

Key Points
-
•
We developed a machine-learning algorithm to guide differential diagnosis of BMF.
-
•
Acquired vs inherited prediction relied on 25 variables recorded through a comprehensive physical and laboratory evaluation at the time of first evaluation.
Visual Abstract
Abstract
The choice to postpone treatment while awaiting genetic testing can result in significant delay in definitive therapies in patients with severe pancytopenia. Conversely, the misdiagnosis of inherited bone marrow failure (BMF) can expose patients to ineffectual and expensive therapies, toxic transplant conditioning regimens, and inappropriate use of an affected family member as a stem cell donor. To predict the likelihood of patients having acquired or inherited BMF, we developed a 2-step data-driven machine-learning model using 25 clinical and laboratory variables typically recorded at the initial clinical encounter. For model development, patients were labeled as having acquired or inherited BMF depending on their genomic data. Data sets were unbiasedly clustered, and an ensemble model was trained with cases from the largest cluster of a training cohort (n = 359) and validated with an independent cohort (n = 127). Cluster A, the largest group, was mostly immune or inherited aplastic anemia, whereas cluster B comprised underrepresented BMF phenotypes and was not included in the next step of data modeling because of a small sample size. The ensemble cluster A–specific model was accurate (89%) to predict BMF etiology, correctly predicting inherited and likely immune BMF in 79% and 92% of cases, respectively. Our model represents a practical guide for BMF diagnosis and highlights the importance of clinical and laboratory variables in the initial evaluation, particularly telomere length. Our tool can be potentially used by general hematologists and health care providers not specialized in BMF, and in under-resourced centers, to prioritize patients for genetic testing or for expeditious treatment.
Gutierrez-Rodrigues et al report on the development of a machine-learning model to distinguish inherited bone marrow failure syndromes from acquired aplastic anemia. The model performed well on a well-defined cohort of patients and is confirmed in an independent cohort with 89% accuracy in predicting inherited vs immune cases. The most important clinical variable in the score is telomere length. The authors have provided a link to an online tool to make the analysis available for practitioners.
Introduction
Bone marrow failure (BMF) syndromes include a spectrum of rare diseases characterized by impaired hematopoiesis and blood cytopenias.1, 2, 3 BMF is caused by different pathophysiologic mechanisms, broadly classified as acquired or inherited. In acquired cases, the hematopoietic failure is caused by an immune-mediated destruction of hematopoietic stem and progenitor cells in the marrow.2 Inherited BMF syndromes (IBMFSs) are a heterogeneous group of diseases caused by pathogenic germ line variants in a variety of genes critical for key pathways in the maintenance, self-renewal, differentiation, and genomic stability of hematopoietic stem and progenitor cells.1,3,4 Telomere biology disorders (TBDs), ribosomopathies, and Fanconi anemia (FA) are examples of classical IBMFSs.3, 4, 5
Treatment decisions and donor selection for hematopoietic cell transplant are dependent on the underlying BMF etiology, making it imperative to distinguish between inherited and acquired diseases. It is often feasible to readily diagnose IBMFSs in children if they present with a family history and typical congenital anomalies. However, many patients with IBMFSs do not have an informative pedigree, lack classical physical characteristics, or present in adulthood. Notably, cytopenias due to various etiologies have similar clinical presentations.1,3,6,7 In contrast, the acquired BMF diagnosis is by exclusion, especially if patients are older or lack a family history and typical features associated with inherited phenotypes. The definitive approach to IBMFS diagnosis is genetic testing, which is expensive and not routinely available worldwide, particularly in low-resource settings.
Machine learning, a subdomain of artificial intelligence, has been increasingly applied in health care to identify patterns and markers of complex diseases toward improved disease classification, risk stratification, and treatment decisions (see supplemental Material for a quick guide to machine learning, available on the Blood website).8, 9, 10, 11, 12 In this field, computer algorithms learn from examples rather than a preestablished set of statistical rules, which can unveil hidden associations and predict outcomes. Among the different types of machine learning, unsupervised algorithms focus on identifying patterns and clusters within a data set, and supervised algorithms learn to automatically predict specific outcomes (labels) based on a set of exemplars.11,13 In hematology, machine learning has been used to improve risk stratification, the diagnosis and prognosis of lymphoid and myeloid malignancies, and mortality prediction in sickle cell disease.8,13, 14, 15, 16, 17, 18 However, no model has yet been developed to improve diagnostic decisions in BMF, likely due to the rarity of these disorders.
We applied machine learning to data from a large historical cohort of patients with BMF in order to develop a model to predict etiology and therefore to guide therapy. The approach was most useful for the differential diagnosis of aplastic anemia (AA) in adults and was based on patients’ clinical and laboratory findings at the initial encounter, in order to facilitate decision making before genetic testing and the initiation of treatment.
Methods
Study cohorts
Clinical records from 2 independent cohorts of consecutive patients with any signs of BMF who were screened for pathogenic variants in IBMFS-associated genes were included in this study (supplemental Table 1): the National Institutes of Health (NIH; 441 patients) and the University of São Paulo (USP; 165 patients) cohorts. For a prediction model interpretation, patients who did not meet the criteria for classical IBMFSs were classified as having moderate AA (MAA), severe AA (SAA), isolated cytopenias (such as thrombocytopenia and neutropenia), myelodysplastic syndromes (MDSs), or hypoplastic MDS based on established criteria (supplemental Table 2). In contrast, classical IBMFS included dyskeratosis congenita (DC) or Hoyeraal-Hreidarsson syndrome, FA, Diamond Blackfan syndrome (DBA), congenital neutropenia, congenital amegakaryocytic thrombocytopenia, and Shwachman Diamond syndrome (SDS).
Written informed consent was obtained from all participants in accordance with the Declaration of Helsinki and under protocols approved by the institutional review boards of National Heart, Lung, and Blood Institute (#NCT00001620, #NCT01623167, #NCT01328587, #NCT01441037, and #NCT00961064), National Cancer Institute (#NCT00027274), and USP (CAAE number, 93617018.0.0000.5440) at initial assessment.
Target classification and data preparation
Germ line variants identified by either whole-exome sequencing, targeted panels, or Sanger sequencing were systematically curated and classified as pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign, or benign based on the Sherloc/American College of Medical Genetics and Genomics (ACMG) criteria (Figure 1).19,20 In a binary target classification, cases were labeled as “inherited” if they had a pathogenic/likely pathogenic disease-associated variant(s), and as “acquired” when they had benign or likely benign variants or a negative genetic test, regardless of the patient’s clinical diagnoses. Next, 30 clinical and laboratory variables consistently available at diagnosis across different institutions were selected for modeling, including a mix of categorical and continuous variables (Table 1; supplemental Table 3). Telomere length (TL) measurement in either peripheral blood lymphocytes or total leukocytes was performed by flow–fluorescent in situ hybridization in the majority of cases by Repeat Diagnostics (Vancouver, Canada) or as previously described.21 Three variables were manually removed because of a high rate of missing values in our cohorts (paroxysmal nocturnal hemoglobinuria [PNH] clone and karyotype; Table 1) and correlation with other variables (white blood cell counts). Records with high ratios of missing variables (NIH, n = 42 and USP, n = 27) or with VUS (NIH, n = 42 and USP, n = 11) were excluded (Figure 1). A final data set included the NIH data, used for training and testing (n = 359), and the USP data set (n = 127), used for independent validation and quantification of the model’s effective generalization.
Figure 1.

Schematic workflow of development of the 2-step machine-learning model. The model was developed with (1) collection of clinical and laboratory data routinely available for patients with BMF from 2 independent cohorts; (2) curation of germ line variants identified by genetic testing in order to assign a label (target classification) for each patient correspondent to BMF etiology: acquired or inherited. All patients identified with pathogenic and likely pathogenic variants were labeled as inherited cases. Patients without germ line variants or with only benign/likely benign variants were labeled as acquired cases. Patients with VUS were not included in the training data set; (3) data preparation; (4) K-means clustering of cases from the training cohort; (5) classification machine-learning algorithm optimized for the cluster with the highest number of cases (cluster A); and (6) validation of the model in an external data set. The predictive model was next applied to predict BMF etiology in patients with VUS.
Table 1.
Clinical and laboratory characteristics of the training and validation data sets
| Training data set (NIH) |
Validation data set (USP) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| All | Labels |
Clustering |
All | Labels |
Clustering |
|||||
| Acquired | Inherited | Cluster A | Cluster B | Acquired | Inherited | Cluster A | Cluster B | |||
| No. of patients (%) | 359 (100) | 232 (65) | 127 (35) | 300 (84) | 59 (16) | 127 (100) | 92 (72) | 35 (28) | 110 (87) | 17 (13) |
| Labels (%) | ||||||||||
| Inherited | 127 (35.3) | 90 (30) | 37 (63) | 35 (27.5) | 29 (26) | 6 (25) | ||||
| Acquired | 232 (64.6) | 210 (70) | 22 (37) | 92 (72.4) | 81 (74) | 11 (65) | ||||
| Sex (%) | ||||||||||
| Female | 174 (48) | 119 (51) | 55 (43) | 140 (47) | 34 (58) | 58 (46) | 49 (53) | 9 (26) | 47 (43) | 11 (65) |
| Male | 185 (52) | 113 (49) | 72 (57) | 160 (53) | 25 (42) | 69 (54) | 43 (47) | 26 (74) | 63 (57) | 6 (35) |
| Median age (range), y | 28 (1-86) | 34 (3-86) | 17 (1-61) | 29 (1-86) | 24 (3-66) | 23 (1-83) | 27 (1-82) | 15 (1-52) | 24 (1-82) | 10 (1-49) |
| Laboratory counts (mean ± SD) | ||||||||||
| Red blood cell counts (103/dL) | 3.06 ± 0.78 | 2.90 ± 0.75 | 3.36 ± 0.78 | 2.9 ± 0.7 | 4.93 ± 0.59 | 2.83 ± 0.97 | 2.7 ± 0.98 | 3.1 ± 0.9 | 2.7 ± 0.8 | 3.9 ± 1.2 |
| Hemoglobin (g/dL) | 9.92 ± 2.29 | 9.2 ± 2.05 | 11.2 ± 2.2 | 9.4 ± 2.1 | 12.3 ± 1.5 | 8.96 ± 2.70 | 8.6 ± 2.7 | 9.9 ± 2.6 | 8.7 ± 2.5 | 10.8 ± 3.0 |
| Mean corpuscular volume (mean ± SD) | 94 ± 11 | 93 ± 11 | 98 ± 11 | 95 ± 11 | 92 ± 10 | 96 ± 12 | 96 ± 12 | 98 ± 12 | 98 ± 11 | 87 ± 12 |
| Platelets (103/dL) | 63 ± 76 | 47 ± 74 | 92 ± 72 | 35 ± 30 | 206 ± 78 | 58 ± 84 | 53 ± 81 | 71 ± 91 | 29 ± 26 | 249 ± 79 |
| Neutrophils (103/dL) | 1.1 ± 1.1 | 0.9 ± 1.1 | 1.5 ± 1.1 | 1.0 ± 1 | 1.63 ± 1.3 | 1 ± 0.86 | 0.9 ± 0.8 | 1.2 ± 1.1 | 1.0 ± 0.9 | 0.9 ± 0.7 |
| Red cell distribution width | 15 ± 3 | 15.7 ± 3.2 | 15 ± 2.9 | 16 ± 3 | 13 ± 1.5 | 16 ± 3 | 16 ± 3.1 | 16 ± 2.9 | 16 ± 3 | 15 ± 2.4 |
| Lymphocytes (103/dL) | 1.48 ± 0.83 | 1.5 ± 0.8 | 1.5 ± 0.9 | 1.46 ± 0.8 | 1.6 ± 0.8 | 1.6 ± 1.1 | 1.6 ± 1.2 | 1.6 ± 0.7 | 1.46 ± 0.9 | 2.6 ± 1.9 |
| Monocytes (103/dL) | 0.21 ± 0.2 | 0.16 ± 0.16 | 0.32 ± 0.22 | 0.19 ± 0.19 | 0.35 ± 0.2 | 0.2 ± 0.16 | 0.2 ± 0.17 | 0.2 ± 0.14 | 0.2 ± 0.15 | 0.3 ± 0.17 |
| Eosinophils (103/dL) | 0.05 ± 0.1 | 0.03 ± 0.06 | 0.07 ± 0.1 | 0.03 ± 0.08 | 0.12 ± 0.1 | 0.05 ± 0.12 | 0.05 ± 0.13 | 0.05 ± 0.08 | 0.04 ± 0.12 | 0.1 ± 0.13 |
| Basophils (103/dL) | 0.01 ± 0.03 | 0.008 ± 0.02 | 0.02 ± 0.04 | 0.01 ± 0.02 | 0.027 ± 0.05 | 0.01 ± 0.03 | 0.01 ± 0.03 | 0.006 ± 0.02 | 0.01 ± 0.03 | 0.02 ± 0.04 |
| Reticulocytes (103/dL) | 44.1 ± 28.2 | 38 ± 27 | 55 ± 27 | 43 ± 29 | 48 ± 24 | 55.1 ± 38 | 52 ± 36 | 63 ± 42 | 53 ± 38 | 71 ± 33 |
| Presence of PNH clones, n (%)∗ | 63 (17) | 62 (27) | 1 (0.7) | 61 (20) | 2 (3) | 15 (12) | 14 (15) | 1 (3) | 15 (14) | 0 |
| (n, % missing values) | 38 (11) | 11 (5) | 27 (21) | 28 (10) | 10 (17) | 11 (9) | 6 (6) | 2 (6) | 5 (4) | 3 (18) |
| Abnormal karyotype, n (%)∗ | 41 (11) | 21 (9) | 20 (16) | 30 (10) | 11 (19) | 9 (7) | 8 (9) | 1 (3) | 7 (6) | 2 (12) |
| Complex or monosomy 7, n (%) | 13 (4) | 7 (3) | 6 (5) | 11 (4) | 2 (3) | 3 (2) | 2 (2) | 1 (3) | 3 (3) | 0 |
| (n, % missing values) | 18 (5) | 4 (2) | 14 (11) | 16 (5) | 2 (3) | 77 (60) | 46 (50) | 23 (65) | 61 (55) | 8 (47) |
| Telomere length, n (%) | ||||||||||
| Normal | 192 (53) | 164 (71) | 28 (22) | 159 (53) | 33 (56) | 79 (62) | 68 (74) | 11 (31) | 67 (61) | 12 (71) |
| <10th percentile | 56 (16) | 39 (17) | 17 (13) | 48 (16) | 8 (14) | 13 (10) | 11 (12) | 2 (6) | 13 (12) | 0 |
| <First percentile | 111 (31) | 29 (13) | 82 (65) | 93 (31) | 18 (31) | 35 (28) | 13 (14) | 22 (63) | 30 (27) | 5 (29) |
| Bone marrow cellularity for age, n (%) | ||||||||||
| Hypocellular | 331 (92.2) | 218 (94) | 113 (90) | 284 (94.7) | 47 (80) | 111 (87) | 79 (86) | 32 (91) | 103 (94) | 8 (47) |
| Normocellular | 24 (6.7) | 10 (4) | 14 (11) | 14 (4.7) | 10 (17) | 16 (13) | 13 (14) | 3 (9) | 7 (6) | 9 (53) |
| Hypercellular | 4 (1.1) | 4 (2) | 0 | 2 (0.7) | 2 (3) | 0 | 0 | 0 | 0 | 0 |
| Dysplasia or increased blasts in bone marrow biopsy, n (%) | 19 (5) | 9 (4) | 10 (8) | 16 (5) | 3 (5) | 16 (13) | 9 (10) | 7 (20) | 13 (12) | 3 (18) |
| Clinical data, n (%) | ||||||||||
| Presence of DC clinical triad | 32 (9) | 4 (2) | 28 (22) | 28 (9) | 4 (7) | 17 (13) | 4 (4) | 13 (37) | 15 (14) | 2 (12) |
| Presence of abnormal cutaneous findings | 44 (12) | 7 (3) | 37 (29) | 27 (9) | 17 (29) | 5 (4) | 2 (2) | 3 (9) | 4 (4) | 1 (6) |
| Presence of physical anomalies | 72 (20) | 12 (5) | 60 (47) | 41 (14) | 31 (53) | 4 (3) | 1 (1) | 3 (9) | 2 (2) | 2 (12) |
| Presence of multiorgan diseases | 87 (24) | 29 (13) | 58 (46) | 66 (22) | 21 (36) | 15 (12) | 3 (3) | 12 (34) | 12 (11) | 3 (18) |
| Long-standing cytopenias or macrocytosis | 66 (30) | 11 (5) | 55 (43) | 44 (15) | 22 (37) | 5 (4) | 1 (1) | 4 (11) | 4 (4) | 1 (6) |
| Long-standing history of recurrent bleeding and infections | 109 (6) | 47 (20) | 62 (49) | 82 (27) | 27 (46) | 6 (5) | 5 (5) | 1 (3) | 5 (5) | 1 (6) |
| Immunodeficiency | 20 (6) | 7 (3) | 13 (10) | 9 (3) | 11 (19) | 1 (1) | 0 | 1 (3) | 1 (1) | 0 |
| Proband with early gray hair | 20 (6) | 9 (4) | 11 (9) | 13 (4) | 7 (12) | 2 (2) | 1 (1) | 1 (3) | 2 (2) | 0 |
| Immediate family members with similar phenotype | 23 (24) | 9 (4) | 14 (11) | 21 (7) | 2 (3) | 0 | 0 | 0 | 0 | 0 |
| Extended family members with similar phenotype | 60 (17) | 31 (13) | 29 (23) | 50 (17) | 10 (17) | 3 (2) | 3 (3) | 0 | 3 (3) | 0 |
| Relatives with early gray hair | 32 (9) | 19 (8) | 13 (10) | 28 (9) | 4 (7) | 0 | 0 | 0 | 0 | 0 |
DC triad is defined by at least 2 of the following: nail dystrophy, skin hyper/hypopigmentation, and leukoplasia.
SD, standard deviation.
Variables excluded from analysis because of a high number of missing values in cases labeled as inherited or in the validation cohort.
K-means clustering
An initial attempt to create a classification model with consecutive BMF cases achieved low accuracy for prediction of presentations that were underrepresented in our cohorts (DBA, FA, SDS, and MDS). The portioning of data into groups that share similarities with an unbiased removal of outliers (which can introduce a noise into the model) aids accurate prediction. K-means clustering was then applied in an attempt to overcome the negative effects of data heterogeneity when only classification modeling was applied. First, K-means clustering calculated and evaluated the ideal number of clusters to partition the NIH data based on similarities of the variables inputted using the Calinski-Harabasz criterion.22 We then applied the same algorithm to cluster the USP data separately. Data were optimally and unbiasedly clustered by the algorithm into 2 major groups, denoted clusters A and B. Only records of cluster A were selected for further processing and classification because data modeling requires large data sets.
Machine-learning classification model selection and optimization
Feature selection was carried out to rank variables by importance via the ReliefF algorithm23 within cluster A from the NIH data set. A final bootstrap aggregation ensemble algorithm model with 25 of the 27 features included in the training data set was selected based on its performance in the validation dataset.
Statistics
Pearson correlation coefficients were computed to evaluate linear correlations between continuous variables as well as the correlations between variables and the outcome of acquired or inherited BMF disease. To evaluate the effects of the predictors and their interactions, a logistic regression model for a binary outcome was established on cluster A using the training data set and tested on the validation datasets.24 Important covariates in this model were chosen by backward stepwise variable selection procedures based on the criteria of classification accuracy. Detailed methods are described in the supplemental Materials.
Results
K-means clustering process structured the data set according to patients’ blood counts and clinical diagnosis
In the NIH data set, median patient age was 28 years (range, 1-86 years), and 52% were male. There were 127 cases labeled as inherited because of the presence of germ line pathogenic variants in IBMFS genes: most commonly in TERT, FANCA, TERC, RTEL1, SBDS, DKC1, TINF2, and MPL (Table 1; Figure 2A). As expected, all pathogenic variants were found in genes known to be linked to IBMFS and correlated with patients’ clinical diagnosis and age of presentation (Figure 2A). The remaining 232 patients were classified as acquired BMF because they had no pathogenic variants. Forty patients with VUS were removed from analysis as they lacked a label (acquired vs inherited) required for data modeling.
Figure 2.

Genetic and clinical characterization of cases from the NIH data set. (A) Germ line variants identified in the NIH data set (n = 399) according to patients’ ages and clinical diagnosis. Variants identified at maximum population frequency of 1% in the general population (gnomAD database) were curated and classified as pathogenic/likely pathogenic (light blue), and as benign, likely benign, or of uncertain significance (VUS; purple). Patients with pathogenic variants in IBMFS genes were labeled as inherited (n = 127). Mutations in genes linked to DBA (n = 9), FA (n = 25), SDS (n = 11), and DC/Hoyeraal-Hreidarsson syndrome (n = 28) were mostly pediatric whereas patients with AA, isolated cytopenias, or MDS/HypoMDS, due to pathogenic variants in telomere biology genes (n = 46) or other genes (RUNX1, n = 1; DDX41, n = 1; and biallelic MPL, n = 1), were in a broader age spectrum. Patients with no variants or with variants classified as benign or likely benign were labeled as acquired (n = 232). In contrast, patients with variants classified as VUS were removed from analysis (n = 40). A final training cohort (n = 359) with 127 labeled as inherited and 232 cases labeled as acquired were used for data modeling. (B) Violin plots of continuous variables in the training cohort (n = 359) according to clusters. Cluster A was enriched for patients who had lower median blood counts, whereas cluster B was enriched for patients with physical anomalies, multiorgan involvement, and long histories of cytopenias or macrocytosis (supplemental Figures 2 and 3). Median ages and blood counts, from both clusters A and B, are shown in the graphic. In general, median blood counts of patients were lower in cluster A than in cluster B and RDW was higher in cluster A than in B, possibly because of enrichment of SAA, which is often transfusion dependent. Within each cluster, inherited cases had lower median ages but higher blood counts. (C) Clinical diagnosis of patients labeled as acquired and inherited in both the training and validation cohorts. Each dot represents a single patient that is colored according to the assigned cluster.
Next, all cases were unbiasedly clustered by an algorithm into 2 main groups based on clinical and laboratory data. In the training cohort, 300 records were assigned to cluster A (90 and 210 cases labeled as inherited and acquired, respectively) and 59 records to cluster B (of which 37 and 22 cases were labeled as inherited and acquired, respectively) (Table 1). Within each cluster, patients labeled as acquired were older, whereas blood counts were higher in cases labeled as inherited (Figure 2B). Most patients in cluster A had hypocellular bone marrow morphology with bilineage or pan-cytopenias (MAA or SAA with or without typical findings of IBMFS), whereas patients in cluster B had single or bilineage cytopenias, with or without bone marrow hypocellularity, as seen in classical IBMFSs such as DBA, SDS, FA, and congenital neutropenia (Figure 2C). These 2 different clusters associated with patients’ blood counts and clinical diagnosis, resulting in an unbiased grouping of patients with BMF likely to share the same pathophysiologic mechanism (Table 1; Figure 2B; supplemental Figure 2).
Clinical and laboratory variables for BMF prediction by machine learning
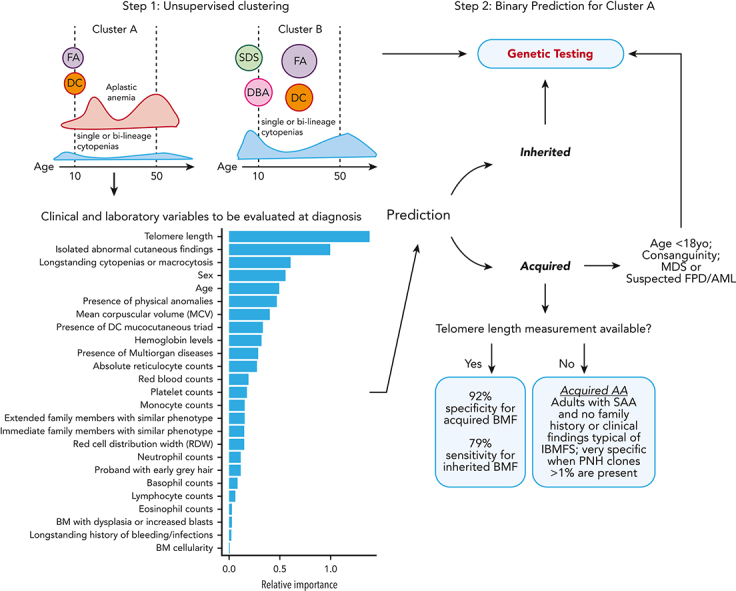
Next, we investigated which variables were most important for the prediction of inherited vs acquired BMF in cluster A using the ReliefF method; 25 of the initial 27 variables from the data set were important (Figure 3A). TL was a top predictor for differential diagnosis of BMF etiology, followed by age, sex, blood counts, and clinical variables, particularly a history of longstanding cytopenias or macrocytosis and mucocutaneous findings (supplemental Table 3). By Pearson correlation analysis, young age and moderate blood counts (excluding lymphocytes) were positively associated with inherited cases (Figure 3B). In addition, continuous variables, mainly represented by patients’ blood counts, were positively intercorrelated, likely because of global hematopoietic failure in patients with AA from cluster A (Figure 3C).
Figure 3.

Classification model for prediction of BMF etiology in cluster A. (A) Top predictors ranked by importance by the ReliefF method. Feature selection ranked 27 variables by importance and the top 25 variables were considered important predictors for the model. (B) Correlation coefficient (R) between a target of prediction (categorical) and continuous variables. R was calculated and plotted in order of a variable’s importance. (C) A heatmap showing correlation among continuous variables. (D) Confusion matrix with prediction results for the validation cohort. The model was validated in the USP data set. Cases labeled or predicted as acquired are represented by “A,” whereas cases labeled or predicted as inherited are represented by “I.” Model sensitivity represents the ability to correctly predict acquired cases, whereas model specificity is the ability of the model to correctly predict inherited cases. (E) Cases from the cluster A of the USP data set that were misclassified by the model. Cases labeled as acquired or inherited that were correctly predicted by the model are represented with purple circles. Cases labeled as acquired that were predicted as inherited, or labeled as inherited and predicted as acquired are indicated with pink triangles. (F) Prediction results of VUS cases. Results are shown according to clinical diagnosis and mutated genes observed in VUS cases. Germ line VUS were mostly found in TERT (n = 10), SAMD9 or SAMD9L (n = 10), RTEL1 (n = 8), SBF2 (n = 6), and GATA2 (n = 3). Cases predicted as inherited or acquired by the model are represented by red and blue circles, respectively. Of note, SAMD9/L variants are often VUSs because in silico tools do not predict the pathogenicity of gain-of-function variants and many cases are de novo without previous family history. ALC, absolute lymphocyte count; ANC, absolute neutrophil count; BM, bone marrow; Hb, hemoglobin level (g/dL); MCV, mean corpuscular volume.
All 25 variables were used to optimize an ensemble an algorithm able to correctly predict acquired vs inherited disease in 89% of cases from the validation cohort, with sensitivity of 79% (correct prediction of inherited cases) and specificity of 92% (correct prediction of acquired cases) (Figure 3D). Clusters A and B in the validation cohort showed similar patterns observed in the training data set; cluster A (n = 110) in the USP cohort was enriched with AA cases whereas cluster B (n = 17) mostly had other IBMFS presentations (Table 1; Figure 2C; supplemental Figure 3). Eleven cases with VUS variants were removed from analysis.
Our model better predicted acquired than inherited AA. In the validation data set, only 12 cases were predicted differently from their labels as determined by genetic sequencing, including 20% (6 of 29) inherited and 7% (6 of 81) acquired cases mispredicted as acquired and inherited, respectively (Figure 3E; Table 2). Among patients mispredicted by the machine, 5 of the 6 labeled as acquired because of a negative genetic test had either DC (n = 3) or MAA with a family history of hematologic malignancies (Table 2; Figure 3E). Therefore, only 1 acquired case (1.2%; USP051) was truly misclassified.
Table 2.
Misclassified cases of cluster A from the validation cohort (n = 12 of 127; 9%): 7% (6 of 81) of acquired and 20% (6 of 29) of inherited cases
| Label | Prediction | Sex | Age | Clinical diagnosis | Pathogenic germ line variant (zygosity) | TL (flow-FISH) | Patient clinical features | |
|---|---|---|---|---|---|---|---|---|
| USP021 | Acquired | Inherited | M | 9 | DC | None∗ | <First | DC clinical triad. |
| USP035 | Acquired | Inherited | F | 18 | DC | None∗ | <First | DC clinical triad since childhood. Sister and cousin with Hodgkin lymphoma. |
| USP036 | Acquired | Inherited | M | 11 | MAA | None∗ | <10th | Chronic pancytopenia and family history of leukemia. |
| USP045 | Acquired | Inherited | F | 7 | MAA | None∗ | Normal | ALL and BMF with café-au-lait spots after chemotherapy. Despite suspicion of FA, DEB in peripheral blood was negative and no variant in FANC-related genes was identified. Patients' uncle also died from ALL. |
| USP051 | Acquired | Inherited | M | 3 | SAA | None∗ | <First | No family history or classical signs of IBMFS. |
| USP159 | Acquired | Inherited | F | 3 | DC | None | <First | DC clinical triad. |
| USP022 | Inherited | Acquired | M | 20 | MAA | TERT: c.2154C>A; p.D718E (het) | <First | No family history or signs of IBMFS. PNH clone of 6%. |
| USP023 | Inherited | Acquired | F | 8 | MAA | TERT: c.1072C>T; p.R358W (hom) | <10th | Consanguinity but no classical signs of inherited disease. |
| USP026 | Inherited | Acquired | F | 38 | MAA | TERT: c.193C>A; p.P65T (hom) | <10th | Pulmonary fibrosis. |
| USP030 | Inherited | Acquired | F | 18 | MAA | 1TINF2:c.844C>T; p.R282C (het) | <First | History of miscarriage. Son with DC and same pathogenic variant in TINF2. |
| USP065 | Inherited | Acquired | M | 24 | HypoMDS | RUNX1: c.497G>C; p.R166P (het) | Normal | Hypocellular bone marrow. Brother died of ALL. |
| USP152 | Inherited | Acquired | F | 1 | SAA | 2MECOM: c.2518delC; p.E841KfsTer3 (het) | Normal | No family history or classical signs of IBMFS. |
ALL, acute lymphocytic leukemia; F, female; FISH, fluorescence in situ hybridization; het, heterozygous; hom, homozygous; HypoMDS, hypoplastic MDS; M, male; <First, TL below the first percentile of age-matched controls; <10th, TL below the tenth percentile of age-matched controls.
These patients were not screened for variants in MECOM, SAMD9, and SAMD9L because these genes were included in the panel after these samples were sequenced.1 Although the TINF2 R282C variant is commonly associated with DC of early onset, USP030 did not have the clinical triad or any classical sign of IBMFS other than a past history of multiple miscarriages. Telomere disease was later suspected after her son was diagnosed with DC at age of 2 years; he was later found to have the same TINF2 pathogenic variant.2MECOM isoform (NM_004991.4).
Among the inherited cases predicted as acquired, 4 had MAA and short telomeres, including 2 with a homozygous TERT variant (USP023 and USP026) (Table 2) and 1 with a TINF2 variant, all classified as pathogenic by the Sherloc/ACMG criteria. Other mispredicted cases included 2 patients with normal TLs; the first had hypoplastic MDS caused by a germ line RUNX1 variant, and the other had SAA at a very early age caused by a MECOM frameshift variant (Table 2). Overall, inherited cases mispredicted as acquired were associated with young age without classical IBMFS features; MAA alone or with other phenotypes was underrepresented in the training cohort.
Logistic regression model
To understand the directionality of prediction of the ensemble model, we attempted to recapitulate the “black-box” machine-learning results by univariable and multivariable logistic regression. In univariable analysis, short or very short telomeres were most predictive of inherited BMF, followed by higher blood counts (particularly basophils and eosinophils), younger age, and presence of clinical signs suggestive of inherited disease (supplemental Table 4). In multivariable analysis, 12 covariates recapitulated the machine-learning results, with the highest accuracy in cluster A from both the NIH and USP data sets (95% and 88%, respectively): short TL, young age, presence of physical anomalies, abnormal mucocutaneous findings, immunodeficiency, multiorgan disease, higher eosinophils and basophils counts, and higher mean corpuscular volume were predictors of inherited disease after full adjustment. Sensitivity and specificity of the multivariable analysis were 76% and 93%, respectively. In both univariable and multivariable analyses, predictors associated with an increased risk of inherited BMF correlated with the machine-learning findings, providing a linear rationale used by the ensemble algorithm for prediction.
Importance of TL measurement and PNH testing in BMF
TL measurement in lymphocytes or total blood was a top predictor in differentiating acquired from inherited BMF by both ensemble and logistic regression models. As expected, short TL implicated a diagnosis of TBDs, whereas a normal TL indicated immune AA in most cases (Figure 3A); short telomeres are also found in some immune AA cases as well as in other IBMFSs. In this study, the majority of patients had TLs measured by flow–fluorescent in situ hybridization, but in some cases, TLs were measured by a Clinical Laboratory Improvement Amendment–certified quantitative polymerase chain reaction assay or in-house Southern blotting; TL measurement technique appeared to have no effect on the model’s predictive value based on the model’s overall performance and interpretation. Regardless of the methodology, obtaining TL measurement may be as challenging as genetic testing in low-resource centers. Therefore, we attempted to develop an alternative algorithm without TL data, but the model underperformed for prediction of inherited cases, especially TBDs without classical signs of DC. Although the overall accuracy of the model without TL was 80%, the specificity was 82% and the sensitivity was only 55%. Nevertheless, clinical variables along with age, sex, higher hemoglobin levels, mean corpuscular volume, reticulocytes, and platelet counts remained top predictors (supplemental Figure 4).
As the presence of a PNH clone is considered an exclusion criterion for the diagnosis of IBMFSs,25,26 in practice, testing for PNH is usually not requested in patients suspected of having an IBMFS, which explains the high rate of missing values for PNH testing in our cohorts. Nevertheless, the presence of a PNH clone strongly correlated with an immune AA pathophysiology. In a subanalysis among patients tested for PNH, 24% (83 of 339) of patients labeled as acquired in both cohorts had a PNH clone >1% either in neutrophils or red blood cells.
In contrast, only 2 of 151 patients labeled as inherited had PNH clones. The first patient predicted as having an acquired AA by the model had the TERT c.2154C>A p.D718E variant, very short TL, and an uncle with pulmonary fibrosis, but a PNH clone of 6% at the age of 20 years (USP030; Table 2). This TERT variant was absent in the general population, predicted to be deleterious in silico, located at the same codon as another variant that had been reported to be pathogenic by reducing telomerase activity to 44%.27 The second patient, labeled and predicted to have an inherited disease by our model, had a DDX41 (c.1016 G>A, p.R339H) variant and a PNH clone of 3% at the age of 12 years (NIH161). This variant was seen in <8 alleles in gnomAD, predicted deleterious in silico, located at the same codon as another variant reported as pathogenic in Clinvar (c.1016G>T, p.R339L), and segregated with disease in the patient’s family. The proband had mild thrombocytopenia but not MDS until the patient lost follow-up; his mother had the same variant and a history of non-Hodgkin lymphoma and melanoma; and his siblings were not tested but had a history of easy bruising and nose bleeding. Nevertheless, because these 2 pathogenic variants were seen in the same context of a PNH, we cannot state that these variants are truly pathogenic in the absence of confirmatory functional assays.
Model applicability in predicting BMF etiology in patients with VUS
After validating the model, we applied it to predict the etiology of BMF cases found with VUS from both the NIH and USP cohorts (n = 51). All VUS cases were assigned to cluster A, and 8 and 43 were predicted to have inherited and acquired IBMFSs, respectively (Figure 3F). With just 1 exception (case NIH214), VUS cases predicted as having inherited disease had clinical features and family histories consistent with IBMFS; patients with TERT and MECOM variants, but not DKC1 and RTEL1 variants, were considered to likely have an IBMFS based on clinical phenotype and disease inheritance (Table 3). In contrast, most of the VUS cases predicted as acquired had no strong clinical evidence of IBMFSs, but 7 had nonspecific findings, such as early gray hair, macrocytic anemia, or relatives with cytopenias. Most VUS cases predicted as acquired had heterozygous variants in SAMD9, SAMD9L, SBF2, and RTEL1, indicating that the interpretation of genetic reports with variants in these genes should be made cautiously (Figure 3F).
Table 3.
Cases with VUS that were predicted as inherited by the algorithm (n = 8 of 51)
| ID | Cluster | Sex | Age | Clinical diagnosis | Zygosity | Germ line variant | TL (flow-FISH) | Patient clinical features | Decision making |
|---|---|---|---|---|---|---|---|---|---|
| NIH024 | 1 | F | 10 | MAA | X-linked | DKC1; c.915+10G>A | <First | Dystrophic nails and abnormal lung testing. Maternal grandfather with thrombocytopenia, pulmonary fibrosis, and early hair graying. Mother with early hair graying. | Variant's pathogenicity needs to be confirmed with functional assays. Extended screening with WES/WGS is also recommended. |
| NIH214 | 1 | M | 8 | MAA | Het | TERT c.1324G>A; p.Asp442Asn | <First | No family history or signs of inherited disease. | The TERT variant is likely the cause of patient’s disease. |
| NIH258 | 1 | M | 17 | Isolated thrombocytopenia | Het | RTEL1: c.2507C>G; p.Pro836Arg | <First | Thrombocytopenia since age 11, development delay, lymphopenia, short tongue frenulum, syndactyly of the second and third digit right foot, microcephaly, shallow forehead, short stature, micrognathia, and almond shaped eyes. Also with splenomegaly, pulmonary obstructive disease, and atrioventricular septal defect repaired as infant. Cousins with thrombocytopenia and enlarged spleen, improved after splenectomy. | Patient’s phenotype is not consistent with an AD telomere disease. This variant is likely benign. Extended screening with WES/WGS is recommended |
| NIH259 | 1 | M | 69 | MAA | Het | MECOM: c.2720A>G; p.Asn907Ser | <First | Long-standing thrombocytopenia and history of non-Hodgkin lymphoma. Both father and 2 sons with thrombocytopenia. | The MECOM variant is likely the cause of patient’s disease. Screening of affected family members can confirm variant's pathogenicity. |
| NIH326 | 1 | M | 32 | MAA | Het |
TERT: c.383C>T; p.T128I |
<First | With splenomegaly. Cousin with thrombocytopenia and uncle died of cirrhosis at age of 60. | The TERT variant is likely the cause of patient’s disease. |
| NCI 456-1 |
2 | F | 5 | Isolated neutropenia | Het |
TERT: c.3158G>A; p.Gly1053Glu |
<First | Cytopenias at age of 2, multiple warts, and mild nail dystrophy. | The TERT variant is likely the cause of patient’s disease. |
| NIH324 | 2 | M | 48 | Isolated anemia | Het | TERT; c.2786 C>T; p.P929L | <First | Cytopenias since age 18, hepatopulmonary syndrome with s/p liver transplant, and interstitial pneumonitis. Father died of cirrhosis at age of 52 and brother had AA at age of 14. | The TERT variant is likely the cause of patient’s disease. |
| USP063 | 2 | M | 1 | Isolated neutropenia | Biallelic |
USB1 c.477A>C; p.Q159H/ c.344G>A; p.R115K |
Normal | Microcytic anemia, and no megakaryocytes and erythroid precursors in bone marrow biopsy. No skin alterations and normal IgG and IgM, but low IgA. Strong family history of immunodeficiency; 3 brothers died months after birth due to severe immunodeficiency. | Patient's family history is not consistent with an AR disease, though disease possible. Extended screening with WES/WGS is recommended |
AD, autosomal dominant; AR, autosomal recessive; Ig, immunoglobulin; WES, whole-exome sequencing; WGS, whole-genome sequencing.
Discussion
In this study, we developed a 2-step data-driven clustering and classification model that reproduced the expert clinicians’ decision-making process for investigating the AA etiology in adults, the most representative phenotype of cluster A. Our model accurately predicted >92% of cases labeled as likely having acquired AA (specificity), with performance approaching >98% when accounting for 5 patients labeled as acquired because of negative genetic testing who were mispredicted by the model; clinically, they were suspected of IBMFSs based on their phenotype. Therefore, specificity of the model should be interpreted with caution as it also reflects a percentage of cases that would benefit from further screening by whole-exome or genome sequencing for unveiling potential uncharacterized mutations (Tables 2 and 3).
In centers specialized in BMF, genetic testing has increasingly been incorporated into standard clinical evaluation. With broader access to next-generation sequencing, the list of genetic loci likely to be involved in IBMFSs and useful in the identification of pathogenic germ line variation is approaching 100 genes. However, genetic testing laboratories lack detailed data from patients with BMF to facilitate variant curation, and functional assays of rare variants are usually not available. In the United States, a high-resource country, the turnaround time for a genomic panel is 6 to 8 weeks, and in developing countries, can approach 12 weeks or longer. By training a predictive model based on routine clinical and laboratory variables, we have created a practical tool that can guide the decision to use expensive genomic assays and expedite initial treatment for hematologists nonspecialized in BMF or with limited resources.
The current lack of prediction algorithms for BMF can be explained by the challenge of data collection; BMF cases are rare and scattered across the world in specialized centers. Our study is limited by the relatively small sample size (although large for BMF) because machine-learning algorithms require a large number of exemplars in a training cohort to accurately identify patterns associated with dichotomous end points.11,13,28 Notably, a machine-learning model trained with >8000 cases underperformed for diseases seen in <10 cases (prevalence below 1/800).14 In our study, we overcame these limitations through a collaborative effort that gathered comprehensive clinical data of >500 patients from different institutions. In addition, we validated the generalizability of the model in a completely independent cohort from a resource-limited country, suggesting that our model will be useful in low-resource centers to prioritize patients who would benefit from genetic testing or who undergo immunosuppressive therapy without awaiting genetic test results, recapitulating the clinical practice of hematologists specialized in BMF. This is particularly important in patients with very low neutrophil counts in whom outcomes are dependent on rapid treatment.
Our model is useful for distinguishing the AA etiology when the chromosome breakage testing is negative and classical IBMFS is not suspected. In cluster A, most patients had AA; TBDs and congenital amegakaryocytic thrombocytopenias were the most challenging inherited diseases to identify without genetic testing or TL measurement. The model performed better for detecting patients with acquired rather than inherited AA. Because no specific diagnostic test is currently available for immune AA, our predictive model should be a valuable clinical tool for these often extremely ill patients. An explanation for underperformance in the identification of IBMFSs is that this group is highly heterogeneous, and many cases did not have clear genotype-phenotype associations, characterized only by genetic testing; in our study, 20% of IBMFS cases confirmed by genetic testing were mispredicted.1,29 Instead, by using variables highly predictive of IBMFSs and blood count thresholds, the algorithm identified a more homogeneous group of patients likely to have immune AA.
Unsurprisingly, TL was a key variable for model accuracy; TBDs were enriched in cluster A because they often present as AA. In practice, our data show that lack of both genetic testing and TL measurement lead to misdiagnosis in ∼45% of patients with IBMFSs. Our model highlights that TL testing can be preferentially incorporated into clinical practice, compared with genomic testing, due to its much lower cost and its importance for prediction; accuracy of IBMFS diagnosis increased from 55% to 79% when TL was available. For centers in which TL measurement is not available, the model still guides physicians toward clinical diagnosis, particularly by identifying patients likely to have immune AA. We found that adult patients with SAA (>18 years old with severe pancytopenia) rarely had an inherited disease without a positive family history and phenotype suggestive of an IBMFS, or consanguinity being present; presence of a PNH clone >1% was a specific marker of immune disease in these patients. Of importance, the specificity of the model for immune AA is 90% vs 92%, absent or with TL, respectively. Therefore, even in the absence of TL, we can identify patients likely to benefit from immunosuppression (Figure 4).
Figure 4.

Two-step clustering and classification model for decision making in BMF. In the first step of the model, K-means clustering grouped cases into clusters A and B, which correlated with clinical diagnosis. Cluster A was enriched for cases of FA and DC, patients who had AA at young ages, and cases with AA and single or bilineage cytopenias over a broad spectrum of age but most frequently 20 and 50 years old. In contrast, cluster B was enriched for classical inherited BMF, including early disease onset DBA and SDS, and cases of FA and DC in middle age. In the second step, a classification model specific to cluster A was developed for binary prediction of cases as acquired and inherited. The cluster A–specific algorithm accurately predicted the BMF etiology in 79% of cases with IBMFS (model sensitivity) and 92% of cases with likely immune BMF (specificity) when TL data were available. The model lost accuracy without TL, a top predictive factor. However, in the absence of TL data, IBMFSs were rarely seen in adults with SAA and no family history or a phenotype suggestive of inherited disease; presence of PNH clone >1% within this group had a specificity of 100% for acquired AA. yo, years old.
To increase access to our model, we have developed a free online R shiny app for clustering and prediction of the etiology of BMF cases, incorporating TL (https://dir.nhlbi.nih.gov/DDxAA). Genetic testing should be considered for patients in cluster A who are predicted to have inherited disease and also for patients in cluster B, for whom no specific model was available, as they were more likely to have an IBMFS in comparison with patients in cluster A (50% vs 30%) (Table 1; Figure 4). Ideally, a new model should be trained and validated with an increased number of cases to accurately predict the BMF etiology of cases from cluster B, enriched for SDS, DBA, and FA. However, most of these cases will be diagnosed on routine chromosome breakage testing or based on classical phenotypes. We also recommend genetic screening in addition to chromosome breakage testing for children predicted to have acquired disease (and often without clinical signs of IBMFS), for patients with consanguinity in the family, or for patients with suspicion of MDS and familial predisposition to myeloid malignancies (all cases in which the model had limited predictive power) (Figure 4).
Our model does not predict the pathogenicity of variants found by genetic testing. Variants need to be carefully curated according to evidence-based data on clinical phenotypes, inheritance patterns, and functional and population data.19 As ongoing work, the model’s prediction could be validated as additional evidence for variant curation using traditional criteria, such as the Sherloc/ACMG criteria (rule PP5).
This machine-learning model is primarily intended for general hematologists and hematologists in training, not experts in BMF, having been created in a multi-institutional collaboration that defined the most important clinical and laboratory data needed for differential diagnosis of BMF at a patient’s first screening. The sample size is very large in the context of low disease prevalence, and the model incorporates clinical data from different health systems, and geographic and cultural backgrounds. The study validation cohort was from the USP BMF clinic, a reference center in a resource-limited country, whereas the training set was from the NIH, a quaternary care center with the ability to undertake extensive evaluations. This approach increases our confidence in the model’s generalizability, especially its utility in low-resource settings.
Our work, to the best of our knowledge, is the first evidence-based, data-driven artificial intelligence approach to the diagnosis of a group of rare, complex, and highly morbid diseases. It incorporates both deep clinical and detailed genomics across a spectrum of rare syndromes, an innovative systems approach. The model can provide a clinical practice guide for management of adult patients with AA, aiming for prospective standardization of data collection and clinical assessment of patients for future studies. All of the selected 25 clinical and laboratory variables were critical for accurate prediction, regardless of the degree of importance (Figure 3A); a comprehensive history, physical examination, and laboratory evaluation encompassing all organ systems are critical, and TL testing is encouraged. Adult patients with inherited AA often had moderate pancytopenia at a younger age in comparison with patients with immune AA, most of whom are severely neutropenic. This practical tool is also part of ongoing research because we will continue accruing to the model in an effort to increase the number of cases to further refine prediction of IBMFS cases that were underrepresented in the current cohort, especially pediatric cases.
Conflict-of-interest disclosure: N.S.Y. received research funding from Novartis by way of a Cooperative Research and Development Agreement. R.M.S. received royalties from cad, Ping An, Philips, Scan Med, and Translation Holdings and his laboratory received research support from Ping An and NVIDIA. Y.T. is currently employed by Ping An. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the study participants and their families for their valuable contributions. Some figures were created with BioRender.com.
This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, the Division of Cancer Epidemiology and Genetics of the National Cancer Institute, the National Human Genome Research Institute, and the Clinical Center, National Institutes of Health. This study was also supported by funding from the Mildred-Scheel-Postdoctoral Fellowship Program by the German Cancer Aid (M.R.N.); in part by grants from the São Paulo Research Foundation (FAPESP) (grants 13/08135-2 and 16/12799-1) (R.T.C.); and scholarships from FAPESP (scholarship numbers 17/09428-4, 16/03620-8, and 14/26379-9) (F.S.D., A.L.P., and G.B., respectively).
Authorship
Contribution: F.G.-R., E.M., X.M., C.O.W., R.T.C., and N.S.Y. made substantial contributions to the conception or design of the work, literature search, figures, data interpretation, and writing; E.M., X.M., and C.O.W. developed the machine learning and logistic regression prediction models; R.M.S. and Y.T. assisted with the conceptual design of the machine-learning model; F.G.-R., E.M.G., B.A.P., Y.T., L.F.B.C., D.V.C., M.R.N., R.M.A.-P., F.S.D., A.L.P., G.B., B.A.S., B.A., L.J.M., N.G., X.F., R.S., C.D., D.Q.R., K.C., R.S., P.L., L.C., D.D.H., C.E.D., R.P., M.M.D.O., E.D.R.P.V., B.P.A., S.A.S., C.B., and R.T.C. contributed to patient care, and acquisition, analysis, or interpretation of data; F.G.-R., E.M., E.M.G., B.A.P., S.K., C.E.D., S.A.S., C.B., C.O.W., R.T.C., and N.S.Y. drafted the work or revised it critically for important intellectual content; and F.G.-R., E.M., C.O.W., R.T.C., and N.S.Y. contributed to the final approval of the version to be published.
Footnotes
∗F.G.-R. and E.M. contributed equally to this study.
All authors were not precluded from accessing data in the study, and they accept responsibility to submit for publication. Data used in this study were retrospectively collected, and requests for deidentified participant data should be made to the corresponding authors, Fernanda Gutierrez-Rodrigues (fernanda.rodrigues@nih.gov) and Rodrigo T. Calado (rtcalado@fmrp.usp.br). The Matlab emsemble code is also available upon request.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
Fernanda Gutierrez-Rodrigues, Email: fernanda.rodrigues@nih.gov.
Rodrigo T. Calado, Email: rtcalado@fmrp.usp.br.
Supplementary Material
References
- 1.Bluteau O, Sebert M, Leblanc T, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. 2018;131(7):717–732. doi: 10.1182/blood-2017-09-806489. [DOI] [PubMed] [Google Scholar]
- 2.Young NS. Aplastic anemia. N Engl J Med. 2018;379(17):1643–1656. doi: 10.1056/NEJMra1413485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wegman-Ostrosky T, Savage SA. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol. 2017;177(4):526–542. doi: 10.1111/bjh.14535. [DOI] [PubMed] [Google Scholar]
- 4.Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124(18):2775–2783. doi: 10.1182/blood-2014-05-526285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361(24):2353–2365. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376(16):1540–1550. doi: 10.1056/NEJMoa1613878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghemlas I, Li H, Zlateska B, et al. Improving diagnostic precision, care and syndrome definitions using comprehensive next-generation sequencing for the inherited bone marrow failure syndromes. J Med Genet. 2015;52(9):575–584. doi: 10.1136/jmedgenet-2015-103270. [DOI] [PubMed] [Google Scholar]
- 8.Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416–1430. doi: 10.1056/NEJMoa1716614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munger E, Choi H, Dey AK, et al. Application of machine learning to determine top predictors of noncalcified coronary burden in psoriasis: an observational cohort study. J Am Acad Dermatol. 2020;83(6):1647–1653. doi: 10.1016/j.jaad.2019.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajkomar A, Oren E, Chen K, et al. Scalable and accurate deep learning with electronic health records. NPJ Digit Med. 2018;1:18. doi: 10.1038/s41746-018-0029-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Libbrecht MW, Noble WS. Machine learning applications in genetics and genomics. Nat Rev Genet. 2015;16(6):321–332. doi: 10.1038/nrg3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajkomar A, Dean J, Kohane I. Machine learning in medicine. N Engl J Med. 2019;380(14):1347–1358. doi: 10.1056/NEJMra1814259. [DOI] [PubMed] [Google Scholar]
- 13.Shouval R, Fein JA, Savani B, Mohty M, Nagler A. Machine learning and artificial intelligence in haematology. Br J Haematol. 2021;192(2):239–250. doi: 10.1111/bjh.16915. [DOI] [PubMed] [Google Scholar]
- 14.Gunčar G, Kukar M, Notar M, Brvar M, Černelč P. An application of machine learning to haematological diagnosis. Sci Rep. 2018;8(1):411. doi: 10.1038/s41598-017-18564-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radakovich N, Nagy M, Nazha A. Machine learning in haematological malignancies. Lancet Haematol. 2020;7(7):e541–e550. doi: 10.1016/S2352-3026(20)30121-6. [DOI] [PubMed] [Google Scholar]
- 16.Sachdev V, Tian X, Gu Y, et al. A phenotypic risk score for predicting mortality in sickle cell disease. Br J Haematol. 2021;192(5):932–941. doi: 10.1111/bjh.17342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abelson S, Collord G, Ng SWK, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559(7714):400–404. doi: 10.1038/s41586-018-0317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagata Y, Zhao R, Awada H, et al. Machine learning demonstrates that somatic mutations imprint invariant morphologic features in myelodysplastic syndromes. Blood. 2020;136(20):2249–2262. doi: 10.1182/blood.2020005488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19(10):1105–1117. doi: 10.1038/gim.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gutierrez-Rodrigues F, Santana-Lemos BA, Scheucher PS, Alves-Paiva RM, Calado RT. Direct comparison of flow-FISH and qPCR as diagnostic tests for telomere length measurement in humans. PLoS One. 2014;9(11):e113747. doi: 10.1371/journal.pone.0113747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calinski T, Harabasz J. A dendrite method for cluster analysis. Commun Stat. 1974:1–27. [Google Scholar]
- 23.Robnik-Šikonja M, Kononenko I. Theoretical and Empirical Analysis of ReliefF and RReliefF. Machine Learning. 2003;53:23–69. [Google Scholar]
- 24.Hastie T, Tibshirani R, Friedman J . Second ed. Springer; 2009. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. [Google Scholar]
- 25.DeZern AE, Symons HJ, Resar LS, Borowitz MJ, Armanios MY, Brodsky RA. Detection of paroxysmal nocturnal hemoglobinuria clones to exclude inherited bone marrow failure syndromes. Eur J Haematol. 2014;92(6):467–470. doi: 10.1111/ejh.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shah YB, Priore SF, Li Y, et al. The predictive value of PNH clones, 6p CN-LOH, and clonal TCR gene rearrangement for aplastic anemia diagnosis. Blood Adv. 2021;5(16):3216–3226. doi: 10.1182/bloodadvances.2021004201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vulliamy TJ, Kirwan MJ, Beswick R, et al. Differences in disease severity but similar telomere lengths in genetic subgroups of patients with telomerase and shelterin mutations. PLoS One. 2011;6(9):e24383. doi: 10.1371/journal.pone.0024383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Ploeg T, Austin PC, Steyerberg EW. Modern modelling techniques are data hungry: a simulation study for predicting dichotomous endpoints. BMC Med Res Methodol. 2014;14:137. doi: 10.1186/1471-2288-14-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang MY, Keel SB, Walsh T, et al. Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica. 2015;100(1):42–48. doi: 10.3324/haematol.2014.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.