

Summary
Restricting caloric intake effectively reduces body weight, but most dieters fail long-term adherence to caloric deficit and eventually regain lost weight. Hypothalamic circuits that control hunger drive critically determine body weight, yet how weight loss sculpts these circuits to motivate food consumption until lost weight is regained remains unclear. Here we probe the contribution of synaptic plasticity in discrete excitatory afferents onto hunger-promoting AgRP neurons. We reveal a crucial role for activity-dependent, remarkably long-lasting, amplification of synaptic activity originating from paraventricular hypothalamus thyrotropin-releasing (PVHTRH) neurons in long-term body weight control. Silencing PVHTRH neurons inhibits potentiation of excitatory input onto AgRP neurons, and diminishes concomitant regain of lost weight. Brief stimulation of the pathway is sufficient to enduringly potentiate this glutamatergic hunger synapse, and triggers an NMDAR-dependent gaining of body weight that enduringly persists. Identification of this activity-dependent synaptic amplifier provides a previously unrecognized target to combat regain of lost weight.
Keywords: Hypothalamus, hunger drive, weight regain, synaptic plasticity, TRH-expressing neurons, AgRP neurons, electrophysiology, optogenetics, chemogenetics, intersectional genetics
Graphical Abstract
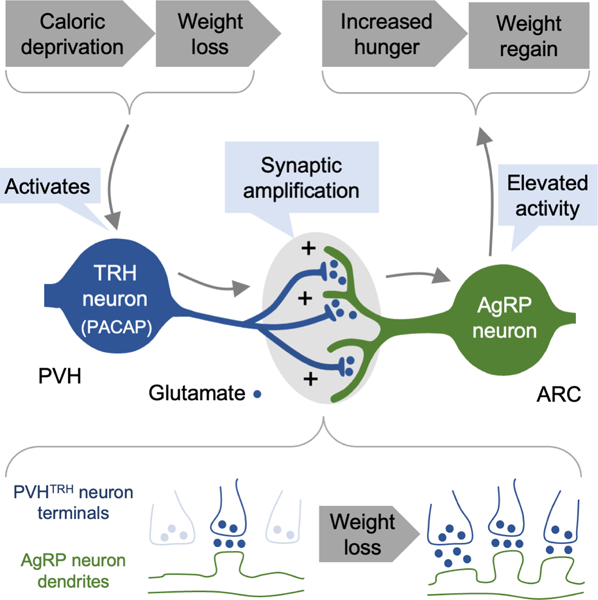
eTOC blurb:
Grzelka et al. reveal that synaptic transmission between excitatory PVHTRH neurons and AgRP ‘hunger’ neurons is distinctively strengthened upon weight loss due to caloric deprivation. Elevated activity of the PVHTRH neurons induces this synaptic amplification, and triggers a long-lasting increase in food intake and body weight gain that enduringly persists.
Introduction
Dieting is the most widely used approach to lose body weight. The vast majority of individuals that initially succeed in reducing weight, fail long-term adherence to a caloric deficit and eventually regain lost weight,1 which poses a major public health issue in humans. The arcuate nucleus of the hypothalamus (ARC) is thought to present an essential part of the neurocircuitry that drives weight regain.2 Indeed, ARC neurons integrate central and circulating signals of energy availability.3 Moreover, distinct ARC neurons are both necessary and sufficient for driving feeding behavior,4–9 and their genetic manipulations result in profound body weight alterations,4,10–14 underscoring a key role for the ARC in long-term control of body weight. However, we have only a rudimentary understanding of how weight loss-induced alterations in ARC neurocircuits relate to enduring coordination of counterregulatory processes, such as increased hunger drive, that may explain why most diets fail in the long-term.
A large body of literature demonstrates that the synaptic inputs to key ARC neurons that control body weight undergo plastic changes in response to caloric deficit.8,15–24 Energy state-dependent adaptations include changes in neurotransmitter release from presynaptic terminals,15,19 formation and elimination of synapses,17,20 as well as recruitment and removal of ion channels from postsynaptic membrane.18,22 Given the ARCs location, adjacent to the median eminence which lacks a blood-brain barrier, ARC neurons and their afferents are subject to modulation by hormonal signals,25 whose circulating level changes are associated with long-term body weight control.26 Indeed, pharmacological studies have implicated leptin, ghrelin, and also estrogen and glucocorticoids, in remodeling ARC synaptic architecture and modifying the strength of synaptic inputs.15–17,19,27,28 Together, these structural and functional synaptic changes in ARC neuron inputs suggest that weight loss fundamentally alters how their activity is coordinated by afferent brain regions. To place the contribution of these circuit refinements within the context of failed dieting, it is essential to understand the circuit-specific plasticity mechanisms in functionally distinct ARC neuron afferents.
A major challenge to understanding the contribution of plastic changes in discrete pathways has been the exceptionally complex neuroanatomy of the hypothalamus. The ARC receives synaptic inputs from numerous brain regions, including the paraventricular hypothalamus (PVH), the dorsal medial hypothalamus (DMH), the bed nucleus of the striae terminalis, and the nucleus of the solitary tract. Each of these regions has been implicated in signaling different facets of energy availability to the ARC.29–32 In addition, ARC neurons and afferent brain regions exhibit considerable cellular heterogeneity, containing multiple molecularly and functionally distinct neuronal populations that play pivotal roles in body weight control.33,34 Moreover, monosynaptic rabies tracing and Channelrhodopsin-2 (ChR2)-assisted circuit mapping (CRACM) studies have established exquisite patterns of synaptic connections to functionally distinct ARC neurons.3,7,29,30 With such a complex wiring diagram in mind, it is of great relevance to identify which synaptic connections are altered by weight loss, and how these discrete circuit adaptations causally relate to long-term body weight regain.
AgRP neurons are the ARC neuron population most strongly associated with driving hunger.3 Caloric deficit, such as during fasting, activates AgRP neurons,35 which, once engaged, cause voracious feeding and weight gain.5,6 Conversely, AgRP neuron inhibition reduces food intake in energy-deprived states,5,36,37 and their ablation leads to starvation and weight loss.10,38 Importantly, energy state-dependent reorganization of the glutamatergic synaptic inputs to AgRP neurons strongly relates to their activity regulation. Food deprivation, high ghrelin, and low leptin increase excitatory synapses, dendritic spines, and the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) in AgRP neurons.17,19–21 This enhanced excitatory drive is thought to promote the activation of AgRP neurons upon caloric deficit, which, in turn, increase hunger drive. However, whether synaptic adaptations in discrete glutamatergic afferents represent a cellular substrate for regaining lost weight remains unknown.
One possibility is that not all glutamatergic inputs onto AgRP neurons are upregulated uniformly, as is often tacitly assumed. Fasting-induced increases in sEPSC frequency and spine number are abolished by genetic deletion of NMDA receptors (NMDARs) from AgRP neurons,20 raising the possibility that excitatory drive may be preferentially enhanced by glutamate release from activated afferent terminals. We therefore hypothesized that weight loss would selectively strengthen synapses whose activity is increased at the time of caloric deficit. To test this hypothesis, we employed a combination of circuit-specific electrophysiological, optogenetic, and chemogenetic approaches to study the functional contribution of synaptic plasticity in two distinct glutamatergic afferents onto AgRP neurons emanating from TRH-expressing PVH (PVHTRH) neurons and Vglut2-expressing DMH (DMHVglut2) neurons (Vglut2: vesicular glutamate transporter 2).29 We found that fasting evokes input-specific forms of potentiation in these excitatory afferents. Moreover, using in vivo manipulations we demonstrate that PVHTRH neuron activity coordinates strength of transmission across PVHTRH➔AgRP synapses, and that activity of this pathway is necessary and sufficient for driving body weight gain. Our results provide a circuit-level understanding of synaptic plasticity in AgRP neurons in response to weight loss and, more specifically, how plastic changes of a defined circuit serve as a control point for long-term maintenance of body weight.
Results
Weight loss evokes input-specific forms of potentiation in discrete excitatory afferents onto AgRP neurons
To characterize plastic changes in discrete glutamatergic afferents onto AgRP neurons in response to weight loss, we optogenetically examined synaptic transmission in fasted or ad libitum fed mice. ChR2 was expressed in PVHTRH or DMHVglut2 neurons (Figure 1A), two distinct neuronal populations which were previously found to provide monosynaptic glutamatergic input to AgRP neurons.29 We injected adeno-associated viruses (AAV) expressing Cre-dependent ChR2 (AAV-FLEX-ChR2-mCherry) into the PVH of Trh-ires-Cre; Npy-hrGFP or into the DMH of Slc17a6-ires-Cre; Npy-hrGFP mice (Figure 1A, Figures S1A, B; Slc17a6 is the official gene symbol for Vglut2). Whole-cell recordings were made in acute brain slices from AgRP neurons, which were identified by their co-expression of neuropeptide Y (NPY, hrGFP-positive; Figure S1B). We note that in the ARC, NPY is expressed exclusively in AgRP neurons.39 AMPAR-mediated, blue light-evoked EPSCs (le-EPSCs) were detected in 43 of 57 PVHTRH➔AgRP neurons, and in 32 of 39 DMHVglut2➔AgRP neurons (Figures S1A, B), confirming the glutamatergic nature of PVHTRH and DMHVglut2 inputs onto AgRP neurons.29
Figure 1: Weight loss evokes input-specific forms of potentiation in discrete excitatory afferents onto AgRP neurons.

A) Schematic of the approach used to characterize plastic changes in distinct glutamatergic afferents onto AgRP neurons. ChR2-mCherry was virally expressed in PVHTRH or DMHVglut2 neurons.
B) Representative traces of light-evoked postsynaptic currents in AgRP neurons under control conditions (black) and after replacing external calcium by strontium (grey), which desynchronizes the timing of vesicle fusion. Blue tics indicate light pulses (473 nm, 5 ms), and arrows indicate single quantal excitatory postsynaptic currents (qEPSCs).
C, D) Representative traces of postsynaptic currents recorded from fed or fasted mice following light stimulation of PVHTRH (C) or DMHVglut2 (D) inputs in presence of strontium (left). Quantification of light-evoked qEPSCs (le-qEPSCs) was performed on a 300 ms time window 25 ms after the light pulse.
The summary of le-qEPSCs obtained from AgRP neurons show that fasting increases the quantal frequency of PVHTRH input (C; N = 4/5 mice, fed/fasted), whereas DMHVglut2 input increases in quantal amplitude (D; N = 5/5 mice).
E, F) Representative traces and summary of paired-pulse ratio (PPR) and coefficient of variation (CV) in PVHTRH (E; N = 7/7 mice) and DMHVglut2 (F; N = 4/3 mice) inputs onto AgRP neurons in fed and fasted mice.
G, H) Representative traces of light-evoked AMPAR- and NMDAR-mediated currents recorded at −70mV and +40mV, respectively (left). PVHTRH (G; N = 7/7 mice) or DMHVglut2 (H; N = 4/3 mice) input was optically stimulated in fed or fasted mice. Summary of AMPAR-mediated le-EPSCs amplitude show that DMHVglut2 input to AgRP neurons is increased by fasting.
All data are presented as mean ± s.e.m.; * p < 0.05, ** p < 0.01; two-tailed unpaired Student’s t-test.
Scale bars: B, 25 pA, 50 ms; C, D, E, and F, 30 pA, 50 ms; G and H, 30 pA, 30 ms.
Previous reports demonstrated that fasting evokes an increase in the frequency, but not amplitude, of sEPSCs in AgRP neurons.19–21 This upregulation of glutamatergic drive has been linked to increased neurotransmitter release from presynaptic terminals 19 as well as postsynaptic adaptations in the number of synapses.20,21 To begin to distinguish between presynaptic and postsynaptic alterations at PVHTRH and DMHVglut2 inputs, we recorded light-evoked quantal EPSCs (le-qEPSCs) in the presence of strontium (Sr2+)(Figures 1B–D), which favors asynchronous release of neurotransmitter vesicles following ChR2-induced stimulation. This circuit-specific optogenetic approach has been demonstrated to allow direct assessment of postsynaptic efficacy (le-qEPSCs amplitude) and indirect assessment of functional synaptic sites (le-qEPSC frequency) in defined circuits.40,41 Recordings from AgRP neurons revealed that substitution of Ca2+ with Sr2+ resulted in decreased peak amplitudes of inward currents immediately following light pulses, and profoundly increased the number of events that continued for approximately 300 milliseconds after the stimulation, confirming effective desynchronization of glutamate release (Figure 1B).
When we examined transmission across PVHTRH➔AgRP neuron synapses, we found that fasting evoked a 2-fold increase in the frequency, but had no effect on the amplitude, of le-qEPSCs (Figure 1C). Since an increase in frequency of le-qEPSCs could reflect an increase of functional connections or an increase in release probability of glutamate from afferent terminals, we applied three additional independent measures to further characterize this synaptic plasticity occurring at PVHTRH synapses: i) paired-pulse ratio (PPR) of two light evoked EPSCs (le-EPSCs) within 50 ms, ii) changes in coefficient of variation (CV) of le-EPSC, and iii) ratio of the amplitude of AMPAR- and NMDAR-mediated le-EPSCs (Figures 1E–H, Figure S1C). We found no alterations in the PPR and CV values between fed and fasted mice, suggesting no changes in glutamate release probability (Figure 1E). We also found no changes in the AMPAR/NMDAR ratio between fed and fasted mice (Figure 1G), indicating no changes in postsynaptic ion conductance at PVHTRH input. Thus, the increase in frequency of le-qEPSCS in the absence of changes in glutamate release probability or ion conductance suggest that fasting increases the number of functional synaptic sites. In support of this possibility, previous work has established that fasting increases the number of dendritic spines in postsynaptic AgRP neurons20,21 – although the source of the glutamatergic input in those studies was unknown.
We next determined alterations of transmission across glutamatergic DMHVglut2➔AgRP synapses. Fasting evoked no changes in the frequency of le-qEPSCs, in PPR, CV, and AMPAR/NMDAR ratio (Figures 1D, F, H). We did observe, however, an increase in the amplitude of le-qEPSCs (Figure 1D) suggesting that transmission across DMHVglut2➔AgRP synapses, unlike PVHTRH➔AgRP synapses, is potentiated through a postsynaptic increase in ion conductance. Consistent with this, fasting augmented the amplitude of le-EPSCs at DMHVglut2 input, but not at PVHTRH input (Figures 1G, H).
PVHTRH neuron activity is required for regaining lost weight
The strikingly different synaptic changes at glutamatergic PVHTRH and DMHVglut2 inputs suggest that increased activity of either excitatory input (or both together) could contribute to AgRP neuron activation and driving in turn feeding behavior after weight loss. To explore this possibility, we employed a chemogenetic approach to silence PVHTRH or DMHVglut2 neurons during fasting (Figure 2A). We injected an AAV expressing Cre-dependent inhibitory chemogenetic receptor construct hGlyR (AAV-FLEX-hGlyR-mCherry)42 into the PVH of Trh-ires-Cre or into the DMH of Slc17a6-ires-Cre mice (Figure 2A). The hGlyR receptor responds to Ivermectin (IVM) instead of glycine,43 and systemic administration of IVM causes reversible silencing of hGlyR-expressing neurons.42 Ex vivo current-clamp recordings confirmed that IVM effectively inhibits hGlyR-expressing neurons (Figure 2B). To determine the functional consequences of chemogenetically silencing PVHTRH or DMHVglut2 neurons, mice were injected IP either with IVM or control vehicle before food removal (Figure 2A, Figure S2A). Notably, the duration of action for a single dose of IVM is reported to be 2–3 days.44 Feeding measurements revealed that inhibition of PVHTRH neurons with hGlyR profoundly reduced increases in food intake at 8- and 24-hour time points after reintroducing food, whereas immediate increase in feeding after ad libitum access to food (hours 1 and 4) was not altered (Figure 2C). Consistent with the reduction in food intake, regain of lost body weight over the following days was decreased (Figure 2C). Thus, PVHTRH neuron activity is of particular importance for feeding adaptations and promoting weight gain over prolonged time periods. By contrast, silencing DMHVglut2 neurons had no significant effects on food intake and body weight regain (Figure 2D).
Figure 2: Activity of PVHTRH neurons is required for regaining body weight after fasting.

A) Experimental schematic: A Cre-dependent viral approach was used to express the Ivermectin (IVM)-responsive glycine receptor hGlyR for selective inhibition of PVHTRH or DMHVglut2 neurons. Mice were injected with vehicle (VEH) or IVM before food removal at the onset of the dark cycle, followed by 16-hours of fasting.
B) Representative trace of current clamp recordings from an hGlyR-expressing neuron. IVM wash-in blocks action-potential firing. Scale bars: 30 mV, 2 min.
C, D) Left: Immunostainings showing site-specific hGlyR-mCherry expression in the PVH of Trh-ires-Cre (C) and in the DMH of Slc17a6-ires-Cre (D) mice. Scale bars: 100 µm. IVM/hGlyR-mediated inhibition of PVHTRH neurons reduces food intake after fasting, and this effect strengthens over time (hours 8 and 24; C). In the same mice, regain of lost body weight is diminished over the week following fasting (C, right).
Selective inhibition of DMHVglut2 neurons does not significantly affect food intake or body weight regain after fasting (D).
All data are presented as mean ± s.e.m.; * p < 0.05, *** p < 0.001, **** p < 0.0001; ordinary two-way ANOVA followed by Sidak’s multiple comparisons test.
PVHTRH neuron activity regulates strength of transmission across glutamatergic PVHTRH➔AgRP synapses
Having established the functional importance of PVHTRH neurons, we next tested for a causal link between PVHTRH neuron activity and increased excitatory drive onto AgRP neurons. We first determined whether fasting activates the PVHTRH neurons which provide synaptic input to AgRP neurons. It is important to note that PVHTRH neurons are heterogeneous. In addition to the Trh-expressing neurons that project to and regulate AgRP neurons,29 there are also median eminence-projecting TRH-releasing neuroendocrine neurons that control thyroid hormone secretion. These neuroendocrine PVHTRH neurons are believed to be inhibited by fasting.45,46 To specifically hone in on the Trh-expressing PVH neurons that regulate AgRP neurons, we focused our analysis on the PVHTRH neurons that co-express pituitary adenylate cyclase-activating polypeptide (PACAP, gene symbol Adcyap1). Trh/Adcyap1 co-expression likely marks the AgRP neuron afferents for the following reasons: a) Trh/Adcyap1-coexpressing neurons are found in the anterior PVH where the AgRP neuron afferents, and not the neuroendocrine Trh neurons are located (as assessed by monosynaptic rabies mapping), b) PVH neurons marked by either Trh or Adcyap1 both provide monosynaptic glutamatergic input to AgRP neurons (as assessed by CRACM), and c) PVH neurons marked by either Trh or Adcyap1 both cause a similar level of hyperphagia when chemogenetically stimulated.29 Using fluorescence in situ hybridization (FISH), we found that overnight fasting profoundly increased the activity of PVHTRH neurons that are PACAP+ as evidenced by Fos expression (Figure 3A).
Figure 3: Potentiation of PVHTRH input to AgRP neurons is driven by PVHTRH neuron activity.
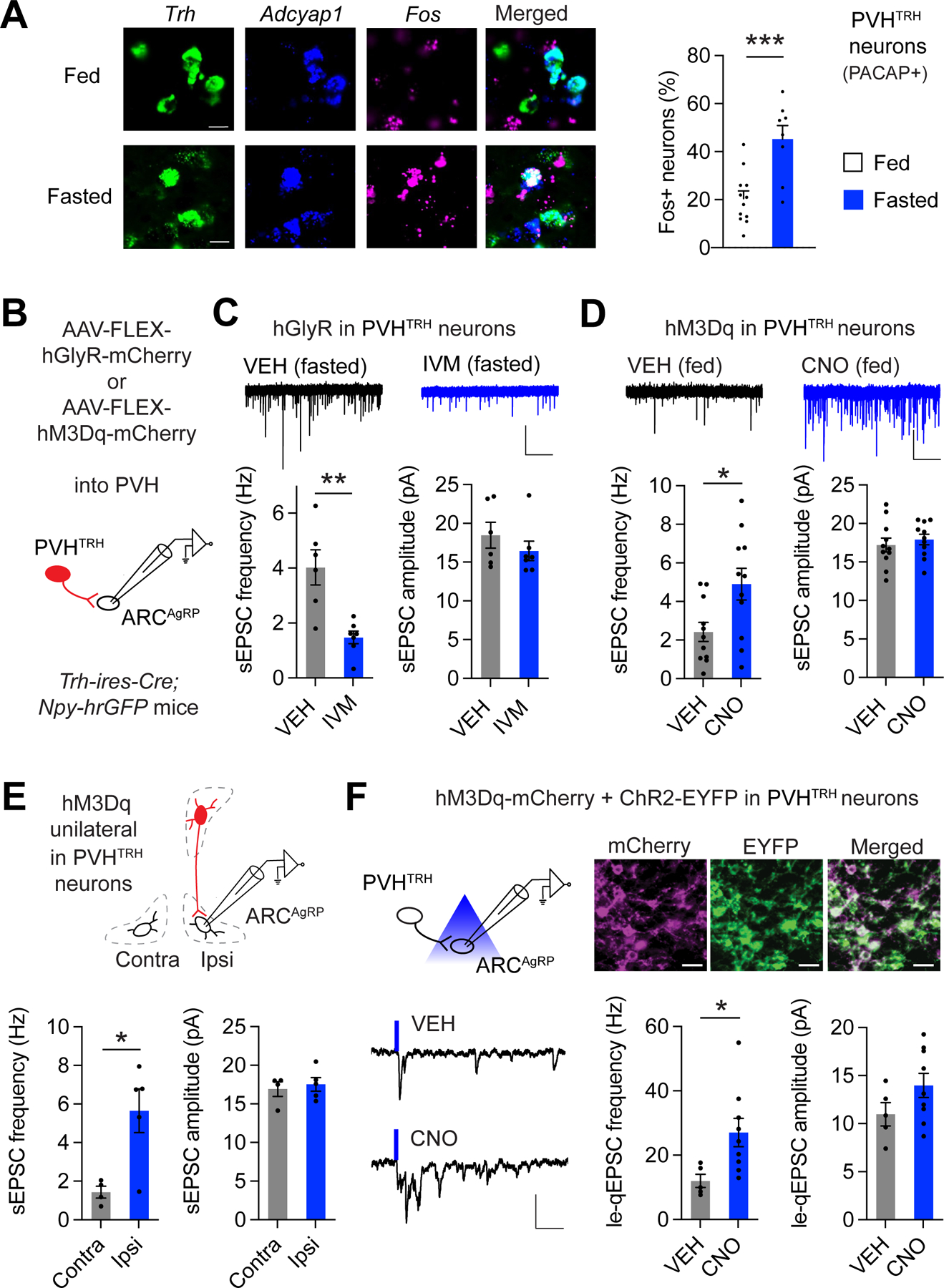
A) Representative histological images and analysis of Fos expression in PVHTRH neurons that express Adcyap1 (PACAP) from fed or fasted mice assessed by FISH. Scale bars represent 10 μm. (N = 4/4 mice).
B) Schematic of recordings from AgRP neurons following in vivo chemogenetic manipulations of PVHTRH neurons. Mice were injected with AAVs to express hGlyR or hM3Dq selectively in PVHTRH neurons.
C) Representative traces (top) of spontaneous excitatory postsynaptic currents (sEPSCs) in mice expressing hGlyR in PVHTRH neurons injected with VEH or IVM before fasting (scale bars: 25 pA, 2 s). Inhibition of PVHTRH neuron activity reduces the frequency, but not the amplitude, of sEPSCs in AgRP neurons in fasted mice (N = 2/2 mice).
D) Fed mice expressing hM3Dq in PVHTRH neurons were administered with vehicle (VEH) or CNO at the onset of the light cycle. Frequency, but not amplitude, of sEPSCs in AgRP neurons is significantly increased in mice treated with CNO 4 hours before brain slices are prepared (N = 2/2 mice; scale bar: 25 pA, 2 s).
E) Because chemogenetic stimulation of PVHTRH neurons acutely increases food intake (Figure S2B and 29), we tested the influence of circuit-specific activation on upregulation of sEPSC frequency. For this, hM3Dq was unilaterally expressed in PVHTRH neurons. In AgRP neurons recorded from the ipsilateral (Ipsi), but not from the contralateral (Contra), site of the stimulated PVHTRH neurons, sEPSC frequency is significantly upregulated, consistent with increased activity of the PVHTRH➔AgRP circuit as the functionally relevant mechanism for the amplification of excitatory drive onto AgRP neurons (N = 2/2 mice).
F) Schematic of the optogenetic approach to assess circuit-specific plasticity upon PVHTRH neuron activation (top left). Representative fluorescence images showing expression of ChR2-EYFP (green) and hM3Dq-mCherry (magenta) in the PVH (top right; scale bars, 20 µm). Note the co-expression of both viruses in PVHTRH neurons.
Representative traces (bottom left) of currents obtained from optical stimulation of PVHTRH afferents in presence of strontium from mice treated with VEH or CNO 4 hours before slice preparation (scale bars, 30 pA, 50 ms).
Summary of le-qEPSC recorded from AgRP neurons show that PVHTRH neuron activation significantly increases quantal frequency, but not amplitude, demonstrating that circuit activation amplifies activity of PVHTRH➔AgRP synapses (N = 5/4 mice).
All data are presented as mean ± s.e.m.; * p < 0.05, ** p < 0.01, ***p < 0.001; two-tailed unpaired Student’s t-test.
To probe whether increased PVHTRH neuron activity upon fasting translates into upregulation of glutamatergic drive onto AgRP neurons, we virally targeted hGlyR into the PVH of Trh-ires-Cre; Npy-hrGFP mice and recorded sEPSCs from AgRP neurons (Figure 3B). Mice were injected with IVM before food removal and brain slices were prepared after overnight fasting. We found that silencing PVHTRH neurons abolished the fasting-induced increase in the frequency of sEPSCs (Figure 3C). These findings demonstrate that increased activity of the upstream PVHTRH neurons is necessary for amplifying glutamatergic synaptic input to AgRP neurons.
Next, we tested whether activation of PVHTRH neurons is sufficient for triggering this synaptic amplification. We injected an AAV expressing Cre-dependent the excitatory chemogenetic receptor hM3Dq (AAV-FLEX-hM3Dq-mCherry) into the PVH of Trh-ires-Cre; Npy-hrGFP mice (Figure 3B). Clozapine-N-oxide (CNO) administration in vivo effectively activated PVHTRH neurons as assessed by Fos (Figure S3A). In addition, CNO administration acutely increased feeding in mice expressing hM3Dq in PVHTRH neurons – as has been previously reported29–but not in control mice expressing mCherry (Figure S3B). We found a significant increase in the frequency, but not amplitude, of sEPSCs in AgRP neurons in brain slices prepared 4 hours after CNO administration in vivo (Figure 3D). The observed upregulation in glutamatergic synaptic input may be either due to the projection-specific activation of PVHTRH➔AgRP synapses, increased feeding in response to PVHTRH neuron activation (Figure S3B and 29), or the release of hormonal or nutrient signals into circulation. To distinguish between these possibilities, we targeted hM3Dq unilaterally to PVHTRH neurons (Figure 3E). sEPSC recordings demonstrated that stimulation of PVHTRH neurons increased the frequency recorded from ipsilateral, but not contralateral, AgRP neurons (Figure 3E). Since PVHTRH neurons project to the ARC unilaterally and engage only ipsilateral AgRP neurons,29 these findings demonstrate that increased excitatory drive arises from activation of PVHTRH➔AgRP synapses and is independent of any secondary systemic signals, which would affect both hemispheres.
To extend these findings, we assessed transmission across PVHTRH➔AgRP synapses upon circuit activation. We targeted AAV-FLEX-hM3Dq together with an AAV expressing Cre-dependent ChR2 (AAV-FLEX-ChR2-EYFP) into the PVH of Trh-ires-Cre; Npy-hrGFP mice (Figure 3F). Histological analysis confirmed efficient co-expression of both viruses in PVHTRH neurons (mCherry- and EYFP-positive; Figure 3F). In vivo chemogenetic stimulation of PVHTRH neurons profoundly increased the frequency, but not amplitude, of le-qEPSCs (Figure 3F), demonstrating activity-dependent potentiation of PVHTRH➔AgRP synapses.
Potentiation of PVHTRH➔AgRP hunger synapses persists in conditions in which weight gain is promoted
We next explored the possibility that strengthening of PVHTRH➔AgRP synapses is long-lasting, and thus could be a cellular mechanism for promoting weight regain, and explain why most diets fail. After overnight fasting, mice exhibited increased feeding for four days after reintroducing food (Figures 4A, B). Recordings from AgRP neurons in brain slices prepared 2 days after reintroducing food displayed increased frequency of le-qEPSCs (Figure 4C). Thus, potentiation of transmission across PVHTRH➔AgRP synapses persists, and is associated with increased feeding and body weight gain after ad libitum access to food (Figure 4B).
Figure 4: Elevated activity of the excitatory PVHTRH➔AGRP circuit persists in circumstances in which weight gain is promoted.

A) Timeline showing the experimental scheme for determination of plasticity at PVHTRH➔ AgRP synapses after an overnight fast. Ad libitum food intake and body weight changes were determined at indicated time points.
B) Summary of food intake (left) show that ad libitum intake was significantly increased for 4 days after an overnight fast. This effect diminished over time (days 2–4) and dissipated at day 7. Right: Body weight development in the same mice.
C) Left: Schematic of recordings from AgRP neurons following optical stimulation of PVHTRH afferents in presence of strontium from mice refed for 2 or 7 days after fasting, or kept on the caloric maintenance paradigm (CMP; D) before slice preparation.
Right: Summary of le-qEPSC recorded from AgRP neurons show that quantal frequency, but not amplitude, is increased on day 2 after refeeding and on day 7 after CMP, demonstrating amplification of PVHTRH➔AgRP synaptic activity in conditions in which weight gain is promoted (N = 4/3/4/3 mice).
D) Timeline showing the experimental scheme for the CMP. After an overnight fast, mice were kept on the CMP for 6 days, during which they were provided the amount of food they consumed on average in ad libitum control conditions (3.8 g per day; bottom).
E) Summary of food intake (left) and body weight (right) show that ad libitum intake and body weight were increased at day 7 when mice were kept on the CMP for 6 days after fasting.
F) Effects of the CMP as well as 7 days of a caloric restriction paradigm (CR 7; Fig. S4) on Fos expression in PVHTRH that express Adcyap1 (PACAP) assessed by FISH. Scale bars represent 10 μm. (N = 4/4 mice).
G) Summary of Fos expression in AgRP neurons in fed mice, in mice that were kept on the CMP after fasting, and in mice that were subjected to the CR for 7 days. (N = 4/4/3 mice)
All data are presented as mean ± s.e.m.; * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001; repeated-measures one-way ANOVA (B and E) and ordinary one-way ANOVA (C, F, and G).
Next, we probed whether this excitatory synaptic increase is causally related to weight gain after a weight maintenance phase, which is commonly seen in human dieters.1 To this end, mice were overnight fasted, and then maintained for one week with chow provided daily with an amount (~4g) equivalent to their 24-hour intake before fasting (Figure 4D). Mice kept on this caloric maintenance paradigm (CMP) exhibited hyperphagia when given ad libitum access to food after the CMP (Figure 4E). Remarkably, AgRP neurons from brain slices of mice kept on CMP for one week showed elevated le-qEPSC frequency that was not significantly different from those observed after one day of refeeding (Figure 4C). As a control, we assessed AgRP neurons in brain slices from mice ad libitum fed for one week after fasting; le-qEPSC frequency in these mice returned to control levels (Figure 4C), consistent with the normalization of food intake and body weight (Figures 4A, B). Fos expression analysis also showed that the activity of PVHTRH neurons that are marked by PACAP expression is increased in mice subjected to the CMP for one week (Figure 4F), as compared with those from fed mice in our earlier study (Figure 3A). Concordant with this elevated neuronal activity as well as strengthening of PVHTRH➔AgRP synapses, Fos expression and sEPSC frequency in AgRP neurons were significantly increased (Figures S4A, B).
Since fasting is accompanied by an acute severe caloric deficit, we additionally determined activity changes of the circuit upon a sustained moderate reduction in energy intake, which is a widely used weight loss strategy in humans.1 Mice were provided daily with reduced amounts of chow (~75% of control intake) for one week (Figure S4C). As expected, mice subjected to this caloric restriction (CR) lost body weight (Figure S4C). Analysis of Fos expression demonstrated that CR significantly increased the activity of both PVHTRH neurons (PACAP+) and AgRP neurons (Figures 4F, G, and S4A). The elevated neuronal activity was paralleled by an increased excitatory input onto AgRP neurons as assessed by sEPSC recordings (Figure S4B). Together, these findings strongly support the view that potentiation of the excitatory PVHTRH➔AgRP circuit is long-lasting and has a key role in promoting regain of lost body weight.
Brief high-frequency bouts potentiate PVHTRH➔AgRP synapses and promote feeding
Activation of NMDARs has a well-established role in mediating activity-dependent plasticity of glutamatergic synapses,47 and is responsible for the fasting-induced increases in sEPSC frequency and spines in AgRP neurons, as well as increased food intake following fasting 20. To probe whether increased transmission across PVHTRH➔AgRP synapses in response to circuit activation requires NMDAR activation, we first employed an ex vivo optogenetic approach (Figure 5A). We virally expressed ChR2 in PVHTRH neurons, prepared brain slices, and photo-stimulated ChR2-expressing axonal terminals in the ARC (Figure 5A). We applied a high frequency (HF; 50 Hz) stimulation protocol, which has been associated with high levels of glutamate release and NMDAR activation.48 Two to three hours later, brain slices were transferred to strontium-containing solution and le-qEPSCs were recorded from AgRP neurons (Figure 5A). Brief 10-minute HF photostimulation (HFpS) of PVHTRH neuron axonal terminals in the ARC increased the frequency, but not the amplitude, of le-qEPSCs (Figure 5B). This strengthening of transmission across PVHTRH➔AgRP synapses could be detected for several hours after photostimulation. Importantly, when the NMDAR antagonist D-AP5 was present during the stimulation period, HFpS failed to increase le-qEPSCs frequency (Figure 5B). Thus, NMDAR activation during circuit stimulation is required for triggering an activity-dependent potentiation of transmission across PVHTRH➔AgRP synapses.
Figure 5: Brief, high-frequency stimulation potentiates PVHTRH➔AgRP synapses and promotes feeding.

A) Schematic of the ex vivo approach used for high frequency photostimulation (HFpS; 10 minutes, 50 Hz) of PVHTRH➔AgRP synapses. The brief HFpS was applied to PVHTRH neuron terminals in the ARC. Two hours later, slices were transferred to strontium-containing ACSF and le-qEPSCs were recorded from AgRP neurons. Representative fluorescence image shows expression of ChR2-mCherry (magenta) in originating from PVHTRH neuron terminals and NPY-hrGFP (green) from AgRP neurons (scale bar, 20 µm).
B) Representative traces of light-evoked currents recorded in strontium-containing solution from unstimulated slices (control), and from slices that were subjected to HFpS in absence or presence of D-AP5 (scale bars, 30 pA, 50 ms).
Summary of le-qEPSCs show that HFpS for 10 minutes increases the quantal frequency, but not the quantal amplitude, of PVHTRH input onto AgRP neurons. When the NMDAR antagonist D-AP5 is present during light illumination, HFpS fails to increase the quantal frequency (N = 4/8/3 mice).
C) Schematic of the in vivo approach. The quantal frequency of PVHTRH input is significantly increased in AgRP neurons when brain slices are prepared 4 hours after the brief HFpS delivered in vivo (N = 2/2 mice).
D) 24-hour food intake was significantly increased in mice that were subjected to HFpS, compared to control condition (no light illumination). This increase was prevented when mice were pretreated with the NMDAR antagonist MK-801 before HFpS, and food intake in these mice did not significantly differ from control condition.
Right: Feeding significantly increased at hour 1 and hours 1–4 following HFpS and tended to increase at hours 8–24.
Data are presented as mean ± s.e.m.; * p < 0.05; ordinary one-way ANOVA followed by Tukey’s multiple comparisons test (B, and D, left), two-tailed unpaired Student’s t-test (C), and two-tailed paired Student’s t-test (D, right).
Together, the above studies show that strengthening of glutamatergic PVHTRH➔AgRP synapses i) is induced by increased activity of PVHTRHneurons, ii) persists for days, ii) requires the activation of NMDARs, and iv) can be triggered by brief HF stimulation, even in absence of circulating signals. To assess the impact of activity-dependent synapse strengthening on feeding behavior, we employed an in vivo optogenetic approach through implanted optical fibers (Figure 5C, Figure S5A). We confirmed that the brief HFpS protocol applied in vivo amplified transmission across PVHTRH➔AgRP synapses as assessed by recordings from brain slices prepared 4 hours after optogenetic stimulation (Figure 5C, Figure S5B). We found that following the 10-minute HFpS, food intake slightly but progressively increased over time, which resulted in a significant increase at 24 hours after stimulation (Figure 5D). When mice received the NMDAR antagonist MK-801 prior to the HFpS, 24-hour food intake was not altered (Figure 5D). Thus, brief circuit stimulation is sufficient for triggering an NMDAR-dependent long-term increase in feeding.
Chemogenetic PVHTRH➔AgRP circuit activation induces long-lasting, NMDAR-dependent hyperphagia
The 24-hour increase in food intake in response to the brief optogenetic activation was less than that observed in mice that were given ad libitum access to food after fasting (Figure 4B). One potential explanation for this difference is that a longer-lasting increase in activity of the circuit is required for driving the full, fasting-induced, feeding response. To test this, we explored the behavioral effects of chemogenetically stimulating the circuit (Figure 6A). le-qEPSC recordings in brain slices prepared 4 hours after CNO administration demonstrated that potentiation of PVHTRH➔AgRP synapses in response to hM3Dq-induced activation of PVHTRH neurons requires NMDAR activation (Figure 6B, Figure S6A). Consistent with the direct, chemogenetic stimulation of the excitatory PVHTRH➔AgRP circuit, CNO injection caused an acute increase in feeding (Figure S3B). Remarkably, food intake continued to increase progressively, such that by 24 hours post a single CNO injection, mice had consumed an amount (~5.5 g, Figure 6C) approximately equivalent to the 24-hour intake after fasting (Figure 4B). When CNO was co-administered with MK-801, increases in feeding up to 8 hours after injections were blunted and, importantly, at later points completely abolished, resulting in a significant reduction in 24-hour intake (Figure 6C). Thus, NMDAR activation during circuit stimulation is of particular importance for the long-term increase in feeding. Administration of CNO as well as MK-801 did not alter food intake in control mice expressing mCherry (Figure S6B).
Figure 6: NMDAR signaling is required for long-lasting hyperphagia upon PVHTRH➔AgRP circuit activation.
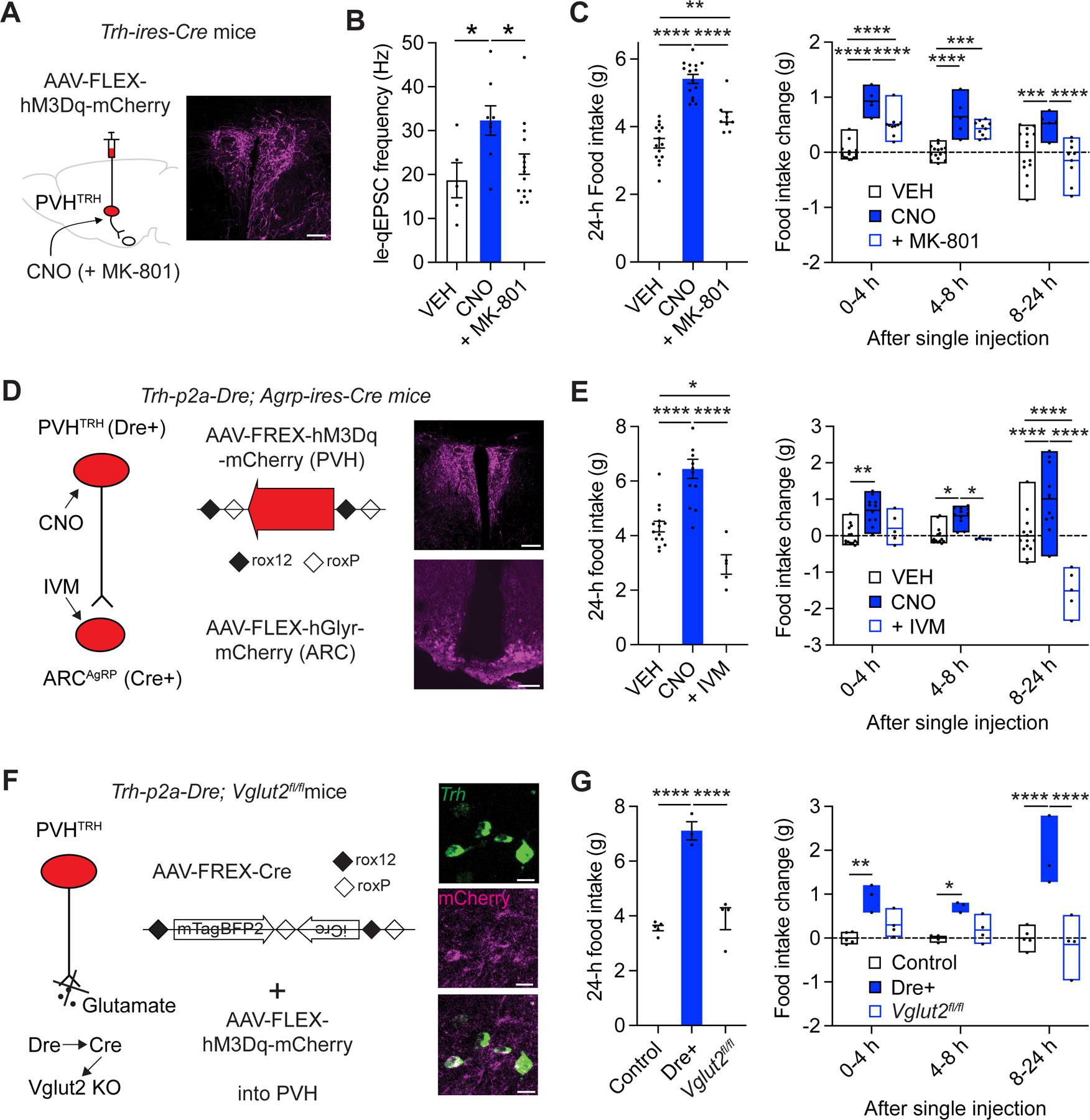
A) Schematic of the chemogenetic approach used for selective stimulation of PVHTRH neurons (left), and representative fluorescence image showing hM3Dq-mCherry (magenta) in the PVH (right; scale bar, 100 µm).
B) Co-administration of the NMDAR antagonist MK-801 significantly reduces the increase in quantal frequency of PVHTRH input onto AgRP neurons upon hM3Dq-induced activation of PVHTRH neurons (N = 4/3/4 mice).
C) 24-hour food intake was increased in hM3Dq-expressing mice that received a single injection of CNO, compared to control conditions (vehicle, VEH injected). Co-administration of MK-801 significantly diminishes the increase in 24-hour food intake (left). Summary of food intake measured at different time points show that the acute increase in feeding (1–4- and 4–8-hours’ time windows) is reduced by MK-801 co-administration, whereas the effect 8–24-hour after CNO administration is completely blocked, consistent with an activity-dependent, NMDAR-dependent signaling that leads to amplification of synaptic excitation in the PVHTRH➔AgRP circuit to promote a long-lasting increase in feeding.
D) Schematic of the genetic approach for simultaneous PVHTRH neuron activation and AgRP neuron inhibition. Mice expressing Dre-recombinase in PVHTRH neurons and Cre-recombinase in AgRP neurons (Trh-p2a-Dre; Agrp-ires-Cre mice) were injected with an AAV to express a Dre-dependent (roxed) hM3Dq-mCherry construct in the PVH, and a Cre-dependent (floxed) hGlyR-mCherry construct in the ARC (left; see methods).
Representative fluorescence images showing hM3Dq-mCherry and hGlyR-mCherry in the PVH and ARC, respectively (magenta; scale bars, 100 µm).
E) Increased food intake upon hM3Dq-induced activation of PVHTRH neurons is blocked in mice whose AgRP neuron activity is simultaneously inhibited through IVM-mediated activation of hGlyR.
F) Schematic of the chemogenetic approach to determine the necessity of glutamatergic transmission in driving the food consumption upon PVHTRH neuron activation. For selective deletion of Vglut2 from PVHTRH neurons, Trh-p2a-Dre mice with loxp-flanked Slc17a6 alleles (Vglut2, Trh-p2a-Dre; Slc17a6fl/fl mice) were injected with a Dre-dependent AAV to express Cre-recombinase. An AAV expressing Cre-dependent-hM3Dq was co-injected. Representative fluorescence images showing co-expression of Trh mRNA (green) and hM3Dq-mCherry in PVHTRH neurons (magenta; scale bars, 50 µm).
G) Food intake is significantly increased in mice expressing hM3Dq in PVHTRH neurons (Dre+) using the genetic approach shown in F, compared to control mice (Dre-recombinase negative). This increase was absent in Trh-p2a-Dre; Slc17a6fl/fl mice Cre-/Dre-dependently lacking Vglut2 in PVHTRH neurons (Slc17a6fl/fl), demonstrating that Vglut2-dependent glutamatergic transmission is necessary for the acute and long-term increases in food consumption upon circuit stimulation.
All mice were co-injected with AAV-FREX-Cre and AAV-FLEX-hM3Dq into the PVH, and received an administration of CNO at the onset of the light cycle.
Data are presented as mean ± s.e.m.; * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001; ordinary one-way ANOVA followed by Tukey’s multiple comparisons test (B, C, E, and G, left), and ordinary two-way ANOVA followed by Tukey’s multiple comparisons test (C, E, and G, right).
To directly show that the long-term increase in food intake upon PVHTRH neuron activation was mediated by activation of downstream AgRP neurons, we chemogenetically silenced AgRP neurons during PVHTRH neuron stimulation with hGlyR (Figure 6D). We employed a Dre- and Cre-recombinase-dependent genetic approach to selectively target hGlyR to AgRP neurons, while avoiding nearby TRH-expressing neurons in the ARC.49 To this end, we generated Trh-p2a-Dre mice that express the Dre-recombinase in TRH+ cells and an AAV expressing Dre-dependent hM3Dq (AAV-FREX-hM3Dq-mCherry, see methods; Figures S7A–D). We crossed Trh-p2a-Dre mice with Agrp-ires-Cre mice and injected the resulting Trh-p2a-Dre; Agrp-ires-Cre mice with AAV-FREX-hM3Dq-mCherry into the PVH and AAV-FLEX-hGlyR-mCherry into the ARC (Figure 6D). Histological analysis confirmed effective stimulation of hM3Dq-expressing PVHTRH neurons using this approach (Figure S7B–D). CNO administration in Dre-/Cre-expressing mice profoundly enhanced feeding, acutely and over 24 hours (Figure 6E). Importantly, this hyperphagia was abolished by co-administration of CNO and IVM, which induces a simultaneous inhibition of hGlyR-expressing AgRP neurons during hM3Dq-induced stimulation of PVHTRH neurons (Figure 6E). Thus, AgRP neuron activation is necessary for driving the acute and long-lasting increase in food intake upon circuit activation.
If glutamate released from activated PVHTRH neurons mediates the NMDAR-dependent feeding response, then stimulating PVHTRH neurons that lack glutamate release (e.g., via deletion of Vglut2) should block the behavioral effects. Notably, Vglut2 is the only vesicular glutamate transporter found in PVHTRH neurons.50 Given the brain-wide expression pattern of TRH during development, we employed a Dre- and Cre-recombinase approach to selectively delete Vglut2 from PVHTRH neurons in adult mice (Figure 6F). Trh-p2a-Dre mice were crossed to Slc17a6flox (Vglut2flox) mice to generate Trh-p2a-Dre; Slc17a6flox/flox mice, which were injected with an AAV expressing Dre-dependent the Cre-recombinase (AAV-FREX-Cre, see methods) together with AAV-FLEX-hM3Dq-mCherry into the PVH (Figure 6F). Histological analysis demonstrated that this intersectional genetic approach caused efficient expression of hM3Dq in PVHTRH neurons (Figure 6F, Figure S7E, F). In agreement with our earlier studies (Figure 6A), chemogenetic stimulation of PVHTRH neurons in control mice (i.e. Trh-p2a-Dre; Slc17a6wt/wt; Dre+) reliably increased feeding, acutely and over prolonged time periods (Figure 6G). In contrast, chemogenetically stimulating PVHTRH neurons in Trh-p2a-Dre; Slc17a6flox/flox (Vglu2 fl/fl) mice, which are notable for their inability to release glutamate, failed to increase food intake (Figure 6G, Figure S7G).
Together, the above data reveal an essential role for glutamate release from upstream PVHTRH neurons, NMDAR activation, and activity of downstream AgRP neurons in the sustained feeding effects in response to circuit activation.
Persistent gaining of body weight following activation of the PVHTRH➔AgRP circuit
The results presented thus far suggest that weight loss increases PVHTRH neuron activity, which strengthens PVHTRH➔AgRP synapses in an activity-dependent manner, and augments concomitant food intake to promote weight gain. To directly test this, we assessed body weight development in response to chemogenetic activation of PVHTRH neurons. Consistent with the substantial increase in 24-hour food intake (Figure 6C), mice increased body weight by ~5% following a single CNO injection after one day (Figure 7A). This increase in body weight was abolished by co-administration of CNO and MK-801 (Figure 7A), demonstrating the requirement of NMDAR activation during circuit activation.
Figure 7: Activation of the PVHTRH➔AgRP circuit evokes persistent gaining of body weight.

A) Mice expressing hM3Dq in PVHTRH neurons significantly increase body weight following a single CNO injection. When the NMDAR antagonist MK-801 is co-administered with CNO, body weight is not affected.
B) Summary of food intake show that food consumption is increased for 2 days following a single injection of CNO. This effect diminishes over time (day 2), and is not followed by reduced food intake over the next 7 days.
C) Body weight change measured 7 days after a single injection. Persistent body weight gain is present after a single CNO injection in mice expressing hM3Dq in PVHTRH neurons, but not in control mice (mCherry-expressing). CNO fails to increase body weight when NMDAR are blocked by co-administration of MK-801 with CNO.
D) Body weight increases further in mice expressing hM3Dq in PVHTRH neurons following single CNO injections with intervals of two weeks. This body weight gain is blocked by co-administration of MK-801.
All data are presented as mean ± s.e.m.; * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001; ordinary one-way ANOVA followed by Tukey’s multiple comparisons test (A, C), repeated-measures one-way ANOVA followed by Tukey’s multiple comparisons test (B), and ordinary two-way ANOVA followed by Tukey’s multiple comparisons test (D).
Further analysis of food intake revealed that increased feeding in response to chemogenetic activation of the circuit was still detectable one day after CNO injection, returning to control levels on day three (Figure 7B). Remarkably, mice increased body weight by ~10% in the week after a single CNO injection (Figure 7C), suggesting that persistent body weight gain was induced by increased caloric intake that was not counterbalanced by hypophagia on the following days. Consistent with this, single CNO injections 2 and 4 weeks later resulted in further weight gain (Figure 7D).
Given that the hM3Dq-mediated neural activation upon CNO administration typically lasts only approximately 8 hours,5,51 the persistent gaining in body weight strongly suggests that long-lasting plasticity in the PVHTRH➔AgRP circuit is induced by a single dose of CNO. Importantly, this long-lasting effect cannot be occurring at the level of AgRP neurons, or downstream of AgRP neurons because it has been shown that direct hM3Dq-induced stimulation of AgRP neurons, unlike stimulation of PVHTRH neurons, loses its stimulatory effect on body weight soon after the last dose of CNO.5,52 One potential explanation is that activity-dependent amplification of synaptic transmission across PVHTRH➔AgRP synapses upon circuit stimulation has long-lasting and slow-to-reverse effects. To directly test whether body weight gain required NMDAR activation during circuit activation, mice that expressed hM3Dq in PVHTRH neurons were co-administered with CNO and MK-801. This completely abolished persistent gaining of body weight (Figure 7D). Thus, NMDAR activation is required for persistent gaining of body weight upon a single episode of activating the PVHTRH➔AgRP circuit.
Discussion
Increased hunger drive is a major component of the adaptive homeostatic response aimed at promoting regain of lost body weight. Although enhanced excitatory drive has been shown to increase activity of hunger-promoting AgRP neuron in response to energy deficit,19 the causal involvement of plastic changes in distinct glutamatergic afferents in body weight control was not known. Previous rabies tracing and optogenetic mapping studies have determined upstream anatomical areas and established functional monosynaptic connectivity between excitatory afferents and AgRP neurons.29,53 Here, using a combination of circuit-specific approaches, we explored the functional contribution of synaptic plasticity in discrete glutamatergic inputs to the regulation of feeding behavior and body weight. Our data support a model in which energy state-dependent synaptic plasticity in AgRP neurons’ glutamatergic afferents is highly selective and pathway-specific. Furthermore, opto- and chemogenetic manipulations reveal that upstream PVHTRH neuron activity bidirectionally controls excitatory synaptic input onto AgRP neurons. Moreover, we show that strength of transmission across PVHTRH➔AgRP synapses is controlled in an activity-dependent manner, and establish a fundamental role for NMDAR-dependent plasticity of this circuit in long-term control of body weight.
We comprehensively interrogated synaptic changes evoked by weight loss following fasting and found that in the same AgRP neurons the frequency of quantal synaptic responses from glutamatergic PVHTRH neurons is increased, whereas neighboring DMHVglut2 synapses increase in synaptic amplitude. These input-specific expression patterns of synapse strengthening point to distinct induction mechanisms that may require activity patterns of the individual upstream neurocircuits coinciding during caloric deficit. Although it is possible that changes in the circulating levels of peripheral hormones, acting on presynaptic19 and/or postsynaptic receptors,54–57 support the induction of synaptic plasticity in AgRP neurons, we demonstrate the necessity of PVHTRH neuron activity in fasting-induced potentiation of excitatory input. Moreover, silencing PVHTRH neurons reduced food intake and diminished regaining of body weight following fasting. These data show that activity of these neurons is critical for driving appropriate hunger responses that might be attributable to a selective increase in synaptic strength of this pathway. What signals tune activity of the PVHTRH neurons during caloric deficit, and what renders the PVHTRH➔AgRP circuit an essential neural substrate for long-term body weight control? The PVH is a key integratory center for the coordination of feeding behavior and multiple brain regions provide synaptic inputs, including cortical sites and brainstem regions that relay nutrient-related signal from the gut.3,34,58 The latter could be of particular interest in light of PVHTRH➔AgRP circuit adaptations as recent imaging studies revealed that AgRP neurons integrate mechanosensory information from the stomach and the intestine related to food consumption.59–62 However, PVHTRH neurons are anatomically and functionally heterogenous, which include neuroendocrine neurons, whose axonal terminals innervate the median eminence and centrally-projecting ones.29,63 Further studies will be required to identify which of these subpopulations synapse onto AgRP neurons and to determine how their activity regulation supports energy state-dependent synaptic plasticity of the PVHTRH➔AgRP circuit.
As evidenced by our opto- and chemogenetic studies, temporally restricted activation of NMDARs during circuit activation is required for amplifying strength of transmission across PVHTRH➔AgRP synapses. Building on these findings, we assessed behavioral changes and demonstrate NMDAR-dependent increase in food intake and body weight gain. Admittedly, systemic administration of the NMDAR antagonist was not circuit specific, but the fact that selective and brief high-frequency optogenetic stimulation of PVHTRH neurons triggers potentiation of excitatory inputs points to a critical contribution of increased glutamate release from axonal terminals and postsynaptic NMDAR activation in long-term synaptic plasticity induction. Furthermore, rendering PVHTRH neurons unable to release glutamate completely abolished the acute and long-lasting increases in feeding. This is consistent with previous results demonstrating that NMDARs on AgRP neurons are required for fasting-induced increase in excitatory drive, and for promoting food intake following fasting.20 Notably, we found that synaptic plasticity can be evoked independently of circulating nutrient and hormonal signals as revealed following in vivo unilateral chemogenetic activation of PVH afferents as well as quantal frequency increase upon ex vivo photostimulation of PVHTRH neuron terminals. Given this, we favor the view that potentiation of PVHTRH➔AgRP synapses is primarily driven in an activity-dependent manner, by increased glutamatergic transmission. This would account for increased hunger drive to counter weight loss.
A key finding of our study is that the reported amplification of transmission across PVHTRH➔AgRP synapses is long-lasting in nature, as it persists during states of energy deficit, namely days after food access and on a caloric restricted diet following fasting. In these conditions, mice exhibited increased feeding and gained body weight. Therefore, such long-term synaptic plasticity may underlie long-lasting facilitation of hunger drive during the weight maintenance phase after dieting and the consequent increased motivation for food consumption and regain of lost body weight in the long-term, which is observed in most human dieters.1,2,64 Full restoration of energy deficit reversed plasticity of PVHTRH➔AgRP synapses, reflected by the normalization of the frequency in quantal synaptic responses to baseline. This suggests that the synaptic amplifier described in this work is actively maintained in the circuit until caloric deficit is restored. The molecular mechanism responsible for these effects are currently unknown but could involve increased Ca2+ concentration following activation of NMDARs in AgRP neurons, and recruitment of calcium-dependent protein kinases.20,21 Ca2+-calmodulin-dependent kinase II (CaMKII) could contribute to this as it plays a crucial role in the maintenance phase of long-term potentiation at excitatory synapses.65–68 Furthermore, CaMKII increases activity of the AMP-activated protein kinase,69 which was previously found responsible for the fasting-induced increases in sEPSC frequency and spines in AgRP neurons.21 In addition, genetic deletion of CaMKII downregulates AgRP gene expression and reduces feeding after fasting, and pharmacological inhibition of CaMKII promotes weight loss.70 Given that cell-type-specific manipulations of both PVHTRH and AgRP neurons are required to determine synaptic plasticity maintenance in a pathway-specific way, the intersectional targeting approach that we describe here provides a selective mean for functional future interrogation of these molecular mechanisms in regaining lost body weight.
In this study, we have demonstrated that a single period of hM3Dq-induced activation of PVHTRH neurons is capable of evoking a long-lasting increase in food consumption that is not counterbalanced by hypophagia, and evokes a persistent body weight gain. Inhibition of NMDARs during stimulation only slightly reduced the immediate increase in feeding, while elevated food consumption at later time points and body weight gain were completely blocked. Inhibiting NMDAR also blocked the increase in frequency of quantal synaptic responses. Although AgRP neuron activity is required for driving food intake following PVHTRH neuron activation (Figure 6E and 29), and direct hM3Dq-induced stimulation of AgRP neurons increases feeding over hours,5,71,72 body weight gain is completely reversed soon after the last dose of CNO.5,72 Therefore, the most plausible explanation of our data is that activity-dependent amplification of synaptic transmission across PVHTRH➔AgRP synapses enhances effectiveness this orexigenic pathway over prolonged time periods, which causes in turn body weight gain that enduringly persists.
In summary, we demonstrate that upregulation of synaptic transmission across PVHTRH➔AgRP synapses causes the previously observed increase in excitatory synaptic input onto AgRP neurons in response to energy deficit. Furthermore, we demonstrate that these pathway-specific changes are closely linked with long-lasting changes in feeding behavior and, importantly, body weight gain. Our findings further illustrate the importance of studying defined hypothalamic neural circuits relevant to hunger regulation. Viewed in the context of the emerging obesity epidemic, our results provide new rationales for developing therapeutic strategies to support long-term maintenance of reduced body weight after dieting.
Limitations of study
In this study, we employed an optogenetic-electrophysiological approach to investigate circuit-specific plasticity in distinct glutamatergic afferents onto AgRP neurons. Limitations of this approach are that synaptic responses evoked by optogenetic stimulation depend on expression levels of ChR2, light intensity, and recording site in the slice. Taking this into account, it is challenging to determine whether the observed increase in le-qEPSC frequency at the PVHTRH➔AgRP circuit could account for an increase in spine number, as previously reported to occur following fasting.20,21 Alternatively, the number of functional release sites of existing synapses could be increased.73,74 In future histological studies, it will be important to explore the structural changes of this defined pathway in conditions, in which body weight gain is promoted.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Henning Fenselau (henning.fenselau@sf.mpg.de).
Materials Availability
No unique reagents were generated in this study.
Data and Code Availability
Source data for the graphs are provided in Data S1.
No original code was generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal care and experiments were conducted in compliance with ethical regulations for animal testing approved by the National Institute of Health, Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee, and government authorities (Bezirksregierung Köln). Mice were monitored for health status daily, housed in a humidity and temperature-controlled environment (22–24 °C) on a 12 h light/12 h dark cycle with water and normal chow diet (NCD; ssniff V1554) provided ad libitum, unless otherwise specified. All experiments were performed in adult mice, 12 weeks (electrophysiology; male and female mice) or 11–18 weeks (behavioral tests; male mice) of age. All Cre- and Dre-driver mice were used in the heterozygous state. Trh-ires-Cre29 (#032468, Jackson Laboratory), Slc17a6-ires-Cre15 (#028863, Jackson Laboratory), Npy-hrGFP39 (#006417, Jackson Laboratory), Agrp-ires-Cre75 (#012899, Jackson Laboratory) and Slc17a6fl 76 (#012898, Jackson Laboratory) mice have been described previously. Slc17a6fl/fl mice were used in the homozygous state. All mice were maintained on a mixed background (129; C57BL/6).
Generation of Trh-p2a-Dre mice
Trh-p2a-Dre mice were made using Easi-CRISPR77 to insert a p2a-Dre recombinase cassette in front of the Trh stop codon TAA in the 2nd exon of the Trh gene. Briefly, CAS9 protein (PNA Bio, Cat. CP01), sgRNA (GGAGGAGUAAGGUUAGAGUC, Synthego) and single-stranded DNA template containing the p2a-Dre cassette and 5’ and 3’ 100nt homology arms (IDT), were injected into mouse fertilized eggs from FVB mice, and founders carrying the inserted sequence were identified by PCR among the live-born pups.
METHOD DETAILS
Stereotaxic surgical procedures
Mice were anesthetized and placed into a stereotaxic frame (David Kopf Instruments). After a local anesthetic agent (Lidocaine) was applied to the skin, the skull surface was exposed through a skin incision and a small drill hole was made. Viruses were delivered through a pulled glass micropipette at speed of 2–5 nl per min and the pipette. AAVs were injected into the PVH (coordinates from bregma AP: −0.75 mm, DV: −4.85 mm, ML: ± 0.25 mm), into the ARC (coordinates from bregma: AP: −1.45 mm, DV: −5.95 mm, ML: ± 0.2 mm), or into the DMH (coordinates from bregma AP: −1.58 mm, DV: −4.75 mm, ML: ± 0.30 mm). After surgery, mice received analgesic treatment for pain relief and were carefully monitored to ensure regain of pre-surgery weight. All animals were allowed 3–4 weeks for virus expression before starting the experiment. Mice with missed injections, incomplete hits or expression outside of the target region were excluded from analysis after post hoc examination of mCherry or EYFP expression.
Viruses
AAV-FLEX-ChR2-mCherry (#20297-AAV1, Addgene), AAV-FLEX-ChR2-EYFP (#20298-AAV1, Addgene), AAV-FLEX-hM3Dq-mCherry (#44361-AAV9, Addgene), AAV-FLEX-mCherry (#50459-AAV8, Addgene), and AAV-FLEX-hGlyR-mCherry42 have been described previously.
AAV-FREX-hM3Dq-mCherry was made by inserting hM3Dq-mCherry (#44361, Addgene) in reverse orientation into the AAV-CAG-FREX vector between the double inverse rox12 and roxP sites at Bmt I and Spe I sites.78 AAV-CAG-FREX vector was kindly provided by T. Badea. The virus was packaged at the viral core of Boston Children’s Hospital with AAV serotype 2/8.
AAV-FREX-Cre was made by modifying the design of a Flp-dependent Cre vector, LF-FdCd.79 Briefly, the mTagBFP2 and iCre sequences were obtained from pCAGGS-mTagBFP2-T2A-iCre (#113837, Addgene), and rearranged by fusion PCR with NLS- and Rox site-containing primers to get the NLS-Frex-mTagBFP-iCre cassette. This NLS-Frex-mTagBFP-iCre cassette was then subcloned into the pAAV-nEF vector backbone (#55644, Addgene), between BamH I and EcoR V sites. The virus was packaged by the Lowell lab with AAV serotype 2/8.
Fiber implantation
Optical fibers (flat tip, diameter core = 200 µm, length = 6 mm, NA = 0.48; Doric Lenses Inc.) were implanted (coordinates from bregma AP: −0.70 mm, DV: −4.35 mm, ML: 0 mm) and anchored to the skull using dental acrylic. The location of the fiber tip was histologically verified after the experiments and animals with fiber placements outside of the target region were excluded from analysis.
Electrophysiology
Mice were deeply anesthetized with isoflurane and decapitated. Brains were quickly removed into ice-cold cutting solution consisting of (in mM): 92 choline chloride, 30 NaHCO3, 25 Glucose, 20 HEPES, 10 MgSO4, 2.5 KCl, 1.25 NaH2PO4, 5 sodium ascorbate, 3 sodium pyruvate, 2 thiourea, 0.5 CaCl2; oxygenated with 95% O2/5% CO2; measured osmolarity 310–320 mOsm/L. 300-µm-thick coronal slices were cut with a Campden vibratome (Model 7000smz-2) and incubated in oxygenated cutting solution at 34 °C for 10 min. Slices were then transferred to oxygenated aCSF consisting of (in mM): 126 NaCl, 21.4 NaHCO3, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 10 glucose at 34 °C for 45 min, and left to recover in the same solution at room temperature (20–24 °C) for at least 60 min prior to recordings. Unless otherwise specified, brain slices were prepared 3 hours into the light cycle.
Whole-cell patch clamp recordings
A single slice was placed in the recording chamber where it was continuously superfused with oxygenated aCSF at a constant rate of 2–3 ml/min. Recordings were obtained at room temperature from GFP-positive AgRP or hGlyR-expressing mCherry-tagged PVH neurons visualized with an upright microscope (SliceScope, Scientifica) equipped with infra-red differential interference contrast and fluorescence optics. For voltage-clamp recordings, borosilicate patch pipettes (3–5 MΩ) were filled with the internal solution consisting of (in mM): 135 CsMeSO3, 10 HEPES, 0.2 EGTA, 10 TEA, 4 MgCl2, 4 Na2-ATP, 0.4 Na2-GTP, 0.1 Spermine (pH 7.3 adjusted with CsOH; 295 mOsm/l). For current-clamp recordings, the internal solution contained (in mM): 135 KMeSO3, 10 HEPES, 1 EGTA, 0.1 CaCl2, 4 MgCl2, 4 Na2-ATP, 0.4 Na2-GTP, 5 Na2-phosphocreatine (pH 7.3 adjusted with KOH; 295 mOsm/l). Voltage-clamp recordings were performed in presence of bicuculline (10 µM) to block GABAergic synaptic transmission. All recordings were obtained with a Multiclamp 700B amplifier, Digidata 1550B converter and pClamp 10.7 software (Axon Instruments), sampled at 10 kHz, and filtered at 2 kHz. Access resistance (<30 MΩ) was continuously monitored by a voltage step and recordings were accepted for analysis if changes were <15%. All recordings were analyzed offline using Clampfit 10.7.
Light-evoked EPSCs
To photostimulate ChR2-expressing fibers, a LED light source (473 nm) was focused onto the back aperture of the microscope objective, producing widefield exposure around recorded cells. The light output was controlled by a programmable pulse stimulator (Master-8, A.M.P.I.) and pClamp 10.7 software. For le-qEPSC recordings, the membrane potential was clamped at Vh = −70 mV and the protocol consisted of one blue light pulse (473-nm wavelength, 5 ms) applied every 30 seconds. le-qEPSCs measurements were performed in aCSF containing 0 mM Ca2+ and 5 mM Sr2+. A 300 ms time window 25 ms after the light pulse was used for analysis of the evoked quantal events. Results from 8–10 sweeps were averaged for analysis. For le-EPSC recordings, the membrane potential was clamped at Vh = −70 mV and the detection protocol consisted of blue light pulses (473-nm wavelength, 5 ms). le-EPSCs with short latency (< 15 ms) upon light stimulation and low jitter were considered light-driven. For paired-pulse ratio (PPR), two light pulses were administered 50 ms apart during every 10 seconds, and the PPR was calculated as the ratio of the peak amplitude of the second le-EPSC divided by the peak amplitude of the first le-EPSC (le-EPSC2/le-EPSC1). For analysis, 8–10 sweeps were averaged. To calculate CV value, the standard deviation of le-EPSC amplitude (from 8–10 sweeps) was divided by the mean le-EPSC amplitude from the same sweeps. To record light-evoked NMDAR-mediated EPSCs, the membrane potential was clamped at Vh = +40 mV and the protocol consisted of one blue light pulse (473-nm wavelength, 5 ms) applied during a 10 s sweep. NMDAR-mediated EPSCs were calculated as the peak of the EPSC at +40 mV at 25–40 ms after light stimulation, at which time the AMPAR-mediated component has decayed. Light-evoked AMPAR/NMDAR ratio was calculated as the ratio of the peak amplitude of the AMPAR-mediated le-EPSCs at Vh = −70 mV to the magnitude of the NMDAR-mediated le-EPSCs recorded at Vh = +40 mV. For analysis, 8–10 sweeps were averaged.
Spontaneous EPSCs
sEPSCs were recorded in whole-cell voltage-clamp mode, with membrane potential clamped at Vh = −70 mV.
Current-clamp recordings
To confirm the hGlyR-induced inhibition, neurons were clamped at Ih = 0 pA. IVM (1 µM) was added to bath solution, and effects on membrane potential and firing rates were determined.
Protocols
To assess the effects of fasting on glutamatergic synaptic activity onto AgRP neurons, fed mice had ad libitum access to food. Fasted mice were overnight food deprived (for 16 hours) and sacrificed on the next morning (fasted group), after ad libitum refeeding for one day (2nd Day Refed group), after ad libitum refeeding for six days (7th Day Refed group), or after six days of the caloric maintenance paradigm (7th Day (CMP) group). To assess the effects of a moderate caloric reduction, mice were provided 75% of the amount of food that they consumed on average in ad libitum control conditions for 7 days (the caloric restriction paradigm (CR) group). To assess the effects of PVHTRH inhibition, hGlyR-expressing mice were injected IP with IVM (5 mg/kg) or VEH (vehicle; propylene glycol:glycerol formal 7:3) before food removal and fasted for 16 hours. Brain slices from all the listed experimental groups were prepared 3 hours into the light cycle. To assess the effects of PVHTRH neuron stimulation, hM3Dq-expressing mice received an IP injection of either CNO (Clozapine N-oxide -dihydrochloride; 3 mg CNO/kg), CNO with (+)-MK-801 maleate (0.5 mg/kg) or vehicle (0.9% saline) 2 hours into the light cycle. Brain slices were prepared 4 hours after the injection.
Ex vivo high frequency photostimulation (HFpS)
A single brain slice was placed in the recording chamber where it was continuously superfused with a standard oxygenated aCSF. The ARC region with ChR2-expressing axonal terminals was focused under the 40x objective and the photostimulation was initiated. Blue light pulses (473-nm wavelength, 5 ms) were applied repeatedly over 10 s followed by a 10 s break, for 10 min at a frequency of 50 Hz. After the stimulation, brain slices were kept in a standard oxygenated aCSF at room temperature for at least 2–3 h prior to recordings. In order to assess effects of NMDA receptors blockade, D-AP5 (50 µM) was added to aCSF during the HFpS. Immediately after the photostimulation, brain slices were moved to drug-free aCSF for 2–3 h.
Behavioral protocols
All mice were singly housed and habituated to food hoppers at least 7 days before the experiment. Animals were handled for 3 consecutive days before the assay to acclimate them to the experimental procedure. Food intake studies were performed in home cages with access to chow ad libitum, unless otherwise specified.
Food intake
The day before the test, mice were provided with fresh cages to avoid leftover food spilling in the bedding. For food intake assessments, animals received an IP injection of either CNO, CNO with MK-801, CNO with IVM, or their vehicle 3 h into the light cycle. Vehicle was delivered at the same volume to maintain consistency in the experiment. Food intake was manually measured after IP injection.
Post-fast refeeding
The day before the experiment, mice were provided with fresh cages without food at the onset of the dark cycle. Food was reintroduced after 16 h of fasting and manually measured. To assess the effects of hGlyR-induced inhibition of PVHTRH or DMHVglut2 neurons, animals received an IP injection of either IVM or vehicle one hour before the onset of the dark cycle. After 16 h of fasting, food was provided and manually measured. In the caloric maintenance paradigm (CMP) experiments, after 16 h of fasting, mice were provided with 3.8 g of NCD per day; the amount of food they consumed on average in ad libitum. After six days, food was provided ad libitum and manually measured. To assess body weight regain, mice were weighted directly before fasting, after 16 h of fasting and on the following days during refeeding (2 – 3 h into the light phase).
In vivo high frequency photostimulation (HFpS)
7 days before the experiment, singly housed mice with implanted optical fibers were connected to a fiber-optic patch cord (diameter core = 200 µm, NA = 0.48; Doric Lenses Inc.) connected to a rotary joint (Doric Lenses Inc.) for adaptation. The day before the test, mice were provided with fresh cages. On the day of the test, 2 h after the beginning of the light phase, the attached fiber-optic patch cord was replaced by a new one and mice received an IP injection of either MK-801 (0.25 mg/kg) or its vehicle (0.9% saline). After 1 hour, the HFpS protocol was started (50-Hz stimulation applied for 10 min, 473-nm wavelength, 5-ms light pulses, 10-s on, 10-s off). The light output power at the tip of the patch cord was adjusted to be 18–20 mW measured with an optical power meter. Food intake was manually measured after the beginning of the HFpS protocol.
Long-term body weight measurements
For long-term body weight assay, mice were injected IP with either CNO or CNO with MK-801every two weeks and received vehicle IP injections every other week for a total period of 6 weeks from the first injection. Body weight was monitored every week prior to injections.
Immunohistochemistry
Mice were deeply anesthetized and perfused transcardially with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA). Brains were removed, postfixed in PFA for 24 hours at 4 °C and transferred into 20% sucrose at 4 °C overnight. Subsequently, brains were cut coronally into 40-μm thick sections on a freezing microtome, collected in bins containing anti-freeze solution and stored at −20 °C until further use. Brain sections were washed (3 × 10 min) in PBS with 0.4% Triton X-100 (PBST) and blocked in 2% normal donkey serum in PBST for 1 h at room temperature. Brain sections were then incubated with the primary antibody overnight at 4 °C. Following primary antibodies were used: monoclonal rat anti-mCherry antibody (1:1000, #M11217, Thermo Fisher/Invitrogen), polyclonal chicken anti-GFP antibody (1:1000, #ab13970, Abcam), polyclonal rabbit anti-cFos antibody (1:1000, #226 003, Synaptic Systems). Of note, anti-GFP antibody was used to detect EYFP. The next morning sections were washed in PBST (3 × 10 min) and incubated with the secondary antibody for 1 h at room temperature, protected from light. Following secondary antibodies were used: Alexa Fluor 594 donkey anti-rat antibody (1:1000, #A21209, Thermo Fisher/Invitrogen), Alexa Fluor 488 goat anti-chicken antibody (1:1000, #A11039, Thermo Fisher/Invitrogen), Alexa Fluor 488 donkey anti-rabbit antibody (1:1000, #A21206, Thermo Fisher/Invitrogen). Brain sections were then washed with PBST (3 × 10 min), mounted using Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories) and stored in the dark at 4°C until imaging.
Fluorescent in situ hybridization (FISH)
Brains were harvested and post-fixed as described above, cut coronally into 20- or 40-μm thick sections on a freezing microtome, and stored in cryoprotectant at −20 °C until further use. For the detection of Trh, Pacap, Fos, and Agrp mRNA, fluorescent RNAscope® (ACD; Advanced Cell Diagnostics Inc., Hayward, CA) was used. The Trh probe targeted region 128–1280, access # NM_009426.3; the Pacap probe targeted region 676 – 1859, access # NM_009625.2; the Fos probe targeted region 407–1427, access # NM_010234.2; the Agrp probe targeted region 11 – 764, access # NM_001271806.1. On the day before the assay, brain slices were mounted onto SuperFrost Plus Gold slides (ThermoFisher), dried at room temperature and incubated at 60 °C overnight. On the next day, slides were incubated in 4% PFA for 15 min at 4 °C. Afterwards slides were dehydrated in Ethanol (50%, 70%, 100% EtOH; 5 min each) and air dried for 5 min. All incubation steps were performed at 40 °C using a humidified chamber and the ACD HybEz oven. Slides were incubated for 7 min in ACD, submerged in Target Retrieval (#322000) at a temperature of 98.5 – 99.5°C for 8 min, and rinsed twice in autoclaved MilliQ-purified water. Slides were then dehydrated in 100% ethanol and allowed to air dry for 5 min. Subsequently, a hydrophobic barrier was created around the brain slices using an ImmEdge hydrophobic barrier pen (#310018). Slides were then incubated with Protease Plus (#322380) for 35 min. Manufacturer’s instructions for the RNAscope Multiplex Fluorescent Detection Kit v2 (#323110) were followed for the hybridization, amplification detection steps. For combining FISH with immunohistochemistry for mCherry, brain slices were moved into the washing phase after the amplification phase, and immunohistochemistry was processed as described above. Sections were counterstained with DAPI, coverslipped with ProLong Gold Antifade Mountant (Cat. No. P36931; ThermoFisher), and stored in dark at 4°C until imaging. For analysis of Fos expression, representative images of PVH and ARC were taken, and processed using ImageJ software. Background was subtracted and images were cropped. In the PVH, the number of Fos-expressing cells that were positive for both Trh and Pacap were counted manually. In the ARC, the number of Fos-expressing cells that were positive for Agrp were counted manually.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism 9.0 software. No statistical method was used to predetermine sample size. For electrophysiology experiments, n values represent the number of recorded neurons and N values represent the number of mice the brain slices were obtained from. For behavioral experiments, n values represent the final number of validated mice. Randomization and blinding methods were not used. Single comparisons between two groups were made by two-tailed unpaired Student’s t-test. Comparisons between multiple groups were performed using ordinary or repeated-measures one-way ANOVA followed by Tukey’s multiple comparisons test. Ordinary two-way ANOVA followed by Sidak’s or Tukey’s multiple comparisons test (as recommended) was applied to determine the statistical differences among groups across different time points. All data presented met the assumptions of the statistical test employed. For all statistical tests, p < 0.05 was considered significant. Calculated values are presented as mean ± s.e.m. (standard error of the mean), unless indicated otherwise. Statistical significance is represented by * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Statistical tests and parameters are reported in figure legends.
Supplementary Material
Key resource table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| rat monoclonal anti-mCherry | Thermofisher | M11217 |
| chicken polyclonal anti-GFP | Abcam | ab13970 |
| rabbit polyclonal anti-c-Fos | Synaptic System | 226 003 |
| donkey anti-rat Alexa594 | Thermofisher | A-21209 |
| goat anti-chicken Alexa488 | Thermofisher | A-11039 |
| donkey anti-rabbit Alexa488 | Thermofisher | A-21206 |
| Bacterial and Virus Strains | ||
| pAAV-EF1a-double floxed-hChR2(H134R)-mCherry-WPRE-HGHpA | Addgene | RRID: Addgene_20297 |
| pAAV-EF1a-double floxed-hChR2(H134R)-EYFP-WPRE-HGHpA | Addgene | RRID: Addgene_20298 |
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Addgene | RRID: Addgene_44361 |
| pAAV-hSyn-DIO-mCherry | Addgene | RRID: Addgene_50459 |
| pAAV-FLEX-hGlyR-mCherry | Todd et al., 2018 | N/A |
| pAAV-FREX-hM3Dq-mCherry | This paper | N/A |
| pAAV-FREX-Cre | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Bicuculline | Sigma-Aldrich | Cat# 14343 |
| Ivermectin | Sigma-Aldrich | I8898 |
| Clozapine N-oxide (CNO) | Hello Bio | Cat#HB6149 |
| (+)-MK 801 maleate | Hello Bio | HB0004 |
| D-AP5 | Alomone labs | Cat# D-145 |
| CNQX disodium salt | Hello Bio | HB0205 |
| Critical commercial assays | ||
| RNAscope Multiplex Fluorescent Detection Kit v2 | ACD bio / Bio-Techne | Cat# 323 110 |
| RNAscope Target Retrieval Reagents | ACD bio / Bio-Techne | Cat# 322 000 |
| Protease Plus | ACD bio / Bio-Techne | Cat# 322 380 |
| TSA PLUS Fluorescence Kits | Perkin-Elmer | Cat# NEL760001KT |
| Experimental models: Organisms/strains | ||
| Mouse: B6; 129S-Trhtm1.1(cre)Mjkr/LowlJ | The Jackson Laboratory | RRID: IMSR_JAX:032468 |
| Mouse: B6J.129S6(FVB)-Slc17a6tm2(cre)Lowl/MwarJ | The Jackson Laboratory | RRID: IMSR_JAX:028863 |
| Mouse: B6.FVB-Tg(Npy-hrGFP)1Lowl/J | The Jackson Laboratory | RRID: IMSR_JAX:006417 |
| Mouse: Agrptm1(cre)Lowl/J | The Jackson Laboratory | RRID: IMSR_JAX:012899 |
| Mouse: B6J.129S6(FVB)-Slc17a6tm2(cre)Lowl/MwarJ | The Jackson Laboratory | RRID: IMSR_JAX:028863 |
| Mouse: Slc17a6tm1Lowl/J | The Jackson Laboratory | RRID: IMSR_JAX:012898 |
| Mouse: Trh-p2a-Dre | This paper | N/A |
| Software & algorithms | ||
| Clampfit | Molecular Devices | https://www.moleculardevices.com |
| Illustrator CC | Adobe Systems | https://www.adobe.com/products/illustrator |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij |
| pCLAMP 10.7 | Molecular Devices | https://www.moleculardevices.com |
| Prism | GraphPad | https://graphpad.com/scientificsoftware/prism |
| Other | ||
| Laser | Doric Lenses Inc. | https://optics.doriclenses.com |
| smFISH probe: Mm-Trh-C1 (probe region: 128 – 1280 (Accession No. NM_009426.3)) | ACD bio / Bio-Techne | Cat# 436 811 |
| smFISH probe: Mm-Adcyap1-C2 (probe region: 676 – 1859 (Accession No. NM_009625.2)) | ACD bio / Bio-Techne | Cat# 405 911-C2 |
| smFISH probe: Mm-Fos-C3 (probe region: 407–1427 (Accession No. NM_010234.2)) | ACD bio / Bio-Techne | Cat# 316921-C3 |
| smFISH probe: Mm-Agrp-C2 (probe region: 11–764 (Accession No. NM_001271806.1)) | ACD bio / Bio-Techne | Cat# 400711-C2 |
| DAPI | Vector Laboratories | Cat# H-1200 |
| ProLong™ Gold Antifade Mountant with DAPI | Thermo Fisher | Cat# P36931 |
Highlights:
Weight loss upon caloric deprivation activates PVHTRH neurons that co-express PACAP
Activated PVHTRH neurons increase the number of active PVHTRH➔AgRP neuron synapses
Potentiation of excitatory PVHTRH➔AgRP synapses lasts until lost weight is regained
PVHTRH➔AgRP circuit activity is necessary and sufficient for driving weight (re)gain
Acknowledgments
We thank Yoav Livneh for comments on the manuscript; all members of the Fenselau lab for helpful discussions; and Ursula Lichtenberg, Karina Schöfisch, Hella Brönneke, and Ramona Braun for their excellent administrative help. Funding: H.F. received funding within the Excellence Initiative by German Federal and State Governments (CECAD) and from a cooperation agreement with Novo Nordisk, Denmark. S.D. received funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; 214362475 / GRK1873/2). B.B.L. received funding from NIH (grants: R01 DK096010, R01 DK075632, R01 DK089044, R01 DK122976, P30 DK046200, P30 DK057521). The Boston Children’s Hospital Viral Core was supported by NEI (grant P30 5P30EY012196).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Inclusion and Diversity Statement
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community.
References
- 1.Nordmo M, Danielsen YS, and Nordmo M (2020). The challenge of keeping it off, a descriptive systematic review of high-quality, follow-up studies of obesity treatments. Obes Rev 21, e12949. 10.1111/obr.12949. [DOI] [PubMed] [Google Scholar]
- 2.Berthoud HR, Seeley RJ, and Roberts SB (2021). Physiology of Energy Intake in the Weight-Reduced State. Obesity (Silver Spring) 29 Suppl 1, S25–S30. 10.1002/oby.23080. [DOI] [PubMed] [Google Scholar]
- 3.Andermann ML, and Lowell BB (2017). Toward a Wiring Diagram Understanding of Appetite Control. Neuron 95, 757–778. 10.1016/j.neuron.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, and van den Pol AN (2016). Hypothalamic arcuate nucleus tyrosine hydroxylase neurons play orexigenic role in energy homeostasis. Nat Neurosci 19, 1341–1347. 10.1038/nn.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 121, 1424–1428. 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aponte Y, Atasoy D, and Sternson SM (2011). AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci 14, 351–355. 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atasoy D, Betley JN, Su HH, and Sternson SM (2012). Deconstruction of a neural circuit for hunger. Nature 488, 172–177. 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fenselau H, Campbell JN, Verstegen AM, Madara JC, Xu J, Shah BP, Resch JM, Yang Z, Mandelblat-Cerf Y, Livneh Y, and Lowell BB (2017). A rapidly acting glutamatergic ARC-->PVH satiety circuit postsynaptically regulated by alpha-MSH. Nat Neurosci 20, 42–51. 10.1038/nn.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jais A, Paeger L, Sotelo-Hitschfeld T, Bremser S, Prinzensteiner M, Klemm P, Mykytiuk V, Widdershooven PJM, Vesting AJ, Grzelka K, et al. (2020). PNOC(ARC) Neurons Promote Hyperphagia and Obesity upon High-Fat-Diet Feeding. Neuron 106, 1009–1025 e1010. 10.1016/j.neuron.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luquet S, Perez FA, Hnasko TS, and Palmiter RD (2005). NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 310, 683–685. 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 11.Zhu C, Jiang Z, Xu Y, Cai ZL, Jiang Q, Xu Y, Xue M, Arenkiel BR, Wu Q, Shu G, and Tong Q (2020). Profound and redundant functions of arcuate neurons in obesity development. Nat Metab 2, 763–774. 10.1038/s42255-020-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Bartolome CL, Low CS, Yi X, Chien CH, Wang P, and Kong D (2018). Genetic identification of leptin neural circuits in energy and glucose homeostases. Nature 556, 505–509. 10.1038/s41586-018-0049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu AW, Kaelin CB, Morton GJ, Ogimoto K, Stanhope K, Graham J, Baskin DG, Havel P, Schwartz MW, and Barsh GS (2005). Effects of hypothalamic neurodegeneration on energy balance. PLoS Biol 3, e415. 10.1371/journal.pbio.0030415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhan C, Zhou J, Feng Q, Zhang JE, Lin S, Bao J, Wu P, and Luo M (2013). Acute and long-term suppression of feeding behavior by POMC neurons in the brainstem and hypothalamus, respectively. J Neurosci 33, 3624–3632. 10.1523/JNEUROSCI.2742-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vong L, Ye C, Yang Z, Choi B, Chua S Jr., and Lowell BB (2011). Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154. 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varela L, Stutz B, Song JE, Kim JG, Liu ZW, Gao XB, and Horvath TL (2021). Hunger-promoting AgRP neurons trigger an astrocyte-mediated feed-forward autoactivation loop in mice. J Clin Invest 131. 10.1172/JCI144239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, and Horvath TL (2004). Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 304, 110–115. 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 18.Suyama S, Ralevski A, Liu ZW, Dietrich MO, Yada T, Simonds SE, Cowley MA, Gao XB, Diano S, and Horvath TL (2017). Plasticity of calcium-permeable AMPA glutamate receptors in Pro-opiomelanocortin neurons. Elife 6:e25755. 10.7554/eLife.25755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Atasoy D, Su HH, and Sternson SM (2011). Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 146, 992–1003. 10.1016/j.cell.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu T, Kong D, Shah BP, Ye C, Koda S, Saunders A, Ding JB, Yang Z, Sabatini BL, and Lowell BB (2012). Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. Neuron 73, 511–522. 10.1016/j.neuron.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong D, Dagon Y, Campbell JN, Guo Y, Yang Z, Yi X, Aryal P, Wellenstein K, Kahn BB, Sabatini BL, and Lowell BB (2016). A Postsynaptic AMPK-->p21-Activated Kinase Pathway Drives Fasting-Induced Synaptic Plasticity in AgRP Neurons. Neuron 91, 25–33. 10.1016/j.neuron.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rau AR, and Hentges ST (2021). Energy state alters regulation of proopiomelanocortin neurons by glutamatergic ventromedial hypothalamus neurons: pre- and postsynaptic mechanisms. J Neurophysiol 125, 720–730. 10.1152/jn.00359.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rau AR, and Hentges ST (2019). GABAergic Inputs to POMC Neurons Originating from the Dorsomedial Hypothalamus Are Regulated by Energy State. J Neurosci 39, 6449–6459. 10.1523/JNEUROSCI.3193-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rau AR, and Hentges ST (2017). The Relevance of AgRP Neuron-Derived GABA Inputs to POMC Neurons Differs for Spontaneous and Evoked Release. J Neurosci 37, 7362–7372. 10.1523/JNEUROSCI.0647-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeltser LM, Seeley RJ, and Tschop MH (2012). Synaptic plasticity in neuronal circuits regulating energy balance. Nat Neurosci 15, 1336–1342. 10.1038/nn.3219. [DOI] [PubMed] [Google Scholar]
- 26.Sumithran P, Prendergast LA, Delbridge E, Purcell K, Shulkes A, Kriketos A, and Proietto J (2011). Long-term persistence of hormonal adaptations to weight loss. N Engl J Med 365, 1597–1604. 10.1056/NEJMoa1105816. [DOI] [PubMed] [Google Scholar]
- 27.Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, Leranth C, Toran-Allerand D, Priest CA, Roberts JL, et al. (2007). Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med 13, 89–94. 10.1038/nm1525. [DOI] [PubMed] [Google Scholar]
- 28.Gyengesi E, Liu ZW, D’Agostino G, Gan G, Horvath TL, Gao XB, and Diano S (2010). Corticosterone regulates synaptic input organization of POMC and NPY/AgRP neurons in adult mice. Endocrinology 151, 5395–5402. 10.1210/en.2010-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krashes MJ, Shah BP, Madara JC, Olson DP, Strochlic DE, Garfield AS, Vong L, Pei H, Watabe-Uchida M, Uchida N, et al. (2014). An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 507, 238–242. 10.1038/nature12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garfield AS, Shah BP, Burgess CR, Li MM, Li C, Steger JS, Madara JC, Campbell JN, Kroeger D, Scammell TE, et al. (2016). Dynamic GABAergic afferent modulation of AgRP neurons. Nat Neurosci 19, 1628–1635. 10.1038/nn.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aklan I, Sayar Atasoy N, Yavuz Y, Ates T, Coban I, Koksalar F, Filiz G, Topcu IC, Oncul M, Dilsiz P, et al. (2020). NTS Catecholamine Neurons Mediate Hypoglycemic Hunger via Medial Hypothalamic Feeding Pathways. Cell Metab 31, 313–326 e315. 10.1016/j.cmet.2019.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith MA, Choudhury AI, Glegola JA, Viskaitis P, Irvine EE, de Campos Silva PCC, Khadayate S, Zeilhofer HU, and Withers DJ (2020). Extrahypothalamic GABAergic nociceptin-expressing neurons regulate AgRP neuron activity to control feeding behavior. J Clin Invest 130, 126–142. 10.1172/JCI130340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jais A, and Bruning JC (2022). Arcuate Nucleus-Dependent Regulation of Metabolism-Pathways to Obesity and Diabetes Mellitus. Endocr Rev 43, 314–328. 10.1210/endrev/bnab025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myers MG Jr., Affinati AH, Richardson N, and Schwartz MW (2021). Central nervous system regulation of organismal energy and glucose homeostasis. Nat Metab 3, 737–750. 10.1038/s42255-021-00408-5. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi KA, and Cone RD (2005). Fasting induces a large, leptin-dependent increase in the intrinsic action potential frequency of orexigenic arcuate nucleus neuropeptide Y/Agouti-related protein neurons. Endocrinology 146, 1043–1047. 10.1210/en.2004-1397. [DOI] [PubMed] [Google Scholar]
- 36.Betley JN, Xu S, Cao ZFH, Gong R, Magnus CJ, Yu Y, and Sternson SM (2015). Neurons for hunger and thirst transmit a negative-valence teaching signal. Nature 521, 180–185. 10.1038/nature14416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vardy E, Robinson JE, Li C, Olsen RHJ, DiBerto JF, Giguere PM, Sassano FM, Huang XP, Zhu H, Urban DJ, et al. (2015). A New DREADD Facilitates the Multiplexed Chemogenetic Interrogation of Behavior. Neuron 86, 936–946. 10.1016/j.neuron.2015.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, et al. (2005). Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci 8, 1289–1291. 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 39.van den Pol AN, Yao Y, Fu LY, Foo K, Huang H, Coppari R, Lowell BB, and Broberger C (2009). Neuromedin B and gastrin-releasing peptide excite arcuate nucleus neuropeptide Y neurons in a novel transgenic mouse expressing strong Renilla green fluorescent protein in NPY neurons. J Neurosci 29, 4622–4639. 10.1523/JNEUROSCI.3249-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacAskill AF, Cassel JM, and Carter AG (2014). Cocaine exposure reorganizes cell type- and input-specific connectivity in the nucleus accumbens. Nat Neurosci 17, 1198–1207. 10.1038/nn.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, and Bonci A (2012). Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76, 790–803. 10.1016/j.neuron.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Todd WD, Fenselau H, Wang JL, Zhang R, Machado NL, Venner A, Broadhurst RY, Kaur S, Lynagh T, Olson DP, et al. (2018). A hypothalamic circuit for the circadian control of aggression. Nat Neurosci 21, 717–724. 10.1038/s41593-018-0126-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lynagh T, and Lynch JW (2010). An improved ivermectin-activated chloride channel receptor for inhibiting electrical activity in defined neuronal populations. J Biol Chem 285, 14890–14897. 10.1074/jbc.M110.107789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lerchner W, Xiao C, Nashmi R, Slimko EM, van Trigt L, Lester HA, and Anderson DJ (2007). Reversible silencing of neuronal excitability in behaving mice by a genetically targeted, ivermectin-gated Cl- channel. Neuron 54, 35–49. 10.1016/j.neuron.2007.02.030. [DOI] [PubMed] [Google Scholar]
- 45.Vella KR, Ramadoss P, Lam FS, Harris JC, Ye FD, Same PD, O’Neill NF, Maratos-Flier E, and Hollenberg AN (2011). NPY and MC4R signaling regulate thyroid hormone levels during fasting through both central and peripheral pathways. Cell Metab 14, 780–790. 10.1016/j.cmet.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lechan RM, and Fekete C (2006). The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res 153, 209–235. 10.1016/S0079-6123(06)53012-2. [DOI] [PubMed] [Google Scholar]
- 47.Holtmaat A, and Svoboda K (2009). Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10, 647–658. 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 48.Luscher C, and Malenka RC (2012). NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol 4. 10.1101/cshperspect.a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Campbell JN, Macosko EZ, Fenselau H, Pers TH, Lyubetskaya A, Tenen D, Goldman M, Verstegen AM, Resch JM, McCarroll SA, et al. (2017). A molecular census of arcuate hypothalamus and median eminence cell types. Nat Neurosci 20, 484–496. 10.1038/nn.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hrabovszky E, Wittmann G, Turi GF, Liposits Z, and Fekete C (2005). Hypophysiotropic thyrotropin-releasing hormone and corticotropin-releasing hormone neurons of the rat contain vesicular glutamate transporter-2. Endocrinology 146, 341–347. 10.1210/en.2004-0856. [DOI] [PubMed] [Google Scholar]
- 51.Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, et al. (2009). Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39. 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakajima K, Cui Z, Li C, Meister J, Cui Y, Fu O, Smith AS, Jain S, Lowell BB, Krashes MJ, and Wess J (2016). Corrigendum: Gs-coupled GPCR signalling in AgRP neurons triggers sustained increase in food intake. Nat Commun 7, 11019. 10.1038/ncomms11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang D, He X, Zhao Z, Feng Q, Lin R, Sun Y, Ding T, Xu F, Luo M, and Zhan C (2015). Whole-brain mapping of the direct inputs and axonal projections of POMC and AgRP neurons. Front Neuroanat 9, 40. 10.3389/fnana.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, et al. (2003). The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37, 649–661. 10.1016/s0896-6273(03)00063-1. [DOI] [PubMed] [Google Scholar]
- 55.Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, et al. (2007). Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab 5, 438–449. 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 56.van den Top M, Lee K, Whyment AD, Blanks AM, and Spanswick D (2004). Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus. Nat Neurosci 7, 493–494. 10.1038/nn1226. [DOI] [PubMed] [Google Scholar]
- 57.Wang P, Loh KH, Wu M, Morgan DA, Schneeberger M, Yu X, Chi J, Kosse C, Kim D, Rahmouni K, et al. (2020). A leptin-BDNF pathway regulating sympathetic innervation of adipose tissue. Nature 583, 839–844. 10.1038/s41586-020-2527-y. [DOI] [PubMed] [Google Scholar]
- 58.Betley JN, Cao ZF, Ritola KD, and Sternson SM (2013). Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell 155, 1337–1350. 10.1016/j.cell.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goldstein N, McKnight AD, Carty JRE, Arnold M, Betley JN, and Alhadeff AL (2021). Hypothalamic detection of macronutrients via multiple gut-brain pathways. Cell Metab 33, 676–687 e675. 10.1016/j.cmet.2020.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bai L, Mesgarzadeh S, Ramesh KS, Huey EL, Liu Y, Gray LA, Aitken TJ, Chen Y, Beutler LR, Ahn JS, et al. (2019). Genetic Identification of Vagal Sensory Neurons That Control Feeding. Cell 179, 1129–1143 e1123. 10.1016/j.cell.2019.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beutler LR, Chen Y, Ahn JS, Lin YC, Essner RA, and Knight ZA (2017). Dynamics of Gut-Brain Communication Underlying Hunger. Neuron 96, 461–475 e465. 10.1016/j.neuron.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Su Z, Alhadeff AL, and Betley JN (2017). Nutritive, Post-ingestive Signals Are the Primary Regulators of AgRP Neuron Activity. Cell Rep 21, 2724–2736. 10.1016/j.celrep.2017.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fekete C, and Lechan RM (2014). Central regulation of hypothalamic-pituitary-thyroid axis under physiological and pathophysiological conditions. Endocr Rev 35, 159–194. 10.1210/er.2013-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anastasiou CA, Karfopoulou E, and Yannakoulia M (2015). Weight regaining: From statistics and behaviors to physiology and metabolism. Metabolism 64, 1395–1407. 10.1016/j.metabol.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 65.Tao W, Lee J, Chen X, Diaz-Alonso J, Zhou J, Pleasure S, and Nicoll RA (2021). Synaptic memory requires CaMKII. Elife 10:e60360. 10.7554/eLife.60360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Incontro S, Diaz-Alonso J, Iafrati J, Vieira M, Asensio CS, Sohal VS, Roche KW, Bender KJ, and Nicoll RA (2018). The CaMKII/NMDA receptor complex controls hippocampal synaptic transmission by kinase-dependent and independent mechanisms. Nat Commun 9, 2069. 10.1038/s41467-018-04439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Herring BE, and Nicoll RA (2016). Long-Term Potentiation: From CaMKII to AMPA Receptor Trafficking. Annu Rev Physiol 78, 351–365. 10.1146/annurev-physiol-021014-071753. [DOI] [PubMed] [Google Scholar]
- 68.Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, and Nicoll RA (1995). Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci U S A 92, 11175–11179. 10.1073/pnas.92.24.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, and Hardie DG (2005). Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2, 9–19. 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 70.Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ, Witters LA, Kemp BE, and Means AR (2008). Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab 7, 377–388. 10.1016/j.cmet.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 71.Krashes MJ, Shah BP, Koda S, and Lowell BB (2013). Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metab 18, 588–595. 10.1016/j.cmet.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakajima K, Cui Z, Li C, Meister J, Cui Y, Fu O, Smith AS, Jain S, Lowell BB, Krashes MJ, and Wess J (2016). Gs-coupled GPCR signalling in AgRP neurons triggers sustained increase in food intake. Nat Commun 7, 10268. 10.1038/ncomms10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brachtendorf S, Eilers J, and Schmidt H (2015). A use-dependent increase in release sites drives facilitation at calretinin-deficient cerebellar parallel-fiber synapses. Front Cell Neurosci 9, 27. 10.3389/fncel.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kohara K, Ogura A, Akagawa K, and Yamaguchi K (2001). Increase in number of functional release sites by cyclic AMP-dependent protein kinase in cultured neurons isolated from hippocampal dentate gyrus. Neurosci Res 41, 79–88. 10.1016/s0168-0102(01)00267-x. [DOI] [PubMed] [Google Scholar]
- 75.Tong Q, Ye CP, Jones JE, Elmquist JK, and Lowell BB (2008). Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11, 998–1000. 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tong Q, Ye C, McCrimmon RJ, Dhillon H, Choi B, Kramer MD, Yu J, Yang Z, Christiansen LM, Lee CE, et al. (2007). Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab 5, 383–393. 10.1016/j.cmet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quadros RM, Miura H, Harms DW, Akatsuka H, Sato T, Aida T, Redder R, Richardson GP, Inagaki Y, Sakai D, et al. (2017). Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol 18, 92. 10.1186/s13059-017-1220-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chuang K, Nguyen E, Sergeev Y, and Badea TC (2015). Novel Heterotypic Rox Sites for Combinatorial Dre Recombination Strategies. G3 (Bethesda) 6, 559–571. 10.1534/g3.115.025841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jung H, Kim SW, Kim M, Hong J, Yu D, Kim JH, Lee Y, Kim S, Woo D, Shin HS, et al. (2019). Noninvasive optical activation of Flp recombinase for genetic manipulation in deep mouse brain regions. Nat Commun 10, 314. 10.1038/s41467-018-08282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for the graphs are provided in Data S1.
No original code was generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.