrosequencing (OCCPI).[44] Eligibility criteria for inclusion in this study were cases whose tumor showed (1) IHC loss of MLH1, or MSI-high if IHC data was missing, and (2) MLH1 methylation, and (3) available genomic DNA from PBL (Columbus) or whole blood (OCCPI) (Figure 1A). Patient specimens and data were deidentified.
Screen for constitutional MLH1 methylation
Bisulfite-conversion was performed on 500ng DNA using the EZ DNA Methylation Gold Kit (Zymo Research) and ~50ng input into each assay. First-pass screening for constitutional MLH1 methylation was performed using two assays previously described for this purpose (Figure 1B), namely quantitative CpG pyrosequencing,[25] and high-sensitivity real-time methylation-specific PCR (qMSP).[12, 18] Samples were considered methylation-positive by pyrosequencing if all five CpG sites interrogated yielded a value ≥1 and the mean methylation was 2.3% (limit of detection) or above (Supplementary information). QMSP was performed on the CFX96 Thermal Cycler (Bio-Rad) using SYBR-Green fluorescein, followed by melt analysis to ensure methylation-specificity of the MLH1 amplicon, alongside MYOD as a control for sample input. A semi-quantitative percentage of methylated reference (PMR) value was calculated with reference to a fully-methylated sample (Universal Methylated DNA Standard, Zymo Research), as previously described.[45] This qMSP enables the detection of low-level mosaicism and samples were considered positive if the PMR was 0.1% (limit of detection) or above (Supplementary information). Both assays were applied to all genomic DNA samples. A follow-up assay, based on direct or clonal bisulfite-sequencing, was performed in one or more tissue samples from methylation-positive patients to confirm the presence of constitutional MLH1 methylation and to determine which allelic methylation patterns in patients heterozygous for the common c.−93G>A (rs1800734) promoter SNP (Figure 1B). The assay selected depended on initial methylation levels detected and c.−93G>A zygosity (Figure 1B). Methodological details are provided in Supplementary Information.
To screen for epimutation-associated genetic variants and promoter SNP genotyping, Sanger sequencing across the MLH1 CpG island was performed in cases with constitutional MLH1 methylation detected, as previously described.[12, 18]
Results
Clinically-ascertained patients
Patients ascertained via cancer clinics were eligible for inclusion if they had presented with EC <60 years and their tumor displayed MMRd (loss of MLH1 expression by IHC, and/or MSI) and MLH1 methylation. Four patients met these inclusion criteria and constitutional MLH1 methylation was detected in three, as follows:
Patient 192, of Native American ancestry, was diagnosed at 57 years with EC, FIGO stage IA (T1a, Nx), histologic grade (G) 1 and cervical clear cell adenocarcinoma, FIGO stage 1A2 (1a2, Nx). There was a wide margin between the two tumors and immunophenotyping of the cervical tumor was consistent with a diagnosis of clear cell adenocarcinoma as a distinct primary cancer (positivity for PAX-8, HNF-1B, CKAE1/AE3, CK7, wildtype TP53, patchy P16, and negativity for ER, PR, Napsin-A, CEA, CK5, P40, CDX2, CK20, OCT3, GATA-3, SOX-10, and CA-IX). MSI testing only was conducted on the EC, which was MSI-high.. MLH1 methylation testing was performed simultaneously on the EC and uninvolved tissue and both were methylation positive. Follow-up testing for constitutional MLH1 methylation in blood was recommended. The patient had a maternal family history that included LS-type cancers, however, age of onset among relatives and paternal history were unknown (Figure 2A). PBL nuclei and saliva tested positive for MLH1 methylation by qMSP (Figure 2B), and pyrosequencing measured methylation levels at 47–48% (Figure 2C). The patient was heterozygous at the c.−93G>A SNP (rs1800734) within the MLH1 promoter and clonal bisulfite-sequencing across the region encompassing this SNP showed monoallelic methylation specifically of the ‘A’ allele (Figure 2D). No other sequence variants were identified within the MLH1 CpG island. This molecular profile was consistent with a “classic” monoallelic constitutional MLH1 epimutation.
Patient 213 is White and was diagnosed at 36 years with EC, FIGO stage IA (T1a, Nx), G1, which showed absence of MLH1/PMS2 and MLH1 methylation. On this basis, no further follow-up was considered necessary at that time. At 41 years, she presented with a poorly differentiated colon (cecum) adenocarcinoma, AJCC stage IIIC (T3, N2b, Mx). Molecular pathology revealed loss of MLH1/PMS2, MLH1 methylation, and absence of the somatic BRAFV600E mutation that is frequently associated with somatic-in-origin MLH1 methylation in sporadic MMRd CRC.[46, 47] Given her personal history of MLH1-methylated metachronous cancers, she was referred for blood-based constitutional MLH1 methylation testing in a CLIA-approved facility (Mayo Clinic) and received a positive result. Patient 213 and her parents joined our study to confirm/determine carrier status and potential inheritance. The mother had no personal history of cancer. The father had prior non-LS cancer diagnoses (Figure 3A). The nuclear trio each provided fresh samples of blood, saliva, and buccal mucosa. MLH1 methylation testing was consistently positive in Patient 213 by qMSP (Figure 3B), and pyrosequencing measured ~50% methylation (Figure 3C). Clonal bisulfite-sequencing confirmed hemiallelic methylation (Figure 3D), however, the patient was homozygous across the MLH1 promoter, so methylation could not be assigned to a particular genetic or parental allele. Both parents were unmethylated in all tissues tested (Figure 3E, F). No sequence variants were identified within the MLH1 CpG island in this nuclear family. This was consistent with a “classic” hemiallelic constitutional MLH1 epimutation, which arose de novo in the proband with no apparent genetic basis.
Patient 177 is White and presented with EC (FIGO stage 1B, G1) at 59 years, with MLH1/PMS2 loss and MLH1 methylation. At 61 years, she presented with metastatic colon cancer, which was MMR-proficient by IHC and BRAFV600E mutant. Her mother developed breast cancer at 35 years. Patient 177 was referred for germline genetic testing (Ambry Genetics 36-gene CancerNext panel) and was negative. PBL DNA tested negative for constitutional MLH1 methylation (Supplementary information).
Patient 166 is White and presented with multiple primary cancers with no remarkable family history. She first presented with skin sebaceous carcinoma at 54 years, then synchronous endometrial and small intestine cancers at 55 years (Figure 4A). Surgical pathology diagnosed EC, FIGO stage IIIC1 (T1a, N1, M0), G1, and small intestine adenocarcinoma, AJCC stage IIB (T4, N0, M0), as distinct primaries. All three cancers showed loss of MLH1/PMS2, and MSI testing of the EC and small intestine tumors showed both were MSI-high. MLH1 methylation testing was performed on the EC, then retrospectively on the sebaceous carcinoma, and both were methylated. Germline genetic testing on Ambry Genetics ColoNext 20-gene panel was negative. Patient 166 provided fresh samples of blood, saliva, and hair follicles. Tumor and accompanying normal FFPE tissue blocks were retrieved. MLH1 methylation testing revealed widespread mosaicism in normal tissues. MLH1 methylation levels were too low in PBL nuclei and saliva to detect by pyrosequencing (Figure 4A), but were both positive by more sensitive qMSP with PMR values of 1.0% (Figure 4B). Hair follicles had no detectable methylation, even by qMSP (Figure 4B). Other histologically normal (resected) tissues showed low-level MLH1 methylation measurable by pyrosequencing, ranging from 2.4% in small intestinal epithelium to 7.6% in uterine epithelium (Figure 4A). Patient 166 was heterozygous for the c.−93G>A promoter SNP. MSP across this SNP followed by direct sequencing of the amplicon revealed the low-level methylation to be monoallelic, linked to the G allele in all normal tissues tested (Figure 4C). To determine if methylation of the “G” allele in such a small proportion of normal cells (~5–15%) had predisposed to the development of multiple primary tumors, we performed extended testing in each tumor to identify potential “second hits” affecting the unmethylated “A” allele. We first measured significant levels of MLH1 methylation in all three tumors by pyrosequencing (Figure 5A). Next, clonal bisulfite-sequencing (Figure 5B), and promoter sequencing (Figure 5C), across the c.−93G>A SNP site in each tumor was performed to trace allelic methylation patterns and detect potential loss-of-heterozygosity (LOH), respectively, with reference to paired normal tissues. The sebaceous carcinoma showed both LOH of the ‘A’ allele, as well as methylation of both the (constitutionally-methylated) G alleles and the remnant A alleles (two second-hits). The EC showed monoallelic methylation of the ‘G’ alleles plus LOH of the ‘A’ allele. The small intestine adenocarcinoma showed methylation of both alleles with retention-of-heterozygosity, indicating somatic methylation of the A allele was the second-hit in this tumor. Collectively, these findings are consistent with constitutional MLH1 methylation of the G allele in a proportion of cells serving as the “first hit”, followed by a somatic second hit affecting the A allele in the development of all three cancers.
Figure 2. Detection of monoallelic constitutional MLH1 epimutation in Proband 192.
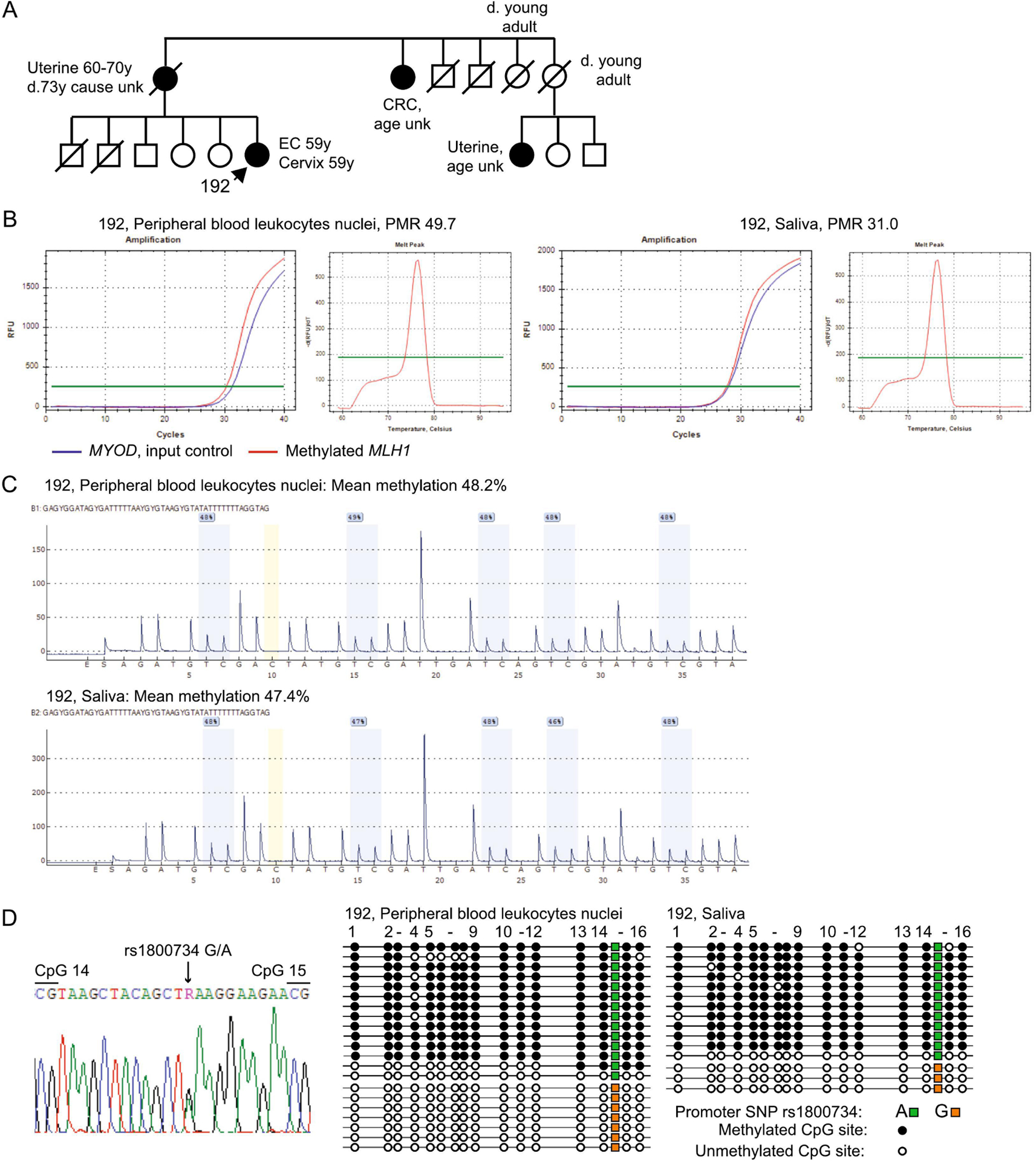
A: Pedigree of Patient 192. EC, endometrial cancer; CRC, colorectal cancer; unk, unknown. For privacy, members of younger generations are not included (none have had a cancer diagnosis). B: Real-time methylation-specific PCR (qMSP) was performed from c.−188 to c.−403within the MLH1 promoter on bisulfite-converted genomic DNA from the nuclei of peripheral blood leukocytes (PBL) and saliva. Amplification curves show methylated MLH1 (red), which amplifies only when methylated DNA is present, run in parallel with MYOD (blue), which serves as a quality control measure for sample input and integrity. The percentage of methylated reference (PMR) value is shown. The high-resolution melt curve of the MLH1 amplicon for each sample indicates specificity for methylated amplicons with melt peak at 76±0.5 °C. C: Pyrosequencing traces are shown for five CpG sites from c.−241 to c.−272 of the MLH1 promoter in bisulfite-converted genomic DNA from peripheral blood leukocyte (PBL) nuclei and saliva. Methylation is detected by the presence of a peak at the cytosine (C) within each CpG site interrogated (gray bars), whereas unmethylated cytosines are detected as thymine (T) peaks within the same CpG sites, due to the conversion of unmethylated cytosines to uracils using bisulfite treatment. The assay measures the relative levels of methylated C against unmethylated cytosines at each CpG site interrogated, and reports these as a percentage of methylation value above. The mean level of methylation across all five CpG sites is calculated and shown above. The yellow bar indicates a non-CpG cytosine used as a quality control measure to ensure complete bisulfite-conversion to T, whereupon this yields a valid test result. Methylation was measured at 48.2% in PBL and 47.4% in saliva. D: Left, Sanger sequencing electropherogram showing Patient 192 was heterozygous for the MLH1 promoter c.−93G>A SNP (rs1800734). Right, pictogram of clonal bisulfite sequencing across a fragment of the MLH1 CpG island promoter region from c.−48 to c. −370 showing the methylation status at 16 individual CpG sites (circles) within individual alleles (horizontal lines). The rs1800734 SNP genotypes are represented by colored squares on each allele. This SNP is flanked by CpG dinucleotides numbered correspondingly in the electropherogram (left) and pictogram (right). Hypermethylation was restricted to alleles bearing the ‘A’ genotype at rs1800734, indicating monoallelic methylation.
Figure 3. Detection of hemiallelic constitutional MLH1 epimutation in EC Proband 213.
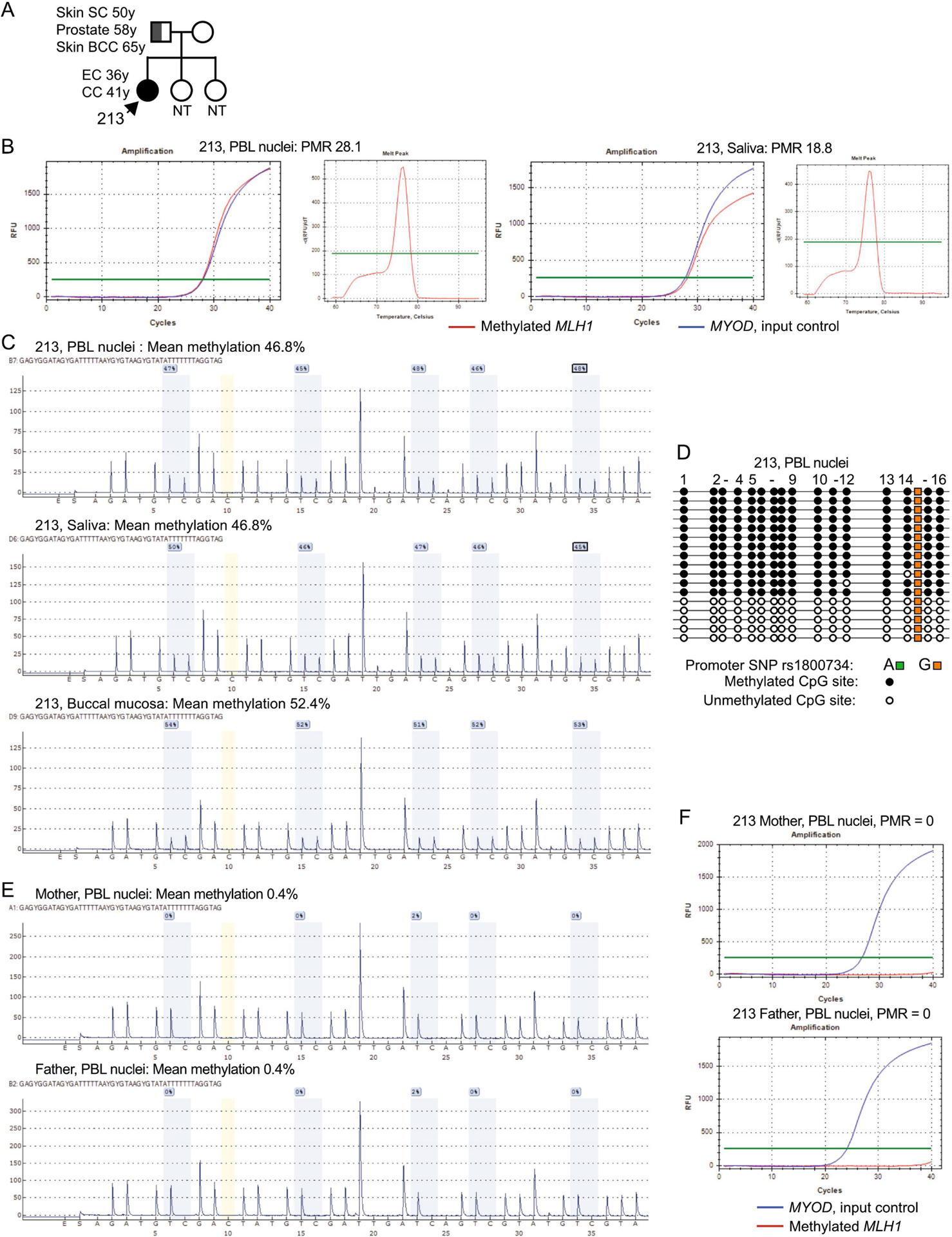
A: Nuclear pedigree of patient 213, showing a non-Lynch syndrome cancer history on the paternal side. B: Left, illustrative amplification curves showing positive amplification of methylated MLH1 by real-time methylation-specific PCR (qMSP) within the MLH1 promoter on bisulfite-converted genomic DNA. Right, melt curve of the amplicons, confirms product specificity at a melt temperature of 76±0.5 °C. C: CpG pyrosequencing traces with legend according to Figure 2. MLH1 methylation measured between 46.8% to 52.4%. D: Clonal bisulfite sequencing of a larger fragment of the MLH1 promoter confirms the presence of methylation in a hemiallelic pattern, consistent with the CpG pyrosequencing result. The patient was homozygous “G” at the rs1800734 SNP, so the parental allele-of-origin of the constitutional MLH1 methylation could not be determined.
Figure 4. Detection of low-level constitutional MLH1 methylation mosaicism in Patient 166.
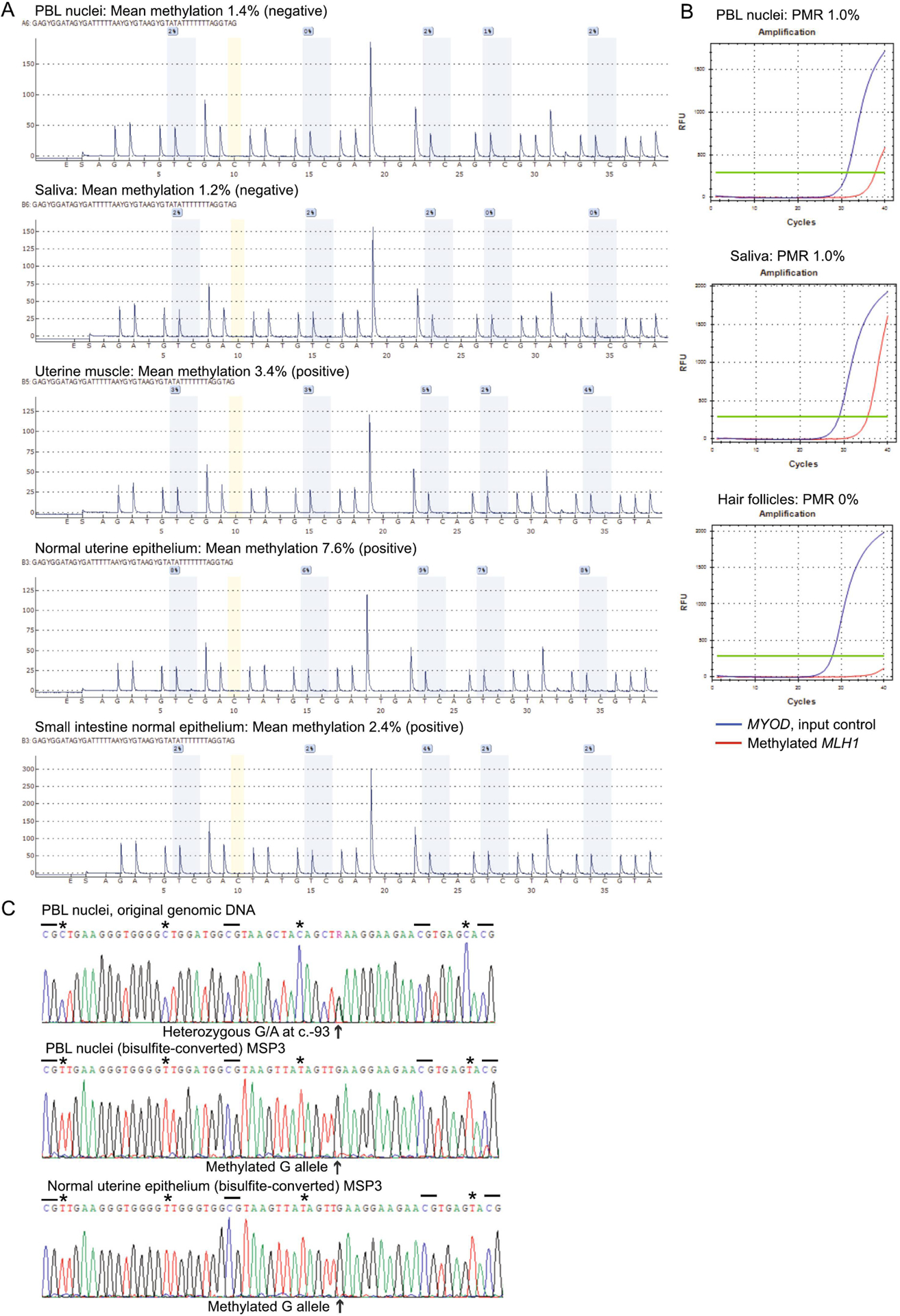
A: CpG pyrosequencing yields negative test results in genomic DNA from peripheral blood leukocytes (PBL) nuclei and saliva, but low levels of methylation are detectable in DNA extracted from other sources of histologically normal tissue samples (macrodissected formalin-fixed paraffin-embedded) from surgically resected organs, as labeled. B: Real-time methylation-specific PCR (qMSP) shows positive amplification of methylated MLH1 templates in PBL and saliva samples with methylation levels too low to be detectable by CpG pyrosequencing, but absence of methylation in hair follicles. C: Top, partial sequence within the MLH1 promoter shows heterozygosity for the c.−93G>A SNP (arrow). Dashed lines show the locations of individual CpG sites flanking the SNP. Beneath, Sanger sequencing of methylation-specific PCR (MSP) products across the same region encompassing the c.−93G>A SNP site after bisulfite-conversion of genomic DNA shows only methylated templates were amplified (as C is retained at CpG sites, whereas isolated Cs at non-CpG sites are converted to T [asterix]) and these are exclusively linked to the G allele at c.−93 SNP position. This indicates the low-level methylation (amplifiable by MSP) is restricted to the G allele.
Figure 5. Low-level mosaic constitutional MLH1 methylation predisposes to multiple MLH1-methylated primary tumors in Patient 166.
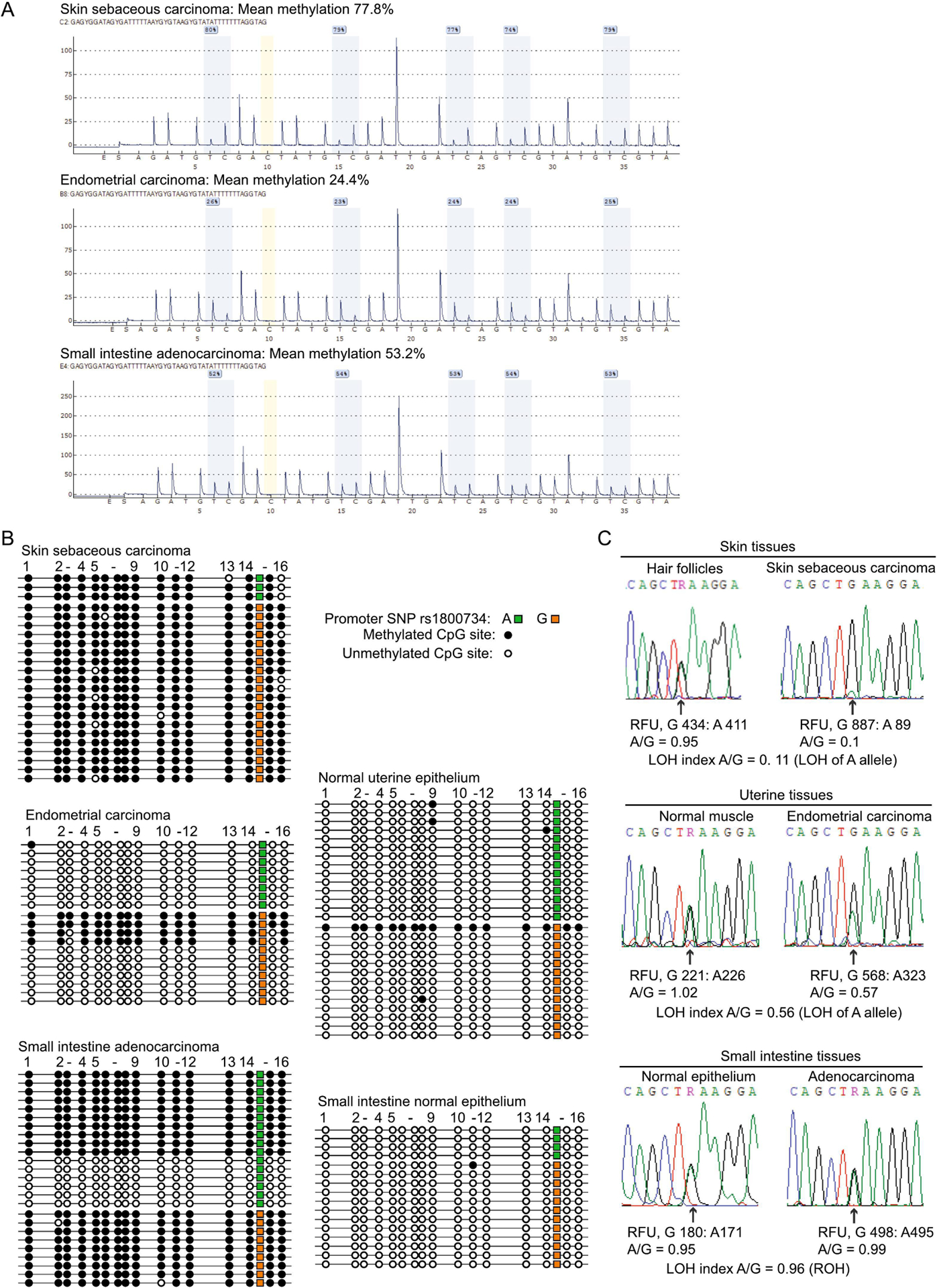
A: CpG pyrosequencing confirms the presence of significant levels of MLH1 methylation in all three primary tumors, including the small intestine adenocarcinoma, which had not previously been assessed for this. B: Clonal bisulfite sequencing within the MLH1 promoter encompassing the c.−93G>A SNP (rs1800734) using primers unbiased with respect to methylation status shows the patterns of methylation in the tumors (left). The skin sebaceous carcinoma and small intestine tumors had methylation on both alleles, although few A alleles at c. −93 remained in the skin tumor. The EC was monoallelically methylated on the G allele. The same assay performed on accompanying normal tissue samples (right) only detected a single methylated molecule in endometrial epithelium, which was the G allele at c. −93G>A. C: The c. −93G>A SNP was used to trace allelic representation in each primary cancer (right) with reference to a paired normal tissue sample (right). Partial Sanger sequences are shown across the c. −93G>A SNP, with measurements of each allele in relative fluorescence units (RFU) taken from the peak for each genotype shown below. Loss-of-heterozygosity (LOH) was assessed by calculating (A/GTumor)/(A/GNormal). If A/G was ≤0.6 in the tumor, this was designated as LOH of the A allele. Retention of heterozygosity (ROH) was designated if A/G >0.6 in the tumor.
Population-based ascertainment
To determine the frequency of constitutional MLH1 methylation among incident EC cases whose tumor was identified as MMRd and MLH1-methylated upon universal screening, we performed a nested retrospective study of EC cases selected by this tumor feature in population-based cohorts (Figure 1A).
Columbus cohort:
Of 543 unselected EC cases, 85 (15.1%) were eligible for inclusion based on IHC loss of MLH1 expression and/or MSI and MLH1 methylation in their tumor, irrespective of age, prior cancer, family history, or prior genetic testing (Figure 1A). PBL DNA was available for 74 cases, median age 64.0 years (range 38–88 years). Of these, 64 tumors (86.5%) were MSI-high, seven MSI-low, and three MSS. Methylation testing in PBL DNA was successful for 68 cases. Test failures did not significantly alter the distribution in age or frequency of MSI within the selected series. All 68 EC cases were negative for constitutional MLH1 methylation, among whom 21 were <60 years and two were <50 years (Figure 1C).
OCCPI cohort:
Given the negative findings in the Columbus cohort and to boost the sample size among younger cases, we limited screening of the OCCPI cohort to EC cases <60 years. Twenty-four, age range 36–59 years, with MLH1 loss and MLH1 methylation in their tumor were included, of which 17 were MSI-high, five MSI-low, and two were MSS. All 24 cases tested negative for MLH1 methylation in whole blood DNA by pyrosequencing, however, low-level MLH1 methylation was detected by qMSP in one patient (4%). Patient EC-32 had a qMSP PMR value of 0.17%, below the pyrosequencing detection threshold (Figure 6A, B). EC-32 was diagnosed with EC(FIGO stage IIIC1), at 36 years and had no family history. To confirm the qMSP methylation-positive signal, we performed conventional (non-fluorescent) MSP in two distinct regions of the MLH1 CpG island (Figure 1B), followed by clonal bisulfite-sequencing of the amplicons. Both confirmed the presence of methylation in blood DNA (Figure 6C). No genetic variants were identified in the MLH1 CpG island.
Figure 6. Detection of low-level constitutional MLH1 methylation in OCCPI patient EC-32.
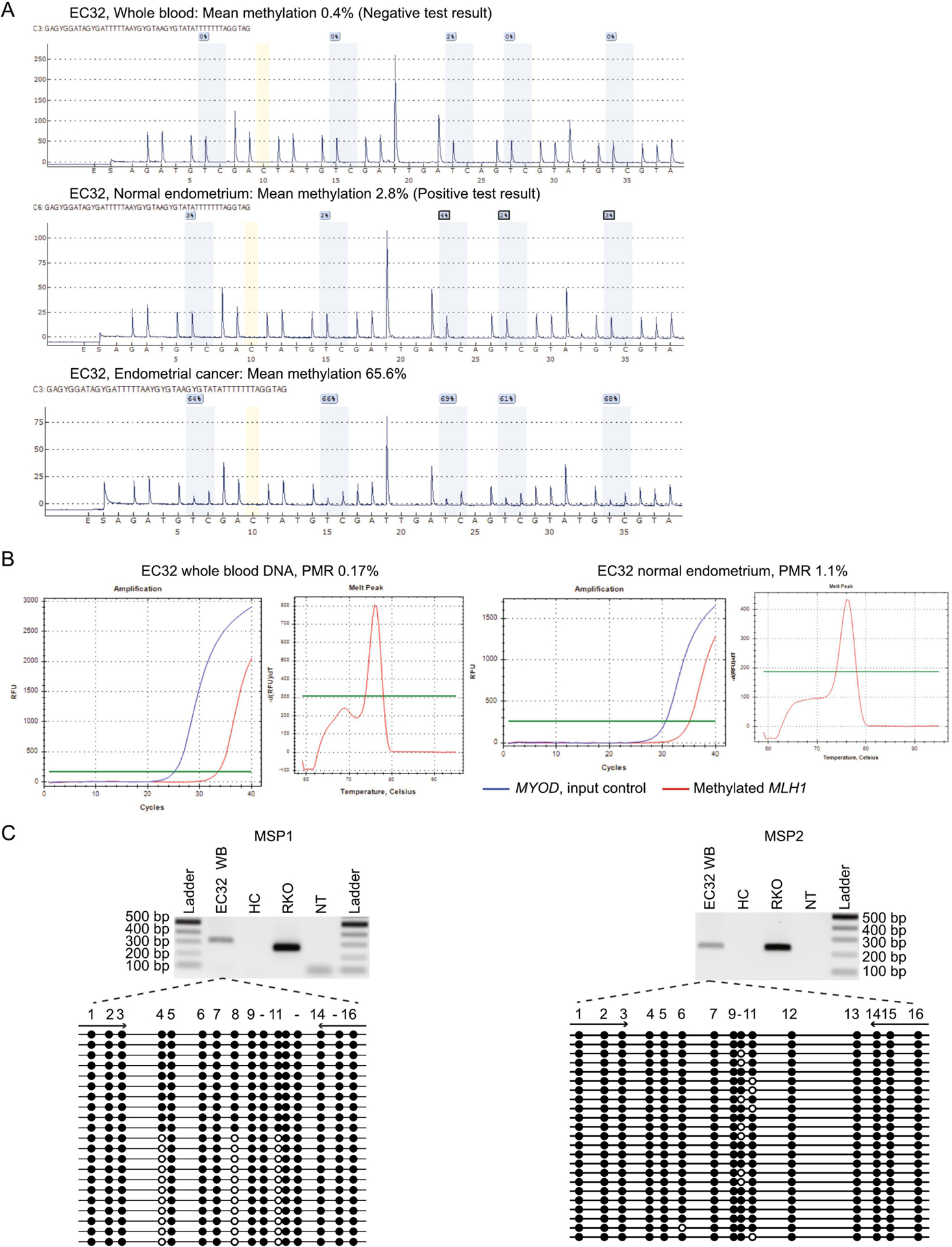
A: CpG pyrosequencing traces are shown DNA from whole blood, uninvolved endometrial tissue, and tumor, as labeled. The mean methylation value is shown above. Methylation was undetectable in whole blood DNA, given this was below the limit of detection (2.3%), but was detectable at low levels (2.8%) in uninvolved normal endometrial tissue and at high levels (65.6%) in the tumor. B. Real-time methylation-specific PCR (qMSP) within the MLH1 promoter on bisulfite-converted genomic DNA from whole blood (left) and confirmed in uninvolved normal endometrial tissue (right). Amplification curves show methylated MLH1 (red), which amplifies only when methylated genomic DNA template is present, run in parallel with MYOD (blue), which serves as a quality control measure for sample input and integrity. An exemplary high resolution melt curve of the MLH1 amplicon is shown for the whole blood sample (middle), indicating specificity for methylated amplicons with melt peak at 76±0.5 °C. C: Electrophoresis gels of traditional MLH1 methylation-specific PCR (MSP) amplification products according to Figure 1. Left, MSP1 used the same primers as the qMSP. Right, MSP2 was conducted in a distinct region of the MLH1 CpG island. Both MSP reactions included DNA from whole blood (WB) of EC-32, a healthy control (HC, negative control), RKO colorectal cancer cell line (positive control), and a sample with no template (NT) added. Beneath, pictograms of clonal bisulfite sequencing of each MSP amplicon to determine the methylation status at 16 individual CpG sites within the respective regions. Black circles show methylated CpG sites and white circles show unmethylated CpG sites on individual molecules (horizontal line).
Since methylation testing in the OCCPI cohort was performed in whole blood DNA, we were cognizant that high-sensitivity qMSP could have detected contaminating circulating tumor DNA (ctDNA) within plasma. However, the blood sample tested was drawn three months post-operatively, therefore, the methylation-positive signal was unlikely generated by ctDNA shed by the primary tumor, although residual disease was possible. Therefore, we tested uninvolved endometrial tissue as an additional source of normal tissue, which was positive for MLH1 methylation both by qMSP with a PMR value of 1.1% and by pyrosequencing at 2.8% (Figure 6A, B). Thus, MLH1 methylation in the tissue-of-origin was slightly higher than in blood. These findings are consistent with Patient EC-32 having low-level mosaicism for constitutional MLH1 methylation, which predisposed to her early-onset EC displaying high-level MLH1 methylation (Figure 6B).
Discussion
Key goals of universal screening of all incident EC (and CRC) for MMRd and LS are to identify patients genetically at high-risk who would benefit from enrollment in life-long cancer surveillance programs and to extend cascade genetic testing to blood relatives. However, current algorithms that utilize tumor MLH1 methylation testing to rule out hereditary risk may result in rare cases with constitutional MLH1 epimutation being misdiagnosed as common sporadic cases. Poor recognition of this mechanism and inconsistent guidelines may result in failure to correctly diagnose it, missing the opportunity to enroll these patients in risk-appropriate surveillance. Consensus, evidence-based guidelines for appropriate triaging of EC patients warranting testing for constitutional MLH1 methylation are needed. Given tumor MLH1 methylation testing is an integral component of the molecular pathology-based algorithm for LS recognition, we aimed to determine the role and frequency of constitutional MLH1 epimutation among patients presenting with MLH1-methylated EC.
Among four index patients referred to our study by a cancer clinic, three (Patients 192, 213, and 166) had constitutional MLH1 methylation. Two (Patients 192 and 213) with mono/hemiallelic epimutation had presented with MLH1-methylated EC as their first or dual-first cancer. Notably, the impetus for referring both Patients 192 and 213 for constitutional MLH1 methylation work-up was not based solely on their initial cancer diagnosis or tumor molecular pathology features. In the case of Patient 192, diagnosed with EC at 57 years, an unspecified sample of resected normal tissue from her TH/BSO had been tested for MLH1 methylation alongside her tumor and both were positive. Serendipitous detection of MLH1 methylation in a normal tissue had prompted the recommendation for follow-up blood-based testing. Parallel testing of uninvolved normal tissue alongside the tumor during routine molecular pathology in early-onset cases could provide a feasible route for the detection of constitutional MLH1 epimutation. In the case of Patient 213, who initially presented with MLH1-methylated EC at 36 years, unfortunately testing for constitutional MLH1 epimutation was not considered until she presented five years later with locally-advanced colon cancer that also displayed MLH1 methylation. A correct molecular diagnosis after her first presentation with EC would have significantly altered her clinical management thereafter.
Patients 166 and OCCPI EC-32 both had low-level mosaic methylation and yet presented either with multiple MLH1-methylated primaries (Patient 166), or EC as the first-presenting cancer at the young age of 36 years (EC-32). In both patients, low-level methylation was confirmed in two or more normal tissue types. In Patient 166, we unraveled clear evidence of the role for low-level, monoallelic methylation in a fraction of cells in predisposing to the development of all three tumors via somatic methylation and/or LOH of the unaffected allele. These two patients illustrate that low-level mosaic constitutional MLH1 methylation can nevertheless confer high risks for LS-type cancers.
To determine if screening for constitutional MLH1 epimutation might be warranted among EC cases identified via universal screening to have MLH1-methylated tumors, we determined the frequency of constitutional MLH1 methylation in retrospective nested studies of population-based cohorts of incidental EC. In the Columbus cohort, EC cases were selected solely based on the tumor molecular features of MMRd and MLH1 methylation, therefore this study was unbiased with respect to age at cancer diagnosis, prior cancer, genetic testing, or family history. Yet, given the negative findings in this cohort, and the small sample size among younger cases, we limited our subsequent study of the OCCPI cohort to EC patients diagnosed <60 years with the same tumor features. By combining the two cohorts, the rate of constitutional MLH1 methylation among incident MLH1-methylated EC cases overall (all ages) was negligible (0–1%) and the rate among cases aged <60 years was 2% (Columbus 0/21, OCCPI 1/24). However, the positive detection rate for constitutional MLH1 methylation in MLH1-methylated EC cases <50 years was 1/6 (17%) in the combined cohorts. If consideration for screening for constitutional MLH1 methylation were limited to cases <50 years of age, only a small proportion of patients with MLH1-methylated tumors overall would warrant testing, rendering this feasible and potentially high-yield. However, limiting screening by age <50 years would miss cases such as Patients 192 and 166, who presented with EC in their fifties.
A key limitation of this study was the small sample size among younger patients, despite their derivation from large population-based series. However, these small case numbers also serve to illustrate that screening among early-onset MLH1-methylated EC cases would be minimal. The rarity of constitutional MLH1 epimutation among incident EC cases with MLH1-methylated tumors overall is unsurprising, given MLH1 methylation is typically somatic-in-origin and accounts for the significant proportion of MMRd EC. However, the finding of constitutional MLH1 epimutation in one patient with incident MLH1-methylated EC out of six (~17%) <50 years, or out of 45 (~2%) <60 years in the combined Ohio cohorts, is non-trivial. Furthermore, EC was the “sentinel” cancer in two clinic-based cases and EC-32. Based on our findings, we proffer that routine screening for constitutional MLH1 methylation is warranted for incident EC cases <50 years whose tumor is MLH1-methylated, as well as patients presenting with synchronous or metachronous LS-type cancers with MLH1-methylation at any age, irrespective of family history or whether prior genetic testing has been completed. Further consideration should also be given to parallel testing for MLH1 methylation in a normal tissue sample from surgical resection specimens in early-onset cases. Gynecologic oncologists, pathologists, and genetic counselors play critical and coordinated roles in identifying patients at high genetic risk for cancer and should also consider referral for constitutional MLH1 epimutation testing in patients displaying these molecular pathology features.
Supplementary Material
Supplementary Figure 1: Illustrative examples of CpG pyrogram traces from positive and negative control samples.
Supplementary Figure 2: Analytical sensitivity and linearity of the quantitative CpG pyrosequencing assay used to detect and measure MLH1 promoter methylation.
Supplementary Figure 3. Illustrative amplification and melt curves for reference, positive, and negative control samples.
Supplementary Figure 4. Analytical sensitivity and linearity of the semi-quantitative real-time methylation specific PCR (qMSP) assay used to screen for constitutional MLH1 methylation, in particular, low-level mosaic methylation.
Supplementary Figure 5. Patient 177 tests negative for constitutional MLH1 methylation.
Research Highlights.
High-risk constitutional MLH1 methylation underlies a significant proportion of early-onset EC with tumor MLH1 methylation
EC with tumor MLH1 methylation is sometimes the ‘sentinel’ cancer in women with constitutional MLH1 methylation
Low-level mosaic constitutional MLH1 methylation confers high-risk for MLH1-methylated cancers including EC
Constitutional MLH1 methylation testing is warranted in cases with early-onset, or prior history of, MLH1-methylated cancer
Acknowledgements
This study was funded by National Institutes of Health/National Cancer Institute R01 grants CA218342 (Megan Hitchins), and CA67941 and CA16058 (Heather Hampel); a Cedars-Sinai Medical Center Precision Health Initiative Award (Megan Hitchins); Spanish Ministry of Science grant SAF2015-68016-R (Gabriel Capellá), PID2019-111254RB-I00 (Gabriel Capellá and Marta Pineda); Travel Fellowships awarded to Estela Damaso from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO, EEBB-I-16-11581) and the RTICC network (RD12/0036/0031).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors have no conflicts of interest to declare
Supplemental Information
Supplementary Methods include detailed descriptions of each assay used, illustrative examples of positive and negative control samples, and the limit of detection of the first-pass screening assays (CpG pyrosequencing and qMSP).
Supplemental Results shows negative findings for Patient 177.
References
- 1.Parsons R, Li GM, Longley MJ, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75(6):1227–36. [DOI] [PubMed] [Google Scholar]
- 2.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–57. [PubMed] [Google Scholar]
- 3.Yamamoto H, Imai K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Seminars in Oncology. 2019;46(3):261–70. [DOI] [PubMed] [Google Scholar]
- 4.Goodfellow PJ, Billingsley CC, Lankes HA, et al. Combined Microsatellite Instability, MLH1 Methylation Analysis, and Immunohistochemistry for Lynch Syndrome Screening in Endometrial Cancers From GOG210: An NRG Oncology and Gynecologic Oncology Group Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(36):4301–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esteller M, Catasus L, Matias-Guiu X, et al. hMLH1 promoter hypermethylation is an early event in human endometrial tumorigenesis. Am J Pathol. 1999;155(5):1767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng G, Peng E, Gum J, et al. Methylation of hMLH1 promoter correlates with the gene silencing with a region-specific manner in colorectal cancer. Br J Cancer. 2002;86(4):574–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peltomaki P Update on Lynch syndrome genomics. Fam Cancer. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynch HT, Snyder CL, Shaw TG, et al. Milestones of Lynch syndrome: 1895–2015. Nat Rev Cancer. 2015;15(3):181–94. [DOI] [PubMed] [Google Scholar]
- 9.Hitchins M, Williams R, Cheong K, et al. MLH1 germline epimutations as a factor in hereditary nonpolyposis colorectal cancer. Gastroenterology. 2005;129(5):1392–9. [DOI] [PubMed] [Google Scholar]
- 10.Gazzoli I, Loda M, Garber J, et al. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002;62(14):3925–8. [PubMed] [Google Scholar]
- 11.Hitchins MP. The role of epigenetics in Lynch syndrome. Fam Cancer. 2013;12(2):189–205. [DOI] [PubMed] [Google Scholar]
- 12.Hitchins MP, Rapkins RW, Kwok CT, et al. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5’UTR. Cancer Cell. 2011;20(2):200–13. [DOI] [PubMed] [Google Scholar]
- 13.Kwok CT, Vogelaar IP, van Zelst-Stams WA, et al. The MLH1 c.−27C>A and c.85G>T variants are linked to dominantly inherited MLH1 epimutation and are borne on a European ancestral haplotype. Eur J Hum Genet. 2014;22(5):617–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leclerc J, Flament C, Lovecchio T, et al. Diversity of genetic events associated with MLH1 promoter methylation in Lynch syndrome families with heritable constitutional epimutation. Genet Med. 2018;20(12):1589–99. [DOI] [PubMed] [Google Scholar]
- 15.Morak M, Schackert HK, Rahner N, et al. Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur J Hum Genet. 2008;16(7):804–11. [DOI] [PubMed] [Google Scholar]
- 16.Morak M, Koehler U, Schackert HK, et al. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J Med Genet. 2011;48(8):513–9. [DOI] [PubMed] [Google Scholar]
- 17.Gylling A, Ridanpaa M, Vierimaa O, et al. Large genomic rearrangements and germline epimutations in Lynch syndrome. Int J Cancer. 2009;124(10):2333–40. [DOI] [PubMed] [Google Scholar]
- 18.Ward RL, Dobbins T, Lindor NM, et al. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med. 2013;15(1):25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cini G, Carnevali I, Quaia M, et al. Concomitant mutation and epimutation of the MLH1 gene in a Lynch syndrome family. Carcinogenesis. 2015;36(4):452–8. [DOI] [PubMed] [Google Scholar]
- 20.Morak M, Ibisler A, Keller G, et al. Comprehensive analysis of the MLH1 promoter region in 480 patients with colorectal cancer and 1150 controls reveals new variants including one with a heritable constitutional MLH1 epimutation. J Med Genet. 2018;55(4):240–8. [DOI] [PubMed] [Google Scholar]
- 21.Crepin M, Dieu MC, Lejeune S, et al. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum Mutat. 2012;33(1):180–8. [DOI] [PubMed] [Google Scholar]
- 22.Hitchins MP, Wong JJ, Suthers G, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. 2007;356(7):697–705. [DOI] [PubMed] [Google Scholar]
- 23.Sloane MA, Nunez AC, Packham D, et al. Mosaic Epigenetic Inheritance as a Cause of Early-Onset Colorectal Cancer. JAMA oncology. 2015;1(7):953–7. [DOI] [PubMed] [Google Scholar]
- 24.Hitchins M, Owens S, Kwok CT, et al. Identification of new cases of early-onset colorectal cancer with an MLH1 epimutation in an ethnically diverse South African cohort(dagger). Clin Genet. 2011. [DOI] [PubMed] [Google Scholar]
- 25.Goel A, Nguyen TP, Leung HC, et al. De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer. 2011;128(4):869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buchanan DD, Tan YY, Walsh MD, et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32(2):90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batte BA, Bruegl AS, Daniels MS, et al. Consequences of universal MSI/IHC in screening ENDOMETRIAL cancer patients for Lynch syndrome. Gynecologic oncology. 2014;134(2):319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stelloo E, Jansen AML, Osse EM, et al. Practical guidance for mismatch repair-deficiency testing in endometrial cancer. Ann Oncol. 2017;28(1):96–102. [DOI] [PubMed] [Google Scholar]
- 29.NCCN. NCCN Clinical Practive Guidelines in Oncology: Uterine Neoplasms. 2021.
- 30.NCCN. Guidelines. Genetic/Familial High-Risk Assessment: Colorectal. 2022. [Google Scholar]
- 31.Niessen RC, Hofstra RM, Westers H, et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer. 2009;48(8):737–44. [DOI] [PubMed] [Google Scholar]
- 32.Valle L, Carbonell P, Fernandez V, et al. MLH1 germline epimutations in selected patients with early-onset non-polyposis colorectal cancer. Clin Genet. 2007;71(3):232–7. [DOI] [PubMed] [Google Scholar]
- 33.Pineda M, Mur P, Iniesta MD, et al. MLH1 methylation screening is effective in identifying epimutation carriers. European journal of human genetics : EJHG. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crucianelli F, Tricarico R, Turchetti D, et al. MLH1 constitutional and somatic methylation in patients with MLH1 negative tumors fulfilling the revised Bethesda criteria. Epigenetics. 2014;9(10):1431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castillejo A, Hernandez-Illan E, Rodriguez-Soler M, et al. Prevalence of MLH1 constitutional epimutations as a cause of Lynch syndrome in unselected versus selected consecutive series of patients with colorectal cancer. J Med Genet. 2015;52(7):498–502. [DOI] [PubMed] [Google Scholar]
- 36.van Roon EH, van Puijenbroek M, Middeldorp A, et al. Early onset MSI-H colon cancer with MLH1 promoter methylation, is there a genetic predisposition? BMC cancer. 2010;10:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pineda M, Mur P, Iniesta MD, et al. MLH1 methylation screening is effective in identifying epimutation carriers. Eur J Hum Genet. 2012;20(12):1256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearlman R, Frankel WL, Swanson BJ, et al. Prospective Statewide Study of Universal Screening for Hereditary Colorectal Cancer: The Ohio Colorectal Cancer Prevention Initiative. JCO Precis Oncol. 2021;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takeda T, Banno K, Yanokura M, et al. Methylation Analysis of DNA Mismatch Repair Genes Using DNA Derived from the Peripheral Blood of Patients with Endometrial Cancer: Epimutation in Endometrial Carcinogenesis. Genes (Basel). 2016;7(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66(15):7810–7. [DOI] [PubMed] [Google Scholar]
- 41.Hampel H, Panescu J, Lockman J, et al. Comment on: Screening for Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) among Endometrial Cancer Patients. Cancer Res. 2007;67(19):9603. [DOI] [PubMed] [Google Scholar]
- 42.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95(12):6870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng G, Chen A, Hong J, et al. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999;59(9):2029–33. [PubMed] [Google Scholar]
- 44.Newton K, Green K, Lalloo F, et al. Colonoscopy screening compliance and outcomes in patients with Lynch syndrome. Colorectal Dis. 2015;17(1):38–46. [DOI] [PubMed] [Google Scholar]
- 45.Ogino S, Kawasaki T, Brahmandam M, et al. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. The Journal of molecular diagnostics : JMD. 2006;8(2):209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGivern A, Wynter CV, Whitehall VL, et al. Promoter hypermethylation frequency and BRAF mutations distinguish hereditary non-polyposis colon cancer from sporadic MSI-H colon cancer. Fam Cancer. 2004;3(2):101–7. [DOI] [PubMed] [Google Scholar]
- 47.Deng G, Bell I, Crawley S, et al. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10(1 Pt 1):191–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Illustrative examples of CpG pyrogram traces from positive and negative control samples.
Supplementary Figure 2: Analytical sensitivity and linearity of the quantitative CpG pyrosequencing assay used to detect and measure MLH1 promoter methylation.
Supplementary Figure 3. Illustrative amplification and melt curves for reference, positive, and negative control samples.
Supplementary Figure 4. Analytical sensitivity and linearity of the semi-quantitative real-time methylation specific PCR (qMSP) assay used to screen for constitutional MLH1 methylation, in particular, low-level mosaic methylation.
Supplementary Figure 5. Patient 177 tests negative for constitutional MLH1 methylation.