
Fig. 3 |. Mechanisms of action of supraphysiological testosterone.
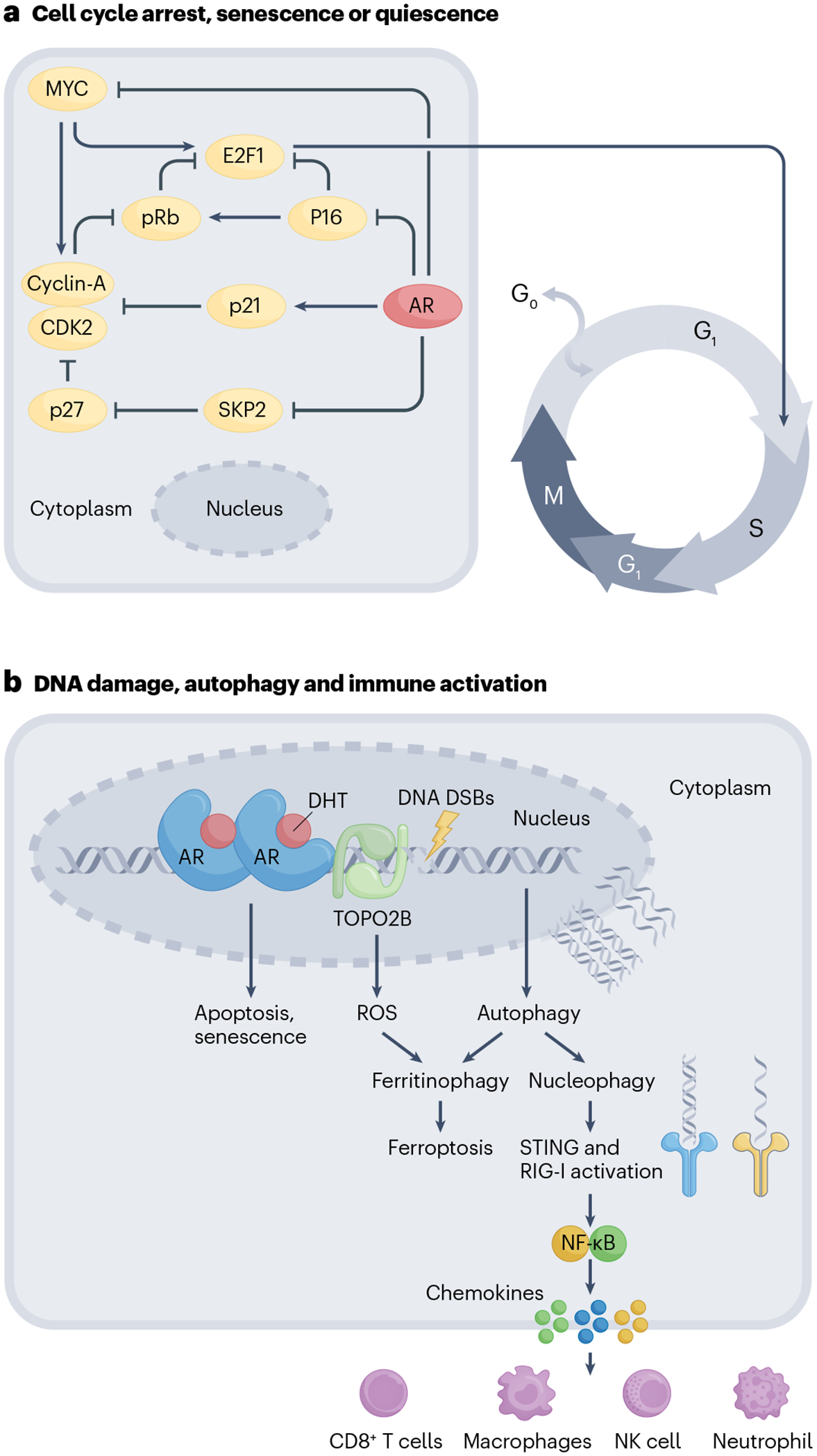
a, Cell-cycle regulation. Supraphysiological testosterone (supraphysiological T) inhibits the transcription of MYC, which is required for cyclin and cyclin-dependent kinase-mediated passage of cells from the G1 to the S phase. Downregulation of MYC suppresses CDK2 and CyclinA activity, which prevents phosphorylation-mediated degradation of RB leading to cell-cycle arrest. Supraphysiological T also increases p21 levels through transcriptional upregulation by the androgen receptor (AR) and inhibits the expression of S-phase kinase-associated protein (SKP2), a subunit of SCF-type cullin ubiquitin ligase. Downregulation of SKP2 by supraphysiological T increases p27, which, in conjunction with p21 and p16 upregulation, causes a G1 phase arrest leading to cell death and quiescence and/or senescence. b, Autophagy and immune activation. Supraphysiological T mediates DNA double-stranded breaks (DSBs) by recruiting TOPO2B to DNA binding sites. Unrepaired DNA lesions cause apoptosis, cell-cycle arrest or senescence. Supraphysiological T also causes an induction of two parallel autophagy-mediated pathways: ferritinophagy and nucleophagy. Ferritinophagy, which involves autophagy-mediated degradation of ferritin, results in increased lipid reactive oxygen species (ROS) and ferroptotic cell death. Supraphysiological T-damaged DNA can be degraded in the autophagosomes by the process of nucleophagy. Cytoplasmic autophagosomal DNA activates a nucleic acid-sensing mechanism through STING and RIG-I. Activated STING and RIG-I signal through NF-κB and cause the release of pro-inflammatory chemokines, including CXCL10, attracting natural killer (NK) cells, T cells, macrophages and neutrophils. DHT, dihydrotestosterone.